

Abstract
Although physical activity and sedentary behavior are moderately heritable, little is known about the mechanisms that influence these traits. Combining data for up to 703,901 individuals from 51 studies in a multi-ancestry meta-analysis of genome-wide association studies yields 99 loci that associate with self-reported moderate-to-vigorous intensity physical activity during leisure time (MVPA), leisure screen time (LST) and/or sedentary behavior at work. Loci associated with LST are enriched for genes whose expression in skeletal muscle is altered by resistance training. A missense variant in ACTN3 makes the alpha-actinin-3 filaments more flexible, resulting in lower maximal force in isolated type IIA muscle fibers, and possibly protection from exercise-induced muscle damage. Finally, Mendelian randomization analyses show that beneficial effects of lower LST and higher MVPA on several risk factors and diseases are mediated or confounded by body mass index (BMI). Our results provide insights into physical activity mechanisms and its role in disease prevention.
Subject terms: Genome-wide association studies, Genetics research, Translational research
Multi-ancestry meta-analyses of genome-wide association studies for self-reported physical activity during leisure time, leisure screen time, sedentary commuting and sedentary behavior at work identify 99 loci associated with at least one of these traits.
Main
Low levels of physical activity have a major effect on disease burden and it is estimated that more than 5 million deaths per year might be prevented by ensuring adequate levels1. Despite efforts to increase physical activity levels2, an estimated 28% of the world's population is insufficiently active, and the prevalence of physical inactivity in high-income countries rose from 31.6% in 2001 to 36.8% in 2016 (ref. 3). Trends of decreasing physical activity levels over time coincide with increases in the time spent sedentary4, which may pose an independent risk for public health5,6.
Physical activity and sedentary behavior are affected by public policy and social support, as well as by cultural, environmental and individual factors7. Factors like socioeconomic status, built environment and media all influence physical activity at a population level7. In parallel, innate biological factors (for example, age, sex hormones, pre-existing medical conditions, epigenetics and genetics) also explain a moderate proportion of the interindividual variability in physical activity and sedentary behavior. Heritability estimates (h2) range from 31% to 71% in large twin studies8,9. Identifying the genetic factors that influence daily physical activity will improve our understanding of this complex behavior, and may (1) facilitate unbiased causal inference; (2) help identify vulnerable subpopulations; and (3) fuel the design of tailored interventions to effectively promote physical activity. A mechanistic understanding of physical activity at a molecular level may even allow its beneficial effects to be attained through pharmacological intervention10.
Genome-wide association studies (GWAS) have identified thousands of loci associated with cardiometabolic risk factors and diseases11. However, similar efforts for physical activity have been sparse and initially had limited success. This likely reflects the comparatively small sample size of these efforts12, along with heterogeneous assessments of physical activity across studies. More recently, GWAS using data from UK Biobank identified nine loci associated with self-reported moderate and/or vigorous intensity physical activity or sports and exercise participation (n ≈ 377,000 individuals) and eight associated with accelerometry-assessed physical activity and sedentary behavior (n ≈ 91,000)13,14. Hence, on the assumption that physical activity is a highly polygenic trait, many common variants influencing physical activity undoubtedly remain to be identified.
Here, we combine data from up to 703,901 individuals (94.0% European, 2.1% African, 0.8% East Asian, 1.3% South Asian ancestries, and 1.9% Hispanic) from 51 studies in a multi-ancestry meta-analysis of GWAS for MVPA, LST, sedentary commuting and sedentary behavior at work. This yields 104 independent association signals in 99 loci, implicating brain and muscle, among others organs. Follow-up analyses improve our understanding of the molecular basis of leisure time physical activity and sedentary behavior, and their role in disease prevention.
Results
Genome-wide analyses yield 99 associated loci
In our primary meta-analysis of European ancestry men and women combined (Supplementary Tables 1, 2), we identify 91 loci that are associated (P < 5 × 10−9) with at least one of four self-reported traits: MVPA (n up to 606,820), LST (n up to 526,725), sedentary commuting (n up to 159,606) and sedentary behavior at work (n up to 372,605) (Supplementary Table 3, Figs. 1 and 2, and Supplementary Fig. 1). The non-European ancestry meta-analyses do not provide new associations themselves and are only used in multi-ancestry meta-analyses. Multi-ancestry and sex-specific meta-analyses yield eight additional loci, resulting in a total of 104 independent association signals in 99 loci (Supplementary Tables 3 and 4). The vast majority of these—89 independent single nucleotide polymorphisms (SNPs) in 88 loci (35 not previously reported13,15)—are associated with LST, explaining 2.75% of its variance. We also identify 11 loci for MVPA (six not previously reported13,15,16, four that overlap with LST) and four loci for sedentary behavior at work (all previously reported13,15; Supplementary Table 3). No loci are identified for sedentary commuting. To increase statistical power for the discovery of new loci, we perform a multi-trait analysis of GWAS (MTAG) using summary statistics of MVPA and LST. This yields 13 additional loci: eight loci for MVPA and eight for LST, with three loci overlapping (Supplementary Table 5)17.
Fig. 1. Overview of the four self-reported physical activity and sedentary traits and correlations with objectively assessed traits.

a, An overview of the four self-reported physical activity and sedentary traits. b, Phenotypic (upper left) and genetic (lower right) correlation coefficients between the four self-reported physical activity and sedentary traits studied here and three accelerometer-assessed traits quantified in UK Biobank participants. AccMod, accelerometer-assessed proportion of time spent in moderate intensity physical activity; AccSed, accelerometer-assessed proportion of time spent sedentary; AccWalking, accelerometer-assessed proportion of time spent walking; SDC, sedentary commuting behavior; SDW, sedentary behavior at work.
Fig. 2. Main results of GWAS and downstream gene prioritization for LST and MVPA.

a, Circular Manhattan plot summarizing the results from European ancestry meta-analyses for LST and MVPA. Outer track, LST; inner track, MVPA. Genome-wide significant variants (P < 5 × 10−9) are highlighted in orange for loci associated with MVPA and in blue for loci associated with LST. b, Dendrogram showing the 101 independent association signals in LST- and MVPA-associated loci from European ancestry or multi-ancestry meta-analyses. Moving outwards from the center are: (1) chromosome; (2) lead SNP identifiers, in orange for loci associated with MVPA, in blue for loci associated with LST; (3) the most promising gene(s) prioritized in the locus (closest genes are highlighted by filled circles); and (4) the approach(es) by which the gene was prioritized, that is, DEPICT gene prioritization (Dg) or tissue enrichment (Dt); SMR of eQTL signals in blood (Sbl), brain (Sbr) or skeletal muscle (Ssm); credible variants identified by FINEMAP that (i) are coding and likely to have a detrimental effect on protein function (Fcadd) or (ii) show evidence of three-dimensional interactions with the candidate gene in central nervous system cell types (Fcrt); activity-by-contact (ABC) in 26 relevant tissues and cell types; a contribution to enrichment for altered expression in skeletal muscle following a resistance training intervention (RTsm); and/or proximity to an association signal for spontaneous running speed (Ms), time run (Mt) or distance run (Md) in a GWAS of 100 inbred mouse strains.
SNP-heritability estimates range from 8% for MVPA to 16% for LST (Supplementary Table 6 and Methods). Genetic correlations between the four traits range from −0.32 for sedentary behavior at work and sedentary commuting, to −0.49 for LST and MVPA (Fig. 1b). To ensure adequate statistical power in instrumental variable and enrichment analyses, we focus on LST and MVPA from here onwards.
Genetic correlations of self-reported LST and MVPA with objective, accelerometry-assessed daily physical activity traits in UK Biobank range from 0.14 to 0.44 (Fig. 1b). Importantly, five of the eight loci previously identified for objectively assessed daily physical activity in UK Biobank data13,14 show directionally consistent associations (P < 0.05) with self-reported LST and/or MVPA in our study (Supplementary Table 7). By contrast, 39 LST- and 4 MVPA-associated loci observed here show directionally consistent associations (P < 0.05) with at least one objectively assessed physical activity and/or sedentary trait (using accelerometry) in UK Biobank (Supplementary Table 8). In line with this, each additional LST-decreasing and MVPA-increasing allele in unweighted genetic predisposition scores of the 88 LST- and 11 MVPA-associated loci, respectively, are associated with higher objectively assessed daily physical activity levels in UK Biobank (P = 5 × 10−23 for LST; P = 2 × 10−3 for MVPA, Supplementary Table 8).
As external validation, we use the European ancestry summary statistics of LST and MVPA to construct polygenic scores (PGSs), and examine their associations with MVPA in 8,195 BioMe BioBank participants of European (n = 2,765), African (n = 2,224) and Hispanic (n = 3,206) ancestry. In general, a higher PGS for MVPA is associated with higher odds of engaging in more than 30 min per week of MVPA, and a higher PGS for LST with lower odds of engaging in MVPA. Individuals at the highest decile of the PGS for LST are 26% less likely to spend more than 30 min per week on MVPA compared with individuals at deciles 4 to 6 (odds ratio (OR) [95% confidence intervals (CI)] = 0.74 [0.55–0.99]) (Fig. 3 and Supplementary Table 9).
Fig. 3. Validation of associations with MVPA and LST using PGSs in BioMe participants of three ancestries.

a,c, The best performing PGSs for MVPA (a) and LST (c) were derived using logistic/linear regression analyses; that is, those with the highest incremental R2 above and beyond models with only sex, age and the top ten principal components. This was accomplished using inclusion thresholds of P < 0.1101 for MVPA and P < 0.14 for LST. b,d, The association—examined using a logistic regression analysis—of MVPA with the PGSs for MVPA (b) and LST (d) in individuals of African (AA, n = 2,224), European (EA, n = 2,765) and Hispanic (HA, n = 3,206) ancestry in data from the BioMe BioBank. Dots and error bars show OR and 95% CI.
Shared genetic architecture
Using linkage disequilibrium (LD) score regression implemented in the LD-Hub18, we observe significant (P < 4.6 × 10−4) genetic correlations of LST and MVPA with adiposity-related traits (r = −0.41 to −0.20), especially with body fat percentage (rg = 0.4 and −0.3, respectively; Fig. 4, Supplementary Fig. 2 and Supplementary Table 10). In line with moderate genetic correlations, 11 of the 99 self-reported loci for physical activity and sedentary behavior have previously been associated with obesity-related traits19–25. In addition, PGSs for lower LST and higher MVPA are associated with lower BMI in up to 23,723 participants from the BioMe BioBank (Supplementary Table 9), and a phenome-wide association study (PheWAS) in 8,959 BioMe European ancestry samples shows a negative association between the PGS for MVPA and morbid obesity (P = 1.1 × 10−5, Supplementary Fig. 3). Strikingly, genetic correlations with body fat percentage are similar for self-reported LST, MVPA (Fig. 4) and accelerometer-assessed physical activity traits13,14 (Supplementary Fig. 2).
Fig. 4. Genetic correlations of four self-reported physical activity traits with complex traits and diseases.

Results are based on published GWAS with P < 4.6 × 10−4 for at least one physical activity or sedentary trait. Darker colors reflect higher negative (purple) or positive (red) correlation coefficients. GC, genomic control; HDL, high-density lipoprotein; HOMA-B, homeostasis model assessment of beta-cell function; HOMA-IR, homeostatasis model assessment of insulin resistance; PGC, psychiatric genomics consortium.
