

Abstract
Estrogen is known to regulate bone metabolism in both women and men, but substantial gaps remain in our knowledge of estrogen and estrogen receptor alpha (ERα) regulation of adult bone metabolism. Studies using global ERα-knock out mice were confounded by high circulating sex-steroid levels, and osteocyte/osteoblast-specific ERα deletion may be confounded by ERα effects on growth versus the adult skeleton. Thus, we developed mice expressing the tamoxifen-inducible CreERT2 in osteocytes using the 8kb Dmp1 promoter (Dmp1CreERT2). These mice were crossed with ERαfl//fl mice to create ERαΔOcy mice, permitting inducible osteocyte-specific ERα deletion in adulthood. After intermittent tamoxifen treatment of adult 4-month-old mice for 1 month, female, but not male, ERαΔOcy mice exhibited reduced spine BV/TV (−20.1%, P = 0.004) accompanied by decreased trabecular bone formation rate (−18.9%, P = 0.0496) and serum PINP levels (−38.9%, P = 0.014). Periosteal (+65.6%, P = 0.004) and endocortical (+64.1%, P = 0.003) expansion were higher in ERαΔOcy mice compared to control (Dmp1CreERT2) mice at the tibial diaphysis, reflecting the known effects of estrogen to inhibit periosteal apposition and promote endocortical formation. Increases in Sost (2.1-fold, P = 0.001) mRNA levels were observed in trabecular bone at the spine in ERαΔOcy mice, consistent with previous reports that estrogen deficiency is associated with increased circulating sclerostin as well as bone SOST mRNA levels in humans. Further, the biological consequences of increased Sost expression were reflected in significant overall downregulation in panels of osteoblast and Wnt target genes in osteocyte-enriched bones from ERαΔOcy mice. These findings thus establish that osteocytic ERα is critical for estrogen action in female, but not male, adult bone metabolism. Moreover, the reduction in bone formation accompanied by increased Sost, decreased osteoblast, and decreased Wnt target gene expression in ERαΔOcy mice provides a direct link in vivo between ERα and Wnt signaling.
Keywords: estrogens and SERMs, sex steroids, genetic animal models, osteoporosis, osteocytes
Introduction
Estrogen deficiency is the major cause of postmenopausal bone loss and also contributes to age-related bone loss in both sexes.(1) Although there are extensive studies on the potential mechanisms of estrogen action on bone in humans and in rodent models (reviewed in Khosla et al.(2)), our understanding of the role of estrogen signaling in specific cell types in the bone microenvironment remains incomplete. It is known that the effects of estrogen on bone (and other tissues) are mediated by two related, but distinct estrogen receptors (ERs): ERα and ERβ. Of the two, it is clear that ERα is the dominant ER mediating the skeletal effects of estrogen.(2,3) However, studies dissecting ERα action on bone are confounded by two fundamental problems. The first is that global deletion of ERα leads, due to loss of hypothalamic-pituitary feedback, to markedly increased estrogen and testosterone levels and a paradoxical increase in bone mass in both sexes, presumably due to activation of ERβ and/or the androgen receptor.(4) This issue can be circumvented by ERα deletion in specific cells in the bone microenvironment (e.g., osteoprogenitors,(5) osteoblasts/osteocytes,(6–8) osteoclasts,(9,10) or immune cells(11)); however, this leads to the second problem potentially confounding each of these models, which is that the deletion of ERα is present from conception onwards. This makes it impossible to dissociate the effects of disrupted ERα signaling on the growth and development of the skeleton versus its effects on the mature, adult skeleton. In addition, due to the prolonged ERα deletion in these constitutive models, it may be difficult to decipher the downstream biological pathways regulated by ERα, as compensatory gene expression changes are likely to occur over time in these mice, particularly during rapid growth.
To address these issues, in the present studies we first developed and validated a new tamoxifen-inducible Cre-recombinase model targeting the osteocyte lineage driven by the 8 kb Dmp1 promoter(12) (Dmp1CreERT2). Although a 10kb Dmp1CreERT2 mouse has been developed and is well characterized,(13) there is evidence that the 8kb promoter may be more specific for osteocytic expression.(12,14) We then used this model to define the consequences of osteocyte-specific ERα deletion on bone in skeletally mature female and male mice without confounding effects of systemic hormonal changes. Further, because we could evaluate the skeletons of adult mice relatively soon after ERα deletion in osteocytes, we were able to identify a number of key downstream biological pathways regulated by estrogen that have previously not been definitively linked to ERα signaling in vivo.
Materials and Methods
ERαfl/fl mice.
ERαfl/fl mice were kindly provided by Dr. Sohaib Khan and have been characterized in detail.(15) Briefly, exon 3 of the mouse ERα is flanked by loxP recombination sites in these mice, leading to a non-functional ERα when crossed with Cre-expressing mice. These mice and all Cre mice used in this study were in the C57BL/6 background.
TdTomato mice.
B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/JAi9 (hereafter referred to as TdTomato) mice(16) were obtained from Jackson Laboratory under stock #007905.
8kb Dmp1-CreERT2 construct design and transgenic mouse production.
The Dmp1-CreERT2 construct was made by PCR amplifying mouse genomic DNA sequences encompassing -8249 to +4479 relative to the transcription start site (TSS) of the Dmp1 promoter(12) using LongAmp Taq DNA Polymerase (New England Biolabs, Ipswich, MA). This product was blunt-end cloned into the Pme1 and HpaI sites of the attB-containing pBT378 plasmid,(17) with a 3’ MluI site incorporated to facilitate the next cloning step. The CreERT2 gene was PCR amplified from pCAG-CreERT2 (Addgene, Watertown, MA) and cloned into this 3’ MluI site to produce the final Dmp1-CreERT2 construct. Transgenic mice were produced through the Stanford Transgenic, Knockout and Tumor Model Center by selectively inserting the Dmp1-CreERT2 construct into the site-specific H11 locus in C57BL/6 mice using integrase-mediated transgenesis.(17) This technique assures a high efficiency of a single-copy transgene insertion into a predetermined and transcriptionally active chromosome locus.
Mouse husbandry and genetic crosses.
Animal studies were conducted in accordance with NIH guidelines and with approval from the Institutional Animal Care and Use Committee (IACUC) at the Mayo Clinic. All assessments were performed in a blinded fashion. Mice were housed in ventilated cages and maintained within a pathogen-free, accredited facility under a 12-hr light/dark cycle with constant temperature (23 °C) and access to food and water ad libitum. Both sexes were studied as specified below and in the figure legends. We used adult mice (treatment starting at 4 months, harvesting at 5 months of age) for our experimental procedures. To generate experimental mice, heterzygous Dmp1CreERT2 sires were bred with homozygous ERαfl/fl dams to generate Dmp1CreERT2+/−/ERαfl/+ heterozygotes. Sires from this generation of mice were then bred to ERαfl/+ mice to generate the experimental Dmp1CreERT2+/−/ERαfl/fl (ERαΔOcy) and Dmp1CreERT2+/− (Control) groups. All mice used in this study were littermates from this generation.
Tamoxifen treatments.
Tamoxifen (Sigma-Aldrich) was dissolved in 98% corn oil, 2% ethanol to a final concentration of 10 mg/mL and delivered subcutaneously at a dose of 50 mg/kg. For reporter studies, 4-month-old Dmp1CreERT2 x TdTomato mice were treated with either corn oil or tamoxifen for 5 consecutive days on days 1 through 5, then again on days 15 and 22 before sacrifice on day 31. Because of the duration of the study (31 days), we were concerned that after the initial pulse of tamoxifen (5 consecutive days), newly formed late osteoblastic cells/osteocytes that began expressing Dmp1 would continue to express ERα. In order to guard against this possibility, additional doses of Tamoxifen were given on days 15 and 22. This same dosing scheme was used for the experimental phenotyping studies. Due to the known effects of tamoxifen on bone metabolism,(18,19) we treated both experimental mice (Dmp1CreERT2 x Erαfl/fl) and control animals (Dmp1CreERT2 only) with tamoxifen.
Tissue collection.
Mice were anesthetized with ketamine/xylazine and blood was collected via cardiac puncture at time of death and stored at −80°C. The L4–6 lumbar vertebrae were dissected, cleaned of soft tissue, and stored in ethanol to be used for microcomputed tomography (μCT). Right femurs were isolated and embedded in methyl methacrylate (MMA) for bone histomorphometry. The left femur and thoracic vertebrae were isolated, cleaned of soft tissue, and osteocyte-enriched fractions were obtained using centrifugation, as previously described.(20) The samples were immediately homogenized in QIAzol Lysis reagent (QIAGEN, Valencia, CA) and stored at −80 °C.
TdTomato histology and staining.
Mice were sacrificed and femur, lumbar spine and soft tissues were harvested and fixed in 4% paraformaldehyde (PFA) at 4°C for 72 h under gentle agitation. Bones were decalcified in 10% EDTA for 2 weeks at 4 °C under gentle shaking agitation, which was followed by incubation in cryopreservation solution (30% sucrose) for 3 days at 4 °C under gentle shaking agitation. Samples were embedded in Cryomatrix (ThermoFisher Scientific, Wilmington, DE) and flash frozen in 2-methylbutane in dry ice. Samples were stored overnight at −80°C. 7 μm-thick cryosections were prepared for fluorescent imaging. The sections were mounted with the ProLong Antifade Kit (ThermoFisher Scientific) and all slides were imaged using a Zeiss Axio Observer Z1 microscope (Carl Zeiss Microscopy, LLC) and ZenPro software (Carl Zeiss Microscopy, LLC).
