

Abstract
Purpose:
Determine whether specific CTNNB1 or APC mutations in patients with desmoid tumor were associated with differences in clinical responses to systemic treatments.
Experimental Design:
We established a multi-institutional dataset of previously treated patients with desmoid tumor across four U.S. sarcoma centers, including demographic and clinicopathologic characteristics, treatment regimens, and clinical and radiographic responses. CTNNB1 or APC mutation status was determined from prior pathology records, or archival tissue was requested and analyzed by Sanger sequencing and/or next-generation sequencing. Evaluable patients with mutation results were analyzed to determine clinical progression-free survival (cPFS), RECIST 1.1 PFS (rPFS), time to next treatment (TTNT), and overall survival (OS). Kaplan–Meier analysis and Cox proportional hazards regression were performed to identify differences in cPFS, rPFS, TTNT, and OS by mutation subtype, desmoid tumor location, and treatment regimen.
Results:
A total of 259 evaluable patients were analyzed for at least one of the survival outcomes, with 177 patients having mutation data. First- and second-line cPFS, rPFS, and TTNT were not significantly affected by mutation subtype; however, APC-mutant desmoid tumors demonstrated nonstatistically significant inferior outcomes. Extremity/trunk desmoid tumor location and treatment with doxorubicin-based, methotrexate/vinca alkaloids and sorafenib regimens were associated with better clinical outcomes compared with surgery or “other” therapies, including estrogen-receptor blockade and imatinib. OS was significantly worse with APC or CTNNB1 negative/other mutations.
Conclusions:
Mutation subtype did not affect responses to specific systemic therapies. APC mutations and nonextremity desmoid tumor locations remain prognostic for worse outcomes, and earlier initiation of systemic therapy for these higher-risk desmoid tumors should be prospectively evaluated.
Translational Relevance.
Desmoid tumors are clonal soft-tissue neoplasms driven by constitutive β-catenin/WNT signaling, either through specific, well-characterized activating mutations in CTNNB1, or loss of suppression of the pathway by APC deletions as occurs in familial adenomatous polyposis. Surgery has been the historical mainstay of desmoid treatment; however, postoperative recurrence is frequent. Systemic therapy is effective but can be associated with significant toxicity. APC deletions and CTNNB1 S45F mutations have been associated with inferior prognosis and more aggressive clinical behavior in some studies, but it is unknown whether individual mutation types are associated with resistance to systemic therapies. We retrospectively examined a desmoid tumor patient cohort and determined that various chemotherapy regimens retain antitumor efficacy independently of the mutation subtype. Thus, our data support the idea that initiation and choice of chemotherapy regimen should be based on symptoms and individual patient risk, rather than the presence of a specific genetic alteration.
Introduction
Desmoid tumors, also known as desmoid fibromatosis, are rare soft-tissue neoplasms driven by aberrant β-catenin signaling, either through activating mutations of the gene encoding β-catenin (CTNNB1) or loss of the pathway suppressor APC (1). Despite a low incidence of approximately two to four cases per million per year (2–4), desmoid tumors can be locally extensive and invasive, and prone to multiple local recurrences, leading to substantial morbidity. Desmoid tumors do not metastasize but can be locally aggressive. When they occur in proximity to major organs and structures, desmoid tumors can lead to significant morbidity and even to death (5). Importantly, clinical behavior of desmoid tumors is highly variable, with up to a 20% spontaneous regression rate, and other patients exhibit stable, asymptomatic disease for years without requiring treatment (6). Conversely, other patients exhibit more rapid growth and symptoms that can severely affect quality of life and threaten life or limb. Surgery is highly effective in treatment of progressive desmoid tumors; however, there remains a high postoperative recurrence rate of up to 39% in some series, even with negative margins (5). Systemic therapy is also active in desmoid tumors, but some regimens impart significant toxicity, which is undesirable for a nonmalignant neoplasm occurring in younger and otherwise healthy individuals. Identifying prognostic and predictive factors to determine which desmoid tumors are likely to behave in an indolent fashion and determining which tumors should be treated more aggressively with specific systemic chemotherapy regimens would greatly impact clinical care for patients with desmoid tumor.
The genetic drivers of desmoid tumors have been well established, and some evidence suggests that specific mutations may be prognostic. A total of 80% to 90% of desmoid tumors carry mutations in CTNNB1, defined as sporadic desmoid tumors, with the most common mutations occurring in exon 3 in codon 41 (T41A), or codon 45 (S45F or S45P; refs. 7, 8), leading to intranuclear accumulation of β-catenin and activation of downstream β-catenin/Wnt signaling (9). Some studies have shown that CTNNB1 codon 45 mutations may have worse progression-free survival (PFS) and higher risk of recurrence (10–12), although other studies suggested a lack of prognostic significance (13, 14). About 10%–20% of desmoid tumors are thought to arise from deletions in APC, with the majority being germline mutations and associated with familial adenomatous polyposis (FAP) syndromes (7). APC negatively regulates β-catenin (15), and loss of APC leads to an increased risk of developing colon polyps, colon cancer, thyroid cancer, as well as desmoid tumors (16). Patients with FAP carry a risk of developing desmoid tumors of about 10% to 30% (17–19). Importantly, FAP patients tend to develop multifocal desmoid tumors, with a predisposition for mesenteric tumors (17). On the basis of the biology of this disease, FAP-associated desmoid tumors have clearly been associated with worse prognosis and risk of death.
While the consensus approach for desmoid tumors has evolved to avoid initial surgical resection, with a “watch and wait” philosophy based on the unpredictable behavior, a variety of systemic therapies have been used to treat progressive or symptomatic desmoid tumors (20, 21). These treatments have included estrogen-receptor (ER) suppressive therapies such as tamoxifen, particularly for desmoid tumors associated with pregnancy or other hormonal exposure (22), and anti-inflammatories such as meloxicam/sulindac. Traditional cytotoxic chemotherapies include doxorubicin/dacarbazine (ADIC), pegylated liposomal doxorubicin, and methotrexate (MTX) with vinca alkaloids, most commonly vinblastine. Newer therapies include tyrosine kinase inhibitors (TKI) such as imatinib, sorafenib, and pazopanib, and multiple novel treatments including gamma-secretase inhibitors, Wnt/β-catenin inhibitors, and even immune checkpoint inhibitors are being studied in clinical trials (23). A few studies have examined the susceptibility of CTNNB1 mutation subtypes to various chemotherapies, with S45F mutations having worse outcomes with meloxicam, and no difference observed in different mutations in patients treated with MTX/vinblastine and imatinib (24–26). However, no data exist for doxorubicin-based treatments and sorafenib. To further address the question of whether mutation data can inform selection of systemic therapy or its optimal timing, we performed a multi-institutional retrospective analysis of patients with desmoid tumor who received systemic chemotherapies, to evaluate the impact of specific mutation subtypes on clinical and radiographic responses.
Materials and Methods
Data collection
We established a deidentified REDCAP database to capture data from patients with desmoid tumor treated at four institutions: The University of Texas MD Anderson Cancer Center (MDACC, Houston, TX), the University of Miami (UM, Miami, FL), the Mount Sinai Medical Center (MSMC, New York, NY), and the Dana-Farber Cancer Institute (DFCI, Boston, MA). Under individual institutionally approved Institutional Review Board protocols providing waiver of informed consent in accordance with the U.S. Common Rule, patients with desmoid tumor were retrospectively identified from each institution by the investigators. Clinical data, including demographics, clinicopathologic features, mutation testing results, treatment regimens, and clinical and radiographic outcomes were abstracted. Surgery, surgery with adjuvant radiation or chemotherapy, radiation/ablation, and the few formally documented observation periods were considered as unique treatment episodes alongside various systemic regimens. Patients were required to have a diagnosis of desmoid tumor previously confirmed by pathologists at the respective institutions. For patients from UM and DFCI with available archival tissue and no prior mutation testing, we performed Sanger sequencing to determine specific CTNNB1 mutations, with next-generation sequencing for APC or CTNNB1 mutations in specimens negative by Sanger sequencing.