Besides adiposity, less sedentary behavior and higher physical activity levels are also genetically correlated with a more favorable cardiometabolic status, including lower triglyceride, total cholesterol, fasting glucose and fasting insulin levels, and lower odds of type 2 diabetes and coronary artery disease; as well as with better mental health outcomes, a lower risk of lung cancer and with longevity (Fig. 4 and Supplementary Fig. 2).
Causal inference
To assess directions of causality between sedentary behavior/physical activity and BMI, we next perform two-sample Mendelian randomization (MR) analyses using multiple MR methods that utilize genome-wide full summary results or genome-wide significant loci (Supplementary Table 11 and Methods)26–30. Causal Analysis Using Summary Effect Estimates (CAUSE)26 as well as traditional MR methods consistently show that LST and BMI causally affect each other, with the causal effect (the per 1 s.d. unit increase in each trait) of higher LST on higher BMI being two- to threefold larger than the effect of BMI on LST (Fig. 5a, Table 1 and Supplementary Table 11). Results are similar for bidirectional causal inference tests using body fat percentage instead of BMI (Table 2). However, CAUSE cannot distinguish a model of causality from horizontal pleiotropy for body fat percentage and LST (Table 2). CAUSE also illustrates a causal effect of higher LST on higher recalled adiposity and height in childhood (Table 2), supporting our hypothesis that a genetic predisposition for higher LST later in life represents a lifelong predisposition that already influences adiposity through sedentary behavior early in life. We observe similar evidence for causal effects between MVPA and adiposity, with smaller effects when compared with LST.
Fig. 5. MR analyses between LST, MVPA, BMI and complex diseases.

a, Median causal estimates for MR analyses using the CAUSE method and causal estimates from the MR-PRESSO method after outlier removal and accounting for horizontal pleiotropy. b, The causal effects of LST on complex risk factors and diseases without (in orange) and with (in blue) adjusting for BMI. Dots and error bars show the estimated causal effect sizes and 95% CI. ADHD, attention deficit hyperactivity disorder; T2D, type 2 diabetes.
Table 1.
Bidirectional MR results for LST and MVPA with BMI or body fat percentage using significant loci only
| Exposure | Outcome | Beta | s.e. | P value | Exposure | Outcome | Beta | s.e. | P value |
|---|---|---|---|---|---|---|---|---|---|
| LST | Body fat % | 0.16 | 0.07 | 0.016 | LST | BMI | 0.40 | 0.04 | 8.4 × 10−14 |
| Body fat % | LST | 0.12 | 0.03 | 0.005 | BMI | LST | 0.16 | 0.01 | 1.4 × 10−74 |
| MVPA | Body fat % | −0.21 | 0.17 | 0.22 | MVPA | BMI | −0.25 | 0.04 | 0.002 |
| Body fat % | MVPA | −0.001 | 0.036 | 0.97 | BMI | MVPA | −0.10 | 0.01 | 5.8 × 10−12 |
We use MR-PRESSO with outliers removed for all pairs of traits except for the causal effect estimation between body fat percentage (body fat %) and MVPA because no outliers were detected by MR-PRESSO. For body fat percentage → MVPA, we reported the causal estimates using an inverse variance-weighted test; for MVPA → body fat percentage, we reported the weighted median method because these two methods were selected by the machine learning framework (Methods) to be the most appropriate approaches for each analysis, respectively. P < 0.0125 indicates significant effects.
Table 2.
Bidirectional MR results for LST and MVPA during leisure time with BMI or body fat percentage using genome-wide summary results (CAUSE method)
| Exposure | Outcome | Gammaa | 95% CI | P valueb | Exposure | Outcome | Gammaa | 95% CI | P valueb |
|---|---|---|---|---|---|---|---|---|---|
| LST | Body fat % | 0.18 | 0.13 to 0.24 | 1.8 × 10−3 | LST | BMI | 0.31 | 0.28 to 0.35 | 6.7 × 10−28 |
| Body fat % | LST | 0.12 | 0.04 to 0.18 | 0.14 | BMI | LST | 0.18 | 0.16 to 0.19 | 1.1 × 10−14 |
| MVPA | Body fat % | −0.12 | −0.20 to −0.04 | 0.07 | MVPA | BMI | −0.14 | −0.20 to −0.07 | 6.0 x 10−3 |
| Body fat % | MVPA | −0.03 | −0.09 to 0.02 | 0.53 | BMI | MVPA | −0.09 | −0.11 to −0.06 | 7.4 x 10−3 |
| LST | Comparative height at age 10 | 0.03 | 0.01 to 0.04 | 0.04 | LST | Comparative body size at age 10 | 0.02 | 0.01 to 0.03 | 0.04 |
aPosterior median of gamma, which can be taken as a point estimate of the causal effect. This estimate tends to be shrunk slightly toward zero compared with other methods. bThe P value for comparing the causal model with the sharing model. P < 0.05 indicates that posteriors estimated under the causal model predict the data significantly better than posteriors estimated under the sharing model.
We next investigate the causal effects of LST and MVPA on common diseases and risk factors, with and without adjusting for BMI (Supplementary Tables 12 and 13). In univariate analyses, we observe effects of lower LST on higher high-density lipoprotein cholesterol levels, higher parental age at death, and on lower odds of type 2 diabetes, attention deficit hyperactivity disorder and depression. The CAUSE model only supports evidence for a causal effect of LST on attention deficit hyperactivity disorder and parental age at death. Importantly, multivariable MR analyses show that all protective causal effects of lower LST are either mediated or confounded by BMI.
Directions of causal effects are consistent across LST and MVPA, but only reach significance for MVPA on parental age at death when using the CAUSE model. As for LST, multivariable MR results suggest that the protective causal effects of higher MVPA are either mediated or confounded by BMI, but results should be interpreted with caution for MVPA because of weak instrument bias (conditional F statistics <10)31 (Fig. 5b and Supplementary Table 13).
Gene expression in skeletal muscle following training
Although behavior is mainly influenced by signals from the brain, in the case of physical activity, characteristics of skeletal muscle can play a facilitating or restricting role32. Therefore, we next examine whether genes in LST- and MVPA-associated loci are enriched for altered messenger RNA expression in skeletal muscle following an acute bout of exercise or a period of training or inactivity33 (Methods). A mild enrichment for transcripts with an altered expression in skeletal muscle after resistance training is observed for genes nearest to lead SNPs in LST-associated loci (P = 0.02) (Extended Data Figs. 1 and 2, and Supplementary Table 14). Of the ten genes driving the enrichment, PDE10A may play a critical role in regulating cyclic AMP and cyclic GMP levels in the striatum, a brain region that harbors the central reward system and is important for physical activity regulation34, and in regulating striatum output35; ILF3 and NECTIN2—near APOE—influence the host response to viral infections36,37; EXOC4 plays a role in insulin-stimulated glucose uptake in skeletal muscle38; and IMMP2L influences the transport of proteins across the inner mitochondrial membrane39 (Supplementary Note).
Extended Data Fig. 1. LST-associated loci are enriched for genes with altered expression in skeletal muscle following resistance training.
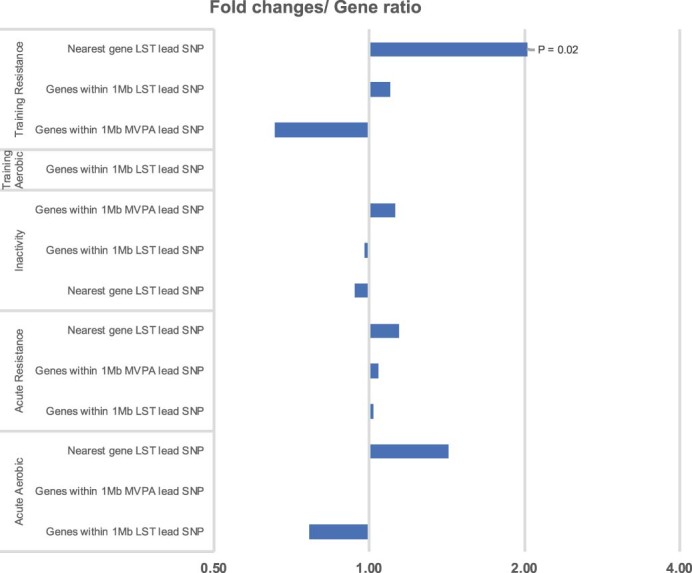
Fold-change plot in log scale for the ratio between: (1) the proportion of genes in physical activity-associated loci that showed an altered expression in skeletal muscle (FDR < 0.01) across five categories: inactivity, acute bout of resistance exercise, acute bout of aerobic exercise, resistance training, or aerobic training; and (2) the proportion of all genes that showed an altered expression following such (in)activity in the MetaMex database (PMID: 31980607). Tested loci were MVPA or LST-associated loci. In a given set of loci, we either considered only the genes nearest to the lead SNP, or all genes within 1 Mb of the lead SNP. Only loci harboring at least five genes with altered gene expression levels after intervention were included in this figure. A one-sided Fisher exact test was used to calculate the P-value for enrichment.
Extended Data Fig. 2. A sensitivity analysis shows the analysis of altered gene expression following resistance training is robust to FDR threshold.

We examined the effect of different FDR thresholds on Fisher’s exact test results for the enrichment analysis of alteration in gene expression in skeletal muscle following resistance training. Red square, genes within 1 Mb of the LST lead SNP; green circle, genes within 1 Mb of the MVPA lead SNP; blue triangle, nearest gene LST lead SNP; purple diamond, nearest gene MVPA lead SNP. The horizonal dotted line indicates nominal significance level (P < 0.05), and the vertical dashed line indicates the FDR threshold that was used. FDR thresholds explored range from 0.001 to 0.5.
Visual information processing and the reward system
To further improve the understanding of the biological factors that influence sedentary behavior and physical activity, we perform a tissue enrichment analysis using DEPICT40. LST- and MVPA-associated loci (P < 1 × 10−5) are most significantly enriched for genes expressed in the retina, visual cortex, occipital lobe and cerebral cortex. This suggests that: (1) possibly subtle differences in the ability to receive, integrate and process visual information influence the likelihood to engage in MVPA; (2) MVPA alters the expression of genes that play a role in visual processes in these tissues; and/or (3) MVPA can slow age-related perceptual and cognitive decline41. The LST-associated loci yield similar tissue enrichment results, with retina having the lowest P value for enrichment. Interestingly, enrichment for genes expressed in retina was also observed in the High Runner mouse model42. Areas related to the reward system (for example, the hippocampus and limbic system) and to memory and navigation (for example, the entorhinal cortex, parahippocampal gyrus, temporal lobe and limbic system) are also enriched in both LST- and MVPA-associated loci (Extended Data Fig. 3 and Supplementary Table 15).
Extended Data Fig. 3. DEPICT-derived tissue enrichment of MVPA and LST.

a, MVPA. b, LST. SNPs with P < 1 x 10−5 for association in the European ancestry GWAS of men and women combined were used as input. The dashed line indicates the FDR corrected significance threshold (FDR < 0.05).
We next use CELLECT43 to identify enriched cell types using single-cell RNA sequencing data from the Tabula Muris and mouse brain projects44. In Tabula Muris data, we observe enrichment in nonmyeloid neurons for MVPA and LST, and of nonmyeloid oligodendrocyte precursor cells for MVPA, possibly highlighting a role for signal transduction (Extended Data Fig. 4 and Supplementary Table 16). In mouse brain data, we identify enrichment for 13 and 45 cell types from 3 and 12 distinct brain regions for MVPA and LST, respectively, including enrichment in dopaminergic neurons (Extended Data Fig. 4 and Supplementary Table 16); a key feature of physical activity regulation in mice45.
