

SUMMARY
The risk of chronic diseases caused by aging is reduced by caloric restriction (CR)-induced immunometabolic adaptation. Here, we found that the matricellular protein, secreted protein acidic and rich in cysteine (SPARC) was inhibited by two-years of 14% sustained CR in humans and elevated by obesity. SPARC converted anti-inflammatory macrophages into a pro-inflammatory phenotype with induction of interferon-stimulated gene (ISG) expression via the transcription factors IRF3/7. Mechanistically, SPARC-induced ISGs were dependent on toll-like receptor-4 (TLR4)-mediated TBK1, IRF3, IFN-β, and STAT1 signaling without engaging the Myd88 pathway. Metabolically, SPARC dampened mitochondrial respiration, and inhibition of glycolysis abrogated ISG induction by SPARC in macrophages. Furthermore, the N-terminal acidic domain of SPARC was required for ISG induction, while adipocyte-specific deletion of SPARC reduced inflammation and extended healthspan during aging. Collectively, SPARC, a CR-mimetic adipokine, is an immunometabolic checkpoint of inflammation and interferon response that may be targeted to delay age-related metabolic and functional decline.
Keywords: SPARC, Matricellular protein, Caloric restriction, Macrophage, Inflammation, TLR4, Interferon-stimulated gene
eTOC blurb
Caloric restriction (CR) reduces inflammation and enhances longevity but the identity of CR-mimetics that improve healthspan is unclear. Ryu et al reveal that sustained CR in humans reduces adipose SPARC. Elevation of SPARC promotes interferon-response and inflammation while reduction of SPARC in adipocytes protects against aging-related inflammation and metabolic dysfunction.
Graphical Abstract
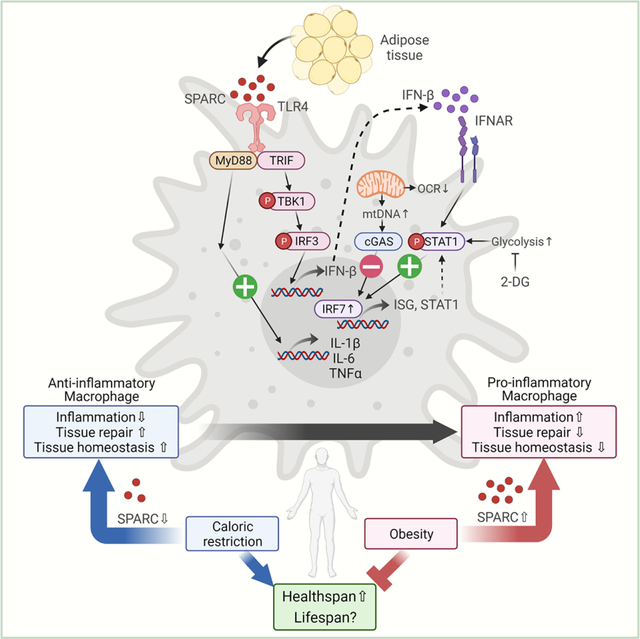
INTRODUCTION
Healthspan, the period of life free of chronic diseases in advanced aging can be extended by multiple mechanisms, including integrative immunometabolic responses (Goldberg and Dixit, 2015). Macrophage activation is recognized to be an essential driver of sterile inflammation in aging as well as obesity (Goldberg and Dixit, 2015; van Beek et al., 2019; Weisberg et al., 2003). The induction of negative energy balance by caloric restriction (CR) inhibits inflammation, reduces disease burden and extends lifespan in mice (de Cabo et al., 2014; Fontana et al., 2010). This suggests that host adaptation to change in energy balance must be sensed and conveyed to macrophages to mount an appropriate pro- or anti-inflammatory response. The precise identity of factor(s) that link alterations in organismal metabolism to macrophage response are unclear. CR in rodents clearly elicits anti-inflammatory effects, but despite several years of research efforts, the endogenous drivers that couple energy balance to inflammatory cellular quiescence or activation remain unclear (Ingram et al., 2006). Notably, 40% model of CR, mostly used in rodent studies, shows lifespan extension but can also reduce longevity in some strains of mice and increase the severity of infections (Gardner et al., 2011; Liao et al., 2010). Importantly, it is now established that humans in free-living conditions can only achieve ~14% CR (Ravussin et al., 2015), which further highlights the artificial and extreme nature of rodent-CR model. Consistent with disposable soma theory, the experimental protocols of 40% CR in rodents causes systems tradeoff such as reproductive stasis, somatopause, and immunopause which likely obfuscates the identification of therapeutically relevant immunometabolic checkpoints (Adler et al., 2013; Kirkwood et al., 2000). Notably, the Comprehensive Assessment of Long-term Effects of Reducing Intake of Energy (CALERIE-II) clinical trial established that CR in humans does not increase risks of infections and lowers inflammatory markers in blood without systems tradeoffs (Kraus et al., 2019; Meydani et al., 2016).
The RNA-sequencing analyses of adipose tissues from CALERIE-II study participants identified that CR in humans activates transcriptional programs implicated in mitochondrial metabolism, anti-inflammatory responses, and longevity (Spadaro et al., 2022). In the current follow-up study, we found Secreted protein acidic and rich in cysteine (SPARC) as a CR-inhibited protein. SPARC is a matricellular protein that is constitutively expressed in many tissues, including adipose tissues. Increased SPARC is associated with obesity, diabetes, coronary artery disease (Kos and Wilding, 2010; Takahashi et al., 2001) and chronic inflammation. SPARC is expressed in adipose tissue in obesity, and is potentially regulated by change in metabolic status (Kos et al., 2009; Lee et al., 2014; Shen et al., 2014). SPARC may also regulate inflammation through interactions with multiple receptors and cell surface matrix associated molecules (Kzhyshkowska et al., 2006; Rivera and Brekken, 2011; Ruan et al., 2010; Shin et al., 2013). Enhanced concentration of SPARC are also associated with inflammation and fibrosis (Ng et al., 2013; Toba et al., 2015; Xu et al., 2013) including age-related cardiac inflammation (Toba et al., 2015). However, SPARC’s mechanism of action on innate immune responses and macrophage derived inflammation remains unclear.
Given SPARC’s established role as a matricellular protein, we hypothesized that reduction of SPARC by negative energy balance signals tissue remodeling to calibrate macrophage activation. The unbiased identification of inhibition of SPARC by CR in human adipose tissue also raised the question of whether it can serve as a CR-mimetic target. Here we show that increased SPARC amounts reversed the established M2-like anti-inflammatory macrophages into an M1-like pro-inflammatory state. The bulk RNA sequencing of macrophages revealed induction of interferon-stimulated genes (ISGs) and upregulation of the transcription factor IRF7. The analysis of mice and macrophages lacking IRF3 and IRF7 revealed that SPARC required these transcription factors for induction of STAT1 and ISGs. Despite an increase in cytosolic mitochondrial Dloop3 and ND4, this was not sufficient to activate cyclic GMP-AMP synthase (cGAS)-mediated interferon response. We found that ISG induction by SPARC required activation of toll-like receptor-4 (TLR4) signaling and glycolysis. It is known that age-related inflammation is also associated with increases in ISGs. Consistent with our hypothesis, the adipocyte-specific reduction of SPARC decreased ISGs, protected against chronic adipose tissue inflammation including systemic ‘inflammaging’, improved metabolic function and enhanced the healthspan of aged mice. Thus, the identification SPARC as an energy-sensing matrikine has broad implications in regulation of sterile inflammation and tissue remodeling.
RESULTS
Regulation of SPARC by caloric restriction and obesity is coupled with inflammation
To identify the endogenous drivers of the beneficial effect of CR in humans, we investigated the adipose tissue transcriptome of participants that underwent two years of sustained CR (CALERIE-II) (Rochon et al., 2011, Spadaro et al 2022) (Figure 1A). Adipose tissue harbors a tissue-resident immune system, including macrophages which regulate lipolysis and insulin sensitivity (Lumeng et al 2007, Camell et al., 2017; Ramkhelawon et al., 2014). Moreover, given that the negative energy balance requires increased lipolysis and fatty acid oxidation from adipocytes, we reasoned that analysis of human adipose tissue might reveal metabolic regulators that act on macrophages to lower inflammation. The RNA sequencing of abdominal subcutaneous adipose tissue biopsies of participants at baseline and one and two years post sustained CR revealed changes in genes associated with regulation of matrisome. Especially, matricellular proteins, including SPARC (SPARC), Tenascin-C (TNC), Lumican (LUM), CCN5 (WISP2), Hemicentin (HMCN2), and Fibulin family (EFEMP1, FBNL2), were downregulated with CR in human adipose tissue (Figure 1B). Prior studies suggest that matricellular proteins are involved in tissue remodeling, inflammation, and neutrophil recruitment (Rienks and Papageorgiou, 2016). Among those, we focused on SPARC, which was significantly downregulated after 1 and 2 years of CR (Figure 1C). Reduction in SPARC was positively correlated with the decreases in BMI and % body fat (Figure S1A), leptin and CRP (Figure 1D), as well as tumor necrosis factor-α (TNFα) and the integrin ICAM-1 (Figure 1D). In contrast to CR, obese individuals had higher concentrations of SPARC compared to lean individuals (Figure 1E), suggesting that SPARC may contribute to the inflammation associated with increased adiposity. Indeed, SPARC induced pro-inflammatory genes (Il1b, Tnf, and Il6) in a dose-dependent manner (Figure 1F) in mouse bone marrow-derived macrophages (BMDMs). Notably, the in vitro concentration of SPARC used to elicit a response from BMDM was in the physiological range observed in local adipose tissue microenvironment (Figure S1B). In addition, SPARC increased Il1b expression in BMDMs primed with TLR4 or TLR2 (Figure S1C). SPARC also increased the pro-IL-1β protein expression in mouse macrophages albeit to a lesser extent compared to LPS (Figure S1D). To investigate whether SPARC affects macrophage inflammation in adipose tissues, we isolated mouse adipose stromal vascular fraction (SVF) cells that contain adipose tissue macrophages (ATMs), and treated them with SPARC ex vivo followed by flow cytometry analysis (Figure 1G). Consistent with the results in BMDMs, we found that SPARC increased the proportion of pro-inflammatory macrophages (CD11c+ CD206−) while decreasing anti-inflammatory macrophages (CD11c− CD206+) (Figure 1H, I) in the SVF. SPARC also enhanced the TNF and NOS2 protein expression in the SVF adipose macrophages (Figure 1I, J). Together, the data show that excess amounts of matricellular protein SPARC promote macrophage-derived inflammation.
Figure 1. Excess adipokine SPARC causes macrophage-derived inflammation.

(A) Schematic of human calorie restriction (CR) study. Clinical data and adipose RNA-sequencing results of baseline, 1 year, and 2 years post CR were used for analysis. (B) Expression pattern of significantly up-and down-regulated genes involved in matrisome post 1 year and 2 years of CR. Arrows indicate matricellular proteins, including SPARC (red). (C) Normalized expression amounts of SPARC from RNA-sequencing analysis before and after CR. (D) Correlation analysis between the percentage of SPARC expression change and percentage of phenotype changes, including Leptin, CRP (upper), TNFα, and ICAM1 (lower) concentration of participants after 1 year CR. (E) SPARC concentrations in the plasma of lean and obese individuals (n=8, 13). (F) Q-PCR analysis for pro-inflammatory gene (Il1b, Tnf, and Il6) expression in M0 BMDMs after SPARC treatment with indicated concentration (1, 5, and 20 μg/ml) for 24 hours (n=5). (G) Schematic of flow cytometry analysis with adipose tissue macrophages (ATMs). (H-J) Quantification of flow cytometry analysis for the macrophages proportion (H, I) and MFI in macrophages (J) with or without ex vivo 20 μg/ml SPARC treatment for 24 hours (n=3). All in vitro or ex vivo experiments were repeated independently at least twice. Error bars represent the mean ± S.E.M. Pearson correlation analysis, two-tailed paired and unpaired t-tests were performed for statistical analysis. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. Please also see Figure S1.