The clinical outcomes under investigation included clinical PFS (cPFS), RECIST 1.1 PFS (rPFS), time to next treatment (TTNT), and overall survival (OS). cPFS was defined as the time from the date of a baseline scan prior to beginning a treatment regimen, or if no baseline scan was performed, from the start date of the treatment regimen, until the date of clinical progression as documented in the treating physician's notes. Determination of clinical progression was abstracted from the physician's notes, and could be based on worsening of symptoms, or general radiographic progression (not necessarily based on RECIST). Patients who did not have documented clinical progression but treatment was stopped or changed for other reasons, including toxicity, investigator or patient preferences, or unknown reasons were censored at the date of last contact or next treatment regimen start date. To determine rPFS, standard calculations were made based on the longest tumor diameter in any plane based on available CT or MRI reports documented in the clinical database. For a subset of patients with scans available, these were rereviewed by fellowship trained radiologists blinded to clinical treatment outcome to determine greatest tumor diameter and response according to RECIST 1.1. TTNT was calculated for each treatment episode as the time elapsed from completion of one treatment episode, until the start date of the next treatment episode for the patient. Patients who did not have a subsequent treatment episode were censored at the date of last follow-up. OS was calculated as the time from desmoid tumor diagnosis by pathology report to death from any cause. In addition to these outcomes, we also evaluated response rates for patients with available radiographic data, with a decrease in maximum tumor diameter of ≥30% considered partial response (PR), −30% to +20% considered stable disease (SD), and increase of ≥20% considered progressive disease (PD).
Statistical analysis
We conducted analysis on two levels, by individual patients, and by treatment episodes. Demographic and clinical data, treatment regimens, and mutation status for evaluable patients within the entire study population were summarized using frequency tables. Continuous variables were reported as median and interquartile range (IQR), and categorical variables were reported as frequencies and percentages of the patient population, and then evaluated by mutation subgroup. Comparisons made across mutation subtype groups were descriptive in nature, with continuous variables tested using Kruskal–Wallis nonparametric test, and categorical variables compared using Fisher exact test.
OS analyses were conducted for the entire patient population incorporating first-line treatments. For cPFS, rPFS, and TTNT, analysis was limited to treatment regimens received in the first-line and second-line setting based on evaluable patient numbers. Survival probabilities for these outcomes were calculated by the Kaplan–Meier (KM) method, and the survival curves and median survival time reported with the corresponding two-sided 95% Brookmeyer–Crowley confidence interval (CI) if feasible. We used the log-rank test to compare survival endpoints by mutation subtype, desmoid tumor anatomic location, and first- and second-line treatment regimens. We then used the Cox proportional hazards model to compare survival endpoints by specific mutation, desmoid tumor anatomic location, and first- and second-line treatment modalities while adjusting for additional covariates as feasible for model convergence, which included age at diagnosis, sex, race/ethnicity, total number of treatments, and history of FAP (for location and treatment analyses only). For second-line survival analyses, the time to second-line therapy was also included as a covariate to correct for biologic variation among patients with desmoid tumor. The adjusted HR with 95% CI was calculated and reported.
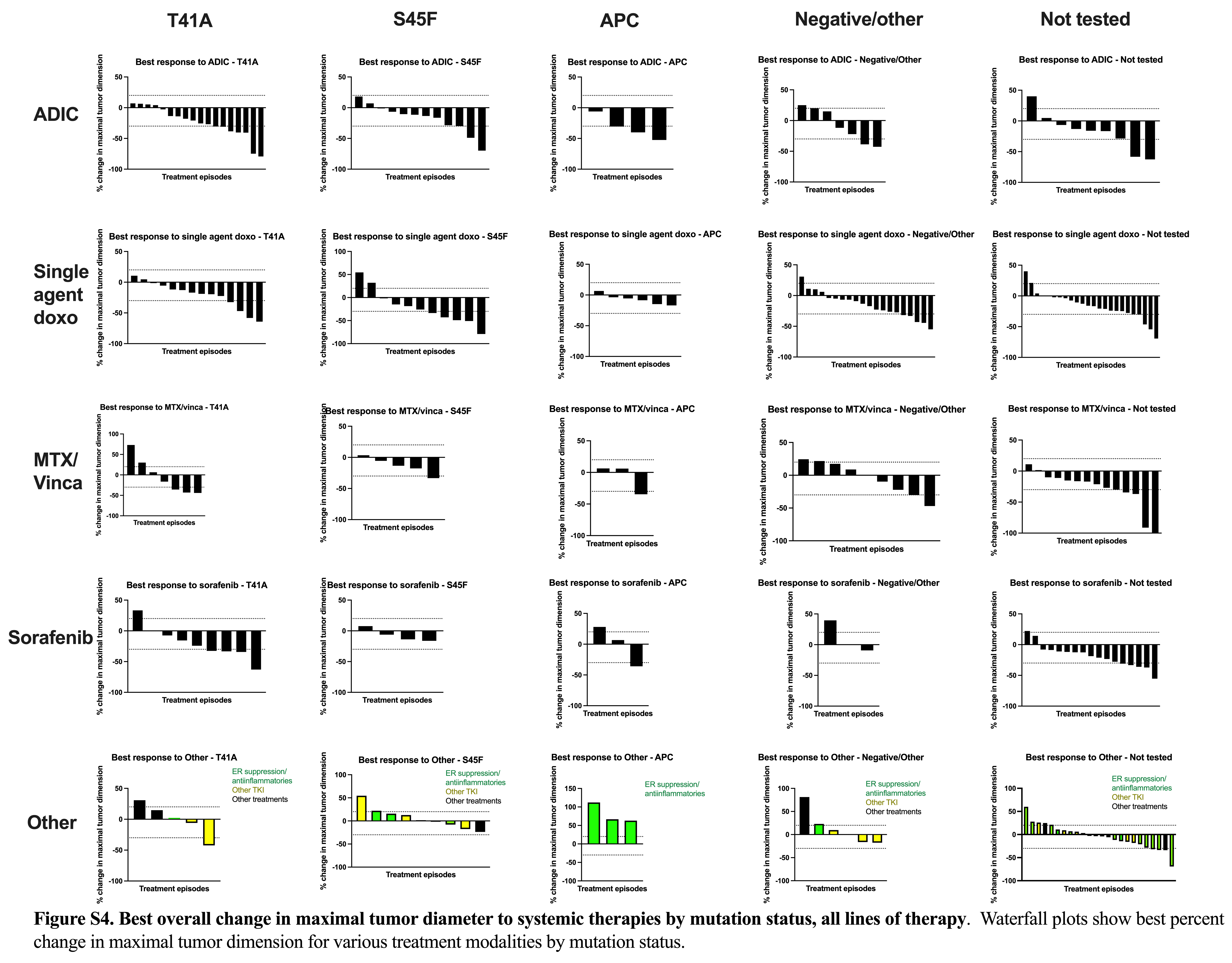
We then conducted a separate analysis to evaluate the impact of mutation subtype, desmoid tumor location, and treatment regimens on cPFS, rPFS, and TTNT of all individual treatment episodes across the evaluable patients without taking into account the line of therapy. Survival probabilities were calculated by the KM method, and the survival curve and the median survival time were reported with the corresponding two-sided 95% Brookmeyer–Crowley CI if feasible. The log-rank test was used to compare the survival endpoints by mutation subtype, desmoid tumor location, and treatment regimen. The log-rank test results were understood to be less robust because multiple treatment episodes associated with the same patient were not independent. Thus, multivariable Cox proportional hazards models incorporating random patient effect (frailty covariate) and the previously mentioned covariates were used to compare survival outcomes by specific mutation, desmoid tumor location, and treatment regimen. The adjusted HRs with 95% CIs were calculated and reported. We also conducted a subgroup analysis to evaluate the effects of mutation subtype on cPFS, rPFS, and TTNT for four key treatment episode types, ADIC, single-agent doxorubicin, MTX/vinca alkaloids, and sorafenib. Survival outcomes in this analysis were compared by KM analysis and Cox proportional hazards models as described above. For episodes with radiographic tumor size data available for the four primary systemic treatments, as well as “other” which primarily included tamoxifen and imatinib, waterfall plots depicting best change in tumor dimensions with these regimens and a summary table of response rates [defined as proportion achieving complete response (CR) or PR], and disease control rates (proportion achieving CR or PR, or SD) were reported.
