

Abstract
Air pollutants are important factors that contribute to the development and/or exacerbation of allergic inflammation accompanied by asthma, but experimental evidence still needs to be collected. Interleukin 33 (IL-33) is closely involved in the onset and progression of asthma. In this study, we examined the effects of particulate matter (PM) on IL-33 expression in macrophages. PM2.5 collected in Yokohama, Japan by the cyclone device significantly induced IL-33 expression in human THP-1 macrophages, and the induction was clearly suppressed by pretreatment with the aryl hydrocarbon receptor (AhR) antagonist CH-223191 or the Toll-like receptor 4 (TLR4) antagonist TAK-242. PM2.5-induced IL-33 expression was significantly attenuated in AhR-knockout or TLR4-mutated macrophages, suggesting an important role of polycyclic aromatic hydrocarbons (PAHs) and endotoxin in IL-33 stimulation. PM samples derived from tunnel dust slightly but significantly induced IL-33 expression, while road dust PM did not affect IL-33 expression. The PAH concentration in tunnel dust was higher than that in road dust. Tunnel dust or road dust PM contained less endotoxin than PM2.5 collected in Yokohama. These data suggest that the potency of IL-33 induction could depend on the concentration of PAHs as well as endotoxin in PMs. Caution regarding PAHs and endotoxin levels in air pollutants should be taken to prevent IL-33-induced allergic inflammation.
Keywords: Particulate matter, PAHs, Endotoxin, IL-33, Macrophages
INTRODUCTION
Airborne fine particulate matter (PM2.5) is the main factor involved in air pollution in the developing world and is considered a great concern to human health. Exposure to PM2.5 is closely related to human respiratory disorders and cardiovascular diseases (Pun et al., 2017; Dominici et al., 2006). Asthma is one of the diseases whose development is affected by PM2.5 exposure (O’Connor et al., 2008). Asthma is a disorder of the conducting airways, which contract too much and too easily spontaneously and in response to a wide range of exogenous and endogenous stimuli (Holgate, 2008). This airway hyperresponsiveness is accompanied by enhanced irritability of the airways and increased mucus secretion. The different clinical manifestations of asthma involve varying environmental factors that interact with the airways to cause acute and chronic inflammation and the varying contributions of smooth muscle contraction, edema and remodeling of the airways. Several studies have been conducted to explain how exposure to PM2.5 could increase the risk of asthma, but the involved mechanisms still need to be addressed.
PM2.5 includes many substances, such as inorganic compounds (i.e., metals and salts), organic compounds (i.e., polycyclic aromatic hydrocarbons (PAHs)) and biological compounds (i.e., endotoxin). These components have been thought to contribute to PM2.5 toxicity. Sulfur oxides such as sulfur dioxide and sulfur trioxide are closely associated with the onset and progression of asthma. The proinflammatory cytokine interleukin 8 (IL-8) is released from airway epithelial cells by sulfur dioxide stimulation and causes the infiltration of inflammatory cells such as neutrophils and eosinophils into the airways (Chanez et al., 1996). Ferrous and ferric iron in PM2.5 reportedly contribute to ferroptosis in human endothelial cells, in which iron overload and subsequent oxidative stress play important roles (Wang and Tang, 2019). Ogino et al. reported that acid-soluble metals in PM2.5 can induce allergic airway inflammation in mice (Ogino et al., 2018). PAHs are well known to induce inflammation in an aryl hydrocarbon receptor (AhR)-dependent manner (Bock, 2020) and are also involved in carcinogenesis (Moorthy et al., 2015). In addition, endotoxin is a strong agonist of Toll-like receptor 4 (TLR4), which increases the expression of inflammatory molecules by activating the transcription factor NF-κB (Ishihara et al., 2015b). Overall, PM2.5 contents exert toxicity via multiple mechanisms (Vogel et al., 2020).
IL-33 is an IL-1 family cytokine, such as IL-1β and IL-18, and was identified in 2005 as a ligand for the receptor ST2 (Dinarello, 2005). IL-33 is localized in the nucleus in epithelial cells, the vascular endothelium and macrophages located in several types of tissue, is released by harmful stimuli and acts as an alarmin. IL-33 can stimulate group 2 innate lymphoid cells to produce Th2 cytokines. Furthermore, IL-33 enhances IL-5/IL-13 production in Th2 cells in cooperation with antigens (Liew et al., 2016). Genome-wide analysis revealed that IL-33 was involved in asthma (Moffatt et al., 2010). The production of IL-33 is largely increased in the lungs of asthma patients (Préfontaine et al., 2010), and this production depends on the severity of asthma (Préfontaine et al., 2009). IL-33 blockade with an IL-33 neutralizing antibody reportedly suppressed inflammation and improved remodeling of both the lung epithelium and lung parenchyma in mice sensitized with house dust mites (Allinne et al., 2019). Therefore, IL-33 could be one of the key molecules in the onset and progression of asthma.
The mechanism of IL-33 expression is not fully understood, but several transcription factors are responsible for its induction. NF-κB, a master regulator of inflammation, increases IL-33 expression in response to lipopolysaccharide (LPS), similar to other inflammatory molecules (Hiraide et al., 2018). We previously reported that ligand-dependent activation of AhR can induce IL-33 expression in macrophages (Ishihara et al., 2019). Dioxin response elements (DREs) were identified upstream of the transcription initiation site of IL-33 in mice and humans. As described previously, PM2.5 exposure is a risk factor for the development and progression of asthma and contains many components, including PAHs and endotoxin, which can induce IL-33 expression. The purpose of this study was to examine the effects of several PM2.5 and related dusts on IL-33 expression in macrophages, focusing on the components, especially PAHs and endotoxin.
MATERIALS AND METHODS
Collection of particulate matter (PM)
Urban aerosol was collected in Yagami Campus, Keio University, Yokohama, Japan, as previously described (Okuda et al., 2015). Briefly, PM2.5 with an aerodynamic diameter of 2.5 μm was separated using an impactor prior to entry into the cyclone device. Subsequently, the cyclone imparted a centrifugal force on the gas stream within a conical-shaped chamber and created a vortex inside the cyclone body.
Tunnel dust was collected in Tokyo, Japan, by a bag filter that was set into the exhaust port of the tunnel. The collected dust was separated by a 100 μm diameter stainless sieve and subsequently the elbow-jet air classifier (Elcan Industries, Tuckahoe, NY, USA) to obtain PM with a diameter less than 5 μm. Road dust was purchased from Sigma-Aldrich (BCR723, Saint Louis, MO, USA) and was classified by the elbow-jet air classifier to prepare PM with diameters less than 5 μm. Polypropylene particles with an average diameter of 2 μm were kind gifts from Seishin Enterprise Co. (Tokyo, Japan).
THP-1 cell culture
The human monocytic cell line THP-1 was obtained from ATCC (TIB-202) and maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum. THP-1 cells were differentiated using 160 nM phorbol 12-myristate 13-acetate (PMA) and then used for experiments (Starr et al., 2018; Tanaka et al., 2021).
Treatment of cells with PM
Collected PM2.5, tunnel dust, road dust and polypropylene particles were suspended into distilled water by sonication for 10 sec at 20 W. The suspension was added to the culture with 100 times dilution.
Total RNA extraction and real-time PCR
mRNA levels were determined according to the protocol described in our previous report (Ishihara et al., 2017). The primer sequences are presented in Table 1. mRNA levels were normalized to the level of β-actin, and the values of the treated samples were divided by those of the untreated samples to calculate the relative mRNA levels.