Extended Data Fig. 4. Cell type prioritization using CELLECT for MVPA and LST.

a, Prioritization of 115 Tabula Muris cell types across 19 tissues identified two cell types from the brain as significantly associated (stratified linkage disequilibrium score regression) with MVPA (left) and LST (right), namely oligodendrocyte precursor cells and neurons (shown in black; Bonferroni-corrected significance threshold, P < 0.05/115). b, Prioritization of 265 mouse nervous system cell types identified 13 and 45 cell types from 12 distinct brain regions as significantly associated (stratified linkage disequilibrium score regression) with MVPA and LST, respectively (highlighted; Bonferroni-corrected significance threshold, P < 0.05/265.
Candidate gene prioritization
To explore mechanisms by which the identified loci may influence LST and MVPA, we next pinpoint genes in GWAS-identified loci: (1) contributing to tissue enrichment or identified by DEPICT’s gene prioritization algorithm (Supplementary Tables 15 and 17); (2) whose expression in brain, blood and/or skeletal muscle is anticipated to mediate the association between locus and outcome based on Summary-based MR46 (SMR; Supplementary Table 18); (3) harboring credible variants with a high posterior probability of being causal (>0.80)47 and a predicted deleterious effect on protein function (Supplementary Table 19)48; (4) showing chromatin–chromatin interactions with credible variants in central nervous system cell types (such genes may be further from lead SNPs, Supplementary Table 19); (5) that—across 26 tissues and cell types—are activated by contact with enhancers presumably affected by causal variants flagged by GWAS hits49 (Supplementary Tables 20–22); (6) associated with physical activity in GWAS in humans and mice and located <100 kb from the lead variant in humans or mice (Supplementary Note, Supplementary Fig. 4 and Supplementary Tables 23 and 24); and (7) driving enrichment of altered expression in skeletal muscle following resistance exercise training (Supplementary Table 14). Twelve (14%) of the LST-associated loci harbor a variant with a high (>80%) posterior probability of being causal, whereas such variants were not identified among the 11 MVPA-associated loci (Supplementary Table 19). Integrating results across approaches yields 268 candidate genes in 70 LST-associated loci and 39 candidate genes in 8 MVPA-associated loci. Forty-six candidate genes are prioritized by multiple approaches (42 for LST and 6 for MVPA; 2 overlap) and point to endocytosis (CNIH2, RAB1B, KLC2, PACS1, REPS1, DNM3, EXOC4), locomotion (CADM2, KLC2) and myopathy (MLF2, HERC1, KLC2, SIL1) as relevant pathways (Supplementary Tables 25 and 26, and Supplementary Note). Seven clusters of protein–protein interactions are predicted, involving 17 of the 46 genes (Extended Data Fig. 5). In vivo perturbation in model systems is required to confirm or refute a role in sedentary behavior and physical activity.
Extended Data Fig. 5. Protein-protein interactions involving 17 of the 46 candidate genes in GWAS-identified loci prioritized by at least two approaches.
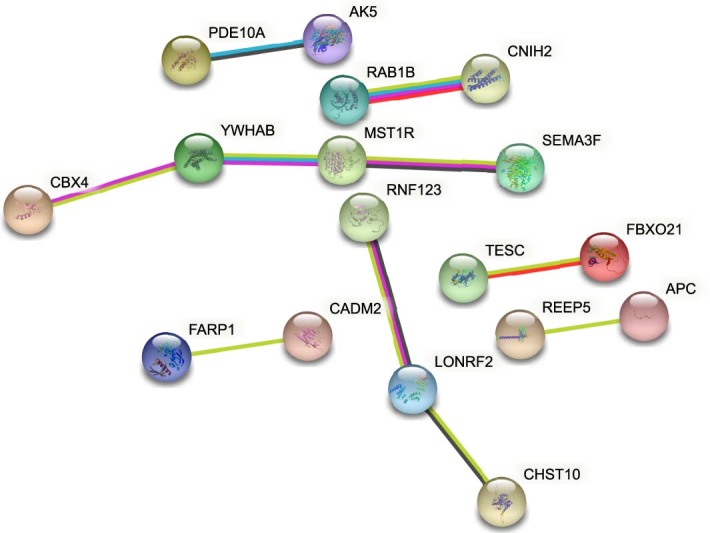
Protein-protein interactions were visualized using String. LONRF2 and CHST10 were prioritized in loci associated with MVPA; the remaining genes were prioritized in loci associated with LST.
Enrichment of previously reported candidate genes
Candidate gene studies in humans have aimed to identify and characterize the role of genes in exercise (physical activity behavior) and fitness (physical activity ability) for decades. We next examine whether variants in genes that have been linked to or associated with exercise and fitness show evidence of associations with self-reported LST and MVPA12,50–54. Of the 58 previously described candidate genes (13 for exercise; 45 for fitness), 56 (13 for exercise and 43 for fitness) harbor variants with P < 0.05 for associations with LST and/or MVPA (Pbinomial = 2.1 × 10−70; Supplementary Fig. 5 and Supplementary Table 27). Associations reach traditional genome-wide significance (P < 5 × 10−8) for variants in three genes: APOE55, PPARD56 and ACTN3 (ref. 57) (Methods).
The SNP in APOE with the lowest P value for association with LST is rs429358, for which the C allele associated with lower LST was previously associated with higher self-reported MVPA13 and forms part of the Ɛ4 risk allele for Alzheimer’s disease (Discussion). The SNP with the lowest P value for association with LST in the locus is rs6857 (D′ = 0.90; r2 = 0.78 with rs429358), in the 3′ untranslated region of NECTIN2. Neither rs429358 (P = 0.16) nor rs6857 (P = 0.18) is associated with MVPA in this study.
The C allele in rs1625595, ~300 kb upstream of ACTN3, is associated with higher MVPA (P = 1.9 × 10−11) as well as with higher ACTN3 expression in skeletal muscle (GTEx, P = 6.6 × 10−5). Alpha-actinin-3 (ACTN3) forms a structural component of the muscle’s Z-disc that is exclusively expressed in type IIA and IIX muscle fibers58. rs1815739, a common ACTN3 variant that introduces a premature stop codon, p.Arg577Ter, also known as p.Arg620Ter, has been extensively studied in the context of exercise performance57. Although we observe little evidence for a role of rs1815739 in leisure time sedentary behavior or physical activity (PLST = 0.017, PMVPA = 0.17), the intronic ACTN3 variants rs679228 (PLST = 4.3 × 10−8) and rs2275998 (PMVPA = 1.8 × 10−7) do show evidence of such associations. Of these, rs2275998—located 646 bp downstream of p.Arg577Ter—is in full LD (r2 = 1.0) with the missense variant rs2229456 (p.Glu635Ala), which likely affects protein function (Combined Annotation Dependent Deletion (CADD) score for the derived, minor, p.635Ala variant =28.6). Each C allele in rs2229456 is associated with less LST (P = 1.4 × 10−4) and higher odds of engaging in MVPA (P = 8.3 × 10−7). Of note, given its downstream location from p.Arg577Ter, a potentially causal effect of rs2229456 on physical activity requires absence of the protein-truncating p.Arg577Ter variant in rs1815739. Haplotype analyses support this (Supplementary Table 28).
Greater ACTN3 flexibility with p.635Ala
Given the striking finding that MVPA and LST are associated with the ACTN3 missense variant rs2229456, but not with the ACTN3-truncating variant rs1815739, we next examine whether rs2229456 (p.Glu635Ala variant) has functional consequences for ACTN3’s mechanistic properties at the molecular level. We add ACTN2 to this comparison because it likely compensates for the loss of ACTN3 in the presence of the truncating p.Arg577Ter variant59. The results of computer-based (steered) molecular dynamics (MD) simulations and umbrella sampling (see Methods and Supplementary Note for more details) show that the ancestral p.Glu635 variant facilitates salt-bridge and hydrogen-bonding interactions at residue 635 with surrounding residues (for example, R638 and Q639; Fig. 6a,b and Supplementary Fig. 6) via its glutamate side chain. Such interactions are not formed in the presence of the ACTN3 p.635Ala product. They are also less likely to be formed in ACTN2, because of a kink that is present at exactly this location in ACTN2 (Fig. 6c and Supplementary Fig. 6). Moreover, p.635Ala and ACTN2 show distinctly different behavior from p.Glu635, with a greater magnitude of root mean squared fluctuations (r.m.s.f.) in the middle section of the spectrin repeats under no-load conditions (Fig. 6d), suggesting a more flexible structural region. When placed under simulated compressive loads that are likely experienced in vivo, p.635Ala shows a more linear force versus distance relationship, with greater variance in the potential of mean force (Fig. 6e and Supplementary Fig. 6). Taken together, these results indicate that the ACTN3 p.635Ala dimer—associated with higher MVPA—exhibits similar flexibility to ACTN2 and greater flexibility than the p.Glu635 dimer.
Fig. 6. Allele p.635Ala in ACTN3 results in a more flexible ACTN3 homodimer.

a, ACTN3 is a homodimer of two antiparallel filaments, with each filament consisting of an N-terminal actin binding domain (ABD, blue), followed by a structural region comprised of four spectrin repeats (gray) with a C-terminal calmodulin (CAM) homology domain (cyan). b, The glutamate residue side chain in position 635 of ACTN3 (p.Glu635) interacts primarily with the arginine in position 638 and the glutamine in position 639. c, The α-helix comprised of residues adjacent to ACTN3 residue 635 (ACTN2 628) exhibits a pronounced kink in ACTN2 (green) at this α-helical turn compared with ACTN3 p.Glu635 (blue) and p.635Ala (orange), decreasing the likelihood of interactions under load with R631, whereas the alanine substitution of ACTN3 p.635Ala precludes any side chain interactions with neighboring residues p.Arg638 or p.Glu639. d, The r.m.s.f. of the spectrin repeat structural region of the ACTN3 dimer for a 150 ns MD simulation for variants p.Glu635 (blue) and p.635Ala (orange, higher MVPA) and ACTN2 (green) (bottom), with the difference in r.m.s.f. between ACTN3 variants shown mapped to the spectrin repeat region (top) with ±0.3 nm difference (red, positive and blue, negative). e, Umbrella sampling of ACTN3 variants p.Glu635 and p.635Ala and ACTN2 with orange, blue and green traces representing the potential of mean force for ACTN3 variants p.635Ala (orange) and p.Glu635 (blue) and ACTN2 (green) ±1 s.d. The reaction coordinate is the distance between the two ABD centers of mass of each dimer, a negative value indicating a shorter distance between the two ABDs. Inset shows the relaxed dimer at reaction coordinate of 0 nm (top) and the direction and effect on the compressive force. f, Single fiber experiments show a higher maximal force and fiber power during isotonic contractions after an eccentric exercise bout in type IIA fibers from an individual homozygous for p.Arg577 and p.Glu635 (blue) compared with type IIA fibers from three p.Arg577 homozygous, p.Glu635Ala heterozygous individuals (orange); and from four p.577Ter homozygous individuals (green).
