

ABSTRACT
Binding to the host cell receptors CD4 and CCR5/CXCR4 triggers conformational changes in the human immunodeficiency virus (HIV-1) envelope glycoprotein (Env) trimer that promote virus entry. CD4 binding allows the gp120 exterior Env to bind CCR5/CXCR4 and induces a short-lived prehairpin intermediate conformation in the gp41 transmembrane Env. Small-molecule CD4-mimetic compounds (CD4mcs) bind within the conserved Phe-43 cavity of gp120, near the binding site for CD4. CD4mcs like BNM-III-170 inhibit HIV-1 infection by competing with CD4 and by prematurely activating Env, leading to irreversible inactivation. In cell culture, we selected and analyzed variants of the primary HIV-1AD8 strain resistant to BNM-III-170. Two changes (S375N and I424T) in gp120 residues that flank the Phe-43 cavity each conferred an ~5-fold resistance to BNM-III-170 with minimal fitness cost. A third change (E64G) in layer 1 of the gp120 inner domain resulted in ~100-fold resistance to BNM-III-170, ~2- to 3-fold resistance to soluble CD4-Ig, and a moderate decrease in viral fitness. The gp120 changes additively or synergistically contributed to BNM-III-170 resistance. The sensitivity of the Env variants to BNM-III-170 inhibition of virus entry correlated with their sensitivity to BNM-III-170-induced Env activation and shedding of gp120. Together, the S375N and I424T changes, but not the E64G change, conferred >100-fold and 33-fold resistance to BMS-806 and BMS-529 (temsavir), respectively, potent HIV-1 entry inhibitors that block Env conformational transitions. These studies identify pathways whereby HIV-1 can develop resistance to CD4mcs and conformational blockers, two classes of entry inhibitors that target the conserved gp120 Phe-43 cavity.
IMPORTANCE CD4-mimetic compounds (CD4mcs) and conformational blockers like BMS-806 and BMS-529 (temsavir) are small-molecule inhibitors of human immunodeficiency virus (HIV-1) entry into host cells. Although CD4mcs and conformational blockers inhibit HIV-1 entry by different mechanisms, they both target a pocket on the viral envelope glycoprotein (Env) spike that is used for binding to the receptor CD4 and is highly conserved among HIV-1 strains. Our study identifies changes near this pocket that can confer various levels of resistance to the antiviral effects of a CD4mc and conformational blockers. We relate the antiviral potency of a CD4mc against this panel of HIV-1 variants to the ability of the CD4mc to activate changes in Env conformation and to induce the shedding of the gp120 exterior Env from the spike. These findings will guide efforts to improve the potency and breadth of small-molecule HIV-1 entry inhibitors.
KEYWORDS: human immunodeficiency virus, envelope, entry inhibitor, CD4, CD4-mimetic compound, BNM-III-170, resistance
INTRODUCTION
The binding of the human immunodeficiency virus (HIV-1) envelope glycoprotein (Env) to the host cell receptors CD4 and CCR5/CXCR4 triggers virus entry into the cell (1–11). The Env trimer consists of three gp120 exterior Envs and three gp41 transmembrane Envs. Prior to receptor engagement, the HIV-1 Env trimer on virions can potentially sample three conformations, a pretriggered “closed” conformation (State 1), the “open” CD4-bound conformation (State 3), and a default intermediate conformation (State 2) (1, 12–15). Primary HIV-1 Envs are maintained in State 1 to various degrees by multiple intramolecular and intersubunit interactions, rendering these Envs relatively resistant to the binding of potentially neutralizing antibodies. CD4 binding drives Env from State 1 to State 2 and then into State 3, the prehairpin intermediate (12–18). In the prehairpin intermediate, the heptad repeat (HR1) region of gp41 forms an exposed coiled coil (16–19). Binding of the State-3 Env to the CCR5 or CXCR4 coreceptor is thought to induce the formation of a highly stable gp41 six-helix bundle, which promotes the fusion of the viral and cell membranes (20–25).
The surface of the HIV-1 Env trimer exhibits significant strain variability and heavy glycosylation, which contributes to the avoidance of potentially neutralizing antibodies (26–29). The binding site for CD4 on gp120 consists of a conserved surface that is conformationally altered by CD4 binding. CD4 binding creates an internal pocket in gp120 called the Phe-43 cavity that is bounded by highly conserved residues from gp120 and a single phenylalanine residue (Phe 43) from CD4 (30). The ~150-Å3 Phe-43 cavity is a target for two classes of small-molecule HIV-1 entry inhibitors, the CD4-mimetic compounds (CD4mcs) and conformational blockers like BMS-806 and BMS-529 (temsavir) (30–40).
CD4-mimetic compounds (CD4mcs) disrupt HIV-1 entry by binding to gp120 in the Phe-43 cavity, directly competing with CD4 but also prematurely triggering Env (31, 40–42). Thus, CD4mcs drive the State-1 Env trimer into downstream conformations (States 2 and 3) that, in proximity to a coreceptor-expressing target cell, can mediate HIV-1 infection (42). These CD4mc-induced Env intermediates are short-lived and, in the absence of a coreceptor-expressing target cell, apparently decay into inactive, dead-end conformations (41, 42). At CD4mc concentrations that do not completely inhibit HIV-1 infection, the induction of State-2 or State-3 Env conformations sensitizes HIV-1 viruses to neutralization by otherwise poorly neutralizing antibodies (43, 44). Such antibodies recognize epitopes induced by CD4, including the V2 and V3 variable regions of gp120, as well as the highly conserved gp120 region that overlaps the coreceptor-binding site (45).
The CD4-bound conformation of the HIV-1 Env on the cell surface can also be targeted by antibody-dependent cell-mediated cytotoxicity (ADCC) (46–48). In HIV-1-infected cells, complexes of Env and CD4 can be recognized by antibodies like 17b against the conserved CD4-induced (CD4i) gp120 region overlapping the coreceptor-binding site (49–52). Binding of CD4i antibodies promotes binding of antibodies like A32 against the highly conserved “cluster A” epitopes in the gp120 inner domain (53). These antibodies cooperate to induce the asymmetric State-2A Env conformation, which is efficiently targeted by ADCC (54). The HIV-1 Nef and Vpu proteins decrease the expression of Env-CD4 complexes on the infected cell surface, reducing the vulnerability of infected cells to ADCC (55–61). CD4mcs induce CD4i conformations of Env but, unlike CD4, are not susceptible to downregulation by Vpu or Nef (62). Thus, CD4mc can sensitize HIV-1-infected cells to ADCC by CD4i and anti-cluster A antibodies that are present at high titer in most HIV-1-infected individuals (49, 62–67). CD4mcs are being evaluated in animal models for their ability to stimulate ADCC as a means to reduce the size of the infected cell reservoir (68).
CD4mcs are also being evaluated for their ability to prevent acquisition of HIV-1. Intravaginal application of a CD4mc, either before or simultaneously with virus challenge, protected bone marrow-liver-thymus humanized mice from HIV-1 infection (69). Although the development of an effective HIV-1 vaccine has been frustrated by the inefficient elicitation of broadly neutralizing antibodies, antibodies that neutralize HIV-1 sensitized by a CD4mc are readily elicited by Env immunogens (43, 44, 62, 63). Antibodies elicited by monomeric gp120 protect monkeys from stringent, heterologous simian-human immunodeficiency virus (SHIV) mucosal challenge, provided the challenge virus is exposed to a CD4mc (43).
Early CD4mcs discovered using a gp120-CD4 screen exhibited weak antiviral potency against a limited range of HIV-1 isolates (31, 40). Iterative cycles of design, guided by CD4mc-gp120 structures and empirical testing, have led to the development of analogues with improved potency (36–38, 70–72). Primary HIV-1 strains exhibit a range of sensitivity to the improved CD4mcs. Progressive increases in CD4mc potency have been accompanied by an increase in the breadth of activity against a wider range of HIV-1 strains (37, 38). BNM-III-170 is currently the most potent and well-studied CD4mc and inhibits approximately 70% of a global panel of multiclade HIV-1 variants (37).
Despite significant improvements in the potency and breadth of CD4mcs, some primary HIV-1 strains remain relatively resistant to their antiviral effects (37). At least two mechanisms underly HIV-1 resistance to the CD4mcs. First, although the gp120 residues lining the Phe-43 cavity are well conserved among group M HIV-1 strains, some variation is tolerated. Of particular importance is variation in gp120 residue 375. In HIV-1 strains that are Clade AE recombinants, the His 375 side chain occupies the Phe-43 cavity, preventing efficient binding of CD4mcs (73–75). For most other group M HIV-1 strains, the Phe-43 cavity is not filled and is therefore hypothetically available for binding of the CD4mc (74, 75). Ser 375 is found in approximately 80% of group M HIV-1; in the remaining HIV-1 strains not in Clade AE, residue 375 is mostly Thr and, much less commonly, Ile or Met (74, 75). Viruses with Thr 375 are even more sensitive to CD4mcs than the corresponding viruses with Ser 375 (73, 76). In contrast, S375I and S375M HIV-1 mutants are more resistant to inhibition by the CD4mcs. Thus, only in a small subset of naturally occurring HIV-1 strains does variation in the gp120 residues lining the Phe-43 cavity account for resistance to the CD4mcs.
A second mechanism of resistance is based on the requirement that to bind and inhibit HIV-1, CD4mcs must induce transitions from State 1 to downstream conformations (26, 31, 35–37, 41, 42, 53, 54, 70–72, 77). Viruses with Envs that have more stable State-1 conformations and therefore are less prone to make transitions from State 1 have been shown to exhibit greater resistance to CD4mcs (26, 78–81). Both primary and laboratory HIV-1 strains exhibit different levels of “triggerability” or intrinsic reactivity, a property that is inversely related to the height of the activation barrier separating State 1 and State 2 (13, 26, 27). Differences in triggerability presumably reflect the different levels of responsiveness to CD4 that are advantageous to HIV-1 in distinct natural or tissue culture circumstances. Such differences in triggerability hypothetically contribute to the wide range of sensitivities to CD4mcs observed for primary HIV-1 strains (37).
To investigate pathways to CD4mc resistance preferred by HIV-1 in the absence of immune selection, we passaged the primary HIV-1AD8 strain in the presence of increasing concentrations of BNM-III-170. We identified three changes in gp120 that contribute individually and additively to the resistance of the selected viruses to BNM-III-170. We evaluated the relative impact of these gp120 changes on CD4 binding and viral fitness. Some of the changes associated with BNM-III-170 resistance also contributed to resistance to BMS-806 and BMS-529, examples of another class of small-molecule entry inhibitors that share part of their gp120 binding site with that of the CD4mcs (33–37, 82–86). We examined the sensitivity of the BNM-III-170-resistant viruses to conformation-dependent Env ligands; the results indicate that BNM-III-170 escape in this case is determined by local changes rather than global effects on Env conformation. These results provide valuable information about HIV-1 pathways to achieve CD4mc resistance and can guide future efforts to develop more potent and broad CD4mcs.
RESULTS
Selection of BNM-III-170-resistant HIV-1AD8.
We sought to evaluate natural pathways that HIV-1 can utilize to achieve resistance to a CD4mc, BNM-III-170, while maintaining replication competence. To this end, we passaged molecularly cloned HIV-1AD8 in C8166-R5 cells in the presence of increasing concentrations of BNM-III-170; starting with 5 μM BNM-III-170, the concentration of the CD4mc was gradually increased to 130 μM until no further effects on virus replication were observed (Fig. 1A). For the first 28 days, the BNM-III-170-treated infected cells produced less virus in the culture medium than the untreated infected cells. The reverse transcriptase levels in the BNM-III-170-treated infected cultures were higher than those in mock-infected cells, suggesting that some level of HIV-1AD8 replication still occurred in the presence of the CD4mc. Virus levels in the medium of the BNM-III-170-treated infected cells increased after day 28, approaching but not reaching the levels in the untreated infected cultures.
FIG 1.

