

ABSTRACT
Noroviruses are a leading cause of gastroenteritis worldwide, yet the molecular mechanisms of how host antiviral factors restrict norovirus infection are poorly understood. Here, we present a CRISPR activation screen that identifies mouse genes which inhibit murine norovirus (MNV) replication. Detailed analysis of the major hit Trim7 demonstrates a potent inhibition of the early stages of MNV replication. Leveraging in vitro evolution, we identified MNV mutants that escape Trim7 restriction by altering the cleavage of the viral NS6-7 polyprotein precursor. NS6, but not the NS6-7 precursor, directly binds the substrate-binding domain of Trim7. Surprisingly, the selective polyprotein processing that enables Trim7 evasion inflicts a significant evolutionary burden, as viruses with decreased NS6-7 cleavage are strongly attenuated in viral replication and pathogenesis. Our data provide an unappreciated mechanism of viral evasion of cellular antiviral factors through selective polyprotein processing and highlight the evolutionary tradeoffs in acquiring resistance to host restriction factors.
IMPORTANCE To maximize a limited genetic capacity, viruses encode polyproteins that can be subsequently separated into individual components by viral proteases. While classically viewed as a means of economy, recent findings have indicated that polyprotein processing can spatially and temporally coordinate the distinct phases of the viral life cycle. Here, we present a function for alternative polyprotein processing centered on immune defense. We discovered that selective polyprotein processing of the murine norovirus polyprotein shields MNV from restriction by the host antiviral protein Trim7. Trim7 can bind the viral protein NS6 but not the viral precursor protein NS6-7. Our findings provide insight into the evolutionary pressures that define patterns of viral polyprotein processing and uncover a trade-off between viral replication and immune evasion.
KEYWORDS: norovirus, polyprotein, antiviral
INTRODUCTION
Noroviruses are nonenveloped, positive-sense RNA viruses that are a leading cause of gastroenteritis worldwide (1). The global economic burden of human norovirus infection is estimated to be more than $60 billion (2). While norovirus gastroenteritis is typically self-limiting, immunocompromised patients can have recurrent and persistent norovirus infections (3, 4). Despite their global health burden, there are currently no approved antivirals or vaccines for noroviruses. This largely stems from a lack of suitable small animal models and difficulties in culturing human noroviruses in vitro (5). While significant progress has been made recently in establishing in vitro culture conditions for human norovirus, these culture systems are cumbersome, can vary between research groups, and in their current state are not amenable to high-throughput screens (6–9). Murine norovirus (MNV) has emerged as the premier model system for investigating norovirus biology. Unlike human noroviruses, MNV grows robustly in established cell lines and is a natural pathogen of mice (10–12). The MNV model system provides a unique opportunity to study host-virus interactions in a natural setting where both host and virus are genetically tractable and phenotypes are robust and reproducible.
We have previously leveraged the robust in vitro replicative capacity of MNV to define fundamental aspects of its interactions with host cells. Using clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 genetic screening tools (13), we have uncovered fundamental details of the MNV life cycle (14–16). Our loss-of-function CRISPR screen revealed CD300lf as the functional receptor for MNV (15, 17), which has been further validated by recent in vivo studies (18, 19). Additionally, our initial screen identified an unappreciated role of sphingolipid biosynthesis in norovirus entry and subversion of the stress granule proteins (14, 20). Critically, both of these pathways are essential for human norovirus infection, highlighting the utility of the MNV screening approach to identify critical features of norovirus biology (20, 21). Additionally, to uncover host antiviral factors that can restrict MNV replication, we used CRISPR activation (CRISPRa), which allows for the overexpression of genes from their native locus (22–24), to perform a gain-of-function screen in human cells expressing CD300lf (16). Tripartite motif-containing protein 7 (Trim7, also known as GNIP), an E3 ubiquitin ligase that was originally described to interact with glycogenin (25), provided the strongest signal in this pooled CRISPRa screen (16). We determined that Trim7 antiviral activity against MNV requires both the E3 ligase activity and a putative substrate binding domain (B30.2, also known as PRY-SPRY) to block MNV replication. Entry bypass experiments demonstrated that Trim7 can inhibit a post-entry stage of the MNV life cycle, although a Trim7-mediated restriction of viral entry cannot be excluded from this experiment (16). Recent work in other viral systems indicates that Trim7 has a complex role in viral regulation, since both proviral and antiviral properties have been observed. Trim7 can ubiquitinate stimulator of interferon genes (STING) and mitochondrial antiviral signaling protein (MAVS), leading to their degradation and a decrease in interferon (IFN) induction (26, 27). Trim7 also ubiquitinates the Zika virus envelope protein E, thereby enhancing the ability of virions to interact with binding receptors on host cells (28). However, Trim7 is antiviral toward enteroviruses, acting to ubiquitinate the enterovirus nonstructural protein 2BC, leading to its proteasome-mediated destruction (29). Whether any of these activities mediate Trim7 inhibition of MNV remains unclear.
Here, we present an orthogonal CRISPRa screen conducted in murine microglial cells, which are naturally susceptible to MNV. Our results expand upon known anti-norovirus host factors and further highlight the potent restriction of MNV by Trim7. Using in vitro evolution, we have determined that Trim7 sensitivity hinges on the selective processing of nonstructural proteins 6 and 7 (NS6-7). We further demonstrate that Trim7 targets the MNV protease NS6 but not the fusion protein of NS6-7. While mutations that alter polyprotein processing provides resistance to Trim7, viruses harboring these mutations are attenuated both in vitro and in vivo. These findings highlight an evolutionary trade-off selective polyprotein processing by noroviruses that balances Trim7 resistance and viral replication.
RESULTS
CRISPRa screening in murine BV2 cells identifies anti-norovirus genes.
To identify murine genes which, when overexpressed, are antiviral in the context of MNV infection, we performed genome-wide positive selection CRISPRa screens in wild-type (WT) BV2 cells. BV2 cells are a mouse microglial cell line that is naturally susceptible and highly permissive to MNV infection (15). We generated BV2-dCas9-VP64 cells and validated their CRISPRa activity prior to screening (Fig. S1 in the supplemental material). BV2-dCas9-VP64 cells were transduced with the Caprano Library, which induces transcriptional activity of over 22,000 mouse genes with each gene targeted by 3 to 6 single guide RNAs (sgRNAs) that are split into two pools (30). After transduction, the library was challenged with MNVCW3 or MNVCR6 at a multiplicity of infection (MOI) of 5. MNVCW3 and MNVCR6 share 86% nucleotide identity, yet cause distinct phenotypes in vivo (31, 32). Both MNVCW3 and MNVCR6 have been used as models for host-viral interactions and human norovirus biology (6, 31, 33–38). Surviving cells infected with MNVCW3 and MNVCR6 were harvested at 10 days postinfection. Screens were performed in duplicate, and analysis compared the relative enrichment of sgRNAs in infected and uninfected populations.
Independent guide RNAs targeting the same gene were averaged together, and their distribution in the ranked list was used to determine a gene-level P value (Fig. 1A and B, Tables S1 and S2 in the supplemental material). Gene set enrichment analysis (GSEA) of screen hits showed an enrichment in known antiviral signaling IFN-gamma and -alpha response pathways (Fig. S2 and Table S3). A total of 38 genes reached statistical significance in the MNVCW3 and MNVCR6 screens (Table S2). Fifteen of the 38 enriched genes are shared between the screens, suggesting that MNVCW3 and MNVCR6 are sensitive to overlapping antiviral mechanisms (Fig. 1C). A modest overlap of enriched genes was identified between the current screen and a previously conducted CRISPRa screen in human, HeLa-CD300lf cells (Fig. S2 and Table S4) (16). A selection of the top antiviral candidate genes from the genome-wide screens—Trim7, Prdm1, Tbx19, Obox6, and Cdkn1a—underwent arrayed validation using two independent sgRNAs per gene. We tested the abilities of cells to survive MNV challenge and to support viral replication. Enhanced cell survival and impaired viral replication were consistently observed with all sgRNAs tested (Fig. 1D and E). Consistent with the screening results, CRISPRa-mediated enhancement of Trim7 and Prdm1 expression exhibited the most potent antiviral activity, while enhancement of Tbx19, Obox6, and Cdkn1a exhibited more modest antiviral activity (Fig. 1D and E). We decided to focus on the mechanism of Trim7 inhibition of MNV due to the potency of Trim7 inhibition, its cell-type independent activity, and the breadth of phenotypes associated with Trim7.