RNAscope analyses.
In situ hybridization of the ERα mRNA transcript in osteocytes was performed on FFPE bone sections (n=4 per group) using the RNAScope 2.5 HD Reagent kit from Advanced Cell Diagnostics (ACD, Newwark, CA). Sections were deparaffinized and antigen retrieval performed using Pepsin Reagent (Sigma) for 30 min at 37°C. An ERα-specific target probe (Catalog #478201, ACD) was used and the RNAScope procedure followed according to the manufacturer’s protocol. Sections were mounted using VectaMount (Vector Laboratories, Burlingame, CA) and visualized using the 40x objective of a EVOS M5000 light microscope (ThermoFisher Scientific). Approximately 200 osteocytes were counted per section (in 10 separate fields of view) and scored for ERα positivity.
Quantitative polymerase chain reaction (qPCR) analysis.
Total RNA was extracted according to the manufacturer’s instructions using RNeasy Mini Columns (QIAGEN). On-column RNase-free DNase solution (QIAGEN), was applied to degrade contaminating genomic DNA. RNA quantity was assessed with Nanodrop spectrophotometry (ThermoFisher Scientific). Standard reverse transcriptase was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems by Life Technologies, Foster City, CA). Transcript mRNA levels were determined by qRT-PCR on the ABI Prism 7900HT Real Time System (Applied Biosystems, Carlsbad, CA), using SYBR green (Qiagen). The mouse primer sequences, designed using Primer Express Software Version 3.0 (Applied Biosystems), for the genes measured by SYBR green are provided in Supplementary Table 1. Input RNA was normalized using reference genes (Actb, Gapdh, Hprt, Tuba1a, Tbp) from which the most stable reference gene was determined by the geNorm algorithm.(21) For each sample, the median cycle threshold (Ct) of each gene (run in triplicate) was normalized to the geometric mean of the median Ct of the most stable reference gene. The delta Ct for each gene was used to calculate the relative mRNA expression changes for each sample. Genes with Ct values > 35 were considered not expressed (NE), as done previously.(22)
Skeletal phenotyping.
All μCT imaging was done in a blinded fashion and performed on a Viva Scan 40 μCT scanner (Scanco Medical AG, Basserdorf, Switzerland) with the following parameters: 55 kVp, 145 mA, high resolution, 21.5 diameter, 10.5-μm voxel size, 300-ms integration time. Longitudinal analysis of bone microarchitecture was performed on the tibial diaphysis with baseline measurements taken before treatment at 4-months of age and endpoint measurements taken the day before sacrifice at 5-months. Animals were anesthetized using 2–4% isoflurane inhalation for induction and 1% isoflurane for maintenance and remained immobilized for the entire scan. Cortical parameters including endocortical (EC) and periosteal cortex (PC) diameters, and cortical thickness (Ct.Th) were assessed at the tibial mid-shaft diaphysis (50 slices). Ex vivo quantitative analysis of the lumbar spine (L5) was performed on dissected tissue after sacrifice. Using two-dimensional (2D) data from scanned slices, 3D analysis was used to calculate morphometric parameters at the lumbar vertebral body (200 slices) defining trabecular bone mass and microarchitecture, including trabecular bone volume fraction (BV/TV; %), trabecular number (Tb.N; 1/mm), trabecular thickness (Tb.Th; mm), trabecular separation (Tb.Sp; mm). All μCT parameters were derived using the manufacturer’s protocols.
Bone histomorphometry.
All histomorphometric analyses were performed in a blinded fashion. For dynamic histomorphometric analyses, mice were injected subcutaneously with Alizarin-3-methyliminodiacetic acid (0.1 ml/animal, 7.5 mg/ml) and calcein (0.1 ml/animal, 2.5 mg/ml) on days 9 and 2, respectively, before euthanasia. The lumbar vertebrae and right femur were isolated from experimental mice and embedded in MMA. Bone sectioning and histomorphometry were performed as previously described from our laboratory.(3)
Bone markers.
Bone marker assays were conducted on cardiac bleed-derived serum from overnight fasted mice for the bone formation serum marker, PINP (amino-terminal propeptide of type I collagen), using the Rat/Mouse P1NP enzyme immunoassay (EIA) kit (Immuno Diagnostic Systems [IDS], Scottsdale, AZ) and the bone resorption serum marker, CTx (cross-linked C-telopeptide of type I collagen), using the RatLaps Rat/Mouse CTx EIA kit (IDS). Serum sclerostin was measured using an ELISA (R and D Systems).
Serum estradiol measurements.
Blood was drawn from cardiac bleeds from overnight fasted mice. Blood was allowed to clot, and serum was collected by centrifugation at 8,000 RPM for 5 min at room temperature. Serum E2 was measured by LC-MS/MS (API 5000, Applied Biosystems-MDS Sciex; interassay CV 8%).
Statistical analyses.
Results were analyzed for statistically significant differences using the Prism 9.3.1 statistical software (GraphPad Software, Inc., La Jolla, CA, USA) and R (4.0.3). All data was tested for normality using the Shapiro-Wilk test. Datasets that passed the Shapiro-Wilk Test (p>0.05, and thus were normally distributed) were assessed with parametric tests, while datasets that did not pass the Shapiro-Wilk Test (p<0.05) were assessed with non-parametric tests. For parametric data, we performed an unpaired t-test and for non-parametric data, the Mann-Whitney test was used. Experimental group numbers (n) are indicated in each figure. Sample sizes were based on pilot or previously conducted and published experiments,(3) in which statistically significant differences were observed on bone. Comparison of pre-specified groups of gene sets was done using a multivariate analysis of variance (MANOVA) as previously validated for such comparisons.(23) Statistical significance set at p < 0.05.
Results
Construction and validation of 8kb Dmp1CreERT2 mice.
To determine if disrupted estrogen signaling in osteocytes influences bone metabolism in adulthood, we constructed Dmp1CreERT2 mice with the 8kb Dmp1 promoter sequence.(12,14) To test cell-specificity, we crossed these mice with the Ai9 TdTomato reporter line and treated with tamoxifen to induce Cre-recombination and fluorescent tracking(16) (Fig 1A). In the Dmp1CreERT2 mice, Cre-recombination occurred in osteocytes embedded in the bone matrix in both the femur and spine and only with treatment of tamoxifen (Fig 1B, C). As shown in higher magnification (Fig 1D), in addition to osteocytes, there was evidence for Cre recombination in a few surface osteoblasts, but not in bone marrow cells. Although we did not directly compare the 8 kb promoter with the 10 kb promoter in our study, the cellular distribution of Cre expression we report for the 8 kb promoter is similar to that reported by Kalajzic et al.;(24) that study also demonstrated more widespread expression of the 10 kb promoter in surface osteoblasts than the 8 kb promoter. Thus, although the 8 kb Dmp1 promoter is relatively more specific for osteocytes, it is still expressed in a subset of late osteoblast cells. As such, when we refer subsequently to “osteocytic” ERα deletion, an implicit caveat is that this deletion is likely also occurring in a subset of late osteoblasts. In observing additional tissues, we found no evidence for recombination in brain, liver kidney, cardiac muscle, or uterus; however, it appeared that the Dmp1CreERT2 line also recombined in small numbers in skeletal muscle (Supplementary Fig 1).
Figure 1.
