

Abstract
Delineating the folding steps of helical-bundle membrane proteins has been a challenging task. Many questions remain unanswered, including the conformation and stability of the states populated during folding, the shape of the energy barriers between the states, and the role of lipids as a solvent in mediating the folding. Recently, theoretical frames have matured to a point that permits detailed dissection of the folding steps, and advances in experimental techniques at both single-molecule and ensemble levels enable selective modulation of specific steps for quantitative determination of the folding energy landscapes. We also discuss how lipid molecules would play an active role in shaping the folding energy landscape of membrane proteins, and how folding of multi-domain membrane proteins can be understood based on our current knowledge. We conclude this review by offering an outlook for emerging questions in the study of membrane protein folding.
Keywords: Membrane protein folding, Folding energy landscape, Single-molecule force spectroscopy, Magnetic tweezers, Steric trapping, Cooperativity, Lipophobic effect
The energetics of membrane protein folding
Membrane proteins carry out essential cellular functions including the synthesis of ATP, material exchange, and eliciting cell signaling, to just name a few. To perform a wide variety of functions, membrane proteins should correctly fold into native three-dimensional structures in the lipid bilayers. Cells invest significant resources for homeostasis of membrane proteins, and failures in their correct folding are implicated in various diseases [1–6]. The folding energy landscape of proteins is generally determined by the amino acid sequence of polypeptide chains and their interaction with the solvent [7]. In the case of globular proteins, the hydrophobic effect provides a critical driving force for folding by inducing the collapse of nonpolar residues and exclusion of water from the protein interior [8]. Due to the distinct environmental constraints provided by the lipid bilayers, the physical principles of folding are expected to be substantially different for the membrane proteins.
A “two-stage” model has been a prominent model for describing the folding of helical-bundle membrane proteins, in which each folding stage is driven by a distinct set of molecular forces [9,10]. In the first stage, polypeptide segments insert into the lipid bilayer as stable transmembrane helices (TMHs), mainly driven by the hydrophobic nature of the amino acids belonging to TMHs and the high desolvation cost of unpaired backbone hydrogen bonding (H-bonding) in the nonpolar bilayer (Figure 1a) [11,12]. In the second stage, the TMHs associate into a compact native structure through side-to-side interactions. The van der Waals (vdW) packing and polar interactions drive this stage largely replacing the hydrophobic effect due to a paucity of water molecules in the bilayer core [13–19]. Interestingly, a recent NMR relaxation study indicates that membrane proteins display more pronounced fast (sub-ns) side-chain motions than globular proteins in the interior. Although the contribution of slower motions to an overall side-chain entropy remains unexplored, this result raises a possibility that the side-chain entropic cost can be relatively small upon folding of membrane proteins in the bilayer [20].
Figure 1 |. Models for the folding of helical-bundle membrane proteins.
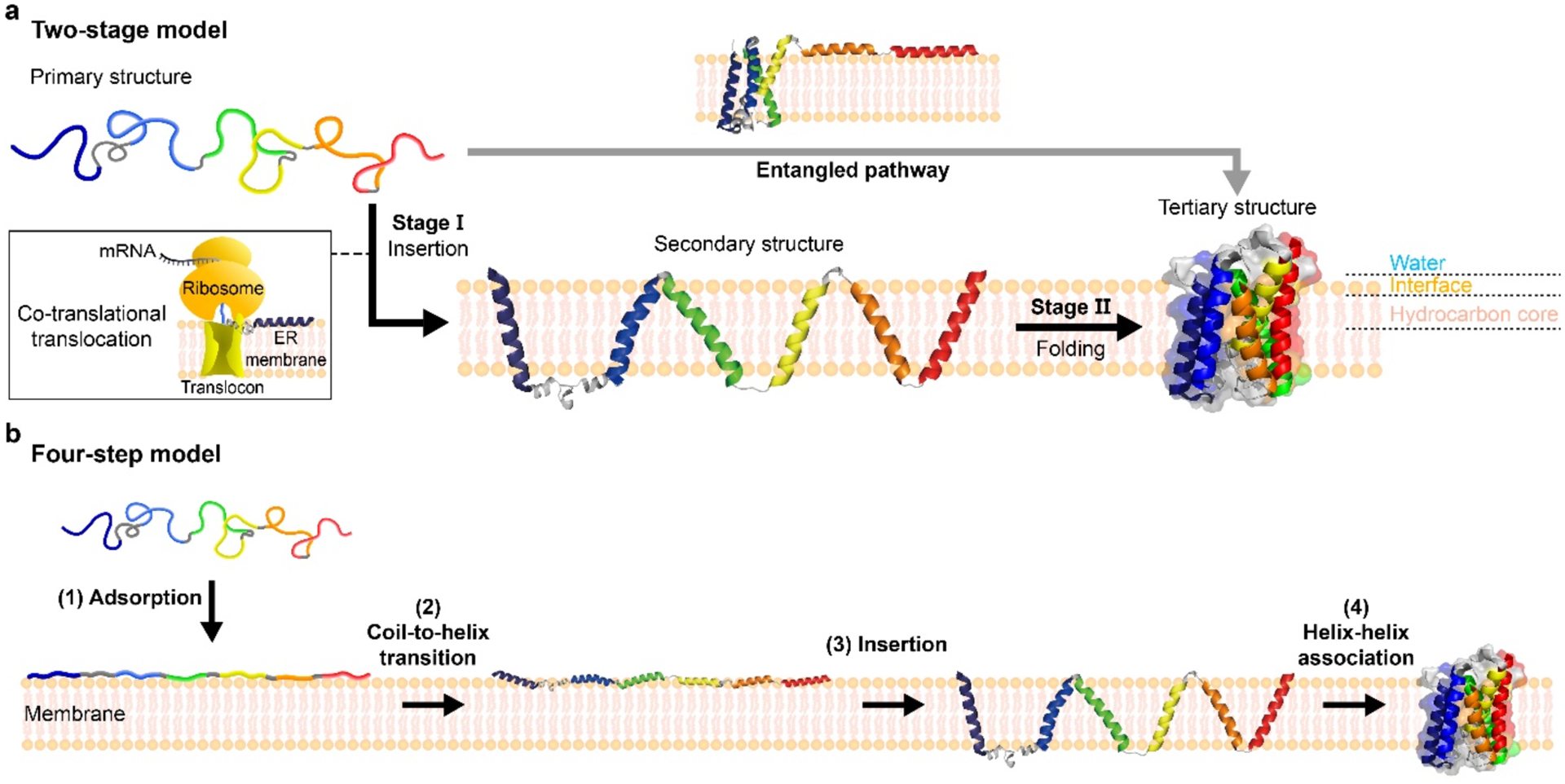
(a) Two-stage model. In Stage Ⅰ, a nascent unstructured polypeptide chain is inserted into a lipid bilayer forming transmembrane helical secondary structures. In the inset, co-translational insertion is described. In Stage Ⅱ, inserted helices laterally associate to form a tertiary structure. Recently, it has been suggested that helices can transiently partition to the membrane surface, or their membrane insertion is coupled to the association (denoted as “entangled pathway”). Adapted and modified from Ref [9]. (b) Four-step model. The overall folding is dissected into (1) adsorption of an unstructured coil onto the membrane surface, (2) coil-to-helix transition, (3) insertion of helices into membrane, and (4) helix-helix association towards its native state. Adapted and modified from Ref [27]. GlpG (2XOV) as the model protein was used to visualized.