FIG 1.

Genome-wide CRISPR activation (CRISPRa) screens in BV2 cells identify TRIM7 as potently antiviral. (A and B) Volcano plot showing CRISPRa screening results in BV2 cells infected with MNVCR6 (A) or MNVCW3 (B) at an MOI of 5. Controls are plotted in gray and relevant hit genes are denoted in color. (C) Venn diagram of significant hits in the MNVCR6 and MNVCW3 CRISPRa screens. (D and E) Results of arrayed validation for MNVCR6 (D) and MNVCW3 (E) with BV2-dCas9-VP64 cells transduced with indicated single guide RNA (sgRNA) construct. Cell viability was measured 24 h after murine norovirus (MNV) infection (MOI = 5). Viral replication was enumerated via plaque assay 24 h after a low MOI infection (0.05). Data are from two independent experiments and presented as mean ± standard error of the mean (SEM). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; one-way analysis of variance (ANOVA) with Tukey’s multiple-comparison test.
Trim7 blocks an early stage in the MNV life cycle.
Previously, we demonstrated that Trim7 blocks a post-entry step in the MNV life cycle, but it remains unclear which post-entry steps are inhibited by Trim7 (16). We longitudinally assessed the production of MNV nonstructural proteins NS1, NS6 (viral protease), and NS7 (RNA-dependent-RNA polymerase) in HeLa cells stably expressing CD300lf (HeLa-CD300lf) and Trim7 isoforms. We previously determined that the longer Trim7 isoform 1 has antiviral activity, but the shorter Trim7 isoform 4 lacks antiviral activity despite having an intact RING domain (16). HeLa-CD300lf cells expressing Trim7 isoform 1 consistently exhibited lower levels of NS1, NS6, and NS7 protein over the duration of MNV infection compared to cells expressing either an empty vector or Trim7 isoform 4 (Fig. 2A and B). This defect in nonstructural protein production was apparent as early as 5 h postinfection. Consistent with decreased levels of the viral polymerase, we observed significant inhibition in the synthesis of viral genomic RNA in cells expressing Trim7 isoform 1 at 8 and 12 h postinfection (Fig. 2C). This block in viral RNA synthesis is not observed with isoform 4 and is dependent upon the catalytic activity of the RING domain of Trim7 (Fig. 2C). These data are consistent with Trim7 inhibiting an early stage in the MNV life cycle, decreasing viral nonstructural protein levels and limiting subsequent viral replication.
FIG 2.

TRIM7 inhibits an early stage in the MNV life cycle. (A and B) Representative Western blots from three independent experiments of HeLa-CD300lf cells expressing an empty vector, Trim7 isoform 1, or Trim7 isoform 4 infected with MNVCW3 (MOI = 25). Cells were lysed at indicated time points and probed with antibodies to MNV nonstructural proteins (panel A, NS1; panel B, NS6-7). Non-specific band is denoted by an asterisk (*). (C) Quantification of MNV genomes in HeLa-CD300lf cells expressing an empty vector, Trim7 isoform 4, Trim7 isoform 1, or Trim7 isoform 1 C29A C32A after infection with MNVCW3 (MOI = 25). Cartoon diagram of the domains of Trim7 isoforms is presented below. Data are from three independent experiments and presented as mean ± SEM. **, P < 0.01; ***, P < 0.001; Kruskal-Wallis test for each time point compared to the empty vector control. Only Trim7 isoform 1 showed significant differences at any time point.
Identification of Trim7 escape mutants.
Trim7-mediated inhibition of MNV replication is not absolute, because viral replication can still be detected in Trim7 expressing cells (Fig. 2). We sought to leverage the ability of viruses to mutate and adapt to determine how Trim7 inhibits MNV replication. We passaged MNVCW3 and MNVCR6 in HeLa-CD300lf and BV2 cells expressing Trim7 (Fig. 3A). By using multiple viral strains and cellular contexts, we aimed to focus on common mechanisms that lead to Trim7 resistance rather than general tissue culture adaptations. We conducted 6 independent passaging experiments. We serially passaged MNV in BV2-Trim7 and HeLa-CD300lf-Trim7 cells for 3 and 4 passages, respectively (Fig. 3A). In all cases, we obtained viral populations that were resistant to Trim7 inhibition (data not shown).
FIG 3.
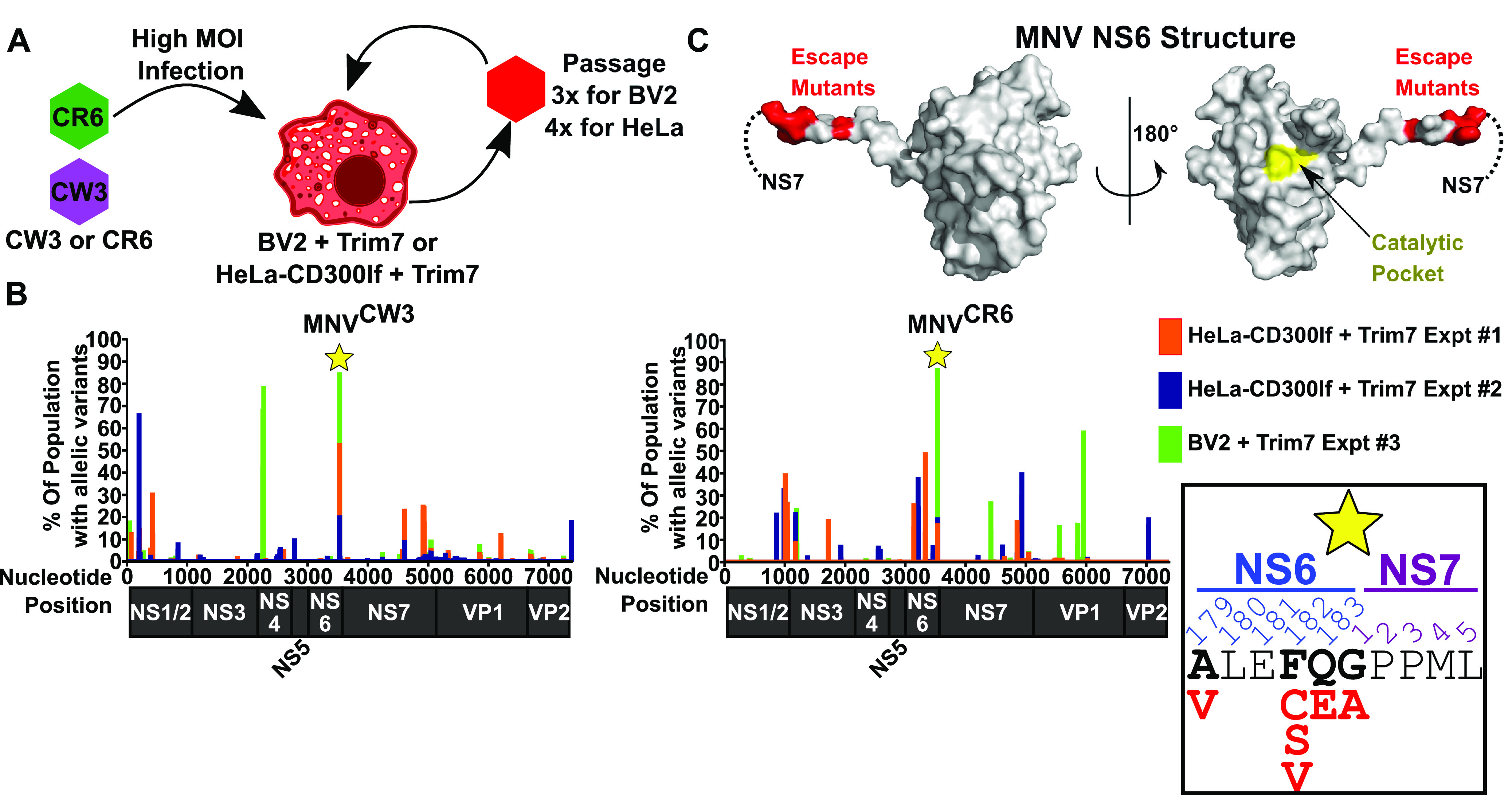
Trim7 resistance mutations map to the NS6-7 cleavage site. (A) Cartoon schematic of strategy to identify Trim7 resistance mutations. BV2 or HeLa-CD300lf cells expressing Trim7 were challenged with MNVCW3 or MNVCR6. After 48 h, virus was passaged onto fresh cells. (B) Summary of the sequencing data with MNVCW3 (left) and MNVCR6 (right). x axis denotes the nucleotide position with a diagram of the protein coding regions below. y axis shows the percentage of the population that contained variants at the indicated position. Both MNVCW3 and MNVCR6 had strong signal in all experiments at the NS6-NS7 junction (indicated by the star). Inset demonstrates the missense variants (shown in red) identified in these populations. (C) Structure of the MNV protease NS6 (PDB: 4X2V). Dashed lines indicate the site where NS7 would be in the NS6-7 precursor protein. Sites with predicted Trim7 resistance mutations are colored red, while the catalytic pocket is shown in yellow.