SPARC converts anti-inflammatory macrophages into a pro-inflammatory phenotype
Macrophages are critical for tissue homeostasis (Okabe and Medzhitov, 2016). Anti-inflammatory macrophages are decreased, while pro-inflammatory macrophages are increased in adipose tissue with aging and obesity (Camell et al., 2017; Lumeng et al., 2007). Although, in vivo, macrophages do not exist in the binary spectrum of an M1- or M2-polarized state (Xue et al., 2014), we wanted to use this established in vitro model to gain mechanistic insights on the role of SPARC on macrophage-derived inflammation (Figure 2A). Thus, mouse BMDMs were polarized into M1 and M2 states and were thereafter exposed to recombinant SPARC at physiologically relevant concentrations. The pro-inflammatory gene expression or cytokine secretion in established M1 macrophages was not affected by SPARC (Figure 2B–D and S1E). SPARC converted the in vitro committed M2 cells into a pro-inflammatory state that showed an increase in Il1b, Tnf, Nos2, Il6, and Il12b (Figure 2B and S1E) and reduction of Retnla, Chil3, and Socs2 (Figure 2C). In addition, compared to untreated M2 cells, SPARC increased the secretion of IL-1β, TNFα, IL-6, and GM-CSF in a dose-related manner (Figure 2D), while decreased secretion of M2 cell-derived, YM1/CHI3L3 proteins (Figure S1F). Next, to confirm whether SPARC similarly impacted in vivo derived macrophages, mice were treated with thioglycollate or IL-4 immune complexes (IL-4c) and activated macrophages were isolated, and treated with SPARC ex vivo (Figure 2E). Consistent with the results in BMDMs, SPARC did not affect pro-inflammatory cytokines in thioglycollate activated cells but increased the markers of M1-like pro-inflammatory state in IL-4c-derived peritoneal macrophages (Figure 2F and S1G) and reduced M2 macrophage genes (Retnla, Chil3, and Arg1) (Figure 2G). Together these data demonstrate that matricellular protein SPARC promotes inflammation by converting M2-like macrophages into an M1-like state.
Figure 2. SPARC switches M2 macrophages to pro-inflammatory macrophages.

(A) Schematic of BMDM experiments with SPARC treatment. (B, C) Q-PCR analysis for pro-inflammatory genes (Il1b, Tnf, and Nos2) (B) and M2 macrophage genes (Retnla, Chil3, and Socs2) (C) in M1 and M2 polarized BMDMs. (D) Luminex assay to detect pro-inflammatory cytokines in supernatants of SPARC treated M1 and M2 polarized BMDMs (n=4). N.D. is non-detected. (E) Schematic of peritoneal macrophage experiments with SPARC treatment. (F, G) Gene expression analysis by Q-PCR for pro-inflammatory genes (F) and M2 macrophage genes (G) in control and ex vivo SPARC treated peritoneal macrophages from thioglycollate (TG) or IL-4 complex injected (IL-4) mice (n=3, 4 each). All in vitro or ex vivo experiments were repeated independently at least twice. Error bars represent the mean ± S.E.M. Two-tailed paired t-tests were performed for statistical analysis. * P < 0.05; ** P < 0.01; *** P < 0.001. Please also see Figure S1.
SPARC reprograms the macrophage transcriptome and induces interferon response genes
Given the effects of SPARC on macrophage activation, we next investigated the transcriptional program and mechanisms that control the pro-inflammatory response driven by this endogenous matrikine. We conducted bulk-RNA sequencing of thioglycollate (TG) and IL-4c (IL-4) activated peritoneal macrophages exposed to SPARC (Figure 3A and S2A). The principal component analysis demonstrated that, compared to controls, SPARC treatment of TG and IL-4c elicited peritoneal macrophages altered the transcriptome that was clearly segregated by activation status (Figure 3B). Analysis of RNA-sequencing data identified significantly upregulated and downregulated genes (Figure 3C and S2B) and validated M1 and M2 macrophage marker gene expression change by SPARC (Figure S2C). We detected marked upregulation of many interferon stimulated genes (ISGs) by SPARC treatment in both M1-like (TG) and M2-like (IL-4) macrophages, while more profound changes were observed in M2-like (IL-4) macrophages (Figure 3C and S2D). Top upregulated ISGs (> 6 log2 fold change) in M2-like macrophage (IL-4) such as Apol9a, Aopl9b, and Ccl5 were reported to have a role in wound healing and tissue repair (Ridiandries et al., 2018; Sun et al., 2015), implicating the function of induced ISGs in tissue homeostasis during the uninfected condition. Further pathway analyses revealed that interferon signaling pathways were significantly upregulated in both M1-like (TG) and M2-like (IL-4) macrophages as well as inflammatory signaling pathways with SPARC treatment (Figure 3D). Additionally, SPARC significantly upregulated TLR signaling pathway and extracellular matrix organization and matrisome-associated pathways, suggesting a potential functional role of SPARC in macrophage-mediated sterile inflammation and tissue remodeling (Figure 3D). Next, we investigated the expression change of transcription factors to find the core factors regulating the ISGs. Since both M1-like (TG) and M2-like (IL-4) peritoneal macrophages induced ISGs, we investigated transcription factors significantly changed in each type of macrophage (Figure S2E) and in both conditions (Figure 3E). IRF7 is the most upregulated transcription factor in both M1-like (TG) and M2-like (IL-4) macrophages with SPARC treatment (Figure 3E). Consistently, many genes regulated by IRF7, including Isg15, Ifit2, and Xaf1, were upregulated in both M1-like (TG) and M2-like (IL-4) macrophages (Figure 3F). The motif enrichment analysis confirmed that IRF7 interacting motif was enriched in significantly upregulated genes in both M1-like (TG) and M2-line (IL-4) macrophages (Figure S2F). Together, RNA-sequencing analysis identified that SPARC causes marked transcriptional reprogramming of committed M2-like macrophages that are characterized by augmentation of ISGs.
Figure 3. Transcriptomic signature of the interferon-stimulated gene is induced by SPARC.

(A) Schematic of experimental design for RNA-sequencing analysis to determine the impact of SPARC on peritoneal macrophages. (B) PCA analysis of RNA-sequencing results of controls and ex vivo SPARC treated peritoneal macrophages from thioglycollate (TG Cont and TG SPARC), or IL-4 complex injected (IL-4 Cont and IL-4 SPARC) mice (n=3 each). (C) Top 20 significantly up-and down-regulated genes in SPARC treated peritoneal macrophages from IL-4 complex injected (IL-4, left), and thioglycollate injected (TG, right) mice compared to each control (Cont). (D) Significantly up-and down-regulated pathways (p < 0.05) in SPARC treated peritoneal macrophages from IL-4 complex injected (IL-4, upper) and thioglycollate injected (TG, lower) mice by GSEA. R, K, P, N, B refer to pathways in Reactome, KEGG, PID, NABA, BioCarta database. (E) Significantly differentially expressed common transcription factors in SPARC treated macrophages from both IL-4 complex injected (IL-4, left) and thioglycollate injected (TG, right) mice. The red arrow indicates the top transcription factor IRF7. (F) IRF7 regulated, significantly changed genes in SPARC treated macrophages from IL-4 complex injected (IL-4, left) and thioglycollate injected (TG, right) mice. Please also see Figure S2.
SPARC induces interferon response via IRF3 and IRF7
We validated the findings that recombinant SPARC activates ISGs by transfecting the macrophage cell line RAW 264.7 with control and SPARC expressing vectors. This led to the successful overexpression of SPARC in RAW 264.7 (Figure 4A). Consistent with our prior results, RAW 264.7 cells overexpressing SPARC had a significant increase in Ifit2, Isg15, Xaf1, and Ifit3 mRNA compared to empty vector-transfected cells (Figure 4B). These data suggest that SPARC, upon secretion, interacts with macrophages to induce its biological effects. The exclusive cell-surface receptor for SPARC has not been established, and it is thought that matricellular SPARC protein may elicit its diverse biological effects via multiple cell-cell and cell-matrix interactions. SPARC has a unique structure that is composed of an N-terminal acidic domain, a follistatin-like domain with epidermal growth factor (EGF)-like module and an extracellular calcium-binding (ECB) domain containing two EF-hands which cooperatively bind two calcium ions with high affinity (Figure 4C) (Kos and Wilding, 2010). We, therefore, next determined which of these functional domains mediated SPARC’s interferon-response in macrophages. The SPARC constructs with deletions in acidic (ΔD1), follistatin-like domain (ΔD2), and ECB domain (ΔD3) were transfected in RAW 264.7 cells (Figure 4C and S4A). Compared to full-length SPARC, ΔD2, or ΔD3, the specific loss of acidic (ΔD1) domain reduced the induction of Irf7 (Figure 4D), ISGs (Figure S3B) as well as Stat1 (Figure S3C), Il1b (Figure S3D), demonstrating the requirement of SPARC’s acidic domain in its immunological effects. Additionally, FSTL1 that shares similar structure with SPARC’s follistatin-like domain (D2) (Li et L 2019) could not induce the upregulation of ISGs as full-length SPARC (Figure S4E).
Figure 4. SPARC upregulates the expression of ISGs via IRF3 and IRF7 signaling.

(A, B) Q-PCR analysis for human SPARC (A) and ISGs (B) in RAW 264.7 cells with mock or SPARC transient transfection. (C) Schematic of constructs used for transfection experiments with intact and deletion of specific domains (D1, D3, D3) of SPARC. (D) Q-PCR analysis for Irf7 in SPARC and domain deleted SPARC (D1, D2, D3) transfected RAW 264.7 cell lines (n=4). Fold changes were calculated based on mock vector-transfected RAW 264.7 cell lines. (E, F) Q-PCR analysis for ISGs (E), Stat1 (F) in M2 polarized BMDMs from wide-type (WT) and Irf3−/− Irf7−/− mice with or without 20 μg/ml SPARC treatment for 24 hours (n=3). (G) Schematic of IL-4c and SPARC co-injection experiments used to perform FACS analysis of peritoneal cells. (H) Quantification of flow cytometry analysis for macrophages (n=4, 8, 8) and proliferating M2 macrophages (n=4, 3, 4) in PBS, IL-4c with PBS, or IL-4c with SPARC injected mice. (I) Schematic of IL-4c and SPARC co-injection experiments used to perform FACS analysis of peritoneal cells from WT controls and Irf3−/− Irf7−/− mice. (J) Quantification of flow cytometry analysis for macrophages and proliferating M2 macrophages in peritoneal cells from IL-4c with PBS or IL-4c with SPARC injected WT and Irf3−/− Irf7−/− mice (n=4). (K) Expression pattern for mitochondrial genes in RNA-sequencing analysis of peritoneal macrophages from IL-4c injected (IL-4), and thioglycollate injected (TG) mice with or without SPARC treatment ex vivo (n=3). (L) Cytosolic mitochondrial DNA abundance measurement in M2 BMDMs with or without SPARC treatment for 6 and 24 hours by Q-PCR for Dloop3 and ND4 (n=3). (M) Q-PCR analysis for Irf7 and ISG Ifit2 in M2 BMDMs from WT and Cgas−/− mice with or without SPARC treatment (20 μg/ml) for 24 hours (n=3, 5). All in vitro or ex vivo experiments were repeated independently at least twice. Error bars represent the mean ± S.E.M. Two-tailed paired and unpaired t-tests were performed for statistical analysis. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. Please also see Figure S3, 4.
Given that transcription factor analysis revealed IRF7 was the top upregulated gene, we next asked if induction of ISGs by SPARC depended on IRF signaling. Indeed, compared to control animals, the M2-like macrophages from IRF3 and IRF7 deficient mice were unable to upregulate Ifit2, Isg15, Xaf1, Ifit3, and Stat1 in response to SPARC (Figure 4E, F and S3F) without affecting M2 genes (Figure S3G). Considering that matricellular proteins increase pro-inflammatory macrophages, we investigated the effect of SPARC on the proportion of macrophages in vivo. As expected, IL-4c injection for four days increased the frequency of total macrophages, including total and proliferating RELMα+ anti-inflammatory macrophages (Figure 4G, H and S3H–K). SPARC injection together with IL-4c reduced peritoneal cells and proportions of macrophages, and total and proliferating M2 macrophages (Ki67+ RELMα+), while pro-inflammatory macrophages (NOS2+) were moderately increased (Figure 4H and S3K). Consistent with our data that SPARC activates IRF3 and IRF7, SPARC’s effects on myeloid cells, including reduction of total M2 and Ki67+ RELMα+ macrophages, were abrogated in mice lacking Irf3 and Irf7 (Figure 4I, J and S3L–N).
We next investigated the mechanism of IRF7 activation by SPARC. Further analysis of RNA sequencing data revealed that SPARC decreased the expression of mitochondria encoded genes in peritoneal macrophages such as mt-Atp8, mt-Nd1, Nd2, Nd4, Nd4l, and Nd5 (Figure 4K and S4A). This raised the possibility that excess SPARC may induce mitochondrial damage or stress that could release mtDNA in the cytosol to activate IRF7 (West et al., 2015; Zhong et al., 2018). Indeed, along with increase of ROS concentrations (Figure S4B), mtDNA (Dloop3 and ND4) concentrations were significantly increased in M2-like peritoneal macrophages (IL-4) after 24 hours of SPARC treatment (Figure S4C). The total and cytosolic mtDNA concentrations were also increased in M2 BMDMs (Dloop3 and ND4) after 24 hours of SPARC treatment (Figure 4L and S4D). Since intracellular double-strand DNA is detected by cGAS and activates downstream IRF3 and IRF7-mediated ISG induction, we tested whether cGAS contributes to SPARC-mediated ISG induction in macrophages. SPARC-induced upregulation of IRF7 and ISGs was unaffected in cGAS deficient macrophages (Figure 4M and S4E). These data suggest that SPARC-induced increase in cytosolic mitochondrial DNA is not sufficient to engage cGAS-mediated ISG response. Mechanistically, while NOS2 (iNOS) induction by SPARC does not affect pro-inflammatory gene expression (Figure S4F), NOS2 was responsible for mitochondrial dysregulation caused by SPARC (Figure S4G, H).