Table 1.
Primers for real-time PCR analysis.
| Target | Forward primer | Reverse primer |
|---|---|---|
| Human IL-33 | TGAATCAGGTGACGGTGTT | TGGTCTGGCAGTGGTTTT |
| Human β-actin | GGACTTCGAGCAAGAGATGG | AGCACTGTGTTGGCGTACAG |
| Mouse IL-33 | GGGCTCACTGCAGGAAAGTA | TCAGGTTTCTCAATACAGACAGGA |
| Mouse β-actin | AGCCATGTACGTAGCCATCC | CTCTCAGCTGTGGTGGTGAA |
Assay for cellular viability
Cellular viability was measured using the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) according to the manufactures’ instruction.
Immunoblotting
Immunoblotting was performed as described previously (Ishihara et al., 2015b). Briefly, cells were collected and lysed with RIPA buffer. Equal amounts of protein were loaded, separated via SDS-PAGE and transferred onto polyvinylidene difluoride membranes. The blocked membranes were incubated with the following primary antibodies: anti-IL-33 (sc-130625, Santa Cruz, Dallas, TX, USA) and anti-β-actin (sc-47778, Santa Cruz). Then, the membrane was incubated with peroxide-conjugated secondary antibodies (Thermo Fisher Scientific, Waltham, MA, USA) and visualized using peroxide substrates (SuperSignal West Dura, Thermo Fisher Scientific). The band intensity was quantified using ImageJ software.
Promoter analysis
Promoter analysis of the 5′-upstream regulatory region of human and mouse IL-33 was performed with the TFSEARCH program (Heinemeyer et al., 1998). Genetic sequence data in GenBank were used for the analysis.
Luciferase assay
The pNL vector (Promega, Madison, WI, USA) containing approximately 1,300 bp of the promoter region flanking human IL-33 exon 1c cloned previously (Ishihara et al., 2019) was transfected into THP-1 cells using jet-PRIME (Polyplus-transfection, Illkirch, France) according to the manufacturer’s instructions. Twenty-four hr after transfection, the cells were treated with PM2.5 and antagonists. Luciferase activity was measured using the Nano-Glo Luciferase Assay System (Promega) with a GloMax 20/20 Luminometer (Promega) (Ishihara et al., 2015a).
Preparation of bone marrow-derived macrophages
All animal procedures were performed in accordance with the Fundamental Guidelines for the Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology, Japan. The Animal Care and Use Committee of Hiroshima University approved the experimental protocols (Approved No. C18–16-4 and C20–33).
AhR-knockout (AhR-KO) mice were generated as described previously (Mimura et al., 1997). Male C57BL/6J, C3H HeJ and C3H HeN mice were purchased from CLEA Japan (Tokyo, Japan). The animals were maintained on a 12:12 hr light/dark cycle and had free access to water and food. The mice were allowed to adapt to the facility for 1 week.
Primary bone marrow progenitor cells were isolated and differentiated as described previously (Ishihara et al., 2019). Briefly, the femurs were isolated under sterile conditions, and bone marrow cells were extracted with an RPMI medium-loaded syringe. The cells were passed through a 30 μm cell strainer, and the supernatant was centrifuged for 5 min at 1000 × g to obtain cells. Bone marrow-derived macrophage differentiation was performed in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF; 20 ng/mL; PeproTech, Cranbury, NJ, USA) for 7 days. Macrophage differentiation was confirmed by F4/80 staining.
Determination of PAH content in PM
PAH concentrations in PM were determined according to a previous report with slight modifications (El-Saeid et al., 2015). One milliliter of dichloromethane and 7.5 ng of the internal standard chrysene-d12 (Wako, Osaka, Japan) were added to the PM (2–5 mg), and then the mixture was sonicated to extract PAHs. After centrifugation, the supernatant was dried under nitrogen flow with heating. Hexane (0.5 mL) was added to the resulting residues. The extracts were passed through a solid column (ISO-LUTE SI SPE, 460–0010-B, BioTage, Uppsala, Sweden) with a mixture of hexane and dichloromethane. The eluents were dried again, and 100 μL of hexane was added to prepare samples for gas chromatography-mass spectrometry (GC-MS).
The samples were analyzed using GC-MS (7890A/5957C, Agilent, Palo Alto, CA, USA) with an HP-5MS capillary column (30 m × 0.25 mm i.d. × 0.25 μm film thickness). One microliter of sample was injected into an autosampler in splitless mode. The GC oven temperature was 60°C for the initial 2 min and was then increased to 300°C from 2 to 32 min and maintained at 300°C from 32 to 47 min. Helium was used as the carrier gas, and the flow rate was 1 mL/min. The qualification and quantification of PAHs was performed in scan mode and selected ion-monitoring mode, respectively. The data were normalized to the area of chrysene-d12. The PAH calibration mix (Sigma-Aldrich), benzo(e)pyrene (Sigma-Aldrich) and coronene Sigma-Aldrich) were used as standards.
The BaP equivalent concentration of each PM was calculated according to the values as previously reported by the CALUX assay (Machala et al., 2001; Boonen et al., 2020; Ciganek et al., 2004).
Measurement of endotoxin levels in PM
PMs were suspended in distilled water and sonicated for 10 min. After centrifugation, endotoxin levels in the supernatant were measured using an Endospecy ES-50M set (Seikagaku Co., Tokyo, Japan) according to the manufacturer’s protocol. Control standard endotoxin derived from E. coli O113 (Seikagaku Co.) was used as a standard.
Statistical analysis
All data are expressed as the mean ± standard error (S.E.). Statistical analyses were performed using one-way analysis of variance (ANOVA), followed by Student’s t test or Dunnett’s test. P values < 0.05 were considered statistically significant.
RESULTS
PM2.5-induced IL-33 expression is mediated by AhR and TLR4 stimulation
THP-1 monocytes were treated with PMA to differentiate into macrophages. When THP-1 macrophages were treated with PM2.5 collected in Yokohama in spring 2020, IL-33 mRNA expression was elevated in a concentration-dependent manner (Fig. 1A). PM2.5 concentration up to 100 μg/mL did not show the toxicity to THP-1 macrophages (Fig. 1B). PM2.5 contains PAHs and endotoxins (Table 2), which are potent inducers of inflammation (Osgood et al., 2017; Monn and Becker, 1999). Therefore, we focused on the PAH receptor AhR and the endotoxin receptor TLR4. Pretreatment with the AhR antagonist CH-223191 moderately but significantly suppressed PM2.5-induced IL-33 expression (Fig. 1C). The TLR4 antagonist TAK-242 largely attenuated IL-33 induction. The combination of these antagonists completely suppressed IL-33 expression in THP-1 macrophages (Fig. 1C). Immunoblotting also showed that IL-33 protein expression was increased by PM2.5 and that this induction was significantly attenuated by pretreatment with CH-223191 and TAK-242 alone or their combination (Fig. 1D and E). Therefore, AhR and TLR4 are involved in the induction of IL-33 expression in macrophages.
Fig. 1.
