

Abstract
Physiologically‐based pharmacokinetic (PBPK) models have an important role in drug discovery/development and decision making in regulatory submissions. This is facilitated by predefined PBPK platforms with user‐friendly graphical interface, such as Simcyp and PK‐Sim. However, evaluations of platform differences and the potential implications for disposition‐related applications are still lacking. The aim of this study was to assess how PBPK model development, input parameters, and model output are affected by the selection of PBPK platform. This is exemplified via the establishment of simvastatin PBPK models (workflow, final models, and output) in PK‐Sim and Simcyp as representatives of established whole‐body PBPK platforms. The major finding was that the choice of PBPK platform influenced the model development strategy and the final model input parameters, however, the predictive performance of the simvastatin models was still comparable between the platforms. The main differences between the structure and implementation of Simcyp and PK‐Sim were found in the absorption and distribution models. Both platforms predicted equally well the observed simvastatin (lactone and acid) pharmacokinetics (20–80 mg), BCRP and OATP1B1 drug–gene interactions (DGIs), and drug–drug interactions (DDIs) when co‐administered with CYP3A4 and OATP1B1 inhibitors/inducers. This study illustrates that in‐depth knowledge of established PBPK platforms is needed to enable an assessment of the consequences of PBPK platform selection. Specifically, this work provides insights on software differences and potential implications when bridging PBPK knowledge between Simcyp and PK‐Sim users. Finally, it provides a simvastatin model implemented in both platforms for risk assessment of metabolism‐ and transporter‐mediated DGIs and DDIs.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Physiologically‐based pharmacokinetic (PBPK) platforms have previously been compared from an oral biopharmaceutics and intestinal absorption perspective, but studies on the potential implications of platform differences for disposition related applications, such as drug–gene interactions and drug–drug interactions, are still lacking.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study aims to assess how PBPK model development, input parameters, and model output might be affected by the selection of the PBPK platform.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study illustrates that in‐depth knowledge of established PBPK platforms is crucial to enable an assessment of what consequences the selection of a specific PBPK platform may bring. In particular, this work provides insights on software differences and potential implications when bridging PBPK knowledge between Simcyp and PK‐Sim platform users.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
This supports an increased emphasis on users knowledge in PBPK methodology as well as possibilities to review model implementations for proper application of PBPK modeling platforms.
INTRODUCTION
The use of physiologically‐based pharmacokinetic (PBPK) modeling has dramatically increased over the last 20 years. 1 Hence, PBPK has become a fundamental tool in drug discovery and development and for regulatory agencies. 2 , 3 The importance of PBPK modeling is emphasized in areas such as first‐in‐human and pediatrics dose predictions, as well as bioequivalence and drug–drug interaction (DDI) risk assessments, and PBPK is used in lieu of clinical studies. 4 , 5 , 6 A crucial factor for the rapid growth of PBPK modeling has been the development of several predefined PBPK platforms with user‐friendly graphical interfaces. These platforms facilitate more convenient and standardized whole‐body PBPK modeling in drug discovery/development and contribute to decision making in regulatory submissions. 7 , 8
The fundamental feature of PBPK models is to describe the drug disposition in a comprehensive structural representation of the organism of interest given its anatomy and physiology. The major organs/tissues are represented by compartments and sets of differential equations linked by representative blood flows. Advanced drug disposition mechanisms, such as transporters, metabolites, absorption, first‐pass, etc., can be added to the model. PBPK modeling thereby integrates species physiology and drug data to describe the drug plasma and tissue concentrations over time. 8 , 9 , 10 , 11 Hence, the parameters in the model are divided into two categories: (1) system‐specific (e.g., tissue volume, blood flow, organ weight, height, genetics, and population variability), which are parameterized with known physiology and anatomy; and (2) drug‐specific parameters (e.g., drug partitioning to tissues, binding to plasma, or affinities toward certain enzymes/transporters) which can be derived by different in vitro, in silico, and in vivo methods. This “bottom‐up” modeling approach is a key strength of PBPK models, but in practice it is common that some drug‐specific parameters need to be estimated based on clinical data (“top‐down” approach). This combined modeling approach is called “middle‐out,” which takes the advantages and strengths of both “bottom‐up” and “top‐down” approaches. 12 Once a model is established, this independent representation of system and drug also facilitates predictions of previously unstudied clinical scenarios (e.g., special populations).
The most commonly used PBPK platforms by industry and regulatory agencies are Simcyp (Certara Simcyp, Sheffield, UK), 13 Gastroplus (Simulations Plus, Lancaster, PA), and PK‐Sim (Open Systems Pharmacology community). 14 The most typical applications in Simcyp and PK‐Sim are study design, biopharmaceutics, enzyme‐mediated DDIs, and special populations, whereas in Gastroplus, biopharmaceutics is the major application. 1 These specialized platforms provide a generic model structure for human (or predefined species) physiology, including large‐scale physiological databases and libraries of model compounds and populations. However, there are differences between these predefined PBPK models/platforms (e.g., system structure, implementation of processes, and parameterization). PBPK models/platforms will consequently not only differ in output but also respond differently to the same input, even though equivalent results can be achieved. PBPK platforms have previously been compared from an oral biopharmaceutics and intestinal absorption perspective, 15 , 16 , 17 but studies on the potential implications of platform differences for disposition related applications, such as DDIs, are still lacking. Thus, there is a need for both structural knowledge of the PBPK methodology as well as detailed insight into the adopted model/platform in order to avoid analysis errors and misinterpretations of the outcome.
Simvastatin is a hypolipidemic agent administered as an inactive prodrug, simvastatin lactone (SVL), which is converted to the active form, simvastatin acid (SVA), via carboxylesterases and paraoxonases mainly expressed in the liver and plasma. 18 , 19 , 20 The re‐conversion of this process (SVA‐to‐SVL) is mainly mediated by paraoxonases and UGT1A1. 19 , 21 Metabolism via CYP3A4 is suggested to be the main elimination pathway for both SVL and SVA, based on in vitro data. 22 , 23 , 24 , 25 , 26 The importance of this pathway is also seen in several clinical DDI studies when simvastatin is co‐administered with CYP3A4 inhibitors/inducers. 27 Additionally, there are DDI and drug–gene interaction (DGI) studies demonstrating SVA influx to hepatocytes via the membrane transporter OATP1B1. 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 Although several DGI studies describe an association between SVL disposition and BCRP transporter polymorphisms, 29 , 30 , 34 , 36 findings are inconsistent and the clinical importance is still highly debatable. In summary, simvastatin was selected for this investigation due to the large number of available clinical studies and the wide range of processes involved in simvastatin disposition (Figure 1), thus enabling the assessment of several aspects of the PBPK platforms.
FIGURE 1.
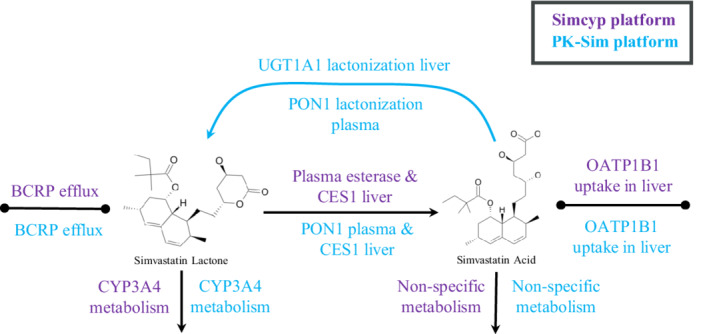
Metabolism and transport pathways included in the PBPK models.