SPARC requires TLR4 signaling to induce inflammation and interferon response
The gene set enrichment analysis (GSEA) identified that TLR4 signaling was among significantly upregulated pathways that were activated by SPARC (Figure 3D). Therefore, we next investigated the SPARC’s upstream signaling mechanism that may control ISGs. Downstream of TLR4, via the TRIF arm of signaling, the activation of TBK1 and IRF3 is implicated in the activation of interferon response (Mlcochova et al., 2020; Sheikh et al., 2014). To determine the downstream signaling, we performed time-course experiments where M2 macrophages were treated with SPARC for 1 to 24 hours. As expected, SPARC treatment caused increased phosphorylation of TBK1, IRF3 after 1 hour, and STAT1, which is downstream of IFNAR after 3 hours (Figure 5A). The amounts of secreted IFN-β were increased between 1 hour and 3 hours of SPARC treatment (Figure 5B), suggesting the activation of IFNAR in an autocrine or paracrine manner. However, low concentrations of IFN-α and IFN- γ were induced at later time points (Figure S5A). These results are consistent with prior data that TBK1 and IRF3 signaling activation produces type I IFNs, which subsequently interact with IFNAR to activate STAT1 and induce ISGs (Mlcochova et al., 2020; Sheikh et al., 2014).
Figure 5. SPARC-mediated ISG induction is through TLR4 activation.

(A) Immunoblot analysis for activated interferon signaling proteins (p-TBK1, p-IRF3, and p-STAT1) in M2 BMDMs with the treatment of 20 μg/ml SPARC for 1, 3, 6, 12, and 24 hours as well as non-treated control. (B) ELISA assay for IFN-β detection in supernatants of SPARC treated M2 BMDMs in (A) (n=3). (C) Solid-phase binding assay to detect the interaction between SPARC and human TLR4 (hTLR), SPARC and human TLR2 (hTLR2), SPARC and human MD-2 (MD-2), SPARC and BSA (BSA). The indicated amount of TLR4s or TLR2s were coated on plates, and 20 μg/ml SPARC, MD-2, or BSA were added for interaction (n=3). Nonlinear regression analyses were performed for the values in the graph. (D, E) Q-PCR analysis for Irf7, Ifit2, Stat1 (D), and pro-inflammatory genes (E) in M2 polarized BMDMs from WT, Tlr4−/−, Myd88−/−, and Ifnar1−/− mice with or without SPARC (20 μg/ml) treatment (n=3). (F) Expression pattern of glycolysis pathway genes in RNA-sequencing analysis of peritoneal macrophages induced by IL-4c (IL-4) and thioglycollate (TG) injection with or without SPARC treatment ex vivo (n=3). (G) Oxygen consumption rate (OCR) for M1 and M2 BMDMs with or without SPARC (20 μg/ml) treatment measured by seahorse mitostress assay (n=3). (H) Oxygen consumption rate (OCR) of M1, M2, and 20 μg/ml SPARC treated M2 BMDMs with or without pre-treatment of TLR4 inhibitor, TAK-242 (2.5 μM). (I, J) Gene expression analysis of Irf7, ISGs, and Stat1 in non-teated and SPARC (20 μg/ml, 24 hours) treated M2 BMDMs with or without pre-treatment of glycolysis inhibitor (2-DG, 10 mM) (n=3). (K) Immunoblot analysis for activated interferon signaling proteins (p-TBK1, p-IRF3, and p-STAT1) in M2 polarized BMDMs treated with SPARC (20 μg/ml) for indicated time with or without pre-treatment of glycolysis inhibitor (2-DG, 10 mM) (n=3). All in vitro or ex vivo experiments were repeated independently at least twice. Error bars represent the mean ± S.E.M. Two-tailed paired and unpaired t-tests were performed for statistical analysis. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. Please also see Figure S5.
The above data raised the possibility that SPARC may interact with TLR4. Therefore, we determined the direct interaction of SPARC with TLR4 using a solid-phase binding assay. Notably, both human TLR4 (hTLR4) and mouse TLR4 (mTLR4) bound to SPARC in a concentration-dependent manner while SPARC is not interacting with TLR2 (hTLR2) (Figure 5C and S5B). Next, we tested whether SPARC requires TLR4 signaling in M2 macrophages for inducing pro-inflammatory genes and IRF7-dependent upregulation of ISGs. We, therefore, treated macrophages with SPARC from control animals and mice lacking TLR4, Myd88, and IFNAR. In SPARC-treated macrophages, Tlr4 ablation completely blocked the induction of ISGs, Stat1, and pro-inflammatory gene expression (Figure 5D, E, and S5C, D). The induction of Irf7, ISGs, and Stat1 was exclusively dependent on IFNAR, while the upregulation of pro-inflammatory genes (Il1b, Tnf, and Il6) was solely dependent on MyD88 (Figure 5D, E and S5C). Furthermore, pre-treatment with the TLR4 specific inhibitor TAK-242, but not TLR1 and TLR2 specific inhibitor (CU-CPT22), recapitulated the gene expression changes detected in SPARC treated TLR4 deficient BMDMs (Figure S5E–H). Together these data demonstrate that SPARC specifically engages TLR4 signaling to control macrophage activation and induction of ISG.
SPARC-mediated ISG induction is dependent on glycolysis
Pro-inflammatory macrophages are preferentially glycolytic with lower capacity for mitochondrial oxidative phosphorylation (O’Neill and Pearce, 2015). Considering that SPARC concentrations are reduced in glucoprivic state, we asked whether excess SPARC causes metabolic reprogramming to activate the macrophages. The RNA-sequencing analysis identified that genes in the glycolysis pathway were upregulated by SPARC treatment in both IL-4c and TG elicited peritoneal macrophages (Figure 5F). Functional metabolic analysis of macrophages revealed that SPARC reduced oxygen consumption rates (OCR) of M2 BMDMs in a dose-dependent manner (Figure 5G and S5I). The dynamics of OCR, including basal respiration, ATP production, maximum respiration, and spare respiration, were calculated (Figure S5J, K). As previously reported, all of these metabolic parameters reported above were higher in M2 macrophages than the M1 macrophages (Tannahill et al., 2013; Vats et al., 2006). SPARC decreased the spare respiratory capacity, suggesting dampened maximum mitochondrial capacity (Figure S5K). Furthermore, we detected glycolysis by ECAR and observed that basal ECAR was increased in M2 BMDMs with SPARC treatment (Figure S5L). Importantly, the SPARC exposed M2 macrophages displayed metabolic characteristics akin to pro-glycolytic M1-like macrophages. Consistent with our prior results, SPARC’s pro-glycolytic effect in macrophages was abrogated by blocking TLR4 (Figure 5H). Additionally, depletion of neither IRF3 and IRF7 nor cGAS affected the OCR reduction by SPARC (Figure S5M, N). As expected, iNOS inhibition restored mitochondrial function dysregulated by SPARC, but glycolysis was not affected (Figure S5O, P). These data suggest specificity of SPARC’s effect on macrophage metabolism depends on TLR4.
To test whether SPARC-induced ISG response is dependent on glycolysis, we pre-treated M2 polarized BMDMs with 2-DG in the presence or absence of SPARC. The glycolysis inhibition blocked the induction of Irf7, ISGs, and Stat1 (Figure 5I, J), without affecting pro-inflammatory gene expression and OCR (Figure S5Q, R). Notably, glycolysis was not required for SPARC-induced activation of TBK1 (IRF3 arm of signaling) and IFN-β upregulation (Figure 5K and S5S), but inhibition of glycolysis reduced the sustained STAT1 activation induced by SPARC (Figure 5K). Given STAT1 controls IRF7 transcription, the data suggest that SPARC-induced glycolysis activates STAT1 to regulate ISGs. Together these data further support the hypothesis that SPARC’s engagement of TLR4 is the limiting signal for macrophage activation, which also induces glycolysis in cells to drive STAT1-dependent IRF7-mediated interferon response.
Reduction of adipocyte-derived SPARC extends healthspan of aged mice
Aging is associated with increased visceral adiposity, which in turn is linked to chronic inflammation, insulin resistance and impaired lipolysis (Kuk et al., 2009; Tchkonia et al., 2010). To determine if decrease in adipocyte-derived SPARC mimics CR’s beneficial effects on age-related inflammation and metabolic dysfunction, we aged littermate control Sparcfl/fl and SPARC adipocyte deficient mice up to 22 months (analogous to approx. 70-year-old human) (Figure 6A and S6A). Aging was associated with increase in circulating SPARC concentrations that were reduced in adipocyte specific SPARC deficient mice. In young animals, ablation of adipocyte derived SPARC is not sufficient to affect the serum SPARC concentrations suggesting additional cell sources can compensate to maintain the circulating concentration (Figure 6B). As expected, the control littermate animals when fed normal chow diet, gained weight, while SPARC adipocyte deficient mice were partially protected from weight gain from about 12 months onwards (Figure 6C). The reduction of body weight was mainly from fat mass with no change in lean mass (Figure 6D). Consistently, 20-month-old SPARC adipocyte deficient mice showed improved glucose tolerance and insulin sensitivity (Figure 6E, F). Importantly, compared to 21-month-old control littermates, the adipocyte-SPARC deficient mice had enhanced grip strength suggesting reduced frailty (Figure 6G). Moreover, the aged, adipocyte-specific SPARC deficient mice displayed reduced latency to fall on a rotating rod (Figure 6H), which is an established test to measure animal’s motor planning, balance, coordination and physical condition (Youm et al., 2013). Together, these data show that reduced SPARC production from adipocytes during aging extends healthspan of aged mice.
Figure 6. Reduction of adipokine SPARC improves healthspan during aging.

(A) Schematic of experiment with Sparcfl/fl (Con) and Sparcfl/fl;Adipoq-Cre+ (Adip-KO) mice with age over 20 months. (B) ELISA assay of circulating SPARC amounts from young (2-month-old) and old (20-month-old) control and Adip-KO mice by ELISA assay (n=12, 12, 10, 10) (C) Weight change of control and Adip-KO mice with age over 20 month (n=13, 15). (D) Body composition of 20-month-old control and Adip-KO mice measured by MRI (n=13, 15). (E) GTT and (F) ITT of 20-month-old control and Adip-KO mice (n=13, 15). (G, H) Healthspan test results of 21-month-old control and Adip-KO mice (n=14, 14). Grip strength test (G) and rotarod test (H) are shown. (I) Glycerol measurement with supernatants of adipose explants (SAT and VAT) derived from 22-month-old control and Adip-KO mice fed or fasted for 24h and stimulated with NE and isoproterenol ex vivo (n=6, 6). (J) Western-blot analysis of p-HSL in VAT explants from 22-month-old control and Adip-KO mice after 24 hour of fasting. (n=3, 6). (K) Q-PCR analysis for Calcrl, Pde3e and Ppara genes in VAT of 22-month-old control and Adip-KO mice (n=6, 5). All ex vivo and in vitro experiments were repeated independently at least twice. Error bars represent the mean ± S.E.M. Two-tailed unpaired and t-tests were performed for statistical analysis. * P < 0.05; ** P < 0.01; *** P < 0.001. Please also see Figure S6.
In addition, the old Sparcfl/fl and SPARC adipocyte deficient mice were tested for their metabolic healthspan using indirect calorimetry during ad libitum feeding, 24 hours of fasting, and refeeding. As predicted before and consistent with lower body mass (Corrigan et al., 2020), measurement of oxygen consumption over 5 days in aged adipocyte-SPARC deficient mice showed reduced energy expenditure (EE) (Figure S6B, C). The ANCOVA analysis of EE with body weight as covariate during feeding, fasting and re-feeding phases showed separation of regression based on genotype but no significant change in EE was detected (Figure S6D). No significant change in RER was detected during feeding, fasting, and refeeding between age matched Sparcfl/fl and 22-month-old SPARC adipocyte deficient mice (Figure S6E). Considering the limitation of ANCOVA analysis when body weight of mice in two groups have already diverged and are not overlapping (Fernández-Verdejo et al., 2019; Speakman, 2013), body weight normalized EE was also incorporated in the analysis. Given the lean mass was same in control and SPARC adipocyte deficient mice, quantification of EE, if normalized to body mass, data suggested that aged SPARC adipocyte deficient mice have increased energy expenditure (Figure S6F–J). Importantly, food intake was not different between groups (Figure S6K), while water intake and locomotive activity was increased in SPARC adipocyte deficient mice (Figure S6L, M). Increased locomotive activity during feeding and refeeding phases in aged adipocyte specific SPARC deficient mice suggested that improved activity and food seeking behavior could partake in anti-adiposity effect.