All statistical analyses were performed by an independent statistician to ensure unbiased data review and conducted on R version 4.1.0. P values less than 0.05 were considered statistically significant. Because of the exploratory and hypothesis-generating nature of this study, no multiple comparison adjustment was implemented.
Data availability
The data collected and genetic sequencing generated in this study are available upon request from the corresponding author.
Results
Patient and treatment characteristics
A total of 309 patients with desmoid tumor were retrospectively identified, with 106 from MDACC, 108 from DFCI, 68 from UM, and 7 from MSMC who were diagnosed and treated between 1985 and 2018 (Fig. 1A). A total of 50 patients were excluded for inadequate data. A total of 259 patients were included in the patient-level analysis for at least one of the survival outcomes. Of the 259 patients in the total evaluable population, 177 (68%) had mutation testing, including CTNNB1 T41A (n = 68, 26%), CTNNB1 S45F (n = 47, 18%), APC (n = 11, 4%), negative or other mutations (n = 51, 20%) which included S45P (n = 4, 8.3%), CTNNB1 p.P16S (n = 1, 2%), or MYH G382D and Y165C (n = 1, 2%), and 32% had no available mutation data, due to either unavailable archival tissue or failure of sequencing (Table 1). Of note, a significant number of patients had been sequenced prior to the development of next-generation sequencing techniques and were recorded as “negative” for CTNNB1 mutations without further information available.
Figure 1.

CONSORT diagrams showing numbers of patients (A) and numbers of episodes (B) included in each analysis. cPFS, clinical progression-free survival; DFCI, Dana-Farber Cancer Institute; MDACC, The MD Anderson Cancer Center; MSMC, Mount Sinai Medical Center; OS, overall survival; rPFS, RECIST 1.1 progression-free survival; TTNT, time to next treatment; UM, University of Miami.
Table 1.
Clinical, demographic, and treatment characteristics for the entire study population.
| Characteristics | Levels | All N (%) |
|---|---|---|
| Sex | Female | 174 (67) |
| Male | 85 (33) | |
| Race | Caucasian | 148 (57) |
| Hispanic | 35 (14) | |
| African American | 21 (8) | |
| Asian | 10 (4) | |
| Other | 10 (4) | |
| (Missing) | 35 (14) | |
| Age at diagnosis | Median (IQR) | 35 (25–46) |
| Mutation type | T41A | 68 (26) |
| S45F | 47 (18) | |
| APC | 11 (4) | |
| Negative/othera | 51 (20) | |
| (Missing) | 82 (32) | |
| Desmoid location | Abdominal/pelvis | 95 (37) |
| Extremity/trunk | 131 (51) | |
| Otherb | 33 (13) | |
| History of FAP | No | 190 (73) |
| Yes | 51 (20) | |
| (Missing) | 18 (7) | |
| Number of treatments | 1 | 72 (28) |
| 2 | 77 (30) | |
| 3+ | 110 (43) | |
| First-line treatment | Surgeryc | 124 (48) |
| ADIC | 18 (7) | |
| Doxorubicin single agentd | 36 (14) | |
| Sorafenib | 11 (4) | |
| MTX/vinca alkaloids | 24 (9) | |
| Othere | 46 (18) | |
| Status at last follow-up | Censored | 245 (95) |
| Died | 14 (5) | |
| Follow-up time in months | Median (IQR) | 64.4 (28–110) |
Abbreviations: IQR, interquartile range; ADIC, doxorubicin/dacarbazine; FAP, familial adenomatous polyposis; MTX, methotrexate.
aNegative/other mutations include CTTNB1 negative, S45P, other CTNNB1, non-CTNNB1/non-APC (Table 2).
bOther location includes head and neck, paraspinal, other location, or multifocal.
cIncludes surgery alone, or surgery with adjuvant radiation or systemic therapies (Supplementary Fig. S1).
dIncludes liposomal and traditional doxorubicin (Supplementary Fig. S1).
ePrimarily estrogen-suppressing therapies, imatinib, and others (Supplementary Fig. S1).
Patients were predominantly female (67%) with a median age of 35 years (IQR, 25–46), and predominantly Caucasian (57%), (Table 1). The majority of desmoid tumors were located in the extremity/trunk (51%), with abdominal/pelvic locations in 37%, and other locations (including head/neck, paraspinal, and multifocal desmoid tumors) making up the remaining 13%. 20% of all patients had a known history of FAP, including 10 of the 11 patients in the APC mutation subgroup, but importantly also 33 patients (65%) in the negative/other mutation subset and 8 (10%) in the untested subset (Table 2). A total of 48% of patients received surgery with or without adjuvant therapy as primary management of the desmoid tumor, whereas 52% received systemic therapies and/or radiotherapy as primary management. The median number of treatment regimens per patient, which included surgical and radiation regimens, was 2 (range, 1–10), with 43% of patients having received three or more treatment regimens. Significant differences seen across mutation subgroups included age, desmoid tumor location, history of FAP, vital status at last follow-up, and median follow-up time (Table 2). Patients with APC or negative/other mutations were younger. Abdominal/pelvis desmoid tumors were seen in 61% of negative/other mutations, compared with 23%–40% in other subgroups, and extremity/flank tumors occurred in only 18% of APC and 24% of negative/other mutant desmoid tumors, compared with 52%–62% in other subgroups. Other/multifocal locations were also most common in APC mutant tumors (45%), compared with 8.8%–16% in other subsets. Patients with APC mutations and negative/other mutations were also more likely to have received three or more treatment lines compared with other subsets (APC 55%, negative/other 61%, compared with 38%–43% in other subsets).
Table 2.
Clinical, demographic, and treatment characteristics stratified by mutation types.
| Characteristics | Levels | T41A | S45F | APC | Negative/Othera | (Missing) | P |
|---|---|---|---|---|---|---|---|
| Sex | Female | 51 (75) | 34 (72) | 5 (46) | 29 (57) | 55 (67) | 0.064 |
| N (%) | Male | 17 (25) | 13 (28) | 6 (55) | 22 (43) | 27 (33) | |
| Race | Caucasian | 47 (69) | 23 (49) | 8 (73) | 36 (71) | 34 (42) | 0.753 |
| N (%) | Hispanic | 7 (10) | 8 (17) | 2 (18) | 5 (10) | 13 (16) | |
| African American | 6 (9) | 5 (11) | 1 (9) | 2 (4) | 7 (9) | ||
| Asian | 5 (7) | 1 (2) | 0 (0) | 1 (2) | 3 (4) | ||
| Other | 1 (2) | 1 (2) | 0 (0) | 1 (2) | 7 (9) | ||
| (Missing) | 2 (3) | 9 (19) | 0 (0) | 6 (12) | 18 (22) | ||
| Age at diagnosis | Median (IQR) | 37 (25–47) | 38 (27–46) | 32 (29–38) | 28 (23–36) | 40 (27–50) | 0.017 |
| Desmoid location | Abdominal/pelvis | 27 (40) | 11 (23) | 4 (36) | 31 (61) | 22 (27) | <0.001 |
| N (%) | Extremity/trunk | 35 (52) | 29 (62) | 2 (18) | 12 (24) | 53 (65) | |
| Otherb | 6 (9) | 7 (15) | 5 (46) | 8 (16) | 7 (9) | ||
| History of FAP | No | 63 (93) | 45 (96) | 0 (0) | 18 (35) | 64 (78) | <0.001 |
| N (%) | Yes | 0 (0) | 0 (0) | 10 (91) | 33 (65) | 8 (10) | |
| (Missing) | 5 (7) | 2 (4) | 1 (9) | 0 (0) | 10 (12) | ||
| Number of treatments | 1 | 12 (18) | 15 (32) | 2 (18) | 9 (18) | 34 (42) | 0.141 |
| N (%) | 2 | 27 (40) | 14 (30) | 3 (27) | 11 (22) | 22 (27) | |
| 3+ | 29 (43) | 18 (38) | 6 (55) | 31 (61) | 26 (32) | ||
| First-line treatment | Surgeryc | 37 (54) | 19 (40) | 4 (36) | 30 (59) | 34 (42) | 0.390 |
| N (%) | ADIC | 7 (10) | 5 (11) | 3 (27) | 1 (2) | 2 (2) | |
| Doxorubicin single agentd | 7 (10) | 8 (17) | 1 (9) | 8 (16) | 12 (15) | ||
| Sorafenib | 3 (4) | 1 (2) | 1 (9) | 1 (2) | 5 (6) | ||
| MTX/vinca alkaloids | 2 (3) | 4 (9) | 0 (0) | 4 (8) | 14 (17) | ||
| Othere | 12 (18) | 10 (21) | 2 (18) | 7 (14) | 15 (18) | ||
| Status at last follow-up | Censored | 68 (100) | 46 (98) | 9 (82) | 43 (84) | 79 (96) | 0.001 |
| N (%) | Died | 0 (0) | 1 (2) | 2 (18) | 8 (16) | 3 (4) | |
| Follow-up time in months | Median (IQR) | 75 (36–107) | 35 (20–92) | 44 (25–90) | 87 (48–157) | 61 (28–108) | 0.006 |
Note: Continuous variables were reported by median and IQR, and compared using Kruskal–Wallis nonparametric test. For categorical variables, the frequencies and percentages were calculated, and compared by Fisher exact test. Patients without mutation information were excluded from testing.