This study aims to assess how PBPK model development, input parameters, and model output are affected by the selection of PBPK platform. This is exemplified via the establishment of simvastatin PBPK models (workflow, final models, and output) in PK‐Sim and Simcyp as representatives of established whole‐body PBPK platforms. Importantly, this work aims not only to compare the performance of the platforms, but also to provide insights on potential platform differences and the implications when bridging knowledge between users of different platforms. Finally, it provides a comprehensive PBPK model for SVL and SVA in both Simcyp and PK‐Sim to predict the implications of enzyme‐ and transporter‐mediated DGIs and DDIs.
METHODS
Data
A total of 30 clinical studies were used for SVL and SVA PBPK model development and validation. Three DGI studies in White subjects, involving BCRP and OATP1B1 transporters, were used for model development and validation of SVL and SVA transporter kinetics. Nine DDI studies with simvastatin as a CYP3A4 and OATP1B1 substrate were used for model validation. Detailed clinical trial information, including original references, the simulated trial designs, and specification of datasets used for model development or validation in the two platforms, are described in Table S3 in Appendix S2. Published plasma concentration–time profiles and variability were digitalized in WebPlotDigitilizer (version 3.6, GitHub).
PBPK platforms
The PBPK models for SVL and SVA were built in two different software platforms: Simcyp version 20.1 population‐based PBPK simulator, and PK‐Sim, and MoBi version 9.1 as part of the Open Systems Pharmacology Suite. Simulations were performed in a virtual healthy population, in Simcyp called “Sim‐Healthy volunteers” and in PK‐Sim “European (P‐pg modified, CYP3A4, 36 h, EHC)” individuals. Several modifications in the healthy populations were performed to implement the BCRP and OATP1B1 polymorphisms (see Section 1 in Appendix S2). The default values/models of CYP3A4 and OATP1B1 inhibitors available in each platform library were applied in the DDI analysis except for the OATP1B1 Ki of clarithromycin in the Simcyp model and OATP1B1 Ki of gemfibrozil in the PK‐Sim model (see Section 2 in Appendix S2). Model parameter estimation (PE) in Simcyp and PK‐Sim was performed by the Nelder–Mead and Monte–Carlo optimization methods, respectively.
In both platforms, a total of 100 individuals were simulated to assess variability across simvastatin pharmacokinetics (PKs). All PK‐Sim population simulations and PK analyses were also carried out in R with the ospsuite‐R package. The specific dosing regimen information was applied as reported in respective clinical trials. The specific demographic simulation settings (age, sex ratio, etc.) used in each simulation can be found in Section 3, Table S1 and S2 in Appendix S2. Graphics and statistical analysis for both platforms were produced in R (version 3.6.0).
Simvastatin model development description
The overall model development strategy in the two platforms is illustrated in Figure 2. In both platforms, the simvastatin full PBPK model, including SVL and SVA, was developed in a similar stepwise manner. The same initial simvastatin model, physicochemical parameters, and information on absorption, distribution, and excretion processes, extracted from literature and internal data, were used in both platforms. Several parameters were estimated by fitting the model to measured plasma concentration–time profiles from the prespecified training datasets (Figure 2 and Table 1). Details on the datasets used for PE and validation in the different platforms can be found in Figure 2 and Table S3 in Appendix S2, and differences on datasets selected for PE in the two platforms are further explained in Section 4 in Appendix S2. In step 1, predicted SVL and SVA PKs were validated against DGI clinical studies. In step 2, the model was validated against another set of clinical PK data in a dose range of 20–80 mg. In step 3, the final model was validated against DDI ratios when simvastatin was co‐administered with strong and moderate CYP3A4 inhibitors/inducers and with an OATP1B1 inhibitor. The pathways included in the two platforms are graphically summarized in the graphical abstract (Figure 1).
FIGURE 2.
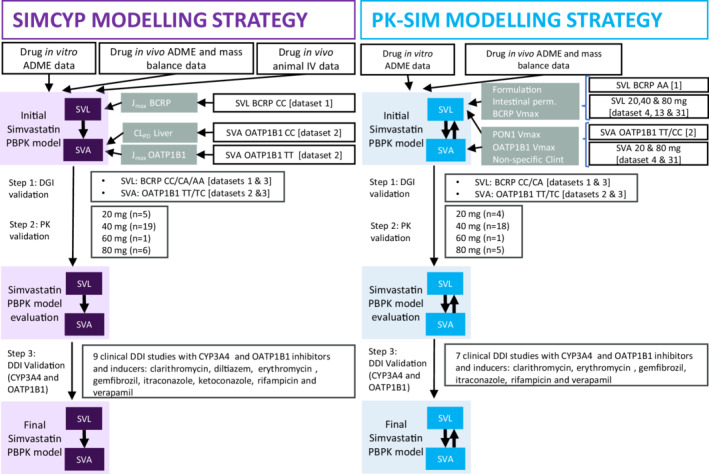
Modeling strategy of simvastatin lactone (SVL) and acid (SVA) in the selected platforms. ADME, absorption, distribution, metabolism, and excretion; DDI, drug–drug interaction; DGI, drug–gene interaction; Jmax, unbound maximum concentration; PBPK, physiologically‐based pharmacokinetic; PK, pharmacokinetic; SVA, simvastatin acid; SVL, simvastatin lactone; Vmax, maximal rate of metabolism.
TABLE 1.
Input parameter for SVL and SVA models
| SVL | ||||
|---|---|---|---|---|
| Simcyp | PK‐Sim | |||
| Parameter | Value | Reference | Value | Reference |
| Molecular weight (g/mol) | 418.57 | Drug bank | 418.57 | Drug bank |
| f u —experimental | 0.017 | (26) | 0.017 | (26) |
| B:P—experimental | 0.57 | Internal database | 0.57 a | Internal database |
| LogPo:w | 4.68 | Drug bank | 4.68 | Drug bank |
| Compound type | Neutral | Drug bank | Neutral | Drug bank |
| Absorption | ||||
| P eff,man (cm/s) | 4.28 10−4 (ADAM) | Predicted based on in vitro Caco‐2 data | ||
| Specific intestinal permeability (cm/s) | 3.2 10−6 | Estimated (PE) | ||
| Formulation | ||||
| Input form | Solid IR | Tablet | ||
| Type of model | DLM | Weibull | ||
| Particle size (radius, μm) | 10.843 | Predicted based on in vitro Dissolution profile (SIVA) (S50) | ‐ | |
| Distribution type | Monodisperse | ‐ | ||
| Particle density (g/ml) | 1.2 | Simcyp default | ‐ | |
| Solubility (mg/L) | 30 | (S49) | 30 | (S49) |
| Dissolution shape | ‐ | 0.48 | Estimated (PE) | |
| Dissolution time (50% dissolved) | ‐ | 10 | (S50) | |
| Intestinal transport | ||||
| V(J)max,BCRP (pmol/min/pmol transporter) | 13302.9 | Estimated (PE) | 338.7 | Estimated (PE) |
| K m (μM) | 5 | Assumed based on other statins | 5 | Assumed based on other statins |
| Distribution | ||||
| Calculation Method | Poulin and Theil | Poulin and Theil | ||
| K p,scalar | 0.2 | Set to match predicted V ss (2 L/kg) from animal i.v. data | ‐ | |
| Cellular permeability (cm/min) | ‐ | 0.26 | Calculated from PK‐Sim Standard method | |
| Elimination | ||||
| Enzyme kinetics | ||||
| Vmax,CYP3A4 (pmol/min/mg prot) | 5895.6 | (24) | 5895.6 | (24) |
| K m,CYP3A4 (μM) | 30.7 | (24) | 30.7 (1.23) | (24) |
| f u,mic | 0.04 | Internally measured | (Applied as K m,u ) | Internally measured |
| Conversion to SVA | ||||
| Plasma | ||||
| Esterase (t 1/2)[min] | 368 | (19) | ‐ | |
| Vmax,PON1 (pmol/min/mg prot) | ‐ | 618.63 | Estimated (PE) | |
| K m,PON1 (μM) | ‐ | 103.1 | (18) | |
| Liver | ||||
| Vmax,CES1 (pmol/min/mg prot) | 500 | (18) | 500 | (18) |
| K m,CES1 (μM) | 87.4 | (18) | 87.4 (3.5) | (18) |