Aging is associated with reduced lipolysis in response to fasting and sympathomimetics (Camell et al., 2017; Gao et al., 2020). We therefore investigated lipolytic response as additional measure of metabolic healthspan in the old mice with 24 hr fasting, and observed that compared to 22-month-old Sparcfl/fl mice, the glycerol and free fatty acid release (Figure 6I and S6O) and activity of lipolysis signaling measured by p-HSL were significantly increased in old SPARC adipocyte deficient mice in both SAT and VAT (Figure 6J). Glucose concentrations after 24 hr fasting in aged female SPARC adipocyte deficient mice were significantly lower than in Sparcfl/fl mice, suggesting better glucose homeostasis (Figure S6P). Similar to CR-induced upregulation of drivers of fatty acid oxidation in human adipose tissue, aged adipocyte SPARC deficient mice had increased expression of Calcrl, Pde3e, and Ppara (Figure 6K), suggesting a CR-mimetic like effect. Overall, in aged mice, SPARC reduction in adipocytes alleviated aging-induced metabolic dysregulation with increased energy expenditure and enhanced healthspan.
Adipokine SPARC controls inflammaging
Inflammaging, characterized by chronic increase in pro-inflammatory cytokines in elderly (Franceschi et al., 2018) and NLRP3 inflammasome activation driven loss of tissue homeostasis is a major driver of age-related chronic diseases (Goldberg and Dixit, 2015; Latz and Duewell, 2018; Youm et al., 2013). Aging is also associated with higher basal type I interferon response in multiple tissues, including adipose (Benayoun et al., 2019; Youm et al., 2013). To investigate how decreased amounts of SPARC affects inflammation in adipose tissue of aged mice, we investigated changes in tissue resident immune cells (Figure 7A). The weight of adipose tissues and cellularity were lower in SPARC adipocyte deficient mice (Figure S7A, B). The proportion of regulatory T cells (Tregs) (Bapat et al., 2015; Raynor et al., 2012), and macrophages, especially CD11c+ CD 206− macrophages (Camell et al., 2017), were reported to be increased in adipose tissue of aged mice. In SPARC adipocyte deficient mice, the proportions of Treg cells, macrophages, and CD11c+ CD206− macrophages were decreased compared to the littermate control Sparcfl/fl mice, while CD11c− CD206+ macrophages were increased (Figure 7B and S7C). We previously reported that aging upregulates the expression of pro-inflammatory proteins GDF3 and GDF15 in adipose tissue macrophages (Camell et al., 2017). Consistent with protection from inflammaging, compared to 22-month-old Sparcfl/fl mice, the SPARC adipocyte deficient mice were protected from age-related increase in Gdf3 and Gdf15 (Figure 7C). Furthermore, aged SPARC adipocyte deficient mice had reduced pro-IL-1β compared to Sparcfl/fl mice, while PPARγ, which lowers adipose inflammation and improves metabolic function, was increased in adipose tissue of aged mice that were deficient in SPARC (Figure 7D, E).
Figure 7. Inhibition of adipocyte-derived SPARC reduces age-related increase in chronic interferon response and inflammation.

(A, B) Schematic and FACS quantification of immune cell population in stromal vascular fraction (SVF) of VAT from 22-month-old control and Sparc Adip-KO mice (n=6, 6). Treg (CD4+CD25+Foxp3+), macrophage, CD11c+ CD206- macrophage, and CD11c- CD206+ macrophage were identified by flow cytometry. (C) Q-PCR analysis for GDF3 and GDF15 from ATMs of 22-month-old control and Adip-KO mice (n=6, 7). (D, E) Immunoblot analysis of pro IL-1β and PPARγ in VATs from 22-month-old control and Adip-KO mice (n=6, 6) and quantification. (F-I) Q-PCR analysis of Irf7 (F), ISGs (G), Stat1 (H), and pro-inflammatory genes (I) in VAT of 22-month-old control and Adip-KO mice (n=6, 5). (J) Serum concentration of inflammatory cytokines in 22-month-old control and Adip-KO mice (n=6, 5). All ex vivo and in vitro experiments were repeated independently at least twice. Error bars represent the mean ± S.E.M. Two-tailed unpaired t-tests were performed for statistical analysis. * P < 0.05; ** P < 0.01. Please also see Figure S7.
Notably, consistent with our data that increased SPARC enhances IRF7-dependent ISGs, compared to littermate controls, the 22-month-old SPARC adipocyte deficient mice had a significant reduction in core transcription factor IRF7 in adipose tissue (Figure 7F). Furthermore, adipocyte-specific reduction of SPARC in visceral adipose tissue of aged mice caused a significant decrease in Ifit2, Isg15, Xaf1, Ifit3, and Stat1 (Figure 7G, H). Moreover, reduction of SPARC in adipocytes protected against the aging-related increase in pro-inflammatory regulators, Il1b and Casp1 as well as Tnf and Nos2, (Figure 7I). This suggests that SPARC-depleted adipose tissue in aged mice mimics several aspects of the integrated immunometabolic response induced by CR in human adipose tissue (Spadaro et al., 2022). Lastly, as further evidence for role of SPARC in regulating age-related systemic inflammation, when compared to control mice, aging-related increase of IL-1β, as well as TNFα, MCP-1 and KC in the serum was significantly lower in SPARC adipocyte deficient KO mice (Figure 7J). Collectively, these findings suggest that adipocyte-derived SPARC acts on macrophages to engage TLR4 signaling, inflammation and interferon-response. Importantly, reduction of ISG in aged mice upon adipose-specific ablation of SPARC supports the hypothesis that negative energy balance regulated secreted proteins link metabolic and immune systems.
DISCUSSION
The immune system is required for the integration of cellular metabolism, tissue repair, and function (Eming et al., 2017; Rankin and Artis, 2018). There is growing evidence that the mechanisms driving chronic low-grade inflammation can be targeted to delay degenerative changes to enhance healthspan and possibly lifespan (Sayed et al., 2021). Moreover, senescent immune cells are now implicated in driving systemic aging (Yousefzadeh et al., 2021). Therefore, the identification of CR-mimetics that can reduce inflammation offers not only opportunities to target chronic diseases but also opens potential therapeutic approaches to extend the healthy lifespan. Our unbiased approach identified SPARC as a CR-inhibited matrikine in human adipose tissue. These findings demonstrate that SPARC acts on macrophages to control inflammation and interferon-response, thus supporting the hypothesis that CR-regulated endogenous immunometabolic checkpoints can be harnessed.
Matricellular proteins such as SPARC not only regulate the ECM as previously reported (Bornstein, 1995), but may also affect the macrophage polarization during the tissue homeostasis response. To efficiently repair and remodel tissues, converting anti-inflammatory macrophages to activated inflammatory macrophages is required to produce distinct ECM modulation factors such as metalloproteases (MMPs) (Huang et al., 2012) and chemokines. In addition, switched M1 macrophages have enhanced the capacity of phagocytosis to clear cell debris during tissue repair. Therefore, fine-tuning of macrophages’ by matricellular proteins may be important for repair and tissue remodeling (Rienks and Papageorgiou, 2016). Indeed, matricellular proteins such as SPARC and CCN1 are induced and required for the wound healing process (Jun et al., 2015; Reed et al., 1993). The transient DAMP response is required for efficient tissue maintenance by homeostatic inflammation (Suganami and Ogawa, 2010). As observed in RNA-sequencing analysis in peritoneal macrophages, SPARC-mediated activation of macrophages upregulates pathways for extracellular matrix organization as well as inflammatory cytokines.
Unlike injury or infection driven tissue remodeling response, the adipose tissue remodeling that involves expansion to store triglycerides during obesity or adipose constriction during weight loss may require distinct immunometabolic mechanism. The identification of a matricellular protein, SPARC, which is highly expressed in adipocytes and controls macrophage activation, may be an endogenous mechanism to control the setpoint of inflammation to restore homeostasis. However, contrary to this hypothesis of immunometabolic specialization, SPARC, an endogenous adipocyte-derived and non-pathogen origin protein, activated TLR4, which is established to regulate host defense against bacterial infection (Medzhitov et al., 1997). This raises the possibility that SPARC represents one of the mechanisms, whereby a host co-opts pathogen-sensing machinery for also sensing cellular energy balance to mount a sterile inflammatory response in specific tissues.
The TLR4 signaling can also be activated by other matricellular proteins like Tenascin C and CCN1 (Jun and Lau, 2020; Midwood et al., 2009). Moreover, the RNA sequencing of SPARC-activated macrophages identified unexpected induction of ISGs that are primarily required to mount an anti-viral immune response. Thus, instead of exclusivity, several aspects of metabolic inflammation are shared by common canonical immunological mechanisms implicated in host defense. Further studies are required to determine the direct consequence of adipose ISG induction in response to SPARC. However, increasing evidence suggests that activated interferon signaling is not only involved in host defense but also regulates response for tissue homeostasis, including brain injury (Khorooshi and Owens, 2010), ischemia (McDonough and Lee, 2017), and skin injury (Zhang et al., 2016). In obese adipose tissue, the cGAS-STING pathway is activated (Bai et al., 2017), IRF3 is upregulated to induce insulin resistance (Kumari et al., 2016), and enhanced type I IFN and IFNAR axis regulates inflammation in both myeloid cells and adipocytes (Chan et al., 2020).
While we validate the ISG expression change by SPARC and identify the specific domain which is responsible for the function, we used mouse RAW 264.7 macrophage cell line due to the technical challenges for transfecting primary bone marrow cell driven BMDMs. Although we observed consistent results in different experiments about SPARC’s effect on ISG induction through TLR4, we need to understand that the cell line does not always reflect classical macrophage phenotypes or gene expression.
The macrophage metabolic status seems to contribute to its activation status in a reversible manner. M1 macrophages are pro-glycolytic, while M2 macrophages engage in high oxidative phosphorylation and fatty acid oxidation (Tannahill et al., 2013; Vats et al., 2006). Blocking glycolysis by 2-DG, hexokinase inhibitor, inhibits LPS induced IL-1β increase in macrophage, but not TNFα and IL-6 (Tannahill et al., 2013), which is consistent with our study that shows SPARC’s requirement of TLR4 signaling. Although IFN induces glycolytic condition (Fritsch and Weichhart, 2016) and IFN-mediated anti-viral activity requires glycolytic condition (Jiang and Shi, 2016), it is not clear how modulation of glycolysis affects ISG activation. It was reported that IFN-γ treated M1 macrophage induced glycolysis and pro-inflammatory activity, and 2-DG treatment blocked the ATP-dependent JAK-STAT1 signaling activation (Wang et al., 2018). We also observed that inhibition of glycolysis by 2-DG reduced ISG activation via inhibition of STAT1 activation, suggesting that glycolysis is required for ISG activation through the JAK-STAT1 signaling pathway. Therefore, SPARC’s sensing of metabolic status such as glycolysis maybe linked with ISG-mediated inflammation in macrophages.
Given that SPARC was reported to be associated with many diseases, including diabetes, cardiovascular disease, and different type of cancers (Kos and Wilding, 2010), the effect of SPARC on macrophages may contribute to the pathogenesis of the diseases. The SPARC deficient mice are protected from age-related cardiac dysfunction and inflammation in parallel to reduced macrophage infiltration and inflammatory cytokine expression (Toba et al., 2015). In cardiovascular disease, pro-inflammatory macrophages are increased to induce plaque formation, recruiting monocyte, and resident M2 macrophages may be converted to pro-inflammatory macrophages. Moreover, increased macrophage-derived SPARC also contributes to myocardial fibrosis (McDonald et al., 2018). In addition, diabetes is also associated with increased inflammation in islets, and pathogenesis was partially attributed to tissue-resident macrophages (Eguchi and Nagai, 2017). Because SPARC is constitutively expressed in multiple cell types, increased SPARC concentrations in multiple tissues may contribute to disease pathogenesis by inducing inflammatory macrophages.
Previously it has been reported that SPARC expression changes according to weight gain or loss in obese individuals (Kos et al., 2009), and SPARC stimulates proinflammatory gene expression in macrophages (Hu et al., 2020; Toba et al., 2015). We found that the matricellular protein SPARC could convert M2 anti-inflammatory macrophages to pro-inflammatory macrophages. We delineated the mechanism that SPARC controls TLR4 downstream signaling to induce both pro-inflammatory cytokines and ISGs. Consistent with our hypothesis that CR-inhibited SPARC may lower inflammation, reduction of adipose SPARC in aged mice extended healthspan and protected the mice from chronic inflammation and interferon response. Collectively, our findings suggest that the matricellular protein SPARC represents a promising therapeutic target to modulate macrophage inflammation during metabolically dysregulated chronic conditions such as obesity and aging.