Abbreviations: ADIC, doxorubicin/dacarbazine; FAP, familial adenomatous polyposis; IQR, interquartile range; MTX, methotrexate.
aNegative/other mutations: CTTNB1 negative (n = 42, 88%), S45P (n = 4, 8%), other CTNNB1 (n = 1, 2%), non-CTNNB1/non-APC (n = 1, 2%).
bOther location includes head and neck, paraspinal, other location, or multifocal.
cIncludes surgery alone, or surgery with adjuvant radiation or systemic therapies (Supplementary Fig. S1).
dIncludes liposomal and traditional doxorubicin (see Supplementary Fig. S1).
ePrimarily estrogen-suppressing therapies, imatinib, and others (see Supplementary Fig. S1).
Across all patients, there were 707 discrete treatment episodes identified, with 624 episodes having adequate data for analysis for at least one of the survival outcomes (Fig. 1B). A total of 374 of these episodes had associated mutation testing data available. Of the total number of treatment episodes administered in the first line, surgery alone was most frequent (41%), followed by single-agent doxorubicin [14%, primarily pegylated liposomal doxorubicin (89%) rather than traditional doxorubicin], ER blockade with or without anti-inflammatories (12%), MTX/vinca alkaloids (9%), and then ADIC (7%; Supplementary Fig. S1). In the second-line setting, ER blockade with/without anti-inflammatories was most common (22%), with surgery alone the next most frequent approach (19%). In third or greater lines, surgery was still commonly utilized (23%); however, more similar distributions were seen between single-agent doxorubicin (14%), ER blockade with/without anti-inflammatory medication (13%), MTX/vinca alkaloids (13%), sorafenib (11%), ADIC (8.8%), and other TKIs (6.9%).
First- and second-line cPFS, rPFS, and TTNT are not affected by mutation subtype, but are impacted by desmoid tumor location and treatment regimen
KM analysis and Cox proportional hazards analysis for cPFS, rPFS, and TTNT were only performed for patients receiving first and second lines of treatment, due to limited numbers of patients treated with three or more lines of treatment. Of patients with mutation testing completed, 122 patients were evaluable for first-line cPFS, and 109 patients were evaluable for second-line cPFS. KM analysis showed no significant differences in first- or second-line cPFS by mutation status (Fig. 2A, P = 0.49; Fig. 2B, P = 0.25). Nonstatistically significant worse outcomes with APC mutations were observed, with first-line median cPFS of 17 months, and second-line median cPFS of 13 months. In addition, S45F mutations had nonstatistically significant inferior cPFS (median PFS of 13 months). However, in Cox proportional hazards analysis, no significant impact was seen from mutation subtype on either first- or second-line cPFS (Tables 3A and 3B). Similar findings were observed for first- and second-line rPFS and TTNT, with no significant differences by KM or Cox multivariable analysis, but nonstatistically significant inferior outcomes for APC mutant tumors (Supplementary Figs. S2 and S3; Supplementary Tables S1–S4).
Figure 2.

KM analysis of first-line clinical PFS (A), second-line clinical PFS (B), and OS (C) by mutation subtype. Survival was calculated for patients with available data and analyzed using the KM method, with the survival curve, median survival time, and proportion free from progression at 1 year (clinical PFS) or 5 years (OS) along with corresponding two-sided 95% Brookmeyer–Crowley CIs. Curves corresponding to specific mutation subtypes were compared using log-rank test, with P < 0.05 considered statistically significant.
Table 3A.
Cox proportional hazards model analysis for first-line clinical PFS by mutation status.
| Variable | All | HR (univariable) (95% CI, P) | HR (multivariable) (95% CI, P) | |
|---|---|---|---|---|
| Sex | Female | 125 (100) | — | — |
| Male | 64 (100) | 0.75 (0.49–1.2, P = 0.19) | 0.83 (0.45–1.5, P = 0.54) | |
| Race | Caucasian | 115 (100) | — | — |
| Hispanic/Latino | 23 (100) | 0.91 (0.48–1.7, P = 0.78) | 1.6 (0.72–3.6, P = 0.25) | |
| African American | 15 (100.0) | 1.08 (0.54–2.2, P = 0.83) | 0.99 (0.45–2.2, P = 0.99) | |
| Asian | 8 (100.0) | 0.77 (0.24–2.5, P = 0.66) | 0.77 (0.17–3.4, P = 0.74) | |
| Other/unknown | 7 (100.0) | 0.94 (0.34–2.6, P = 0.91) | 0.78 (0.09–7.0, P = 0.83) | |
| Age | Mean (SD) | 36.9 (14.9) | 0.99 (0.97–1.0, P = 0.096) | 0.99 (0.97–1.0, P = 0.42) |
| No. of treatment lines | 1 | 73 (100.0) | — | — |
| 2 | 45 (100.0) | 6.7 (3.4–13, P < 0.001) | 7.9 (2.2–28, P = 0.001) | |
| 3+ | 71 (100.0) | 8.6 (4.5–16.4, P < 0.001) | 11 (3.2–35, P < 0.001) | |
| Mutation subtype | T41A | 45 (100.0) | — | — |
| S45F | 36 (100.0) | 1.1 (0.61–2.1, P = 0.68) | 1.0 (0.50–2.0, P = 0.99) | |
| APC | 10 (100.0) | 1.8 (0.75–4.1, P = 0.20) | 1.2 (0.49–2.9, P = 0.69) | |
| Other | 31 (100.0) | 0.89 (0.49–1.6, P = 0.70) | 0.68 (0.32–1.4, P = 0.30) | |
Table 3B.