Although the two-stage model still serves as a useful, conceptual framework, there is an accumulating body of evidence that the folding of helical membrane proteins is more complex than expected from the canonical two-stage model [21–26]. Wimley and White further dissected the helical membrane protein folding into four steps (Figure 1b), each consisting of an experimentally accessible sequential step with a corresponding free energy contribution [27]:
| (1) |
Here, ΔG0 denotes the total free energy difference between an unfolded coil state in water and the final folded state in the membrane. and reflect the free energy differences caused upon transfer of an unfolded coil from the water phase to a water–membrane interface and the coil–helix transition at the interface, respectively. and denote the respective changes accompanying insertion of the interfacial helix to the membrane, and the helix–helix association to make helical bundles in the membrane. These free-energy changes have been extensively characterized using model peptides [27–29].
A multitude of studies have determined the range of free energy changes in each step. By tracking partitioning of unstructured peptides and formation of the backbone hydrogen bonds, and have been estimated as approximately −0.4 kBT/residue [30,31] and −0.7 to −0.2 kBT/residue [32], respectively. The late-stage values of and are alleged to span −0.8 to −0.6 kBT/residue [33–35] and −0.4 to −0.1 kBT/residue from TM helix dimerization of glycorphorin A [13,36,37], respectively. Notably, in calculating , the membrane insertion of a polar H-bonded peptide backbone is unfavorable with a penalty of ~2.0 kBT/residue, causing a struggle that has to be compensated by the favorable membrane partitioning of hydrophobic side chains [38]. The membrane insertion of helices also depends on the bilayer properties including thickness and area-compressibility modulus [35]. Collectively, a back-of-envelope calculation yields −2.3 to −1.3 kBT/residue for ΔG0, indicating that membrane protein folding is a spontaneous, thermodynamically down-hill process.
Emerging tools for dissecting membrane protein folding
Delineating the folding energy landscape of helical membrane proteins is a daunting task because a reversible control of their unfolding and refolding is notoriously difficult while maintaining the intact bilayer environments. For example, sodium dodecylsulfate (SDS) – a strong anionic detergent used as a denaturant of membrane proteins – inevitably disrupt the bilayer structure at high concentrations, yielding denatured states with non-native topology and interactions [39–43]. Recently, novel methods have been devised for selective control of membrane protein unfolding/refolding, enabling dissection of the folding steps and underlying energetics in unprecedented details.
For water-soluble proteins, methods such as hydrogen/deuterium (H/D) exchange and NMR relaxation dispersion have revealed the dynamic and multistate nature of the native state, which is essential for folding, function and quality control [44–48]. Steric trapping delves into membrane protein folding by coupling spontaneous denaturation of a double biotin-tagged protein to competitive binding of bulky monovalent streptavidin (mSA, ~52 kDa [49]), retaining native lipid–protein and water–protein interactions (Figure 2a) [36,37,50–52]. When biotin tags are attached to two specific residues that are spatially close in the folded state but distant in the amino acid sequence, a first mSA molecule binds either of the biotin tags with an intrinsic binding affinity of ΔGoBind. Due to steric hindrance, a second mSA binds only when the membrane protein is transiently denatured. Steric trapping thus selectively captures disruption of the tertiary interactions in the region between two biotin tags minimally affecting their secondary structures. By adjusting the biotin affinity of mSA by mutation, the denaturation and binding can be reversibly controlled analogous to the EX2 condition in H/D exchange (i.e., the exchange is limited by the equilibrium constant between the native and denatured states). Protein stability (ΔGoN-D) is obtained from the attenuated second binding of mSA, which is coupled to denaturation (ΔGoN-D + ΔGoBind). Analogous to the EX1 condition in H/D exchange (i.e., the exchange is limited by the denaturation rate of a protein), wild-type mSA with a high biotin binding affinity and slow dissociation (Kd = ~10−14 M and τoff = ~days) can irreversibly trap the denatured state, allowing the determination of a spontaneous denaturation rate [53–55].
Figure 2 |. Emerging techniques for resolving membrane protein folding.

(a) Illustration of steric trapping method for studying membrane protein folding under native conditions. The principle of the method is described in the main text. ΔGoBinding: the free energy change upon binding of mSA to biotin; ΔGoN-D: the free energy change upon denaturation (i.e., thermodynamic stability); kon: on-rate constant of mSA binding to biotin; koff: off-rate constant of mSA-biotin complex; kU: spontaneous denaturation rate; kF: spontaneous folding rate. Adapted and modified from Refs [52,53] (b) Schematic diagram of single-molecule mangetic tweezer experiments for examining folding of single GlpG proteins (2XOV). Adapted and modified from Ref [61]. (c) Folding energy landscape of GlpG obtained from steric trapping in dodecylmaltoside micelles. ΔGoN-D and activation energy of denaturation (Ea,D) of each subdomain were determined using the biotin pair conjugated to each subdomain. ‡: transition state. Adapted and modified from Ref [53]. (d) The cooperative interaction map of GlpG obtained by the steric trapping-based cooperativity profiling method. The degree of cooperativity or localization of each residue interaction is color-coded. Notably, the residues at the TMH4 TMH6 interface are located at C-subdomain but the disruption of these residue interactions more substantially destabilize N-subdomain (i.e., highly overpropagated interactions). Absolutely conserved residues among the rhomboid protease family are marked with asterisks. ΔΔΔG = ΔΔGoWT-Mut (N-subdomain) – ΔGoN-D (C-subdomain). Here, ΔΔΔG, the cooperativity parameter, is defined as the difference between the stability changes induced by a specific point mutation (ΔΔGoWT-Mut) measured at the biotin pairs in N- and C-subdomains. Adapted and modified from Ref [52]. (e) Reconstructed folding energy landscapes (black for Bell-Zhrukov model, red for Dudko model) for E. coli GlpG at zero force. Errors in free energy are from the errors in prefactors (for Bell-Zhrukov) and rates (for Dudko model), and errors in reaction coordinate are from the errors in distances between equilibrium states and transition states. Adapted and modified from Ref [61].
