

Abstract
Purpose:
PD-L1 expression on tumor cells (TC) is associated with response to anti-PD-1-based therapies in some tumor types, but its significance in ccRCC is uncertain. We leveraged tumor heterogeneity to identify molecular correlates of TC PD-L1 expression in ccRCC and assessed their role in predicting response to anti-PD-1 monotherapy.
Experimental Design:
RNA-sequencing was performed on paired TC PD-L1 positive and negative areas isolated from 8 ccRCC tumors and transcriptomic features associated with PD-L1 status were identified. A cohort of 232 metastatic ccRCC patients from the randomized CheckMate (CM)-025 trial was used to confirm the findings and correlate transcriptomic profiles with clinical outcomes.
Results:
In both the paired samples and the CM-025 cohort, TC PD-L1 expression was associated with combined overexpression of immune- and cell proliferation-related pathways, upregulation of T-cell activation signatures, and increased tumor-infiltrating immune cells. In the CM-025 cohort, TC PD-L1 expression was not associated with clinical outcomes. A molecular RCC subtype characterized by combined overexpression of immune- and cell proliferation-related pathways (previously defined by unsupervised clustering of transcriptomic data) was enriched in TC PD-L1 positive tumors and displayed longer progression free survival (HR 0.32; 95% CI 0.13–0.83) and higher objective response rate (30% versus 0%, p=0.04) on nivolumab compared to everolimus.
Conclusions:
Both tumor cell-extrinsic (immune-related) and -intrinsic (cell proliferation-related) mechanisms are likely intertwined in the regulation of TC PD-L1 expression in ccRCC. The quantitation of these transcriptional programs may better predict benefit from anti-PD-1-based therapy compared to TC PD-L1 expression alone in ccRCC.
Keywords: Gene expression, gene pathways, immune checkpoints, biomarker, cell proliferation, immune cells, PD-1
Introduction
Immune checkpoint inhibitors (ICI) targeting the PD-1/PD-L1 pathway have become a standard of care in multiple cancer types, including clear cell renal cell carcinoma (ccRCC), which represents the most common type of kidney cancer in adults (1,2).
PD-L1 expression is a physiological immunomodulatory mechanism involved in the regulation of inflammation that is hijacked by tumor cells to escape the immune system. PD-L1 expression on tumor cells (TC) correlates with response to ICI-based therapies in some tumor types, particularly non-small cell lung cancer (NSCLC). In ccRCC, however, correlation with clinical outcome has been inconsistent, but more evident in the first-line setting (3). Tumor heterogeneity (intratumoral as well as between primary tumor and metastases) (4), poor tissue quality and possibly suboptimal assay sensitivity, likely contribute to this lack of association. The identification of signaling pathways that are associated and possibly regulate tumor cell PD-L1 expression in ccRCC might not only shed light on ccRCC biology but could also provide more robust predictors that might help identify patients that respond to PD-1/PD-L1 targeting.
The genomic and transcriptomic determinants of PD-L1 expression are poorly understood in the context of ccRCC. Positive regulators identified in other tumor types include pro-inflammatory cytokines and signaling pathways such as interferon IFN-γ (5–7), IL-6 (8), IL-8 (9), IL-17, TNF-α (10), IL-27 (11,12), STAT3 (13,14), NFkB/RELA (6,15,16), JUN (13,17), TGF-β via SMAD2 (18) and oncogenic actors such as MYC (19–21), HIF-1α (22,23), YAP (24), PTEN loss and PI3K-AKT-mTOR pathway (6,13,25–27), CUL3 via KEAP1 and NRF2 (28), RAS (23,29–31), EGFR (27,30,32–34), ALK (35–41). Moreover, genetic amplification at 9p24.1 involving the CD274 gene (coding for PD-L1) has been identified in Hodgkin lymphoma (42), B cell lymphoma (42,43), NSCLC (44), gastric cancer (45), where it has been linked to increased PD-L1 protein expression but its prevalence in ccRCC is very low (46). MEK-ERK signaling was found to be an activator in some cancers (13,29,47) and a negative regulator in others (26,48). Numerous micro RNAs were also found to be either positive or negative regulators of PD-L1 expression in various cancer types (39). Methylation of the PD-L1 promoter has been associated with decreased PD-L1 expression in various cancer types (49). Finally, PD-L1 expression and tumor mutational burden were shown to be independent in most tumor types (50).
In RCC cell lines, treatment with IL-4 and TNF-α was accompanied by STAT6 phosphorylation and activation of NFkB, and had a synergistic effect on the induction of PD-L1 expression (51). PD-L1 was also shown to be upregulated by HIF-2α (52,53). A recent analysis of tumor samples from patients with advanced RCC, metastatic urothelial carcinomas, and advanced/metastatic NSCLC showed that tumors positive for PD-L1 on either the TC or immune cells were enriched for immune signatures including IFN-ɣ, immune checkpoints, and cytotoxicity. PD-L1 negative tumors presented with an increase in metabolic pathways involved in ATP biosynthesis and oxidative phosphorylation/amino acid biosynthesis (54).
In this study, we leveraged tumor heterogeneity to understand the molecular underpinning of PD-L1 expression in ccRCC by analyzing paired samples with either positive or negative TC PD-L1 expression, isolated from individual tumors. We then confirmed our findings in a cohort of patients treated with an immune checkpoint inhibitor (nivolumab) or an mTOR inhibitor (everolimus) in the context of a clinical trial (CheckMate-025) (55).
Materials and Methods
Patients
Formalin-fixed paraffin-embedded (FFPE) tumor tissue samples from eight patients with ccRCC were selected from the archives of Brigham and Women’s Hospital and Beth Israel Deaconess Medical Center. Each sample had highly heterogeneous expression of PD-L1 in tumor cells and showed the presence of discrete tumor regions characterized by either positive or negative PD-L1 expression. Informed written consent’ was obtained from each subject or each subject’s guardian Institutional Review Board approval was obtained locally before tissue collection and immunohistochemical staining.