Maximal force and fiber power lower with ACTN3 p.635Ala
We next examine whether a higher predicted ACTN3 dimer flexibility in the presence of p.635Ala has functional consequences in isolated human skeletal muscle fibers. To this end, we compare functional readouts in 298 isolated type I and IIA fibers from vastus lateralis biopsies obtained from eight healthy, young, untrained male participants before and after an eccentric exercise bout60,61. Results from a 15,000 iteration Markov chain Monte Carlo model show that stable maximal force—with fibers submerged in activating solution—and fiber power during isotonic load clamps are similar in 32 ± 7 fibers (mean ± s.d.) from three p.Arg577 homozygous, p.Glu635Ala heterozygous individuals compared with 39 ± 6 fibers from four individuals homozygous for the p.577Ter variant; and lower in both groups when compared with 46 fibers from an individual that is homozygous for both the p.Arg577 and p.Glu635 variants (Fig. 6f and Methods). Associations are most striking after an eccentric exercise intervention and are, as expected, more pronounced in type IIA than in type I fibers (Supplementary Fig. 7). Taken together, these results suggest that a more flexible ACTN dimer with lower peak performance (ACTN3 p.635Ala or ACTN2) may be less susceptible to exercise-induced muscle damage than the ancestral ACTN3 p.Glu635, thereby facilitating a more active lifestyle.
Discussion
By doubling the sample size compared with earlier GWAS, we identify 104 independent association signals in 99 loci, including 42 newly identified loci, for self-reported traits reflecting MVPA and sedentary behavior during leisure time. Around half of these also show evidence of directionally consistent associations with objectively assessed physical activity traits. Genetic correlations and two-sample MR analyses show that lower LST results in lower adiposity. Protective causal effects of higher MVPA and lower LST—acting through or confounded by BMI—are observed for longevity. Tissue and cell-type enrichment analyses suggest a role for visual information processing and the reward system in MVPA and LST, including enrichment for dopaminergic neurons. Loci associated with LST are enriched for genes whose expression in skeletal muscle is altered by resistance training. Forty-six candidate genes are prioritized by more than one approach and point to pathways related to endocytosis, locomotion and myopathy. Finally, results from MD simulations, umbrella sampling and single fiber experiments suggest that a missense variant (rs2229456 encoding ACTN3 p.Glu635Ala) likely increases MVPA, at least in part by reducing susceptibility to exercise-induced muscle damage.
Recent MR studies reported causal protective effects of self-reported and objectively assessed physical activity on breast and colorectal cancer62,63. One study concluded that a 1 s.d. increase in self-reported MVPA was associated with lower odds of colorectal cancer (OR = 0.56), with BMI only mediating 2% of the protective effect63. Our results—on lung cancer rather than colorectal cancer—show that instrumental variables of MVPA in multivariable MR are weak, and results should be interpreted with caution. Furthermore, a causal effect of objectively assessed, but not self-reported physical activity (MVPA) on depression has been reported64. Our MR results for LST on depression show that although the physical activity trait matters, the self-reported nature of it seems inconsequential. According to an earlier study, TV viewing has an attenuated effect but still causes coronary artery disease when adjusting for BMI15. The discrepancy with our results—suggesting mediation or confounding by BMI—highlights the importance of including physical activity, as well as BMI-associated variants in multivariable MR analysis, to prevent loss of precision and potentially even biased estimates31.
It is of interest that a proxy of rs429358, part of the established APOE Ɛ4 risk allele for Alzheimer’s disease, is associated with lower LST. Klimentidis et al. previously showed that the association of rs429358 with MVPA was stronger in those reporting a family history of Alzheimer’s disease, and among older individuals13. Based on the direction of the association, it was hypothesized that individuals at higher risk of developing Alzheimer’s disease may adopt a healthy lifestyle to mitigate their risk, especially later in life13. However, our MR analyses show no evidence of a causal role of MVPA or LST in Alzheimer’s disease, and lower average physical activity levels in individuals with a first-degree family history of Alzheimer’s disease or dementia13 suggest other explanations are more likely, although a role for survival bias cannot be ruled out13. For example, APOE Ɛ4 carriers have a greater increase in aerobic capacity following exercise training65, which may reinforce a physically active lifestyle independently of Alzheimer’s risk. Furthermore, several studies have investigated the moderating role of the APOE Ɛ4 allele in the relationship between physical activity and Alzheimer prevention66. Although more studies are needed to resolve inconsistencies in the literature, Ɛ4 carriers seem to benefit more from physical activity in terms of reducing the risk of dementia and brain pathology66.
To investigate the molecular basis for the association of ACTN3 with MVPA, we compare the ACTN3 p.Glu635 and p.635Ala variants (rs2229456) with each other and with ACTN2—as a functional proxy for ACTN3 p.577Ter—using MD simulations and single fiber experiments. Previous studies using normal mode analysis of alpha-actinin show that several of the natural frequencies have bending flexibility near residue 635. This is interesting because ACTN3’s residue 635—the 356th residue of the spectrin repeat region (Fig. 6)—lies outside the linkers between the α-helices of the spectrin repeats, where most flexibility is expected and observed67. The absence of salt-bridge and hydrogen-bonding interactions between position 635 (628 in ACTN2) and surrounding residues—due to either the presence of the alanine substitution at ACTN3’s residue 635, or a kink in the α-helix at ACTN2’s residue 628—increases the flexibility of the dimer under a compressive load, with far less work required to deform the homodimer beyond a compressive distance of 1.2 nm. The p.635Ala substitution may reduce the stiffness of the muscle fiber while undergoing elastic deformation during exercise to a level that is comparable with ACTN2. Although at the expense of the maximal force that single fibers can generate, this may reduce exercise-induced microtrauma caused by Z-disc rupture or streaming1, alleviating delayed onset muscle soreness2 and risk of injuries3, enabling a more active lifestyle. Our results suggest it would be interesting to revisit the plethora of data on p.Arg577Ter, and differentiate between effects of the p.Arg577Ter and p.Glu635Ala variants.
In conclusion, our results shed light on genetic variants and molecular mechanisms that influence physical activity and sedentary behavior in daily life. As would be expected for complex behaviors that involve both motivation and physical ability, these mechanisms occur in multiple organs and organ systems. In addition, our causal inference supports the important public health message that a physically active lifestyle mitigates the risk of multiple diseases, in major part through or confounded by an effect on BMI.
Methods
Each study (Supplementary Table 2) obtained informed consent from participants and approval from the appropriate institutional review boards or committees.
Samples and study design
We conducted a large meta-analysis for physical activity traits, including results from up to 703,901 individuals (including nearly half-a-million from the UK Biobank) to identify genetic loci associated with physical activity and sedentary behavior across different ancestries. We first examined genome-wide, ancestry- and sex-stratified associations in 51 studies with questionnaire-based data on: (1) MVPA; (2) LST; (3) sedentary commuting behavior; and/or (4) sedentary behavior at work, using study-specific, tailored analysis plans (Supplementary Table 2, see Supplementary Note for rationale). Next, we performed ancestry-specific, inverse variance-weighted fixed-effects meta-analyses of summary statistics for each of the four self-reported traits (Fig. 1a), including data from up to 703,901 individuals consisting of European (94.0%), African (2.1%), East Asian (0.8%) and South Asian (1.3%) ancestries; as well as Hispanics (1.9%) (Supplementary Table 1). Our primary meta-analyses were restricted to 661,399 European ancestry participants. Secondary meta-analyses were also conducted for: (1) all ancestries (European + other ancestries), (2) European ancestry men, (3) European ancestry women, and (4) each non-European ancestry separately. Details of participating studies are described in Supplementary Tables 1 and 2. Although modest genomic inflation68 was observed (lambda 1.2–1.4) (Supplementary Fig. 1), LD score regression analyses indicated this reflects true polygenic architecture rather than cryptic population structure69.
Self-reported physical activity and sedentary behavior traits
The self-reported outcomes in this study are domain- and intensity-specific physical activity and sedentary traits that, unlike accelerometry-based outcomes, are subject to misclassification and bias by recall and awareness of the beneficial effects of physical activity, among others. Furthermore, different studies used different questionnaires to capture physical activity, and so we defined cohort-specific traits that make optimal use of the available data, while striving for consistency across studies (Supplementary Table 2). As a result, and based on the zero-inflated negative binomial nature of the distribution of MVPA in most studies, we had to analyze MVPA as a dichotomous outcome, which had a negative impact on statistical power. Descriptive information of these four outcomes is reported by study in Supplementary Table 1.
Genotyping, imputation and quality control
Detailed information about the genotyping platform used, and quality control measures applied within each study are presented in Supplementary Table 2. Quality control following study level analyses was conducted using standard procedures70.
GWAS and meta-analyses
GWAS were performed within each study in a sex- and ancestry-specific manner. Additive genetic models accounting for family relatedness (where appropriate) were adjusted for age, age-squared, principal components reflecting population structure and additional study-specific covariates as presented in Supplementary Table 2. Analyses were limited to genotyped and imputed variants with minor allele frequency >0.1% in UK Biobank, and minor allele count >3 in other studies. Study-, sex- and ancestry-specific GWAS results were meta-analyzed using the fixed-effects, inverse variance-weighted method implemented in METAL71, for 19.1 to 22.5 million SNPs per trait. Because we did not include a replication stage and given the high SNP density, we applied a stricter than usual Bonferroni correction and considered associations with P < 5 × 10−9 statistically significant72.
To identify genome-wide significant loci, we defined a distance criterion of ±1 Mb surrounding each genome-wide significant peak (P < 5 × 10−9). We extracted previously reported genome-wide significant associations within 1 Mb of any index variants we identified from the NHGRI-EBI GWAS Catalog11 and PhenoScanner V2 (ref. 73). A locus is considered previously reported if any variant we extracted at that locus was in LD (r2 > 0.1) with a lead variant that has been associated with objectively assessed or self-reported physical activity and sedentary traits previously. To identify physical activity- and sedentary behavior-associated loci that were previously associated with obesity-related traits, we performed a look up for each lead variant (and their proxies with LD r2 > 0.2) in the GWAS catalog and PhenoScanner V2.
SNP-based heritability estimation
To estimate the heritability explained by genotyped SNPs for each physical activity and sedentary trait, we used BOLT-REML variance components analysis74, a Monte Carlo average information restricted maximum likelihood algorithm implemented in the BOLT-LMM v.2.3.3 software. As in most GWAS for complex traits, the SNP heritability (up to 16%) was lower than the heritability estimates from twin studies (31%–71%)8,9, likely at least in part due to the absence of rare variants in GWAS75.
Although we performed a multi-ancestry meta-analysis, data from relatively few individuals of non-European ancestries were available to us, and our functional follow-up analyses were conducted based on the European ancestry results. Studies with data from more individuals of non-European ancestry will no doubt further increase the understanding of physical activity etiology.
Joint and conditional analyses
To identify additional independent signals in associated loci, we performed approximate joint and conditional SNP association analyses in each locus, using GCTA76. Any lead SNPs identified in known long-range high-LD regions77 were treated as a single large locus in the GCTA analysis. We used unrelated European ancestry participants from the UK Biobank as the reference sample to acquire conditional P values for association.
MTAG
MTAG results were calculated using the European ancestry meta-analysis results of LST and MVPA, using standard settings17. Because MTAG's estimates are biased away from zero when SNPs are null for one trait but non-null for other traits, we applied it to only the two outcomes that were most strongly genetically correlated: MVPA and LST (absolute value of genetic correlation 0.49).