To render this method broadly applicable, novel thiol-reactive biotin probes have been developed combining all essential features for steric trapping [52]. These probes contain a fluorophore to detect binding of quencher-labeled mSA or a spin label to measure interspin distances in the native and denatured states using double electron-electron resonance spectroscopy [52]. Steric trapping can be transformed into various formats for investigating several key elements in membrane protein folding [52]: 1) The method captures transient disruption of the tertiary contacts at a specific biotin pair. Thus, by moving the location of the biotin pair, local thermodynamic and kinetic stability can be measured, 2) By introducing a point mutation that perturbs local residue interaction and measuring the mutation-induced stability changes at different biotin pairs, it can be evaluated whether the perturbed residue interactions are localized or cooperatively propagated through the surrounding (called “cooperativity profiling”), and 3) Detailed conformational features (e.g., the flexibility and compactness) of the trapped denatured state can be studied under native conditions.
Single-molecule force spectroscopy is a unique tool for studying membrane protein folding because its signals can be directly translated into structural information. This salient feature was well demonstrated in the pioneering works by Müller, Gaub and co-workers, in which atomic force microscopy (AFM) was used to pull single membrane proteins to induce their unfolding [56–58]. Tracking trajectories of the AFM tip as a function of applied force directly reveals the numbers of amino acid residues in unfolded parts of single membrane proteins, allowing to trace unfolding and refolding events in a time-resolved manner. Recently, Perkins and co-workers realized 1 μs resolution in their AFM measurement, which led to unveiling of unfolding intermediates of bateriorhodopsin at a resolution of 3 amino acid residues [59,60]. However, the loading rates – by which applied force increases and decreases with time – typically reach a few nN/s in AFM studies, driving unfolding and refolding events to occur in a deep non-equilibrium regime. In addition, the unfolded parts are inevitably pulled out of lipid bilayers while losing their secondary structures, making it difficult to resolve respective contributions to the total free energy differences.
Single-molecule magnetic tweezer technique has recently been used in an effort to observe the late-stage folding events (Figure 2b) [61]. Because magnetic tweezing and biochemical processes in question can be completely decoupled, unfolding and refolding events of single membrane proteins can be observed under essentially constant force in a magnetic tweezer setup. Helical structures were observed to be restored in a force range from 20 to 10 pN for all membrane proteins examined, confirming the validity of the two- and four-stage models, in which the coil-to-helix transition (e.g., and precedes membrane insertion of TMHs and tertiary structure formation. When the magnetic force was further relaxed down to ~5 pN, the single membrane proteins showed discrete transitions between intermediates before reaching the native folded states, revealing crucial on-pathway intermediates of membrane protein folding (Figure 2b). Of note, these transitions are found to involve both membrane insertion and tertiary structure formation that are intricately mixed with one another (e.g., and ).
One emerging question is which energetics out of membrane insertion or tertiary contact formation drives these late-stage transitions. The magnetic tweezers data yielded mixed implications to this question. On one hand, single-point mutations – out of more than two hundreds of amino acid residues and presumably affecting membrane insertion to a marginal extent – still markedly changed the kinetics of state transitions, an indicative that tertiary contact formation is a determining factor in membrane protein folding. On the other hand, adopting vesicle membranes, instead of more permeable bicelles, seriously impedes the observed structural transitions to native folded states. Also, for a transporter protein with a weaker folding propensity, dedicated insertase complexes need to be added to facilitate their folding (H.-K.C. and T.-Y.Y., unpublished data). These latter observations strongly point to a hypothesis that membrane insertion also presents serious energy barriers, at least, for a certain class of membrane proteins exhibiting higher insertion energy costs for some of their TMHs.
Native nano-electrospray ionization ion-mobility/mass spectrometry is also addressing several key problems in membrane protein folding, including identification of the lipids that tightly bind to proteins [62], the affinity and cooperativity of lipid-protein interactions [63,64], and the effect of bound lipids on protein stability. Single-molecule photobleaching is especially powerful for measuring strengths of protein-protein interactions in a lipid bilayer [65]. This method has been applied for determining the dimer affinity of the H+/Cl− antiporter of E. coli (CLC-ec1) in the bilayer [65], revealing one of the strongest membrane protein interactions ever measured and the lipid effect on the dimerization equilibrium [66].
Dissecting the folding pathways of helical membrane proteins: Lessons from GlpG
One of the major questions regarding the folding of helical-bundle membrane proteins is whether these proteins fold cooperatively or via partially folded intermediates. Cooperativity links the behavior of different residues within a protein [67], manifested as all-or-none (i.e., two-state) folding and a single predominant free energy barrier separating the native and the denatured states [68]. While theoretical tests with membrane proteins have reached diverse conclusions, with some supporting cooperative folding to attain the native state as the global free energy minima, and others predicting multiple or more rugged folding pathways with local energy minima [69,70]. Recently, the TM domain of GlpG of E. coli, a six-transmembrane helical bundle membrane protein that belongs to the rhomboid protease family, has emerged as a tractable model due to its folding reversibility [71], monomeric character [72], and biological importance of the protein family [73]. Multiple experimental [43,52,53,61,71,74,75] and computational [69,70,76,77] studies with GlpG have provided a wholistic picture, pointing to a complex folding energy landscape even for the single-domain membrane protein.
With the ability to control folding and denaturation in a native environment, steric trapping has resolved two subdomains with distinct folding properties in micelles (Figure 2c) [52,53]. A N-terminal subdomain consisting of TMHs 1–3 has the higher thermodynamic (ΔGN-D = 9.8 kBT) and kinetic stability (Ea,D = 62.0 kBT ; Ea,D denotes the activation energy of denaturation), whose disruption leads to global denaturation. The remaining subdomain (TMHs 4–6), harboring the catalytic dyad, is less stable (ΔGN-D = 7.9 kBT and Ea,D = 48.5 kBT) and undergoes subglobal denaturation. Cooperativity profiling based on steric trapping reveals multiple clusters of cooperative interactions in GlpG (Figure 2d) [52]. Notably, the cooperative clusters overlap with the stability hotspots determined by thermal denaturation [71], as well as the folding nucleus (a membrane-buried packing core consisting of N-terminal regions) suggested from the ϕ-value analysis [43]. This distributive nature of cooperative interactions in GlpG is markedly distinguished from folding of soluble, globular proteins that often involves cooperative interactions in the hydrophobic core working as a single folding nucleus [44,67,78,79].
Single-molecule force spectroscopy using magnetic tweezers have also resolved detailed conformational and energetic features of the intermediates populated during GlpG folding in the lipid bilayer environment [61,74]. By relaxing force to low-pN levels, Choi, Yoon and co-workers were able to observe two intermediates repeatedly populated during folding of single GlpG proteins [61]. While the first intermediate (I1) consists of TMHs 1 and 2 connected by a long linker region, the second intermediate (I2) involves TMHs 3 and 4 in the growing structure, indicating helical hairpins as a basic folding unit. Because the magnetic tweezer experiments were conducted under essentially constant force levels, the observed kinetics of state transition was directly converted to parameters describing the energy landscape governing the late-stage folding steps [61] (Figure 2e). Different models yielded consistent results, indicating the robustness of the energy landscape reconstruction.