| f u,mic | 0.04 | Internally measured | (Applied as K m,u ) | Internally measured |
| SVA | ||||
|---|---|---|---|---|
| Simcyp | PK‐Sim | |||
| Parameter | Value | Reference | Value | Reference |
| Molecular weight (g/mol) | 436.6 | Drug bank | 436.6 | Drug bank |
| f u —experimental | 0.057 | (26) | 0.057 | (26) |
| B:P—experimental | 0.75 | Internal database | 0.75 | Internal database |
| LogPo:w | 4.235 | Drug bank | 2.21 | Estimated (PE) |
| Compound type | Acid | Drug bank | Acid | Drug bank |
| pKa | 4.31 | Drug bank | 4.31 | Drug bank |
| Solubility (mg/L) | 11 (pH = 7) | Predicted Phytia tool | ||
| Distribution | ||||
| Calculation method | Rodger | PK‐Sim Standard | ||
| K p,scalar | 2 | Set to match predicted Vss (0.25 L/kg) from animal i.v. data | ‐ | |
| Cellular permeability (cm/min) | ‐ | 6.79 E‐4 | Calculated from charge dependent Schmitt normalized to PK‐Sim | |
| Elimination | ||||
| Enzyme kinetics | ||||
| Non‐specific liver CLint (μl/min/mg. prot) | 55 | (25) | 1.22 | Estimated (PE) |
| Conversion to SVL | ||||
| Plasma | ||||
| Vmax,PON1 (pmol/min/mg prot) | ‐ | 618.63 | Co‐estimated with Vmax PON1 SVL | |
| K m,PON1 (μM) | ‐ | 103.1 | Li et al. 2019 | |
| Reference concentration | ‐ | 0.125 | Assumed based on in vitro data (19) | |
| Liver | ||||
| CLint,UGTA1 (μl/min/mg. prot) | ‐ | 0.4 | (21) | |
| Reference concentration (pmol/mg prot) | ‐ | 34.3 | (S58) | |
| Transporter | ||||
| CLPD (ml/min/mill.hep) | 0.005 | Estimated (PE) | ‐ | |
| Liver permeability | ‐ | 1.23 E‐6 | Calculated based on cellular permeability | |
| V(J)max,OATP1B1 (pmol/min/pmol transp) | 9.16 | Estimated (PE) | 2.82 | Estimated (PE) |
| K m,OATP1B1 (μM) | 2 | (31) | 2 | (31) |
Abbreviations: ADAM, advanced dissolution absorption metabolism model; B:P, blood‐to‐plasma ratio; CLint, intrinsic clearance; CLPD, tissue passive permeability; f u,mic, fraction unbound drug in microsomal incubation; f u , fraction unbound in plasma; K m,u , unbound Michaelis constant; K m , Michaelis constant; K p,scalar, tissue‐plasma partition coefficient scalar; LogPo:w, the octanol/water partition coefficient; PE, parameter estimation; P eff,man, effective intestinal permeability in man; prot, protein; SIVA, Simcyp In vitro data Analysis; SVA, simvastatin acid; SVL, simvastatin lactone; t 1/2, terminal half‐life; Vmax, maximum velocity of the metabolic reaction; V ss, volume of distribution at steady state.
Set by the type value of hematocrit and blood cell to plasma partition coefficient (see Appendix S2).
Simcyp platform
The advanced dissolution absorption metabolism model (ADAM) was applied to describe the drug absorption process. Simvastatin was modeled as a solid immediate release formulation including particle dissolution. Tissue‐plasma partition coefficients (K p ) and the volume of distribution (V ss) were calculated by Poulin and Theil for SVL and the Rodgers and Rowland method for SVA. The K p scalar was set to match the human predicted V ss values for SVL and SVA which were scaled from animal intravenous (i.v.) data. Elimination of SVL was described by CYP3A4 enzyme kinetics, whereas SVA elimination was described by non‐specific hepatic clearance. Conversion of SVL to SVA in plasma was captured by a default Simcyp plasma esterase route. The conversion in the liver was described by CES1 esterase. Re‐conversion of SVA to SVL was not included in the Simcyp model, as this mechanism is not currently possible to incorporate in UGTs given the version 20 platform configuration. The parameters used in the Simcyp platform are listed in Table 1. The estimation of parameters in Simcyp was done in a stepwise manner by fitting the model to one dataset at a time, as illustrated in Figure 2. The Simcyp developed models of clarithromycin, itraconazole, verapamil, erythromycin, diltiazem, ketoconazole, and gemfibrozil were used together with the simvastatin model to perform the DDI model validation.
PK‐Sim and MoBi platforms
Intestinal absorption of simvastatin in PK‐Sim was modeled by adopting a solid formulation with Weibull release and the default multicompartmental transit and absorption model. The K p values were calculated by Poulin and Theil for SVL and PK‐Sim standard method for SVA. Additionally, cellular permeabilities were calculated based on PK‐Sim standard method for SVL and charge dependent Schmitt normalized to PK‐Sim method for SVA. Elimination of SVL was described by CYP3A4 enzyme kinetics, whereas SVA elimination was described by a nonspecific enzyme expressed in the liver. Conversion of SVL to SVA was assigned to PON1 esterase in plasma and CES1 esterase in the liver. 18 The re‐conversion (SVA‐to‐SVL) was described by PON1 esterase‐mediated clearance in plasma, assuming an eight‐fold lower capacity compared to the conversion of SVL to SVA, 19 and by UGT1A1 enzyme kinetics in the liver. 21 The parameters used in the PK‐Sim platform are listed in Table 1. The estimation of parameters in PK‐Sim was done simultaneously by fitting the model to several datasets, as illustrated in Figure 2. The previously developed models of clarithromycin, itraconazole, verapamil, erythromycin, and gemfibrozil were used together with the simvastatin model to perform the DDI validation. Models for diltiazem and ketoconazole, required to simulate DDI of dataset 36 and 40, were not available in PK‐Sim.
More detailed descriptions of the model development in both platforms are provided in Section 5 of Appendix S2.
Model evaluation
The predictive performance of each model was evaluated by overlaying the observed concentration–time data with the model predicted profile (geometric mean and 90% prediction interval). The PK parameters assessed were the area under the curve (AUC) and peak plasma concentration (Cmax). The predictive performance of PK metrics (AUC and Cmax parameters), DGI ratios (AUC and Cmax reduced/normal function), and DDI ratios (AUC and Cmax with/without inhibitor) were assessed based on a predefined criterion of twofold range of the predicted/observed ratio. Population geometric mean of the concentration–time profiles and PK metrics were predicted and compared to observed data. For statistical model evaluation, the geometric mean fold error (GMFE) was calculated for PK metrics, DGI, and DDI ratios. Additionally, goodness‐of‐fit plots were performed for concentration–time profiles at a dose range of 20–80 mg and f1 method developed by Marston and Polli 37 was calculated to measure the prediction accuracy, as previously described. 38
RESULTS
DGI validation (step 1)
DGI study: BCRP and SVL PKs
Simulated and observed plasma concentration–time profiles for SVL after 40 mg single dose administration in healthy volunteers with normal (CC), intermediate (CA), and poor (AA) BCRP transporter function were well‐captured in both platforms (Figure 3 and Figure S1 in Appendix S2). The final PBPK models predicted the DGI ratios within twofold of the observed for poor or intermediate function 29 , 36 .