Selection and characterization of BNM-III-170-resistant HIV-1AD8. (A) The replication of HIV-1AD8 in C8166-R5 cells in the absence and presence of increasing concentrations (5 to 130 μM) of BNM-III-170 is shown. The results from mock-infected cultures are also shown. Virus produced from HEK 293T cells transfected with the pNL4-3-AD8 proviral construct was incubated with C8166-R5 cells. Virus replication was monitored by measuring reverse transcriptase in the culture medium by quantitative real-time PCR. (B) A schematic representation of the wild-type (WT) HIV-1AD8 Env is shown, with the changes in two different cloned Envs (130-C and 130-3) derived from the viruses from the BNM-III-170-treated cells. In the bottom diagram, the changes common to both cloned Envs are shown. The V1/V2, V3, V4, and V5 variable gp120 regions, gp120/gp41 cleavage site and transmembrane region (TM) are shown. The gp41 cytoplasmic tail of the virus is a chimera of HIV-1AD8 and HIV-1NL4-3 Env sequences. (C) The expression level, processing and virion incorporation of the indicated Env variants are shown. HEK 293T cells were transfected with pNL4-3-AD8 proviral constructs expressing the Env variants. Cell lysates and viruses were harvested 48 h later and analyzed by Western blotting for the indicated proteins. A minor form of gp41 resulting from cleavage of the cytoplasmic tail (114) is designated by an asterisk. (D) The infectivity of viruses with the indicated Envs was measured on Cf2Th-CD4/CCR5 cells. HEK 293T cells were transfected with plasmids encoding the indicated Envs and HIV-1 packaging proteins and a luciferase-expressing HIV-1 vector. Forty-eight h later, cell supernatants were cleared and added to Cf2Th-CD4/CCR5 cells. Two days later, the cells were lysed and the luciferase activity was measured. The relative infectivity of viruses with the mutant Envs is compared with that of viruses with wild-type (WT) HIV-1AD8 Env. The results shown are representative of those obtained in three or more independent experiments.
To characterize HIV-1AD8 Env variants that were potentially selected during passage in the presence of BNM-III-170, env DNA was amplified by PCR and sequenced. The predicted amino acid sequences of two representative Envs (130-C and 130-3) from the BNM-III-170-treated culture are compared with that of the parental HIV-1AD8 Env in Fig. 1B. Three amino acid changes (E64G, S375N and I424T) were found in both Envs derived from the BNM-III-170-treated culture. All three changes in the gp120 glycoprotein are unusual in natural HIV-1 strains (74, 75). Glutamic acid 64 is nearly invariant in HIV-1 strains. Approximately 80% of HIV-1 strains have a serine residue at 375, and the remainder have threonine, histidine, isoleucine, or methionine residues at this position; Asn 375 is very rare in natural HIV-1 strains. Isoleucine (46%) and valine (54%) predominate at residue 424 in natural HIV-1 variants; Thr 424 is highly unusual (74, 75).
The three gp120 changes associated with BNM-III-170 selection were introduced individually or in combination into the HIV-1AD8 Env. The processing and incorporation into virion particles of these Env variants were comparable to those of the wild-type (WT) HIV-1AD8 and 130-C Envs (Fig. 1C). All Env variants detectably supported the entry of pseudotyped viruses into Cf2Th-CD4/CCR5 cells, although the infectivity of the viruses with the E64G change was significantly lower than that of WT HIV-1AD8 (Fig. 1D). Apparently, the E64G change in Env decreases the fitness of HIV-1, whereas the S375N and I424T changes are functionally well tolerated.
Effects of Env changes on resistance to HIV-1 entry inhibitors.
We examined the sensitivity of viruses pseudotyped with the Env variants to inhibition by BNM-III-170 and other gp120-directed entry blockers (Fig. 2 and Table 1). No toxicity of BNM-III-170 was detected at concentrations up to 300 μM (data not shown). Viruses with the 130-C and 130-3 Envs were completely resistant to high concentrations (300 μM) of BNM-III-170, whereas the WT HIV-1AD8 was inhibited with a 50% inhibitory concentration (IC50) of 0.3 μM (Fig. 2A, left). Approximately 5-fold higher concentrations of BNM-III-170 were required to inhibit the S375N and I424T viruses (Fig. 2A, middle). Viruses with the E64G Env exhibited even greater (approximately 100-fold) resistance to BNM-III-170. In combination, the gp120 amino acid changes contributed additively to BNM-III-170 resistance (Fig. 2A, right). Viruses with the S375N + I424T changes could still be inhibited at high concentrations of BNM-III-170, whereas viruses with E64G in combination with either S375N or I424T were resistant to the CD4mc. Viruses with all three Env changes were as resistant to BNM-III-170 as viruses with the 130-C Env. We conclude that all three gp120 changes contribute to BNM-III-170 resistance, with E64G exerting the largest effect. The other changes observed in the 130-C Env (colored gray in Fig. 1B) were also individually tested for potential effects on HIV-1AD8 sensitivity to BNM-III-170. No differences in BNM-III-170 inhibition of WT HIV-1AD8 and the G398S, D674N and Y802V Env mutants were noted (data not shown).
FIG 2.

Effect of changes in HIV-1AD8 Env on virus sensitivity to BNM-III-170, sCD4-Ig, and BMS-806. Recombinant luciferase-expressing viruses pseudotyped with the indicated Envs were produced as described in the Fig. 1D legend. Viruses were incubated with the indicated concentrations of BNM-III-170 (A), sCD4-Ig (B), and BMS-806 (C) for 1 h at 37°C. The viruses were then added to Cf2Th-CD4/CCR5 cells. After 2 days of culture, the cells were lysed and the luciferase activity was measured. The relative infectivity represents the luciferase activity obtained at each concentration of inhibitor compared with that obtained in the absence of inhibitor. The results shown are representative of those obtained in three or more independent experiments.
TABLE 1.
Sensitivity of HIV-1AD8 Env variants to inhibition by BNM-III-170, sCD4-Ig, BMS-806, and BMS-529a
| IC50 (μM) | BNM-III-170 | sCD4-Ig | BMS-806 | BMS-529 |
|---|---|---|---|---|
| AD8-WT | 0.23 ± 0.07 | 0.10 ± 0.08 | 0.02 ± 0.01 | 0.003 ± 0.001 |
| E64G | 25 ± 3.1 | 0.23 ± 0.19 | 0.02 ± 0.01 | 0.003 ± 0.001 |
| S375N | 1.2 ± 0.09 | 0.10 ± 0.09 | 0.30 ± 0.11 | 0.005 ± 0.002 |
| I424T | 1.2 ± 0.08 | 0.08 ± 0.07 | 0.03 ± 0.01 | 0.005 ± 0.002 |
| E64G + S375N | >300 | 0.12 ± 0.80 | 0.20 ± 0.1 | 0.006 ± 0.002 |
| E64G + I424T | >300 | 0.21 ± 0.16 | 0.03 ± 0.01 | 0.005 ± 0.002 |
| S375N + I424T | 48 ± 4.92 | 0.04 ± 0.02 | >2.4 | 0.1 ± 0.02 |
| E64G + S375N + I424T | >300 | 0.10 ± 0.07 | >2.4 | 0.1 ± 0.06 |
| 130-C | >300 | 0.07 ± 0.07 | >2.4 | 0.1 ± 0.06 |
The sensitivity of viruses pseudotyped with the WT HIV-1AD8 and variant Envs to inhibition by BNM-III-170, sCD4-Ig, BMS-806, and BMS-529 was determined as described above. The IC50 values for each inhibitor are reported as means and standard deviations derived from at least three independent experiments.
We examined the sensitivity of viruses pseudotyped by the HIV-1AD8 Env variants to soluble CD4-Ig (sCD4-Ig). Most of the variants, including 130-C, were inhibited by sCD4-Ig comparably to WT HIV-1AD8 (Fig. 2B and Table 1). The E64G and E64G + I424T viruses required 2- to 3-fold more sCD4-Ig for neutralization than the WT HIV-1AD8. The S375N + I424T virus was neutralized by sCD4-Ig slightly more efficiently than WT HIV-1AD8. Thus, in the context of some Envs, slight decreases or increases in sensitivity to sCD4-Ig were associated with the E64G and S375N changes, respectively. Overall, small differences in the sensitivity of these variant viruses to sCD4-Ig were observed, relative to the profound differences seen for BNM-III-170.
BMS-806 and BMS-529 (temsavir) are potent small-molecule entry inhibitors that bind the pretriggered (State 1) conformation of Env and block CD4-induced conformational changes required for HIV-1 entry (12, 15, 19, 32, 83–86). The gp120 binding site of BMS-806 and BMS-529 overlaps that of BNM-III-170 and other CD4mcs (33–37, 82) (see below). We evaluated the effect of the gp120 changes implicated in BNM-III-170 resistance on virus sensitivity to BMS-806 and BMS-529. The WT HIV-1AD8 was inhibited completely at a BMS-806 concentration of 40 nM, whereas the 130-C virus was not inhibited even at a BMS-806 concentration of 2.4 μM (Fig. 2C and Table 1). The S375N change alone resulted in an approximately 15-fold reduction in sensitivity to BMS-806. The I424T change minimally affected virus sensitivity to BMS-806. The E64G change had no effect on virus inhibition by BMS-806. The combination of S375N + I424T changes was sufficient to account for the full (>100-fold) resistance of the 130-C virus to BMS-806. Thus, the I424T change synergizes with the S375N substitution to increase HIV-1AD8 resistance to BMS-806.
We tested the sensitivity of the HIV-1AD8 Env variants to a more potent conformational blocker, BMS-529 (temsavir) (12, 34, 82, 85, 86). Although the S375N and I424T changes individually altered virus sensitivity to BMS-529 less than 2-fold, together these changes accounted for the 33-fold resistance observed for the 130-C virus (Table 1). As was seen for BMS-806, the E64G change exerted no significant effect on virus sensitivity to BMS-529.
Env structures.
We located the resistance-associated changes on structures of HIV-1 Env bound to the relevant ligands. In the process of binding CD4, gp120 components (the inner domain, outer domain and bridging sheet) are thought to rearrange around the internal Phe-43 cavity (30). Both Asn 375 and Thr 424 line the Phe-43 cavity but do not directly contact CD4 in structures of gp120-CD4 complexes (Fig. 3A) (30). In the gp120-CD4 complex, Gly 64 is located in “layer 1” of the gp120 inner domain and is distant from CD4 (87). During the process of gp120 binding to CD4, the formation of the α0 helix in layer 1 allows His 66 and Trp 69 of gp120 to associate with the back of the Phe-43 cavity, decreasing the off-rate of CD4 (80, 87). The alteration of the highly conserved Glu 64 to glycine may decrease the efficiency with which the α0 helix forms.
FIG 3.