Cox proportional hazards model analysis for second-line clinical PFS by mutation status.
| Variable | All | HR (univariable) (95% CI, P) | HR (multivariable) (95% CI, P) | |
|---|---|---|---|---|
| Sex | Female | 99 (100) | — | — |
| Male | 46 (100) | 0.90 (0.54–1.5, P = 0.69) | 1.1 (0.59–2.2, P = 0.71) | |
| Age | Mean (SD) | 35.3 (14) | 0.98 (0.97–1.0, P = 0.07) | 0.99 (0.97–1.0, P = 0.38) |
| Time to second-line treatment | Mean (SD) | 16.3 (21) | 1.0 (0.98–1.01, P = 0.57) | 1.0 (0.98–1.01, P = 0.77) |
| No. of treatment lines | 2 | 68 (100) | — | — |
| 3+ | 77 (100) | 5.4 (3.0–9.7, P < 0.001) | 6.0 (2.5–14.4, P < 0.001) | |
| Mutation subtype | T41A | 45 (100) | — | — |
| S45F | 28 (100) | 1.7 (0.92–3.3, P = 0.09) | 1.8 (0.89–3.8, P = 0.10) | |
| APC | 7 (100) | 2.0 (0.78–5.6, P = 0.14) | 1.59 (0.57–4.5, P = 0.38) | |
| Other | 29 (100) | 1.2 (0.62–2.4, P = 0.57) | 1.1 (0.50–2.3, P = 0.85) | |
We then evaluated the impact of desmoid tumor location on first- and second-line cPFS, hypothesizing that cPFS may be driven by high-risk sites such as the mesentery. The median first-line cPFS of abdominal/pelvic desmoid tumors was only 18 months, less than extremity/trunk (33 months) and other locations (20 months), and second-line cPFS also showed nonstatistically significant inferior outcomes with “other” locations (13 months), compared with extremity/trunk location (25 months; Supplementary Tables S5 and S6). Cox multivariable analysis confirmed superior cPFS in extremity/trunk desmoid tumor location relative to abdominal/pelvis location in the first line [HR, 0.59 (95% CI, 0.35–0.99), P = 0.047], but was not significant in the second line. Similar findings were observed in the first- and second-line rPFS analysis, with superior outcomes with extremity/trunk location, but we found no significant differences in TTNT by desmoid tumor location (data not shown).
Next, we analyzed the effects of specific first- or second-line treatments on cPFS. Treatment choice did significantly impact first-line cPFS, with notably worse outcomes with surgery (median PFS 13 months) and “other” treatments [primarily ER blockade/anti-inflammatories (Supplementary Fig. S1), median PFS 23 months], compared with treatment with ADIC, single-agent doxorubicin, sorafenib, and MTX/vinca alkaloids (Supplementary Table S7, P = <0.001). Cox multivariable analysis confirmed significant improvement in first-line cPFS with ADIC chemotherapy [HR, 0.30 (95% CI, 0.11–0.77), P = 0.013], with nonstatistically significant improvement with single-agent doxorubicin [HR, 0.50 (95% CI, 0.24–1.1), P = 0.075] and MTX/vinca alkaloids [HR, 0.50 (95% CI, 0.21–1.2, P = 0.13] (Supplementary Table S7C). Analysis of second-line cPFS by treatment type also did not reach statistical significance (Supplementary Table S8). Analysis of first- and second-line rPFS and TTNT showed similarly superior outcomes with doxorubicin-based regimens and sorafenib, compared with surgery and “other” treatments (data not shown).
In summary, cPFS, rPFS, and TTNT were not significantly impacted by mutation subtype; however, trends were observed toward worse outcomes with APC mutant tumors. Extremity/trunk desmoid tumor location and treatment with doxorubicin-based, MTX/vinca alkaloids and sorafenib chemotherapy regimens were associated with better clinical outcomes compared with other treatment alternatives.
OS is significantly affected by APC or negative/other mutation subtype and abdominal/pelvis desmoid tumor location
Out of 259 patients included, 14 patients died and 245 were censored. Six patients died from desmoid tumor complications, 6 died from colorectal cancer, and 2 had an unknown cause of death. Of the patients who died from desmoid tumors, 5 had known FAP and mesenteric desmoid tumor location, and 1 had a retroperitoneal desmoid tumor with an S45F mutation. The median OS time was not reached with a median follow-up time of 64 months. Among the 177 patients with mutation testing, negative impact of mutation on OS was seen in patients with APC mutations and negative/other mutations (Fig. 2C, P = 0.015). OS was also significantly worse in patients with abdominal/pelvis desmoid tumor, with 5-year OS rate of 91% compared with 99% in extremity/trunk, and 95% in other sites. There were no effects on OS by type of first-line treatment.
Episode-level analysis demonstrates no significant impact on clinical PFS, RECIST PFS, or TTNT by mutation status or desmoid tumor location, but is affected by treatment regimen
Because of the number of patients in our cohort and the heterogeneity of treatment patterns, we were unable to evaluate activity of systemic treatments used in later lines of therapy. Thus, we performed a second analysis of treatment episodes, to capture activity of multiple treatments utilized across the same patients. Recognizing that different treatments applied to the same patient would not be independent observations, and that patients with higher numbers of treatment lines were likely to have more biologically aggressive disease, we included additional covariates including the number of prior treatments as well as a frailty covariate. Analysis at the episode level showed no significant differences in cPFS, rPFS, or TTNT by mutation subtype or desmoid tumor location. Non-statistically significant inferior outcomes were associated with APC and negative/other mutations, as well as abdominal/pelvic location. However, cPFS was significantly affected by type of treatment, with favorable outcomes of ADIC chemotherapy [HR, 0.50 (95% CI, 0.30–0.82), P = 0.006], single-agent doxorubicin [HR, 0.38 (0.24–0.59), P < 0.001], and sorafenib [HR, 0.33 (95% CI, 0.17–0.61), P = 0.001], relative to surgery (Supplementary Table S9). Similar findings were observed with inferior rPFS with “other” treatments [HR, 3.41 (95% CI, 1.2–9.9), P = 0.024], (Supplementary Table S10), and significantly improved TTNT with single-agent doxorubicin [HR, 0.56 (95% CI, 0.38–0.80), P = 0.0002] and sorafenib [HR, 0.49 (95% CI, 0.29–1.2), P = 0.010] relative to surgery, and statistically insignificant benefit with ADIC chemotherapy [HR, 0.69 (95% CI, 0.46–1.0), P = 0.075] relative to surgery (Supplementary Table S11).
Given our findings that the use of chemotherapy was the strongest factor impacting our clinical outcomes, we again reviewed the impact of mutation status on cPFS for ADIC, single-agent doxorubicin, MTX/vinca alkaloids, and sorafenib treatment episodes. No significant differences were observed by KM analysis among these four therapies by mutation status (Fig. 3). In Cox multivariable analysis, APC and other/negative mutations tended to do worse regardless of specific therapy. We did note that no clinical progression occurred in patients treated with sorafenib who had S45F mutations (Fig. 3D). For episodes with radiographic tumor measurements, we created waterfall plots by mutation for treatment episodes from the four main therapies as well as the “other” therapy group to look at activity from ER blockade/anti-inflammatories and imatinib. Overall, ADIC, single-agent doxorubicin, MTX/vinca alkaloids, and sorafenib all had consistent antitumor activity for desmoid tumors, with no obvious differences across mutation subtypes (Supplementary Fig. S4; Supplementary Table S12). Response rates ranged from 24% to 34% across the different treatments, with the “other” treatment cohort showing a lower response rate at 11%. Disease control rates were overall high with the “other” therapy cohort achieving 72%, doxorubicin-based regimens at greater than 90%, and MTX/vinca alkaloids and sorafenib at 87% and 89%, respectively. Overall, we did not see convincing evidence of resistance to chemotherapies across the various mutations.
Figure 3.

KM analysis of clinical PFS for treatment episodes by mutation status. Survival curves and median clinical PFS shown for ADIC (A), single-agent doxorubicin (primarily liposomal doxorubicin (B), MTX/vinca alkaloids (C), and sorafenib (D). Survival was calculated for patients with available data and analyzed using the KM method, with the survival curve, median survival time, and proportion free from progression at 1 year (clinical PFS) or 5 years (OS) along with corresponding two-sided 95% Brookmeyer-Crowley CIs. Curves corresponding to specific mutation subtypes were compared using log-rank test, with P < 0.05 considered statistically significant.
Discussion
The treatment paradigm for desmoid tumors has evolved considerably in recent years, with adoption of a “watch and wait” approach based on variable biology in individual desmoid tumors and patients (20, 27). However, the recommendation to monitor a large desmoid tumor with uncertain potential for future problems can be difficult for both patients and clinicians to accept. Prognostic factors for desmoid tumors have largely been based on postoperative recurrences, including clinical factors such as large size (greater than 5 cm), extraabdominal tumor location, superficial and deep fascial locations, and controversially, margin status at surgery (14, 28, 29). In addition, a recent study reported on 64 cases of desmoid tumors and showed that a three gene risk score including IFI6, LGMN, and CKLF was a strong prognostic indicator for recurrence-free survival (30). The specific mutation leading to desmoid tumor formation has been suggested in some, but not all series to be a marker for recurrence risk, with CTNNB1 S45F and APC mutations previously being ascribed with more aggressive behavior (10–12, 17). A prognostic system for FAP-related desmoid tumors has been derived by investigators at the Cleveland Clinic to predict 5-year mortality rates encompassing desmoid tumor–related death, secondary neoplasms, and surgical and supportive care complications (31). Unfortunately, there have not yet been data to link these prognostic factors to desmoid tumor treatment outcomes, to guide timing for initiation or to predict response to particular therapies. An ongoing clinical trial of patients with desmoid tumor includes mutation testing during the “watch and wait” period and will hopefully offer some prospective guidance on these issues (NCT02547831).