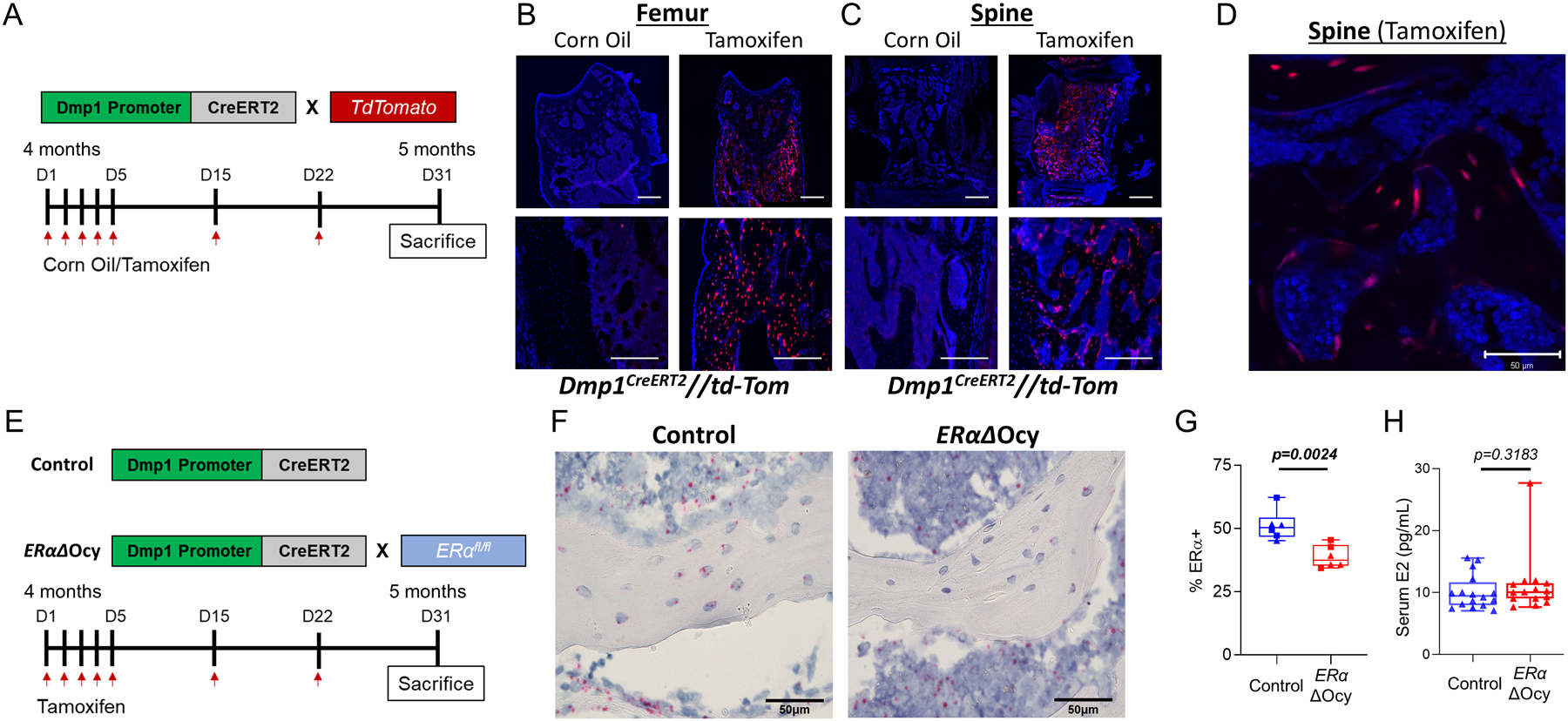
Dmp1-CreERT2 targets osteocytes. (A) Experimental outline for reporter induction in Dmp1CreERT2 x TdTomato mice. (B) Representative images of induced TdTomato expression in osteocytes in the femur and (C, D) spine of corn oil- versus tamoxifen-treated Dmp1CreERT2 x TdTomato mice at low (10X, 20X, C) and high (40X, D) magnification. Black areas are bone, blue areas show extensive DAPI staining in the bone marrow. Scale bars = 10μm for 10X, 20X and 50μm for 40X. ;(E) Experimental outline for inducible deletion of ERα in osteocytes. (F) Images of RNAscope for Esr1 mRNA (encoding ERα) performed on spine bone sections from tamoxifen-treated Dmp1CreERT2 (Control) or ERαΔOcy mice. Scale bars = 50μm (G) Quantification of percentage of osteocytes positive for ERα observed from RNAscope (n = 6 per group [3 males (squares), 3 females (triangles)]). (H) Measurement of serum estradiol (E2) from cardiac blood (n = 15–16 per group). Boxes represent median and interquartile range and whiskers represent minimum and maximum values. Datasets were analyzed by unpaired t-test (G) or Mann-Whitney test (H).
Effects of osteocyte-specific ERα deletion on bone microarchitecture in adult mice.
Using the validated Dmp1CreERT2 line, the effects of osteocyte-specific ERα deletion on adult bone were then tested. Dmp1CreERT2 mice was crossed with mice homozygous for a floxed allele of the gene encoding estrogen receptor α (ERαfl/fl) (see methods for breeding strategy). This permits conditional knockout of ERα in osteocytes (Dmp1CreERT2 x ERαfl/fl [ERαΔOcy]) through administration of tamoxifen (Fig 1E), as demonstrated by assessing the ERα transcript in osteocytes using in situ hybridization (RNAScope; Fig 1F). This was quantified and demonstrated a significant, albeit modest, reduction in osteocytes expressing ERα mRNA (51.1% ERα+ to 38.8% ERα+, P = 0.029; Fig 1G), with similar reductions observed in male and female mice. Serum estradiol (E2) levels did not change following osteocytic ERα deletion in female mice (Fig 1G), indicating that this cell-specific knockout did not lead to a rise in circulating estrogen levels, which is observed in global ERα knockout models due to loss of hypothalamic-pituitary feedback.(4)
We next assessed the skeletal phenotype of the ERαΔOcy mice by comparing them to Dmp1CreERT2 (Control) littermates who were also administered the identical regimen of tamoxifen, given the known effects of tamoxifen on bone.(18,19) ERαΔOcy mice exhibited significantly reduced trabecular bone volume fraction and trabecular thickness at the lumbar spine in female mice (Fig 2A, B), although trabecular number or separation did not change (Fig 2C, D). Similar findings were noted for trabecular bone changes at the femur metaphysis in the female mice (Supplementary Fig 2A–D). The trabecular changes were restricted to female mice, as we did not observe these in male mice (Supplementary Fig 3). Next, we assessed changes in cortical bone parameters; because these were at a distal skeletal site, we could assess these longitudinally before and after ERα deletion (see Methods). The physiological action of estrogen through ERα inhibits periosteal apposition and endocortical resorption, and loss of estrogen in postmenopausal women(25) and rodents(26) reverses this action, leading to expansion of both periosteal and endocortical diameters. Interestingly, in ERαΔOcy mice we observed this similar phenomenon. Loss of ERα in osteocytes led to a blunted contraction of the endocortical diameter and enhanced periosteal expansion in the tibial diaphyses (Fig 2E, F, demonstrated in 2H), mimicking the physiology observed in women and rodents with estrogen deficiency(26–28) and mice with global loss of ERα.(29) We did not observe any longitudinal changes in cortical thickness (Fig 2G).
Figure 2.
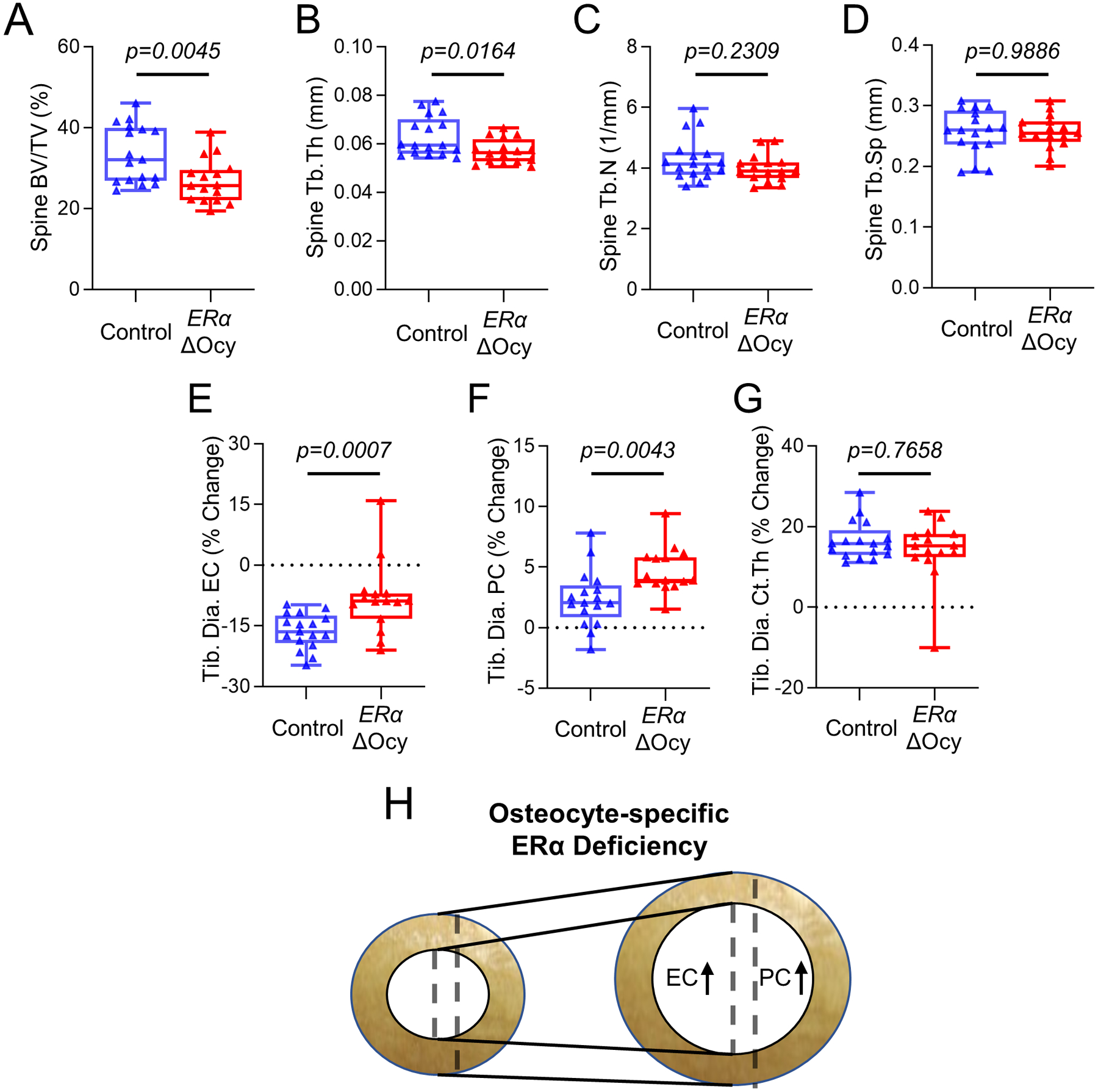
Deletion of osteocyte ERα in adult female mice leads to reduced bone mass and expansion of endosteal and periosteal surfaces. (A-D) Micro-CT analyses of the lumbar spine of tamoxifen-treated Control or ERαΔOcy mice: (A) bone volume fraction (BV/TV), (B) trabecular thickness (Tb.Th), (C) trabecular number (Tb.N), and (D) trabecular spacing (Tb.Sp). (E-H) Longitudinal micro-CT analyses of cortical bone at the tibial diaphysis (Tib. Dia.) showing percent (%) change between baseline and endpoint. (E) Endocortical circumference (EC), (F) periosteal circumference (PC), and (G) cortical thickness (Ct.Th).. (H) Diagram of changes in tibial cortical bone with osteocyte-specific ERα knockout. n=16–17 mice per group. Boxes represent median and interquartile range and whiskers represent minimum and maximum values. Datasets analyzed by unpaired t-test (A, D, F) or Mann-Whitney test (B, C, E, G).