FIGURE 3.
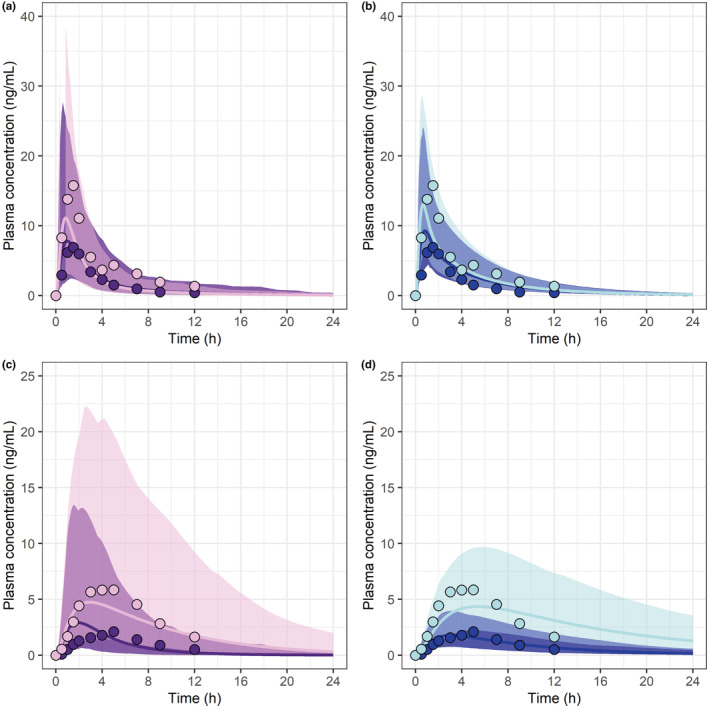
Predicted simvastatin lactone PK profiles (lines) in Simcyp (a) and PK‐Sim (b) versus clinical observations reported as mean (circles) for BCRP CC (dark colors) and AA (light colors) genotypes (31). Predicted simvastatin acid PK profiles (lines) in Simcyp (c) and PK‐Sim (d) versus observed (circles) for OATP1B1 TT and CC genotypes (25). The solid lines represent the predicted geometric mean and the shaded area the predicted 5%–95% quantiles for virtual populations. The normal genotypes (BCRP‐CC and OATP1B1‐TT) are represented with dark colors and the genotypes with reduced function of transporter (BCRP‐AA and OATP1B1‐CC) with light colors. PK, pharmacokinetic.
DGI study: OATP1B1 and SVA PKs
Simulated and observed plasma concentration–time profiles for SVA after 40 mg single dose administration in healthy volunteers with normal (TT), intermediate (CA), and poor (CC) OATP1B1 transporter function were in good agreement for both platforms (Figure 3 and Figure S2 in Appendix S2). The final PBPK models predicted the DGI ratios within twofold of the observed for poor or intermediate function 29 , 33 .
PK validation (step 2)
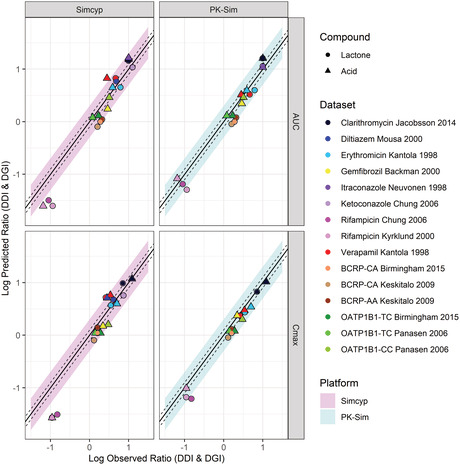
Simulated and observed plasma concentration–time profiles for SVL and SVA after 20, 40, 60, and 80 mg simvastatin single dose administration were in good agreement in both platforms (Figure 4 [40 mg], Figures S3–S5 in Appendix S2 [additional doses]). The f1 value for SVL and SVA plasma concentration–time profiles across doses was 44 and 78% in Simcyp compared to 38 and 37% in PK‐Sim. The observed SVL and SVA PK variability was also well‐captured by the two platforms. The final PBPK model in each platform predicted the PK metrics within twofold of the observed clinical data at all doses and 50% were within 1.25‐fold. Predicted and observed PK metrics are summarized in Figure 5 and GMFE in Table S4 in Appendix S2.
FIGURE 4.
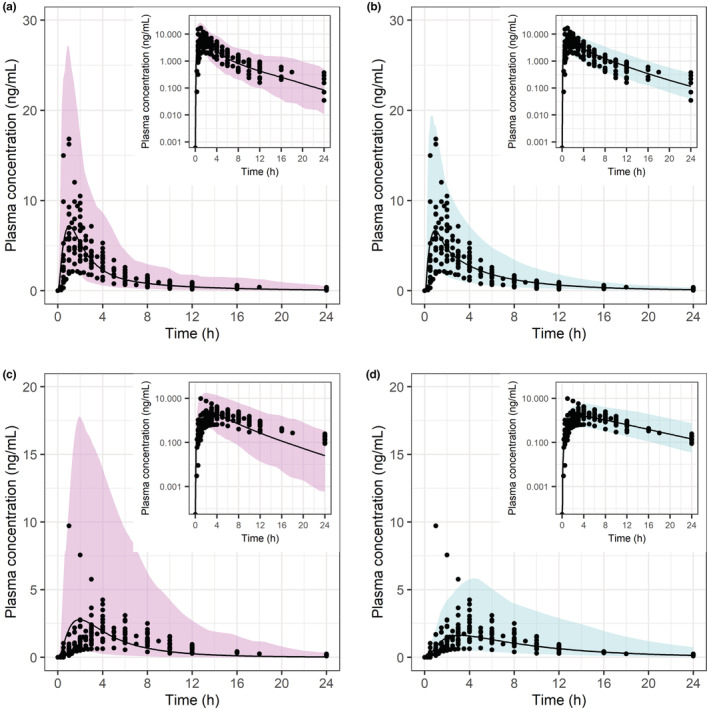
Predicted simvastatin lactone (a, b) and acid (c, d) PK profiles in Simcyp (pink/left) and PK‐Sim (blue/right) versus clinical observations reported as study mean (dots) for 40 mg dose (S19–S37). The black line represents the predicted geometric mean and the shaded area represents the predicted 5%–95% quantiles for virtual populations. AUC, area under the curve; Cmax, maximum plasma concentration; Obs, observed; PK, pharmacokinetic; Pred, predicted.
FIGURE 5.