A subset of patients included in the CheckMate-025 (CM-025) randomized open label trial of nivolumab versus everolimus in metastatic ccRCC, previously analyzed by our group (56,57), was used as a validation cohort. Specifically, a total of 232 patients with available WHO/ISUP grade evaluation, TC PD-L1 immunohistochemistry results (positivity defined as ≥1% positive TC), and RNA-sequencing data were analyzed.
The study was performed according to the principles of the Declaration of Helsinki.
Pathological evaluation and immunohistochemistry (IHC) analysis for PD-L1 and CD8 expression in paired samples
Nuclear grade was evaluated according to the 2016 WHO/ISUP classification (58). Sarcomatoid differentiation was defined according to criteria discussed at the International Society of Urological Pathology 2012 Consensus Conference.
IHC studies in the 8 paired samples were performed on FFPE tissue sections. Staining was performed using an extensively validated antibody against PD-L1 [405. 9A11 mouse monoclonal antibody, 1:100 of 1.3 mg/mL, Dr. G. Freeman laboratory, Dana-Farber Cancer Institute, Boston, MA, and commercially available through Cell Signaling Technology (CST); (4,59–61)] and an antibody against CD8 [C8/144B, 1:100 Agilent (Cat#M710301–2, RRID:AB_2075537)]. Tumor sections were stained with Bond Rx Autostainer (Leica Biosystems) using the Bond Polymer Refine Detection Kit (DS9800; Leica Biosystems). Antigen retrieval was performed with Bond Epitope Retrieval Solution 2 (EDTA, pH = 9.0) for 30 minutes for the anti-PD-L1 antibody and 20 minutes for the anti-CD8 antibody. All slides were counterstained with hematoxylin, dehydrated in graded ethanol and xylene, mounted, and coverslipped. Available tissue section from 6 paired samples were stained for CD8. The slides immunostained for CD8 were subsequently scanned using the Leica XT scanner (Leica Biosystems) and the density of CD8 positive cells was measured using the multiplex IHC algorithm v2.3.4 of the HALO software (Indica Labs).
RNA sequencing (RNA-seq)
PD-L1 positive and negative areas to be microdissected were visualized by a light microscope and circled with a marker (on the IHC-stained slides) by an expert pathologist (S. Signoretti). The areas of interest were identified on de-paraffinized 4-micron-thick unstained slides by comparing the tissue with the circled areas on the corresponding IHC-stained slide. Microdissection was performed by scraping tissue of interest from the slide using a sterile syringe needle and/or a scalpel blade. The microdissected tissue was then placed in a sterile tube containing RNA extraction buffer. RNA extraction was performed using an AllPrep DNA/RNA FFPE Mini Kit (Qiagen Cat# 80234), according to the manufacturer instructions.
As previously described (56) RNA quality was assessed with Caliper LabChip GX2 (Perkin Elmer). The percentage of fragments with a size greater than 200 nt (DV200) was calculated. The RNA-Seq libraries were prepared using a transcriptome capture approach (TruSeq RNA Access Library Prep Kit (Illumina)) following a validated SOP. Briefly, total RNA samples are fragmented, randomly primed for first and second strand cDNA synthesis ensuring strandedness, and then enriched into indexed double-stranded cDNA libraries. Indexed libraries are then subsequently enriched for coding RNA using hybrid capture probes specific for coding RNA. After enrichment, the libraries were quantified with qPCR using the KAPA Library Quantification Kit for Illumina Sequencing Platforms, followed by equimolar pooling. Flowcell cluster amplification and sequencing were performed according to the manufacturer’s protocols using HiSeq 2000 or 2500 (Illumina). Each run was a 76 bp paired-end.
Transcriptomic analysis
RNA-seq data from the CM-025 cohort (56) and 8 pairs were aligned using STAR (62), quantified using RSEM (63), and evaluated for quality using RNA-seqQC2 (64). Samples with low quality were excluded according to (56). Estimated counts per transcript data were imported from RSEM and aggregated to the gene-level with DESeqDataSetFromTximport() function from tximport method (65). Differential gene expression analysis on PD-L1 effect was performed using DESeq2 package (DESeq2, RRID:SCR_015687) (66) between PD-L1 positive and PD-L1 negative samples. For the cohort of 8 paired samples, a paired Wald test was performed by adding a pair level factor in the design model. For the CM-025 cohort, only PD-L1 status was included in the design model. The “normal” type shrunken log2 fold change for each sample was ranked and used for the GSEA prerank analysis (67) with fgsea package (68) and MSigDB’s hallmark gene set (69). All p values were corrected by Benjamini–Hochberg false discovery rate correction with q<0.25 considered significant.
Single sample GSEA (ssGSEA) score was computed using the R package ‘GSVA’ to obtain sample-level GSEA scores for MSigDB’s hallmark gene sets. Only ssGSEA scores from the 29 commonly upregulated pathways were plotted according to the percentage of positive TC PD-L1 for CM-025 cohort. ssGSEA scores for each hallmark pathways were normalized to mu=0 and variance=1.
Inflammatory signature analysis
Inflammatory signature analysis was performed using 6 immune-related signatures listed below:
IMmotion150_Angio:VEGFA,KDR,ESM1,PECAM1,ANGPTL4,CD34 (70).
IMmotion150_Teff:CD8A,EOMES,PRF1,IFNG,CD274 (70).
IMmotion150_Myeloid:IL6,CXCL1,CXCL2,CXCL3,CXCL8,PTGS2 (70).
JAVELIN:CD3G,CD3E,CD8B,THEMIS,TRAT1,GRAP2,CD247,CD2,CD96,PRF1,CD6,IL7R, ITK,GPR18,EOMES,SIT1,NLRC3,CD244,KLRD1,SH2D1A,CCL5,XCL2,CST7,GFI1,KCNA3, PSTPIP1 (71).
Merck18 :PSMB10,HLA-DQA1,HLA-DRB1,CMKLR1,HLA-E, NKG7, CD8A,CCL5,CXCL9, CD27,CXCR6,IDO1,STAT1,TIGIT,LAG3,CD274,PDCD1LG2,CD276 (72).