Assessment of bone histomorphometry.
To determine if the effects we observed were driven by bone formation or resorption, we performed static and dynamic bone histomorphometry in the ERαΔOcy compared to the Control mice, focusing on the female mice where the skeletal consequences of ERα deletion were most evident. In the ERαΔOcy mice, there were significantly reduced rates of bone formation (Fig 3A) and mineral apposition (Fig 3B) in trabecular bone at the spine. This was accompanied by an increase in osteoclast numbers (Fig 3C) and a trend for reduced osteoblast numbers (Fig 3D, P = 0.12). We also measured serum bone turnover markers and, reflecting the histologically observed reduction in bone formation rate, there was a significant reduction in serum PINP levels in the ERαΔOcy mice (Fig. 3E). By contrast, the increase in osteoclast numbers was not accompanied by a significant increase in serum CTx levels (Fig. 3F). Finally, osteocytic ERα deletion had no effect on osteocyte numbers or empty lacunae in trabecular bone (Supplementary Fig. 2E, F).
Figure 3.
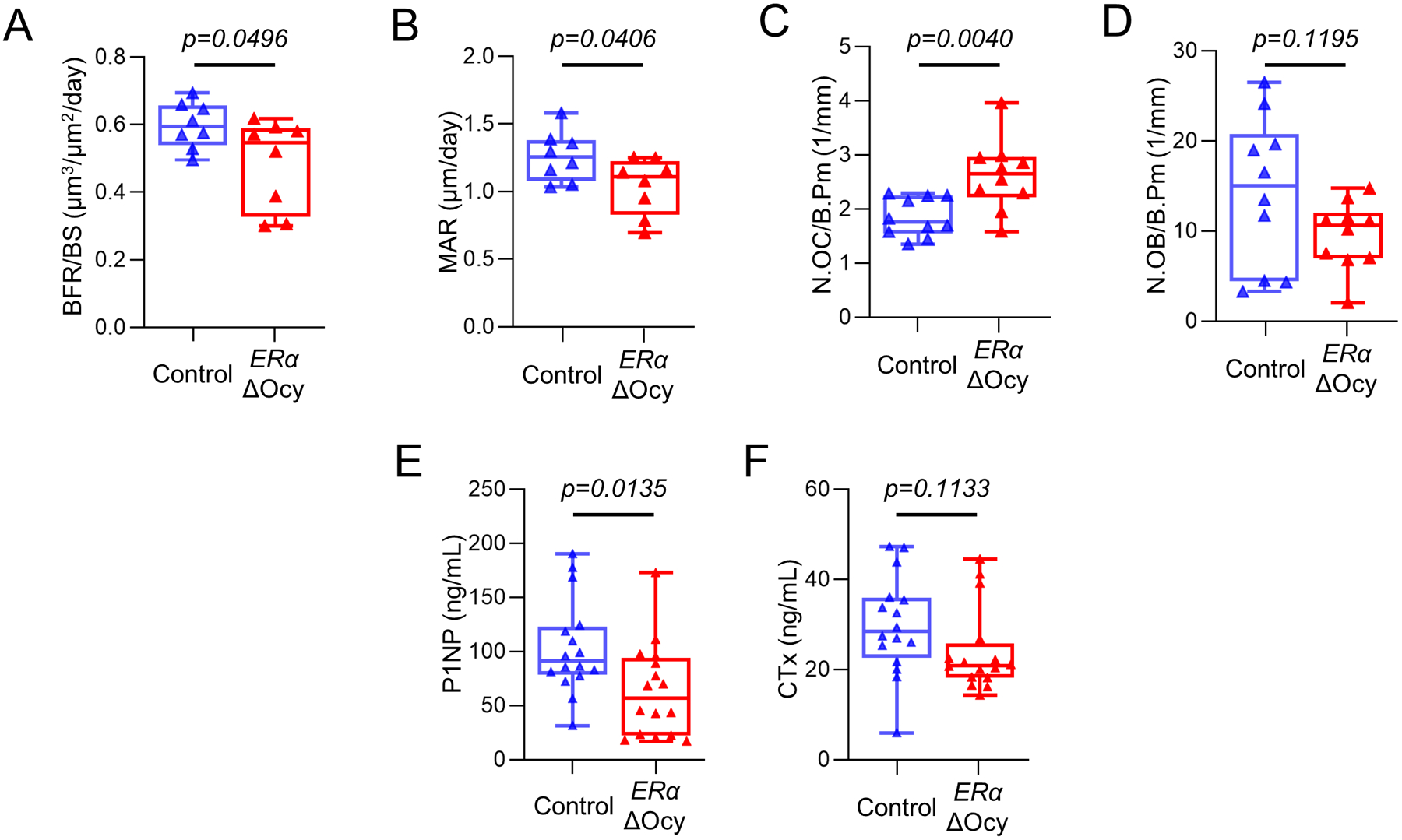
ERα deletion in osteocytes leads to reduced bone formation and increased osteoclast numbers. (A) Measurement of bone formation rate per bone surface (BFR/BS) and (B) mineral apposition rates (MAR) in the lumbar spines of tamoxifen-treated Control or ERαΔOcy mice using double-label dynamic histomorphometry. (C) Counted number of osteoclasts (N.OC/B.Pm) and (D) osteoblasts (N.OB/B.Pm) normalized to bone perimeter using static histomorphometry. (E) PINP and (F) CTx levels measured from endpoint cardiac serum. n=8–16 per group. Boxes represent median and interquartile range and whiskers represent minimum and maximum values. Datasets analyzed by unpaired t-test.
Effects of osteocyte-specific ERα deletion on key skeletal biological pathways.
Next, we attempted to uncover the effects of osteocyte-specific ERα deletion on established skeletal biological pathways. We reasoned that, in contrast to constitutive, long-term ERα deletion from conception onwards, the relatively short duration of inducible ERα deletion in our study may uncover pathways obscured in the previous, longer term studies confounded by compensatory mechanisms, especially during rapid growth. For this, we first examined expression of the Sost gene, which encodes sclerostin, a potent inhibitor of Wnt signaling(30) and, as reviewed previously,(2) is perhaps the most consistently regulated factor in bone and peripheral circulation by estrogen in humans. Indeed, as shown in Fig 4, Sost mRNA levels were significantly higher in both the spines (Fig 4A) and femurs (Fig 4B) of ERαΔOcy as compared to Control mice; however, serum sclerostin levels were not different between the two groups (Supplementary Fig 4A). In more detailed pathway-based gene analyses (see Methods), the changes in Sost mRNA expression were accompanied by a significant downregulation of osteoblast (Fig 4C) and Wnt target (Fig 4D) genes. Thus, osteocytic loss of ERα resulted in decreased bone formation, increased Sost mRNA expression, and significant reductions in osteoblast and Wnt target genes in osteocyte-enriched bone samples.
Figure 4.
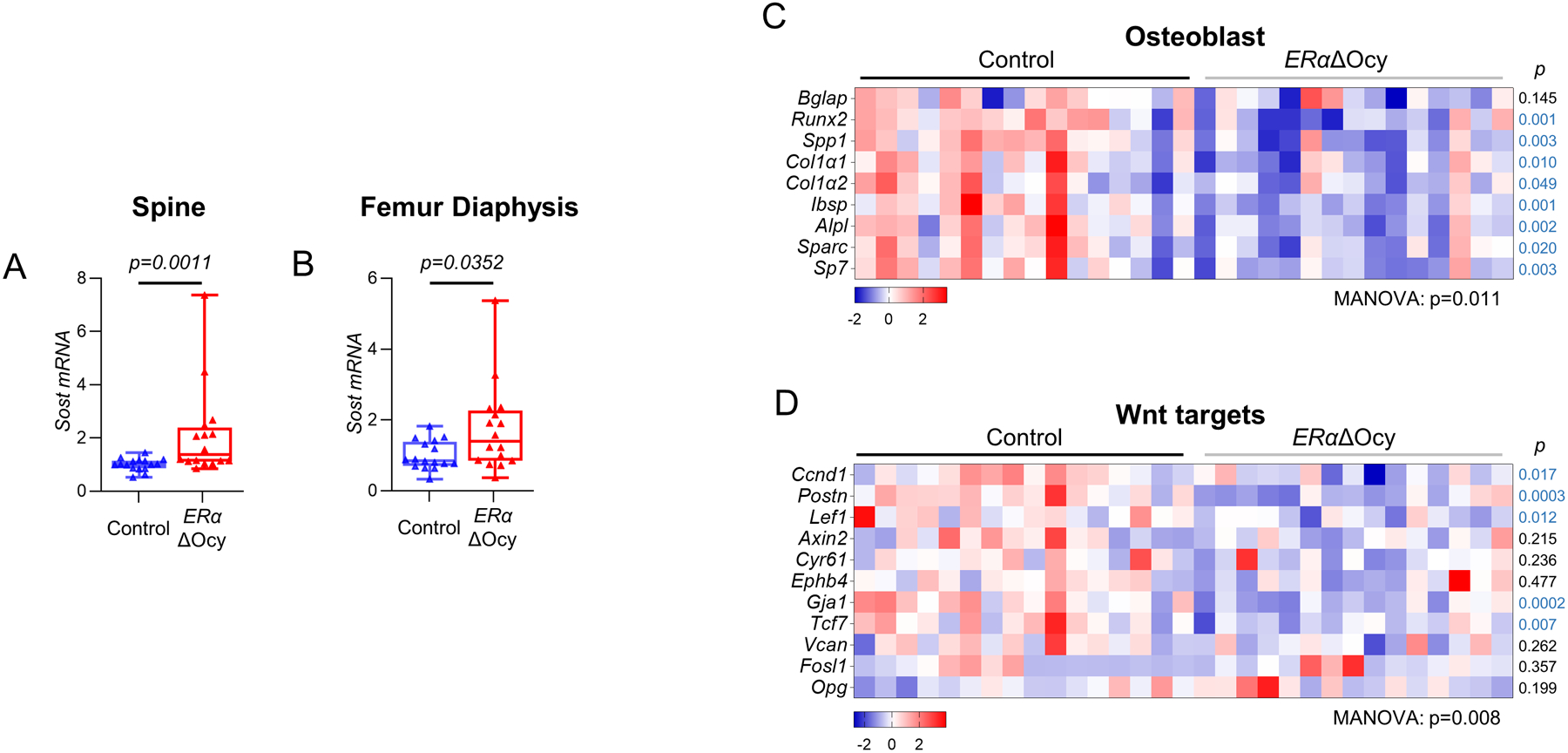
ERαΔOcy mice display increased Sost expression and a reduction in bone anabolic pathways. (A) mRNA expression of Sost in Dmp1CreERT2 and ERαΔOcy mice at the spine and (B) femur diaphysis by qRT-PCR. (C) Heatmap of mRNA expression changes in osteoblast and (D) Wnt signaling pathways from spines of Control and ERαΔOcy mice by qRT-PCR. n=16–20 mice per group. Boxes represent median and interquartile range and whiskers represent minimum and maximum values. Datasets analyzed by Mann-Whitney test (A, B) or MANOVA (C, D) with pathway-level p-values listed below each heatmap and individual gene p-values beside each row (blue font p-value indicates downregulated gene in the ERαΔOcy mice).