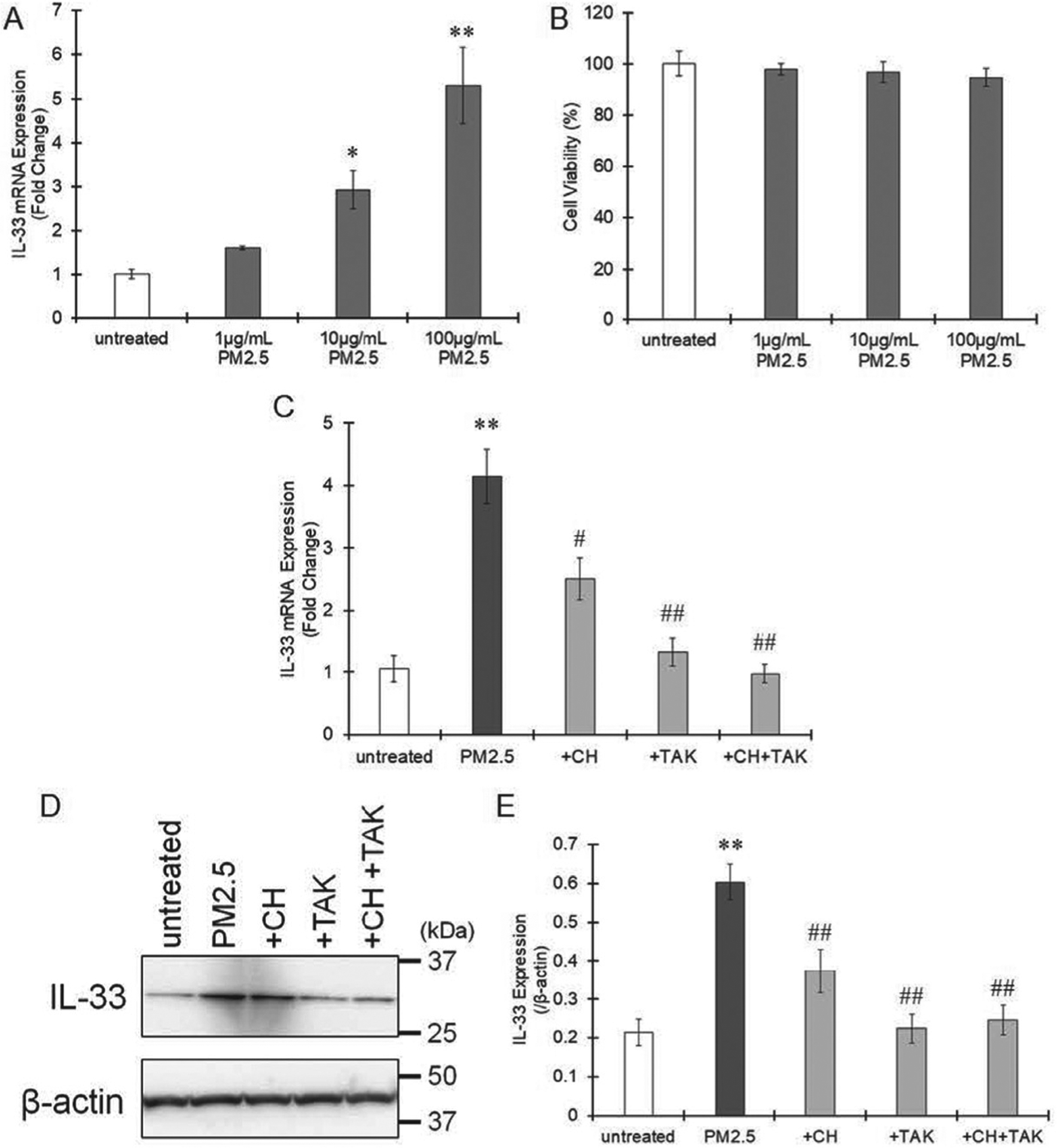
Effects of AhR and TLR4 antagonism on IL-33 expression induced by PM2.5 exposure. A. THP-1 macrophages were treated with different concentrations (0, 1, 10 and 100 μg/mL) of PM2.5 collected in Yokohama, Japan, in spring 2020 for 6 hr and then IL-33 mRNA expression was measured by real-time PCR. The values represent the mean ± S.E. (n = 6). B. Cellular viability was measured after treatment of THP-1 macrophages with PM2.5 for 24 hr. The values represent the mean ± S.E. (n = 4). C. THP-1 macrophages were pretreated with the AhR antagonist CH-223191 (10 μM) or the TLR4 antagonist TAK-242 (1 μM) and then treated with PM2.5 (100 μg/mL) for 6 hr. Total RNA was extracted, and the mRNA levels of IL-33 were measured by real-time PCR. The values represent the mean ± S.E. (n = 4). D and E. IL-33 protein expression was evaluated by immunoblotting 24 hr after treatment with PM2.5 (100 μg/mL). Representative immunoblot images were shown as panel D. The band intensity was measured, and the protein levels of IL-33 were divided by those of β-actin to calculate the relative protein levels shown as panel E. The values represent the mean ± S.E. (n = 3). The data were analyzed using one-way ANOVA, followed by Student’s t test or Dunnett’s test. Holm’s corrections were used for multiple comparisons. *P < 0.05, **P < 0.01 vs. the untreated group. #P < 0.05, ##P < 0.01 vs. the PM2.5 group.
Table 2.
PAH and endotoxin contents in PM.
| Yokohama 2018 Winter | Yokohama 2018 Spring | Yokohama 2020 Spring | Tunnel Dust | Road Dust | |
|---|---|---|---|---|---|
| Phenanthrene | 1.38 | 0.82 | 0.73 | 2.20 | 2.07 |
| Anthracene | 0.29 | 0.18 | 0.21 | 0.38 | 0.11 |
| Fluoranthene | 1.81 | 1.04 | 0.94 | 3.89 | 5.40 |
| Pyrene | 1,75 | 1.14 | 0.74 | 5.33 | 3.02 |
| Benz[a]anthracene | 4.95 | 2.53 | 1.87 | 3.16 | 2.62 |
| Chrysene | 4,78 | 2.18 | 2.37 | 4.61 | 3.62 |
| Benzo[b+j]fluoranthene | 11.5 | 8.19 | 4.10 | 4.75 | 2.80 |
| Benzo[k]fluoranthene | 9.52 | 2.82 | 2.86 | 3.98 | 2.08 |
| Benzo(e)pyrene | 12.6 | 9.02 | 3.55 | 8.01 | 4.17 |
| Benzo(a)pyrene | 6.88 | 5.43 | 1.81 | 5.15 | 1.24 |
| Indeno[1,2,3-cd]pyrene | 3.43 | 2.44 | 1.84 | 1.78 | 2.32 |
| Dibenz[a,h]anthracene | 0.44 | 0.42 | 0.42 | 0.45 | 0.32 |
| Benzo[ghi]perylene | 3.32 | 2.84 | 2.06 | 5.09 | 8.43 |
| Coronene | 3.41 | 2.90 | 2.01 | 2.05 | 6.11 |
| Total PAHs | 66.1 | 42.0 | 25.5 | 50.9 | 44.3 (ng/mg PM) |
| Endotoxin | 0.83 | 0.39 | 0.97 | 0.07 | 0.001 (ng/mg PM) |
We have identified 2 dioxin response elements (DREs) upstream of human IL-33 exon 1c previously (Ishihara et al., 2019). In addition, 2 NF-κB response elements, a RelB/AhR response element (RelBAhRE) and a 12-O-tetradecanoylphorbol-13-acetate response element (TRE) were found by the TFSEARCH program in human IL-33 promoter region (Fig. 2A). When the THP-1 macrophages transfected with pNL including the IL-33 promoter were treated with PM2.5, luciferase activity was significantly higher than that of the untreated cells (Fig. 2B). This increment of luciferase activity was significantly suppressed by CH-223191 and TAK-242 alone or their combination (Fig. 2B), suggesting IL-33 promoter activation by AhR and TLR4.