CytolyticScore: PRF1, GZMA (73).
The signature score was calculated as the arithmetic mean of TPM of all genes in that signature for each sample. Comparisons of each signature score between groups (PD-L1 positive vs. PD-L1 negative) were done with the non-parametric Wilcoxon rank-sum test for the CM-025 cohort and paired Wilcoxon test for the 8 pairs cohort, respectively. All comparisons were two-sided with an alpha level of 0.05. All p values were corrected by Benjamini–Hochberg false discovery rate correction with q<0.25 considered significant.
CibersortX analysis
As previously described (56) the CIBERSORTx deconvolution algorithm (74) was used to infer immune cell infiltration from RNA-seq data, in absolute mode, using the LM22 signature, with B mode batch correction, quantile normalization disabled, and 1,000 permutations. All samples which had a p-value for deconvolution >0.05 were considered to have failed deconvolution and were therefore discarded from all downstream analyses. Relative cell proportions were obtained by normalizing the CIBERSORTx output to the sample-level sum of cell counts (to obtain percentages of immune infiltration). A constant of 10−6 was added to all proportions in order to allow the computation of immune cell ratios. All immune cell proportions and ratios were compared using a non-parametric Wilcoxon rank-sum test with Benjamini–Hochberg correction. Due to the low number of samples, we did not detect any immune cells in 8 pairs cohort that was significant at q<0.25. We instead used a p-value threshold of 0.05 for assessing statistical significance. Significant results, with q-values at the 0.05 and 0.25 thresholds in the CM-025 cohort are indicated by ** and * symbols, respectively.
Molecular subtypes analysis
Non-negative Matrix Factorization (NMF) on IMmotion 151 cohort: According to Motzer et al. (75), we selected 3072 genes (top 10%) with the highest variability across 823 tumors from IMmotion 151 cohort, using Median Absolute Deviation (MAD) analysis. Subclasses were then computed using consensus NMF clustering method (76) with k=7 factors to decompose the log2-transformed TPM matrix and evaluate the stability of the solutions using a cophenetic coefficient.
Random Forest Validation on CM-025 Cohort: To validate molecular subtypes derived in IMmotion151, we used the random forest machine learning algorithm (R package random- Forest) to derive a classifier and then predict the NMF clusters in an independent data set (CM-025). Before learning the random forest classifier, we preprocessed the data to generate the training set. We limited the gene expression matrix to genes common between CM-025 and the top 10% most variable genes in IMmotion151 (n = 2790). Next, we log2-transformed and z-score normalized the TPM matrix in each set to ensure that the test and training set were on the same scale. Finally, we trained the random forest classifier on the IMmotion151 cohort and then applied the training classifier to predict the NMF classes in CM-025 cohort. Since the 2790 genes common between IMmotion151 and CM-025 cohort did not include cluster 7 genes (SNORD-family genes), our CM-025 cohort did not identify the 7th cluster as described in (67).
For categorical variable analysis, Pearson’s Chi-squared test with continuity correction was used. Survival analyses were conducted using Cox-proportional hazard models, and p values were calculated using the log-rank test.
Endogenous Retrovirus (ERV) analysis
As previously described (56) we implemented a pipeline to quantify the expression of ERVs in RNA-seq data. Briefly, a reference set of 3,173 hERV sequences was first obtained from Vargiu et al. (77). Bowtie2 v2.3.4.3 (78) (Bowtie 2, RRID:SCR_016368) was used to align RNA-Seq FASTQ files for pairs to hg38 human transcriptome. All unmapped, single-end mapped, and ill-formed pair-end alignments to the human transcriptome were selected and aligned to hERV reference transcriptome using bowtie2. Mapped reads to hERV reference were kept, followed by filtering of single-end perfect matches to the human transcriptome. Finally, pair-end alignments with no more than one mismatch and single-end alignments with perfect matches to the hERV reference but not to human transcriptome were preserved. Duplicates in the final kept reads were removed using MarkDuplicates tool in Picard v2.19.0 (http://broadinstitute.github.io/picard) (Picard, RRID:SCR_006525) and then quantified using HTSeq (79) v0.11.0 (HTSeq, RRID:SCR_005514) with the settings (htseq-count --stranded=no --mode=union --secondary-alignments=score--supplementary-alignments=score --nonunique=all -a 0). Raw counts of paired-end and single-end alignments were added with different weights (pair-counts×2+single-counts) and then normalized to TPM. Differential expression analysis was performed on TPM values for each ERV between PD-L1 positive and PD-L1 negative samples using a non-parametric Wilcoxon rank-sum test for CM-025 and paired Wilcoxon test for 8 pairs. A p-value threshold of 0.05 was used for statistical significance.
Data availability statement
Relevant data are available from the corresponding authors and/or are included in the manuscript. Gene signature scores are available in Supplementary Table 1 for the 8 pairs and in Supplementary Table 4 for the CheckMate 025 cohort. Clinical data for the CheckMate 025 cohort are available in Supplementary Table 4. Differentially expressed genes between PD-L1 positive and negative samples, gene set enrichment scores, and immune deconvolution data (by CIBERSORTx), are available in Supplementary Tables 2, 3, 5, respectively, for both the 8 pairs and the CheckMate 025 cohort. Normalized RNA-seq expression data are available in Supplementary Table 6 for the 8 pairs and in Supplementary Table 7 for the CheckMate 025 cohort. ERV expression (inferred from RNA-seq data) are available in Supplementary Table 8 for the 8 pirs and in Supplementary Table 9 for the CheckMate 025 cohort.
Results
Pathologic features associated with tumor cell PD-L1 expression in paired samples
To identify potential drivers of PD-L1 expression in human ccRCC, we selected 8 patients with tumors displaying a highly heterogeneous expression of PD-L1 in tumor cells (TC), as assessed by immunohistochemistry. Specifically, these tumors were characterized by the coexistence of areas containing PD-L1-positive tumor cells with areas in which tumor cells were consistently PD-L1-negative. In the PD-L1-positive areas, the percentage of PD-L1-positive cells ranged from 15 to 95% (Figure 1).