Others have shown that the mesenchymal-expressed Cxcl12 (encoding for Stromal cell-derived factor 1) contributes to the loss of cortical bone with estrogen deficiency.(31) Consistent with this, we found increased levels of Cxcl12 in the ERαΔOcy mice at the spine (Fig 5A), with a similar trend at the femur diaphysis (Fig 5B, P = 0.12). Gene expression of Rankl, Opg (also a Wnt target, Fig 4D), or Sostdc1, which have been shown to be regulated by estrogen or following ERα deletion in other studies in mice and humans,(6,32,33) was not altered in the ERαΔOcy mice (Fig 5C–E). Expression of additional osteocyte genes is shown in Supplementary Fig 4B–F. As is evident, changes in these genes were inconsistent, with Dmp1 and Pdpn being reduced in the ERαΔOcy as compared to the Control mice without significant changes in Mepe, Fgf23, or Phex.
Figure 5.
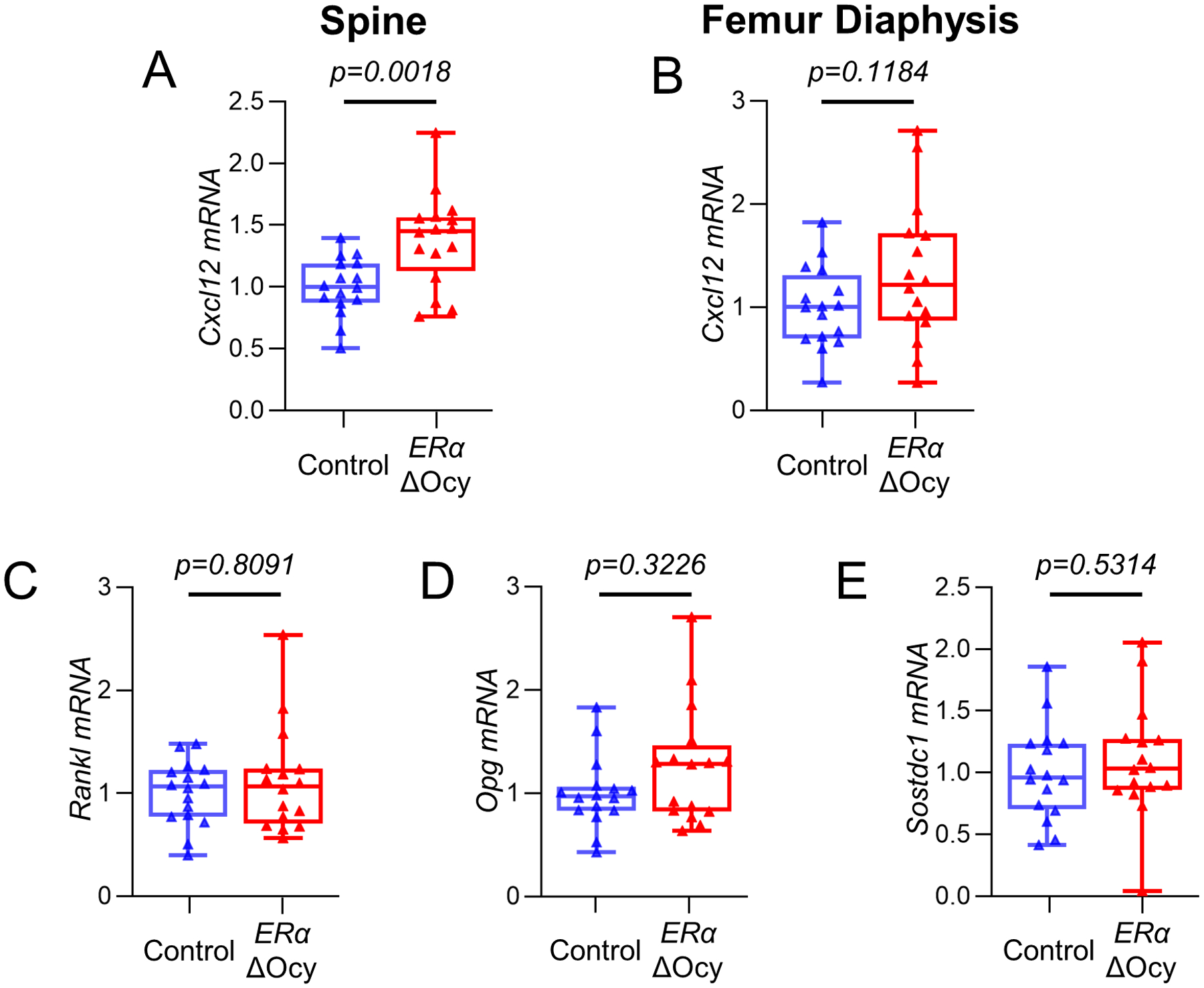
Cxcl12, but not other markers of bone turnover are altered in ERαΔOcy mice. (A) mRNA expression of Cxcl12 at the spine and (B) femur diaphysis of Control and ERαΔOcy mice by qRT-PCR. (C) Rankl, (D) Opg, and (E) Sostdc1 mRNA expression by qRT-PCR at the spine. Boxes represent median and interquartile range and whiskers represent minimum and maximum values. Datasets analyzed by unpaired t-test (A, B, E) or Mann-Whitney test (C, D).
In addition to these selected genes based on the literature and Wnt target genes, we examined other skeletal pathways for possible changes following osteocytic ERα deletion (Supplementary Table 1). As shown in Fig. 6, ERαΔOcy mice exhibited significant reductions in BMP (Fig 6A) as well as Notch (Fig 6B) signaling, whereas proliferation- and apoptosis-related genes increased in the ERαΔOcy versus the control mice (Fig 6C, D). There were no significant changes in senescence, oxidative stress, or stem cell genes (Supplementary Fig 5A–C). These data thus demonstrate widespread effects of osteocytic ERα loss not only in Wnt, but also in other key skeletal biological pathways.
Figure 6.