We deep sequenced the viral populations using our recently developed method and computational pipeline, obtaining robust coverage of the viral genomes (Fig. S3) (39). Under all conditions, variants were detected at multiple sites throughout the MNV genome (Table S6). To aid in visualization, the percentage of the population with any variation at a given nucleotide position was plotted (Fig. 3B). In doing so, we consolidated mis-sense variants, same-sense variants, insertions, and deletions into a single variable. In all six experiments, there was a clustering of variants at the junction of NS6 and NS7 (Fig. 3B). More specifically, the major mutations detected were within the cleavage site of NS6 and NS7 and distal from the active site of the NS6 protease (Fig. 3C). This region of the MNV polyprotein is highly conserved among sequenced MNV strains (Fig. S4). Variation at this site is surprising given that the cleavage of NS6-7 is required for MNV and that MNV does not tolerate large changes at the NS6-7 junction, even though it is variably cleaved in related Caliciviridae (40, 41). We focused on amino acid 182 in NS6 since multiple variants were detected at this site (Fig. 3B). In nearly all MNV strains, a phenylalanine is present at this position (Fig. S4). Our data suggest that a cysteine, serine, or valine in this position can confer resistance to Trim7 (Fig. 3B). We generated these three variants using MNVCW3 molecular clones and confirmed that MNVCW3 NS6F182C was not sensitive to Trim7 inhibition (Fig. 4A). However, MNVCW3 molecular clones containing NS6F182S and NS6F182V did not yield infectious virus. We obtained similar results with other mutations near the NS6-7 cleavage site (data not shown). These observations suggest a severe fitness disadvantage for these individual mutations and the possibility that other linked variants may be necessary to allow viral replication. Therefore, we focused our remaining efforts on understanding how the NS6F182C substitution contributes to Trim7 resistance.
FIG 4.
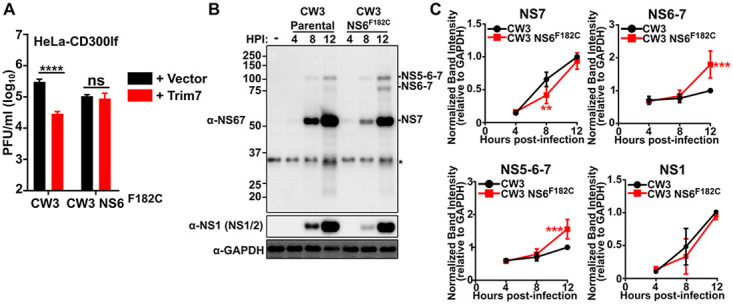
NS6 F182C substitution provides Trim7 resistance and decreased NS6-7 processing. (A) HeLa-CD300lf cells expressing either a vector control or Trim7 were infected with the parental or MNVCW3 NS6F182C mutant derived from CW3 molecular clone (MOI = 0.05). Viral titers were calculated after 24 h postinfection. Data are shown as mean ± SEM from three independent experiments. ****, P < 0.0001; one-way ANOVA with Tukey’s multiple-comparison test. (B) Representative Western blot from BV2 cells infected with either the CW3 parental or CW3 NS6F182C derived from MNV molecular clones (MOI = 5) and lysed at indicated hours postinfection (hpi). NS1/2, NS6, precursor bands were identified based upon specific antibody staining and expected molecular weights of polyprotein products. Non-specific band is denoted by an asterisk (*). (C) Quantification of the band intensities from panel B. Intensities were normalized to 1 for 12 h postinfection for MNVCW3 for NS7, NS6-7, and NS-5-6-7 relative to GAPDH (glyceraldehyde-3-phosphate dehydrogenase). Data are shown as mean ± standard deviation (SD) from three independent experiments. **, P < 0.01; ***, P < 0.0001; one-way ANOVA with Tukey’s multiple-comparison test.
Trim7 targets NS6 but not the NS6-7 precursor.
The clustering of mutations at the protease cleavage site of NS6-7 suggests that altered polyprotein processing of the NS6-7 junction could confer resistance to MNV. To test whether this was the case for the NS6F182C mutation, we quantified the production of NS6-7 containing polyproteins over time during infection of wild-type BV2 cells. Using a polyclonal antibody raised against the NS6-7 precursor, we could readily detect NS7, NS6-7, and NS5-6-7 in infected cell lysates (Fig. 4B). NS6 was not included in our analysis because detection of the NS6 band was faint, variable, and tended to migrate near a contaminating background band. The emergence of NS7 indicates that polyprotein processing occurs in both MNVCW3 parental- and MNVCW3 NS6F182C-infected cellular lysates (Fig. 4B). However, MNVCW3 NS6F182C consistently exhibited an increase in precursor proteins NS6-7 and NS5-6-7 compared to the parental virus (Fig. 4B and C). MNVCW3 NS6F182C also showed a delay in the accumulation of NS7 at earlier time points (Fig. 4B and C). These data indicate that processing of the NS6-7 cleavage site is altered during MNVCW3NS6F182C infection.
This alteration in the cleavage of NS6-7 suggests that Trim7 targets NS6 or NS7 when they are liberated, but not when they are in their uncleaved, precursor state (NS6-7). Indeed, we detected a physical interaction via coimmunoprecipitation between full-length Trim7 and NS6, but not NS7 (Fig. 5A). To further define the mechanism of Trim7 restriction, we investigated these interactions using recombinant proteins. A direct interaction between NS6 and the putative substrate binding domain, B30.2, of Trim7 was detected using an in vitro glutathione S-transferase (GST) pulldown assay (Fig. 5B). We did not detect interactions between the B30.2 domain of Trim7 and NS7 (Fig. 5B). Furthermore, an NS6-7 recombinant protein lacking protease activity to prevent autoprocessing was unable to interact with the B30.2 domain of Trim7 (Fig. 5B). These data suggest that Trim7 targets NS6 but not the NS6-7 precursor protein. Our data does not exclude the possibility that Trim7 could target other NS6 precursors that accumulate during infection such as NS5-6 or NS4-6 (42).
FIG 5.

Trim7 binds NS6 but not NS7 nor the NS6-7 precursor protein. (A) Western blot from coimmunoprecipitation experiment conducted in HEK 293T cells cotransfected with Ty1-tagged Trim7 (T7) or control Trim protein (C) with Flag-tagged GFP, NS6, or NS7 (n = 3). (B) Coomassie brilliant blue stain of a representative glutathione S-transferase (GST) pulldown assay with recombinant GST- or MBP-tagged proteins purified from E. coli. The GST-NS67 protein represents a fusion of NS6-7 that lacks autoprocessing due to mutations in the NS6 protease catalytic site. The B30.2 domain of Trim7 (MBP-B30.2) binds NS6 but not NS7 or NS6-7 (n = 4).