Remarkably, despite subtle differences in intermediate conformations, the steric trapping and the single-molecule force spectroscopy methods agree on many observations: 1) GlpG has moderate thermodynamic stability with a large unfolding barrier. 2) GlpG has a modular architecture consistent with predictions from the AWSEM (Associative Memory, Water Mediated, Structure and Energy Model)-membrane molecular dynamics simulation [70], in which the more stable N-terminal region serves as a keystone for the global conformational integrity whereas the folding of the C-terminal region is rather dispensable [52,61,70,74,76]. Taken together, these findings suggest that the single-domain helical-bundle membrane protein GlpG has a rugged folding energy landscape during folding within the bilayer. The high kinetic barrier between the native and unfolded state and the strong foldability of the N-terminal region may be beneficial for avoiding misfolding or aggregation in the crowded cellular environments [61,80].
Does a “lipophobic effect” promote protein folding in the membrane?
In the second stage of the two-step model, individual TMHs associate into a compact native structure while overcoming their favorable interactions with lipids (Figure 1a). An emerging question is if there is a lipophobic effect that induces coalescence of unassembled TMHs in the lipid bilayers [81,82], analogous to the way the hydrophobic effect drives collapse of hydrophobic residues repelled by water molecules. On the bright side, such lipophobic effect could serve as a folding driving force that can compensate the lack of the hydrophobic effect within the membrane. On the dark side, the same force would facilitate nonspecific collapse of TMHs in the membrane, increasing a chance for misfolding. Interestingly, a number of evidence imply that membrane proteins are vulnerable to misfolding and nonspecific protein-protein interactions in the cell membranes: 1) The cellular folding efficiency of membrane proteins is typically low, ranging from only 20 to 50 % [83,84]; 2) Proper membrane protein biogenesis requires an assistance of quality control machineries (e.g., chaperones and proteases [2,83,85–90]; 3) In the liposomes, the affinity of native TM helix-helix interactions dramatically decreases when the total membrane protein extracts are co-reconstituted [37].
At the molecular level, the lipophobic effect may be manifested as a way to relieve the energetic penalty caused by unfavorable protein-lipid interactions in the denatured state, such as a hydrophobic mismatch between a protein and the bilayer leading to lipid deformation at the protein surface [66,91] and an unfavorable exposure of bulky polar/charged residues to the bilayer core that results in ineffective lipid solvation and burial of the exposed residues into the protein interior [92,93] (Figure 3a). Another possible scenario for the lipophobic effect is that inserted TMHs perturb favorable lipid-lipid interactions [94], increasing the overall chemical potential of the denatured state relative to the native state (Figure 3b). If so, the tendency to maximal restoration of the perturbed lipid-lipid interactions may provide a driving force for protein compaction within the bilayer regardless of the hydrophobic mismatch and amino acid sequence of the TMHs. Notably, an all-atom MD simulation study predicts that glycorphorin A monomers collapse with non-native contacts in the bilayer as an obligatory step prior to the formation of the native dimer [95].
Figure 3 |. Proposed modes of the lipophobic effect.

(a) Illustration of the monomer-dimer equilibrium of E. coli H+/Cl− antiporter (ClC-ec1; 1OTS). In the monomeric state of ClC-ec1, the hydrophobic mismatch near the protein-lipid interfacial regions (green and blue) can occur, causing the unfavorable lipid deformation at the dimer interfaces and the penetration of water molecules (inset). These unfavorable lipid-protein interactions can drive dimerization. Adapted and modified from Ref [66]. (b) Illustration of the general lipophobic effect. Relative to the native state, the lipid-lipid interactions in the lipid bilayer are perturbed by the lipid solvation of individual TM segments (i.e., unfavorable lipid-lipid interactions: the chemical potential contribution from this interaction is positive, ΔμLipid-Lipid > 0, red upward arrow). Here, the favorable intramolecular protein interactions are not yet fully realized (i.e., unfavorable protein-protein interactions: ΔμProtein-Protein > 0, red upward arrow), and the TM segments are highly solvated by lipids (i.e., favorable lipid-protein interaction: ΔμLipid-Lipid < 0, blue downward arrow). Summed up, the chemical potential of the system in the denatured state increases (Δμ > 0) relative to the native state. These energetic factors in the denatured state can serve as driving forces for folding into the compact native state.
Folding of multi-domain membrane proteins
Many membrane proteins are characterized by higher-order symmetry properties in their structures, with the pseudo-C2 and -C4 symmetry of transporter and channel proteins being outstanding examples [96]. The symmetry planes divide the tertiary and quaternary structures of membrane proteins (or complexes) into subdomains and subunits, each defining basic units in function and folding. For example, secondary transporters belonging to the major facilitator superfamily (MFS) have two highly symmetric domains with conserved structures (referred to as the MFS fold) [70,97] (Figure 4a). These two domains serve as principal units in the transporters’ rocker switch motions, creating conformational states with alternating access to extra- and intra-cellular spaces [97].
Figure 4 |. Folding of multi-domain membrane proteins.

(a) Structure of human glucose transporter 3 with peusdo-C2 symmetry (GLUT3; 4ZW9). Left and right insets show the projected structures from the extracellular view and intracellular view, respectively. (b) A representative schematic for a possible folding pathway of the MFS fold. The N-terminal domain of human GLUT3 is used as a model. MFS; major facilitator superfamily.
One interesting observation is many membrane proteins, including the MFS transporters, have evolved to form a connected structure via duplication or fusion of the genes of primordial transporters [98,99]. Such connected structures, however, seemingly pose a higher risk in terms of foldability because a failure in folding of either one domain would jeopardize folding of the entire protein, raising a question about the advantages of the connected structure despite the increased risk for misfolding. Indeed, it is recently suggested that gene duplication likely increases the chance for misfolding by forming a swapped structure between duplicated domains [100]. One possible hypothesis is specialization of each domain in its role, with one domain evolving to contend with functional requirements (but becoming less foldable) and the other domains providing the structural stability of the complex. It is observed that the C-domain of E. coli ClC-ec1 is unstable by itself and requires its N-terminal counterpart for maintaining its tertiary structure and likely, proper folding in the endoplasmic reticulum (ER) membrane [101]. The differentiated folding propensity of the N- and C-terminal domains has been observed for GlpG using the steric trapping method (Figure 2). This strict hierarchy in the folding order of respective domains may account for how membrane proteins structure its folding and prevent a domain-swapped structure. It remains to be examined whether this specialization of different domains in foldability and functionality is observed for a broader class of multi-domain membrane proteins.