Figure 1. Clear cell RCC with heterogeneous tumor cell PD-L1 expression and CD8 positive cell infiltration.
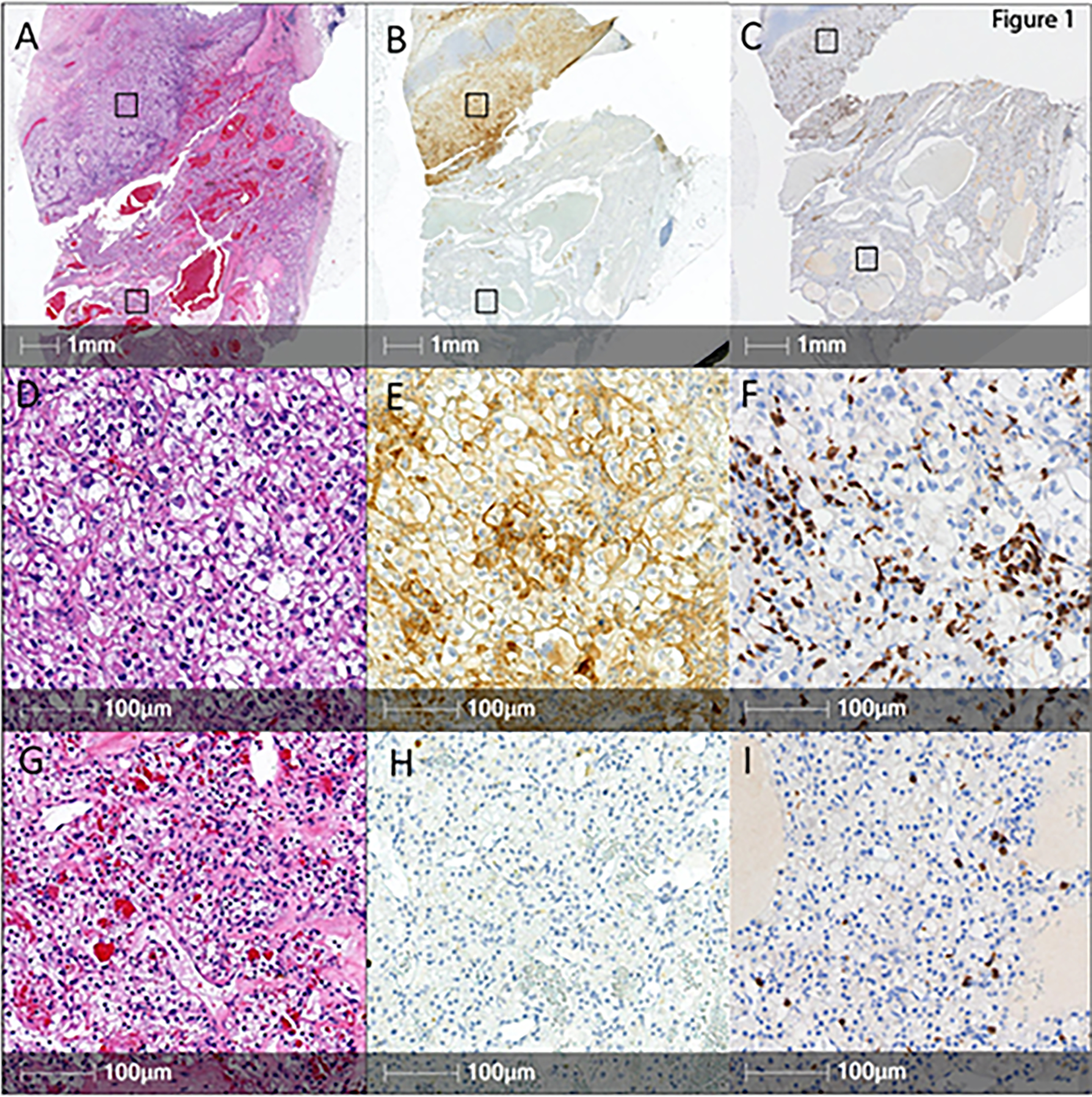
Representative low magnification images of tissue sections from a ccRCC stained with hematoxylin & eosin (A) or immunostained for PD-L1 (B) or for CD8 (C) (scale bars: 1mm). Higher magnifications of the selected PD-L1 positive (D, E, F) and PD-L1 negative (G, H, I) areas are also shown (scale bars: 100μm).
By analyzing individual tumors, we observed that in PD-L1-expressing areas, the WHO/ISUP nuclear grade was similar or higher compared to corresponding PD-L1-negative areas. Overall, 6/8 (75%) of tumors were WHO/ISUP grade 4 in the PD-L1 positive areas versus 1/8 (12.5%) in the PD-L1 negative areas. A sarcomatoid component was identified in 3 patients and was restricted to the PD-L1 positive area of the tumor in all cases (Table S1).
Analysis of tumor-infiltrating lymphocytes (TILs) revealed that the mean density of CD8+ TILs was 238 cells/mm2 in the PD-L1 positive areas (0–754 cells/mm2) and 81 cells/mm2 in the PD-L1 negative areas (5–395 cells/mm2). Paired sample comparison highlighted a strong trend in favor of a higher CD8+ TIL infiltration in PD-L1 positive areas (p= 0.063 with Wilcoxon).
Tumor cell PD-L1 expression is associated with combined overexpression of immune- and cell proliferation-related pathways
To better understand the mechanisms of PD-L1 regulation in tumor cells of ccRCC, we performed RNA sequencing and identified differentially expressed genes between TC PD-L1 positive and negative areas in the paired samples (Table S2). The analysis of the transcriptomic data using paired gene set enrichment analysis and the 50 Hallmark genes set from the Molecular Signatures Database showed a significant (FDR q value <0.25) overexpression of multiple oncogenic and inflammatory pathways (Figure 2A and Table S3). Most pathways (72%) upregulated in the PD-L1 positive regions continued to be upregulated on repeated analysis after removing the 3 pairs with sarcomatoid component (Figure S1A, B). Five pathways (45%) upregulated in the PD-L1 negative regions were shared between the 8 pairs and the 5 non-sarcomatoid pairs (Figure S1A, C); these included pathways involved in fatty acid metabolism, oxidative phosphorylation, NOTCH and WNT signaling, and a gene set correlated with inactive KRAS state (KRAS_DN).