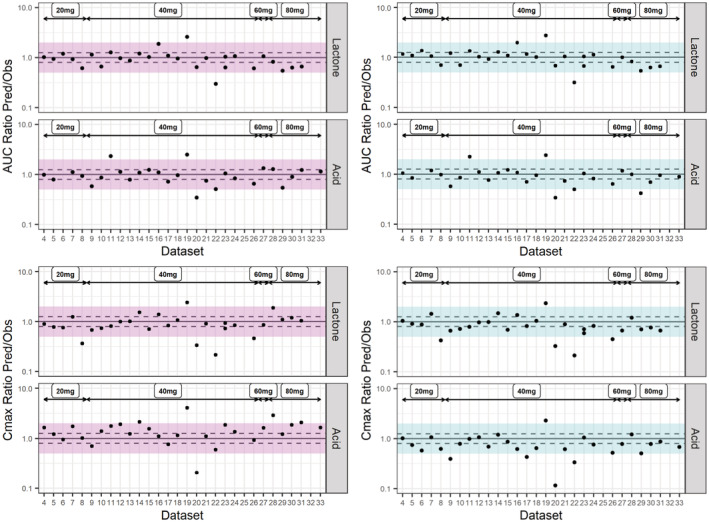
Predicted versus observed simvastatin lactone and acid AUC and Cmax for different datasets at a dose range 20–80 mg (S15–S44). Each study is represented by a dot. The solid line represents the line of unity, the dashed lines represent the 1.25‐fold error and the shaded area represents the twofold error in Simcyp (pink/left) and PK‐Sim (blue/right). AUC, area under the curve; Cmax, maximum plasma concentration; DDI, drug–drug interaction; DGI, drug–gene interaction.
DDI validation (step 3)
The predicted DDI ratios for the interaction between simvastatin (SVL and SVA) and strong and moderate CYP3A4 inhibitors/inducers and OATP1B1 inhibitors were overall within twofold of the observed values (Figure 6). Predicted and observed DGI and DDI ratios are summarized in Figure 6 and the GMFE in Table S5 in Appendix S2. Additionally, the observed SVL and SVA plasma concentration–time profiles with and without inhibitors fell within the 90% predictive interval (Figures S6–S18 in Appendix S2).
FIGURE 6.
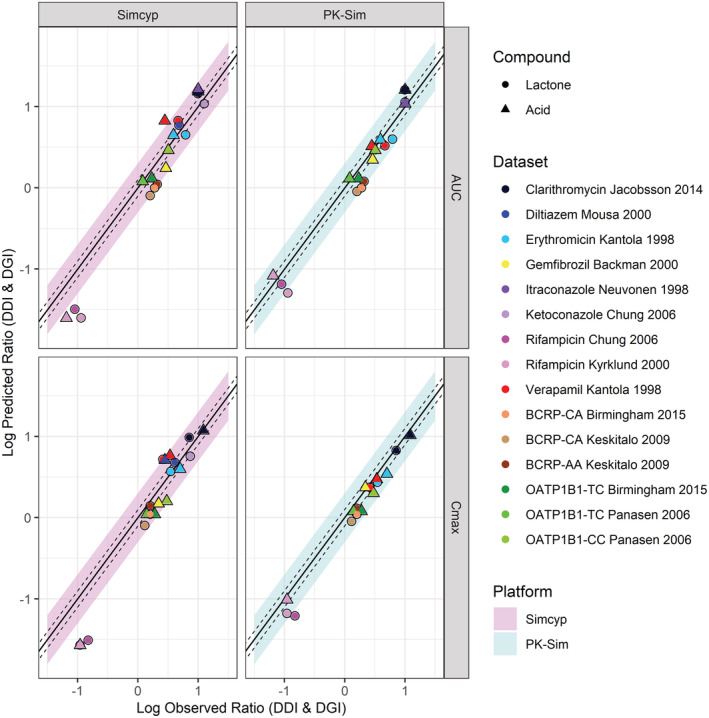
Predicted versus observed simvastatin lactone and acid AUC and Cmax ratio when coadministered with CYP3A4 and OATP1B1 inhibitors for nine clinical DDI studies (S17;S25;S28;S45‐S48) and for different BCRP and OATP1B1 genotypes for three clinical DGI studies (S1–S3). Each study is represented by a circle (lactone) or triangle (acid); details of the study design can be found in Table S2 in Appendix S2. The solid line represents the line of unity, the dashed lines represent the 1.25‐fold error and the shaded area represents the twofold error in Simcyp (pink/left) and PK‐Sim (blue/right). AUC, area under the curve; Cmax, maximum plasma concentration
DISCUSSION
The major finding of the present study was that the choice of PBPK platform clearly influenced both the model development strategy and the final model input parameters, however, the predictive performance of the simvastatin models was still comparable between the platforms. The modeling development strategies differed between the platforms as a consequence of available functionalities (illustrated in Figure 2 and Section 4 in Appendix S2). Model input parameters (see Table 1) were dependent on model structure and implementation in each platform.
Both platforms predicted the observed SVL and SVA PK (dose range 20–80 mg), DGI (BCRP and OATP1B1), and DDI when co‐administered with CYP3A4 and OATP1B1 inhibitors/inducers equally well (Figures 5 and 6). Moreover, the overall precision and bias was similar for predictions of SVL and SVA PK metrics (Table S4 in Appendix S2). However, the dynamics of SVA plasma concentration–time profiles were more accurately captured by PK‐Sim (f1 = 78%) than by Simcyp (f1 = 37%; Figure S19 in Appendix S2). A possible explanation could be the differences in the implementation of the distribution processes between the two platforms. Overall, the DDIs for both SVL and SVA were well‐predicted with DDI ratios within twofold error in both platforms. A minor discrepancy was the overprediction of the induction effect mediated by rifampicin in Simcyp (Figure 6), which has also been reported previously for SVL and midazolam as victim drugs. 39 An additional difference in output was that Simcyp predicted higher population variability in SVA plasma concentration–time profiles than PK‐Sim (Figure 4). Our investigations could not elucidate the source of this difference. It could not be explained by the different population variability in enzyme and transporter‐related parameters or due to allele frequencies in the populations (only present in Simcyp platform). Nevertheless, the population variability in SVL plasma concentration–time profiles was similar between the platforms. Therefore, additional case studies are needed to determine if this is a systematic difference and to delineate the nature of this discrepancy. Further elaborations on virtual populations are summarized in Section 6 in Appendix S2.
The platform differences identified as having the largest impact on the simvastatin case study were the implementation of the absorption and distribution models, as outlined in Table 2.
TABLE 2.
Major platform differences of relevance for simvastatin case study
| Model | Aspect | Simcyp | PK‐Sim |
|---|---|---|---|
| Absorption | Model structure | ADAM Model (9 compartments) | Physiological GIT model (12 compartments + mucosa) |
| Input of Passive permeability | Built‐in IVIVC of several in vitro systems (Caco‐2, MDCK, LLC‐PK1, PAMPA) and in silico (user defined IVIVC and PSA) | Semi‐empirical method based on Thelen et al. 2011, informed by in vitro measurements or estimated | |
| Input formulation | Several in vitro alternatives for solubility and dissolution rate input parameters including DLM and SIVA | Several alternatives for in vitro solubility but dissolution rate by Weibull function or particle‐based | |
| Distribution | Model structure | Perfusion‐limited distribution | Permeability‐limited distribution |
| K p calculation methods | M1: Poulin and Theil/Berezhkovskiy; M2: Rodgers & Rowland; M3: M2 + ion permeability | M1: Poulin and Theil; M2: Berezhkovskiy; M3: Rodgers and Rowland; M4: PK‐Sim Standard; M5: Schmitt | |
| Passive permeability in tissues | Upon user selection of predefined tissues: liver, kidney, brain, lung, tumor, and additional organ (placenta/lactation) | All | |
| Input tissue passive permeability | CLPD parameter separately for each tissue informed from in vitro system or estimated | Calculated for all tissues based on M1: PK‐Sim Standard; M2: Charge depend Schmitt; M3: Charge dependent Schmitt normalized to PK‐Sim | |
| Transporters | Transporters are added separately for each predefined tissue | Transporters are added to all tissues according to the gene expression database |
Abbreviations: ADAM, advanced dissolution absorption metabolism model; CLPD, tissue passive diffusion clearance; DLM, diffusion layer model; GIT, gastrointestinal tract; IVIVC, in vitro in vivo correlation; K p , tissue‐plasma partition coefficient; SIVA, Simcyp In Vitro data Analysis.