PheWAS with physical activity PGSs
To assess the out-of-sample predictive power of the variants associated with self-reported sedentary behavior and physical activity, we constructed two PGSs—for LST and for MVPA—in up to 23,723 Mount Sinai BioMe BioBank participants, using summary statistics of the primary European ancestry meta-analyses and PRSice software78. We subsequently assessed the association of MVPA and BMI with the PGSs in individuals of European and African ancestry, as well as in Hispanic participants, within the BioMe BioBank. Among the 2,765 European ancestry individuals with physical activity measurements and genotypes, the PGSs were calculated on common variants (minor allele frequency >1%) using P value thresholds from 5 × 10−8 to 1 (all variants) in the LST and MVPA GWAS, and clumping parameters of r2 < 0.5 over a 250-kb window. Logistic regression models were used to examine the associations between MVPA (defined as at least 30 min per week of MVPA yes/no in BioMe) and the PGSs in European ancestry participants of BioMe. In each analysis, we estimated the variance in MVPA explained by the PGS, adjusting for age, sex and the top ten principal components for population structure. For both LST and MVPA, the P value threshold resulting in the best performing PGS was defined based on the highest R2 increase upon adding the PGS to the regression model. To examine the generalizability of the two PGSs, we next examined their associations with MVPA in 3,206 Hispanic individuals and 2,224 African ancestry participants of BioMe. We then tested each PGS for classification performance and examined whether the generated PGS was associated with any other trait by performing a PheWAS. Briefly, International Classification of Diseases 9 and 10 codes from electronic health records were mapped to phecodes using the PheWAS package79. Among 8,959 BioMe European ancestry participants, the 1,039 disease outcomes with at least ten cases were analyzed. We used logistic regression to separately model each phecode as a function of the two PGSs, adjusting for age, age-squared, sex and the top ten principal components. Interpretation of results was restricted to outcomes with more than ten cases. Multiple testing thresholds for statistical significance were set to P < 4.8 × 10−5 (0.05/1,039).
Genetic correlations
To explore a possibly shared genetic architecture, we next estimated genetic correlations of the four self-reported traits examined in this study and five accelerometry-assessed physical activity traits assessed in UK Biobank14 with relevant complex traits and diseases based on established associations at the trait level using LD score regression implemented in the LD-Hub web resource18. To define significance, we applied a Bonferroni correction for the 108 selected phenotypes available on LD-Hub (P < 4.6 × 10−4). Supplementary Table 10 shows the complete set of pairwise genetic correlations of the four self-reported physical activity traits with relevant complex traits and diseases. Next, we prioritized traits and diseases showing evidence of genetic overlap (associated with at least one of the physical activity traits). These can be divided into six categories: lifestyle traits, anthropometric traits, psychiatric diseases, other diseases (cardiometabolic diseases and cancer), biomarkers and others (Fig. 4). Using objectively assessed physical activity traits (accelerometry) instead of self-reported traits yielded similar results (Supplementary Fig. 2).
Two-sample MR
We performed MR analyses to disentangle the causality between LST and MVPA, on the one hand, and BMI, on the other hand. We further investigated the causal effects of LST and MVPA on common diseases and risk factors, while considering BMI through multivariable MR. For multivariable MR, we used BMI (exposure 2) summary statistics based on UK Biobank data, and summary statistics for disease outcomes and other relevant traits based on data from the largest publicly available GWAS without data from UK Biobank participants on the MR-Base platform and OpenGWAS database80,81. This way, we aimed to minimize bias due to sample overlap in the two-sample MR analysis82. The source of each of the instruments is presented in Supplementary Table 12. Genetic instrumental variables for each of the traits and diseases consisted of genome-wide significant (P < 5 × 10−8) index SNPs. Index SNPs were LD clumped (r2 > 0.001 within a 10-Mb window) to remove any correlated variants. In the multivariable MR that evaluates the independent effects of each risk factor, the genetic instrumental variables from two risk factors were combined. For both LST and MVPA, independent loci associated with physical activity or BMI were used as instrumental variables.
We followed several steps to evaluate potential causality. Because MR results can be severely biased if instrumental SNPs show horizontal pleiotropy and violate the instrumental variable assumptions28, we prioritized methods that are robust to horizontal pleiotropy when calculating causal estimates. We did not use the MR-Egger intercept test to identify the presence of potential pleiotropy, because the MR-Egger intercept parameter estimate is positively biased when the NO Measurement Error assumption is violated, as indicated by lower values of I2GX in our two-sample MR setting83. Instead, we applied MR-PRESSO (pleiotropy residual sum and outlier)27, which removes pleiotropy by identifying and discarding influential outlier predictors from the standard inverse variance-weighted test28. For analyses with evidence of no distortion due to pleiotropy (MR-PRESSO Global test P > 0.05), we considered other robust methods, for instance fixed- and random-effect inverse variance-weighted, weighted- or simple- median and mode methods. We also conducted Steiger filtering to remove variants likely influenced by reverse causation and used Cook’s distance filtering to remove outlying heterogeneous variants as deemed necessary. To select the most appropriate approach, we implemented a machine learning framework30. Finally, we performed a leave-one-out analysis to identify potential outliers among the variants included in the instrumental variables tested. We set the multiple testing significance threshold for MR analyses with disease outcomes at 1.9 × 10−3, that is, Bonferroni correction for 13 disease outcomes and 2 types of risk factors: physical activity or sedentary behavior and adiposity (0.05/(13 × 2)).
We also applied the recently published Bayesian-based MR method CAUSE, which accounts for both correlated and uncorrelated pleiotropy26, in evaluating bidirectional causal effects between physical activity and adiposity. Compared with the other two-sample MR methods, CAUSE calculates the posterior probabilities of the causal effect and the shared effect, and tests whether the causal model fits the data better than the sharing model. That is, it examines whether the association between the traits is more likely to be explained by causality than horizontal pleiotropy. In addition, CAUSE improves the power of MR analysis by using full genome-wide summary results (LD pruned at r2 < 0.1 with P < 1 × 10−3, as recommended by the CAUSE authors). In addition, we took advantage of the robustness of the CAUSE method—which allows overlapping GWAS samples—to test the assumption that a genetic predisposition for LST assessed later in life reflects a lifetime liability. Using the summary statistics of SNPs for childhood adiposity (comparative body size at age 10) and height (comparative height at age 10) in UK Biobank84, we examined bidirectional causal effects between LST and these two recalled childhood traits.
Enrichment for genes with altered expression in skeletal muscle after an intervention
A high degree of physical fitness and a strong adaptive response to exercise interventions facilitate a physically active lifestyle. To identify plausible candidate genes in GWAS-identified loci, we examined enrichment for transcripts whose expression in skeletal muscle was changed after an acute bout of aerobic exercise, aerobic training, an acute bout of resistance exercise, resistance training and inactivity33. We excluded individuals with pre-existing conditions such as chronic kidney disease, chronic obstructive pulmonary disease, frailty, metabolic syndromes and obesity. We also excluded athletes because in this subgroup, transcripts with differential expression in response to (in)activity interventions are likely not representative for the general population85. Enrichment was examined for genes nearest to, or within 1 Mb of lead variants for LST- and MVPA-associated loci. We used false discovery rate <0.01 as the threshold for altered expression after intervention. A sensitivity analysis with a series of different false discovery rate cut-offs (0.001 to 0.5) showed that results were robust.
Gene, tissue and cell-type prioritization
We used DEPICT40 to identify enriched gene sets and tissues, as well as to prioritize candidate genes in the identified loci, using variants with P < 1 × 10−5 in the primary meta-analysis of European ancestry men and women combined as input. We also used CELLECT43 to identify enriched cell types for physical activity, by combining MVPA and LST GWAS summary statistics with single-cell RNA sequencing data. We sought to further refine the set of prioritized candidate genes using SMR and HEIDI tests46. Briefly, this approach integrates summary-level data from GWAS and expression quantitative trait loci (eQTL) studies to test whether a transcript and phenotype are likely associated because of a shared causal variant (pleiotropy). We considered genes candidates if they had a Bonferroni-corrected PSMR < 1.02 × 10−5 and showed no evidence of heterogeneity (PHEIDI > 0.05), as in earlier studies46. Based on tissue enrichment results from DEPICT, the SMR analyses were performed using brain eQTL information obtained from GTEx-brain (n = 72)86,87, CommonMind Consortium (n = 467)88, ROSMAP (n = 494)89, and Brain-eMeta (n = 1,194)87; blood eQTL summary information obtained from the eQTLGen Consortium90, which is based on peripheral blood samples from 31,684 individuals; and skeletal muscle eQTL information from the GTEx project (n = 803)91.
To identify variants in GWAS-identified loci with a high posterior probability of being causal, we used LST and MVPA summary statistics as input for FINEMAP47. We used default parameters and selected a maximum of ten putative causal variants per locus. The output variants identified as credible were mapped to genes using tissue-specific HiC chromatin conformation capture data92. We integrated all HiC data in the brain (dorsolateral prefrontal cortex, hippocampus, neural progenitor cell, and adult and fetal cortex) available on FUMA v.1.3.5, using the same approach. Genes in GWAS-identified loci containing FINEMAP-identified credible coding variants with a CADD score >12.33 were also prioritized. Finally, we used data from 26 of the 131 available tissues and cell types deemed relevant for sedentary behavior and physical activity (Supplementary Table 20) to identify genes that are contacted by enhancers affected by causal variants flagged by GWAS lead SNPs, using the recently described activity-by-contact model49.
Enrichment for previously reported candidate genes
We next conducted a literature review of previously reported genes with evidence of a role in exercise (physical activity behavior) and fitness (physical activity ability) and identified 58 such candidate genes (13 for exercise; 45 for fitness)12,50–53. For each gene, we identified all variants within the gene, examined their associations with LST and MVPA in our meta-analysis of European ancestry individuals and, for each gene–trait combination, retained the summary statistics for the variant with the lowest P value for association. Variants in three genes reached the traditional threshold for genome-wide significance (PPARD, APOE and ACTN3). Based on LD and predicted effects on protein function, rs2229456 in ACTN3 (encoding p.Glu635Ala) may have a causal effect.
MD simulation for p.Glu635Ala
Because no structure for human ACTN3 has yet been experimentally determined, we constructed a homology model of the p.Glu635 variant monomeric filament using the fully annotated protein (UniProt ID Q08043) using Phyre2 (ref. 93), with the p.635Ala variant mutated in silico. Residue 635 of ACTN3 resides in the 356th residue of the spectrin repeat region and corresponds with residue 628 in ACTN2 (see the Supplementary Methods for more information). For each variant, the spectrin repeats of the ACTN3 monomer were aligned with the crystal structure of the rod domain of alpha-actinin (PDB ID 1HCI), to give the dimeric form of ACTN3. MD system preparation and simulation was conducted with GROMACS 2020.1 (ref. 94) and using mdanalysis v.2.0. The MD topology was created with GROMACS pdb2gmx using the ACTN2 and ACTN3 dimer models and parameterized with the CHARMM36 all-atom force field95. The ACTN2 and ACTN3 dimers were placed in a rectangular simulation box with a 1.0-nm buffer between the protein and the box extent, with periodic boundary conditions in all three spatial axes. The system was solvated with TIP3P water molecules and using GROMACS genion, random solvent molecules were replaced with K+ and Cl− to a concentration of 150 mM with additional K+ ions added to provide an electrostatically neutral system. Energy minimization was accomplished using the steepest descent algorithm. To equilibrate the system, two 100-ps simulations were conducted using a constant temperature ensemble (NVT, that is, a constant number of particles [N], volume [V] and temperature [T]) at 310 K via a Berendsen thermostat, followed by a constant pressure ensemble (NPT, that is, a constant number of particles [N], pressure [P] and temperature [T]) at 1 bar with a Parinello–Rahman barostat. MD simulation parameters were set in accordance with the recommendations for the CHARMM36 force field in GROMACS. A short production run of 1 ns without position restraints was followed by a full simulation of 150 ns with weak position restraints on the ABD of chain B to prevent self-interaction across the periodic boundaries.