The NS6F182C substitution attenuates viral replication in vitro and in vivo.
We were surprised to find that a single amino acid substitution could render MNV resistant to Trim7 restriction (Fig. 4A), and we wished to understand the consequences of this change on the fitness of MNV. In a multistep growth curve of virally infected wild-type BV2 cells, MNVCW3 NS6F182C exhibits a significant replication defect (Fig. 6A). To test whether this defect was dependent on MNV strain or cellular context, we tested the ability of MNVCR6 NS6F182C to grow in HeLa-CD300lf cells. Consistent with our work with MNVCW3, the NS6F182C substitution provided the MNVCR6 strain with a fitness advantage compared to the parental virus when cells expressed Trim7 (Fig. 6B). However, MNVCR6 NS6F182C was attenuated in its ability to replicate in control HeLa-CD300lf cells (Fig. 6B). Thus, the NS6F182C substitution attenuates the ability of multiple MNV strains to replicate in vitro.
FIG 6.
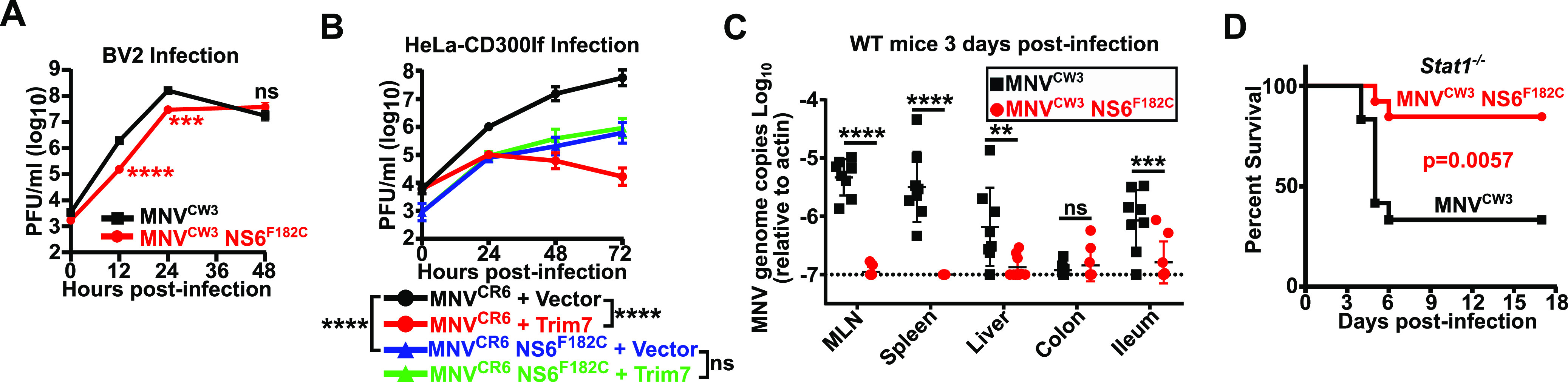
The NS6 F182C mutation leads to viral attenuation in vitro and in vivo. (A) BV2 cells were infected with MNVCW3 or MNVCW3 NS6F182C (MOI = 0.05). Viral production was measured using plaque assays at indicated time points. Data are shown as mean ± SEM from three independent experiments. (B) HeLa-CD300lf cells expressing either an empty vector or Trim7 isoform 1 were infected with MNVCR6 or MNVCR6 NS6F182C (MOI = 0.05). Viral production was measured using plaque assays at indicated time points. Data are shown as mean ± SEM from three independent experiments. Statistics shown are comparing viral titers at 72 h postinfection. ****, P < 0.0001; one-way ANOVA with Tukey’s multiple-comparison test. (C) C57BL/6 wild-type (WT) mice were inoculated with 5 × 106 PFU of MNVCW3 or MNVCW3 NS6F182C, euthanized 3 days postinfection, and the indicated tissues were collected. MNV genomes were enumerated via quantitative PCR. Data are shown as mean ± SD from two independent experiments with 8 to 9 mice per group. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; one-way ANOVA with Tukey’s multiple-comparison test. (D) Stat1–/– mice were inoculated with 1 × 104 PFU of MNVCW3 or MNVCW3 NS6F182C and monitored twice daily for survival for 17 days postinfection (12 to 13 mice per group). Data were analyzed using log-rank Mantel-Cox test.
We next tested the fitness of the MNVCW3 NS6F182C in vivo in two independent assays. First, we orally infected C57BL/6 WT mice with MNVCW3 parental or MNVCW3 NS6F182C virus. Three days postinfection, mice were euthanized, tissues were collected, and viral genomes enumerated. In mice infected with MNVCW3, MNV genomes were readily detected in the mesenteric lymph nodes, spleen, liver, and ileum (Fig. 6C). In contrast, mice infected with MNVCW3 NS6F182C had significantly lower levels of viral replication in these tissues (Fig. 6C). These data indicate a severe attenuation of the NS6F182C substitution in WT mice. Next, we tested whether the NS6F182C substitution modulated the pathogenesis of MNVCW3 in Stat1–/– mice, since Stat1–/– mice are exceptionally sensitive to MNVCW3 infection (12, 33). Indeed, we observed significantly reduced lethality in Stat1–/– mice when infected with MNVCW3 NS6F182C compared to that in mice infected with parental MNVCW3 (Fig. 6D). Taken together, these data indicate that Trim7 resistance can evolve in vitro, but it is accompanied by a trade-off in viral fitness both in vivo and in vitro (Fig. 7).
FIG 7.

Model of Trim7 mechanism and the trade-off for MNV to acquire resistance. Wild-type MNV separates NS6 from NS7, which allows for robust replication unless Trim7 is present, which can bind NS6. Viruses with decreased NS6-7 processing have attenuated viral replication in cells without Trim7 expression. However, the NS6-7 fusion protein enables MNV to replicate in Trim7 expressing cells as Trim7 cannot bind to the NS6-7 precursor protein.
DISCUSSION
Genome-wide CRISPR/Cas9 screens have uncovered numerous fundamental aspects of host-viral interactions, including for noroviruses (13). Here, we revealed numerous antiviral host factors utilizing a CRISPRa screen conducted in murine microglial cells. Importantly, in concordance with our previously conducted CRISPRa screen in HeLa cells ectopically expressing the MNV receptor CD300lf, Trim7 emerged as one of the most potent regulators of MNV in this system as well (16). While several genes were identified by both screens as antiviral, a number of unique antiviral factors were identified in this distinct system (Fig. S2). These findings add to the growing literature that similar screens conducted using distinct cellular models can reveal novel insights (43). In combination, these CRISPRa screens serve as a rich resource for those studying norovirus biology.
While Trim7 inhibition of viral replication is significant, it is not absolute. We identified a single nucleotide change leading to a coding variant at the NS6-7 junction that is sufficient to evade Trim7 restriction (Fig. 3). Interestingly, this virus exhibits significantly attenuated replication in vitro and in vivo and has reduced in vivo pathogenesis (Fig. 6). It is possible that additional mutations outside of the NS6-7 cleavage site we identified through our passaging studies may compensate for the replication defect of the NS6F182C mutation. These data highlight the tradeoffs viruses make in acquiring fitness in one environment compared to another. Previous work has indicated that the cleavage of NS6-7 is required for MNV replication (40) and that the virus does not tolerate large changes to the NS6-7 junction sequence (41). Surprisingly, cleavage of NS6-7 (also called Pro-Pol) is variable in the Caliciviridae family. For example, the NS6-7 precursor is stably expressed during feline calicivirus (FCV) infection (44). It is not clear why these closely related viruses differ in their polyprotein processing, and why MNV is attenuated when the NS6-7 precursor is inefficiently processed. It is tempting to speculate that Trim7 or a similar restriction factor has influenced the evolution of the viral polyprotein processing for viruses such as FCV. Whether other viruses acquire resistance to Trim7 or other host restriction factors through alterations in polyprotein processing is an exciting possibility.