In Simcyp, different models for absorption with a wide range of complexity are available. 40 In this study, the ADAM model was applied, including intestinal metabolism and transporters, where the gastrointestinal tract (GIT) is divided into nine anatomically defined segments. In PK‐Sim, absorption is described by the physiological multicompartmental transit and absorption model, including intestinal metabolism, transporters, and enterohepatic re‐circulation. The GIT is divided into 12 anatomically defined segments with an elaborate representation of the mucosa. 41 Additionally, platform differences on how absorption input parameters are informed, conditioned model development. In general, Simcyp provides a variety of options (alternative to user defined/estimated) for the input of absorption parameters including several built‐in correlation methods to scale in vitro measured values as well as the Simcyp In Vitro data Analysis (SIVA) toolkit, 40 whereas in PK‐Sim, these methods are not present. In this study, the “bottom‐up” functionalities were applied in the Simcyp model for the input of the intestinal passive permeability and formulation dissolution parameters (Section 5 in Appendix S2). In PK‐Sim, however, these parameters were optimized toward clinical data for this study. These platform differences are expected to have substantial implications on absorption‐related investigations, handling of input, and interpretation of outcome. As an example, despite a large difference in passive permeability input values, simvastatin is predicted to be readily absorbed over the investigated dose range (20–80 mg) in both platforms (i.e., defined as highly permeable; Table 1). This can be explained by differences in parameterization and how the permeability input value is translated in the models.
In Simcyp, applying the full PBPK option, the distribution to tissues is described as perfusion limited distribution, implemented by K p values. 13 The K p values for each tissue can be calculated by three mechanistic models (Table 2). 42 , 43 , 44 When transporters need to be included, the permeability limited model is implemented solely for the tissue of interest (limited to predefined tissues, see Table 2) and the parameter of passive permeability (passive diffusion clearance [CLPD]) is incorporated for each tissue separately. In PK‐Sim, distribution to all tissues is implemented by the permeability limited distribution model. 8 In addition to the mechanistic models available in Simcyp for estimation of K p values, the PK‐Sim standard 45 and Schmitt 46 models can be applied in PK‐Sim. Furthermore, cellular permeability can be automatically calculated with three different methods (see Table 2). In contrast to Simcyp, transporters in PK‐Sim can be implemented in any compartments/organs. 8 In addition, PK‐Sim has a functionality to identify the optimal distribution method by comparing the performance of several methods in a single run. In this case study, the Poulin and Theil method was identified to best describe the SVL PK profile in both platforms. For the description of the SVA PK profile, the combination of PK‐Sim Standard/Schmitt normalized to PK‐Sim Standard was identified as optimal in PK‐Sim, whereas the Rodgers and Rowland method was the best alternative in Simcyp. The PK profile of SVA was better captured in PK‐Sim than in Simcyp. This may be because PK‐Sim considers the influence of cell wall permeability in all tissues, whereas this is only implemented for the liver in Simcyp.
The largest discrepancy in input parameters was observed in the transporter kinetics values for V(J)max, despite it was estimated from same clinical study in both cases (Table 1). This could be mainly explained by differences in transporter abundancy, Simcyp platform (informed based on measured abundancy), and PK‐Sim (default reference concentration of 1 μM), but also by differences in other important parameters, such as intestinal or tissue (cellular) permeability, calculated free fraction in the tissue, tissue vascularity, and specification of the surface area.
The final models in the two platforms differed in several PK processes. The PK‐Sim final model includes SVA‐SVL reconversion, and renal and biliary excretion, which are not present in the Simcyp model. This was a consequence of higher flexibility in PK‐Sim to include user‐defined processes and differences on the implementation of renal and biliary excretion between the platforms (details in Section 7 in Appendix S2). However, these processes are not highly relevant for the simvastatin disposition. 22 , 27 Therefore, in this case study, these differences did not influence model performance or final model input parameters between the platforms. This was reflected in the PK‐Sim model where (1) the same parameters values were estimate (PE) with or without SVA‐SVL reconversion and model performance did not improve by the addition of reconversion; (2) the fraction of dose renally or biliary eliminated was lower than 0.05 having no impact on the predicted SVL and SVA PKs. Nevertheless, these platform differences could be significantly important for the disposition of other compounds. Additional details in differences of implementations and handling of input parameters encountered during this investigation are summarized in Section 7 in Appendix S2.
Considering that each platform presents its own advantages, the choice of platform is ultimately dependent on the scope of the modeling and the available input data. The major strengths of Simcyp are: (1) it is an established and widely used PBPK platform with a large publication and regulatory track record; (2) it has numerous in‐built in vitro‐in vivo extrapolation/correlation (IVIVE/C) possibilities including the SIVA toolkit, making a “bottom‐up” modeling approach easily accessible; (3) it has an elaborate population generation, including a population library for several diseases and ethnicities. In contrast, the major strengths of the PK‐Sim platform are: (1) the Open Systems Pharmacology (OSP) platform (including PK‐Sim) is open source and models can be examined for details on model implementation and parameterization; (2) it provides extensive flexibility (e.g., inclusion of an unlimited number of metabolites and the possibility of back‐conversion in all processes). Via MoBi it also allows modelers with expertise to access equations and to include additional processes to the backbone model structure; (3) PK‐Sim includes a sophisticated and flexible PE functionality making it easy to use for “middle‐out” modeling. For example, it allows for simultaneous PE of multiple parameters, including simulations with different populations.
There are several previously published PBPK models for simvastatin: one in Simcyp, 39 two in PK‐Sim 47 , 48 and another semimechanistic PBPK model in NONMEM. 49 The models presented herein differ to previously reported PBPK models in several aspects (Section 8 in Appendix S2). Overall, two aspects of simvastatin PK processes that would benefit from further investigation are: (1) the importance of BCRP for SVL PKs, and (2) the CYP3A4 contribution to SVA elimination.
Although our presented models include BCRP‐mediated cell membrane translocation, informed by the PK data reported by Keskitalo, 36 these do not predict significant changes in AUC for the different BCRP genotypes (Figure S20 in Appendix S2). The models are not sensitive to inhibition or activity reduction of BCRP, suggesting that the contribution of BCRP to simvastatin disposition is not of major importance. In line with this, Wojtiniak 48 reported that only when the BCRP transporter was included in the blood cells were they able to recover the AUC changes observed for the poor transporter phenotype. However, there is limited availability of pharmacogenetic information, with only a few studies comprising a small number of subjects (4 to 15 individuals) for each transporter genotype. 29 , 36 Considering the intrinsic variability in SVL PKs, these studies were not sufficiently powered to confidently conclude the implication of BCRP phenotype on SVL PKs. Additional clinical pharmacogenetic studies on the BCRP polymorphism with larger cohorts are needed to fill this knowledge gap.