Figure 2. Immune- and cell proliferation-related pathways are enriched in TC PD-L1 positive samples.
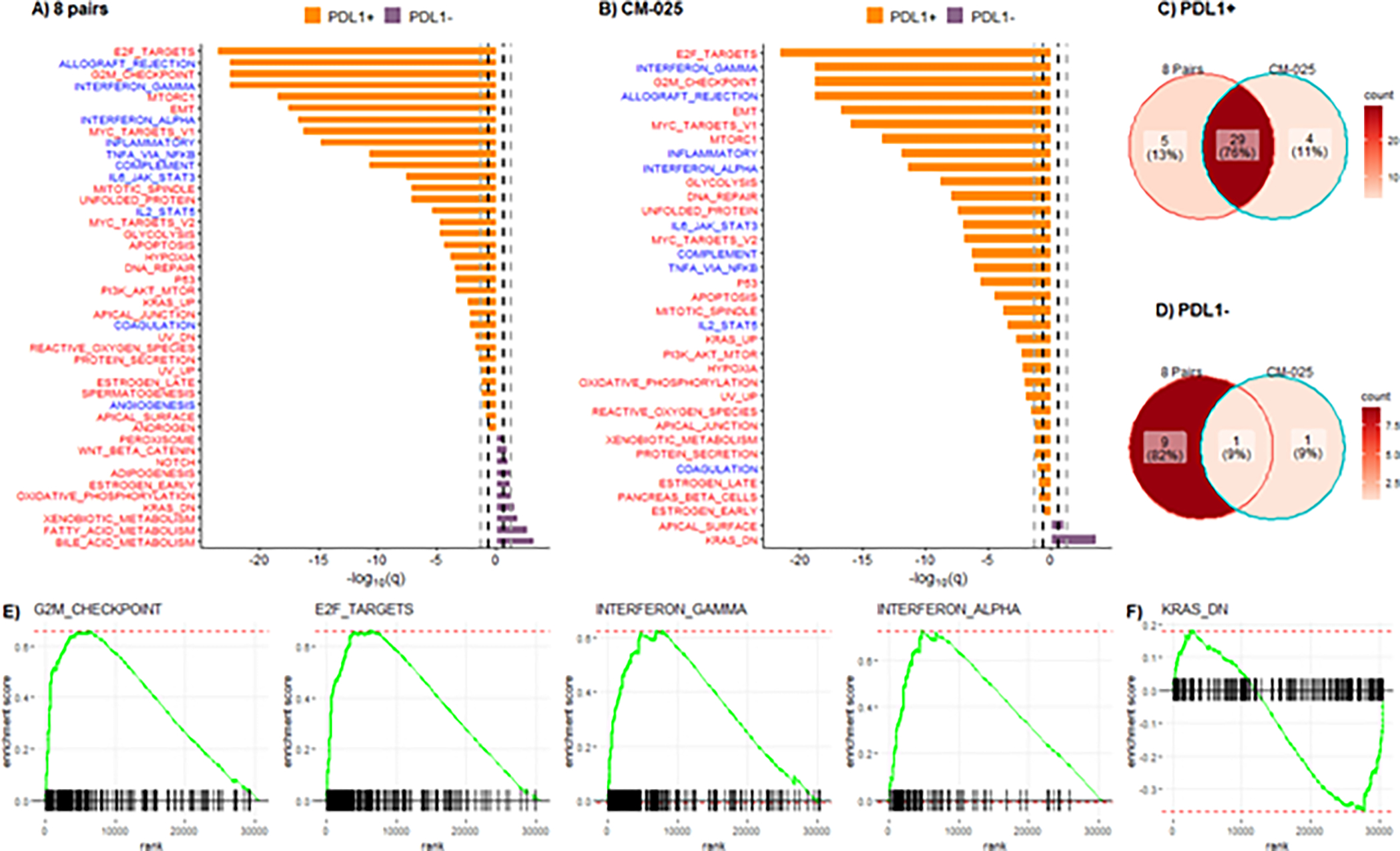
Hallmark pathways differentially expressed (q<0.25) between PD-L1 positive and PD-L1 negative samples in (A) 8 pairs and (B) CM-025 cohort. Red color pathways are tumor intrinsic and blue color pathways are tumor extrinsic. Bold black dashed lines indicate q<0.25 and gray dashed lines indicate q<0.05. Venn diagram of significant pathways (q<0.25) in the 8 pairs and CM-025 cohort that are enriched in (C) PD-L1 positive and (D) PD-L1 negative samples. Representative GSEA plots for common pathways between the 8 pairs and CM-025 cohort that are enriched in (E) PD-L1 positive and (F) PD-L1 negative samples.
To assess the robustness of our findings in an external dataset, we conducted the same analysis on a cohort of 232 pre-treatment tumors with known TC PD-L1 status (52 PD-L1 positive [16 WHO/ISUP grade 2/3, 36 grade 4] and 180 TC PD-L1 negative [136 WHO/ISUP grade 2/3, 44 grade 4]) from patients with metastatic ccRCC who received either the anti-PD-1 antibody nivolumab or the mTOR inhibitor everolimus (Table S4), as part of the CM-025 trial (Figure 2B). Twenty-nine Hallmark pathways were upregulated in both the PD-L1 positive areas of the discovery cohort and the PD-L1 positive tumors of the CM-025 validation cohort (Figure 2C). These pathways could broadly be classified into two classes: (i) immune-related pathways (e.g. IFN-α and IFN-γ responses, allograft rejection, TNF-α), and (ii) cell-intrinsic pathways involved in cell proliferation (e.g., G2M checkpoint, E2F targets, MYC targets), and other oncogenic processes (e.g. mTOR signaling and the epithelial-mesenchymal transition) (Figure 2A, B). Only 5 pathways upregulated in the context of TC PD-L1 expression were unique to the 8 paired samples, and four others were unique to the CM-025 samples (Figure 2C). Downmodulation of KRAS signaling (KRAS_DN) was the only Hallmark pathway significantly upregulated in PD-L1 negative samples of both patient cohorts (Figures 2D). Representative GSEA plots for common pathways between the two cohorts are shown in Figures 2E–F.