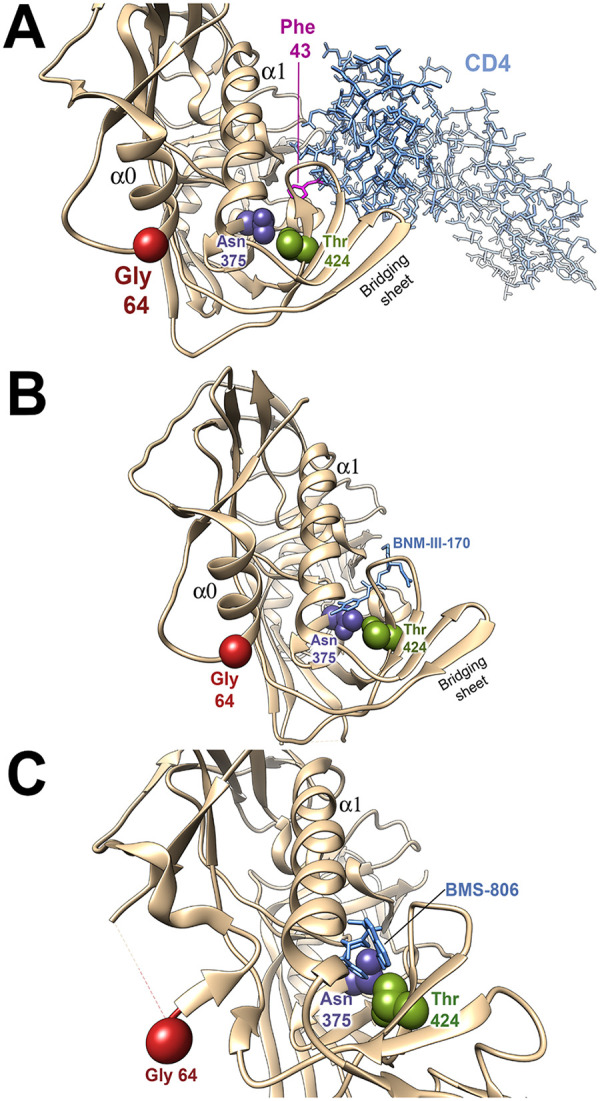
Location of amino acid residues implicated in virus resistance in Env structural models. The gp120 amino acid residues implicated in resistance to the inhibitors are shown on structures of HIV-1 Env-inhibitor complexes. In all structures, the resistance-associated changes have been introduced by the swapaa function in Chimera (115). (A) The HIV-1 gp120 core with N/C termini (tan ribbon) complexed with two-domain CD4 (light blue sticks) (PDB: 3JWO) is shown (87). Phe 43 of CD4 is colored magenta. (B) The HIV-1 gp120 core with N/C termini (tan ribbon) complexed with BNM-III-170 (light blue sticks) (PDB: 5F4P) is shown (87). (C) The HIV-1 sgp140 SOSIP.664 trimer (tan ribbon) complexed with BMS-806 (light blue sticks) (PDB: 6MTJ) is shown (82). The Envs are oriented with the viral membrane at the top of the figure and the target cell membrane at the bottom. The α0 and α1 helices of gp120 are shown. Note that in panel C, the α0 helix is not present. Part of the gp120 layer 1 loop between residues 59 and 64 is disordered in 6MTJ, and therefore the location of Gly 64 is an approximation.
In contrast to CD4, CD4-mimetic compounds like BNM-III-170 penetrate into the Phe-43 cavity (Fig. 3B) (35–37, 77). The S375N and I424T changes are expected to alter the shape of the Phe-43 cavity, explaining their individual and additive effects on BNM-III-170 antiviral potency. Although Gly 64 cannot contact BNM-III-170, the E64G change potentially decreases the layer 1-Phe-43 cavity interaction, as discussed above for CD4. This interaction is also important for the binding of CD4mcs; for example, changes in His 66 in layer 1 have been shown to decrease the antiviral efficacy of CD4mcs and sCD4 (78).
BMS-806 binds HIV-1 gp120 by occupying the Phe-43 cavity and adjacent water-filled channel (near Thr 424) (Fig. 3C) (34, 79). The alterations in the shape of these gp120 elements resulting from the S375N and I424T changes likely explain the observed decreases in BMS-806 potency. Gly 64 is distant from the BMS-806 binding site and since the formation of the α0 helix is not critical for BMS-806 binding (34, 82), the E64G change exerts little effect on its antiviral activity.
Mechanisms of escape from BNM-III-170.
The CD4-bound conformation of the HIV-1 Env trimer is more susceptible to disruption, resulting in the shedding of gp120 (88). We examined the effect of the Env changes on susceptibility of the virion spike to BNM-III-170-induced gp120 shedding at 37°C. The WT HIV-1AD8 Env shed nearly all of the gp120 after a 2.5-h incubation with 10 μM BNM-III-170 (Fig. 4A and B). In contrast, the 130-C- and the BNM-III-170-resistant Envs (E64G + I424T, E64G + S375N, and E64G + S375N +I424T) were relatively unaffected by incubation with up to 100 μM BNM-III-170. The Env mutants (S375N, I424T, E64G, and S375N + I424T) with intermediate levels of BNM-III-170 resistance exhibited gp120 shedding at high doses of BNM-III-170. Thus, there is a strong correlation between the sensitivity of the functional Env variant to BNM-III-170 inhibition of infection and susceptibility to BNM-III-170-induced gp120 shedding from the Env trimer (Spearman rank-order correlation coefficient, rS = −0.97, P < 0.05) (Fig. 4C).
FIG 4.

Mechanisms of HIV-1AD8 escape from BNM-III-170 and sCD4-Ig. (A) BNM-III-170-induced shedding of gp120 from virions with the indicated Envs was measured. Virions produced transiently from HEK 293T cells transfected with pNL4-3-AD8 proviral constructs were harvested, clarified by low-speed centrifugation, filtered (0.45-μm) and pelleted at 14,000 × g for 1.5 h at 4°C. The virus pellet was resuspended in 1× PBS and incubated with the indicated concentrations of BNM-III-170 for 2.5 h at 37°C. The viruses were then pelleted; the virus pellet was lysed and the supernatant containing shed gp120 was incubated with Galanthus nivalis lectin (GNL) beads. The viral lysates and the protein captured on the GNL beads were Western blotted. (B) gp120 shedding in the presence of BNM-III-170 was measured by quantifying the gp120 bands in the Western blots shown in A. The shed gp120 was calculated according to the formula: gp120 supernatant/(gp120 supernatant + gp120 virions). (C) The correlation between BNM-III-170 inhibition of pseudovirus infection and BNM-III-170-induced gp120 shedding is shown. The amount of gp120 shedding at a BNM-III-170 concentration of 50 μM, measured as in panel B, was used in the correlation analysis. The Spearman rank-order correlation coefficient (rS) and P value are shown. (D) Precipitation of virion Env by sCD4-Ig was evaluated as an indication of the affinity of sCD4-Ig for the Env variants. Virion pellets prepared as in panel A were resuspended in buffer containing the indicated concentrations of sCD4-Ig and incubated for 1.5 h at room temperature. After pelleting, the viruses were washed, lysed and incubated with protein A-agarose beads. The proteins captured on the beads were Western blotted. The results shown are typical of those obtained in at least two independent experiments. (E) The ratio of the virion gp120 glycoproteins precipitated by sCD4-Ig to the input gp120 glycoproteins was calculated from the experiment shown in panel D. The ratio is shown as a function of the sCD4-Ig concentration used for the immunoprecipitation assay.
To evaluate the binding of sCD4-Ig to the Env variants, virions with the different Envs were incubated with sCD4-Ig at room temperature; under these conditions, sCD4-Ig-induced gp120 shedding is relatively inefficient (89). The virions were washed and the virion-associated sCD4-Ig was used to precipitate the bound Envs. The sCD4-Ig precipitated the WT HIV-1AD8 Env at a half-maximal concentration of approximately 25 nM (Fig. 4D and E). No sCD4-Ig binding was detected to virions with the 130-C Env. The sCD4-Ig bound to virions with the other Env variants with intermediate levels of efficiency. The E64G change apparently makes a major contribution to the decrease in the efficiency of sCD4-Ig binding observed for the 130-C Env on virions.
Activation of HIV-1 infection of CD4-negative, CCR5-expressing cells by BNM-III-170 and sCD4-Ig.
We tested the ability of BNM-III-170 and sCD4-Ig to activate infection of CD4-negative, CCR5-expressing cells by the HIV-1AD8 Env variants (31, 41, 42, 90, 91). Infection of Cf2Th-CCR5 cells by viruses with WT HIV-1AD8 Env was activated by BNM-III-170 and sCD4-Ig (Fig. 5A and B). Activation by BNM-III-170 was less efficient for viruses with the 130-C and other variant Envs. This was also true for sCD4-Ig activation, with one exception; infection of Cf2Th-CCR5 cells by viruses with the S375N + I424T Env was activated by sCD4-Ig more efficiently than infection by viruses with the WT HIV-1AD8 Env. The susceptibility of the Env variants to BNM-III-170 and sCD4-Ig activation correlated with their sensitivity to inhibition by these Env ligands (Spearman rank-order correlation coefficients, rS = 0.94 and 0.81, respectively; P < 0.05 for both correlations) (Fig. 5C).
FIG 5.

Activation of HIV-1 infection by BNM-III-170 and sCD4-Ig. Activation of HIV-1 infection of CD4-negative, CCR5-expressing cells by BNM-III-170 (A) and sCD4-Ig (B) was evaluated. HEK 293T cells were transfected with plasmids expressing the indicated Envs and HIV-1 packaging proteins and a luciferase-expressing HIV-1 vector. After 48 h, pseudoviruses were harvested and incubated with Cf2Th-CCR5 cells in 96-well plates. The plates were centrifuged at 600 × g for 30 min at 21°C. Medium containing serial dilutions of BNM-III-170 or sCD4-Ig was then added. After incubation at 37°C in a CO2 incubator for 48 h, the cells were lysed and luciferase activity was measured. RLU, relative light units. (C) The relationship between inhibition of infection of Cf2Th-CD4/CCR5 cells (on the x axis) and activation of infection of Cf2Th-CCR5 cells (on the y axis) by BNM-III-170 and sCD4-Ig is shown for the indicated pseudovirus variants. The y axis values represent the concentrations of BNM-III-170 (in μM) and sCD4-Ig (in nM) required to achieve a level of infection in the Cf2Th-CCR5 cells of 0.5 × 104 RLU, determined as in A and B, respectively. The Spearman rank-order correlation coefficients (rS) and P values are shown.
The inhibition and activation of the functional Env trimer reflect both the ability of sCD4-Ig to bind Env and to induce concomitant conformational changes. We did not observe a significant correlation between the ability of the virion Envs to bind sCD4-Ig in our assay and the sensitivity of the Env variants to sCD4-Ig inhibition or activation (data not shown). As an example, the S375N + I424T Env bound sCD4-Ig less efficiently than WT HIV-1AD8 Env but was inhibited and activated more efficiently than WT HIV-1AD8 Env. Apparently, the assay measuring sCD4-Ig interaction with the virion Env trimer does not capture all of the factors that influence sCD4-Ig inhibition and activation of infection. Likewise, only modest (<4-fold) differences in the affinity of monomeric gp120 from the different Env variants for sCD4 were observed (Table 2), and these did not significantly correlate with sCD4-Ig activation and inhibition of infection by these variants (data not shown).
TABLE 2.
Affinity of monomeric gp120 from the different Env variants for sCD4a
| gp120 | KD (nM) | ka (1/Ms) | kdis (1/s) |
|---|---|---|---|
| AD8-WT | 13.46 ± 0.05 | 5E+04 ± 5E+03 | 7E-04 ± 7E-05 |
| E64G | 11.04 ± 1.93 | 7E+04 ± 2E+04 | 7E-04 ± 7E-05 |
| S375N | 17.85 ± 3.63 | 1E+05 ± 2E+03 | 2E-03 ± 3E-04 |
| I424T | 4.04 ± 0.02 | 6E+04 ± 3E+03 | 2E-04 ± 1E-05 |
| E64G + S375N | 8.83 ± 3.89 | 7E+04 ± 3E+04 | 6E-04 ± 3E-05 |
| E64G + I424T | 17.73 ± 6.65 | 1E+05 ± 8E+03 | 2E-03 ± 9E-04 |
| S375N + I424T | 25.51 ± 11.10 | 8E+04 ± 2E+04 | 2E-03 ± 3E-04 |
| E64G + S375N + I424T | 48.54 ± 1.56 | 6E+04 ± 5E+03 | 3E-03 ± 3E-04 |
The means and standard deviations derived from two independent experiments are shown. Association of sCD4 with monomeric gp120 from the different variants was estimated by biolayer interferometry, with the calculated on-rates (ka), off-rates (kdis), and equilibrium dissociation constants (KD). E represents 10 raised to the given exponential.
Effects of Env changes on virus sensitivity to conformation-dependent ligands.