To address this deficiency, we conducted a large, retrospective analysis to evaluate potential links between desmoid tumor mutation subtypes and clinical responses to surgery and chemotherapy regimens chosen in standard clinical practice at four large U.S. sarcoma centers. The patients included in this analysis were managed prior to the development of modern consensus recommendations that suggest the following: (i) active surveillance is most appropriate for primary/recurrent desmoid tumors given variable biologic behavior, (ii) surgical resection should be reserved for sporadic desmoid tumors of the abdominal wall that progress on observation, and (iii) medical therapies should be utilized instead of surgery for nonabdominal wall desmoid tumors that progress on observation (20). Importantly, our data did not permit reliable assessment of clinical outcomes from the “watch and wait” observation approach and our analysis was focused on patients who actually received systemic treatments.
Overall, we could not conclusively identify worse clinical outcomes with CTNNB1 S45F mutations compared with T41A mutations in the first-line treatment setting, which is in accord with more recent prognostic studies (13, 14). We did note trends suggesting that APC and other/negative mutations (which were largely patients with clinical history of FAP) had worse outcomes with treatment, but few of these analyses were statistically significant. Despite these findings, we feel that mutation testing still has important roles in the management of patients with desmoid tumor. The numbers of patients in this study are small, and prospective evaluation of mutation subtype, clinical course, and response to treatment is warranted to confirm our findings and to settle the conflicting reports on prognostic impact of S45F CTNNB1 mutations. In addition, performing mutation testing helps to confirm the desmoid tumor diagnosis when histologic analysis can be misleading, and identification of an APC deletion in a desmoid tumor prompts germline testing for FAP which has important connotations for risk of other cancers.
Importantly, the strongest impact on clinical outcomes was observed from systemic therapies, especially doxorubicin-containing regimens, which were associated with longer cPFS, rPFS, and TTNT compared with surgery or “other” systemic therapies, primarily ER blockade and imatinib. This finding supports future testing of the hypothesis that desmoid tumors who progress on “watch and wait” periods will have better outcomes with doxorubicin-based, MTX/vinca alkaloid, or sorafenib chemotherapy compared with surgery or ER blockade/imatinib therapy. While patients with APC and negative/other mutations tended to have worse outcomes with these therapies compared with T41A and S45F mutations, our subgroup analysis did not suggest a superior choice of therapy for mutation subtypes. Our data suggest that at least in the first- and second-line settings, a clinically meaningful likelihood of desmoid tumor regression or prolonged stability can be achieved regardless of mutation type with the use of doxorubicin-based, MTX/vinca alkaloid, or sorafenib therapy.
Despite the superior results of doxorubicin-containing therapies, ADIC is associated with potential long-term toxicities, including loss of fertility, cardiomyopathy, and development of secondary cancers. Pegylated liposomal doxorubicin is better tolerated, but the duration of dosing is often limited by significant skin toxicity, at least when given at the approved dose and schedule. We were unable to perform a direct comparison to determine whether the addition of dacarbazine in ADIC significantly improved outcomes over single-agent doxorubicin to warrant the additional toxicity. With the emergence of better tolerated treatments with very promising activity, such as sorafenib (32), pazopanib (33), and nirogacestat (34), or novel interventional strategies such as cryoablation and high-intensity frequency ultrasound procedures (35), the future of doxorubicin-containing regimens and MTX/vinca alkaloids for desmoid tumors is uncertain, and likely will continue to be reserved for desmoid tumors where a rapid decrease in tumor bulk or severe symptoms is needed for life or limb threats. Importantly, our study also contained relatively few patients treated with radiation, which has historically had excellent results for management of desmoid tumors, albeit with significant long-term toxicity (36).
Promising signals of activity were seen in our study with sorafenib across all mutation subtypes, with none of the S45F patients progressing on sorafenib, though the small numbers of patients treated in this series limits statistical significance. We found this finding interesting, given that previous preclinical data suggested a mechanism for resistance to sorafenib in S45F-mutant desmoid tumor cell lines (37). However, the positive results from a pivotal phase III trial of sorafenib versus placebo for desmoid tumors support the hypothesis that sorafenib maintains activity across all mutation subgroups. The results from this trial showed 2-year PFS of 81% in the sorafenib group (95% CI, 69–96) versus 36% in the placebo group (95% CI, 22–57), with an objective response rate of 33% (95% CI, 20–48) in the sorafenib group compared with 20% in the placebo group (95% CI, 8–38; ref. 32). Correlative biopsies from patients treated on this study would be an ideal platform to further confirm any impact of mutation status in sorafenib-treated patients.
We noted that at times, the differences in outcomes between rPFS and cPFS or TTNT were striking; the overall first-line median rPFS for all patients was 168 months, compared with median cPFS of 31 months and median TTNT of 18 months. While we ensured that RECIST measurements were performed by musculoskeletal radiologists, scans were not available for all patients, and were not done at consistent times across the patient cohort. Despite this, the observed differences demonstrate how RECIST could be misleading for clinical outcomes for patients with desmoid tumor. Slow growing, large desmoid tumors are well known to display unusual growth patterns that may not lend themselves to longest tumor diameter measurements. In addition, when becoming dormant, desmoid tumors may not change size but become more collagenized. Even if active, large desmoid tumors may not reach the RECIST PD threshold (20% growth failure cutoff in longest tumor diameter from nadir) for a very long time, and the RECIST PR criterion (30% decrease in longest tumor diameter from baseline) may also underestimate benefit of potentially effective therapies. The inefficiency of RECIST is further evidenced in our review of the waterfall plots for various treatment regimens, that show very high disease control rates that incorporate SD, but relatively modest response rates, usually less than 30%. Our findings support continued exploration of alternative response criteria for desmoid tumors, including three-dimensional volumetrics and MRI radiomics to better assess changes in response to therapies (38–41), which are being incorporated in ongoing clinical trials (NCT03785964).
Overall, our study represents one of the largest retrospective series and one of the few studies to examine the effect of desmoid tumor mutation status on response to systemic therapies in desmoid tumors. However, this study has several critical limitations. All retrospective studies are subject to reporting and follow-up biases and given that patients were studied from tertiary referral centers, these patients likely represent more refractory cases that were more heavily treated than would be in lower volume centers and certainly in the community (referral bias). In addition, we relied heavily on clinical impression and decision-making for outcomes, which can be biased by subjective rather than objective assessments, as evidenced by the differences observed between cPFS and rPFS. Over the timeframe in this study, there was significant variation in practice patterns, without standardization of the number of cycles or doses used for systemic chemotherapy, or timing of imaging, which can affect assessment of PFS. Despite the large numbers of patients, we were still limited in statistical power by the numbers of patients with mutation data within various treatment subsets, due to lack of next-generation sequencing at the time of original testing, inability to obtain archival samples, or failure of DNA extraction from very old or poorly preserved materials. After subdividing our patient population by treatments, lines of therapy, and mutation subtypes, patients were lumped into categories that may dilute a signal of activity. Finally, there were patients with clinically documented FAP, but where an APC mutation could not be confirmed, and thus, included in the negative/other mutation category.