Fig. 2.
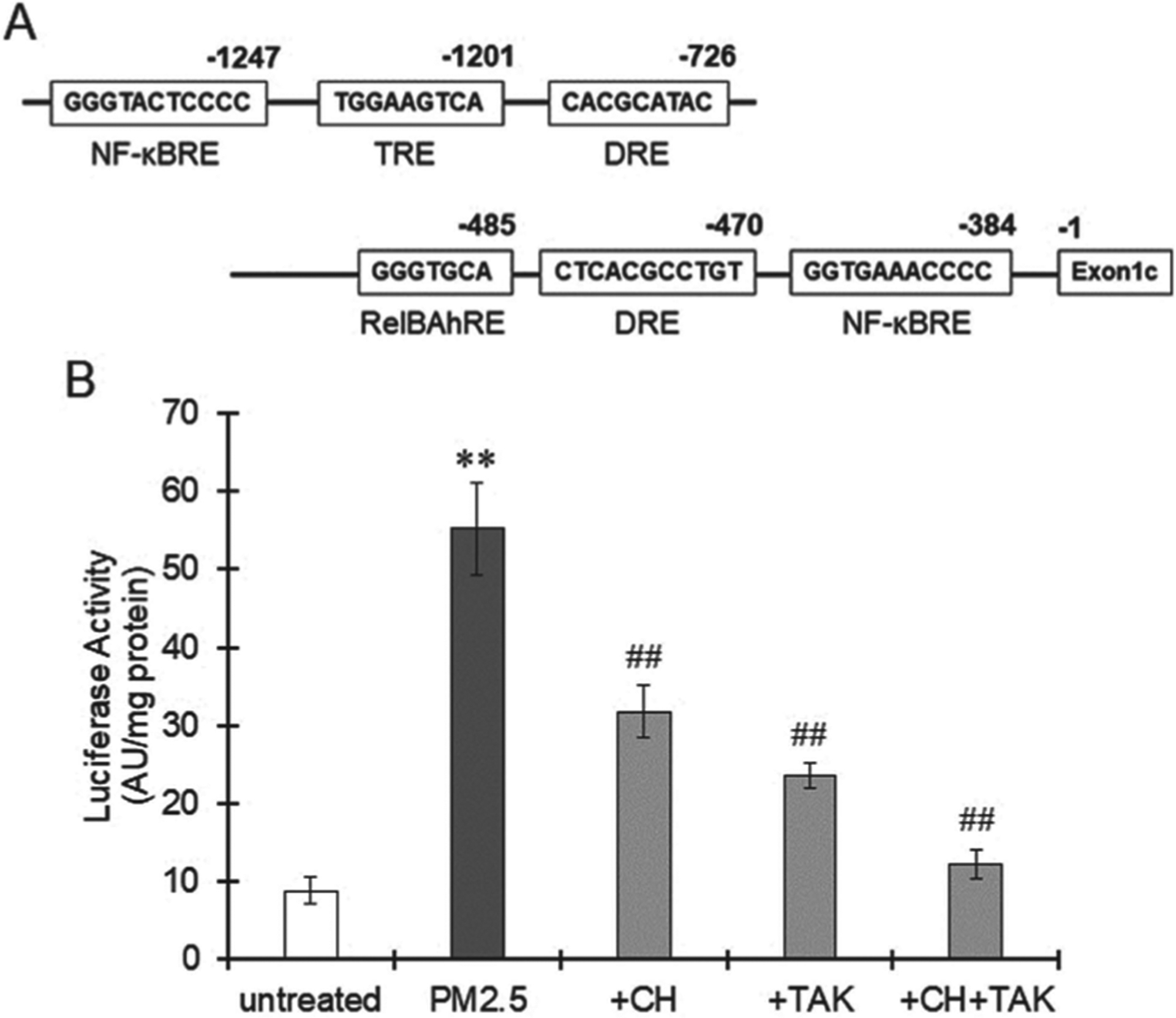
IL-33 promoter activation induced by PM2.5 exposure. A. Several transcription factor binding sites in the human IL-33 promoter region identified by the TFSEARCH program. B. THP-1 macrophages were transfected with pNL containing the human IL-33 promoter region and then pretreated with CH-223191 (10 μM) or TAK-242 (1 μM). Three hours after treatment with PM2.5 (100 μg/mL) collected in Yokohama, Japan, in spring 2020, the cells were collected and lysed to measure luciferase activity. The values represent the mean ± S.E. (n = 6). Data were analyzed using oneway ANOVA, followed by Student’s t test or Dunnett’s test. Holm’s corrections were used for multiple comparisons. **P < 0.01 vs. the untreated group. ##P < 0.01 vs. the PM2.5 group.
To confirm the role of AhR and TLR4 in the increase in IL-33 in macrophages, we used bone marrow macrophages isolated from AhR-KO mice and C3H HeJ mice. A spontaneous mutation in the TLR4 gene is present in C3H/HeJ mice, causing TLR4 dysfunction (Poltorak et al., 1998). AhR-KO macrophages showed low sensitivity to PM2.5 exposure, as indicated by IL-33 induction, compared with wild-type cells (Fig. 3A). Pretreatment with TAK-242 completely suppressed PM2.5-induced IL-33 expression in AhR-KO macrophages (Fig. 3A). IL-33 mRNA was induced in TLR4-mutated macrophages, but the level was largely decreased compared with that in control macrophages isolated from C3H HeN mice (Fig. 3B). Pretreatment with CH-223191 completely suppressed IL-33 induction by PM2.5 in TLR4-mutated macrophages (Fig. 3B). Based on these results, the potency of IL-33 induction mediated by TLR4 was stronger than that mediated by AhR. Stimulation of both AhR and TLR4 potently induced IL-33 expression in macrophages.
Fig. 3.