LIMITATIONS OF THE STUDY
In this study, we demonstrated that increased SPARC concentrations are associated with obesity. The SPARC adipocyte deficient mice were protected from weight gain in response to normal diet. However, it is not clear if SPARC deficiency will confer protection from weight gain in response to high-fat diet. Future work will be needed to determine whether reduction or neutralization of SPARC impacts obesity-associated metabolic inflammation and insulin resistance. Our data shows that reduction of SPARC production from adipocytes can lower age-related inflammation and improve health of mice, but whether SPARC mimics caloric restriction’s pro-longevity effect was not determined.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vishwa Deep Dixit (vishwa.dixit@yale.edu).
Materials Availability
Materials used in this study are available upon request.
Data and Code Availability
The RNA-sequencing data for human CR has been uploaded at SYNAPSE (www.synapse.org) with Synapse ID syn23667189. The RNA-sequencing data for peritoneal macrophages has been uploaded to Gene Expression Omnibus (GSE167950).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Samples
The human CR study included data from CALERIE Phase 2 (Rochon et al., 2011), a multicenter, parallel-group, randomized controlled trial with non-obese healthy individuals. More than two hundred participants were recruited at Pennington Biomedical Research Center (Baton Rouge, LA), Washington University (St. Louis, MO), and Tufts University (Boston, MA) (NCT00427193). A coordinating center was at Duke University (Durham, NC). The effects of up to 2 years of CR on health, including immune and metabolic phenotypes, were investigated in the study. The study was designed to achieve and maintain a weight loss of 25% by decreased caloric intake, but they, in fact, reached 14% of CR (Ravussin et al., 2015). Participants were randomly assigned to 25% calorie restriction (CR group) or calorie intake ad libitum (Control group) for two years after baseline assessments. Men participants were between 20 and 50 years old, and women participants were between 20 and 47 years old. At the initial visit, their body mass index (BMI) was between 22.0 and 27.9 kg/m2. The samples and clinical data collected at baseline, after 1 year and 2 years of intervention were used for experiments and analysis. For RNA-sequencing in this study, abdominal subcutaneous adipose tissue biopsy was performed in part of CR group participants. The plasma samples from obese and lean individuals were from the BARIA study (NCT00936130). The studies were performed under an approved protocol by the Pennington institutional review board with informed consent from participants.
Animal Care
All mice used in this study were housed in specific pathogen-free facilities in ventilated cage racks that deliver HEPA-filtered air to each cage with free access to sterile water through a Hydropac system at Yale School of Medicine. Sentinel mice in our animal rooms were negative for currently tested standard mouse pathogens (Ectromelia, EDIM, LCMV, Mycoplasma pulmonis, MHV, MNV, MPV, MVM, PVM, REO3, TMEV, and Sendai virus) while the studies were performed (Research Animal Diagnostic Laboratory). Mice were fed a standard vivarium chow (Harlan 2018s) unless a special diet was provided and housed under 12 h light/dark cycles. All experiments and animal use were conducted in compliance with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee (IACUC) at Yale University. C57BL/6 (WT) mice were used for experiments. They were bred from our lab colony or purchased from Jackson Laboratories.
Mouse Model
SPARC adipocyte deficient mice were generated and bred in the Dixit lab. ES cells of Sparctm1a(EUCOMM)Wtsi transgenic mice with C57BL/6 background were purchased from European Mouse Mutant Cell Repository (EuMMCR) and injected at Yale Genome Editing Center to make transgenic construct harboring heterozygote mice. Then, the mice were bred with flippase transgenic mice to remove reporter and selection marker of transgenic construct, followed by crossing with Adiponectin-Cre+ mice to generate adipocyte-specific SPARC deficient mice. Littermate control mice (Sparcfl/fl) were used for WT controls in all experiments.
BMDM Culture
Femurs and tibias from WT mice were collected in complete media containing RPMI (Thermo Fischer Scientific), 10% FBS (Omega Scientific), and 1% antibiotics/antimycotic (Thermo Fischer Scientific). Bone marrow was flushed into new complete media by a needle and syringe. Red blood cells were lysed by ACK lysis buffer (Quality Biological) followed by neutralization with the collection media. The collected cells were seeded in 6 well plates and differentiated into macrophages with M-CSF (10 ng/ml, R&D) and L929 (ATCC) conditioned media. Non-adherent cells were harvested on day 7 and seeded as 1 × 106 cell/well in 24 well plate. Cells were treated with recombinant SPARC protein (PeproTech, 120–36, lot 0510518 and 0510518-1, or BioVision, 7204, lot 7E28L72040) for the indicated time with or without M1 and M2 polarization. Recombinant human FSTL1 (PeproTech, 120–51) was treated to M2 polarized BMDMs. M1 polarized BMDMs were incubated with LPS (1 μg/ml, Sigma) and IFNγ (20 ng/ml, Thermo Fischer Scientific), and M2 polarized BMDMs were incubated with IL-4 (10 ng/ml, BD) for 24 hours. For treatment of inhibitors, inhibitors were pre-incubated for 1 hour before SPARC treatment. Inhibitors used for BMDM experiments are the following. 2-DG for glycolysis inhibitor (10 mM, Sigma), TAK-242 for TLR4 inhibitor (2.5 μM, Cayman), CU-CPT22 for TLR1 and TLR2 inhibitor (8 μM, Emd millipore), and L-NIL (40 μM, Cayman) for iNOS inhibitor were used. BMDMs from IRF3 and IRF7 and, cGAS deficient mice (were bred in Dr. Akiko Iwasaki’s lab), TLR4, MyD88 and IFNAR deficient mice (were bred in Dr. Andrew Wang’s lab) and have been described previously.
Cell-line Culture and Transfection
RAW 264.7 cell line was purchased from ATCC (ATCC® TIB-71™). The cells were cultured in DMEM with high glucose (Thermo Fischer Scientific), including 10% FBS (Omega Scientific) and 1% antibiotics/and antimycotic (Thermo Fischer Scientific), and plated in 24 well plates as 1×106 cells per well. The plated cells were transfected with a vector containing human SPARC ORF or a mock vector (GeneCopoeia) with Lipofectamine 3000 (Thermo Fisher Scientific) following the manufacturer’s instruction. After 4 hours of transfection, the media was replaced, and cells were collected with RLT buffer (Qiagen) for Q-PCR analysis after 48 hours of transfection.
Peritoneal Cell Isolation and Culture
Thioglycollate or IL-4 complex (IL-4c) were intraperitoneally injected for inducing in vivo model of classically activated and alternatively activated peritoneal macrophages. 3% thioglycollate 1ml was injected once 4 days before peritoneal cell collection. IL-4c were injected twice, the first injection was done 4 days before peritoneal cell collection, and the second injection was done 2 days after the first injection. IL-4 (5ug, Peprotech) and IL-4 antibody (25ug, BioXcell) were incubated as 1:5 ratio in PBS for 1–2 min right before each injection. Peritoneal cells were collected by injecting 7 ml of PBS into the peritoneum, and the isolated peritoneal cells were seeded as 3×106 per well in 24 well plates. After 2 hours of incubation, cells were washed with PBS three times to keep the only attached peritoneal macrophages. The peritoneal macrophages were non-treated or SPARC (20 μg/ml) treated for 24 hours.
Adipose tissue processing and stromal vascular fraction (SVF) collection
After euthanization of mice, visceral and subcutaneous adipose were collected. Each adipose tissue was enzymatically digested in 0.1% collagenase I or II (Worthington Biochemicals) in HBSS (Life Technologies) for 45 min at 37°C. The SVF was collected by centrifugation at 1500 rpm for 10 min, then washed and filtered by 100 μm and 70 μm strainers. Red blood cells were lysed using ACK lysis buffer (Quality Biological). Cells in SVF were resuspended in 1 ml of RPMI (Thermo Fischer Scientific) with 10% FBS and 1% antibiotics/antimycotic (Thermo Fischer Scientific) for counting before further experiments.
Macrophage isolation
For positive selection of macrophages, SVF from SAT or VAT was incubated with biotinylated anti-mouse F4/80 antibody (eBioscience, 13-4801-85) in isolation buffer (PBS, 0.1% BSA, 2mM EDTA, pH 7.4). Then, cells were washed and incubated with pre-washed magnetic beads (Dynabeads Biotin binder, Life Technologies, 11047) to isolate the F4/80+ fraction.
METHOD DETAILS
Q-PCR
RNA was extracted and purified using the RNeasy Plus micro kit (Qiagen) or Direct-zol RNA Miniprep Plus kit (Zymo Research) according to the manufacturer’s instructions. RNA concentration of the samples was measured, and cDNA was synthesized using the iScript cDNA synthesis kit (Bio-Rad). Real-time quantitative PCR was performed with the cDNA, gene-specific primers, and Power SYBR Green detection reagent (Thermo Fischer Scientific) in the LightCycler 480 II (Roche). Measured values from specific genes were analyzed by ΔCt method normalized with Gaphd or 18S rRNA as an endogenous control. Gene primer sequences are in Table S1.
Western Blotting
Cell lysates were prepared by harvesting cells in RIPA buffer with protease inhibitors. After measuring protein concentration with DC protein assay (Bio-Rad), an equal amount of protein was run on SDS-PAGE gel and transferred to the nitrocellulose membrane. Primary antibodies and appropriate secondary antibodies (Thermo Fisher Scientific) were used to probe blots. The bands were detected by chemiluminescent visualization (PI32209; Pierce). The following primary antibodies were used for experiments. Antibodies to β-actin (1:1,000, 4967L; Cell Signaling), IL-1β (1:1000, GTX74034; Genetex), STAT1 (1:1000, 9172S; Cell Signaling), p-STAT1 (1:1000, 9167S; Cell Signaling), p-TBK1 (1:1,000, 5483S; Cell Signaling), TBK (1:1,000, 38066S; Cell Signaling), p-IRF3 (1:1,000, 4947S; Cell Signaling), IRF3 (1:1,000, 4302S; Cell Signaling), PPARγ (1:200, sc-7273; Santa Cruz), p-HSL (S563) (1:1000, 4139; Cell Signaling), HSL (1:1000, 4107; Cell Signaling).
ELISA and Luminex
ELISA was performed following the manufacturer’s instruction. 100 μl of diluted human plasma was used for the human SPARC ELISA kit (R&D), 100 μl of undiluted mouse serum or 1/10 diluted VAT or SAT lysates sonicated in 500 μl PBS was used for the mouse SPARC ELISA kit (LifeSpan BioSciences), and 50 μl of cell culture supernatant was used for mouse IFN-β, IFN-α, IFN-γ, and YM1 ELISA kits (R&D). For the Luminex assay, 50 μl of collected cell supernatant or 25 μl of collected serum was used, and the assay was prepared following the manufacture’s instruction. Mouse inflammatory 4-plex Magnetic Bead Panel (Thermo Fisher Scientific) was used to detect IL-1β, TNFα, IL-6, and GM-CSF. Customized assay (Thermo Fisher Scientific) to detect IL-1β, TNFα, IL-6, Gro-α/ and KC, and MCP-1 was used for serum. The assay was performed on Luminex xPONENT system. For normalization of SPARC concentrations in VAT or SAT lysate, SPARC concentrations in VAT or SAT lysates were multiplied by the ratio calculated by protein concentrations (Protein concentrations in serum / Protein concentrations in VAT or SAT).
mtDNA Quantification
For each condition, two wells of BMDMs in 24 well plates (1 × 106 per well) were prepared. The detached cells were combined and divided into two equal aliquots. The first aliquots were used for total genomic DNA preparation, and the other aliquot was used for cytosolic DNA preparation. Whole genomic DNA, including mtDNA, was isolated from BMDMs by incubation with 200 μl of 50 mM NaOH for 10 min at 95°C followed by neutralization with 20 μl of 1 M Tris (pH 8.0). Cytosolic DNA was isolated as follows. The second equal aliquots were resuspended in 100 μl buffer containing 150 mM NaCl, 50 mM HEPES pH 7.4, and 25 μg/ml digitonin (Sigma). The homogenates were incubated end over end for 10 min, then centrifuged at 1000g for 10 min to get cell pellets. The cytosolic supernatants were transferred to new tubes and centrifuged at 17000g for 10 min to pellet any remaining cellular debris. The cytosolic DNA was isolated from the supernatants using QIAQuick Nucleotide Removal Columns (QIAGEN). The isolated DNA was used as a template for Q-PCR. As an endogenous control, the nucleus gene Tert was used, and the DNA amounts of Dloop3 and ND4 gene region was measured to detect the mtDNA amount. For both samples isolated total and cytosolic DNA, the quantification of detected values for Dloo3 and ND4 genes was done by ΔCt method normalized with values detected for Tert gene from total DNA samples.