The results from our study are descriptive and hypothesis-generating and highlight important questions for the next generation of prospective clinical trials. First, we look forward to the results of ongoing prospective observational studies to analyze mutation subtype and desmoid tumors location for patients on active observation, who are then initiated on traditional and novel systemic therapies. We also noted improved clinical outcomes with treatment for extremity/trunk desmoid tumor locations, which contrasts with the higher risk of recurrence that has been reported in extremity desmoid tumors even over intraabdominal tumors postoperatively (14). Our data suggest that extremity tumors may have a higher response rate overall when treatment is needed and argues for future prospective studies to evaluate whether earlier initiation of systemic treatments should be adopted for extremity desmoid tumors. On the basis of our observations and outcomes from recently reported clinical trials of desmoid tumor treatment, prospective evaluation of sorafenib or pazopanib compared with pegylated liposomal doxorubicin would formally compare the activity, toxicity, and quality-of-life measures of these commonly used regimens in modern practice. In addition, FAP-associated desmoid tumors continue to be a significant challenge with worse outcomes regardless of systemic therapy, in need of further clinical trials. With the emergence of novel treatments including gamma secretase inhibitors and Wnt/β-catenin inhibitors, and increasing use of targeted approaches such as high frequency ultrasound and cryoablation, it is likely that the treatment landscape will continue to evolve over the next years for this rare and challenging group of neoplasms.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This research was supported by a grant from the Desmoid Tumor Research Foundation, including direct support to T. Subhawong and B. Wilky. Additional support from NCI Cancer Center Support Grant, 5P30CA046934, to J. Hu, J. Sheren, Y. Caldas, C. Moreno Tellez, and B.A. Wilky. M.J. Nathenson was also supported by a Willie Tichenor Fellowship in Sarcoma Medical Oncology from the QuadW Foundation.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
M.J. Nathenson reports other support from Quad W Foundation during the conduct of the study; this work was completed when M.J. Nathenson was a sarcoma medical oncologist the Dana-Farber Cancer Institute, now M.J. Nathenson is employed by GlaxoSmithKline. R. Ratan reports other support from Springworks Therapeutics and Ayala Pharmaceuticals outside the submitted work. N. Somaiah reports other support from Deciphera, Aadi Biosciences, Epizyme, Boehringer Ingelhein, Blueprint, and Bayer outside the submitted work. T.K. Subhawong reports grants from Toshiba America Medical Systems/RSNA Research Seed Grant #RSD1635 during the conduct of the study as well as personal fees from Arog Pharmaceuticals outside the submitted work. R.G. Maki reports personal fees from Bayer, Deciphera, Karyopharm Therapeutics, Peel Therapeutics, Tracon Pharmaceuticals, American Society for Clinical Oncology, and UpToDate outside the submitted work as well as clinical trials support to institution from Amgen, Astex, Boeringher Ingelheim, Bioatla, C4 Therapeutics, Exelexis, InhibRx, Janssen/PharmaMar, Peel Therapeutics, Rain Therapeutics, Regeneron, Presage, Sarcoma Alliance for Research through Collaboration (SARC), Springworks, SynOx, and Tracon. J. Cohen reports grants from Desmoid Tumor Research Foundation during the conduct of the study. J.C. Trent reports other support from Deciphera, Blueprint Medicine, Eisai, Daiichi Sankyo, C4 Therapeutics, Foghorn Therapeutics, AADI Pharmaceuticals, and Cogent Pharmaceuticals outside the submitted work. B.A. Wilky reports personal fees from Springworks, Deciphera, Epizyme, Adaptimmune, Daiichi Sankyo, Adcendo, GlaxoSmithKline, and Polaris outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
M.J. Nathenson: Conceptualization, data curation, formal analysis, investigation, methodology, writing–original draft, writing–review and editing. J. Hu: Formal analysis, methodology, writing–original draft, writing–review and editing. R. Ratan: Data curation. N. Somaiah: Data curation. R. Hsu: Data curation, writing–review and editing. P.J. DeMaria: Data curation, writing–review and editing. H.W. Catoe: Data curation. A. Pang: Data curation. T.K. Subhawong: Resources, data curation, supervision, visualization, methodology, writing–review and editing. B. Amini: Resources, data curation, visualization, writing–review and editing. K. Sweet: Data curation, visualization, writing–review and editing. K. Feister: Data curation, visualization. K. Malik: Data curation, visualization, methodology. J. Jagannathan: Data curation, visualization, methodology. M. Braschi-Amirfarzan: Data curation, visualization. J. Sheren: Resources, data curation, formal analysis, methodology. Y. Caldas: Data curation. C. Moreno Tellez: Data curation. A.E. Rosenberg: Resources, data curation, writing–review and editing. A.J. Lazar: Resources, data curation, writing–review and editing. R.G. Maki: Conceptualization, resources, data curation, supervision, writing–review and editing. P. Benedetto: Data curation. J. Cohen: Data curation. J.C. Trent: Data curation. V. Ravi: Conceptualization, resources, software, methodology. S. Patel: Resources, data curation. B.A. Wilky: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, investigation, methodology, writing–original draft, writing–review and editing.
References
- 1. WHO classification of tumours of soft tissue and bone. 4th ed.Lyon, France: WHO, International Agency for Research on Cancer IARC; 2013. [Google Scholar]
- 2. Reitamo JJ, Häyry P, Nykyri E, Saxen E. The desmoid tumor. I. Incidence, sex-, age- and anatomical distribution in the finnish population. Am J Clin Pathol 1982;77:665–73. [DOI] [PubMed] [Google Scholar]
- 3. Reitamo JJ, Scheinin TM, Hayry P. The desmoid syndrome. New aspects in the cause, pathogenesis and treatment of the desmoid tumor. Am J Surg 1986;151:230–7. [DOI] [PubMed] [Google Scholar]
- 4. Nieuwenhuis MH, Casparie M, Mathus-Vliegen LMH, Dekkers OM, Hogendoorn PCW, Vasen HFA. A nation-wide study comparing sporadic and familial adenomatous polyposis-related desmoid-type fibromatoses. Int J Cancer 2011;129:256–61. [DOI] [PubMed] [Google Scholar]
- 5. Salas S, Dufresne A, Bui B, Blay J-Y, Terrier P, Ranchere-Vince D, et al. Prognostic factors influencing progression-free survival determined from a series of sporadic desmoid tumors: a wait-and-see policy according to tumor presentation. J Clin Oncol 2011;29:3553–8. [DOI] [PubMed] [Google Scholar]
- 6. Bonvalot S, Ternès N, Fiore M, Bitsakou G, Colombo C, Honoré C, et al. Spontaneous regression of primary abdominal wall desmoid tumors: more common than previously thought. Ann Surg Oncol 2013;20:4096–102. [DOI] [PubMed] [Google Scholar]
- 7. Fallen T, Wilson M, Morlan B, Lindor NM. Desmoid tumors – a characterization of patients seen at Mayo Clinic 1976–1999. Fam Cancer 2006;5:191–4. [DOI] [PubMed] [Google Scholar]
- 8. Salas S, Chibon F, Noguchi T, Terrier P, Ranchere-Vince D, Lagarde P, et al. Molecular characterization by array comparative genomic hybridization and DNA sequencing of 194 desmoid tumors. Genes Chromosomes Cancer 2010;49:560–8. [DOI] [PubMed] [Google Scholar]
- 9. Crago AM, Chmielecki J, Rosenberg M, O'Connor R, Byrne C, Wilder FG, et al. Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid-type fibromatosis by whole-exome sequencing and genomic analysis. Genes Chromosomes Cancer 2015;54:606–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lazar AJF, Tuvin D, Hajibashi S, Habeeb S, Bolshakov S, Mayordomo-Aranda E, et al. Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol 2008;173:1518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Colombo C, Miceli R, Lazar AJ, Perrone F, Pollock RE, Le Cesne A, et al. CTNNB1 45F mutation is a molecular prognosticator of increased postoperative primary desmoid tumor recurrence: an independent, multicenter validation study. Cancer 2013;119:3696–702. [DOI] [PubMed] [Google Scholar]