The sensitivity of HIV-1 infection to inhibition by specific Env ligands or by exposure to cold can provide an indication of global changes in Env conformation (26, 78, 79, 92, 93). Therefore, we examined the susceptibility of the virus variants to inhibition by conformation-sensitive broadly neutralizing antibodies and poorly neutralizing antibodies, the T20 peptide corresponding to the gp41 heptad repeat (HR2) region (94, 95) and cold incubation. The sensitivities of the viruses pseudotyped by the WT HIV-1AD8 and variant Envs to these treatments were similar, with two exceptions (Table 3). Viruses pseudotyped by Envs with the E64G change were approximately 15-fold more sensitive to inhibition by T20 than the other viruses. Viruses with the 130-C Env were slightly less sensitive to cold inhibition than the WT HIV-1AD8 or other viruses. These results indicate that BNM-III-170 escape is not accompanied by global changes in Env conformation.
TABLE 3.
Sensitivity of HIV-1AD8 Env variants to antibody neutralization and cold inactivationa
| IC50 ligand/antibody (μg/ml) | PGT145 | PGT121 | PGT151 | VRC03 | T20 | 4E10 | 17b | 19b | Half-life of infectivity at 0°C (h) |
|---|---|---|---|---|---|---|---|---|---|
| AD8-WT | 0.10 ± 0.07 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.03 ± 0.08 | 2.5 ± 0.35 | >10 | >10 | >10 | 18.5 ± 6 |
| E64G | 0.21 ± 0.05 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.03 ± 0.09 | 0.16 ± 0.03 | >10 | >10 | >10 | 18.8 ± 8 |
| S375N | 0.21 ± 0.05 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.03 ± 0.1 | 2.0 ± 0.35 | >10 | >10 | >10 | 18.5 ± 6 |
| E64G+S375N | 0.11 ± 0.06 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.03 ± 0.08 | 0.16 ± 0.03 | >10 | >10 | >10 | 18.4 ± 6 |
| 130-C | 0.21 ± 0.04 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.03 ± 0.08 | 0.16 ± 0.04 | >10 | >10 | >10 | 40.9 ± 8 |
The means and standard deviations derived from three independent experiments are shown. The sensitivity of viruses pseudotyped with the indicated HIV-1AD8 Env variants to poorly and broadly neutralizing antibodies was evaluated. HEK 293T cells were transfected with plasmids encoding the indicated Envs and HIV-1 packaging proteins as well as a luciferase-expressing HIV-1 vector. Forty-eight h later, cell supernatants were clarified and incubated with different antibodies for 1 h at 37°C before the mixture was added to Cf2Th-CD4/CCR5 target cells. The concentration of antibody required to inhibit 50% of virus infection (IC50) was calculated using the GraphPad Prism program. In the cold sensitivity assay, viruses were incubated on ice for the indicated times, after which the virus infectivity was measured.
Susceptibility of Env variants to BNM-III-170 induction of Env epitopes for ADCC.
In HIV-1-infected cells, the formation of Env-CD4 complexes potentially exposes CD4-induced (CD4i) Env epitopes that can be recognized by ADCC-mediating antibodies (46–48). These Env-CD4 complexes are downregulated from the infected cell surface by the HIV-1 Vpu and Nef proteins (55–61). CD4mcs like BNM-III-170 induce conformational changes in HIV-1 Env that expose CD4-induced epitopes on cell-surface Env, which can then be targeted by ADCC responses (50, 53, 54, 62–64). To evaluate the effect of the Env changes on this process, cells infected with molecularly cloned viruses containing the Env variants were incubated with BNM-III-170 or the dimethyl sulfoxide (DMSO) control and tested for the ability to be recognized by antibodies. Figure 6 shows that, although the WT HIV-1AD8 Env responds to BNM-III-170 by exposing epitopes recognized by the 17b CD4i antibody and antibodies from the plasma of an HIV-1-infected individual, the variant Envs tested did not. The cells with the WT HIV-1AD8 Env and variant Envs downregulated CD4 and BST-2 comparably; this was expected, as these functions are mediated by the viral Vpu and Nef proteins produced by the integrated proviruses (55–61). These results are consistent with the relatively low ability of the 130-C, E64G + S375N, and E64G + S375N + I424T Envs to bind and respond to BNM-III-170.
FIG 6.

Effect of HIV-1AD8 Env changes on BNM-III-170-induced exposure of ADCC epitopes. Activated primary CD4+ T cells were infected with the molecularly cloned viruses expressing the indicated Env variants. Two days after infection, cells were incubated with BNM-III-170 (50 μM) or DMSO and stained with anti-CD4, anti-BST-2 or 17b, 2G12, and A32 antibodies or HIV+ plasma followed by detection of intracellular p24. (A and B) The binding of 17b (A) or HIV+ plasma (B) relative to the binding of 2G12 is shown. Similar results were obtained with the A32 antibody (not shown). (C and D) The mean fluorescence intensity (MFI) for the staining of cells with antibodies against CD4 (C) and BST-2 (D) is shown. Means and standard deviations from two independent experiments are shown.
DISCUSSION
CD4mcs compete with CD4 for binding to HIV-1 gp120 (40, 42, 91). In the process of binding, CD4mcs induce conformational changes in Env similar to those induced by CD4 (31, 41, 42). These conformational changes ultimately contribute to the binding affinity of the CD4mcs. Thus, resistance to CD4mcs can involve changes in HIV-1 Env near the binding site or changes outside the binding site that decrease the ability of Env to undergo the appropriate conformational changes (35, 67–69, 76, 79).
Selection of HIV-1 variants resistant to CD4mc can provide insights into preferred viral pathways to achieve resistance while minimizing fitness costs. BNM-III-170 is currently the CD4mc with the highest potency and greatest breadth against HIV-1 (37). The viruses selected for resistance to BNM-III-170 exhibited a modest decrease in the ability of their Envs to support virus infection. Most of this fitness cost resulted from the substitution of a glycine residue for the highly conserved Glu 64 in layer 1 of the gp120 inner domain. Modest decreases in the binding and antiviral potency of sCD4-Ig were associated with the E64G change. Conformational transitions in layer 1 of gp120 are thought to contribute to the binding of CD4 and CD4mcs, and the E64G change likely decreases the efficiency of this process (78, 80, 87). A large gp120 ligand like CD4 is more able to overcome the resistance to conformational change imposed by E64G than a small molecule like BNM-III-170. Indeed, the E64G change resulted in only 2- to 3-fold resistance to sCD4-Ig but approximately 100-fold resistance to BNM-III-170. We note that a gp120 region, including Glu 64, has been proposed as a second contact site for CD4 on the adjacent Env protomer (96, 97). However, such quaternary contacts, which are not relevant to the binding of a small molecule like a CD4mc, do not explain the significantly greater impact of the E64G change on susceptibility to BNM-III-170 compared with sCD4-Ig. We also note the proximity of the gp120 inner domain layer 1 to the gp41 heptad repeat (HR1) region in structural models of Env trimers (34, 77, 82, 98–100). The CD4-induced formation and exposure of the HR1 coiled coil on the gp41 prehairpin intermediate creates a target for antiviral peptides like T20 that mimic the HR2 region of gp41 (94, 95, 101, 102). Slowing layer 1 conformational transitions may increase the window of exposure of the gp41 HR1 region, accounting for the observed increase in sensitivity of the variants with Gly 64 to T20 inhibition. We did not detect changes in the sensitivity of the BNM-III-170-resistant viruses to neutralization by the antibodies tested, suggesting that resistance is not accompanied by global changes in Env architecture.
The CD4-binding site on gp120 is one of the few conserved protein surfaces exposed on the pretriggered (State 1) Env trimer (30, 103). The associated Phe-43 cavity, although largely inaccessible to protein ligands, allows the binding of small molecules and is well conserved among HIV-1 group M viruses. Thus, it is no coincidence that multiple small molecules that inhibit the entry of a variety of HIV-1 strains utilize the Phe-43 cavity for binding Env (33–37, 70–72, 76, 77, 82). However, as we observed in this study, HIV-1 can exploit differences in the requirements for CD4 and CD4mc binding by altering residues like Ser 375 or Ile 424 that line the walls of the Phe-43 cavity. The resulting changes in the shape of the Phe-43 cavity exert only modest effects on CD4 binding affinity and are well tolerated with respect to HIV-1 replication. However, the S375N and the I424T changes individually result in approximately 5-fold effects on virus sensitivity to BNM-III-170; when combined, these changes lower the inhibitory potency of BNM-III-170 by approximately 200-fold. Thus, the different dependence of CD4mc and CD4 binding on the specific shape of the Phe-43 cavity creates opportunities for HIV-1 to escape the CD4mcs. We note that changes in Ser 375 and other residues lining the Phe-43 cavity have been shown to confer resistance to less potent CD4mcs (35, 44). The gp120 changes that resulted in HIV-1 resistance to the direct antiviral effect of BNM-III-170 also negated the ability of the CD4mc to enhance the binding of antibodies that potentially mediate ADCC lysis of infected cells.
BMS-806 analogues can bind a pretriggered (State 1) conformation of HIV-1 Env and block some of the conformational changes resulting from CD4 binding (19, 32, 83–86). In the course of binding Env, BMS-806 analogues do not need to induce major conformational changes in Env; therefore, they bind with higher affinity and achieve greater potency than the CD4mcs (12, 15, 31, 34, 82). Nonetheless, as for the CD4mcs, changes in the residues lining the Phe-43 cavity can result in significant HIV-1 resistance to BMS-806 analogues (33, 104, 105). In our study, the S375N change had the biggest impact on HIV-1 sensitivity to BMS-806, and the combined S375N + I424T changes resulted in profound levels of resistance to both BMS-806 and the more potent BMS-529 (temsavir). Of interest, changes in Ser 375, including substitution of an asparagine residue, have been observed in resistant HIV-1 variants derived from individuals treated with BMS-806 analogues (104, 105). Like the I424T change, these alterations of the Phe-43 cavity likely affect the accommodation of the BMS-806 benzoyl group (34, 82). Changes in Met 426 and Met 434, which flank the methoxy pyrrolopyridine group residing in the gp120 water-filled channel, have also been associated with resistance in HIV-1-infected individuals treated with these conformational blockers (104, 105). Consistent with the expectation that CD4-induced conformational changes are not required for BMS-806 and BMS-529 binding (12, 14, 34, 82), the E64G change had no impact on their potency. Despite the opposing mechanisms of Env inhibition by conformational blockers like BMS-806/BMS-529 and CD4mcs, shared elements of their binding sites in the Phe-43 cavity of gp120 create opportunities for HIV-1 to develop resistance simultaneously to both classes of entry inhibitors.
Clinical applications of HIV-1 entry inhibitors targeting the gp120 Phe-43 cavity may be affected by the potential of the virus to develop resistance. Indeed, primary HIV-1 strains exhibit a wide range of sensitivities to CD4mcs and conformational blockers (37, 83–85). Variation of Ser 375 in natural HIV-1 strains can influence the efficacy of CD4mcs, either positively or negatively (73, 76, 106). Viruses with Thr 375 are more susceptible to inhibition by CD4mc than matched viruses with Ser 375 (73, 76). On the other hand, the His 375 residue in Clade AE HIV-1 fills the Phe-43 cavity and renders these viruses resistant to CD4mc (73, 76). Consideration of the impact of natural HIV-1 strain variation on CD4mc efficacy will be important for their successful application in therapy or prophylaxis.
MATERIALS AND METHODS
Ethics statement.
Written informed consent was obtained from all study participants and research adhered to the ethical guidelines of Centre de Recherche du CHUM (CRCHUM) and was reviewed and approved by the CRCHUM Institutional Review Board (Ethics Committee, approval no. CE 16.164 - CA). Research adhered to the standards indicated by the Declaration of Helsinki. All participants were adult and provided informed written consent prior to enrollment in accordance with Institutional Review Board approval.
Cell lines and primary cells.
293T cells were grown in Dulbecco’s modified Eagle’s medium (Life Technologies, Wisent Inc.) supplemented with 10% fetal bovine serum (FBS) (Life Technologies, VWR) and 100 μg/ml of penicillin-streptomycin (Life Technologies, Wisent Inc.). Cf2Th-CD4/CCR5 cells stably expressing the human CD4 and CCR5 coreceptors for HIV-1 were grown in the same medium supplemented with 0.4 mg/ml of G418 and 0.2 mg/ml of hygromycin. Cf2Th-CCR5 cells stably expressing the CCR5 coreceptor for HIV-1 were grown in the same medium supplemented with 0.4 mg/ml of G418. C8166-R5 cells were generously provided by Seth Pincus, Montana State University (107). The C8166-R5 cells were grown in RPMI 1640 medium (Life Technologies) with 10% FBS. Puromycin (Life Technologies) (1 μg/ml) was used for selection and was added every fifth passage.