Previously, we determined that Trim7 blocks a post-entry step in the MNV replication cycle (16). Here, we discovered that Trim7 can bind to NS6, but not to the NS6-7 precursor protein, via its putative substrate-binding domain, and that a virus with decreased processing of NS6 and NS7 (NS6-7) is resistant to Trim7 antagonism (Fig. 4 and 5). Our findings thus provide a mechanism for viral evasion. However, it is also possible that an unappreciated function of the NS6-7 precursor, but not the cleaved proteins, provides resistance. A recent report demonstrated that NS6-containing precursor proteins exhibit differential localizations and protease activities during infection (42). The NS6-7 precursor, while produced at low levels, remains localized at the MNV replication complex while NS6 is able to cleave other cellular substrates, including poly(A)-binding protein (PABP) (42, 45). These data suggest that in addition to differential binding of Trim7 to NS6-containing precursors, viral resistance to Trim7 may rely on the alternative activities of the precursors or on differential localization that sequesters NS6-containing precursors away from Trim7. Recent studies have placed Trim7 at the nexus of host-viral interactions. While Trim7 has been shown to downregulate IFN production through the ubiquitination and degradation of STING and MAVS (26, 27), we found an antiviral role of Trim7 for MNV. Similarly, a recent study determined that Trim7 inhibits diverse enteroviruses from replicating by ubiquitinating the viral 2BC protein (29). Interestingly, Trim7 has also been reported to ubiquitinate the Zika virus envelope protein E to promote Zika virus entry (28). Our findings reveal a new mechanism for Trim7 regulation of viral replication through the selective targeting of a viral protease that is dependent upon specific polyprotein processing. How Trim7 recognizes such diverse viral and host substrates in the cell remains an outstanding question. Furthermore, how these seemingly distinct proviral and antiviral functions of Trim7 are regulated in vivo will be an exciting area for future investigations.
MATERIALS AND METHODS
Cell culture.
BV2 (Yuanan Lu, University of Hawaii at Manoa), 293T (ATCC), and HeLa cells (ATCC) were cultured in Dulbecco’s modified Eagle medium (DMEM) with 5% fetal bovine serum (FBS) and 1% HEPES. During CRISPRa screening, cells were supplemented with 10% FBS, 1% HEPES, and 1% penicillin and streptomycin. For antibiotic selection, BV2 medium was supplemented with 4 μg/mL puromycin (Gibco) and 5 μg/mL blasticidin (Gibco), and HeLa medium with 1 μg/mL of puromycin and 5 μg/mL blasticidin (Gibco) when required.
Stable cell lines were generated via lentivirus transduction. Lentivirus was generated by transfecting lentiviral vectors with packaging vector (psPax2, a gift from Didier Trono; Addgene plasmid no. 12260) and pseudotyping vector (pCMV-VSV-G was a gift from Bob Weinberg; Addgene plasmid no. 8454) into 293T cells using TransIT-LT1 (Mirus). Then, 48 h later, supernatants were filtered through a 0.45-μm pore filter and added to the indicated cells. At 48 h after transduction, cells were selected with the appropriate antibiotic.
MNV assays.
MNV stocks were generated from plasmids encoding parental MNVCR6 (GenBank ID JQ237823) or parental MNVCW3 (GenBank ID EF014462.1) as described previously (46). Briefly, MNV molecular clone plasmids were transfected into 293T cells and frozen 48 h posttransfection. After thawing and centrifugation at 3,000 × g, virus was passaged onto BV2 cells. When 50% of the cells exhibited cytopathic effects, cultures were frozen, thawed, and clarified by centrifugation (3,000 × g) to form P1 stocks. For animal experiments, viral stocks were passaged an additional time at an MOI of 0.05. After clarification, viral P2 stocks were concentrated by filtration over a 100,000-kDa filter (Sartorius catalog no. VF20P4) by tangential flow (Sartorius catalog no. VFP002). Viral titers were enumerated by plaque assay on BV2 cells as described previously (15). Genetic identity of viral stocks was confirmed by targeted sequencing. MNV growth assays were performed as previously described (16). For viral protease processing studies, BV2 cells were seeded at 5 × 105 per well of a six-well plate. After adhering overnight, cells were infected with the indicated viruses at an MOI of 5.0. After incubating for 30 min, the cells were washed and fresh medium was added. Cells were lysed at indicated time points in Laemmli Buffer (Bio-Rad). Lysates were resolved on SDS-PAGE gels and transferred to polyvinylidene difluoride (PVDF) membranes to be probed with the indicated antibodies. Quantification was performed in Image J and the expression of each nonstructural protein was normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase). Furthermore, the relative expression of indicated proteins is relative to the levels at 12 h postinfection for the parental MNVCW3 infection.
CRISPRa screening.
BV2-dCas9-VP64 cells were generated from the plasmid lenti dCas9-VP64, a gift from Feng Zhang (Addgene plasmid no. 61425). CRISPRa activity in BV2-dCas9-VP64 cells was validated through the introduction of sgRNAs targeting CD4 cloned in the pXPR_502 vector. Cells transduced with sgRNA constructs were selected for 5 to 7 days with puromycin, and CD4 expression was assessed via flow cytometry.
BV2-dCas9-VP64-expressing cells were transduced separately with Set A and Set B of the Caprano CRISPRa library at an MOI of 0.3 to 0.5 to achieve a 500× library representation (30). Each experimental arm was performed in duplicate, with each replicate consisting of 3.0 × 107 BV2 cells expressing the complete CRISPRa system. Cells were infected with either MNVCW3 or MNVCR6 24 h post-seeding at an MOI of 5. Mock-infected cells were harvested 48 h after seeding. Ten days postinfection, cells were washed extensively and DNA from surviving cells was isolated using the QIAamp DNA Blood Midi kit (Qiagen). PCR, next generation Illumina sequencing, and analysis were performed as previously described (30). Briefly, harvested experimental and mock screen arms underwent DNA extraction, followed by PCR sample barcoding of guide sequences and next-generation Illumina sequencing. Sequencing was demultiplexed and mapped using Poolq, and the difference between the log-normalized sgRNA abundances of the mock-infected cells and survival populations yielded the overall log-fold change (LFC) of each guide. Further analysis was carried out using a Python hypergeometric script that calculates P values based on the consistency of ranks of guides targeting the same gene. The overall analysis was output in a volcano plot displaying log10 (P value) versus average LFC (log2) ranking the top enriched and depleted genes in the screen (https://github.com/mhegde). GSEA of screen data was performed using the Molecular Signatures Database (MSigDB): https://www.gsea-msigdb.org/gsea/msigdb/index.jsp. Z-score was calculated according to the formula ([X – μ]/σ)/γ, where X is the observed LFC of a gene, μ is the mean of the sample, σ is the standard deviation of the sample, and γ is the number of perturbations per an individual gene.
Arrayed screen validation.
Arrayed CRISPRa pXPR_502 constructs were delivered via lentivirus into BV2-dCas9-VP64 cells. For cell viability assays, cells were seeded at 1 × 105 cells per well of a white-walled glass-bottomed 96-well plate (Corning) and allowed to adhere to the plate overnight before viral infection. BV2s were infected with indicated MNV strains at an MOI of 5 and returned to the incubator for 24 h before viability was assessed via CellTiter-Glo (Promega) according to the manufacturer’s protocol and read out on a plate reader (Synergy II). The viability for each CRISPR cell line was normalized to a paired mock-infected sample of that cell line. To assess MNV growth, arrayed BV2 CRISPRa cell lines were seeded at 5 × 104 per well of a flat-bottomed 96-well plate and infected in suspension with MNVCW3 or MNVCR6 at an MOI of 0.05. At 24 h postinfection, plates were frozen at –80°C. Titers were enumerated via PFU as described previously (47). Experiments were performed in two independent biological replicates containing three technical replicates for viability measurements and two technical replicates for enumerating viral growth.
Plasmids.