In vitro studies suggest that the elimination of SVA is predominantly (>90%) mediated by CYP3A4. 23 , 25 In the initial model, the in vitro enzyme kinetic values for SVA were implemented. However, despite the overall good performance in predicting the SVL response to CYP3A4 inhibition, there was simultaneously a trend to overpredict SVA exposure in both platforms. The CYP3A4 DDI ratio of SVL:SVA (1:2) predicted by these initial models (data not shown) was not in line with the observed ratio (1:1). Thus, in vitro calculated CYP3A4 contribution to SVA elimination may be overestimated, or there could be alternative routes of elimination in vivo which are not captured by in vitro assays. When SVA elimination was attributed to a nonspecific metabolic pathway, we were able to recover the observed SVA DDI ratio in CYP3A4 DDI studies as well as keeping the observed SVL:SVA ratio (1:1). The CYP3A4 contribution to SVA elimination has not been reported in previous published simvastatin PBPK models. 48 , 49 Additional in vitro and/or clinical studies could help clarify the routes of SVA metabolism to further inform elimination of SVA in PBPK models.
The purpose of this study was to assess the implications of PBPK platform selection with simvastatin disposition and DDIs as a case study in Simcyp and PK‐Sim. Accordingly, it was beyond the scope of this investigation to include other PBPK platforms or additional compounds. In addition, applications of the presented models, such as efficacy or safety assessments of simvastatin, were not included, but could be part of future studies.
In conclusion, this study illustrates that in‐depth knowledge of established PBPK platforms is crucial to enable an assessment of what consequences the selection of a specific PBPK platform may bring. In particular, this work provides insights on software differences and potential implications when bridging PBPK knowledge between Simcyp and PK‐Sim platform users. Finally, it provides a simvastatin model implemented in both platforms for risk assessment of metabolism‐ and transporter‐mediated DGIs and DDIs.
AUTHOR CONTRIBUTIONS
L.P.G. and E.S. wrote the manuscript. L.P.G., C.A., H.L., and E.S. designed the research. L.P.G., A.L., and E.S. performed the research. L.P.G., A.V., and E.S. analyzed the data.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
Supporting information
Appendix S1
Appendix S2
Appendix S3
ACKNOWLEDGEMENTS
The authors are grateful to Dr. Jin Dong for the discussions and input on IVIVE of the formulation and distribution parameters of simvastatin disposition in Simcyp.
Prieto Garcia L, Lundahl A, Ahlström C, Vildhede A, Lennernäs H, Sjögren E. Does the choice of applied physiologically‐based pharmacokinetics platform matter? A case study on simvastatin disposition and drug–drug interaction. CPT Pharmacometrics Syst Pharmacol. 2022;11:1194‐1209. doi: 10.1002/psp4.12837
REFERENCES
- 1. el‐Khateeb E, Burkhill S, Murby S, Amirat H, Rostami‐Hodjegan A, Ahmad A. Physiological‐based pharmacokinetic modeling trends in pharmaceutical drug development over the last 20‐years; in‐depth analysis of applications, organizations, and platforms. Biopharm Drug Dispos. 2021;42:107‐117. doi: 10.1002/bdd.2257 [DOI] [PubMed] [Google Scholar]
- 2. Jamei M. Recent advances in development and application of physiologically‐based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep. 2016;2:161‐169. doi: 10.1007/s40495-016-0059-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jones HM, Chen Y, Gibson C, et al. Physiologically based pharmacokinetic modeling in drug discovery and development: a pharmaceutical industry perspective. Clin Pharmacol Ther. 2015;97:247‐262. doi: 10.1002/cpt.37 [DOI] [PubMed] [Google Scholar]
- 4. EMEA (ed Committee for Medical Products for Human Use [CHMP]) . 2018.
- 5. FDA (ed U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research [CDER]) . 2018.
- 6. FDA (ed U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research [CDER]) . 2020.
- 7. Bouzom F, Ball K, Perdaems N, Walther B. Physiologically based pharmacokinetic (PBPK) modelling tools: how to fit with our needs? Biopharm Drug Dispos. 2012;33:55‐71. doi: 10.1002/bdd.1767 [DOI] [PubMed] [Google Scholar]
- 8. Kuepfer L, Niederalt C, Wendl T, et al. Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacometrics Syst Pharmacol. 2016;5:516‐531. doi: 10.1002/psp4.12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jones H, Rowland‐Yeo K. Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development. CPT Pharmacometrics Syst Pharmacol. 2013;2:e63. doi: 10.1038/psp.2013.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peters SA. Physiologically‐Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry. John Wiley & Sons; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rowland M, Peck C, Tucker G. Physiologically‐based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol Toxicol. 2011;51:45‐73. doi: 10.1146/annurev-pharmtox-010510-100540 [DOI] [PubMed] [Google Scholar]
- 12. Rostami‐Hodjegan A. Reverse translation in PBPK and QSP: going backwards in order to go forward with confidence. Clin Pharmacol Ther. 2018;103:224‐232. doi: 10.1002/cpt.904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jamei M, Marciniak S, Feng K, Barnett A, Tucker G, Rostami‐Hodjegan A. The Simcyp population‐based ADME simulator. Expert Opin Drug Metab Toxicol. 2009;5:211‐223. doi: 10.1517/17425250802691074 [DOI] [PubMed] [Google Scholar]
- 14. Stefan Willmann JL, Sevestre M, Solodenko J, Fois F, Schmitt W. PK‐Sim®: a physiologically based pharmacokinetic ‘whole‐body’ model. BIOSILICO. 2003;1:121‐124. [Google Scholar]
- 15. Ahmad A, Pepin X, Aarons L, et al. IMI ‐ Oral biopharmaceutics tools project ‐ evaluation of bottom‐up PBPK prediction success part 4: prediction accuracy and software comparisons with improved data and modelling strategies. Eur J Pharm Biopharm. 2020;156:50‐63. doi: 10.1016/j.ejpb.2020.08.006 [DOI] [PubMed] [Google Scholar]
- 16. Parrott N, Lave T. Prediction of intestinal absorption: comparative assessment of GASTROPLUS and IDEA. Eur J Pharm Sci. 2002;17:51‐61. doi: 10.1016/s0928-0987(02)00132-x [DOI] [PubMed] [Google Scholar]
- 17. Sjogren E, Thorn H, Tannergren C. In silico modeling of gastrointestinal drug absorption: predictive performance of three physiologically based absorption models. Mol Pharm. 2016;13:1763‐1778. doi: 10.1021/acs.molpharmaceut.5b00861 [DOI] [PubMed] [Google Scholar]
- 18. Li Z, Zhang J, Zhang Y, Zuo Z. Role of esterase mediated hydrolysis of simvastatin in human and rat blood and its impact on pharmacokinetic profiles of simvastatin and its active metabolite in rat. J Pharm Biomed Anal. 2019;168:13‐22. doi: 10.1016/j.jpba.2019.02.004 [DOI] [PubMed] [Google Scholar]
- 19. Prueksaritanont T, Qiu Y, Mu L, et al. Interconversion pharmacokinetics of simvastatin and its hydroxy acid in dogs: effects of gemfibrozil. Pharm Res. 2005;22:1101‐1109. doi: 10.1007/s11095-005-6037-2 [DOI] [PubMed] [Google Scholar]