We then calculated ssGSEA scores for each of the 29 pathways commonly upregulated in PD-L1 positive samples to evaluate the relative level of activity of each pathway in individual tumors of the CM-025 cohort. A graphical representation of the ssGSEA scores sorted by TC PD-L1 levels showed that most TC PD-L1 positive tumors displayed combined overexpression of both immune-related and cell proliferation-related pathways (Figure S2).
Since both this study and prior investigations by our group and others demonstrated that TC PD-L1 expression is positively associated with tumor grade in ccRCC, we evaluated differential gene expression between PD-L1 negative and positive samples without the interference of grade, by repeating the analysis in the subset of WHO/ISUP grade 4 tumors from the CM-025 cohort (44 PD-L1 negative, 36 PD-L1 positive) (Table S4). Twenty-seven pathways that were found to be upregulated in PD-L1 positive tumors within the grade 4 subset were consistent with those obtained by the analysis of the entire CM-025 cohort (Figure S3A, S3C). In addition, 2 pathways upregulated in PD-L1 negative tumors (including downmodulation of KRAS signaling) were identified in both the grade 4 subset and the entire CM-025 cohort (Figure S3A, S3D). We also studied the subset of tumors with WHO/ISUP grade 2 or 3 (16 PD-L1 positive, 136 PD-L1 negative), although the small number of PD-L1 positive samples in this group likely limited the power of the analysis. Nevertheless, we found that signatures of mTOR signaling and immune response were still significantly overexpressed in the PD-L1 positive tumors. Downmodulation of KRAS signaling pathway remained significantly overexpressed in the PD-L1 negative tumors (Figure S3B, S3D).
A molecular RCC subtype characterized by combined overexpression of immune- and cell proliferation-related pathways shows improved response to anti-PD-1 monotherapy
A recent analysis of the IMmotion 151 trial in patients with advanced ccRCC identified transcriptionally defined ccRCC molecular subtypes (clusters) with improved outcomes to the anti-PD-L1 antibody atezolizumab in combination with the anti-VEGF antibody bevacizumab relative to the VEGF receptor inhibitor sunitinib (75). Of note, one of the subtypes associated with an improved outcome to ICI, the T effector/proliferative subtype (cluster 4), was characterized by high levels of PD-L1 expression on immune cells and showed overexpression of inflammatory and cell cycle pathways (75) significantly overlapping with those found to be upregulated in association with TC PD-L1 expression in our datasets (Figure 2). When we applied the same clustering algorithm to the CM-025 cohort, we observed that tumors expressing PD-L1 on tumor cells were enriched in the T effector/proliferative subtype (cluster 4) (Figure 3A, B). Moreover, patients in cluster 4 had improved PFS on nivolumab relative to everolimus (HR 0.32; 95% CI 0.13–0.83) (Figure 3C, D). In addition, 30% of patients in cluster 4 treated with nivolumab achieved CR or PR, compared to 0% of the patients treated with everolimus (p=0.04) (Figure S6). It should be noted that TC PD-L1 expression was not by itself significantly associated with clinical outcome in the CM-025 cohort (Figure S4).