CRISPR sgRNA constructs for arrayed validation were cloned into pXPR_502 (Addgene no. 96923). A list of sgRNAs used can be found in Table S5. Trim7 cDNA sequences were cloned into pCDH-MCS-T2A-Puro-MSCV (Systems Biosciences, CD522A-1) as described previously (16). pcDNA5/FRT/TO CBP-3×Flag-GFP, pcDNA5/FRT/TO CBP-3×Flag-NS6, and pcDNA5/FRT/TO CBP-3×Flag-NS7 were kind gifts of Seungmin Hwang. Trim7 and Trim47 (control plasmid) were cloned into a pcDNA 3.1 vector containing a GST-3×Ty1 sequence. The B30.2 domain of Trim7 was cloned into pET28B-His-MBP (a kind gift from Neal Alto). NS6, NS7, and NS6-7 (with NS6C139A eliminating protease activity and processing and NS7D346A D347A eliminating polymerase activity for better expression) were cloned into pGEX-4T1 vector. Mutations in MNV molecular clones were introduced through splicing by overlap extension PCR. All plasmid sequences were verified through sequencing prior to use.
Antibodies and Western blotting.
Cells were placed on ice and washed once in phosphate-buffered saline (PBS) prior to lysis in ice-cold mammalian lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 5 mM EDTA, 0.5% Triton X-100) supplemented with HALT protease and phosphatase inhibitors (Thermo Fisher Scientific). Lysates were incubated on ice for 30 min with periodic vortexing prior to clarification by centrifugation. Lysates were resolved on SDS-PAGE gels and transferred to PVDF membranes.
The following antibodies were used for Western blotting: rabbit polyclonal anti-NS67 (a kind gift from Kim Green), mouse monoclonal anti-NS1 (a kind gift from Sanghyun Lee), mouse anti-Flag M2 HRP (horseradish peroxidase; Sigma-Aldrich), mouse anti-GAPDH HRP (Sigma-Aldrich), anti-Ty1 clone BB2 (Thermo Fisher Scientific), anti-Rabbit IgG HRP (Sigma-Aldrich), and anti-mouse IgG (Sigma-Aldrich).
Viral evolution.
We performed six independent viral passaging experiments. We performed viral passaging in BV2-Trim7 isoform 1 cells with MNVCW3 and MNVCR6, with one replicate of each (two independent). We passaged MNVCW3 and MNVCR6 in HeLa-CD300lf Trim7 isoform 1 cells with two independent experiments for each strain (four independent experiments). BV2-Trim7 isoform 1 and HeLa-CD300lf-Trim7 isoform 1 cells (106) were seeded in a 10-cm2 plate and subsequently infected with MNVCW3 or MNVCR6 at an MOI of 5. At 48 h later, supernatants from the cultures were harvested and clarified (10 min at 3,000 × g). One mL of the clarified supernatant was added onto the respective BV2 and HeLa-CD300lf Trim7-expressing cell lines. This passaging was performed three times for BV2 cells and four times for HeLa cells. One mL of the final clarified viral supernatant was used to isolate total RNA using the Direct-zol kit (Zymo Research).
Coimmunoprecipitation.
293T cells were seeded at 5 × 105 per well of a six-well plate and, after overnight adherence, were transfected with indicated plasmid constructs. At 48 h posttransfection, cells were lysed on ice with mammalian lysis buffer as indicated above. Clarified lysates were incubated with anti-Flag-M2 agarose beads (Sigma-Aldrich) for 2 h with gentle rocking on a nutator at 4°C. Samples were washed four times in mammalian lysis buffer. Proteins were eluted from beads with Laemmli buffer and subjected to SDS-PAGE and Western blotting.
Protein purification.
Recombinant proteins were produced in BL21-DE3 Escherichia coli strains (Thermo Fisher Scientific). Protein expression was induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) when cells reached an optical density at 600 nm (OD600) of 0.6 to 0.8. Bacterial pellets were resuspended in either His buffer (100 mM HEPES [pH 7.5], 300 mM NaCl) or GST buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 2 mM DTT) supplemented with HALT protease inhibitor cocktail (Thermo Fisher Scientific). Bacteria were lysed using an EmulsiFlex C5 (Avestin) following the manufacturer’s protocol. Lysates were clarified by centrifugation at 12,000 × g for 30 min prior to adding to nickel agarose (Thermo Fisher Scientific) or glutathione Sepharose (Thermo Fisher Scientific) beads. Proteins were purified following manufacturer’s instructions. Eluted proteins were buffer-exchanged into PBS using concentration centrifugal columns (Millipore). Glycerol was added to 10% and the proteins were then stored at –80°C.
In vitro GST pulldowns.
A total of 30 μg of recombinant GST proteins was immobilized to glutathione Sepharose and incubated with 50 μg of 6×His-MalE fusion proteins for 1 h at 4°C. Samples were washed three times in TBS (Tris-buffered saline) supplemented with 0.5% Triton X-100. Proteins were eluted from beads with Laemmli buffer and subjected to SDS-PAGE. Gels were analyzed with Coomassie blue staining.
Mouse lines and infections.
All mouse experiments were conducted at University of Texas Southwestern Medical Center and approved by the University of Texas Southwestern Medical Center’s Institutional Animal Care and Use committees. C57BL/6J wild-type and Stat1–/– [B6.129S(Cg)-Stat1tm1Div/J] (48) mice were originally purchased from Jackson Laboratories and bred and housed in University of Texas Southwestern Medical Center animal facilities under specific pathogen-free conditions, including devoid of murine norovirus. A total of 1 × 104 PFU of MNVCW3 or MNVCW3NS6F182C was inoculated per orally to Stat1–/– mice. Stat1–/– mice were singly housed immediately after inoculation and monitored daily for survival for 17 days. A total of 5 × 106 PFU MNVCW3 or MNVCW3NS6F182C was inoculated per orally to WT mice. Immediately after inoculation, mice were singly housed. At 3 days postinfection, mice were euthanized and the indicated tissues harvested for RNA isolation. All experiments were performed three independent times with gender-balanced littermate controls.
RNA extraction and MNV qPCR.
Quantification of MNV genomes from infected tissues was performed as previously described (15). Briefly, RNA from infected tissues was isolated using TRI Reagent (Sigma-Aldrich) with a Direct-zol kit (Zymo Research) following the manufacturers’ protocols. One μg of RNA was used for cDNA synthesis using a High-Capacity cDNA Reverse Transcription kit, following the manufacturer’s protocols (Thermo Fisher Scientific). TaqMan quantitative PCR (qPCR) for MNV was performed in triplicate on each sample and standard with forward primer 5′-GTGCGCAACACAGAGAAACG-3′, reverse primer 5′-CGGGCTGAGCTTCCTGC-3′, and probe 5′-6FAM-CTAGTGTCTCCTTTGGAGCACCTA-BHQ1-3′. TaqMan qPCR for Actin was performed in triplicate on each sample and standard with forward primer 5′-GATTACTGCTCTGGCTCCTAG-3′, reverse primer 5′-GACTCATCGTACTCCTGCTTG-3′, and probe 5′-6FAM-CTGGCCTCACTGTCCACCTTCC-6TAMSp-3′.
Data availability.
Sequencing of MNV populations was carried out as we have recently described (39). Sequencing data have been uploaded to the European Nucleotide Archive under accession no. PRJEB47213.
ACKNOWLEDGMENTS
We thank John Schoggins, Wenchun Fan, Craig Wilen, and Moiz Munir for helpful discussions. S.C. was supported by a UT Southwestern Immunology T32 Training Grant (AI005284). R.C.O. was supported by NIH grants (R00DK116666 and R35GM142684) and by the Welch Foundation (I-2006-20190330). M.T.B. was supported by NIH grants (R01AI139314 and R01AI127552), the Pew Biomedical Scholars Program of the Pew Charitable Trusts, the Mathers Foundation, and the Burroughs Wellcome Fund.
M.E.S. and L.R.P. designed the project, performed experiments, and helped to draft the paper. M.A.S., S.L.C., B.F.D. R.R., L.A.S., E.A.K., B.M.S., and J.G.D. performed experiments. R.C.O. and M.T.B. conceptualized the project, provided supervision, and wrote the paper. All authors read and edited the manuscript.