Steered MD and umbrella sampling for p.Glu635Ala
We next compared the properties of ACTN2 and of ACTN3 p.635Ala and p.Glu635 when placed under the simulated compressive loads that are likely experienced in vivo. The final frame of the 1-ns MD production run was used as the starting topology for steered MD simulations using fully relaxed dimers. Steered MD simulations were run for 2 ns with a pulling rate of 0.005 nm ps−1 and a harmonic potential of 50 kJ mol−1 nm−2. Center-of-mass pull groups were defined as the ABD of each respective monomer, with a weak position restraint placed on the Cα atom of threonine 52 (ACTN3) or threonine 45 (ACTN2)—a centrally located residue in the core of the ABD—on one ABD, enabling full rotational freedom of each ABD during the course of the steered MD simulations. The pulling vector was oriented along the axis on which the spectrin repeats were initially aligned. Suitable frames from each steered MD simulation were selected that differed by no more than 0.2 nm from 0 to −5.5 nm (a contraction of the dimer by 5.5 nm or ~18%) and were used as the starting topology for a series of 10-ns umbrella sampling simulations. Analysis of the umbrella sampling simulations was conducted using g_wham, to yield the potential of mean force versus reaction coordinate for each variant.
Single skeletal muscle fiber functional characteristics in relation to p.Glu635Ala
Single muscle fibers from eight nonathletic young men in which contractile and morphological properties were previously characterized in vastus lateralis biopsies obtained before and after an eccentric exercise bout60,61 were genotyped for rs2229456. A hierarchical linear mixed effects model was constructed for each fiber type and time point using rstanarm96 to test the genotype fixed effect, with muscle fibers nested within each of the eight individuals as random factors for each contractile and morphological variable. Genotypes at p.Arg577Ter and p.Glu635Ala were clustered into three groups: RR-AA (n = 1 individual, 46 fibers, reference group); RR-AC (n = 3 individuals, 32 ± 5 fibers); and XX-AA (n = 4 individuals, 39 ± 6 fibers). Using weakly informative priors, the posterior distribution was estimated with Markov chain Monte Carlo sampling (20,000 samples total with 5,000 sample burn-in). We calculated 90% credible intervals of the posterior density and distribution-free overlapping indices97 to compare single fiber properties between genotypes.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41588-022-01165-1.
Supplementary information
Supplementary Results, Discussion, Methods, Figs. 1–7, Box 1, study-specific acknowledgments and references.
Supplementary Tables 1–29.
Acknowledgements
T.S.A. is supported by the Steno Diabetes Center Copenhagen, Copenhagen, Denmark and the Novo Nordisk Foundation Grant NNF18OC0052457. J.W.C. was supported by grants from the National Institutes of Health (NIH) (R01-NS100178; R01-NS105150), the US Department of Veterans Affairs and the American Heart Association (AHA) (15GPSPG23770000; 17IBDG33700328). B.F. was supported by the Oak Foundation. T.O.K. was supported by the Novo Nordisk Foundation (grant numbers NNF17OC0026848 and NNF18CC0034900). N.G.M. is funded by a National Health and Medical Research Council (NHMRC) Investigator Grant (APP1172990). S.E.M. is funded by NHMRC Investigator Grant (APP1172917). D.M. is supported by a Canada Research Chair in Genetics of Obesity. R.C.R. is a de Pass Vice Chancellor's Research Fellow at the University of Bristol. S.R.-d.-P was supported by the Heart and Stroke Foundation of Ontario (grant number NA 7293). N.J.S. holds a National Institute for Health and Care Research (NIHR) Senior Investigator award. N.J.T. is a Wellcome Trust (WT) Investigator (202802/Z/16/Z), is the principal investigator of the Avon Longitudinal Study of Parents and Children (Medical Research Council (MRC) & WT 217065/Z/19/Z), is supported by the University of Bristol NIHR Biomedical Research Centre (BRC-1215-2001), the MRC Integrative Epidemiology Unit (MC_UU_00011) and works within the Cancer Research UK Integrative Cancer Epidemiology Programme (C18281/A19169). X.Z. is supported by China Scholarship Council 201406220101. T.P. is supported by the Bio-Synergy Research Project (2013M3A9C4078158) of the Ministry of Science, ICT and Future Planning through the National Research Foundation of Korea. S.S. is supported by the Swedish Research Council (grant numbers 2016-06264, 2018-05946 and 2018-05498). J.W.C. was partially supported by an AHA-Bayer Discovery Grant (grant 17IBDG33700328), the AHA Cardiovascular Genome–Phenome Study (grant-15GPSPG23770000), NIH (grants R01-NS114045, R01-NS100178, R01-NS105150), and the US Department of Veterans Affairs. H.X. was supported by AHA grant 19CDA34760258 and NIH grants R01-NS114045, R01-NS100178 and R01-NS105150. K.E.N. is funded by AHA grants 13GRNT16490017 and 15GRNT25880008, and by NIH grants R01DK089256, R01DK101855, R01HD057194, R01DK122503, 01HG010297, R01HL142302, R01HL143885 and R01HG009974. L.F.-R. is supported by an AHA grant (13PRE16100015). C.P.N. is funded by the British Heart Foundation (SP/16/4/32697). L.M.H., C.P.N., P.S.B. and N.J.S. are supported by the NIHR Leicester Cardiovascular Biomedical Research Centre (BRC-1215-20010). T.G. Jr and D.H. were supported by US National Science Foundation grant IOS-2038528. R.J.F.L. is supported by the NIH (R01DK110113, R01DK075787, R01DK107786, R01HL142302, R01HG010297, R01DK124097, R01HL151152). M.d.H. is a fellow of the Swedish Heart–Lung Foundation (20170872, 20200781) and a Kjell and Märta Beijer Foundation researcher. He is further supported by project grants from the Swedish Heart–Lung Foundation (20140543, 20170678, 20180706, 20200602) and the Swedish Research Council (2015-03657, 2019-01417).
Extended data
Author contributions
M.C., R.J.F.L. and M.d.H. conceived the study. M.C., S. Brage, U.E., R.J.F.L. and M.d.H. conceived trait definitions. M.C. and M.d.H. generated study-specific analysis plans. Z.W. and M.d.H. performed GWAS QC and meta-analyses. Z.W. performed SNP-based heritability estimations. Z.W. performed joint and conditional analyses. Z.W. performed PheWAS with physical activity polygenic scores. Z.W. performed the genetic correlation analysis. Z.W. performed the Mendelian randomization analysis. N.J.P., A.K., J.R.Z. and Z.W. analyzed enrichment of altered gene expression in skeletal muscle following intervention. D.H. performed biological annotation using DEPICT, SMR and FINEMAP. A.M.J. and T.P. performed cell-type enrichment using CELLECT. Z.W. and M.d.H. integrated results across gene prioritization approaches. T.M. and A.L.H. performed the GWAS for physical activity in 100 mouse strains. M.d.H. compared GWAS results across mice and humans. C.M. and A.A. explored expression of a 4930413E15Rik ortholog in humans. M.d.H. examined overlap between physical activity loci and loci showing evidence of selection. Z.W. performed the candidate gene analysis for exercise and fitness. E.M. constructed a homology model of the ACTN3 p.Glu635 variant. A.E. performed (steered) molecular dynamics simulations and umbrella sampling for p.Glu635Ala. S. Broos, L.D. and M.A.I.T. performed single skeletal muscle fiber experiments. A. Pacolet, M.V. and M.A.I.T. performed de novo genotyping for functional characterization of p.Glu635Ala. A.E. analyzed the isolated skeletal muscle fiber data. Z.W., A.E., R.J.F.L. and M.d.H. wrote the manuscript. T.S.A., T.M.B., A.R.B., L.F.B., M.C., A.Y.C., D.B., R.D., N.D.D., K.E., B.F., M.F.F., C.G., M.G., L.M.H., T. Haller, F.P.H., D.A.H., V.H., N.H.-C., C. Holzapfel, A.U.J., Å.J., M.A.K., R.K., K.F.K., B.K., C.M.K., Z.K., V.L., P.A.L., M. Lorentzon, L.-P.L., M. Mangino, C.M., K.R.M., A. Pacolet, L.P., R.P., R.C.R., N.V.R., S.R.-d.-P., K.E.S., C.-A.S., H.M.S., T.T., A. Tremblay, A. Tönjes, C.T., P.J.v.d.M., M.V., F.J.A.v.R., J.V.v.V.-O., X.Z., J.-H.Z., W.Z., Z.B., M.N.B.-H., S.E.B., J. Beilby, J. Blangero, D.I.B., S. Brage, P.S.B., J.A.B., M. Boehnke, C.L., J.W.C., F.S.C., L.A.C., T.E., S.E., J.D.F., L.F.-R., A.E.F., O.H.F., T.E.G., S.D.G., N.G., C.A.H., G.H., J.H., T.I., R.J., A.J., P.K.J., T.J., M.K., T.O.K., W.-P.K., I.K., P.P.K., J.K., L.J.L., A. Li, A. Linneberg, J.L., P.M.V., S.E.M., Y.M., A. Moscati, B.M., C.P.N., I.M.N., N.L.P., A. Peters, P.A.P., C.P., O.T.R., M.R., A.P.R., P.M.R., I.R., K.R., M.A.S., L.J.S., R.A.S., S.S., K.S., A.V.S., J.A.S., E.S., M.S., E.S.T., K.K.T., B.T., A. Tönjes, A. Tremblay, A.G.U., J.V., N.v.S., U.V., G. Waeber, K.W., Q.W., H.X., K.L.Y., J.M.Y., M.C.Z., A.B.Z., A.A., S. Broos, J.C.B., M. Bruinenberg, C.B., D.I.C., G.D.S., E.J.C.d.G., L.D., M.D., M.K.E., L.F., M.F., C.F., T.G., V.G., U.G., T. Hansen, C. Hayward, B.L.H., E.H., M.-R.J., W.C.J., S.L.R.K., L.A.K., M. Laakso, C.L., T.L., L.L.M., P.K.E.M., N.G.M., M. Melbye, A. Metspalu, D.M., K.E.N., C.O., A.J.O., M.O.-M., G.P., T.P., O. Pedersen, B.W.J.H.P., O. Polasek, I.P., C.N.R., N.J.S., X.S., H.S., T.I.A.S., T.D.S., N.J.T., R.M.v.D., N.v.d.V., C.M.v.D., P.V., H.V., T.V., G. Willemsen, N.J.W., D.R.W., H.-E.W., and J.F.W. contributed cohort study level results (concept, design, data acquisition, QC, and analysis. Z.W., A.E., N.J.P., T.M., D.H., M.C.C., E.M., S. Brage, T.S.A., T.M.B., A.R.B., L.F.B., M.C., A.Y.C., D.B., R.D., N.D.D., K.E., B.F., M.F.F., C.G., M.G., L.M.H., T. Haller, F.P.H., D.A.H., V.H., N.H.-C., C. Holzapfel, A.U.J., Å.J., A.M.J., M.A.K., R.K., K.F.K., B.K., C.M.K., Z.K., V.L., P.A.L., M. Lorentzon, L.-P.L., M. Mangino, C.M., K.R.M., A. Pacolet, L.P., R.P., R.C.R., N.V.R., S.R.-d.-P., K.E.S., C.-A.S., H.M.S., T.T., A. Tremblay, A. Tönjes, C.T., P.J.v.d.M., M.V., F.J.A.v.R., J.V.v.V.-O., X.Z., J.-H.Z., W.Z., Z.B., M.N.B.-H., S.E.B., J. Beilby, J. Blangero, D.I.B., P.S.B., J.A.B., M. Boehnke, U.E., C.-T.L., J.W.C., F.S.C., L.A.C., T.E., S.E., J.D.F., L.F.-R., A.E.F., O.H.F., T.E.G., S.D.G., N.G., C.A.H., G.H., J.H., T.I., R.J., A.J., P.K.J., T.J., M.K., T.O.K., W.-P.K., I.K., P.P.K., J.K., L.J.L., A. Li, A. Linneberg, J.L., P.M.V., S.E.M., Y.M., A. Moscati, B.M., C.P.N., I.M.N., N.L.P., A. Peters, P.A.P., C.P., O.T.R., M.R., A.P.R., P.M.R., I.R., K.R., M.A.S., L.J.S., R.A.S., S.S., K.S., A.V.S., J.A.S., E.S., M.S., E.S.T., K.K.T., B.T., A. Tönjes, A. Tremblay, A.G.U., J.V., N.v.S., U.V., G. Waeber, K.W., Q.W., H.X., K.L.Y., J.M.Y., M.C.Z., A.B.Z., A.A., S. Broos, J.C.B., M. Bruinenberg, C.B., D.I.C., G.D.S., E.J.C.d.G., L.D., M.D., M.K.E., L.F., M.F., C.F., T.G., V.G., U.G., T. Hansen, C. Hayward, B.L.H., E.H., M.-R.J., W.C.J., S.L.R.K., L.A.K., M. Laakso, C.-T.L., T.L., L.L.M., P.K.E.M., N.G.M., M. Melbye, A. Metspalu, D.M., K.E.N., C.O., A.J.O., M.O.-M., G.P., T.P., O. Pedersen, B.W.J.H.P., T.H.P., O. Polasek, I.P., C.N.R., N.J.S., X.S., H.S., T.I.A.S., T.D.S., N.J.T., R.M.v.D., N.v.d.V., C.M.v.D., P.V., H.V., T.V., G. Willemsen, N.J.W., D.R.W., H.-E.W., J.F.W., A.L.H., A.K., J.R.Z., M.A.I.T., R.J.F.L. and M.d.H. provided input and feedback to the final manuscript.