Figure 3. A molecular subtype of RCC characterized by overexpression of immune- and cell proliferation-related pathways (cluster 4) shows significantly improved PFS on nivolumab relative to everolimus in the CM-025 cohort.
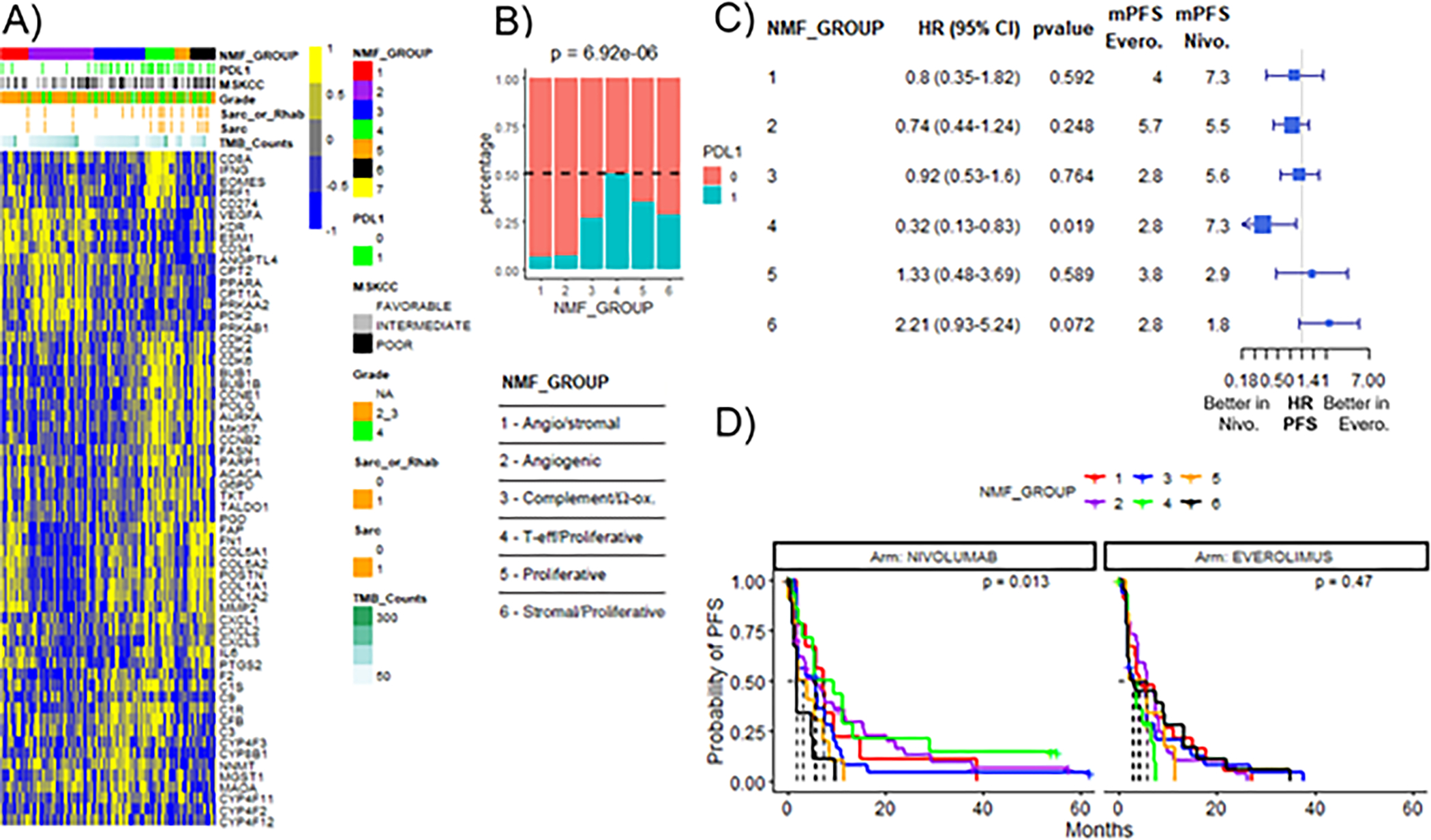
A) Heatmap of genes included in transcriptional signatures. Samples are grouped by molecular subtypes (NMF_GROUP) determined by NMF clustering according to Motzer et al (75). The number of patients in each NMF group is: 1=31; 2=72; 3=56; 4=32; 5=17; 6=28. B) TC PD-L1 expression by immunohistochemistry in each NMF_GROUP. p value was obtained from Pearson’s chi-square test. C) Forest plots for PFS hazard ratios by NMF_GROUP between patients treated with everolimus (Evero) versus nivolumab (Nivo). mPFS: median PFS. D) Kaplan-Meier curves of PFS by NMF_GROUP in the nivolumab or everolimus treatment arm. p value was calculated using log-rank test.
Tumor cell PD-L1 expression is associated with higher levels of T-cell activation
Recent studies of ccRCC transcriptomic datasets have identified gene signatures of T-cell activation that predict favorable outcomes to ICI relative to targeted therapies and an angiogenesis signature associated with improved outcome in patients treated with a VEGFR-tyrosine kinase inhibitor (i.e., sunitinib). Here we investigated the link between T cell activity and tumor cell PD-L1 expression by comparing signature scores in PD-L1 positive and negative samples.
In the 8 paired samples, levels of T-cell activation signatures (IMmotion 150 Teff signature, the JAVELIN signature, the Merck18 signature, and the Cytolytic signature) were numerically higher in the PD-L1 positive tumor areas with statistical significance (q<0.25). The IMmotion 150 angiogenesis signature was significantly enriched in the PD-L1 negative tumor areas (q<0.25) (Figure 4A). In the CM-025 cohort, all four T-cell activation signatures were significantly upregulated in the PD-L1 positive tumors (q<0.25). No signature was significantly upregulated in the PD-L1 negative tumors (Figure 4A).
Figure 4. TC PD-L1 positive samples are characterized by higher levels of both T-cell activity and tumor-infiltrating immune cells.
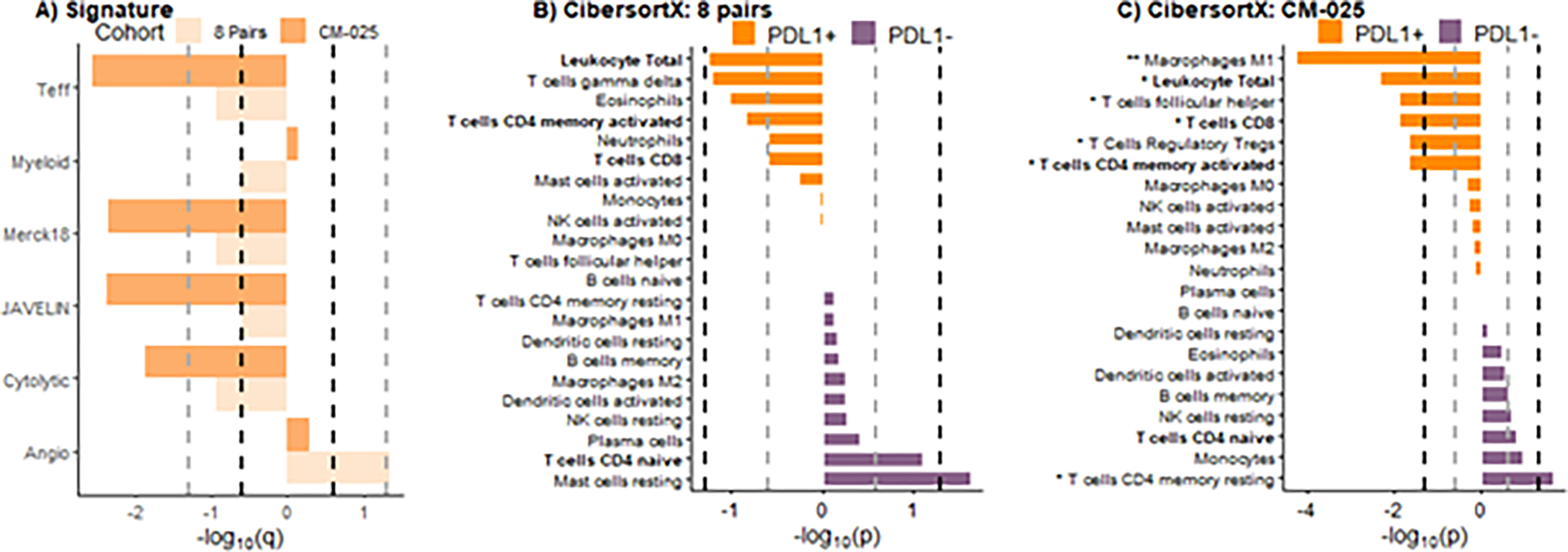
A) Barplot of gene signatures between two cohorts (8 pairs: light orange bars; CM-025 cohort: orange bars). Bold black dashed lines indicate statistical significance with q<0.25 and gray dashed lines indicate q<0.05. CibersortX p values of all immune cells distribution between PD-L1 positive and PD-L1 negative samples for (B) 8 pairs and (C) CM-025 cohort. Bold black dashed lines indicate statistical significance with p<0.05 and gray dashed lines indicate p<0.25. ** indicates q<0.05, * indicates q<0.25.