J.G.D. consults for Microsoft Research, Abata Therapeutics, Servier, Maze Therapeutics, BioNTech, Sangamo, and Pfizer. J.G.D. consults for and has equity in Tango Therapeutics. J.G.D. serves as a paid scientific advisor to the Laboratory for Genomics Research, funded in part by GlaxoSmithKline. J.G.D. receives funding support from the Functional Genomics Consortium: Abbvie, Bristol Myers Squibb, Janssen, Merck, and Vir Biotechnology. J.G.D.’s interests were reviewed and are managed by the Broad Institute in accordance with its conflict of interest policies.
Footnotes
Supplemental material is available online only.
Contributor Information
Megan T. Baldridge, Email: mbaldridge@wustl.edu.
Robert C. Orchard, Email: Robert.Orchard@utsouthwestern.edu.
Colin R. Parrish, Cornell University
REFERENCES
- 1.Glass RI, Parashar UD, Estes MK. 2009. Norovirus gastroenteritis. N Engl J Med 361:1776–1785. 10.1056/NEJMra0804575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartsch SM, Lopman BA, Ozawa S, Hall AJ, Lee BY. 2016. Global economic burden of norovirus gastroenteritis. PLoS One 11:e0151219. 10.1371/journal.pone.0151219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartz S, Vergoulidou M, Schreier E, Loddenkemper C, Reinwald M, Schmidt-Hieber M, Flegel WA, Thiel E, Schneider T. 2011. Norovirus gastroenteritis causes severe and lethal complications after chemotherapy and hematopoietic stem cell transplantation. Blood 117:5850–5856. 10.1182/blood-2010-12-325886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siebenga JJ, Beersma MF, Vennema H, van Biezen P, Hartwig NJ, Koopmans M. 2008. High prevalence of prolonged norovirus shedding and illness among hospitalized patients: a model for in vivo molecular evolution. J Infect Dis 198:994–1001. 10.1086/591627. [DOI] [PubMed] [Google Scholar]
- 5.Ludwig-Begall LF, Mauroy A, Thiry E. 2021. Noroviruses: the state of the art, nearly fifty years after their initial discovery. Viruses 13:1541. 10.3390/v13081541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinjé J, Tibbetts SA, Wallet SM, Karst SM. 2014. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346:755–759. 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng XL, Qu L, Kou B, Opekun AR, Burrin D, Graham DY, Ramani S, Atmar RL, Estes MK. 2016. Replication of human noroviruses in stem cell-derived human enteroids. Science 353:1387–1393. 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones MK, Grau KR, Costantini V, Kolawole AO, de Graaf M, Freiden P, Graves CL, Koopmans M, Wallet SM, Tibbetts SA, Schultz-Cherry S, Wobus CE, Vinjé J, Karst SM. 2015. Human norovirus culture in B cells. Nat Protoc 10:1939–1947. 10.1038/nprot.2015.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Estes MK, Ettayebi K, Tenge VR, Murakami K, Karandikar U, Lin SC, Ayyar BV, Cortes-Penfield NW, Haga K, Neill FH, Opekun AR, Broughman JR, Zeng XL, Blutt SE, Crawford SE, Ramani S, Graham DY, Atmar RL. 2019. Human norovirus cultivation in nontransformed stem cell-derived human intestinal enteroid cultures: success and challenges. Viruses 11:638. 10.3390/v11070638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baldridge MT, Turula H, Wobus CE. 2016. Norovirus regulation by host and microbe. Trends Mol Med 22:1047–1059. 10.1016/j.molmed.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wobus CE, Karst SM, Thackray LB, Chang KO, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, Virgin HW. 2004. Replication of norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol 2:e432. 10.1371/journal.pbio.0020432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW. 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299:1575–1578. 10.1126/science.1077905. [DOI] [PubMed] [Google Scholar]
- 13.Puschnik AS, Majzoub K, Ooi YS, Carette JE. 2017. A CRISPR toolbox to study virus-host interactions. Nat Rev Microbiol 15:351–364. 10.1038/nrmicro.2017.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orchard RC, Wilen CB, Virgin HW. 2018. Sphingolipid biosynthesis induces a conformational change in the murine norovirus receptor and facilitates viral infection. Nat Microbiol 3:1109–1114. 10.1038/s41564-018-0221-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orchard RC, Wilen CB, Doench JG, Baldridge MT, McCune BT, Lee YC, Lee S, Pruett-Miller SM, Nelson CA, Fremont DH, Virgin HW. 2016. Discovery of a proteinaceous cellular receptor for a norovirus. Science 353:933–936. 10.1126/science.aaf1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orchard RC, Sullender ME, Dunlap BF, Balce DR, Doench JG, Virgin HW. 2019. Identification of antinorovirus genes in human cells using genome-wide CRISPR activation screening. J Virol 93:e01324-18. 10.1128/JVI.01324-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haga K, Fujimoto A, Takai-Todaka R, Miki M, Doan YH, Murakami K, Yokoyama M, Murata K, Nakanishi A, Katayama K. 2016. Functional receptor molecules CD300lf and CD300ld within the CD300 family enable murine noroviruses to infect cells. Proc Natl Acad Sci USA 113:E6248–E6255. 10.1073/pnas.1605575113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graziano VR, Walker FC, Kennedy EA, Wei J, Ettayebi K, Strine MS, Filler RB, Hassan E, Hsieh LL, Kim AS, Kolawole AO, Wobus CE, Lindesmith LC, Baric RS, Estes MK, Orchard RC, Baldridge MT, Wilen CB. 2020. CD300lf is the primary physiologic receptor of murine norovirus but not human norovirus. PLoS Pathog 16:e1008242. 10.1371/journal.ppat.1008242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graziano VR, Alfajaro MM, Schmitz CO, Filler RB, Strine MS, Wei J, Hsieh LL, Baldridge MT, Nice TJ, Lee S, Orchard RC, Wilen CB. 2021. CD300lf conditional knockout mouse reveals strain-specific cellular tropism of murine norovirus. J Virol 95:e01652-20. 10.1128/JVI.01652-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hosmillo M, Lu J, McAllaster MR, Eaglesham JB, Wang X, Emmott E, Domingues P, Chaudhry Y, Fitzmaurice TJ, Tung MK, Panas MD, McInerney G, Locker N, Wilen CB, Goodfellow IG. 2019. Noroviruses subvert the core stress granule component G3BP1 to promote viral VPg-dependent translation. Elife 8:e46681. 10.7554/eLife.46681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murakami K, Tenge VR, Karandikar UC, Lin SC, Ramani S, Ettayebi K, Crawford SE, Zeng XL, Neill FH, Ayyar BV, Katayama K, Graham DY, Bieberich E, Atmar RL, Estes MK. 2020. Bile acids and ceramide overcome the entry restriction for GII.3 human norovirus replication in human intestinal enteroids. Proc Natl Acad Sci USA 117:1700–1710. 10.1073/pnas.1910138117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS. 2013. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154:442–451. 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS. 2014. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159:647–661. 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517:583–588. 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skurat AV, Dietrich AD, Zhai L, Roach PJ. 2002. GNIP, a novel protein that binds and activates glycogenin, the self-glucosylating initiator of glycogen biosynthesis. J Biol Chem 277:19331–19338. 10.1074/jbc.M201190200. [DOI] [PubMed] [Google Scholar]