Peer review
Peer review information
Nature Genetics thanks Yann Klimentidis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Funding
Open access funding provided by Uppsala University
Data availability
European and multi-ancestry meta-analyses summary statistics for the genome-wide association study are available through the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics, GCP ID: GCP000358). UK Biobank individual-level data can be obtained through a data access application available at https://www.ukbiobank.ac.uk/. In this study we made use of data made available by: MetaMex https://www.metamex.eu/; Tabula Muris https://www.czbiohub.org/tabula-muris/; Open GWAS https://gwas.mrcieu.ac.uk/; MR Base https://www.mrbase.org/; GTEx Consortium https://gtexportal.org/home/; eQTLGen Consortium https://www.eqtlgen.org/; CommonMind Consortium https://www.synapse.org/#!Synapse:syn2759792/wiki/69613; Brain zQTLServe http://mostafavilab.stat.ubc.ca/xqtl/; MetaBrain https://www.metabrain.nl/.
Code availability
We made use of publicly available software and tools such as METAL (https://genome.sph.umich.edu/wiki/METAL), GCTA (https://yanglab.westlake.edu.cn/software/gcta/), LD score regression (https://github.com/bulik/ldsc), SMR (https://cnsgenomics.com/software/smr/) and PLINK (www.cog-genomics.org/plink/).
Competing interests
C.F. is Vice President and Head at Genetics and Pharmacogenomics, Merck labs. M. Lorentzon has received lecture or consulting fees from Amgen, Lilly, UCB Pharma, Radius Health, Meda, GE-Lunar and Santax Medico/Hologic. P.V. received an unrestricted grant from GlaxoSmithKline to build the CoLaus study. These authors played a role in individual studies that contributed to the meta-analysis, but not to the meta-analysis of GWAS studies, downstream experiments and analyses, or interpretation of the data. Hence, it is highly unlikely to have influenced the results of this study. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Deceased: Bill Musk.
A list of authors and their affiliations appears at the end of the paper.
Contributor Information
Zhe Wang, Email: zhe.wang@mssm.edu.
Marcel den Hoed, Email: marcel.den_hoed@igp.uu.se.
Lifelines Cohort Study:
Behrooz Z. Alizadeh, H. Marike Boezen, Lude Franke, Morris Swertz, Cisca Wijmenga, Pim van der Harst, Gerjan Navis, Marianne Rots, and Bruce H. R. Wolffenbuttel
Extended data
is available for this paper at 10.1038/s41588-022-01165-1.
Supplementary information
The online version contains supplementary material available at 10.1038/s41588-022-01165-1.
References
- 1.Lee IM, et al. Effect of physical inactivity on major non-communicable diseases worldwide: an analysis of burden of disease and life expectancy. Lancet. 2012;380:219–229. doi: 10.1016/S0140-6736(12)61031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Global Action Plan for the Prevention and Control of Noncommunicable Diseases 2013–2020 (World Health Organization, 2013).
- 3.Guthold R, Stevens GA, Riley LM, Bull FC. Worldwide trends in insufficient physical activity from 2001 to 2016: a pooled analysis of 358 population-based surveys with 1.9 million participants. Lancet Glob. Health. 2018;6:e1077–e1086. doi: 10.1016/S2214-109X(18)30357-7. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, et al. Secular trends in sedentary behaviors and associations with weight indicators among Chinese reproductive-age women from 2004 to 2015: findings from the China Health and Nutrition Survey. Int J. Obes. (Lond.) 2020;44:2267–2278. doi: 10.1038/s41366-020-00684-3. [DOI] [PubMed] [Google Scholar]
- 5.Wijndaele K, et al. Television viewing time independently predicts all-cause and cardiovascular mortality: the EPIC Norfolk study. Int J. Epidemiol. 2011;40:150–159. doi: 10.1093/ije/dyq105. [DOI] [PubMed] [Google Scholar]
- 6.Wijndaele K, Sharp SJ, Wareham NJ, Brage S. Mortality risk reductions from substituting screen time by discretionary activities. Med Sci. Sports Exerc. 2017;49:1111–1119. doi: 10.1249/MSS.0000000000001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauman AE, et al. Correlates of physical activity: why are some people physically active and others not? Lancet. 2012;380:258–271. doi: 10.1016/S0140-6736(12)60735-1. [DOI] [PubMed] [Google Scholar]
- 8.den Hoed M, et al. Heritability of objectively assessed daily physical activity and sedentary behavior. Am. J. Clin. Nutr. 2013;98:1317–1325. doi: 10.3945/ajcn.113.069849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stubbe JH, et al. Genetic influences on exercise participation in 37,051 twin pairs from seven countries. PLoS ONE. 2006;1:e22. doi: 10.1371/journal.pone.0000022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan W, et al. PPARδ promotes running endurance by preserving glucose. Cell Metab. 2017;25:1186–1193.e4. doi: 10.1016/j.cmet.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buniello A, et al. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarzynski MA, et al. Advances in exercise, fitness, and performance genomics in 2015. Med. Sci. Sports Exerc. 2016;48:1906–1916. doi: 10.1249/MSS.0000000000000982. [DOI] [PubMed] [Google Scholar]
- 13.Klimentidis YC, et al. Genome-wide association study of habitual physical activity in over 377,000 UK Biobank participants identifies multiple variants including CADM2 and APOE. Int. J. Obes. 2018;42:1161–1176. doi: 10.1038/s41366-018-0120-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doherty A, et al. GWAS identifies 14 loci for device-measured physical activity and sleep duration. Nat. Commun. 2018;9:5257. doi: 10.1038/s41467-018-07743-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van de Vegte YJ, Said MA, Rienstra M, van der Harst P, Verweij N. Genome-wide association studies and Mendelian randomization analyses for leisure sedentary behaviours. Nat. Commun. 2020;11:1770. doi: 10.1038/s41467-020-15553-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kilpeläinen TO, et al. Multi-ancestry study of blood lipid levels identifies four loci interacting with physical activity. Nat. Commun. 2019;10:376. doi: 10.1038/s41467-018-08008-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turley P, et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 2018;50:229–237. doi: 10.1038/s41588-017-0009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng J, et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics. 2016;33:272–279. doi: 10.1093/bioinformatics/btw613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shungin D, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518:187–196. doi: 10.1038/nature14132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kichaev G, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 2019;104:65–75. doi: 10.1016/j.ajhg.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Astle WJ, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016;167:1415–1429.e19. doi: 10.1016/j.cell.2016.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pulit SL, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum. Mol. Genet. 2019;28:166–174. doi: 10.1093/hmg/ddy327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winkler TW, et al. The influence of age and sex on genetic associations with adult body size and shape: a large-scale genome-wide interaction study. PLoS Genet. 2015;11:e1005378. doi: 10.1371/journal.pgen.1005378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Locke AE, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Justice AE, et al. Genome-wide meta-analysis of 241,258 adults accounting for smoking behaviour identifies novel loci for obesity traits. Nat. Commun. 2017;8:14977. doi: 10.1038/ncomms14977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrison J, Knoblauch N, Marcus JH, Stephens M, He X. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat. Genet. 2020;52:740–747. doi: 10.1038/s41588-020-0631-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verbanck M, Chen C-Y, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 2018;50:693–698. doi: 10.1038/s41588-018-0099-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013;37:658–665. doi: 10.1002/gepi.21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 2017;46:1985–1998. doi: 10.1093/ije/dyx102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemani, G. et al. Automating Mendelian randomization through machine learning to construct a putative causal map of the human phenome. Preprint at bioRxiv10.1101/173682 (2017).
- 31.Sanderson E, Smith GD, Windmeijer F, Bowden J. An examination of multivariable Mendelian randomization in the single-sample and two-sample summary data settings. Int. J. Epidemiol. 2019;48:713–727. doi: 10.1093/ije/dyy262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lightfoot JT, et al. Biological/genetic regulation of physical activity level: consensus from GenBioPAC. Med. Sci. Sports Exerc. 2018;50:863–873. doi: 10.1249/MSS.0000000000001499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pillon NJ, et al. Transcriptomic profiling of skeletal muscle adaptations to exercise and inactivity. Nat. Commun. 2020;11:470. doi: 10.1038/s41467-019-13869-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saul MC, et al. High motivation for exercise is associated with altered chromatin regulators of monoamine receptor gene expression in the striatum of selectively bred mice. Genes Brain Behav. 2017;16:328–341. doi: 10.1111/gbb.12347. [DOI] [PubMed] [Google Scholar]
- 35.Threlfell S, Sammut S, Menniti FS, Schmidt CJ, West AR. Inhibition of phosphodiesterase 10A increases the responsiveness of striatal projection neurons to cortical stimulation. J. Pharmacol. Exp. Ther. 2009;328:785–795. doi: 10.1124/jpet.108.146332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harashima A, Guettouche T, Barber GN. Phosphorylation of the NFAR proteins by the dsRNA-dependent protein kinase PKR constitutes a novel mechanism of translational regulation and cellular defense. Genes Dev. 2010;24:2640–2653. doi: 10.1101/gad.1965010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu Y, et al. Identification of CD112R as a novel checkpoint for human T cells. J. Exp. Med. 2016;213:167–176. doi: 10.1084/jem.20150785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inoue M, Chang L, Hwang J, Chiang SH, Saltiel AR. The exocyst complex is required for targeting of Glut4 to the plasma membrane by insulin. Nature. 2003;422:629–633. doi: 10.1038/nature01533. [DOI] [PubMed] [Google Scholar]
- 39.Burri L, et al. Mature DIABLO/Smac is produced by the IMP protease complex on the mitochondrial inner membrane. Mol. Biol. Cell. 2005;16:2926–2933. doi: 10.1091/mbc.e04-12-1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pers, T. H. et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat. Commun.6, 5890 (2015). [DOI] [PMC free article] [PubMed]
- 41.Muiños, M. & Ballesteros, S. Does physical exercise improve perceptual skills and visuospatial attention in older adults? A review. Eur. Rev. Aging Phys. Act.15, 2 (2018).