Mitochondria functional assay
MitoSpy™ Green FM (Biolegend) or Image-iT™ TMRM Reagent (Thermo Fisher Scientific) was used to measure mitochondrial mass and membrane potential each. The assays were done following manufacturer’s instructions. For fluorescence plate reader experiments, cells were seeded as 1 × 105 per well on black 96 well plate (Nunc). The plates were read at designated wavelength (Ex 490nm /Em 516nm for MitoSpy and Ex 548nm /Em 574 nm for TMRM) in a plate reader (Thermo Fischer Scientific). Average of three technical replicates were used for analysis. For fluorescence microscope experiments, cells were seeded as 1 × 106 per well on 24 well plate, and culture media was replaced with Live Cell Imaging Solution (Thermo Fischer Scientific) before taking imaging by BZ-X800 Keyence fluorescence microscope. ROS concentrations were measured by CM-H2DCFDA assay (Thermo Fischer Scientific) following manufacturer’s instruction with cells seeded 1 × 105 per well on black 96 well plate (Nunc). The plates were read at designated wavelength (Ex 492nm /Em 517nm) in a plate reader (Thermo Fischer Scientific). Average of three technical replicates were used for analysis.
Solid-Phase Binding Assay
Nunc Maxisorp 96-well plates (Thermo Fischer Scientific) were coated with 100 μl solution containing 0.25, 2.5, and 25 μg/ml of each hTLR4 (R&D), hTLR2 (R&D), human MD-2 (R&D), mTLR4 (CUSABIO), and BSA (Bio-Rad) in PBS and incubated overnight at 4 °C. After washing with washing buffer (PBST, Tween 20 0.05%) three times, unspecific binding was blocked with 10% BSA in PBST (0.05% Tween 20) for 1 hour at room temperature. Following three times of washing, 20 μg/ml SPARC recombinant proteins (Peprotech) were added in 2% BSA/PBST, incubated for 2 hours at room temperature, and washed three times. The SPARC binding was detected by 1-hour incubation of 1 μg/ml SPARC antibody (Cell Signaling, #5420) in 2% BSA/PBST at RT, followed by three times of washing, 1-hour incubation of secondary antibody anti-rabbit IgG-HRP (1:1000, Thermo Fischer Scientific) in 2% BSA/PBST at RT, and three times of washing. TMB substrate (Thermo Fischer Scientific) was added to start the colorimetric reaction and incubated for 10 min in the dark. The reaction was stopped by adding a stop solution (Thermo Fischer Scientific), and the plate was read at OD 450 nm in a plate reader (Thermo Fischer Scientific).
Seahorse Assay
1.5 × 105 BMDMs were seeded on Seahorse XFe96 Cell Culture Microplates (Agilent), and BMDMs were non-polarized or polarized to M1 and M2 BMDMs the next day for 24 hours. SPARC (1, 5, 20 μg/ml) was treated to non-polarized or polarized BMDMs for additional 24 hours. Inhibitors were pre-treated before SPARC treatment. Seahorse XF Cell Mito Stress Test was performed following manufacturer’s instruction adding 1.5 μM Oligomycin, 0.75 μM FCCP, and 3 μM Rotenone with 3 μM Antimycin A at three-time points. The assay was run on Seahorse XFe96 Analyzer (Agilent).
Flow Cytometry
The peritoneal cells and cells from adipose tissues were collected as described previously. The cells were incubated with Fc Block CD16/32 antibodies (Thermo Fisher Scientific) followed by surface antibodies for 30 min on ice in the dark. RELMα, Ki67, NOS2, TNFα, and CD206 were stained using Foxp3 Transcription Factor Staining (Thermo Fisher Scientific) or BD Cytofix/Cytoperm™ Fixation/Permeabilization Kit (BD). The cells were stained with LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (Thermo Fisher Scientific) to detect live cells. Flow Cytometry was performed on a BD LSRII, and results were analyzed by the FlowJo program. The following antibodies were used for flow cytometry analysis to detect each experiment. For peritoneal cells, PE-CD11b, eFluor450-F4/80, Alexa488-NOS2, PerCP-Cy5.5-Ki67, and Biotinylated RELMα with Alexa 647-Streptavidin were used. The following antibodies were used for flow cytometry analysis to detect Treg cells and macrophages: BV711-CD45, eFluor450-B220, FITC-CD25, PE-F4/80, eFluor450-F4/80, APC-eF780-CD11c, APC-Foxp3, PE-Cy7-CD206, PerCP-Cy5.5-CD11b, BV605-CD4, PE-NOS2, and Alexa488-TNFα.
GTT and ITT
For the Glucose Tolerance Test (GTT), mice were fasted 14 hours and 10% glucose solution Sigma Aldrich, G8270) was delivered by intraperitoneal injection based on body weight (0.4 g/kg). For Insulin Tolerance Test (ITT), mice were fasted for 4 hours and insulin was given by intraperitoneal injection (0.8 U/kg). For both GTT and ITT, the concentration of blood glucose was measured by handheld glucometer (Breeze, Bayer Health Care) at baseline and multiple time points.
MRI
The parameters of body composition were measured in vivo by magnetic resonance imaging (EchoMRI; Echo Medical Systems). The amount of fat mass, lean mass and free water were measured by the analysis. For the analysis, each mouse was placed in an acrylic tube with breathing holes and the tube was inserted in the MRI machine. The analysis per mouse takes approximately 90 sec and automatically calculated numerical results were analyzed.
Metabolic cage
The energy expenditure (EE), activity, food intake and water consumption of mice were monitored using the TSE PhenoMaster System (V3.0.3) Indirect Calorimetry System. Each mouse was located in individual chambers for indicated time and the parameters were measured every 30 min. EE was calculated based on the oxygen consumption (O2) and carbon dioxide production (CO2). The EE was unnormalized or normalized by body weight to be used for analysis. Mouse activity was detected by infrared sensors, and food intake and water consumption were measured via weight sensors on food and water dispensers located in the cage.
Behavioral test
Grip strength test was performed using the grip strength meter (TSE system). Maximum strength (gram force, gf) of forelimb of mice holding a bar in the meter was measured 3 times and average of them was used for the analysis. Rotarod test was performed with accurotor rotarod (AccuScan Instruments). One day after a training, the time (sec) mice can stay on the rotarod was measured for a total of 3 tests, and the average of results was used for analysis. The setting used were max speed as 400 (× 0.1 RPM) and time to max speed as 300 (sec).
Lipolysis
For ex vivo lipolysis assay, mice were either fed or fasted for 24 hours until they were euthanized for collecting adipose tissues. 15 mg of VAT and SAT was incubated in 100 μl lipolysis buffer (Krebs buffer with 0.1% glucose and 3.5% fatty-acid free BSA; Sigma) in a 96-well plate for 2 hours at 37°C at 450 rpm. Tissues from fasted mice were incubated without stimulating reagents, while tissues from fed mice were incubated with stimulating reagents such as with 1 μM noradrenaline or 2 mM Isoproterenol. With supernatants from lipolysis assay, glycerol assay (Sigma) and FFA assay (WAKO) was performed following manufacturer’s instruction. For SPARC treatment, adipose tissues from fed mice were pre-incubated with 20 μg/ml SPARC for 24 hours before lipolysis assay.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-sequencing and Analysis
The quality of raw reads was assessed with FastQC (de Sena Brandine and Smith, 2019). Raw reads were mapped to the GENCODE vM9 mouse reference genome using STAR aligner (Dobin et al., 2013) with the following options: --outFilterMultimapNmax 15 --outFilterMismatchNmax 6 --outSAMstrandField All --outSAMtype BAM SortedByCoordinate --quantMode TranscriptomeSAM. Expression quantification was performed using RSEM (Li and Dewey, 2011). PCA was performed in R. Gene differential expression was calculated using DESeq2 (Love et al., 2014). In the analysis of the differential expression of transcription factors, we used the TRANSFAC Curated Transcription Factor Targets dataset from Harmonizome (https://maayanlab.cloud/Harmonizome/). Pathway analysis was done using fgsea (fast GSEA) R-package (Sergushichev, 2016) with a minimum of 15 and a maximum of 500 genes in a pathway and with 1 million permutations. For the pathway analysis, we used the following pathway sets from MSigDB, v6.1: Canonical pathways (CP) from C2 and transcription factor targets (TFT) from C3.
Experimental Design
It was not possible to perform experiments with the blinding of investigators. For studies using mice, experimental groups and matched control groups were randomly assigned by the cage.
Statistical Analysis
A two-tailed Student’s t-test was used for calculating statistical significance. For correlation analysis between human phenotypes and RNA-seq results, Pearson correlation analysis was used. The significance was indicated as follows. *p < 0.05; **p < 0.005; ***p < 0.001; ****p < 0.0001. If needed to determine exclusion criteria, outliers were statistically defined by GraphPad Prism (GraphPad Software, San Diego, United States of America), then excluded from data analysis. A 95% confidence interval was used for all statistical tests, and data was assumed to be normally distributed. Determination of sample size in each experiment was by previously published experiments. Numbers of biological replication and the independent experiment repeat were indicated in each figure legend corresponding to each experiment. Data were shown as mean ± S.E.M. GraphPad Prism was used for all statistical tests for analysis of experimental results.