- 26.Yang B, Zhang G, Qin X, Huang Y, Ren X, Sun J, Ma S, Liu Y, Song D, Liu Y, Cui Y, Wang H, Wang J. 2021. Negative regulation of RNF90 on RNA virus-triggered antiviral immune responses targeting MAVS. Front Immunol 12:730483. 10.3389/fimmu.2021.730483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang B, Liu Y, Cui Y, Song D, Zhang G, Ma S, Liu Y, Chen M, Chen F, Wang H, Wang J. 2020. RNF90 negatively regulates cellular antiviral responses by targeting MITA for degradation. PLoS Pathog 16:e1008387. 10.1371/journal.ppat.1008387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giraldo MI, Xia H, Aguilera-Aguirre L, Hage A, van Tol S, Shan C, Xie X, Sturdevant GL, Robertson SJ, McNally KL, Meade-White K, Azar SR, Rossi SL, Maury W, Woodson M, Ramage H, Johnson JR, Krogan NJ, Morais MC, Best SM, Shi PY, Rajsbaum R. 2020. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 585:414–419. 10.1038/s41586-020-2457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan W, Mar KB, Sari L, Gaszek IK, Cheng Q, Evers BM, Shelton JM, Wight-Carter M, Siegwart DJ, Lin MM, Schoggins JW. 2021. TRIM7 inhibits enterovirus replication and promotes emergence of a viral variant with increased pathogenicity. Cell 184:3410.e17–3425.e17. 10.1016/j.cell.2021.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanson KR, Hanna RE, Hegde M, Donovan KF, Strand C, Sullender ME, Vaimberg EW, Goodale A, Root DE, Piccioni F, Doench JG. 2018. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat Commun 9:5416. 10.1038/s41467-018-07901-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nice TJ, Strong DW, McCune BT, Pohl CS, Virgin HW. 2013. A single-amino-acid change in murine norovirus NS1/2 is sufficient for colonic tropism and persistence. J Virol 87:327–334. 10.1128/JVI.01864-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thackray LB, Wobus CE, Chachu KA, Liu B, Alegre ER, Henderson KS, Kelley ST, Virgin HW. 2007. Murine noroviruses comprising a single genogroup exhibit biological diversity despite limited sequence divergence. J Virol 81:10460–10473. 10.1128/JVI.00783-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mumphrey SM, Changotra H, Moore TN, Heimann-Nichols ER, Wobus CE, Reilly MJ, Moghadamfalahi M, Shukla D, Karst SM. 2007. Murine norovirus 1 infection is associated with histopathological changes in immunocompetent hosts, but clinical disease is prevented by STAT1-dependent interferon responses. J Virol 81:3251–3263. 10.1128/JVI.02096-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilen CB, Lee S, Hsieh LL, Orchard RC, Desai C, Hykes BL, McAllaster MR, Balce DR, Feehley T, Brestoff JR, Hickey CA, Yokoyama CC, Wang YT, MacDuff DA, Kreamalmayer D, Howitt MR, Neil JA, Cadwell K, Allen PM, Handley SA, van Lookeren Campagne M, Baldridge MT, Virgin HW. 2018. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 360:204–208. 10.1126/science.aar3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsuzawa-Ishimoto Y, Shono Y, Gomez LE, Hubbard-Lucey VM, Cammer M, Neil J, Dewan MZ, Lieberman SR, Lazrak A, Marinis JM, Beal A, Harris PA, Bertin J, Liu C, Ding Y, van den Brink MRM, Cadwell K. 2017. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J Exp Med 214:3687–3705. 10.1084/jem.20170558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, Virgin HW. 2010. Virus-plus-susceptibility gene interaction determines Crohn's disease gene Atg16L1 phenotypes in intestine. Cell 141:1135–1145. 10.1016/j.cell.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, Diamond MS, Ivanova Y, Artyomov M, Virgin HW. 2015. Commensal microbes and interferon-λ determine persistence of enteric murine norovirus infection. Science 347:266–269. 10.1126/science.1258025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, Diamond MS, Virgin HW. 2015. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science 347:269–273. 10.1126/science.1258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Walker FC, Hassan E, Peterson ST, Rodgers R, Schriefer LA, Thompson CE, Li Y, Kalugotla G, Blum-Johnston C, Lawrence D, McCune BT, Graziano VR, Lushniak L, Lee S, Roth AN, Karst SM, Nice TJ, Miner JJ, Wilen CB, Baldridge MT. 2021. Norovirus evolution in immunodeficient mice reveals potentiated pathogenicity via a single nucleotide change in the viral capsid. PLoS Pathog 17:e1009402. 10.1371/journal.ppat.1009402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ward VK, McCormick CJ, Clarke IN, Salim O, Wobus CE, Thackray LB, Virgin HW, Lambden PR. 2007. Recovery of infectious murine norovirus using pol II-driven expression of full-length cDNA. Proc Natl Acad Sci USA 104:11050–11055. 10.1073/pnas.0700336104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emmott E, Sweeney TR, Goodfellow I. 2015. A cell-based fluorescence resonance energy transfer (FRET) sensor reveals inter- and intragenogroup variations in norovirus protease activity and polyprotein cleavage. J Biol Chem 290:27841–27853. 10.1074/jbc.M115.688234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emmott E, de Rougemont A, Hosmillo M, Lu J, Fitzmaurice T, Haas J, Goodfellow I. 2019. Polyprotein processing and intermolecular interactions within the viral replication complex spatially and temporally control norovirus protease activity. J Biol Chem 294:4259–4271. 10.1074/jbc.RA118.006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rebendenne A, Roy P, Bonaventure B, Chaves VA, Desmarets L, Rouille Y, Tauziet M, Arnaud-Arnould M, Giovannini D, Lee Y, DeWeirdt P, Hegde M, Garcia de Gracia F, McKellar J, Wencker M, Dubuisson J, Belouzard S, Moncorge O, Doench JG, Goujon C. 2021. Bidirectional genome-wide CRISPR screens reveal host factors regulating SARS-CoV-2, MERS-CoV and seasonal coronaviruses. bioRxiv. 10.1038/s41588-022-01110-2. [DOI] [PMC free article] [PubMed]
- 44.Ward JM, Wobus CE, Thackray LB, Erexson CR, Faucette LJ, Belliot G, Barron EL, Sosnovtsev SV, Green KY. 2006. Pathology of immunodeficient mice with naturally occurring murine norovirus infection. Toxicol Pathol 34:708–715. 10.1080/01926230600918876. [DOI] [PubMed] [Google Scholar]
- 45.Emmott E, Sorgeloos F, Caddy SL, Vashist S, Sosnovtsev S, Lloyd R, Heesom K, Locker N, Goodfellow I. 2017. Norovirus-mediated modification of the translational landscape via virus and host-induced cleavage of translation initiation factors. Mol Cell Proteomics 16:S215–S229. 10.1074/mcp.M116.062448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strong DW, Thackray LB, Smith TJ, Virgin HW. 2012. Protruding domain of capsid protein is necessary and sufficient to determine murine norovirus replication and pathogenesis in vivo. J Virol 86:2950–2958. 10.1128/JVI.07038-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hwang S, Alhatlani B, Arias A, Caddy SL, Christodoulou C, Cunha JB, Emmott E, Gonzalez-Hernandez M, Kolawole A, Lu J, Rippinger C, Sorgeloos F, Thorne L, Vashist S, Goodfellow I, Wobus CE. 2014. Murine norovirus: propagation, quantification, and genetic manipulation. Curr Protoc Microbiol 33:15K.2.1-61. 10.1002/9780471729259.mc15k02s33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Durbin JE, Hackenmiller R, Simon MC, Levy DE. 1996. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84:443–450. 10.1016/S0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Download jvi.00707-22-s0001.xlsx, XLSX file, 11.5 MB (11.5MB, xlsx)
Table S2. Download jvi.00707-22-s0002.xlsx, XLSX file, 0.02 MB (20KB, xlsx)
Table S3. Download jvi.00707-22-s0003.xlsx, XLSX file, 0.02 MB (20.5KB, xlsx)
Table S4. Download jvi.00707-22-s0004.xlsx, XLSX file, 10.5 MB (10.5MB, xlsx)
Table S5. Download jvi.00707-22-s0005.xlsx, XLSX file, 0.01 MB (9.7KB, xlsx)
Table S6. Download jvi.00707-22-s0006.xlsx, XLSX file, 0.03 MB (33.4KB, xlsx)
Supplemental material. Download jvi.00707-22-s0007.pdf, PDF file, 3.2 MB (3.2MB, pdf)
Data Availability Statement
Sequencing of MNV populations was carried out as we have recently described (39). Sequencing data have been uploaded to the European Nucleotide Archive under accession no. PRJEB47213.