Most multi-domain membrane protein families are epitomized by the structural folds that have been conserved over a long evolutionary distance from E. coli to human. This would suggest that the folding pathway of these structural folds are essentially shared across the family members. One interesting finding is that many of these conserved folds have entangled topologies in their TMH alignment, defying a folding pathway in which TMHs are added onto one another in a simple, interchangeable manner. In the case of MSF folds, TM helix 1 is flanked by TMHs 5 and 6 (Figure 4b). This entanglement suggests that TMHs 5 and 6 should follow TM helix 1 in the folding order, likely but not the other way around. Likewise, for the partial structure composed of TMHs 1 to 4, TMHs 1 and 2 in turn wrap around TMH 4, which would force folding of TMHs 1 and 2 only after that of TMH 4. Thus, by only considering the topology of the structure, one would be able to roughly delineate the folding order of the MFS fold. Presumably, TMHs 4 (and 3) form the inner core of the MFS fold, onto which TMHs 1 and 2 are added. TMHs 5 and 6 follow to wrap around the growing structure to complete folding of the MFS fold. This folding order remains to be confirmed by experimental observations.
Conclusion
Despite the daunting complexity of membrane protein biogenesis, theoretical frames have evolved to permit detailed dissection of membrane protein folding into elementary steps. Advances in experimental platforms now allow us to observe and modulate different steps of membrane protein folding in a highly selective manner. These collective efforts suggest what appears to be overarching principles for biogenesis of diverse classes of membrane proteins. One prominent example is an overall hierarchical N- to C-terminal folding pathway. During the translation of membrane proteins, the more stable N-terminal segments are favorably inserted into the membrane and partially folded, protecting the nascent chain from misfolding and aggregation. Upon emergence of more C-terminal TMHs from the ribosome vestibule, the partially folded N-terminal region works as a structural template for subsequent folding guided by the funneled energy landscape. Once folded, the protein is kinetically stabilized to carry out function. This scenario may be adopted by other types of membrane proteins since a similar N-to-C directional folding has been observed for GPCRs such as β2-adrenergic receptor [61] and rhodopsin [80], which are evolutionary distant from GlpG. Finally, we note that cryo-electron microscopy techniques are unveiling the tertiary and quaternary structures of membrane proteins as well as interactions with lipids with unprecedented speed [102], which will serve as bases for elucidation of their folding pathways and underlying physical principles. The new computational platforms that permit direct structural predictions from sequence information [103,104] will also expedite the discovery of new membrane protein folds and the design and analysis of membrane protein folding experiments. Thus, we expect the coming decades will be an exciting era for the field of membrane protein folding to make previously inconceivable discoveries and chart new territories.
Acknowledgement
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2021R1A3B1071354 to T.-Y.Y.), Postdoctoral Fellowship Program granted by National Research Foundation of Korea government (2021R1A6A3A0303838211 to H.-K.C.) and the National Institute of Health (USA) grant (NIH R01GM118684 to H.H.).
References
- 1.Winklhofer KF, Tatzelt J, Haass C: The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J 2008, 27:336–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marinko JT, Huang H, Penn WD, Capra JA, Schlebach JP, Sanders CR: Folding and Misfolding of Human Membrane Proteins in Health and Disease: From Single Molecules to Cellular Proteostasis. Chem Rev 2019, 119:5537–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lukacs GL, Mohamed A, Kartner N, Chang XB, Riordan JR, Grinstein S: Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. The EMBO journal 1994, 13:6076–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, Sandberg MA, Berson EL: A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990, 343:364–366. [DOI] [PubMed] [Google Scholar]
- 5.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B: The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325:733–736. [DOI] [PubMed] [Google Scholar]
- 6.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M: alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 1998, 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy Y, Onuchic JN: Water mediation in protein folding and molecular recognition. Annu Rev Biophys Biomol Struct 2006, 35:389–415. [DOI] [PubMed] [Google Scholar]
- 8.Dill KA: Dominant forces in protein folding. Biochemistry 1990, 29:7133–7155. [DOI] [PubMed] [Google Scholar]
- 9.Popot JL, Engelman DM: Membrane protein folding and oligomerization: the two-stage model. Biochemistry 1990, 29:4031–4037. [DOI] [PubMed] [Google Scholar]
- 10.Engelman DM, Chen Y, Chin CN, Curran AR, Dixon AM, Dupuy AD, Lee AS, Lehnert U, Matthews EE, Reshetnyak YK, et al. : Membrane protein folding: beyond the two stage model. FEBS Lett 2003, 555:122–125. [DOI] [PubMed] [Google Scholar]
- 11.Cao Z, Hutchison JM, Sanders CR, Bowie JU: Backbone hydrogen bond strengths can vary widely in transmembrane helices. J Am Chem Soc 2017, 139:10742–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Using Hydrogen-deuterium exchange, the free energy changes upon formation of the backbone hydrogen bonds were first determined in the hydrophobic environment provided by micelles.
- 12.Engelman DM, Steitz TA: The spontaneous insertion of proteins into and across membranes: the helical hairpin hypothesis. Cell 1981, 23:411–422. [DOI] [PubMed] [Google Scholar]
- 13.Fleming KG, Engelman DM: Specificity in transmembrane helix-helix interactions can define a hierarchy of stability for sequence variants. Proceedings of the National Academy of Sciences of the United States of America 2001, 98:14340–14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joh NH, Oberai A, Yang D, Whitelegge JP, Bowie JU: Similar energetic contributions of packing in the core of membrane and water-soluble proteins. J Am Chem Soc 2009, 131:10846–10847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mravic M, Thomaston JL, Tucker M, Solomon PE, Liu L, DeGrado WF: Packing of apolar side chains enables accurate design of highly stable membrane proteins. Science 2019, 363:1418–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work first demonstrated that the geometric van der Waals packing between side chains is enough to drive the association of transmembrane helices.
- 16.Joh NH, Min A, Faham S, Whitelegge JP, Yang D, Woods VL, Bowie JU: Modest stabilization by most hydrogen-bonded side-chain interactions in membrane proteins. Nature 2008, 453:1266–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou FX, Merianos HJ, Brunger AT, Engelman DM: Polar residues drive association of polyleucine transmembrane helices. Proc Natl Acad Sci U S A 2001, 98:2250–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tatko CD, Nanda V, Lear JD, Degrado WF: Polar networks control oligomeric assembly in membranes. J Am Chem Soc 2006, 128:4170–4171. [DOI] [PubMed] [Google Scholar]
- 19.Hong H: Toward understanding driving forces in membrane protein folding. Arch Biochem Biophys 2014, 564:297–313. [DOI] [PubMed] [Google Scholar]
- 20.O’Brien ES, Fuglestad B, Lessen HJ, Stetz MA, Lin DW, Marques BS, Gupta K, Fleming KG, Wand AJ: Membrane Proteins Have Distinct Fast Internal Motion and Residual Conformational Entropy. Angew Chem Int Ed Engl 2020, 59:11108–11114. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This NMR relaxation study shows that methyl-bearing side chains of membrane proteins are more dynamic in the sub-ns timescale than water-soluble proteins, providing a new insight into the internal organization of membrane proteins.