Human peripheral blood mononuclear cells (PBMCs) from HIV-negative individuals were obtained by leukapheresis and Ficoll-Paque density gradient isolation and were cryopreserved in liquid nitrogen until use. CD4+ T lymphocytes were purified from resting PBMCs by negative selection using immunomagnetic beads according to the manufacturer’s instructions (StemCell Technologies). The cells were activated with phytohemagglutinin-L (10 μg/ml) for 48 h and then maintained in RPMI 1640 complete medium supplemented with recombinant interleukin-2 (rIL-2) (100 U/ml).
CD4-mimetic compound.
The small-molecule CD4-mimetic compound (CD4mc) BNM-III-170 was synthesized as described previously (108). The compounds were dissolved in DMSO at a stock concentration of 10 mM and diluted to 50 μM in phosphate-buffered saline (PBS) for cell-surface staining or in RPMI 1640 complete medium for ADCC assays.
Antibodies and plasma.
Antibodies and sCD4-Ig used in neutralization assays are described in reference 109. BMS-806 and BMS-529 were purchased from Selleckchem and APExBIO, respectively. The anti-coreceptor binding site 17b monoclonal antibody (MAb) (NIH HIV Reagent Program) and plasma from HIV-1-infected individuals were used to assess cell-surface Env conformation. Plasma from HIV-infected individuals was collected, heat-inactivated, and conserved at −80°C until use. The conformation-independent anti-gp120 outer-domain 2G12 MAb (NIH HIV Reagent Program) was used to normalize Env expression. Mouse anti-human CD4 (clone OKT4; Thermo Fisher Scientific) and mouse anti-human BST-2 (clone RS38E, PE-Cy7-conjugated; Biolegend, USA) were also used as primary antibodies for cell-surface staining. Goat anti-mouse IgG (H+L) and goat anti-human IgG (H+L) (Thermo Fisher Scientific) antibodies precoupled to Alexa Fluor 647 were used as secondary antibodies in flow cytometry experiments.
Selection of BNM-III-170-resistant HIV-1AD8.
293T cells were transfected with the pNL4-3-AD8 provirus construct using effectene transfection reagent (Qiagen). Forty-eight hours afterwards, the virus-containing cell medium was collected, clarified by low-speed centrifugation (600 × g for 10 min), and filtered through a 0.45-μm membrane. C8166-R5 cells were then incubated with the virus preparation. Twenty-four hours later, cells were washed and fresh medium was added. In the first two passages of the cells, the medium was supplemented with 5 μM BNM-III-170. The concentration of BNM-III-170 in the culture medium was gradually increased with every passage. Virus replication was monitored by a quantitative real-time PCR assay measuring the viral reverse transcriptase activity (110). Briefly, a small amount of cell supernatant was collected and lysed (lysis buffer: 50 mM Tris HCl pH 7.4, 25 mM KCl, 0.15% Triton X-100, 20% glycerol, and RNase inhibitor) as a source of reverse transcriptase. MS2 RNA (Roche Diagnostics) was used as the template. RT-PCR master mix and RNase inhibitor were obtained from Life technologies. The oligonucleotides (5′-tcctgctcaacttcctgtcga, 5′-cacaggtcaaacctcctaggaatg, and 6[FAM]cgagacgctaccatggctatcgctgtag[TAMsp]) for the reverse transcriptase assay were synthesized by Integrated DNA technologies.
RNA isolation and cloning.
Viral RNA was isolated from the supernatant of BNM-III-170-treated C8166-R5 cells using the QIAamp viral RNA minikit (Qiagen). cDNA was synthesized using Superscript III reverse transcriptase (Thermo Fisher Scientific) with gene-specific primer (5′-tcgtctcattctttcccttacagcaggccat). Using cDNA as the template, the HIV-1 env sequence was amplified using primers (forward primer: 5′-atctagaattcgatatgacaaaagccttaggcatctccta; reverse primer: 5′-tcaggcggccgcttatagcaaaatcctttccaagccct) flanking the Env region and cloned into pcDNA3.1 vector. The amplified envs were sequenced to identify changes potentially responsible for BNM-III-170 resistance.
The env sequence changes potentially associated with viral resistance to BNM-III-170 were introduced individually or in combination into the pSVIIIenv AD8 plasmid (to produce pseudoviruses in conjunction with a packaging plasmid and HIV-1 vector) or the pNL4-3-AD8 provirus plasmid (to produce infectious virions) using a Q5 site-directed mutagenesis kit (New England BioLabs).
The presence of the desired mutations was confirmed by DNA sequencing.
Expression and processing of the HIV-1AD8 Env variants associated with BNM-III-170 resistance.
HEK 293T cells were transfected transiently with pNL4-3-AD8 proviral plasmids with env variants. Forty-eight hours later, the cell supernatant was cleared (600 × g for 10 min) followed by filtration through a 0.45-μm membrane. The viruses were pelleted by centrifugation at 14,000 × g for 1.5 h at 4°C and lysed. The cell and virus lysates were Western blotted and probed with a goat polyclonal anti-gp120 antibody (Invitrogen), the 4E10 anti-gp41 antibody (NIH HIV Reagent Program), or a rabbit anti-Gag antibody (Abcam). The cell lysates were also Western blotted and probed with a mouse anti-β-actin antibody (Invitrogen).
His6-tagged HIV-1AD8 gp120 glycoproteins were produced by introducing the His6 tag at the gp120-gp41 junction. The gp120 glycoproteins with the BNM-III-170-resistance-associated changes and the corresponding WT HIV-1AD8 gp120 glycoprotein were produced transiently in 293T cells and purified by Ni-NTA affinity chromatography.
Production of recombinant pseudoviruses expressing luciferase.
As described previously (26), 293T cells were transfected with pSVIIIenv-AD8 plasmids expressing Env variants, the pCMVΔP1Δenv HIV-1 Gag-Pol packaging construct and the firefly luciferase-expressing HIV-1 vector at a 1:1:3 μg DNA ratio using effectene transfection reagent (Qiagen). Recombinant, luciferase-expressing viruses capable of a single round of replication were released into the cell medium and were harvested 48 h later. The virus-containing supernatants were clarified by low-speed centrifugation (600 × g for 10 min) and used for single-round infections.
Virus infectivity, neutralization, and cold sensitivity.
Single-round virus infection assays were used to measure the ability of the Env variants to support virus entry, as described previously (26). To measure the infectivity of the Env pseudotypes, recombinant viruses were added to Cf2Th-CD4/CCR5 target cells expressing CD4 and CCR5. Forty-eight hours later, the target cells were lysed and the luciferase activity was measured.
For neutralization assays, the compounds/antibodies to be tested were incubated with pseudoviruses for 1 h at 37°C. The mixture was then added to Cf2Th-CD4/CCR5 target cells expressing CD4 and CCR5. Forty-eight hours later, the target cells were lysed and the luciferase activity was measured.
To evaluate the cold sensitivity of the Env variants, pseudotyped recombinant viruses were incubated on ice for various lengths of time prior to measuring their infectivity, as described previously (26, 27, 79, 93).
gp120 shedding.
To measure BNM-III-170-induced gp120 shedding, 293T cells were transfected transiently with pNL4-3-AD8 provirus constructs. The transfected cell supernatants were harvested 48 h later, clarified by low-speed centrifugation (600 × g for 10 min) and filtered through a 0.45-μm membrane. The viruses were pelleted by centrifugation at 14,000 × g for 1.5 h at 4°C and resuspended in 100 μL of 1× PBS. The viruses were incubated with different concentrations of BNM-III-170 for 2.5 h at 37°C, followed by centrifugation at 14,000 × g for 1.5 h at 4°C. The virus pellets were lysed 1× LDS buffer and analyzed as described below. The supernatants containing shed gp120 were bound to Galanthus nivalis lectin (GNL) beads (Thermo Fisher Scientific). The glycoproteins captured on the beads and the lysates of virus pellets were Western blotted and probed with a goat anti-gp120 antibody, the 4E10 anti-gp41 antibody or a rabbit anti-Gag antibody. The Western blots were quantified using ImageJ software (111).
Immunoprecipitation of virion Envs with sCD4-Ig.
Virions produced following transfection with pNL4-3-AD8 proviral plasmids expressing Env variants were pelleted as described above. The pellets were incubated with sCD4-Ig at the indicated concentrations for 1.5 h at 37°C. After being washed with 1× PBS, viruses were lysed using buffer (5 M NaCl, 1.5 M Tris-HCl pH 7.4,1:20 dilution of 10% NP-40, and 1× protease inhibitor [Sigma-Aldrich]) and incubated with protein A-agarose beads (Thermo Fisher Scientific). The proteins captured on the beads were Western blotted and probed with a goat anti-gp120 antibody. The Western blots were quantified using ImageJ software (111).
Activation of virus infection by BNM-III-170 and sCD4-Ig.
Pseudoviruses were incubated with CD4-negative, CCR5-expressing Cf2Th-CCR5 cells in 96-well plates. The plates were centrifuged at 600 × g for 30 min at 21°C. Medium containing serial dilutions of sCD4-Ig or BNM-III-170 was then added. Forty-eight hours later, cells were lysed, and luciferase activity was measured.
Biolayer interferometry.
Binding kinetics were performed on an Octet RED96e system (FortéBio) at 25°C with shaking at 1,000 rpm. Amine-Reactive Second-Generation (AR2G) biosensors were hydrated in water and then activated for 300 s with an S-NHS/EDC solution (Fortébio) prior to amine coupling. WT and mutant HIV-1AD8 gp120 glycoproteins were loaded into the activated AR2G biosensor at 12.5 μg/ml in 10 mM acetate solution pH 5 (Fortébio) for 600 s and then quenched into 1 M ethanolamine solution pH 8.5 (Fortébio) for 300 s. Baseline equilibration was collected for 120 s in 10× kinetics buffer. Association of sCD4 (in 10× kinetics buffer) to the different gp120 glycoproteins was carried out for 180 s at various concentrations in a 2-fold dilution series from 250 nM to 15.625 nM prior to dissociation for 300 s. The data were baseline subtracted prior to fitting, which was performed using a 1:1 binding model and the FortéBio data analysis software. On-rates (ka), off-rates (kdis), and equilibrium dissociation constants (KD) were computed using a global fit applied to all data.
Establishment of primary CD4+ T cells infected with Env variants.
The vesicular stomatitis virus G (VSV-G)-encoding plasmid was previously described (112). Vesicular stomatitis virus G (VSV-G)-pseudotyped HIV-1 vectors were produced by cotransfection of 293T cells with HIV-1 proviral constructs encoding the variant Envs and a VSV-G-encoding plasmid using the calcium phosphate method. Two days later, cell supernatants were harvested, clarified by low-speed centrifugation (300 × g for 5 min), and concentrated by ultracentrifugation at 4°C (100,605 × g for 1 h) through a 20% sucrose cushion. Pellets were resuspended in fresh RPMI medium, and aliquots were stored at −80°C until use. Viruses were then used to infect activated primary CD4+ T cells from healthy HIV-1-negative donors by spin infection at 800 × g for 1 h in 96-well plates at 25°C. Viral preparations were titrated directly on primary CD4+ T cells to achieve similar levels of infection among the different proviruses used.
Flow cytometry analysis of cell-surface staining.