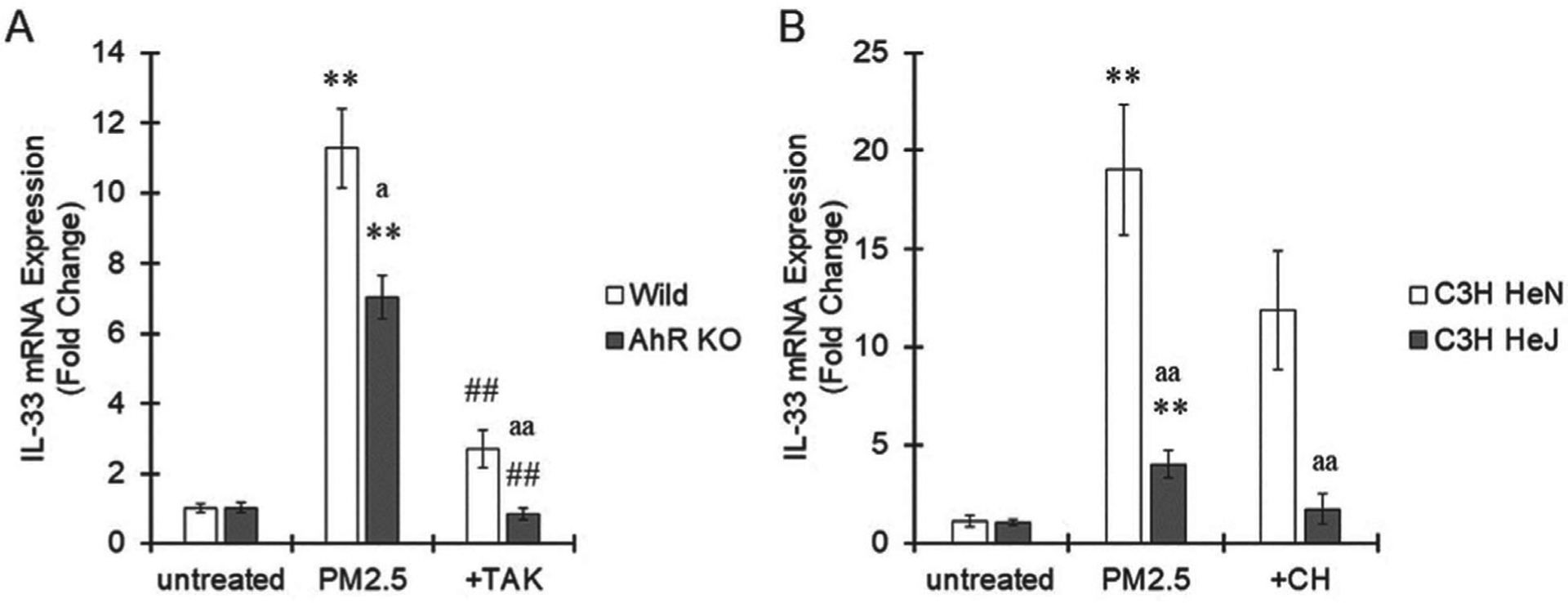
Contribution of AhR and TLR4 to IL-33 induction in macrophages. A. Bone marrow-derived macrophages were isolated from wild-type or AhR-KO mice, pretreated with TAK-242 (1 μM) and then treated with PM2.5 (100 μg/mL) collected in Yokohama, Japan, in spring 2020 for 6 hr. Cells were collected, and IL-33 mRNA expression was measured by real-time PCR. B. Bone marrow-derived macrophages were isolated from C3H HeN or C3H HeJ mice, pretreated with CH-223191 (10 μM) and then treated with PM2.5 (100 μg/mL) collected in Yokohama, Japan, in spring 2020 for 6 hr. Cells were collected, and IL-33 mRNA expression was measured by real-time PCR. The values represent the mean ± S.E. (n = 4). the data were analyzed using one-way ANOVA, followed by Student’s t test. Holm’s corrections were used for multiple comparisons. **P < 0.01 vs. the untreated group. ##P < 0.01 vs. the PM2.5 group. aP < 0.05, aaP < 0.01 vs. the wild-type or C3H HeN group.
Effects of several types of PM on IL-33 induction
PM2.5 collected in Yokohama in spring or winter 2018 significantly induced IL-33 expression in THP-1 macrophages, similar to the effect of PM2.5 collected in 2020 (Fig. 1A and 4A). Thus, PM2.5 in Yokohama exhibits a certain level of IL-33 induction regardless of the year or season. Next, we used commercial road dust (BCR-723) and tunnel dust that was collected by the filter setting in a tunnel in Tokyo, Japan. Particulates less than 5 μm were isolated by the elbow-jet air classifier for cell-based exposure experiments. Road dust did not induce IL-33 expression in THP-1 macrophages (Fig. 3B). Exposure to tunnel dust significantly increased IL-33 expression, but the levels were lower than those induced by PM2.5 collected in Yokohama (Fig. 1A, 4A and 4B). Control polypropylene particles showed no effect on IL-33 expression in macrophages (Fig. 4B).
Fig. 4.
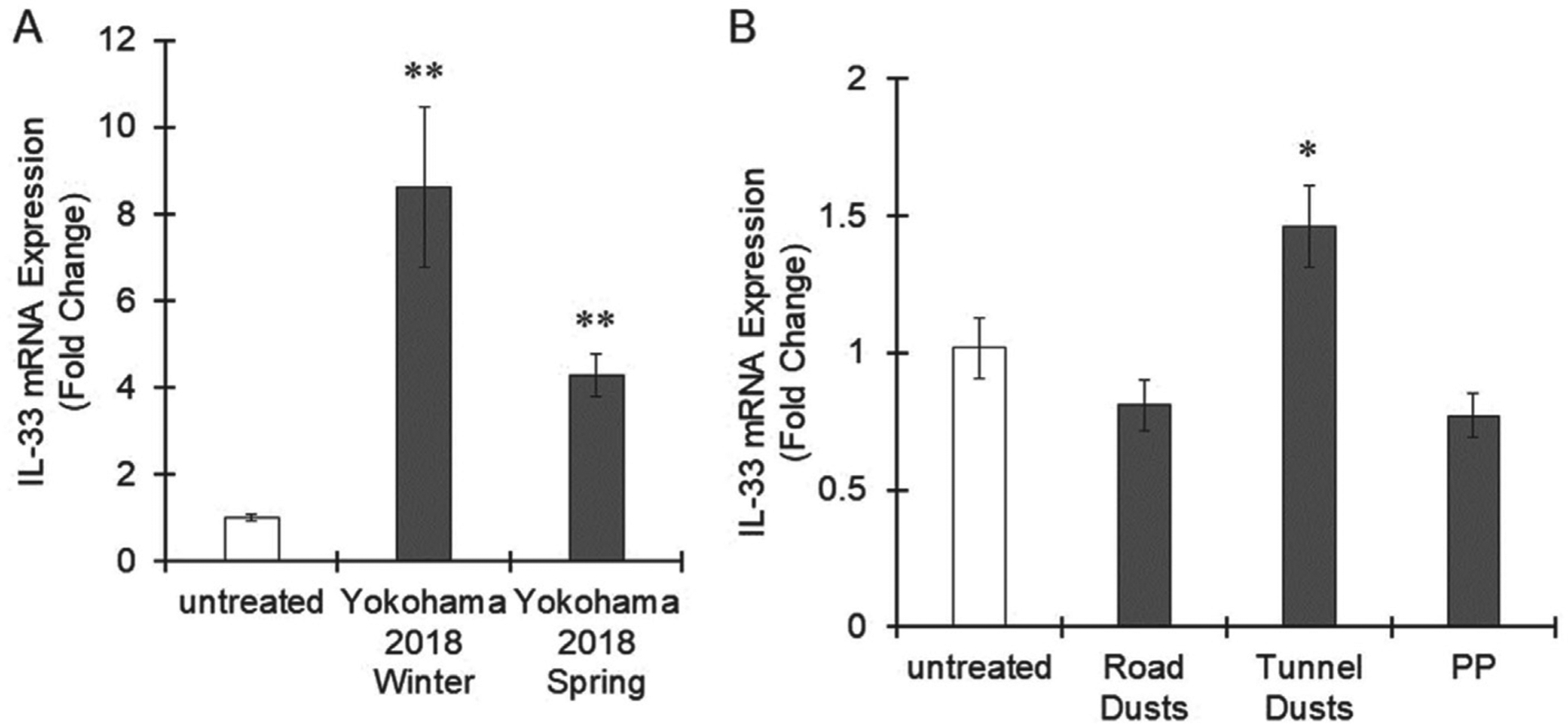
Effects of several kinds of PM on IL-33 expression in macrophages. A. THP-1 macrophages were treated with PM2.5 (100 μg/mL) collected in Yokohama, Japan, in spring 2018 or winter 2018 for 6 hr. B. THP-1 macrophages were exposed to road dust (100 μg/mL), tunnel dust (100 μg/mL) or polypropylene (PP) particles (100 μg/mL) for 6 hr. IL-33 expression was evaluated by real-time PCR. The values represent the mean ± S.E. (n = 4). The data were analyzed using one-way ANOVA, followed by Dunnett’s test. *P < 0.05, **P < 0.01 vs. the untreated group.