DATA AND CODE AVAILABILITY
Data Resources
The RNA-sequencing data for human CR has been uploaded at SYNAPSE (www.synapse.org) with Synapse ID syn23667189. The RNA-sequencing data for peritoneal macrophages has been uploaded to Gene Expression Omnibus (GSE167950).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-β actin | Cell Signaling Technology | Cat#4967L; RRID:AB_330288 |
| Rabbit polyclonal anti-IL-1β | GeneTex | Cat#GTX74034; RRID:AB_378141 |
| Rabbit monoclonal anti-IRF-3 (D83B9) | Cell Signaling Technology | Cat# 4302; RRID:AB_1904036 |
| Rabbit monoclonal anti-Phospho-IRF-3 (Ser396) (4D4G) | Cell Signaling Technology | Cat# 4947; RRID:AB_823547 |
| Rabbit monoclonal anti-TBK1/NAK (E8I3G) | Cell Signaling Technology | Cat# 38066; RRID:AB_2827657 |
| Rabbit monoclonal anti-Phospho-TBK1/NAK (Ser172) (D52C2) | Cell Signaling Technology | Cat# 5483; RRID:AB_10693472 |
| Rabbit polyclonal anti-STAT1 | Cell Signaling Technology | Cat# 9172; RRID:AB_2198300 |
| Rabbit polyclonal anti-Phospho-Stat1 (Tyr701) (58D6) | Cell Signaling Technology | Cat# 9167; RRID:AB_561284 |
| Rabbit polyclonal anti-SPARC | Cell Signaling Technology | Cat# 5420; RRID:AB_10692794 |
| Mouse monoclonal anti-PPAR | Santa Cruz | Cat# sc-7273; RRID:AB_628115 |
| Rabbit polyclonal anti-Phospho-HSL (Ser563) | Cell Signaling Technology | Cat# 4139; RRID:AB_2135495 |
| Rabbit polyclonal anti-HSL | Cell Signaling Technology | Cat# 4107; RRID:AB_2296900 |
| PE Rat monoclonal anti-CD11b (M1/70) | Thermo Fisher Scientific | Cat# 12-0112-82; RRID:AB_2734869 |
| eFluor 450 Rat monoclonal anti-F4/80 (BM8) | Thermo Fisher Scientific | Cat# 48-4801-82; RRID:AB_1548747 |
| Alexa 488 Rat monoclonal anti-iNOS (CXNFT) | Thermo Fisher Scientific | Cat# 53-5920-80; RRID:AB_2574422 |
| PerCP-Cy5.5-Rat monoclonal anti-Ki67 (16A8) | BioLegend | Cat# 652424 RRID: AB_2629531 |
| Biotinylated polyclonal anti-Murine RELMα | Peprotech inc | Cat# 500-P214BT |
| Streptavidin, Alexa Fluor 647 conjugate | Thermo Fisher Scientific | Cat# S21374 |
| BV711 Rat monoclonal anti-CD45 (30-F11) | BioLegend | Cat#103147; RRID:AB_2564383 |
| eFluor 450 Rat monoclonal anti-B220 (RA3-6B2) | Thermo Fisher Scientific | Cat# 48-0452-82; RRID:AB_1548761 |
| FITC Rat monoclonal anti-CD25 (3C7) | Biolegend | Cat# 101908; RRID:AB_961212 |
| PE Rat monoclonal anti-F4/80 (BM8) | Thermo Fisher Scientific | Cat# 12-4801-82; RRID:AB_465923 |
| eFluor 450 Rat monoclonal anti-F4/80 (BM8) | Thermo Fisher Scientific | Cat# 48-4801-82; RRID:AB_ 1548747 |
| APC-eFluor 780-Hamster monoclonal anti-CD11c (N418) | Thermo Fisher Scientific | Cat# 47-0114-82; RRID:AB_1548652 |
| APC Rat monoclonal anti-Foxp3 (FJK-16s) | Thermo Fisher Scientific | Cat# 17-5773-82; RRID:AB_469457 |
| PE/Cy7-Rat monoclonal anti-CD206 (C068C2) | Biolegend | Cat# 141720; RRID:AB_2562248 |
| PerCP-Cy5.5-Rat monoclonal anti-CD11b (M1/70) | Thermo Fisher Scientific | Cat# 45-0112-82; RRID:AB_953558 |
| BV605-Rat monoclonal anti-CD4 (RM4-5) | Biolegend | Cat# 100548; RRID:AB_2563054 |
| PE Rat monoclonal anti-iNOS (CXNFT) | Thermo Fisher Scientific | Cat# 12-5920-80; RRID:AB_2572641 |
| APC Rat monoclonal anti-TNF alpha (MP6-XT22) | Thermo Fisher Scientific | Cat# 17-7321-81; RRID:AB_10115438 |
| InVivoPlus anti-mouse IL-4 | BioXCell | Cat# BP0045-A005mg RRID:AB_1107707 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Mouse M-CSF Protein | R&D Systems | Cat#416-ML-050 |
| Human SPARC recombinant protein | Peprotech inc | Cat#120-36 (lot 0510518 and 0510518-1) |
| Human SPARC recombinant protein | BioVision | Cat#7204 (lot 7E28L72040) |
| Recombinant Human TLR4 Protein, CF | R&D Systems | Cat#1478-TR-050 |
| Recombinant Mouse Toll-like receptor 4(Tlr4), partial, Yeast | CUSABIO | Cat#CSB-YP023603MO |
| Recombinant Human TLR2 Protein, CF | R&D Systems | Cat# 2616-TR-050 |
| Recombinant Human MD-2 Protein, CF | R&D Systems | Cat# 1787-MD-050/CF |
| Recombinant Human FSTL1 protein | PeproTech | Cat#120-51 |
| TAK-242 | Cayman Chemical | Cat#13871 |
| 2-DG | Sigma | Cat# D6134 |
| CU-CPT22 | EMD milipore | Cat# 614305 |
| L-NIL | Cayman Chemical | Cat# 80310 |
| L-(−)-Norepinephrine-(+)-bitartrate (Noradrenaline) | Abcam | Cat#ab120717 |
| Isoproterenol hcl | Millipore | Cat#420355 |
| Recombinant Mouse M-CSF Protein | R&D Systems | Cat#416-ML-050 |
| Recombinant mouse IL-4 | Peprotech inc | Cat#13871 |
| Recombinant Mouse IL-4 | BD Biosciences | Cat# 550067 |
| Mouse IFN gamma Recombinant Protein | Thermo Fisher Scientific | Cat# PMC4034 |
| Critical Commercial Assays | ||
| Fixation/Permeablization Kit | BD Biosciences | Cat# 554714 |
| Foxp3 / Transcription Factor Staining Buffer Set | Thermo Fisher Scientific | Cat# 00-5523-00 |
| Human SPARC Quantikine ELISA Kit | R&D Systems | Cat#DSP00 |
| Mouse IFN-alpha ELISA Kit | R&D Systems | Cat#42120-1 |
| Mouse IFN-beta ELISA Kit | R&D Systems | Cat#42120-1 |
| Mouse Osteonectin / SPARC (Sandwich ELISA) ELISA Kit | LifeSpan BioScience | Cat#LS-F21843 |
| Mouse IFN-gamma Quantikine ELISA Kit | R&D Systems | Cat# MIF00 |
| Mouse YM1/Chitinase 3-like 3 DuoSet ELISA | R&D Systems | Cat# DY2446 |
| DuoSet ELISA Ancillary Reagent Kit 2 | R&D Systems | Cat# DY008R&D |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | Cat#L34966 |
| Lipofectamine™ 3000 Transfection Reagent | Thermo Fisher Scientific | Cat#L3000008 |
| RNeasy Plus micro kit | Qiagen | Cat#74034 |
| Direct-zol RNA Miniprep Plus kit | Zymo Research | Cat#R2072 |
| iScript cDNA synthesis kit | Bio-Rad | Cat#1708891 |
| Power SYBR Green PCR Master Mix | Thermo Fisher Scientific | Cat#4367659 |
| Mouse inflammatory 4-plex Magnetic Bead Panel | Thermo Fisher Scientific | Cat#LMC0003M |
| ProcartaPlex multiplex assay | Thermo Fisher Scientific | Cat#PPX |
| Glycerol assay kit | Sigma | Cat#MAK117 |
| NEFA-HR2 (FFA assay) | WAKO | Cat#999-34691, 991-34891, 995-34791, 993-35191, 276-76491 |
| MitoSpy™ Green FM | Biolegend | Cat#424805 |
| Image-iT TMRM assay | Thermo Fisher Scientific | Cat#I34361 |
| CM-H2DCFDA | Thermo Fisher Scientific | Cat#C6827 |
| Deposited Data | ||
| Raw data | This paper | SYNAPSE: syn23667189 |
| Raw data | This paper | Gene Expression Omnibus (GSE167950) |
| Experimental Models: Cell Lines | ||
| Mouse: RAW 264.7 | ATCC | ATCC® TIB-71™ |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | JAX: 000644 |
| Mouse: B6.Cg-Tg(ACTFLPe)9205Dym/J | The Jackson Laboratory | JAX: 005703 |
| Mouse: B6;FVB-Tg(Adipoq-cre)1Evdr/J | The Jackson Laboratory | JAX: 010803 |
| Mouse: Sparcfl/fl:Adipoq-Cre+ | This paper | N/A |
| Mouse: C57BL/6 Tlr4−/− | Dr. Andrew Wang Lab | N/A |
| Mouse:C57BL/6 Myd88−/− | Dr. Andrew Wang Lab | N/A |
| Mouse: C57BL/6 Ifnar −/− | Dr. Andrew Wang Lab | N/A |
| Mouse: C57BL/6 Irf3−/− Irf7−/− | Dr. Akiko Iwasaki Lab | N/A |
| Mouse: C57BL/6 Cgas−/− | Dr. Akiko Iwasaki Lab | N/A |
| Oligonucleotides | ||
| qPCR primers, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| Empty control vector for pReceiver-M02 | GeneCopoeia | EX-NEG-M02 |
| ORF expression clone for SPARC (NM_003118.3) | GeneCopoeia | EX-D0051-M02-H |
| Software and Algorithms | ||
| Image J | NIH | https://imagej.nih.gov/ij/ |
| Prism 7 | Graphpad | https://www.graphpad.com/ |
| FlowJo | Treestar | https://www.flowjo.com/ |
| STAR aligner (v2.5.3a) | http://code.google.com/p/rna-star/ | |
| FastQC (v0.11.3) | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | |
| Picard tools (v2.18.4) | https://broadinstitute.github.io/picard/ | |
| RSEM | https://github.com/deweylab/RSEM | |
| DeSeq2 R package (v1.24.0) | http://www-huber.embl.de/HTSeq | |
| fast GSEA | https://github.com/ctlab/fgsea | |
Highlight.
Caloric restriction in humans decreases matricellular protein SPARC in adipose.
SPARC stimulates interferon response in macrophage via glycolysis and IRF7.
SPARC converts M2 macrophages to M1 macrophage by activating TLR4.
Depletion of SPARC in adipocyte decreases ‘inflammaging’ and extends healthspan.
ACKNOWLEDGMENTS
We thank all investigators and staff involved in coordinating and executing CALERIE-II clinical trial. We thank Dr. Gerald Shadel for helpful discussion and protocols to quantify mtDNA. SR is a recipient of AFAR postdoctoral fellowship. This research was supported in part by NIH grants AG031797, AG045712, P01AG051459, AR070811, Glenn Foundation for Medical Research, and Cure Alzheimer’s Fund (CAF) to VDD and Aging Biology Foundation grant to MNA; CALERIE study was funded by the National Institute on Aging grants U01AG022132, U01AG020478, U01AG020487, and U01AG020480.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adler MI, Cassidy EJ, Fricke C, and Bonduriansky R (2013). The lifespan-reproduction trade-off under dietary restriction is sex-specific and context-dependent. Exp Gerontol 48, 539–548. [DOI] [PubMed] [Google Scholar]
- Bai J, Cervantes C, Liu J, He S, Zhou H, Zhang B, Cai H, Yin D, Hu D, Li Z, et al. (2017). DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS-cGAMP-STING pathway. Proceedings of the National Academy of Sciences 114, 12196–12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A, Zhou C, Liang Y, LeBlanc M, Liddle C, et al. (2015). Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature 528, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Singh PP, Mahmoudi S, Harel I, Casey KM, Dulken BW, Kundaje A, and Brunet A (2019). Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses. Genome Res 29, 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein P (1995). Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol 130, 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, Goldberg EL, Youm YH, Brown CW, Elsworth J, et al. (2017). Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 550, 119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CC, Damen MSMA, Moreno-Fernandez ME, Stankiewicz TE, Cappelletti M, Alarcon PC, Oates JR, Doll JR, Mukherjee R, Chen X, et al. (2020). Type I interferon sensing unlocks dormant adipocyte inflammatory potential. Nature Communications 11, 2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan JK, Ramachandran D, He Y, Palmer CJ, Jurczak MJ, Chen R, Li B, Friedline RH, Kim JK, Ramsey JJ, et al. (2020). A big-data approach to understanding metabolic rate and response to obesity in laboratory mice. Elife 9, e53560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cabo R, Carmona-Gutierrez D, Bernier M, Hall MN, and Madeo F (2014). The search for antiaging interventions: from elixirs to fasting regimens. Cell 157, 1515–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sena Brandine G, and Smith AD (2019). Falco: high-speed FastQC emulation for quality control of sequencing data. F1000Res 8, 1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi K, and Nagai R (2017). Islet inflammation in type 2 diabetes and physiology. The Journal of clinical investigation 127, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eming SA, Wynn TA, and Martin P (2017). Inflammation and metabolism in tissue repair and regeneration. Science 356, 1026–1030. [DOI] [PubMed] [Google Scholar]
- Fernández-Verdejo R, Ravussin E, Speakman JR, and Galgani JE (2019). Progress and challenges in analyzing rodent energy expenditure. Nature methods 16, 797–799. [DOI] [PubMed] [Google Scholar]
- Fontana L, Partridge L, and Longo VD (2010). Extending healthy life span--from yeast to humans. Science 328, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Garagnani P, Parini P, Giuliani C, and Santoro A (2018). Inflammaging: a new immune–metabolic viewpoint for age-related diseases. Nature Reviews Endocrinology 14, 576–590. [DOI] [PubMed] [Google Scholar]
- Fritsch SD, and Weichhart T (2016). Effects of Interferons and Viruses on Metabolism. Front Immunol 7, 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Arner P, Beauchef G, Guéré C, Vie K, Dahlman I, Mejhert N, and Rydén M (2020). Age-Induced Reduction in Human Lipolysis: A Potential Role for Adipocyte Noradrenaline Degradation. Cell Metabolism 32, 1–3. [DOI] [PubMed] [Google Scholar]
- Gardner EM, Beli E, Clinthorne JF, and Duriancik DM (2011). Energy intake and response to infection with influenza. Annu Rev Nutr 31, 353–367. [DOI] [PubMed] [Google Scholar]
- Goldberg EL, and Dixit VD (2015). Drivers of age-related inflammation and strategies for healthspan extension. Immunological reviews 265, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Ma Y, Ma J, Chen S, Zhang X, Guo S, Huang Z, Yue T, Yang Y, Ning Y, et al. (2020). Macrophage-derived SPARC Attenuates M2-mediated Pro-tumour Phenotypes. J Cancer 11, 2981–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W-C, Sala-Newby GB, Susana A, Johnson JL, and Newby AC (2012). Classical Macrophage Activation Up-Regulates Several Matrix Metalloproteinases through Mitogen Activated Protein Kinases and Nuclear Factor-κB. PLOS ONE 7, e42507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram DK, Zhu M, Mamczarz J, Zou S, Lane MA, Roth GS, and deCabo R (2006). Calorie restriction mimetics: an emerging research field. Aging Cell 5, 97–108. [DOI] [PubMed] [Google Scholar]