Cell-surface staining was performed 48 h after infection of primary CD4+ T cells. Mock-infected or HIV-1-infected primary CD4+ T cells were incubated for 45 min at 37°C with anti-CD4 (0.5 μg/ml), anti-BST-2 (2 μg/ml), anti-Env MAbs (5 μg/ml) or plasma (1:1000 dilution) in the presence or absence of the CD4mc BNM-III-170 (50 μM) or equivalent volume of DMSO. Cells were then washed twice with PBS and stained with the appropriate Alexa Fluor 647-conjugated secondary antibody (2 μg/ml), when needed, for 20 min at room temperature. After two more PBS washes, cells were fixed in a 2% PBS-formaldehyde solution. Infected cells were then permeabilized using the Cytofix/Cytoperm Fixation/Permeabilization kit (BD Biosciences) and stained intracellularly using PE-conjugated mouse anti-p24 MAb (clone KC57; Beckman Coulter, Brea, CA, USA; 1:100 dilution). The percentage of infected cells (p24+) was determined by gating on the living cell population according to staining with a viability dye (Aqua Vivid; Thermo Fisher Scientific). Data were acquired on an LSR II cytometer (BD Biosciences), and data analysis was performed using FlowJo v10.5.3 (Tree Star).
Statistics.
The concentrations of antibodies and other inhibitors that inhibit 50% of infection (IC50 values) were determined by fitting the data in five-parameter dose-response curves using GraphPad Prism 8. Spearman rank-order correlation coefficients (rS) and P values were calculated using VassarStats (113).
ACKNOWLEDGMENTS
We thank Elizabeth Carpelan for manuscript preparation. Antibodies against HIV-1 were kindly supplied by Dennis Burton (Scripps), Peter Kwong and John Mascola (Vaccine Research Center NIH), Barton Haynes (Duke University), Hermann Katinger (Polymun), James Robinson (Tulane University), and Marshall Posner (Mount Sinai Medical Center). We thank the NIH HIV Reagent Program for providing additional reagents. We thank Mario Legault from the FRQS AIDS and Infectious Diseases network for cohort coordination and clinical samples.
This work was supported by grants from the National Institutes of Health (grants AI145547, AI124982, AI129017, AI164562, AI150471, AI148379, AI150322, and AI129769), HIV Cure Research Grant from Gilead Sciences, and a gift from the late William F. McCarty-Cooper. This study was partially supported by a Canadian Institutes of Health Research (CIHR) foundation grant no. 352417, Team grant no. 422148 to A.F., and a Canada Foundation for Innovation (CFI) grant no. 41027 to A.F. A.F. is the recipient of a Canada Research Chair on Retroviral Entry no. RCHS0235 950-232424.
We declare no conflicts of interest.
Contributor Information
Joseph Sodroski, Email: Joseph_Sodroski@dfci.harvard.edu.
Viviana Simon, Icahn School of Medicine at Mount Sinai.
REFERENCES
- 1.Wyatt R, Sodroski J. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280:1884–1888. 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 2.Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. 1984. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature 312:767–768. 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 3.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763–767. 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 4.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. 1996. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 384:179–183. 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 5.Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, Cheng-Mayer C, Robinson J, Maddon PJ, Moore JP. 1996. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature 384:184–187. 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- 6.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. 1996. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85:1135–1148. 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 7.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661–666. 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 8.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381:667–673. 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 9.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. 1996. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85:1149–1158. 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 10.Feng Y, Broder CC, Kennedy PE, Berger EA. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872–877. 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 11.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. 1996. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955–1958. 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 12.Munro JB, Gorman J, Ma X, Zhou Z, Arthos J, Burton DR, Koff WC, Courter JR, Smith AB, III, Kwong PD, Blanchard SC, Mothes W. 2014. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 346:759–763. 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herschhorn A, Ma X, Gu C, Ventura JD, Castillo-Menendez L, Melillo B, Terry DS, Smith AB, III, Blanchard SC, Munro JB, Mothes W, Finzi A, Sodroski J. 2016. Release of gp120 restraints leads to an entry-competent intermediate state of the HIV-1 envelope glycoproteins. mBio 7:e01598-16. 10.1128/mBio.01598-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma X, Lu M, Gorman J, Terry DS, Hong X, Zhou Z, Zhao H, Altman RB, Arthos J, Blanchard SC, Kwong PD, Munro JB, Mothes W. 2018. HIV-1 Env trimer opens through an asymmetric intermediate in which individual protomers adopt distinct conformations. Elife 7:e34271. 10.7554/eLife.34271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu M, Ma X, Reichard N, Terry DS, Arthos J, Smith AB, III, Sodroski JG, Blanchard SC, Mothes W. 2020. Shedding-resistant HIV-1 envelope glycoproteins adopt downstream conformations that remain responsive to conformation-preferring ligands. J Virol 94:e00597-20. 10.1128/JVI.00597-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furuta RA, Wild CT, Weng Y, Weiss CD. 1998. Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Biol 5:276–279. 10.1038/nsb0498-276. [DOI] [PubMed] [Google Scholar]
- 17.Koshiba T, Chan DC. 2003. The prefusogenic intermediate of HIV-1 gp41 contains exposed C-peptide regions. J Biol Chem 278:7573–7579. 10.1074/jbc.M211154200. [DOI] [PubMed] [Google Scholar]
- 18.He Y, Vassell R, Zaitseva M, Nguyen N, Yang Z, Weng Y, Weiss CD. 2003. Peptides trap the human immunodeficiency virus type 1 envelope glycoprotein fusion intermediate at two sites. J Virol 77:1666–1671. 10.1128/jvi.77.3.1666-1671.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Si Z, Madani N, Cox JM, Chruma JJ, Klein JC, Schon A, Phan N, Wang L, Biorn AC, Cocklin S, Chaiken I, Freire E, Smith AB, III, Sodroski JG. 2004. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc Natl Acad Sci USA 101:5036–5041. 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. 10.1016/S0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 21.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430. 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 22.Lu M, Blacklow SC, Kim PS. 1995. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat Struct Biol 2:1075–1082. 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 23.Wilen CB, Tilton JC, Doms RW. 2012. Molecular mechanisms of HIV entry. Adv Exp Med Biol 726:223–242. 10.1007/978-1-4614-0980-9_10. [DOI] [PubMed] [Google Scholar]
- 24.Melikyan GB, Markosyan RM, Hemmati H, Delmedico MK, Lambert DM, Cohen FS. 2000. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J Cell Biol 151:413–423. 10.1083/jcb.151.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuhmann SE, Platt EJ, Kozak SL, Kabat D. 2000. Cooperation of multiple CCR5 coreceptors is required for infections by human immunodeficiency virus type 1. J Virol 74:7005–7015. 10.1128/jvi.74.15.7005-7015.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haim H, Strack B, Kassa A, Madani N, Wang L, Courter JR, Princiotto A, McGee K, Pacheco B, Seaman MS, Smith AB, III, Sodroski J. 2011. Contribution of intrinsic reactivity of the HIV-1 envelope glycoproteins to CD4-independent infection and global inhibitor sensitivity. PLoS Pathog 7:e1002101. 10.1371/journal.ppat.1002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haim H, Salas I, McGee K, Eichelberger N, Winter E, Pacheco B, Sodroski J. 2013. Modeling virus- and antibody-specific factors to predict human immunodeficiency virus neutralization efficiency. Cell Host Microbe 14:547–558. 10.1016/j.chom.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guttman M, Cupo A, Julien JP, Sanders RW, Wilson IA, Moore JP, Lee KK. 2015. Antibody potency relates to the ability to recognize the closed, pre-fusion form of HIV Env. Nat Commun 6:6144. 10.1038/ncomms7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, Steenbeke TD, Venturi M, Chaiken I, Fung M, Katinger H, Parren PW, Robinson J, Van Ryk D, Wang L, Burton DR, Freire E, Wyatt R, Sodroski J, Hendrickson WA, Arthos J. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:678–682. 10.1038/nature01188. [DOI] [PubMed] [Google Scholar]
- 30.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659. 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schon A, Madani N, Klein JC, Hubicki A, Ng D, Yang X, Smith AB, III, Sodroski J, Freire E. 2006. Thermodynamics of binding of a low-molecular-weight CD4 mimetic to HIV-1 gp120. Biochemistry 45:10973–10980. 10.1021/bi061193r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herschhorn A, Gu C, Espy N, Richard J, Finzi A, Sodroski JG. 2014. A broad HIV-1 inhibitor blocks envelope glycoprotein transitions critical for entry. Nat Chem Biol 10:845–852. 10.1038/nchembio.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Madani N, Perdigoto AL, Srinivasan K, Cox JM, Chruma JJ, LaLonde J, Head M, Smith AB, III, Sodroski JG. 2004. Localized changes in the gp120 envelope glycoprotein confer resistance to human immunodeficiency virus entry inhibitors BMS-806 and #155. J Virol 78:3742–3752. 10.1128/JVI.78.7.3742-3752.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pancera M, Lai Y-T, Bylund T, Druz A, Narpala S, O'Dell S, Schön A, Bailer RT, Chuang G-Y, Geng H, Louder MK, Rawi R, Soumana DI, Finzi A, Herschhorn A, Madani N, Sodroski J, Freire E, Langley DR, Mascola JR, McDermott AB, Kwong PD. 2017. Crystal structures of trimeric HIV envelope with entry inhibitors BMS-378806 and BMS-626529. Nat Chem Biol 13:1115–1122. 10.1038/nchembio.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madani N, Schon A, Princiotto AM, Lalonde JM, Courter JR, Soeta T, Ng D, Wang L, Brower ET, Xiang SH, Kwon YD, Huang CC, Wyatt R, Kwong PD, Freire E, Smith AB, III, Sodroski J. 2008. Small-molecule CD4 mimics interact with a highly conserved pocket on HIV-1 gp120. Structure 16:1689–1701. 10.1016/j.str.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwon YD, LaLonde JM, Yang Y, Elban MA, Sugawara A, Courter JR, Jones DM, Smith AB, III, Debnath AK, Kwong PD. 2014. Crystal structures of HIV-1 gp120 envelope glycoprotein in complex with NBD analogues that target the CD4-binding site. PLoS One 9:e85940. 10.1371/journal.pone.0085940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melillo B, Liang S, Park J, Schon A, Courter JR, LaLonde JM, Wendler DJ, Princiotto AM, Seaman MS, Freire E, Sodroski J, Madani N, Hendrickson WA, Smith AB, III. 2016. Small-molecule CD4-mimics: structure-based optimization of HIV-1 entry inhibition. ACS Med Chem Lett 7:330–334. 10.1021/acsmedchemlett.5b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Curreli F, Kwon YD, Belov DS, Ramesh RR, Kurkin AV, Altieri A, Kwong PD, Debnath AK. 2017. Synthesis, antiviral potency, in vitro ADMET, and X-ray structure of potent CD4 mimics as entry inhibitors that target the Phe43 cavity of HIV-1 gp120. J Med Chem 60:3124–3153. 10.1021/acs.jmedchem.7b00179. [DOI] [PubMed] [Google Scholar]