- 21.Seurig M, Ek M, von Heijne G, Fluman N: Dynamic membrane topology in an unassembled membrane protein. Nat Chem Biol 2019, 15:945–948. [DOI] [PubMed] [Google Scholar]; •• This work experimentally demonstrates that the less hydrophobic transmembrane segments can undergo topology changes partitioning into the membrane surface in the timescale of minutes.
- 22.Van Lehn RC, Zhang B, Miller TF 3rd: Regulation of multispanning membrane protein topology via post-translational annealing. Elife 2015, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Using coarse-grained molecular dynamic simulation, this work suggests that individual transmembrane segments of multispanning membrane proteins can undergo topology changes even after translation and membrane-insertion are completed.
- 23.Woodall NB, Hadley S, Yin Y, Bowie JU: Complete topology inversion can be part of normal membrane protein biogenesis. Protein Sci 2017, 26:824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vitrac H, MacLean DM, Jayaraman V, Bogdanov M, Dowhan W: Dynamic membrane protein topological switching upon changes in phospholipid environment. Proc Natl Acad Sci U S A 2015, 112:13874–13879. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work reports that the dramatic toplogy changes of multidomain membrane proteins can occur even at the domain level depending on the lipid composition.
- 25.Ojemalm K, Halling KK, Nilsson I, von Heijne G: Orientational preferences of neighboring helices can drive ER insertion of a marginally hydrophobic transmembrane helix. Mol Cell 2012, 45:529–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cymer F, von Heijne G: Cotranslational folding of membrane proteins probed by arrest-peptide–mediated force measurements. Proceedings of the National Academy of Sciences 2013, 110:14640–14645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.White SH, Wimley WC: Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct 1999, 28:319–365. [DOI] [PubMed] [Google Scholar]
- 28.Hristova K, White SH: An experiment-based algorithm for predicting the partitioning of unfolded peptides into phosphatidylcholine bilayer interfaces. Biochemistry 2005, 44:12614–12619. [DOI] [PubMed] [Google Scholar]
- 29.Wimley WC, Gawrisch K, Creamer TP, White SH: Direct measurement of salt-bridge solvation energies using a peptide model system: implications for protein stability. Proc Natl Acad Sci U S A 1996, 93:2985–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ladokhin AS, White SH: Folding of amphipathic alpha-helices on membranes: energetics of helix formation by melittin. J Mol Biol 1999, 285:1363–1369. [DOI] [PubMed] [Google Scholar]
- 31.Andersson M, Ulmschneider JP, Ulmschneider MB, White SH: Conformational states of melittin at a bilayer interface. Biophys J 2013, 104:L12–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wieprecht T, Apostolov O, Beyermann M, Seelig J: Thermodynamics of the alpha-helix-coil transition of amphipathic peptides in a membrane environment: implications for the peptide-membrane binding equilibrium. J Mol Biol 1999, 294:785–794. [DOI] [PubMed] [Google Scholar]
- 33.Soekarjo M, Eisenhawer M, Kuhn A, Vogel H: Thermodynamics of the membrane insertion process of the M13 procoat protein, a lipid bilayer traversing protein containing a leader sequence. Biochemistry 1996, 35:1232–1241. [DOI] [PubMed] [Google Scholar]
- 34.White SH, Ladokhin AS, Jayasinghe S, Hristova K: How membranes shape protein structure. J Biol Chem 2001, 276:32395–32398. [DOI] [PubMed] [Google Scholar]
- 35.Ulmschneider JP, Smith JC, White SH, Ulmschneider MB: In silico partitioning and transmembrane insertion of hydrophobic peptides under equilibrium conditions. J Am Chem Soc 2011, 133:15487–15495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong H, Blois TM, Cao Z, Bowie JU: Method to measure strong protein-protein interactions in lipid bilayers using a steric trap. Proc Natl Acad Sci U S A 2010, 107:19802–19807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong H, Bowie JU: Dramatic destabilization of transmembrane helix interactions by features of natural membrane environments. J Am Chem Soc 2011, 133:11389–11398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jayasinghe S, Hristova K, White SH: Energetics, stability, and prediction of transmembrane helices. J Mol Biol 2001, 312:927–934. [DOI] [PubMed] [Google Scholar]
- 39.Lau FW, Bowie JU: A method for assessing the stability of a membrane protein. Biochemistry 1997, 36:5884–5892. [DOI] [PubMed] [Google Scholar]
- 40.Otzen DE: Protein unfolding in detergents: effect of micelle structure, ionic strength, pH, and temperature. Biophys J 2002, 83:2219–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faham S, Yang D, Bare E, Yohannan S, Whitelegge JP, Bowie JU: Side-chain contributions to membrane protein structure and stability. J Mol Biol 2004, 335:297–305. [DOI] [PubMed] [Google Scholar]
- 42.Schlebach JP, Woodall NB, Bowie JU, Park C: Bacteriorhodopsin folds through a poorly organized transition state. Journal of the American Chemical Society 2014, 136:16574–16581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paslawski W, Lillelund OK, Kristensen JV, Schafer NP, Baker RP, Urban S, Otzen DE: Cooperative folding of a polytopic alpha-helical membrane protein involves a compact N-terminal nucleus and nonnative loops. Proc Natl Acad Sci U S A 2015, 112:7978–7983. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work reports the transition state model of the multispanning membrane protein GlpG in detergent micelles, identifies the folding nucleus, and suggests a novel back-tracking folding mechanism.
- 44.Bai Y, Sosnick TR, Mayne L, Englander SW: Protein folding intermediates: native-state hydrogen exchange. Science 1995, 269:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baldwin AJ, Kay LE: NMR spectroscopy brings invisible protein states into focus. Nat Chem Biol 2009, 5:808–814. [DOI] [PubMed] [Google Scholar]
- 46.Lee C, Schwartz MP, Prakash S, Iwakura M, Matouschek A: ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol Cell 2001, 7:627–637. [DOI] [PubMed] [Google Scholar]
- 47.Motlagh HN, Wrabl JO, Li J, Hilser VJ: The ensemble nature of allostery. Nature 2014, 508:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Best RB, Lindorff-Larsen K, DePristo MA, Vendruscolo M: Relation between native ensembles and experimental structures of proteins. Proc Natl Acad Sci U S A 2006, 103:10901–10906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Howarth M, Chinnapen DJ, Gerrow K, Dorrestein PC, Grandy MR, Kelleher NL, El-Husseini A, Ting AY: A monovalent streptavidin with a single femtomolar biotin binding site. Nat Methods 2006, 3:267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Blois TM, Hong H, Kim TH, Bowie JU: Protein unfolding with a steric trap. J Am Chem Soc 2009, 131:13914–13915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang YC, Bowie JU: Measuring membrane protein stability under native conditions. Proc Natl Acad Sci U S A 2014, 111:219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo R, Gaffney K, Yang Z, Kim M, Sungsuwan S, Huang X, Hubbell WL, Hong H: Steric trapping reveals a cooperativity network in the intramembrane protease GlpG. Nat Chem Biol 2016, 12:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work reports novel steric trapping-based methods for characterizing thermodynamic stability, compactness of denatured states and the degree of cooperativity of individual residues of a multispanning membrane protein.