- Jiang H, and Shi H (2016). PFKFB3-Driven Macrophage Glycolytic Metabolism Is a Crucial Component of Innate Antiviral Defense. 197, 2880–2890. [DOI] [PubMed] [Google Scholar]
- Jun JI, Kim KH, and Lau LF (2015). The matricellular protein CCN1 mediates neutrophil efferocytosis in cutaneous wound healing. Nat Commun 6, 7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun JI, and Lau LF (2020). CCN1 is an opsonin for bacterial clearance and a direct activator of Toll-like receptor signaling. 11, 1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorooshi R, and Owens T (2010). Injury-induced type I IFN signaling regulates inflammatory responses in the central nervous system. J Immunol 185, 1258–1264. [DOI] [PubMed] [Google Scholar]
- Kirkwood TL, Kapahi P, and Shanley DP (2000). Evolution, stress, and longevity. J Anat 197 Pt 4, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kos K, and Wilding JP (2010). SPARC: a key player in the pathologies associated with obesity and diabetes. Nat Rev Endocrinol 6, 225–235. [DOI] [PubMed] [Google Scholar]
- Kos K, Wong S, Tan B, Gummesson A, Jernas M, Franck N, Kerrigan D, Nystrom FH, Carlsson LM, Randeva HS, et al. (2009). Regulation of the fibrosis and angiogenesis promoter SPARC/osteonectin in human adipose tissue by weight change, leptin, insulin, and glucose. Diabetes 58, 1780–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus WE, Bhapkar M, Huffman KM, Pieper CF, Krupa Das S, Redman LM, Villareal DT, Rochon J, Roberts SB, Ravussin E, et al. (2019). 2 years of calorie restriction and cardiometabolic risk (CALERIE): exploratory outcomes of a multicentre, phase 2, randomised controlled trial. Lancet Diabetes Endocrinol 7, 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuk JL, Saunders TJ, Davidson LE, and Ross R (2009). Age-related changes in total and regional fat distribution. Ageing Res Rev 8, 339–348. [DOI] [PubMed] [Google Scholar]
- Kumari M, Wang X, Lantier L, Lyubetskaya A, Eguchi J, Kang S, Tenen D, Roh HC, Kong X, Kazak L, et al. (2016). IRF3 promotes adipose inflammation and insulin resistance and represses browning. J Clin Invest 126, 2839–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kzhyshkowska J, Workman G, Cardó-Vila M, Arap W, Pasqualini R, Gratchev A, Krusell L, Goerdt S, and Sage EH (2006). Novel Function of Alternatively Activated Macrophages: Stabilin-1-Mediated Clearance of SPARC. The Journal of Immunology 176, 5825–5832. [DOI] [PubMed] [Google Scholar]
- Latz E, and Duewell P (2018). NLRP3 inflammasome activation in inflammaging. Semin Immunol 40, 61–73. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Heo YS, Park HS, Lee SH, Lee SK, and Jang YJ (2014). Serum SPARC and matrix metalloproteinase-2 and metalloproteinase-9 concentrations after bariatric surgery in obese adults. Obes Surg 24, 604–610. [DOI] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Li L, Chang Y, Ning W, and Liu X (2019). Structural and functional study of FK domain of Fstl1. Protein Sci 28, 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CY, Rikke BA, Johnson TE, Diaz V, and Nelson JF (2010). Genetic variation in the murine lifespan response to dietary restriction: from life extension to life shortening. Aging Cell 9, 92–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, and Saltiel AR (2007). Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald LT, Zile MR, Zhang Y, Laer AOV, Baicu CF, Stroud RE, Jones JA, LaRue AC, and Bradshaw AD (2018). Increased macrophage-derived SPARC precedes collagen deposition in myocardial fibrosis. American Journal of Physiology-Heart and Circulatory Physiology 315, H92–H100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough A, and Lee RV (2017). Ischemia/Reperfusion Induces Interferon-Stimulated Gene Expression in Microglia. 37, 8292–8308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, and Janeway CA (1997). A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397. [DOI] [PubMed] [Google Scholar]
- Meydani SN, Das SK, Pieper CF, Lewis MR, Klein S, Dixit VD, Gupta AK, Villareal DT, Bhapkar M, Huang M, et al. (2016). Long-term moderate calorie restriction inhibits inflammation without impairing cell-mediated immunity: a randomized controlled trial in non-obese humans. Aging (Albany NY) 8, 1416–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, et al. (2009). Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 15, 774–780. [DOI] [PubMed] [Google Scholar]
- Mlcochova P, Winstone H, Zuliani-Alvarez L, and Gupta RK (2020). TLR4-Mediated Pathway Triggers Interferon-Independent G0 Arrest and Antiviral SAMHD1 Activity in Macrophages. Cell reports 30, 3972–3980.e3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng YL, Klopcic B, Lloyd F, Forrest C, Greene W, and Lawrance IC (2013). Secreted protein acidic and rich in cysteine (SPARC) exacerbates colonic inflammatory symptoms in dextran sodium sulphate-induced murine colitis. PLoS One 8, e77575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LAJ, and Pearce EJ (2015). Immunometabolism governs dendritic cell and macrophage function. Journal of Experimental Medicine 213, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe Y, and Medzhitov R (2016). Tissue biology perspective on macrophages. Nat Immunol 17, 9–17. [DOI] [PubMed] [Google Scholar]
- Ramkhelawon B, Hennessy EJ, Ménager M, Ray TD, Sheedy FJ, Hutchison S, Wanschel A, Oldebeken S, Geoffrion M, Spiro W, et al. (2014). Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nature Medicine 20, 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin LC, and Artis D (2018). Beyond Host Defense: Emerging Functions of the Immune System in Regulating Complex Tissue Physiology. Cell 173, 554–567. [DOI] [PubMed] [Google Scholar]
- Ravussin E, Redman LM, Rochon J, Das SK, Fontana L, Kraus WE, Romashkan S, Williamson DA, Meydani SN, Villareal DT, et al. (2015). A 2-Year Randomized Controlled Trial of Human Caloric Restriction: Feasibility and Effects on Predictors of Health Span and Longevity. The journals of gerontology Series A, Biological sciences and medical sciences 70, 1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynor J, Lages CS, Shehata H, Hildeman DA, and Chougnet CA (2012). Homeostasis and function of regulatory T cells in aging. Current opinion in immunology 24, 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed MJ, Puolakkainen P, Lane TF, Dickerson D, Bornstein P, and Sage EH (1993). Differential expression of SPARC and thrombospondin 1 in wound repair: immunolocalization and in situ hybridization. J Histochem Cytochem 41, 1467–1477. [DOI] [PubMed] [Google Scholar]
- Ridiandries A, Tan JTM, and Bursill CA (2018). The Role of Chemokines in Wound Healing. International Journal of Molecular Sciences 19, 3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rienks M, and Papageorgiou A-P (2016). Novel regulators of cardiac inflammation: Matricellular proteins expand their repertoire. Journal of Molecular and Cellular Cardiology 91, 172–178. [DOI] [PubMed] [Google Scholar]
- Rivera LB, and Brekken RA (2011). SPARC promotes pericyte recruitment via inhibition of endoglin-dependent TGF-β1 activity. Journal of Cell Biology 193, 1305–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochon J, Bales CW, Ravussin E, Redman LM, Holloszy JO, Racette SB, Roberts SB, Das SK, Romashkan S, Galan KM, et al. (2011). Design and conduct of the CALERIE study: comprehensive assessment of the long-term effects of reducing intake of energy. J Gerontol A Biol Sci Med Sci 66, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan BH, Li X, Winkler AR, Cunningham KM, Kuai J, Greco RM, Nocka KH, Fitz LJ, Wright JF, Pittman DD, et al. (2010). Complement C3a, CpG Oligos, and DNA/C3a Complex Stimulate IFN-α Production in a Receptor for Advanced Glycation End Product-Dependent Manner. The Journal of Immunology 185, 4213–4222. [DOI] [PubMed] [Google Scholar]
- Sayed N, Huang Y, Nguyen K, Krejciova-Rajaniemi Z, Grawe AP, Gao T, Tibshirani R, Hastie T, Alpert A, Cui L, et al. (2021). An inflammatory aging clock (iAge) based on deep learning tracks multimorbidity, immunosenescence, frailty and cardiovascular aging. Nature Aging 1, 598–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergushichev AA (2016). An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv, 060012. [Google Scholar]
- Sheikh F, Dickensheets H, Gamero AM, Vogel SN, and Donnelly RP (2014). An essential role for IFN-β in the induction of IFN-stimulated gene expression by LPS in macrophages. J Leukoc Biol 96, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Zhao Y, Yuan L, Yi W, Zhao R, Yi Q, and Yong T (2014). SPARC is over-expressed in adipose tissues of diet-induced obese rats and causes insulin resistance in 3T3-L1 adipocytes. Acta Histochem 116, 158–166. [DOI] [PubMed] [Google Scholar]
- Shin M, Mizokami A, Kim J, Ofude M, Konaka H, Kadono Y, Kitagawa Y, Miwa S, Kumaki M, Keller ET, et al. (2013). Exogenous SPARC suppresses proliferation and migration of prostate cancer by interacting with integrin β1. Prostate 73, 1159–1170. [DOI] [PubMed] [Google Scholar]
- Spadaro O, Youm Y, Shchukina I, Ryu S, Sidorov S, Ravussin A, Nguyen K, Aladyeva E, Predeus AN, Smith SR, et al. (2022). Caloric restriction in humans reveals immunometabolic regulators of health span. Science 375, 671–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speakman JR (2013). Measuring energy metabolism in the mouse - theoretical, practical, and analytical considerations. Frontiers in physiology 4, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganami T, and Ogawa Y (2010). Adipose tissue macrophages: their role in adipose tissue remodeling. Journal of Leukocyte Biology 88, 33–39. [DOI] [PubMed] [Google Scholar]
- Sun L, Miyoshi H, Origanti S, Nice TJ, Barger AC, Manieri NA, Fogel LA, French AR, Piwnica-Worms D, Piwnica-Worms H, et al. (2015). Type I interferons link viral infection to enhanced epithelial turnover and repair. Cell Host Microbe 17, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Nagaretani H, Funahashi T, Nishizawa H, Maeda N, Kishida K, Kuriyama H, Shimomura I, Maeda K, Hotta K, et al. (2001). The expression of SPARC in adipose tissue and its increased plasma concentration in patients with coronary artery disease. Obes Res 9, 388–393. [DOI] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchkonia T, Morbeck DE, Von Zglinicki T, Van Deursen J, Lustgarten J, Scrable H, Khosla S, Jensen MD, and Kirkland JL (2010). Fat tissue, aging, and cellular senescence. Aging Cell 9, 667–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toba H, de Castro Brás LE, Baicu CF, Zile MR, Lindsey ML, and Bradshaw AD (2015). Secreted protein acidic and rich in cysteine facilitates age-related cardiac inflammation and macrophage M1 polarization. Am J Physiol Cell Physiol 308, C972–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beek AA, Van den Bossche J, Mastroberardino PG, de Winther MPJ, and Leenen PJM (2019). Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends in Immunology 40, 113–127. [DOI] [PubMed] [Google Scholar]
- Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, and Chawla A (2006). Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, Folmes CD, Dzeja PD, and Herrmann J (2018). Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. EBioMedicine 30, 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, and Ferrante AW Jr. (2003). Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112, 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Ping F, Yin J, Xiao X, Xiang H, Ballantyne CM, Wu H, and Li M (2013). Elevated plasma SPARC levels are associated with insulin resistance, dyslipidemia, and inflammation in gestational diabetes mellitus. PLoS One 8, e81615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, et al. (2014). Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Grant RW, McCabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, et al. (2013). Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab 18, 519–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefzadeh MJ, Flores RR, Zhu Y, Schmiechen ZC, Brooks RW, Trussoni CE, Cui Y, Angelini L, Lee K-A, McGowan SJ, et al. (2021). An aged immune system drives senescence and ageing of solid organs. Nature 594, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LJ, Sen GL, Ward NL, Johnston A, Chun K, Chen Y, Adase C, Sanford JA, Gao N, Chensee M, et al. (2016). Antimicrobial Peptide LL37 and MAVS Signaling Drive Interferon-β Production by Epidermal Keratinocytes during Skin Injury. Immunity 45, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin X. j., Wong J, Ding S, Seki E, Schnabl B, et al. (2018). New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing data for human CR has been uploaded at SYNAPSE (www.synapse.org) with Synapse ID syn23667189. The RNA-sequencing data for peritoneal macrophages has been uploaded to Gene Expression Omnibus (GSE167950).
Data Resources
The RNA-sequencing data for human CR has been uploaded at SYNAPSE (www.synapse.org) with Synapse ID syn23667189. The RNA-sequencing data for peritoneal macrophages has been uploaded to Gene Expression Omnibus (GSE167950).