- 39.Herschhorn A, Gu C, Moraca F, Ma X, Farrell M, Smith AB, III, Pancera M, Kwong PD, Schön A, Freire E, Abrams C, Blanchard SC, Mothes W, Sodroski JG. 2017. The β20-β21 of gp120 is a regulatory switch for HIV-1 Env conformational transitions. Nat Commun 8:1049. 10.1038/s41467-017-01119-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. 2005. Identification of N-phenyl-N'-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 339:213–225. 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 41.Haim H, Si Z, Madani N, Wang L, Courter JR, Princiotto A, Kassa A, DeGrace M, McGee-Estrada K, Mefford M, Gabuzda D, Smith AB, III, Sodroski J. 2009. Soluble CD4 and CD4-mimetic compounds inhibit HIV-1 infection by induction of a short-lived activated state. PLoS Pathog 5:e1000360. 10.1371/journal.ppat.1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madani N, Princiotto AM, Zhao C, Jahanbakhshsefidi F, Mertens M, Herschhorn A, Melillo B, Smith AB, III, Sodroski J. 2017. Activation and inactivation of primary human immunodeficiency virus envelope glycoprotein trimers by CD4-mimetic compounds. J Virol 91:e01880-16. 10.1128/JVI.01880-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madani N, Princiotto AM, Mach L, Ding S, Prévost J, Richard J, Hora B, Sutherland L, Zhao CA, Conn BP, Bradley T, Moody MA, Melillo B, Finzi A, Haynes BF, Smith AB, III, Santra S, Sodroski J. 2018. A CD4-mimetic compound enhances vaccine efficacy against stringent immunodeficiency virus challenge. Nat Commun 9:2363. 10.1038/s41467-018-04758-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoshimura K, Harada S, Shibata J, Hatada M, Yamada Y, Ochiai C, Tamamura H, Matsushita S. 2010. Enhanced exposure of human immunodeficiency virus type 1 primary isolate neutralization epitopes through binding of CD4 mimetic compounds. J Virol 84:7558–7568. 10.1128/JVI.00227-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madani N, Princiotto AM, Schon A, LaLonde J, Feng Y, Freire E, Park J, Courter JR, Jones DM, Robinson J, Liao HX, Moody MA, Permar S, Haynes B, Smith AB, III, Wyatt R, Sodroski J. 2014. CD4-mimetic small molecules sensitize human immunodeficiency virus to vaccine-elicited antibodies. J Virol 88:6542–6555. 10.1128/JVI.00540-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Veillette M, Désormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, Haynes BF, Robinson JE, Kaufmann DE, Bonsignori M, Sodroski J, Finzi A. 2014. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J Virol 88:2633–2644. 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veillette M, Coutu M, Richard J, Batraville LA, Dagher O, Bernard N, Tremblay C, Kaufmann DE, Roger M, Finzi A. 2015. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J Virol 89:545–551. 10.1128/JVI.02868-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding S, Gasser R, Gendron-Lepage G, Medjahed H, Tolbert WD, Sodroski J, Pazgier M, Finzi A. 2019. CD4 incorporation into HIV-1 viral particles exposes envelope epitopes recognized by CD4-induced antibodies. J Virol 93:e01403-19. 10.1128/JVI.01403-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Richard J, Veillette M, Ding S, Zoubchenok D, Alsahafi N, Coutu M, Brassard N, Park J, Courter JR, Melillo B, Smith AB, III, Shaw GM, Hahn BH, Sodroski J, Kaufmann DE, Finzi A. 2016. Small CD4 mimetics prevent HIV-1 uninfected bystander CD4 + T cell killing mediated by antibody-dependent cell-mediated cytotoxicity. EBioMedicine 3:122–134. 10.1016/j.ebiom.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richard J, Nguyen DN, Tolbert WD, Gasser R, Ding S, Vézina D, Yu Gong S, Prévost J, Gendron-Lepage G, Medjahed H, Gottumukkala S, Finzi A, Pazgier M. 2021. Across functional boundaries: making nonneutralizing antibodies to neutralize HIV-1 and mediate Fc-mediated effector killing of infected cells. mBio 12:e0140521. 10.1128/mBio.01405-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thali M, Moore JP, Furman C, Charles M, Ho DD, Robinson J, Sodroski J. 1993. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J Virol 67:3978–3988. 10.1128/JVI.67.7.3978-3988.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rizzuto C, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong PD, Hendrickson WA, Sodroski J. 1998. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280:1949–1953. 10.1126/science.280.5371.1949. [DOI] [PubMed] [Google Scholar]
- 53.Richard J, Pacheco B, Gohain N, Veillette M, Ding S, Alsahafi N, Tolbert WD, Prévost J, Chapleau JP, Coutu M, Jia M, Brassard N, Park J, Courter JR, Melillo B, Martin L, Tremblay C, Hahn BH, Kaufmann DE, Wu X, Smith AB, III, Sodroski J, Pazgier M, Finzi A. 2016. Co-receptor binding site antibodies enable CD4-mimetics to expose conserved anti-cluster A ADCC epitopes on HIV-1 envelope glycoproteins. EBioMedicine 12:208–218. 10.1016/j.ebiom.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alsahafi N, Bakouche N, Kazemi M, Richard J, Ding S, Bhattacharyya S, Das D, Anand SP, Prévost J, Tolbert WD, Lu H, Medjahed H, Gendron-Lepage G, Ortega Delgado GG, Kirk S, Melillo B, Mothes W, Sodroski J, Smith AB, III, Kaufmann DE, Wu X, Pazgier M, Rouiller I, Finzi A, Munro JB. 2019. An asymmetric opening of HIV-1 envelope mediates antibody-dependent cellular cytotoxicity. Cell Host Microbe 25:578–587.e5. 10.1016/j.chom.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pham TN, Lukhele S, Hajjar F, Routy JP, Cohen ÉA. 2014. HIV Nef and Vpu protect HIV-infected CD4+ T cells from antibody-mediated cell lysis through down-modulation of CD4 and BST2. Retrovirology 11:15. 10.1186/1742-4690-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wildum S, Schindler M, Münch J, Kirchhoff F. 2006. Contribution of Vpu, Env, and Nef to CD4 down-modulation and resistance of human immunodeficiency virus type 1-infected T cells to superinfection. J Virol 80:8047–8059. 10.1128/JVI.00252-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kwon Y, Kaake RM, Echeverria I, Suarez M, Karimian Shamsabadi M, Stoneham C, Ramirez PW, Kress J, Singh R, Sali A, Krogan N, Guatelli J, Jia X. 2020. Structural basis of CD4 downregulation by HIV-1 Nef. Nat Struct Mol Biol 27:822–828. 10.1038/s41594-020-0463-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prévost J, Anand SP, Rajashekar JK, Richard J, Goyette G, Medjahed H, Gendron-Lepage G, Chen H-C, Chen Y, Horwitz JA, Grunst MW, Zolla-Pazner S, Haynes BF, Burton DR, Flavell RA, Kirchhoff F, Hahn BH, Smith AB, III, Pazgier M, Nussenzweig MC, Kumar P, Finzi A. 2022. HIV-1 Vpu restricts Fc-mediated effector functions in vivo. bioRxiv. 10.1101/2022.02.21.481308. [DOI] [PMC free article] [PubMed]
- 59.Chen BK, Gandhi RT, Baltimore D. 1996. CD4 down-modulation during infection of human T cells with human immunodeficiency virus type 1 involves independent activities of vpu, env, and nef. J Virol 70:6044–6053. 10.1128/JVI.70.9.6044-6053.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arias JF, Heyer LN, von Bredow B, Weisgrau KL, Moldt B, Burton DR, Rakasz EG, Evans DT. 2014. Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc Natl Acad Sci USA 111:6425–6430. 10.1073/pnas.1321507111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.von Bredow B, Arias JF, Heyer LN, Gardner MR, Farzan M, Rakasz EG, Evans DT. 2015. Envelope glycoprotein internalization protects human and simian immunodeficiency virus-infected cells from antibody-dependent cell-mediated cytotoxicity. J Virol 89:10648–10655. 10.1128/JVI.01911-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richard J, Veillette M, Brassard N, Iyer SS, Roger M, Martin L, Pazgier M, Schon A, Freire E, Routy JP, Smith AB, III, Park J, Jones DM, Courter JR, Melillo BN, Kaufmann DE, Hahn BH, Permar SR, Haynes BF, Madani N, Sodroski JG, Finzi A. 2015. CD4 mimetics sensitize HIV-1-infected cells to ADCC. Proc Natl Acad Sci USA 112:E2687–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ding S, Verly MM, Princiotto A, Melillo B, Moody AM, Bradley T, Easterhoff D, Roger M, Hahn BH, Madani N, Smith AB, III, Haynes BF, Sodroski J, Finzi A. 2017. Short Communication: small-molecule CD4 mimetics sensitize HIV-1-infected cells to antibody-dependent cellular cytotoxicity by antibodies elicited by multiple envelope glycoprotein immunogens in nonhuman primates. AIDS Res Hum Retroviruses 33:428–431. 10.1089/AID.2016.0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ding S, Grenier MC, Tolbert WD, Vezina D, Sherburn R, Richard J, Prévost J, Chapleau JP, Gendron-Lepage G, Medjahed H, Abrams C, Sodroski J, Pazgier M, Smith AB, III, Finzi A. 2019. A new family of small-molecule CD4-mimetic compounds contacts highly conserved aspartic acid 368 of HIV-1 gp120 and mediates antibody-dependent cellular cytotoxicity. J Virol 93:e01325-19. 10.1128/JVI.01325-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Prévost J, Richard J, Gasser R, Medjahed H, Kirchhoff F, Hahn BH, Kappes JC, Ochsenbauer C, Duerr R, Finzi A. 2022. Detection of the HIV-1 accessory proteins Nef and Vpu by flow cytometry represents a new tool to study their functional interplay within a single infected CD4+ T cell. J Virol 96:e0192921. 10.1128/jvi.01929-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prévost J, Richard J, Medjahed H, Alexander A, Jones J, Kappes JC, Ochsenbauer C, Finzi A. 2018. Incomplete downregulation of CD4 expression affects HIV-1 Env conformation and antibody-dependent cellular cytotoxicity responses. J Virol 92:e00484-18. 10.1128/JVI.00484-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alsahafi N, Ding S, Richard J, Markle T, Brassard N, Walker B, Lewis GK, Kaufmann DE, Brockman MA, Finzi A. 2015. Nef proteins from HIV-1 elite controllers are inefficient at preventing antibody-dependent cellular cytotoxicity. J Virol 90:2993–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rajashekar JK, Richard J, Beloor J, Prévost J, Anand SP, Beaudoin-Bussières G, Shan L, Herndler-Brandstetter D, Gendron-Lepage G, Medjahed H, Bourassa C, Gaudette F, Ullah I, Symmes K, Peric A, Lindemuth E, Bibollet-Ruche F, Park J, Chen H-C, Kaufmann DE, Hahn BH, Sodroski J, Pazgier M, Flavell RA, Smith AB, Finzi A, Kumar P. 2021. Modulating HIV-1 envelope glycoprotein conformation to decrease the HIV-1 reservoir. Cell Host Microbe 29:904–916. 10.1016/j.chom.2021.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Princiotto AM, Vrbanac VD, Melillo B, Park J, Tager AM, Smith AB, III, Sodroski J, Madani N. 2018. A small-molecule CD4-mimetic compound protects bone marrow-liver-thymus humanized mice from HIV-1 infection. J Infect Dis 218:471–475. 10.1093/infdis/jiy174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Curreli F, Kwon YD, Zhang H, Yang Y, Scacalossi D, Kwong PD, Debnath AK. 2014. Binding mode characterization of NBD series CD4-mimetic HIV-1 entry inhibitors by X-ray structure and resistance study. Antimicrob Agents Chemother 58:5478–5491. 10.1128/AAC.03339-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.LaLonde JM, Kwon YD, Jones DM, Sun AW, Courter JR, Soeta T, Kobayashi T, Princiotto AM, Wu X, Schon A, Freire E, Kwong PD, Mascola JR, Sodroski J, Madani N, Smith AB, III. 2012. Structure-based design, synthesis, and characterization of dual hotspot small-molecule HIV-1 entry inhibitors. J Med Chem 55:4382–4396. 10.1021/jm300265j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Courter JR, Madani N, Sodroski J, Schön A, Freire E, Kwong PD, Hendrickson WA, Chaiken IM, LaLonde JM, Smith AB, III. 2014. Structure-based design, synthesis and validation of CD4-mimetic small molecule inhibitors of HIV-1 entry: conversion of a viral entry agonist to an antagonist. Acc Chem Res 47:1228–1237. 10.1021/ar4002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zoubchenok D, Veillette M, Prévost J, Sanders-Buell E, Wagh K, Korber B, Chenine AL, Finzi A. 2017. Histidine 375 modulates CD4 binding in HIV-1 CRF01_AE envelope glycoproteins. J Virol 91:e02151-16. 10.1128/JVI.02151-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Foley BLT, Apetrei C, Hahn B, Mizrachi I, Mullins J, Rambaut A, Wolinsky S, Korber B, ed 2014. HIV sequence compendium 2014. Los Alamos National Laboratory, Theoretical Biology and Biophysics, Los Alamos, NM; LA-UR-14–26717. [Google Scholar]