- 42.Hillis DA, et al. Genetic basis of aerobically supported voluntary exercise: results from a selection experiment with house mice. Genetics. 2020;216:781–804. doi: 10.1534/genetics.120.303668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Timshel, P. N., Thompson, J. J. & Pers, T. H. Genetic mapping of etiologic brain cell types for obesity. eLife9, e55851 (2020). [DOI] [PMC free article] [PubMed]
- 44.Schaum N, et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. 2018;562:367–372. doi: 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roberts MD, Ruegsegger GN, Brown JD, Booth FW. Mechanisms associated with physical activity behavior: insights from rodent experiments. Exerc. Sport Sci. Rev. 2017;45:217–222. doi: 10.1249/JES.0000000000000124. [DOI] [PubMed] [Google Scholar]
- 46.Zhu, Z. et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet.48, 481 (2016). [DOI] [PubMed]
- 47.Benner C, et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics. 2016;32:1493–1501. doi: 10.1093/bioinformatics/btw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nasser J, et al. Genome-wide enhancer maps link risk variants to disease genes. Nature. 2021;593:238–243. doi: 10.1038/s41586-021-03446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bray MS, et al. The human gene map for performance and health-related fitness phenotypes: the 2006–2007 update. Med. Sci. Sports Exerc. 2009;41:35–73. doi: 10.1249/MSS.0b013e3181844179. [DOI] [PubMed] [Google Scholar]
- 51.de Geus EJ, Bartels M, Kaprio J, Lightfoot JT, Thomis M. Genetics of regular exercise and sedentary behaviors. Twin Res. Hum. Genet. 2014;17:262–271. doi: 10.1017/thg.2014.42. [DOI] [PubMed] [Google Scholar]
- 52.Weyerstraß J, Stewart K, Wesselius A, Zeegers M. Nine genetic polymorphisms associated with power athlete status – a meta-analysis. J. Sci. Med. Sport. 2018;21:213–220. doi: 10.1016/j.jsams.2017.06.012. [DOI] [PubMed] [Google Scholar]
- 53.Moir HJ, et al. Genes and elite marathon running performance: a systematic review. J. Sports Sci. Med. 2019;18:559–568. [PMC free article] [PubMed] [Google Scholar]
- 54.Kim DS, Wheeler MT, Ashley EA. The genetics of human performance. Nat. Rev. Genet. 2021;23:40–54. doi: 10.1038/s41576-021-00400-5. [DOI] [PubMed] [Google Scholar]
- 55.Hagberg JM, et al. Apolipoprotein E genotype and exercise training-induced increases in plasma high-density lipoprotein (HDL)- and HDL2-cholesterol levels in overweight men. Metabolism. 1999;48:943–945. doi: 10.1016/S0026-0495(99)90185-3. [DOI] [PubMed] [Google Scholar]
- 56.Gielen, M. et al. Heritability and genetic etiology of habitual physical activity: a twin study with objective measures. Genes Nutr.9, 415, 1–12 (2014). [DOI] [PMC free article] [PubMed]
- 57.Pickering, C. & Kiely, J. ACTN3: more than just a gene for speed. Front. Physiol.8, 1080 (2017). [DOI] [PMC free article] [PubMed]
- 58.Vincent B, et al. ACTN3 (R577X) genotype is associated with fiber type distribution. Physiol. Genomics. 2007;32:58–63. doi: 10.1152/physiolgenomics.00173.2007. [DOI] [PubMed] [Google Scholar]
- 59.Norman B, et al. Strength, power, fiber types, and mRNA expression in trained men and women with different ACTN3 R577X genotypes. J. Appl. Physiol. (1985) 2009;106:959–965. doi: 10.1152/japplphysiol.91435.2008. [DOI] [PubMed] [Google Scholar]
- 60.Broos, S. et al. Evidence for ACTN3 as a speed gene in isolated human muscle fibers. PLoS ONE11, e0150594 (2016). [DOI] [PMC free article] [PubMed]
- 61.Broos S, et al. The stiffness response of type IIa fibres after eccentric exercise-induced muscle damage is dependent on ACTN3 r577X polymorphism. Eur. J. Sport Sci. 2019;19:480–489. doi: 10.1080/17461391.2018.1529200. [DOI] [PubMed] [Google Scholar]
- 62.Papadimitriou, N. et al. Physical activity and risks of breast and colorectal cancer: a Mendelian randomisation analysis. Nat. Commun.11, 597 (2020). [DOI] [PMC free article] [PubMed]
- 63.Zhang X, et al. Genetically predicted physical activity levels are associated with lower colorectal cancer risk: a Mendelian randomisation study. Br. J. Cancer. 2021;124:1330–1338. doi: 10.1038/s41416-020-01236-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Choi KW, et al. Assessment of bidirectional relationships between physical activity and depression among adults: a 2-sample Mendelian randomization study. JAMA Psychiatry. 2019;76:399–408. doi: 10.1001/jamapsychiatry.2018.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thompson PD, et al. Apolipoprotein E genotype and changes in serum lipids and maximal oxygen uptake with exercise training. Metabolism. 2004;53:193–202. doi: 10.1016/j.metabol.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 66.de Frutos-Lucas, J. et al. Does APOE genotype moderate the relationship between physical activity, brain health and dementia risk? A systematic review. Ageing Res. Rev.64, 101173 (2020). [DOI] [PubMed]
- 67.Golji, J., Collins, R. & Mofrad, M. R. Molecular mechanics of the alpha-actinin rod domain: bending, torsional, and extensional behavior. PLoS Comput. Biol.5, e1000389 (2009). [DOI] [PMC free article] [PubMed]
- 68.Yang J, et al. Genomic inflation factors under polygenic inheritance. Eur. J. Hum. Genet. 2011;19:807–812. doi: 10.1038/ejhg.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet.47, 291 (2015). [DOI] [PMC free article] [PubMed]
- 70.Winkler, T. W. et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc.9, 1192 (2014). [DOI] [PMC free article] [PubMed]
- 71.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pulit SL, de With SA, de Bakker PI. Resetting the bar: statistical significance in whole-genome sequencing-based association studies of global populations. Genet. Epidemiol. 2017;41:145–151. doi: 10.1002/gepi.22032. [DOI] [PubMed] [Google Scholar]
- 73.Kamat MA, et al. PhenoScanner V2: an expanded tool for searching human genotype–phenotype associations. Bioinformatics. 2019;5:4851–4853. doi: 10.1093/bioinformatics/btz469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loh PR, et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet. 2015;47:1385–1392. doi: 10.1038/ng.3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wainschtein, P. et al. Assessing the contribution of rare variants to complex trait heritability from whole-genome sequence data. Nat. Genet.54, 263–273 (2022). [DOI] [PMC free article] [PubMed]
- 76.Yang J, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 2012;44:369–375. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Price AL, et al. Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet. 2008;83:132–139. doi: 10.1016/j.ajhg.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Choi, S. W. & O'Reilly, P. F. PRSice-2: Polygenic Risk Score software for biobank-scale data. GigaScience8, giz082 (2019). [DOI] [PMC free article] [PubMed]
- 79.Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics. 2014;30:2375–2376. doi: 10.1093/bioinformatics/btu197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife7, e34408 (2018). [DOI] [PMC free article] [PubMed]
- 81.Elsworth, B. et al. The MRC IEU OpenGWAS data infrastructure. Preprint at bioRxiv10.1101/2020.08.10.244293 (2020).
- 82.Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet. Epidemiol. 2016;40:597–608. doi: 10.1002/gepi.21998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bowden J, et al. Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int. J. Epidemiol. 2016;45:1961–1974. doi: 10.1093/ije/dyw252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lyon, M. S. et al. The variant call format provides efficient and robust storage of GWAS summary statistics. Genome Biol.22, 32 (2021). [DOI] [PMC free article] [PubMed]
- 85.Koch LG, et al. Test of the principle of initial value in rat genetic models of exercise capacity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005;288:R466–R472. doi: 10.1152/ajpregu.00621.2004. [DOI] [PubMed] [Google Scholar]
- 86.Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qi T, et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat. Commun. 2018;9:2282–2282. doi: 10.1038/s41467-018-04558-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fromer M, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016;19:1442–1453. doi: 10.1038/nn.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ng B, et al. An xQTL map integrates the genetic architecture of the human brain's transcriptome and epigenome. Nat. Neurosci. 2017;20:1418–1426. doi: 10.1038/nn.4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Võsa U, et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021;53:1300–1310. doi: 10.1038/s41588-021-00913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barbeira, A. N. et al. Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol.22, 49 (2021). [DOI] [PMC free article] [PubMed]
- 92.Belton JM, et al. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–276. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abraham MJ, et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 95.Huang J, MacKerell AD., Jr. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goodrich, B., Gabry, J., Ali, I. & Brilleman, S. rstanarm: Bayesian applied regression modeling via Stan. R package version 2.14.1 https://mc-stan.org/rstanarm (2016).
- 97.Pastore, M. & Calcagnì, A. Measuring distribution similarities between samples: a distribution-free overlapping index. Front. Psychol.10, 1089 (2019). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Results, Discussion, Methods, Figs. 1–7, Box 1, study-specific acknowledgments and references.
Supplementary Tables 1–29.
Data Availability Statement
European and multi-ancestry meta-analyses summary statistics for the genome-wide association study are available through the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/downloads/summary-statistics, GCP ID: GCP000358). UK Biobank individual-level data can be obtained through a data access application available at https://www.ukbiobank.ac.uk/. In this study we made use of data made available by: MetaMex https://www.metamex.eu/; Tabula Muris https://www.czbiohub.org/tabula-muris/; Open GWAS https://gwas.mrcieu.ac.uk/; MR Base https://www.mrbase.org/; GTEx Consortium https://gtexportal.org/home/; eQTLGen Consortium https://www.eqtlgen.org/; CommonMind Consortium https://www.synapse.org/#!Synapse:syn2759792/wiki/69613; Brain zQTLServe http://mostafavilab.stat.ubc.ca/xqtl/; MetaBrain https://www.metabrain.nl/.
We made use of publicly available software and tools such as METAL (https://genome.sph.umich.edu/wiki/METAL), GCTA (https://yanglab.westlake.edu.cn/software/gcta/), LD score regression (https://github.com/bulik/ldsc), SMR (https://cnsgenomics.com/software/smr/) and PLINK (www.cog-genomics.org/plink/).