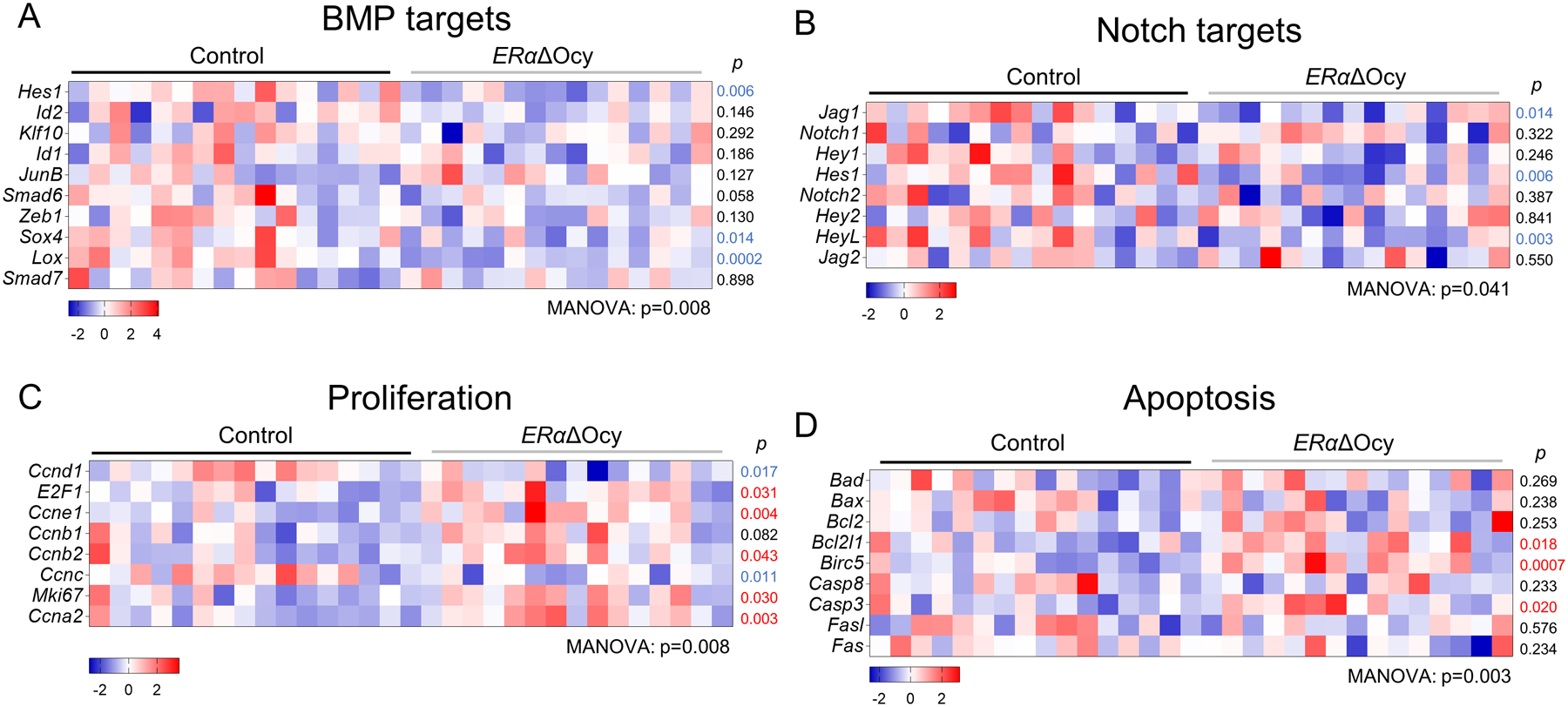
Osteocyte ERαΔOcy deletion suppresses osteogenic pathways, while promoting proliferation and apoptosis gene expression. (A-D) Heatmaps of mRNA expression changes in (A) BMP signaling, (B) Notch signaling, (C) proliferation, and (D) apoptosis pathways from spines of tamoxifen-treated Control and ERαΔOcy mice by qRT-PCR. Datasets analyzed by MANOVA with pathway-level significance-values listed below each heatmap and individual gene p-values beside each row. Blue font p-value represent significantly downregulated expression in ERαΔOcy mice, while red font p-values represent upregulated expression in ERαΔOcy mice.
Discussion
In the present study, we first developed and validated a new tamoxifen-inducible Cre model targeting the osteocyte lineage (the 8kb Dmp1CreERT2 mouse). This model showed no “leakiness” in the absence of tamoxifen, was relatively specific for osteocytes, and had none/minimal expression in non-skeletal tissues, with the exception of skeletal muscle. As such, although a 10kb Dmp1CreERT2 mouse is available,(13) the 8kb Dmp1CreERT2 mouse provides an alternate model for inducible osteocytic gene deletions perhaps with greater promoter specificity.(12,14)
As previously recognized by Kedlaya et al. using the 10kb Dmp1CreERT2 mouse,(34) demonstrating deletion of target genes in these inducible osteocytic Cre models is not trivial. Thus, as we also did here, one can demonstrate activity of the Cre in osteocytes using reporter mice; but because gene deletions with a given Cre can vary greatly from one floxed gene to another, perhaps due to the local chromatin structure around different floxed alleles,(35) reliance solely on reporter mice is problematic. Simply measuring mRNA levels of the deleted gene in whole bone lacks sufficient sensitivity due to contamination by cells not expressing the Cre and the fact that gene deletion is likely incomplete in these models due to the known stochastic nature of gene expression,(36) and thus dependent upon whether a particular cell is expressing Dmp1 during the intermittent tamoxifen dosing. Likely due to these issues, Kedlaya et al. had to use a complex strategy where they generated triple transgenic mice harboring the 10kb Dmp1CreERT2, their floxed allele (β-catenin), and the floxed Ai9 allele. They then flow sorted Tdtomato− (non-rearranged) and Tdtomato+ (rearranged) cells and were able to demonstrate depletion of the β-catenin mRNA in the Tdtomato+ as compared to the Tdtomato− cells. Given the laborious nature of this approach, we used an alternate approach of in situ hybridization (RNAscope) for the ERα transcript and demonstrated a modest (from 51.1% to 38.8% osteocytes positive for ERα), but significant, reduction in the ERα mRNA in the ERαΔOcy compared to the Control mice. As noted above, this modest reduction may be due to the known stochastic nature of gene expression,(36) whereby ERα gene deletion would be dependent upon whether a particular cell is expressing Dmp1 during the intermittent tamoxifen dosing.
Importantly, even with this relatively modest deficit in ERα induced during adulthood, we found that ERαΔOcy mice had a significant reduction in trabecular bone mass in female, but not male, mice. This was due to an unequivocal reduction in bone formation using both histomorphometry and serum PINP levels. In terms of bone resorption, although osteoclast numbers increased significantly in the ERαΔOcy mice, this was not accompanied by an increase in serum CTx levels.
These data using inducible osteocytic ERα deletion in adult mice help resolve currently conflicting findings using the constitutive Dmp1Cre mice: consistent with our work, Kondoh et al found that osteocytic ERα deletion resulted in trabecular osteopenia in female, but not male, mice.(6) In contrast to these findings, Windahl et al. used the same Cre and found trabecular osteopenia in male, but not female, mice following osteocytic ERα deletion.(8) Because these conflicting findings may, in part, be due to developmental effects of the constitutive osteocytic ERα deletion, our data using an inducible osteocytic Cre in adult mice help resolve this issue and the majority of the evidence now points to an important role for the osteocytic ERα in regulating trabecular bone in female, but not male, mice.
In the previous studies described above with the constitutive Dmp1Cre, bone histomorphometry revealed a decrease in bone formation without a change in osteoclast numbers.(6,8) We also noted a reduction in bone formation in the inducible ERαΔOcy mice, but in contrast to the previous data, we found that inducible osteocytic ERα deletion also resulted in a significant increase in osteoclast numbers. The lack of a concomitant increase in serum CTx levels may reflect a modest effect on bone resorption following osteocytic ERα deletion that contrasts with the more dramatic increases in osteoclast number and activity observed following constitutive ERα deletion in osteoclasts.(9,10) Thus, although our data indicate some effect of osteocytic ERα deletion on osteoclasts, this is likely much smaller than that observed following non-inducible deletion of ERα directly in osteoclasts, at least during our relatively short treatment period.
In cortical bone, the ERαΔOcy mice had preserved cortical thickness but a relative increase in both endocortical and periosteal diameters. These findings are consistent with existing data in humans(25) as well as rodents,(26) demonstrating that estrogen inhibits periosteal bone formation and endocortical bone resorption; indeed, there is also evidence that estrogen may stimulate endocortical bone formation.(37) Our data indicate that these effects of estrogen on endocortical and periosteal surfaces are mediated directly through the osteocytic ERα.
A key finding of our study is perhaps the most direct in vivo link to date between ERα and Wnt signaling in osteocytes. As noted above, a reduction in bone formation in mice with osteocytic ERα deletion has been consistent across models. In our study, this reduction was associated with increased Sost mRNA levels, decreased expression of osteoblast genes, and decreased expression of Wnt target genes. This makes a strong argument that the impairment in bone formation induced by osteocytic ERα deletion is due to upregulated Sost production by osteocytes, which inhibits Wnt signaling and overall bone formation by osteoblasts. Although our study represents the most direct demonstration of this link between ERα and Wnt signaling, it is consistent with at least indirect evidence in mice and humans linking these pathways. For example, Niziolek et al.(38) found that mice carrying two different Lrp5 mutations that conferred resistance to sclerostin were protected against ovariectomy-induced bone loss. In human studies, we(39–41) and others(42) have identified sclerostin as perhaps the most consistent bone-regulatory factor suppressed by estrogen at both the mRNA and protein levels. In contrast to these findings, studies examining circulating sclerostin or bone Sost levels in mice following ovariectomy have generally been inconsistent.(43) The reasons for these differences may have to do with the specifics of the models. Thus, following ovariectomy, there is a marked increase in bone turnover, particularly in osteoclast numbers and bone resorption;(44) this likely leads to secondary changes in gene expression independent of the direct effects of estrogen signaling in osteocytes. By contrast, our model of inducible ERα deletion in adult mice clearly had reduced bone formation with no change bone resorption (serum CTx), although a small increase in osteoclast numbers was noted. In addition, unlike the previous studies using constitutive osteocytic ERα deletion from conception onwards, we studied our mice relatively early after ERα deletion rather than months later as in the constitutive models, which were further confounded by the effects of growth and development. As such, our specific model of adult osteocytic ERα deletion was perhaps better able to uncover the link between ERα and Wnt signaling in vivo than was possible with the previous osteocytic ERα deletion models.
We should note that the effects we observed on osteoclast numbers in the ERαΔOcy mice are consistent with known effects of sclerostin and Wnt signaling on osteoclasts. Indeed, canonical Wnt receptors are expressed on osteoclasts; Wnt3a treatment reduces osteoclast formation in vitro, and reduction of Lrp5/6 expression in vivo in osteoclast precursors leads to reduced bone mass.(45) As such, the observed increase in osteoclast numbers without a detectable increase in CTx would be consistent with downstream effects of increased sclerostin production in the bone microenvironment in the ERαΔOcy mice, but with the restraining effects of direct estrogen signaling in osteoclasts intact.(9,10) Although Opg is a known Wnt target gene,(46) we did not observe changes Opg expression in the osteocyte-enriched bones of ERαΔOcy mice.