- 75.Davey NE, Satagopam VP, Santiago-Mozos S, Villacorta-Martin C, Bharat TA, Schneider R, Briggs JA. 2014. The HIV mutation browser: a resource for human immunodeficiency virus mutagenesis and polymorphism data. PLoS Comput Biol 10:e1003951. 10.1371/journal.pcbi.1003951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Prévost J, Tolbert WD, Medjahed H, Sherburn RT, Madani N, Zoubchenok D, Gendron-Lepage G, Gaffney AE, Grenier MC, Kirk S, Vergara N, Han C, Mann BT, Chenine AL, Ahmed A, Chaiken I, Kirchhoff F, Hahn BH, Haim H, Abrams CF, Smith AB, III, Sodroski J, Pazgier M, Finzi A. 2020. The HIV-1 Env gp120 inner domain shapes the Phe43 cavity and the CD4 binding site. mBio 11:e00280-20. 10.1128/mBio.00280-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jette CA, Barnes CO, Kirk SM, Melillo B, Smith AB, III, Bjorkman PJ. 2021. Cryo-EM structures of HIV-1 trimer bound to CD4-mimetics BNM-III-170 and M48U1 adopt a CD4-bound open conformation. Nat Commun 12:1950. 10.1038/s41467-021-21816-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kassa A, Madani N, Schon A, Haim H, Finzi A, Xiang SH, Wang L, Princiotto A, Pancera M, Courter J, Smith AB, III, Freire E, Kwong PD, Sodroski J. 2009. Transitions to and from the CD4-bound conformation are modulated by a single-residue change in the human immunodeficiency virus type 1 gp120 inner domain. J Virol 83:8364–8378. 10.1128/JVI.00594-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nguyen HT, Qualizza A, Anang S, Zhao M, Zou S, Zhou R, Wang Q, Zhang S, Deshpande A, Ding H, Chiu TJ, Smith AB, III, Kappes JC, Sodroski JG. 2022. Functional and highly cross-linkable HIV-1 envelope glycoproteins enriched in a pretriggered conformation. J Virol 96:e0166821. 10.1128/jvi.01668-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Finzi A, Xiang S-H, Pacheco B, Wang L, Haight J, Kassa A, Danek B, Pancera M, Kwong PD, Sodroski J. 2010. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol Cell 37:656–667. 10.1016/j.molcel.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pacheco B, Alsahafi N, Debbeche O, Prévost J, Ding S, Chapleau JP, Herschhorn A, Madani N, Princiotto A, Melillo B, Gu C, Zeng X, Mao Y, Smith AB, III, Sodroski J, Finzi A. 2017. Residues in the gp41 ectodomain regulate HIV-1 envelope glycoprotein conformational transitions induced by gp120-directed inhibitors. J Virol 91:e02219-16. 10.1128/JVI.02219-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lai YT, Wang T, O'Dell S, Louder MK, Schon A, Cheung CSF, Chuang GY, Druz A, Lin B, McKee K, Peng D, Yang Y, Zhang B, Herschhorn A, Sodroski J, Bailer RT, Doria-Rose NA, Mascola JR, Langley DR, Kwong PD. 2019. Lattice engineering enables definition of molecular features allowing for potent small-molecule inhibition of HIV-1 entry. Nat Commun 10:47. 10.1038/s41467-018-07851-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. 2003. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci USA 100:11013–11018. 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang T, Zhang Z, Wallace OB, Deshpande M, Fang H, Yang Z, Zadjura LM, Tweedie DL, Huang S, Zhao F, Ranadive S, Robinson BS, Gong YF, Ricarrdi K, Spicer TP, Deminie C, Rose R, Wang HG, Blair WS, Shi PY, Lin PF, Colonno RJ, Meanwell NA. 2003. Discovery of 4-benzoyl-1-[(4-methoxy-1H- pyrrolo[2,3-b]pyridin-3-yl)oxoacetyl]-2- (R)-methylpiperazine (BMS-378806): a novel HIV-1 attachment inhibitor that interferes with CD4-gp120 interactions. J Med Chem 46:4236–4239. 10.1021/jm034082o. [DOI] [PubMed] [Google Scholar]
- 85.Wang T, Ueda Y, Zhang Z, Yin Z, Matiskella J, Pearce BC, Yang Z, Zheng M, Parker DD, Yamanaka GA, Gong YF, Ho HT, Colonno RJ, Langley DR, Lin PF, Meanwell NA, Kadow JF. 2018. Discovery of the human immunodeficiency virus type 1 (HIV-1) attachment inhibitor temsavir and its phosphonooxymethyl prodrug fostemsavir. J Med Chem 61:6308–6327. 10.1021/acs.jmedchem.8b00759. [DOI] [PubMed] [Google Scholar]
- 86.Zou S, Zhang S, Gaffney A, Ding H, Lu M, Grover JR, Farrell M, Nguyen HT, Zhao C, Anang S, Zhao M, Mohammadi M, Blanchard SC, Abrams C, Madani N, Mothes W, Kappes JC, Smith AB, III, Sodroski J. 2020. Long-acting BMS-378806 analogues stabilize the State-1 conformation of the human immunodeficiency virus type 1 envelope glycoproteins. J Virol 94:e00148-20. 10.1128/JVI.00148-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pancera M, Majeed S, Ban YE, Chen L, Huang CC, Kong L, Kwon YD, Stuckey J, Zhou T, Robinson JE, Schief WR, Sodroski J, Wyatt R, Kwong PD. 2010. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc Natl Acad Sci USA 107:1166–1171. 10.1073/pnas.0911004107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moore JP, McKeating JA, Weiss RA, Sattentau QJ. 1990. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science 250:1139–1142. 10.1126/science.2251501. [DOI] [PubMed] [Google Scholar]
- 89.Fu YK, Hart TK, Jonak ZL, Bugelski PJ. 1993. Physicochemical dissociation of CD4-mediated syncytium formation and shedding of human immunodeficiency virus type 1 gp120. J Virol 67:3818–3825. 10.1128/JVI.67.7.3818-3825.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Clapham PR, McKnight A, Weiss RA. 1992. Human immunodeficiency virus type 2 infection and fusion of CD4-negative human cell lines: induction and enhancement by soluble CD4. J Virol 66:3531–3537. 10.1128/JVI.66.6.3531-3537.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schenten D, Marcon L, Karlsson GB, Parolin C, Kodama T, Gerard N, Sodroski J. 1999. Effects of soluble CD4 on simian immunodeficiency virus infection of CD4-positive and CD4-negative cells. J Virol 73:5373–5380. 10.1128/JVI.73.7.5373-5380.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Medjahed H, Pacheco B, Désormeaux A, Sodroski J, Finzi A. 2013. The HIV-1 gp120 major variable regions modulate cold inactivation. J Virol 87:4103–4111. 10.1128/JVI.03124-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kassa A, Finzi A, Pancera M, Courter JR, Smith AB, III, Sodroski J. 2009. Identification of a human immunodeficiency virus (HIV-1) envelope glycoprotein variant resistant to cold inactivation. J Virol 83:4476–4488. 10.1128/JVI.02110-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med 4:1302–1307. 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 95.Chen CH, Matthews TJ, McDanal CB, Bolognesi DP, Greenberg ML. 1995. A molecular clasp in the human immunodeficiency virus (HIV) type 1 TM protein determines the anti-HIV activity of gp41 derivatives: implication for viral fusion. J Virol 69:3771–3777. 10.1128/JVI.69.6.3771-3777.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu Q, Zhang P, Lusso P. 2021. Quaternary interaction of the HIV-1 envelope trimer with CD4 and neutralizing antibodies. Viruses 13:1405. 10.3390/v13071405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu Q, Acharya P, Dolan MA, Zhang P, Guzzo C, Lu J, Kwon A, Gururani D, Miao H, Bylund T, Chuang GY, Druz A, Zhou T, Rice WJ, Wigge C, Carragher B, Potter CS, Kwong PD, Lusso P. 2017. Quaternary contact in the initial interactionof CD4 with the HIV-1 envelope trimer. Nat Struct Mol Biol 24:370–378. 10.1038/nsmb.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, Ward AB, Wilson IA. 2013. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 342:1477–1483. 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee JH, Ozorowski G, Ward AB. 2016. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 351:1043–1048. 10.1126/science.aad2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang GY, Ofek G, Stewart-Jones GB, Stuckey J, Bailer RT, Joyce MG, Louder MK, Tumba N, Yang Y, Zhang B, Cohen MS, Haynes BF, Mascola JR, Morris L, Munro JB, Blanchard SC, Mothes W, Connors M, Kwong PD. 2014. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature 514:455–461. 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang X, Ding X, Zhu Y, Chong H, Cui S, He J, Wang X, He Y. 2019. Structural and functional characterization of HIV-1 cell fusion inhibitor T20. AIDS 33:1–11. 10.1097/QAD.0000000000001979. [DOI] [PubMed] [Google Scholar]
- 102.Ding X, Zhang X, Chong H, Zhu Y, Wei H, Wu X, He J, Wang X, He Y. 2017. Enfuvirtide (T20)-based lipopeptide is a potent HIV-1 cell fusion inhibitor: implications for viral entry and inhibition. J Virol 91:e00831-17. 10.1128/JVI.00831-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG. 1998. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393:705–711. 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 104.Zhou N, Nowicka-Sans B, Zhang S, Fan L, Fang J, Fang H, Gong YF, Eggers B, Langley DR, Wang T, Kadow J, Grasela D, Hanna GJ, Alexander L, Colonno R, Krystal M, Lin PF. 2011. In vivo patterns of resistance to the HIV attachment inhibitor BMS-488043. Antimicrob Agents Chemother 55:729–737. 10.1128/AAC.01173-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lataillade M, Zhou N, Joshi SR, Lee S, Stock DA, Hanna GJ, Krystal M, AI438011 study team . 2018. Viral drug resistance through 48 weeks, in a phase 2b, randomized, controlled trial of the HIV-1 attachment inhibitor prodrug, fostemsavir. J Acquir Immune Defic Syndr 77:299–307. 10.1097/QAI.0000000000001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Richard J, Ding S, Finzi A. 2017. Unlocking HIV-1 Env: implications for antibody attack. AIDS Res Ther 14:42. 10.1186/s12981-017-0168-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Krowicka H, Robinson JE, Clark R, Hager S, Broyles S, Pincus SH. 2008. Use of tissue culture cell lines to evaluate HIV antiviral resistance. AIDS Res Hum Retroviruses 24:957–967. 10.1089/aid.2007.0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen J, Park J, Kirk SM, Chen HC, Li X, Lippincott DJ, Melillo B, Smith AB, III. 2019. Development of an effective scalable enantioselective synthesis of the HIV-1 entry inhibitor BNM-III-170 as the bis-trifluoroacetate salt. Org Process Res Dev 23:2464–2469. 10.1021/acs.oprd.9b00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang Q, Esnault F, Zhao M, Chiu T-J, Smith AB, III, Nguyen HT, Sodroski JG, Simon V. 2022. Global increases in human immunodeficiency virus neutralization sensitivity due to alterations in the membrane-proximal external region of the envelope glycoprotein can be minimized by distant State 1-stabilizing changes. J Virol 96:e0187821. 10.1128/jvi.01878-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vermeire J, Naessens E, Vanderstraeten H, Landi A, Iannucci V, Van Nuffel A, Taghon T, Pizzato M, Verhasselt B. 2012. Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS One 7:e50859. 10.1371/journal.pone.0050859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Emi N, Friedmann T, Yee JK. 1991. Pseudotype formation of murine leukemia virus with the G protein of vesicular stomatitis virus. J Virol 65:1202–1207. 10.1128/JVI.65.3.1202-1207.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lowry R. 2004. VassarStats: website for statistical computation. http://vassarstats.net/.
- 114.Waheed AA, Ablan SD, Roser JD, Sowder RC, Schaffner CP, Chertova E, Freed EO. 2007. HIV-1 escape from the entry-inhibiting effects of a cholesterol-binding compound via cleavage of gp41 by the viral protease. Proc Natl Acad Sci USA 104:8467–8471. 10.1073/pnas.0701443104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]