- 53.Yang Y, Guo R, Gaffney K, Kim M, Muhammednazaar S, Tian W, Wang B, Liang J, Hong H: Folding-Degradation Relationship of a Membrane Protein Mediated by the Universally Conserved ATP-Dependent Protease FtsH. J Am Chem Soc 2018, 140:4656–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jefferson RE, Blois TM, Bowie JU: Membrane proteins can have high kinetic stability. Journal of the American Chemical Society 2013, 135:15183–15190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Srisa-Art M, Dyson EC, deMello AJ, Edel JB: Monitoring of real-time streptavidin- biotin binding kinetics using droplet microfluidics. Analytical chemistry 2008, 80:7063–7067. [DOI] [PubMed] [Google Scholar]
- 56.Oesterhelt F, Oesterhelt D, Pfeiffer M, Engel A, Gaub H, Müller D: Unfolding pathways of individual bacteriorhodopsins. Science 2000, 288:143–146. [DOI] [PubMed] [Google Scholar]
- 57.Kedrov A, Ziegler C, Janovjak H, Kühlbrandt W, Müller DJ: Controlled unfolding and refolding of a single sodium-proton antiporter using atomic force microscopy. Journal of molecular biology 2004, 340:1143–1152. [DOI] [PubMed] [Google Scholar]
- 58.Müller DJ, Kessler M, Oesterhelt F, Möller C, Oesterhelt D, Gaub H: Stability of bacteriorhodopsin α-helices and loops analyzed by single-molecule force spectroscopy. Biophysical journal 2002, 83:3578–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu H, Siewny MG, Edwards DT, Sanders AW, Perkins TT: Hidden dynamics in the unfolding of individual bacteriorhodopsin proteins. Science 2017, 355:945–950. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work uncovered partial refolding and unfolding of membrane protein during unfolding pathway and observed numerous intermediate states with three-amino acids resolution using high-temporal resolution AFM.
- 60.Heenan PR, Yu H, Siewny MG, Perkins TT: Improved free-energy landscape reconstruction of bacteriorhodopsin highlights local variations in unfolding energy. The Journal of chemical physics 2018, 148:123313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Choi HK, Min D, Kang H, Shon MJ, Rah SH, Kim HC, Jeong H, Choi HJ, Bowie JU, Yoon TY: Watching helical membrane proteins fold reveals a common N-to-C-terminal folding pathway. Science 2019, 366:1150–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work first developed the method to capture every four-step in fout-step model using single-molecule force spectroscopy, and first reported the folding pathway of alpha-helical membrane proteins within the membrane.
- 62.Laganowsky A, Reading E, Allison TM, Ulmschneider MB, Degiacomi MT, Baldwin AJ, Robinson CV: Membrane proteins bind lipids selectively to modulate their structure and function. Nature 2014, 510:172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This is a landmark study applying native nano-electrospray ion-mobility/mass spectrometry for indentifying tightly bound lipids and their effect on the stability of membrane proteins.
- 63.Cong X, Liu Y, Liu W, Liang X, Russell DH, Laganowsky A: Determining Membrane Protein-Lipid Binding Thermodynamics Using Native Mass Spectrometry. J Am Chem Soc 2016, 138:4346–4349. [DOI] [PubMed] [Google Scholar]; •• This is the first study that quantitatively measure the affinity of specifically bound lipids to a membrane protein.
- 64.Patrick JW, Boone CD, Liu W, Conover GM, Liu Y, Cong X, Laganowsky A: Allostery revealed within lipid binding events to membrane proteins. Proc Natl Acad Sci U S A 2018, 115:2976–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work reports the novel cooperativity phenomenon in lipid binding to membrane proteins using thermodynamic cycles and native mass spectrometry.
- 65.Chadda R, Krishnamani V, Mersch K, Wong J, Brimberry M, Chadda A, Kolmakova-Partensky L, Friedman LJ, Gelles J, Robertson JL: The dimerization equilibrium of a ClC Cl(−)/H(+) antiporter in lipid bilayers. Elife 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work reports the novel single-molecule photobleaching method to determine the strong dimerization affinity of a dimeric membrane transporter.
- 66.Chadda R, Bernhardt N, Kelley EG, Teixeira SCM, Griffith K, Gil-Ley A, Ozturk TN, Hughes LE, Forsythe A, Krishnamani V, et al. : Membrane transporter dimerization driven by differential lipid solvation energetics of dissociated and associated states. Elife 2021, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work demonstrates that membrane protein-protein interaction can be strongly modulated by the lipid deformation energy caused by the hydrophobic mistmach in both the dissociated and assembled states.
- 67.Hilser VJ, Dowdy D, Oas TG, Freire E: The structural distribution of cooperative interactions in proteins: analysis of the native state ensemble. Proc Natl Acad Sci U S A 1998, 95:9903–9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan HS, Zhang Z, Wallin S, Liu Z: Cooperativity, local-nonlocal coupling, and nonnative interactions: principles of protein folding from coarse-grained models. Annu Rev Phys Chem 2011, 62:301–326. [DOI] [PubMed] [Google Scholar]
- 69.Kim BL, Schafer NP, Wolynes PG: Predictive energy landscapes for folding alpha-helical transmembrane proteins. Proc Natl Acad Sci U S A 2014, 111:11031–11036. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work suggests that membrane protein folding can be described using the minimally-frustrated, funneled folding energy landscape.
- 70.Schafer NP, Truong HH, Otzen DE, Lindorff-Larsen K, Wolynes PG: Topological constraints and modular structure in the folding and functional motions of GlpG, an intramembrane protease. Proc Natl Acad Sci U S A 2016, 113:2098–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This is the first comprehensive computational study describing how the folding energy landscape of a specific membrane protein can change depending on the geometry of the hydrophobic environment.
- 71.Baker RP, Urban S: Architectural and thermodynamic principles underlying intramembrane protease function. Nat Chem Biol 2012, 8:759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kreutzberger AJB, Urban S: Single-Molecule Analyses Reveal Rhomboid Proteins Are Strict and Functional Monomers in the Membrane. Biophys J 2018, 115:1755–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Freeman M: The rhomboid-like superfamily: molecular mechanisms and biological roles. Annu Rev Cell Dev Biol 2014, 30:235–254. [DOI] [PubMed] [Google Scholar]
- 74.Min D, Jefferson RE, Bowie JU, Yoon TY: Mapping the energy landscape for second-stage folding of a single membrane protein. Nat Chem Biol 2015, 11:981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work first reports the application of single-molecule force spectroscopy to studying the lateral transmembrane helix-helix interactions of multispanning membrane proteins.