The PAH contents and endotoxin levels in the 5 kinds of PM used in this study were measured. The total PAH level was highest in tunnel dust, followed by road dust (Table 2). Individual PAH compositions were similar among the 3 types of PM2.5 collected in Yokohama, but the benzo[k]fluoranthene concentration in Yokohama 2018 winter was higher than that in the other 2 PM2.5 collected in Yokohama (Table 2, Fig. 5). Endotoxin levels in PM2.5 collected in Yokohama were much higher than those in tunnel dust (Table 2). Endotoxin was rarely detected in road dust (Table 2).
Fig. 5.
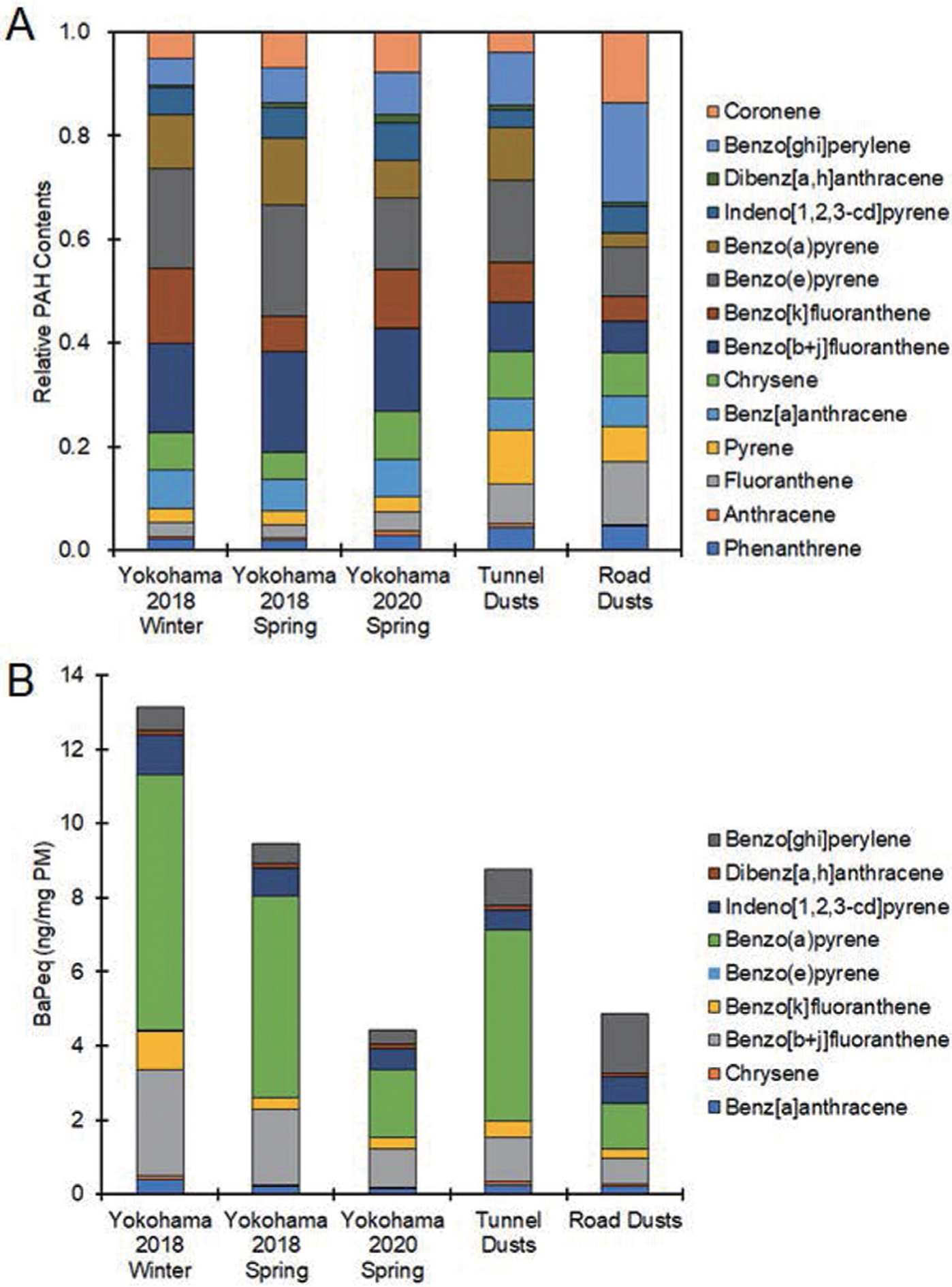
Relative PAH contents and benzo[a]pyrene equivalent values in PMs. PAHs were extracted from five samples of PM with dichloromethane, and PAH concentrations were determined by GC-MS. The data were averaged from 3 independent experiments. A. Relative PAH concentrations are shown as total PAH amounts in each PM set to 1. B. Benzo[a]pyrene equivalent values (BaPeq) of PAHs in PM were calculated according to previous reports that described CALUX analysis of PAHs. The exact procedure is described in the Materials and Methods.
DISCUSSION
In this study, we evaluated the ability of PM to induce IL-33 expression in macrophages, focusing on PAHs and endotoxin in PM. Several PAHs are known to be potent ligands of AhR. Ligand stimulation activates AhR, which translocates into the nucleus and binds to the DRE upstream of the transcription start site of several genes, including those of proinflammatory molecules (Vogel et al., 2020). We previously reported that AhR could induce IL-33 expression because there are 2 DREs in the promoter region of IL-33 (Ishihara et al., 2019). PM2.5 collected in Yokohama contained many PAHs, including those that can stimulate AhR, such as benzo[a]pyrene. IL-33 induction by PM2.5 was significantly antagonized by the AhR antagonist CH-223191. Therefore, PAHs in PM2.5 are involved in IL-33 expression in macrophages. On the other hand, IL-33 is also reported to be induced by endotoxin (Hiraide et al., 2018). Endotoxin binding to TLR4 leads to activation of the transcription factor NF-κB, which upregulates proinflammatory molecules, including IL-33 (Hiraide et al., 2018). IL-33 has 2 NF-κBREs at the promoter region (Fig. 2A). Because PM2.5 collected in Yokohama contained high levels of endotoxin and the TLR4 antagonist TAK-242 suppressed IL-33 expression, it is thought that endotoxin in PM2.5 contributes to IL-33 induction.