- 12. van Broekhoven DLM, Verhoef C, Elias SG, Witkamp AJ, van Gorp JMHH, van Geel BAN, et al. Local recurrence after surgery for primary extra-abdominal desmoid-type fibromatosis. Br J Surg 2013;100:1214–9. [DOI] [PubMed] [Google Scholar]
- 13. Mullen JT, DeLaney TF, Kobayashi WK, Szymonifka J, Yeap BY, Chen Y-L, et al. Desmoid tumor: analysis of prognostic factors and outcomes in a surgical series. Ann Surg Oncol 2012;19:4028–35. [DOI] [PubMed] [Google Scholar]
- 14. Crago AM, Denton B, Salas S, Dufresne A, Mezhir JJ, Hameed M, et al. A prognostic nomogram for prediction of recurrence in desmoid fibromatosis. Ann Surg 2013;258:347–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li C, Bapat B, Alman BA. Adenomatous polyposis coli gene mutation alters proliferation through its beta-catenin-regulatory function in aggressive fibromatosis (desmoid tumor). Am J Pathol 1998;153:709–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lynch HT, Snyder C, Davies JM, Lanspa S, Lynch J, Gatalica Z, et al. FAP, gastric cancer, and genetic counseling featuring children and young adults: a family study and review. Fam Cancer 2010;9:581–8. [DOI] [PubMed] [Google Scholar]
- 17. Nieuwenhuis MH, Lefevre JH, Bülow S, Järvinen H, Bertario L, Kernéis S, et al. Family history, surgery, and APC mutation are risk factors for desmoid tumors in familial adenomatous polyposis: an international cohort study. Dis Colon Rectum 2011;54:1229–34. [DOI] [PubMed] [Google Scholar]
- 18. Lefevre JH, Parc Y, Kernéis S, Goasguen N, Benis M, Parc R, et al. Risk factors for development of desmoid tumours in familial adenomatous polyposis. Br J Surg 2008;95:1136–9. [DOI] [PubMed] [Google Scholar]
- 19. Bertario L, Russo A, Sala P, Eboli M, Giarola M, D'amico F, et al. Genotype and phenotype factors as determinants of desmoid tumors in patients with familial adenomatous polyposis. Int J Cancer 2001;95:102–7. [DOI] [PubMed] [Google Scholar]
- 20. Desmoid Tumor Working Group. The management of desmoid tumours: a joint global consensus-based guideline approach for adult and paediatric patients. Eur J Cancer 2020;127:96–107. [DOI] [PubMed] [Google Scholar]
- 21. de Camargo VP, Keohan ML, D'Adamo DR, Antonescu CR, Brennan MF, Singer S, et al. Clinical outcomes of systemic therapy for patients with deep fibromatosis (desmoid tumor). Cancer 2010;116:2258–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bocale D, Rotelli MT, Cavallini A, Altomare DF. Anti-oestrogen therapy in the treatment of desmoid tumours: a systematic review. Colorectal Dis 2011;13:e388–95. [DOI] [PubMed] [Google Scholar]
- 23. Napolitano A, Mazzocca A, Spalato Ceruso M, Minelli A, Baldo F, Badalamenti G, et al. Recent advances in desmoid tumor therapy. Cancers 2020;12:2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hamada S, Futamura N, Ikuta K, Urakawa H, Kozawa E, Ishiguro N, et al. CTNNB1 S45F mutation predicts poor efficacy of meloxicam treatment for desmoid tumors: a pilot study. PLoS One 2014;9:e96391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kasper B, Gruenwald V, Reichardt P, Bauer S, Hohenberger P, Haller F. Correlation of CTNNB1 mutation status with progression arrest rate in RECIST progressive desmoid-type fibromatosis treated with imatinib: translational research results from a phase 2 study of the german interdisciplinary sarcoma group (GISG-01). Ann Surg Oncol 2016;23:1924–7. [DOI] [PubMed] [Google Scholar]
- 26. Nishida Y, Tsukushi S, Urakawa H, Hamada S, Kozawa E, Ikuta K, et al. Low-dose chemotherapy with methotrexate and vinblastine for patients with desmoid tumors: relationship to CTNNB1 mutation status. Int J Clin Oncol 2015;20:1211–7. [DOI] [PubMed] [Google Scholar]
- 27. Fiore M, Rimareix F, Mariani L, Domont J, Collini P, Le Péchoux C, et al. Desmoid-type fibromatosis: a front-line conservative approach to select patients for surgical treatment. Ann Surg Oncol 2009;16:2587–93. [DOI] [PubMed] [Google Scholar]
- 28. Zeng W-G, Zhou Z-X, Liang J-W, Hou H-R, Wang Z, Zhou H-T, et al. Prognostic factors for desmoid tumor: a surgical series of 233 patients at a single institution. Tumour Biol 2014;35:7513–21. [DOI] [PubMed] [Google Scholar]
- 29. Kamali F, Wang W-L, Guadagnolo BA, Fox PS, Lewis VO, Lazar AJ, et al. MRI may be used as a prognostic indicator in patients with extra-abdominal desmoid tumours. Br J Radiol 2016;89:20150308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kohsaka S, Hirata M, Ikegami M, Ueno T, Kojima S, Sakai T, et al. Comprehensive molecular and clinicopathological profiling of desmoid tumours. Eur J Cancer 2021;145:109–20. [DOI] [PubMed] [Google Scholar]
- 31. Quintini C, Ward G, Shatnawei A, Xhaja X, Hashimoto K, Steiger E, et al. Mortality of intra-abdominal desmoid tumors in patients with familial adenomatous polyposis: a single center review of 154 patients. Ann Surg 2012;255:511–6. [DOI] [PubMed] [Google Scholar]
- 32. Gounder MM, Lefkowitz RA, Keohan ML, D'Adamo DR, Hameed M, Antonescu CR, et al. Activity of Sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res 2011;17:4082–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Toulmonde M, Pulido M, Ray-Coquard I, Andre T, Isambert N, Chevreau C, et al. Pazopanib or methotrexate-vinblastine combination chemotherapy in adult patients with progressive desmoid tumours (DESMOPAZ): a non-comparative, randomised, open-label, multicentre, phase 2 study. Lancet Oncol 2019;20:1263–72. [DOI] [PubMed] [Google Scholar]
- 34. Kummar S, O'Sullivan Coyne G, Do KT, Turkbey B, Meltzer PS, Polley E, et al. Clinical activity of the γ-secretase inhibitor PF-03084014 in adults with desmoid tumors (aggressive fibromatosis). J Clin Oncol 2017;35:1561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ilaslan H, Schils J, Joyce M, Marks K, Sundaram M. Radiofrequency ablation: another treatment option for local control of desmoid tumors. Skeletal Radiol 2010;39:169–73. [DOI] [PubMed] [Google Scholar]
- 36. Guadagnolo BA, Zagars GK, Ballo MT. Long-term outcomes for desmoid tumors treated with radiation therapy. Int J Radiat Oncol Biol Phys 2008;71:441–7. [DOI] [PubMed] [Google Scholar]
- 37. Shang H, Braggio D, Lee Y‐J, Al Sannaa GA, Creighton CJ, Bolshakov S, et al. Targeting the Notch pathway: a potential therapeutic approach for desmoid tumors. Cancer 2015;121:4088–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sheth PJ, del Moral S, Wilky BA, Trent JC, Cohen J, Rosenberg AE, et al. Desmoid fibromatosis: MRI features of response to systemic therapy. Skeletal Radiol 2016;45:1365–73. [DOI] [PubMed] [Google Scholar]
- 39. Subhawong TK, Feister K, Sweet K, Alperin N, Kwon D, Rosenberg A, et al. MRI volumetrics and image texture analysis in assessing systemic treatment response in extra-abdominal desmoid fibromatosis. Radiol Imaging Cancer 2021;3:e210016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Crombé A, Kind M, Ray-Coquard I, Isambert N, Chevreau C, André T, et al. Progressive desmoid tumor: radiomics compared with conventional response criteria for predicting progression during systemic therapy-a multicenter study by the french sarcoma group. AJR Am J Roentgenol 2020;215:1539–48. [DOI] [PubMed] [Google Scholar]
- 41. Timbergen MJM, Starmans MPA, Padmos GA, Grünhagen DJ, van Leenders GJLH, Hanff DF, et al. Differential diagnosis and mutation stratification of desmoid-type fibromatosis on MRI using radiomics. Eur J Radiol 2020;131:109266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data collected and genetic sequencing generated in this study are available upon request from the corresponding author.