- 20. Garcia MJ, Reinoso RF, Sanchez Navarro A, Prous JR. Clinical pharmacokinetics of statins. Methods Find Exp Clin Pharmacol. 2003;25:457‐481. [PubMed] [Google Scholar]
- 21. Prueksaritanont T, Subramanian R, Fang X, et al. Glucuronidation of statins in animals and humans: a novel mechanism of statin lactonization. Drug Metab Dispos. 2002;30:505‐512. doi: 10.1124/dmd.30.5.505 [DOI] [PubMed] [Google Scholar]
- 22. Cheng H, Schwartz MS, Vickers S, et al. Metabolic disposition of simvastatin in patients with T‐tube drainage. Drug Metab Dispos. 1994;22:139‐142. [PubMed] [Google Scholar]
- 23. Fujino H, Saito T, Tsunenari Y, Kojima J, Sakaeda T. Metabolic properties of the acid and lactone forms of HMG‐CoA reductase inhibitors. Xenobiotica. 2004;34:961‐971. doi: 10.1080/00498250400015319 [DOI] [PubMed] [Google Scholar]
- 24. Prueksaritanont T, Gorham LM, Ma B, et al. In vitro metabolism of simvastatin in humans [SBT]identification of metabolizing enzymes and effect of the drug on hepatic P450s. Drug Metab Dispos. 1997;25:1191‐1199. [PubMed] [Google Scholar]
- 25. Prueksaritanont T, Ma B, Yu N. The human hepatic metabolism of simvastatin hydroxy acid is mediated primarily by CYP3A, and not CYP2D6. Br J Clin Pharmacol. 2003;56:120‐124. doi: 10.1046/j.1365-2125.2003.01833.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vickers S, Duncan CA, Chen IW, Rosegay A, Duggan DE. Metabolic disposition studies on simvastatin, a cholesterol‐lowering prodrug. Drug Metab Dispos. 1990;18:138‐145. [PubMed] [Google Scholar]
- 27. Elsby R, Hilgendorf C, Fenner K. Understanding the critical disposition pathways of statins to assess drug–drug interaction risk during drug development: it's not just about OATP1B1. Clin Pharmacol Ther. 2012;92:584‐598. doi: 10.1038/clpt.2012.163 [DOI] [PubMed] [Google Scholar]
- 28. Backman J. Plasma concentrations of active simvastatin acid are increased by gemfibrozil. Clin Pharmacol Ther. 2000;68:122‐129. doi: 10.1067/mcp.2000.108507 [DOI] [PubMed] [Google Scholar]
- 29. Birmingham BK, Bujac SR, Elsby R, et al. Impact of ABCG2 and SLCO1B1 polymorphisms on pharmacokinetics of rosuvastatin, atorvastatin and simvastatin acid in Caucasian and Asian subjects: a class effect? Eur J Clin Pharmacol. 2015;71:341‐355. doi: 10.1007/s00228-014-1801-z [DOI] [PubMed] [Google Scholar]
- 30. Choi HY, Bae KS, Cho SH, et al. Impact of CYP2D6, CYP3A5, CYP2C19, CYP2A6, SLCO1B1, ABCB1, and ABCG2 gene polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid. Pharmacogenet Genomics. 2015;25:595‐608. doi: 10.1097/FPC.0000000000000176 [DOI] [PubMed] [Google Scholar]
- 31. Huang, H. Characterization of in vitro systems for transporter studies. PhD thesis thesis, Uppsala University; 2010.
- 32. Jiang F, Choi JY, Lee JH, et al. The influences of SLCO1B1 and ABCB1 genotypes on the pharmacokinetics of simvastatin, in relation to CYP3A4 inhibition. Pharmacogenomics. 2017;18:459‐469. doi: 10.2217/pgs-2016-0199 [DOI] [PubMed] [Google Scholar]
- 33. Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics. 2006;16:873‐879. doi: 10.1097/01.fpc.0000230416.82349.90 [DOI] [PubMed] [Google Scholar]
- 34. Tsamandouras N, Dickinson G, Guo Y, et al. Identification of the effect of multiple polymorphisms on the pharmacokinetics of simvastatin and simvastatin acid using a population‐modeling approach. Clin Pharmacol Ther. 2014;96:90‐100. doi: 10.1038/clpt.2014.55 [DOI] [PubMed] [Google Scholar]
- 35. Wagner JB, Abdel‐Rahman S, van Haandel L, et al. Impact of SLCO1B1 genotype on pediatric simvastatin acid pharmacokinetics. J Clin Pharmacol. 2018;58:823‐833. doi: 10.1002/jcph.1080 [DOI] [PubMed] [Google Scholar]
- 36. Keskitalo JE, Pasanen MK, Neuvonen PJ, Niemi M. Different effects of the ABCG2 c.421C>a SNP on the pharmacokinetics of fluvastatin, pravastatin and simvastatin. Pharmacogenomics. 2009;10:1617‐1624. doi: 10.2217/pgs.09.85 [DOI] [PubMed] [Google Scholar]
- 37. Marston SA, Polli JE. Evaluation of direct curve comparison metrics applied to pharmacokinetic profiles and relative bioavailability and bioequivalence. Pharm Res. 1997;14:1363‐1369. doi: 10.1023/a:1012160419520 [DOI] [PubMed] [Google Scholar]
- 38. Prieto Garcia L, Janzén D, Kanebratt KP, Ericsson H, Lennernäs H, Lundahl A. Physiologically based pharmacokinetic model of itraconazole and two of its metabolites to improve the predictions and the mechanistic understanding of CYP3A4 drug–drug interactions. Drug Metab Dispos. 2018;46:1420‐1433. doi: 10.1124/dmd.118.081364 [DOI] [PubMed] [Google Scholar]
- 39. Almond LM, Mukadam S, Gardner I, et al. Prediction of drug–drug interactions arising from CYP3A induction using a physiologically based dynamic model. Drug Metab Dispos. 2016;44:821‐832. doi: 10.1124/dmd.115.066845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jamei M, Turner D, Yang J, et al. Population‐based mechanistic prediction of oral drug absorption. AAPS J. 2009;11:225‐237. doi: 10.1208/s12248-009-9099-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thelen K, Coboeken K, Willmann S, Burghaus R, Dressman JB, Lippert J. Evolution of a detailed physiological model to simulate the gastrointestinal transit and absorption process in humans, part 1: oral solutions. J Pharm Sci. 2011;100:5324‐5345. doi: 10.1002/jps.22726 [DOI] [PubMed] [Google Scholar]
- 42. Berezhkovskiy LM. Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J Pharm Sci. 2004;93:1628‐1640. doi: 10.1002/jps.20073 [DOI] [PubMed] [Google Scholar]
- 43. Poulin P, Theil FP. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism‐based prediction of volume of distribution. J Pharm Sci. 2002;91:129‐156. doi: 10.1002/jps.10005 [DOI] [PubMed] [Google Scholar]
- 44. Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res. 2007;24:918‐933. doi: 10.1007/s11095-006-9210-3 [DOI] [PubMed] [Google Scholar]
- 45. Willmann S, Lippert J, Schmitt W. From physicochemistry to absorption and distribution: predictive mechanistic modelling and computational tools. Expert Opin Drug Metab Toxicol. 2005;1:159‐168. doi: 10.1517/17425255.1.1.159 [DOI] [PubMed] [Google Scholar]
- 46. Schmitt W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol In Vitro. 2008;22:457‐467. doi: 10.1016/j.tiv.2007.09.010 [DOI] [PubMed] [Google Scholar]
- 47. Lippert J, Brosch M, von Kampen O, et al. A mechanistic, model‐based approach to safety assessment in clinical development. CPT Pharmacometrics Syst Pharmacol. 2012;1:e13. doi: 10.1038/psp.2012.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wojtyniak JG, Selzer D, Schwab M, Lehr T. Physiologically based precision dosing approach for drug–drug–gene interactions: a simvastatin network analysis. Clin Pharmacol Ther. 2021;109:201‐211. doi: 10.1002/cpt.2111 [DOI] [PubMed] [Google Scholar]
- 49. Tsamandouras N, Dickinson G, Guo Y, et al. Development and application of a mechanistic pharmacokinetic model for simvastatin and its active metabolite simvastatin acid using an integrated population PBPK approach. Pharm Res. 2015;32:1864‐1883. doi: 10.1007/s11095-014-1581-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Appendix S2
Appendix S3