Although we found a clear increase in bone Sost mRNA levels in the ERαΔOcy versus the Control mice, this was not reflected by parallel changes in serum sclerostin levels in these mice. The reasons for this discrepancy are unclear, but previous studies in mice have demonstrated that circulating sclerostin levels may not always reflect changes in sclerostin protein or mRNA levels in the bone microenvironment related perhaps to altered sclerostin binding proteins in bone or the contribution of non-skeletal sources of sclerostin to circulating levels.(47)
In addition to Wnt signaling, we also found reductions in BMP and Notch target genes in the osteocyte-enriched bones of the ERαΔOcy mice. These findings are consistent with previous work showing that estrogen stimulates BMP-2(48) as well as BMP-6(49) production by osteoblastic cells and increases their sensitivity to BMP-4.(50) Moreover, estrogen also has been shown to enhance differentiation of osteoblastic cells by activating Notch signaling.(51) Finally, the effects we observed of osteocytic ERα deletion on increasing proliferation and apoptosis genes and lack of an effect of osteocytic ERα deletion on senescence genes are entirely consistent with previous studies in other models on these pathways.(52–54) As such, our gene expression studies are very consistent with data from other systems and help establish the validity of our in vivo targeted pathway analyses.
Our study, and that of Kondoh et al.,(6) found that osteocytic ERα (inducible or constitutive) only altered bone mass in female, but not male, mice. These findings are in marked contrast to extensive data in humans that estrogen, rather than testosterone, is the dominant sex steroid regulating bone metabolism in men (reviewed by Khosla(55)). Although these discrepant findings may be due to species differences, a plausible explanation is that in human males, estrogen may predominantly regulate intra-cortical osteonal bone remodeling, which is absent in rodents.(56)
As noted earlier, we recognize a limitation of our study is that tamoxifen does have independent effects on bone.(18,19) We attempted to control for this by treating both experimental mice (Dmp1CreERT2 x Erαfl/fl) and control animals (Dmp1CreERT2 only) with tamoxifen. However, additional studies using different tamoxifen dosing regimens, as demonstrated by Jardi et al.,(57) should be performed using this Dmp1CreERT2 model to define whether a tamoxifen regimen can be identified that does not affect bone metabolism. In addition, given the other analyses conducted in these mice, including histomorphometry and gene expression, we did not have additional bones remaining for mechanical testing so are unable to provide this data. We should note, in general, that unless there is a change in bone material properties (which would not be anticipated in this model), changes in bone microarchitecture/mass generally correlate with changes in bone biomechanical strength.(58)
In summary, we have developed and validated a new inducible 8 kb Dmp1CreERT2 mouse line for the osteocytic lineage. Using this model, we demonstrate a clear role for osteocytic ERα in regulating bone metabolism in female, but not male, mice. Our skeletal and gene expression analyses collectively provide perhaps the first direct in vivo link between impaired osteocytic ERα signaling leading to reductions in bone formation and Wnt signaling mediated, at least in part, through increases in Sost expression. We also identify a number of other pathways regulated by osteocytic ERα signaling in vivo that are generally consistent with previous in vitro studies. Collectively, these studies provide new insights into ERα action via osteocytes and some of the key downstream pathways regulated by this receptor in vivo in bone.
Supplementary Material
Acknowledgments:
This work was supported by NIH Grants P01 AG004875, AG062413 (SK/DGM), R01 AR068275 (DGM), R01 AG063707 (DGM), R01 DK128552.
Footnotes
Disclosure: The authors have no relevant financial disclosures.
Data Availability:
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1.Khosla S, Melton LJ 3rd, Riggs BL. The unitary model for estrogen deficiency and the pathogenesis of osteoporosis: is a revision needed? J Bone Miner Res. Mar 2011;26(3):441–51. Epub 2010/10/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khosla S, Monroe DG. Regulation of Bone Metabolism by Sex Steroids. Cold Spring Harb Perspect Med. Jan 2 2018;8(1). Epub 2017/07/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicks KM, Fujita K, Fraser D, McGregor U, Drake MT, McGee-Lawrence ME, et al. Deletion of Estrogen Receptor Beta in Osteoprogenitor Cells Increases Trabecular but Not Cortical Bone Mass in Female Mice. J Bone Miner Res. Mar 2016;31(3):606–14. Epub 2015/09/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sims NA, Dupont S, Krust A, Clement-Lacroix P, Minet D, Resche-Rigon M, et al. Deletion of estrogen receptors reveals a regulatory role for estrogen receptors-beta in bone remodeling in females but not in males. Bone. Jan 2002;30(1):18–25. Epub 2002/01/17. [DOI] [PubMed] [Google Scholar]
- 5.Almeida M, Iyer S, Martin-Millan M, Bartell SM, Han L, Ambrogini E, et al. Estrogen receptor-alpha signaling in osteoblast progenitors stimulates cortical bone accrual. J Clin Invest. Jan 2013;123(1):394–404. Epub 2012/12/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kondoh S, Inoue K, Igarashi K, Sugizaki H, Shirode-Fukuda Y, Inoue E, et al. Estrogen receptor alpha in osteocytes regulates trabecular bone formation in female mice. Bone. Mar 2014;60:68–77. Epub 2013/12/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melville KM, Kelly NH, Khan SA, Schimenti JC, Ross FP, Main RP, et al. Female mice lacking estrogen receptor-alpha in osteoblasts have compromised bone mass and strength. J Bone Miner Res. Feb 2014;29(2):370–9. Epub 2013/09/17. [DOI] [PubMed] [Google Scholar]
- 8.Windahl SH, Borjesson AE, Farman HH, Engdahl C, Moverare-Skrtic S, Sjogren K, et al. Estrogen receptor-alpha in osteocytes is important for trabecular bone formation in male mice. Proc Natl Acad Sci U S A. Feb 5 2013;110(6):2294–9. Epub 2013/01/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, et al. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. Sep 7 2007;130(5):811–23. Epub 2007/09/07. [DOI] [PubMed] [Google Scholar]
- 10.Martin-Millan M, Almeida M, Ambrogini E, Han L, Zhao H, Weinstein RS, et al. The estrogen receptor-alpha in osteoclasts mediates the protective effects of estrogens on cancellous but not cortical bone. Mol Endocrinol. Feb 2010;24(2):323–34. Epub 2010/01/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujiwara Y, Piemontese M, Liu Y, Thostenson JD, Xiong J, O’Brien CA. RANKL (Receptor Activator of NFkappaB Ligand) Produced by Osteocytes Is Required for the Increase in B Cells and Bone Loss Caused by Estrogen Deficiency in Mice. J Biol Chem. Nov 25 2016;291(48):24838–50. Epub 2016/10/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalajzic I, Braut A, Guo D, Jiang X, Kronenberg MS, Mina M, et al. Dentin matrix protein 1 expression during osteoblastic differentiation, generation of an osteocyte GFP-transgene. Bone. Jul 2004;35(1):74–82. Epub 2004/06/23. [DOI] [PubMed] [Google Scholar]
- 13.Powell WF, Barry KJ, Tulum I, Kobayashi T, Harris SE, Bringhurst FR, et al. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J Endocrinol. Apr 2011;209(1):21–32. Epub 2011/01/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bivi N, Condon KW, Allen MR, Farlow N, Passeri G, Brun LR, et al. Cell autonomous requirement of connexin 43 for osteocyte survival: consequences for endocortical resorption and periosteal bone formation. J Bone Miner Res. Feb 2012;27(2):374–89. Epub 2011/10/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng Y, Manka D, Wagner KU, Khan SA. Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc Natl Acad Sci U S A. Sep 11 2007;104(37):14718–23. Epub 2007/09/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. Jan 2010;13(1):133–40. Epub 2009/12/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tasic B, Hippenmeyer S, Wang C, Gamboa M, Zong H, Chen-Tsai Y, et al. Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proc Natl Acad Sci U S A. May 10 2011;108(19):7902–7. Epub 2011/04/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z, Park JW, Ahn IS, Diamante G, Sivakumar N, Arneson D, et al. Estrogen receptor alpha in the brain mediates tamoxifen-induced changes in physiology in mice. Elife. Mar 1 2021;10. Epub 2021/03/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie Z, McGrath C, Sankaran J, Styner M, Little-Letsinger S, Dudakovic A, et al. Low-Dose Tamoxifen Induces Significant Bone Formation in Mice. JBMR Plus. Mar 2021;5(3):e10450. Epub 2021/03/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly NH, Schimenti JC, Patrick Ross F, van der Meulen MC. A method for isolating high quality RNA from mouse cortical and cancellous bone. Bone. Nov 2014;68:1–5. Epub 2014/07/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. Jun 18 2002;3(7):RESEARCH0034. Epub 2002/08/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, et al. Identification of Senescent Cells in the Bone Microenvironment. J Bone Miner Res. Nov 2016;31(11):1920–9. Epub 2016/10/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai CA, Chen JJ. Multivariate analysis of variance test for gene set analysis. Bioinformatics. Apr 1 2009;25(7):897–903. Epub 2009/03/04. [DOI] [PubMed] [Google Scholar]