Interestingly, while TLR4 mutations almost completely attenuate IL-33 induction by PM2.5, the suppressive effects of AhR KO on IL-33 stimulation by PM2.5 were partial. A similar trend was observed in experiments using CH-223191 and TAK-242 in THP-1 macrophages. These data show that endotoxin in PM has a critical role in IL-33 induction and that importantly, signals induced by PAHs could overlap with endotoxin signaling in macrophages. In other words, in the presence of endotoxin, AhR signaling pathway may interact with endotoxin signaling. AhR has been reported to interact with NF-κB RelA to induce the expression of several genes, including proinflammatory molecules (Vogel et al., 2014). In addition, the NF-κB subunit RelB reportedly associates with AhR and mediates the transcription of proinflammatory proteins such as IL-8. The AhR/RelB dimer can bind to the RelBAhRE to enhance gene expression (Vogel et al., 2007). Importantly, IL-33 has a RelBAhRE in its promoter region (Fig. 2A). Taken together, these findings suggest that PAHs and endotoxin in PM might induce IL-33 expression via crosstalk between AhR and NF-κB. Our previous report showed that LPS enhanced AhR-dependent IL-33 expression in macrophages (Ishihara et al., 2019), supporting this hypothesis. However, AhR has been reported to negatively regulate the expression of some cytokines depending on the experimental conditions. Kimura et al. showed that AhR forms a complex with Stat1 and NF-κB, which inhibits the promoter activity of IL-6 (Kimura et al., 2009). In addition, AhR activation reportedly enhances NF-κB RelA subunit degradation by the ubiquitin-proteasome system and lysosomes, and subsequently down-regulates cytokine expression (Domínguez-Acosta et al., 2018). The interaction between AhR and NF-κB may be more complex than we have considered.
Tunnel dust and road dust rarely contained endotoxins compared with PM2.5 collected in Yokohama. Tunnel dust contained PAH levels that were twice as high as those in road dust. When benzo[a]pyrene equivalent values of PM were calculated based on the data obtained by the CALUX assay, which is a DRE-based luciferase assay and thus is considered an index of AhR activation potency, tunnel dust was twice as robust in activating AhR as road dust. IL-33 was slightly but significantly induced by exposure to tunnel dust but not to road dust. Therefore, the PAH contents might contribute to the differences in IL-33 induction between tunnel and road dust.
Among the 3 types of PM2.5 collected in Yokohama, PM2.5 collected in spring 2018 had the lowest IL-33 inducibility. This is probably because endotoxin levels in PM2.5 in spring 2018 the lowest among the 3 PM2.5 collected in Yokohama. PM2.5 in winter 2018 showed high potency to induced IL-33 compared with PM2.5 in spring 2020. While endotoxin amounts in PM2.5 in winter 2018 were almost similar to those in PM2.5 in spring 2020, benzo[k]fluoranthene concentration of PM2.5 in winter 2018 was approximately 3 times higher than that of PM2.5 in spring 2020. Since benzo[k]fluoranthene is reported to strongly activate AhR (Machala et al., 2001), benzo[k]fluoranthene levels in PM2.5 could contribute to the difference in IL-33 inducibility among the 3 types of PM2.5 collected in Yokohama.
PM contains several factors other than PAHs and endotoxin, such as metals. Transition metals, especially iron and copper, induce oxidative stress via the Fenton reaction. We have shown that organotin compounds induce oxidative stress through glutathione S transferase inhibition and subsequent reactive metabolite accumulation. Importantly, oxidative stress activates the transcription factor Nrf-2, which exerts anti-inflammatory effects (Jin et al., 2012), while oxidative stress also activates NF-κB by suppressing the degradation of Ik-B, which binds to NF-κB to prevent the nuclear translocation of NF-κB (Morgan and Liu, 2011). It is very important to further examine the interaction between oxidative stress induced by PM and inflammatory reactions in macrophages.
In conclusion, PAHs and endotoxin in PM are involved in IL-33 induction in macrophages via AhR and NF-κB signaling, respectively. Considering the ability of PM to induce IL-33 in AhR-KO or TLR4-mutated macrophages, endotoxin plays a major role in IL-33 expression, but an interaction between AhR and TLR4 signaling might also occur. Caution regarding PAHs and endotoxin levels in air pollutants should be taken to prevent IL-33-induced allergic inflammation.
ACKNOWLEDGMENTS
The authors thank Ms. Kaede Namba for technical assistance during the course of this study. This work was supported by KAKENHI grants from the Japan Society for the Promotion of Science (grant numbers 20H04341, 17H04714 and 15KK0024 to YI), the Environment Research and Technology Development Funds of the Environmental Restoration and Conservation Agency of Japan (JPMEERF20165051 to TO, and JPMEERF20215003 to HO, and JPMEERF20205007 to YI) and the National Institute of Environmental Health Sciences (R01ES029126 to CFAV).
Footnotes
Conflict of interest---- The authors declare that there is no conflict of interest.
REFERENCES
- Allinne J, Scott G, Lim WK, Birchard D, Erjefält JS, Sandén C, Ben L-H, Agrawal A, Kaur N, Kim JH, Kamat V, Fury W, Huang T, Stahl N, Yancopoulos GD, Murphy AJ, Sleeman MA and Orengo JM (2019): IL-33 blockade affects mediators of persistence and exacerbation in a model of chronic airway inflammation. J. Allergy Clin. Immunol, 144, 1624–1637 e1610. [DOI] [PubMed] [Google Scholar]
- Bock KW (2020): Aryl hydrocarbon receptor (AHR)-mediated inflammation and resolution: non-genomic and genomic signaling. Biochem. Pharmacol, 182, 114220. [DOI] [PubMed] [Google Scholar]
- Boonen I, Van Heyst A, Van Langenhove K, Van Hoeck E, Mertens B, Denison MS, Elskens M and Demaegdt H (2020): Assessing the receptor-mediated activity of PAHs using AhR-, ERα- and PPARγ- CALUX bioassays. Food Chem. Toxicol, 145, 111602. [DOI] [PubMed] [Google Scholar]
- Chanez P, Enander I, Jones I, Godard P and Bousquet J (1996): Interleukin 8 in bronchoalveolar lavage of asthmatic and chronic bronchitis patients. Int. Arch. Allergy Immunol, 111, 83–88. [DOI] [PubMed] [Google Scholar]
- Ciganek M, Neca J, Adamec V, Janosek J and Machala M (2004): A combined chemical and bioassay analysis of traffic-emitted polycyclic aromatic hydrocarbons. Sci. Total Environ, 334–335, 141–148. [DOI] [PubMed] [Google Scholar]
- Dinarello CA (2005): An IL-1 family member requires caspase-1 processing and signals through the ST2 receptor. Immunity, 23, 461–462. [DOI] [PubMed] [Google Scholar]
- Domínguez-Acosta O, Vega L, Estrada-Muñiz E, Rodríguez MS, Gonzalez FJ and Elizondo G (2018): Activation of aryl hydrocarbon receptor regulates the LPS/IFNγ-induced inflammatory response by inducing ubiquitin-proteosomal and lysosomal degradation of RelA/p65. Biochem. Pharmacol, 155, 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominici F, Peng RD, Bell ML, Pham L, McDermott A, Zeger SL and Samet JM (2006): Fine particulate air pollution and hospital admission for cardiovascular and respiratory diseases. JAMA, 295, 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Saeid MH, Al-Turki AM, Nadeem ME, Hassanin AS and Al-Wabel MI (2015): Photolysis degradation of polyaromatic hydrocarbons (PAHs) on surface sandy soil. Environ. Sci. Pollut. Res. Int, 22, 9603–9616. [DOI] [PubMed] [Google Scholar]