- 75.Panigrahi R, Arutyunova E, Panwar P, Gimpl K, Keller S, Lemieux MJ: Reversible Unfolding of Rhomboid Intramembrane Proteases. Biophys J 2016, 110:1379–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu W, Schafer NP, Wolynes PG: Energy landscape underlying spontaneous insertion and folding of an alpha-helical transmembrane protein into a bilayer. Nat Commun 2018, 9:4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang Z, Jumper JM, Freed KF, Sosnick TR: On the Interpretation of Force-Induced Unfolding Studies of Membrane Proteins Using Fast Simulations. Biophys J 2019, 117:1429–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This work reports the first successful application of the novel computational strategy, Upside, to the folding problem of membrane proteins.
- 78.Bedard S, Mayne LC, Peterson RW, Wand AJ, Englander SW: The foldon substructure of staphylococcal nuclease. J Mol Biol 2008, 376:1142–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Whitten ST, Garcia-Moreno EB, Hilser VJ: Local conformational fluctuations can modulate the coupling between proton binding and global structural transitions in proteins. Proc Natl Acad Sci U S A 2005, 102:4282–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roushar FJ, Gruenhagen TC, Penn WD, Li B, Meiler J, Jastrzebska B, Schlebach JP: Contribution of Cotranslational Folding Defects to Membrane Protein Homeostasis. J Am Chem Soc 2019, 141:204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Korendovych IV, DeGrado WF: De novo protein design, a retrospective. Q Rev Biophys 2020, 53:e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jahnig F: Thermodynamics and kinetics of protein incorporation into membranes. Proc Natl Acad Sci U S A 1983, 80:3691–3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schlebach JP, Narayan M, Alford C, Mittendorf KF, Carter BD, Li J, Sanders CR: Conformational Stability and Pathogenic Misfolding of the Integral Membrane Protein PMP22. J Am Chem Soc 2015, 137:8758–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sanders CR, Myers JK: Disease-related misassembly of membrane proteins. Annu Rev Biophys Biomol Struct 2004, 33:25–51. [DOI] [PubMed] [Google Scholar]
- 85.Schulze RJ, Komar J, Botte M, Allen WJ, Whitehouse S, Gold VA, Lycklama ANJA, Huard K, Berger I, Schaffitzel C, et al. : Membrane protein insertion and proton-motive-force-dependent secretion through the bacterial holo-translocon SecYEG-SecDF-YajC-YidC. Proc Natl Acad Sci U S A 2014, 111:4844–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shurtleff MJ, Itzhak DN, Hussmann JA, Schirle Oakdale NT, Costa EA, Jonikas M, Weibezahn J, Popova KD, Jan CH, Sinitcyn P, et al. : The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Braunger K, Pfeffer S, Shrimal S, Gilmore R, Berninghausen O, Mandon EC, Becker T, Forster F, Beckmann R: Structural basis for coupling protein transport and N-glycosylation at the mammalian endoplasmic reticulum. Science 2018, 360:215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nagamori S, Smirnova IN, Kaback HR: Role of YidC in folding of polytopic membrane proteins. J Cell Biol 2004, 165:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Avci D, Lemberg MK: Clipping or Extracting: Two Ways to Membrane Protein Degradation. Trends Cell Biol 2015, 25:611–622. [DOI] [PubMed] [Google Scholar]
- 90.Brodsky JL: Cleaning up: ER-associated degradation to the rescue. Cell 2012, 151:1163–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Andersen OS, Koeppe RE 2nd: Bilayer thickness and membrane protein function: an energetic perspective. Annu Rev Biophys Biomol Struct 2007, 36:107–130. [DOI] [PubMed] [Google Scholar]
- 92.Adamian L, Nanda V, DeGrado WF, Liang J: Empirical lipid propensities of amino acid residues in multispan alpha helical membrane proteins. Proteins: Structure, Function, and Bioinformatics 2005, 59:496–509. [DOI] [PubMed] [Google Scholar]
- 93.Ballweg S, Sezgin E, Doktorova M, Covino R, Reinhard J, Wunnicke D, Hänelt I, Levental I, Hummer G, Ernst R: Regulation of lipid saturation without sensing membrane fluidity. Nature communications 2020, 11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This work reports a novel “hellix rotation” phenomenon that the interface of a transmembrane helix-helix interaction is modulated by the lateral lipid-lipid packing interactions.
- 94.Duneau JP, Khao J, Sturgis JN: Lipid perturbation by membrane proteins and the lipophobic effect. Biochim Biophys Acta Biomembr 2017, 1859:126–134. [DOI] [PubMed] [Google Scholar]
- 95.Domański J, Sansom MS, Stansfeld PJ, Best RB: Atomistic mechanism of transmembrane helix association. PLoS computational biology 2020, 16:e1007919. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Using all-atom MD simulation, this work reports a detailed mechanism of TMH–TMH association in a lipid bilayer involving an ensemble of non-native dimeric intermediates prior to the formation of the native dimer.
- 96.Forrest LR: Structural symmetry in membrane proteins. Annual review of biophysics 2015, 44:311–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yan N: Structural advances for the major facilitator superfamily (MFS) transporters. Trends in biochemical sciences 2013, 38:151–159. [DOI] [PubMed] [Google Scholar]
- 98.Lolkema JS, Dobrowolski A, Slotboom D-J: Evolution of antiparallel two-domain membrane proteins: tracing multiple gene duplication events in the DUF606 family. Journal of molecular biology 2008, 378:596–606. [DOI] [PubMed] [Google Scholar]
- 99.Västermark Å, Lunt B, Saier M: Major facilitator superfamily porters, LacY, FucP and XylE of Escherichia coli appear to have evolved positionally dissimilar catalytic residues without rearrangement of 3-TMS repeat units. Journal of molecular microbiology and biotechnology 2014, 24:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Andersen KK, Vad B, Omer S, Otzen DE: Concatemers of outer membrane protein A take detours in the folding landscape. Biochemistry 2016, 55:7123–7140. [DOI] [PubMed] [Google Scholar]
- 101.Min D, Jefferson RE, Qi Y, Wang JY, Arbing MA, Im W, Bowie JU: Unfolding of a ClC chloride transporter retains memory of its evolutionary history. Nat Chem Biol 2018, 14:489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This is the first report applying single-molecule force spectropy to a multidomain membrane protein.
- 102.Cheng Y: Membrane protein structural biology in the era of single particle cryo-EM. Current opinion in structural biology 2018, 52:58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lu P, Min D, DiMaio F, Wei KY, Vahey MD, Boyken SE, Chen Z, Fallas JA, Ueda G, Sheffler W: Accurate computational design of multipass transmembrane proteins. Science 2018, 359:1042–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Žídek A, Bridgland A, Cowie A, Meyer C, Laydon A: Highly accurate protein structure prediction for the human proteome. Nature 2021, 596:590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]