- 24.Kalajzic I, Matthews BG, Torreggiani E, Harris MA, Divieti Pajevic P, Harris SE. In vitro and in vivo approaches to study osteocyte biology. Bone. Jun 2013;54(2):296–306. Epub 2012/10/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahlborg HG, Johnell O, Turner CH, Rannevik G, Karlsson MK. Bone loss and bone size after menopause. N Engl J Med. Jul 24 2003;349(4):327–34. Epub 2003/07/25. [DOI] [PubMed] [Google Scholar]
- 26.Turner RT, Wakley GK, Hannon KS. Differential effects of androgens on cortical bone histomorphometry in gonadectomized male and female rats. J Orthop Res. Jul 1990;8(4):612–7. Epub 1990/07/01. [DOI] [PubMed] [Google Scholar]
- 27.Szulc P, Seeman E, Duboeuf F, Sornay-Rendu E, Delmas PD. Bone fragility: failure of periosteal apposition to compensate for increased endocortical resorption in postmenopausal women. J Bone Miner Res. Dec 2006;21(12):1856–63. Epub 2006/09/28. [DOI] [PubMed] [Google Scholar]
- 28.Seeman E Bone quality: the material and structural basis of bone strength. J Bone Miner Metab. 2008;26(1):1–8. Epub 2007/12/21. [DOI] [PubMed] [Google Scholar]
- 29.Syed FA, Fraser DG, Monroe DG, Khosla S. Distinct effects of loss of classical estrogen receptor signaling versus complete deletion of estrogen receptor alpha on bone. Bone. Aug 2011;49(2):208–16. Epub 2011/04/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D’Agostin D, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. Jun 2008;23(6):860–9. Epub 2008/02/14. [DOI] [PubMed] [Google Scholar]
- 31.Ponte F, Kim HN, Iyer S, Han L, Almeida M, Manolagas SC. Cxcl12 Deletion in Mesenchymal Cells Increases Bone Turnover and Attenuates the Loss of Cortical Bone Caused by Estrogen Deficiency in Mice. J Bone Miner Res. Aug 2020;35(8):1441–51. Epub 2020/03/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eghbali-Fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. Apr 2003;111(8):1221–30. Epub 2003/04/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Spelsberg TC, Riggs BL. Estrogen stimulates gene expression and protein production of osteoprotegerin in human osteoblastic cells. Endocrinology. Sep 1999;140(9):4367–70. Epub 1999/08/28. [DOI] [PubMed] [Google Scholar]
- 34.Kedlaya R, Kang KS, Hong JM, Bettagere V, Lim KE, Horan D, et al. Adult-Onset Deletion of beta-Catenin in (10kb)Dmp1-Expressing Cells Prevents Intermittent PTH-Induced Bone Gain. Endocrinology. Aug 2016;157(8):3047–57. Epub 2016/06/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vooijs M, Jonkers J, Berns A. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep. Apr 2001;2(4):292–7. Epub 2001/04/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raj A, van Oudenaarden A. Nature, nurture, or chance: stochastic gene expression and its consequences. Cell. Oct 17 2008;135(2):216–26. Epub 2008/10/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frame B The Earlier Gain and the Later Loss of Cortical Bone in Nutritional Perspective. Radiology. 1971;98(2):310-. [Google Scholar]
- 38.Niziolek PJ, Bullock W, Warman ML, Robling AG. Missense Mutations in LRP5 Associated with High Bone Mass Protect the Mouse Skeleton from Disuse- and Ovariectomy-Induced Osteopenia. PLoS One. 2015;10(11):e0140775. Epub 2015/11/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Modder UI, Clowes JA, Hoey K, Peterson JM, McCready L, Oursler MJ, et al. Regulation of circulating sclerostin levels by sex steroids in women and in men. J Bone Miner Res. Jan 2011;26(1):27–34. Epub 2010/05/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Modder UI, Roforth MM, Hoey K, McCready LK, Peterson JM, Monroe DG, et al. Effects of estrogen on osteoprogenitor cells and cytokines/bone-regulatory factors in postmenopausal women. Bone. Aug 2011;49(2):202–7. Epub 2011/05/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujita K, Roforth MM, Demaray S, McGregor U, Kirmani S, McCready LK, et al. Effects of estrogen on bone mRNA levels of sclerostin and other genes relevant to bone metabolism in postmenopausal women. J Clin Endocrinol Metab. Jan 2014;99(1):E81–8. Epub 2013/10/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chung YE, Lee SH, Lee SY, Kim SY, Kim HH, Mirza FS, et al. Long-term treatment with raloxifene, but not bisphosphonates, reduces circulating sclerostin levels in postmenopausal women. Osteoporos Int. Apr 2012;23(4):1235–43. Epub 2011/06/11. [DOI] [PubMed] [Google Scholar]
- 43.Jastrzebski S, Kalinowski J, Stolina M, Mirza F, Torreggiani E, Kalajzic I, et al. Changes in bone sclerostin levels in mice after ovariectomy vary independently of changes in serum sclerostin levels. J Bone Miner Res. Mar 2013;28(3):618–26. Epub 2012/10/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Most W, van der Wee-Pals L, Ederveen A, Papapoulos S, Lowik C. Ovariectomy and orchidectomy induce a transient increase in the osteoclastogenic potential of bone marrow cells in the mouse. Bone. Jan 1997;20(1):27–30. Epub 1997/01/01. [DOI] [PubMed] [Google Scholar]
- 45.Weivoda MM, Ruan M, Hachfeld CM, Pederson L, Howe A, Davey RA, et al. Wnt Signaling Inhibits Osteoclast Differentiation by Activating Canonical and Noncanonical cAMP/PKA Pathways. J Bone Miner Res. Aug 2019;34(8):1546–8. Epub 2019/08/16. [DOI] [PubMed] [Google Scholar]
- 46.Kobayashi Y, Thirukonda GJ, Nakamura Y, Koide M, Yamashita T, Uehara S, et al. Wnt16 regulates osteoclast differentiation in conjunction with Wnt5a. Biochem Biophys Res Commun. Aug 7 2015;463(4):1278–83. Epub 2015/06/21. [DOI] [PubMed] [Google Scholar]
- 47.Chang M-K, Kramer I, Huber T, Kinzel B, Guth-Gundel S, Leupin O, et al. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proceedings of the National Academy of Sciences. 2014;111(48):E5187–E95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou S, Turgeman G, Harris SE, Leitman DC, Komm BS, Bodine PV, et al. Estrogens activate bone morphogenetic protein-2 gene transcription in mouse mesenchymal stem cells. Mol Endocrinol. Jan 2003;17(1):56–66. Epub 2003/01/04. [DOI] [PubMed] [Google Scholar]
- 49.Rickard DJ, Hofbauer LC, Bonde SK, Gori F, Spelsberg TC, Riggs BL. Bone morphogenetic protein-6 production in human osteoblastic cell lines. Selective regulation by estrogen. J Clin Invest. Jan 15 1998;101(2):413–22. Epub 1998/02/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsumoto Y, Otsuka F, Takano-Narazaki M, Katsuyama T, Nakamura E, Tsukamoto N, et al. Estrogen facilitates osteoblast differentiation by upregulating bone morphogenetic protein-4 signaling. Steroids. May 2013;78(5):513–20. Epub 2013/03/19. [DOI] [PubMed] [Google Scholar]
- 51.Fan JZ, Yang L, Meng GL, Lin YS, Wei BY, Fan J, et al. Estrogen improves the proliferation and differentiation of hBMSCs derived from postmenopausal osteoporosis through notch signaling pathway. Mol Cell Biochem. Jul 2014;392(1–2):85–93. Epub 2014/04/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jilka RL, Takahashi K, Munshi M, Williams DC, Roberson PK, Manolagas SC. Loss of estrogen upregulates osteoblastogenesis in the murine bone marrow. Evidence for autonomy from factors released during bone resorption. J Clin Invest. May 1 1998;101(9):1942–50. Epub 1998/06/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Plotkin LI, Aguirre JI, Kousteni S, Manolagas SC, Bellido T. Bisphosphonates and estrogens inhibit osteocyte apoptosis via distinct molecular mechanisms downstream of extracellular signal-regulated kinase activation. J Biol Chem. Feb 25 2005;280(8):7317–25. Epub 2004/12/14. [DOI] [PubMed] [Google Scholar]
- 54.Farr JN, Rowsey JL, Eckhardt BA, Thicke BS, Fraser DG, Tchkonia T, et al. Independent Roles of Estrogen Deficiency and Cellular Senescence in the Pathogenesis of Osteoporosis: Evidence in Young Adult Mice and Older Humans. J Bone Miner Res. Aug 2019;34(8):1407–18. Epub 2019/03/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khosla S New Insights Into Androgen and Estrogen Receptor Regulation of the Male Skeleton. J Bone Miner Res. Jul 2015;30(7):1134–7. Epub 2015/04/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jilka RL. The relevance of mouse models for investigating age-related bone loss in humans. J Gerontol A Biol Sci Med Sci. Oct 2013;68(10):1209–17. Epub 2013/05/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jardí F, Laurent MR, Dubois V, Khalil R, Deboel L, Schollaert D, et al. A shortened tamoxifen induction scheme to induce CreER recombinase without side effects on the male mouse skeleton. Mol Cell Endocrinol. Sep 5 2017;452:57–63. Epub 2017/05/16. [DOI] [PubMed] [Google Scholar]
- 58.Ward WE, Piekarz AV, Fonseca D. Bone mass, bone strength, and their relationship in developing CD-1 mice. Can J Physiol Pharmacol. Feb 2007;85(2):274–9. Epub 2007/05/10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.