- Heinemeyer T, Wingender E, Reuter I, Hermjakob H, Kel AE, Kel OV, Ignatieva EV, Ananko EA, Podkolodnaya OA, Kolpakov FA, Podkolodny NL and Kolchanov NA (1998): Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res, 26, 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraide S, Yanagawa Y and Iizuka K (2018): Tranilast inhibits interleukin-33 production by macrophages. Eur. J. Pharmacol, 818, 235–240. [DOI] [PubMed] [Google Scholar]
- Holgate ST (2008): Pathogenesis of asthma. Clin. Exp. Allergy, 38, 872–897. [DOI] [PubMed] [Google Scholar]
- Ishihara Y, Haarmann-Stemmann T, Kado NY and Vogel CF (2019): Interleukin 33 Expression Induced by Aryl Hydrocarbon Receptor in Macrophages. Toxicol. Sci, 170, 404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara Y, Itoh K, Ishida A and Yamazaki T (2015a): Selective estrogen-receptor modulators suppress microglial activation and neuronal cell death via an estrogen receptor-dependent pathway. J. Steroid Biochem. Mol. Biol, 145, 85–93. [DOI] [PubMed] [Google Scholar]
- Ishihara Y, Itoh K, Tanaka M, Tsuji M, Kawamoto T, Kawato S, Vogel CF and Yamazaki T (2017): Potentiation of 17β-estradiol synthesis in the brain and elongation of seizure latency through dietary supplementation with docosahexaenoic acid. Sci. Rep, 7, 6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara Y, Takemoto T, Itoh K, Ishida A and Yamazaki T (2015b): Dual role of superoxide dismutase 2 induced in activated microglia: oxidative stress tolerance and convergence of inflammatory responses. J. Biol. Chem, 290, 22805–22817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin GH, Park SY, Kim E, Ryu EY, Kim YH, Park G and Lee SJ (2012): Anti-inflammatory activity of Bambusae Caulis in Taeniam through heme oxygenase-1 expression via Nrf-2 and p38 MAPK signaling in macrophages. Environ. Toxicol. Pharmacol, 34, 315–323. [DOI] [PubMed] [Google Scholar]
- Kimura A, Naka T, Nakahama T, Chinen I, Masuda K, Nohara K, Fujii-Kuriyama Y and Kishimoto T (2009): Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J. Exp. Med, 206, 2027–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew FY, Girard JP and Turnquist HR (2016): Interleukin-33 in health and disease. Nat. Rev. Immunol, 16, 676–689. [DOI] [PubMed] [Google Scholar]
- Machala M, Vondrácek J, Bláha L, Ciganek M and Neca JV (2001): Aryl hydrocarbon receptor-mediated activity of mutagenic polycyclic aromatic hydrocarbons determined using in vitro reporter gene assay. Mutat. Res, 497, 49–62. [DOI] [PubMed] [Google Scholar]
- Mimura J, Yamashita K, Nakamura K, Morita M, Takagi TN, Nakao K, Ema M, Sogawa K, Yasuda M, Katsuki M and Fujii-Kuriyama Y (1997): Loss of teratogenic response to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells, 2, 645–654. [DOI] [PubMed] [Google Scholar]
- Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, von Mutius E, Farrall M, Lathrop M and Cookson WO; GABRIEL Consortium. (2010): A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med, 363, 1211–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monn C and Becker S (1999): Cytotoxicity and induction of pro-inflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10–2.5) in outdoor and indoor air. Toxicol. Appl. Pharmacol, 155, 245–252. [DOI] [PubMed] [Google Scholar]
- Moorthy B, Chu C and Carlin DJ (2015): Polycyclic aromatic hydrocarbons: from metabolism to lung cancer. Toxicol. Sci, 145, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MJ and Liu ZG (2011): Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res, 21, 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor GT, Neas L, Vaughn B, Kattan M, Mitchell H, Crain EF, Evans R 3rd, Gruchalla R, Morgan W, Stout J, Adams GK and Lippmann M (2008): Acute respiratory health effects of air pollution on children with asthma in US inner cities. J. Allergy Clin. Immunol, 121, 1133–1139.e1. [DOI] [PubMed] [Google Scholar]
- Ogino K, Nagaoka K, Ito T, Takemoto K, Okuda T, Nakayama SF, Ogino N, Seki Y, Hamada H, Takashiba S and Fujikura Y (2018): Involvement of PM2.5-bound protein and metals in PM2.5-induced allergic airway inflammation in mice. Inhal. Toxicol, 30, 498–508. [DOI] [PubMed] [Google Scholar]
- Okuda T, Isobe R, Nagai Y, Okahisa S, Funato K and Inoue K (2015): Development of a high-volume PM2.5 particle sampler using impactor and cyclone techniques. Aerosol Air Qual. Res, 15, 759–767. [Google Scholar]
- Osgood RS, Upham BL, Bushel PR, Velmurugan K, Xiong KN and Bauer AK (2017): Secondhand Smoke-Prevalent Polycyclic Aromatic Hydrocarbon Binary Mixture-Induced Specific Mitogenic and Pro-inflammatory Cell Signaling Events in Lung Epithelial Cells. Toxicol. Sci, 157, 156–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B and Beutler B (1998): Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science, 282, 2085–2088. [DOI] [PubMed] [Google Scholar]
- Préfontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, Lemière C, Martin JG and Hamid Q (2009): Increased expression of IL-33 in severe asthma: evidence of expression by airway smooth muscle cells. J. Immunol, 183, 5094–5103. [DOI] [PubMed] [Google Scholar]
- Préfontaine D, Nadigel J, Chouiali F, Audusseau S, Semlali A, Chakir J, Martin JG and Hamid Q (2010): Increased IL-33 expression by epithelial cells in bronchial asthma. J. Allergy Clin. Immunol, 125, 752–754. [DOI] [PubMed] [Google Scholar]
- Pun VC, Kazemiparkouhi F, Manjourides J and Suh HH (2017): Long-Term PM2.5 Exposure and Respiratory, Cancer, and Cardiovascular Mortality in Older US Adults. Am. J. Epidemiol, 186, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T, Bauler TJ, Malik-Kale P and Steele-Mortimer O (2018): The phorbol 12-myristate-13-acetate differentiation protocol is critical to the interaction of THP-1 macrophages with Salmonella Typhimurium. PLoS One, 13, e0193601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Fujikawa M, Oguro A, Itoh K, Vogel CF and Ishihara Y (2021): Involvement of the Microglial Aryl Hydro-carbon Receptor in Neuroinflammation and Vasogenic Edema after Ischemic Stroke. Cells, 10, 718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CF, Khan EM, Leung PS, Gershwin ME, Chang WL, Wu D, Haarmann-Stemmann T, Hoffmann A and Denison MS (2014): Cross-talk between aryl hydrocarbon receptor and the inflammatory response: a role for nuclear factor-κB. J. Biol. Chem, 289, 1866–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CF, Sciullo E, Li W, Wong P, Lazennec G and Matsumura F (2007): RelB, a new partner of aryl hydrocarbon receptor-mediated transcription. Mol. Endocrinol, 21, 2941–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel CF, Van Winkle LS, Esser C and Haarmann-Stemmann T (2020): The aryl hydrocarbon receptor as a target of environmental stressors - Implications for pollution mediated stress and inflammatory responses. Redox Biol, 34, 101530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y and Tang M (2019): PM2.5 induces ferroptosis in human endothelial cells through iron overload and redox imbalance. Environ. Pollut, 254 (Pt A), 112937. [DOI] [PubMed] [Google Scholar]