Tumor cell PD-L1 expression is associated with higher levels of tumor-infiltrating immune cells
To gain insights into the composition of the tumor microenvironment of ccRCCs expressing PD-L1 on cancer cells, we performed RNAseq data deconvolution using CibersortX (Table S5). In the 8 paired samples, we observed that levels of total leukocytes, gamma delta T cells, eosinophils, CD4 memory activated T cells, neutrophils, and CD8 T cells were numerically overrepresented in the TC PD-L1 positive areas but without reaching the statistical significance (p<0.05, q<0.25). Levels of T cells CD4 naïve (p<0.25) and mast cells resting (p<0.05) were numerically more prominent in the PD-L1 negative areas (Figure 4B).
In the CM-025 cohort, M1 macrophages, total leukocytes, follicular helper T cell, CD8 T cell, regulatory T cells (Tregs), and CD4 memory activated T cells were significantly (p<0.05) overrepresented in the TC PD-L1 positive tumors. CD4 memory resting T cells were significantly overrepresented in the PD-L1 negative tumors (p<0.05) (Figure 4C).
Endogenous Retroviruses (ERVs) are differentially expressed between PD-L1 positive and negative tumor samples
There is evidence that ERVs can elicit an anti-tumor immune response and may predict response to ICI in ccRCC (57,80,81). Using RNA sequencing data, we explored the expression of endogenous retroviruses in relation to the TC PD-L1 status.
Analysis of the 8 paired samples revealed that 34 ERVs were differentially expressed between PD-L1 positive and negative regions (p<0.05 and log2 fold change >1) with nine ERVs overexpressed in the PD-L1 positive areas and 25 in the PD-L1 negative ones (Figure S5A, S5C).
In the CM-025 cohort, 55 ERVs were significantly overexpressed in the PD-L1 positive tumors and 190 in the PD-L1 negative ones (Figure S5B, S5D). Only one ERV was found to be overexpressed in the PD-L1 positive samples of both the 8 pairs and the CM-025 cohort (ERVH-7 or 4185_chr14:76044307–76053991) (Figure S5C). Six ERVs were overexpressed in PD-L1 negative samples in both cohorts (ERVH-6 or 1313_chr4:9640061–9648771; ERVFRD-2 or 1812_chr5:37086510–37094092; ERVH-1 or 3250_chr10:53788498–53799321; ERV9-1 or 3199_chr10:17052206–17060764; ERVH-4 or 3081_chr9:82752251–82760393; ERVH-2 or 505_chr2:35026065–35031792) (Figure S5D).
Discussion
Prior studies have demonstrated that several signal transduction pathways can individually upregulate PD-L1 expression in mouse models and human cell lines of multiple tumor types, including melanoma, prostate, colon, pancreas, breast, and sarcomas (39–41). Through the analysis of patient samples and paired tumor areas, we have shown that both tumor cell-intrinsic and -extrinsic mechanisms are likely intertwined in the regulation of TC PD-L1 expression in ccRCC.
By conducting a controlled study focused on the comparison of transcriptomic data obtained from PD-L1 positive and PD-L1 negative areas isolated from individual ccRCC tumors, we found that TC PD-L1 expression was associated with increased inflammatory signals mediated by cytokines (e.g. IFN-α and IFN-γ responses, allograft rejection, TNF-α) as well as enhanced cell cycle and oncogenic pathways (e.g. G2M checkpoint, E2F targets, MYC targets, mTOR signaling). Importantly, these findings were confirmed through the analysis of an independent cohort of patients from a phase III clinical trial of nivolumab versus everolimus in metastatic ccRCC (CM-025). In line with the observation that cytokine signaling correlated with upregulation of PD-L1 in cancer cells, we found that T cell activation signature levels as well as levels of tumor infiltrating immune cells were higher in TC PD-L1 positive samples. One possible limitation of these findings is that in ccRCC, high nuclear grade is known to be associated with both TC PD-L1 expression (4) and increased levels of inflammation (82) and cell proliferation (83) (found to be associated with TC PD-L1 expression in this study), raising the concern that it might represent a confounding variable. However, by performing a sensitivity analysis in the grade 4 or grade 2/3 tumor subsets of the CM-025 cohort, we obtained results largely overlapping with those observed in the entire cohort, suggesting that the correlates of TC PD-L1 expression identified by our investigation are independent of tumor grade.
There is substantial evidence that signaling through several cytokines (e.g. IFN-ɣ, IFN-α, TNF-α, IL-6 and others) can induce expression of PD-L1 in cancer cells (39–41). In line with recent findings from Banchereau and colleagues (54), our results suggest that cell-extrinsic immune-related signals are likely critical for TC PD-L1 regulation in ccRCC tumors. On the other hand, several tumor cell-intrinsic oncogenic signals that were found to be consistently associated with TC PD-L1 expression in our study (e.g. MYC and PI3K-AKT-mTOR) have also been shown to regulate PD-L1 protein levels in preclinical models of various tumor types (39–41). Of note, Messai and colleagues recently reported that PD-L1 represents a direct target of HIF-2α in ccRCC cell lines (52). In our study, the hypoxia pathway was generally upregulated in TC PD-L1 positive samples (with the exception of the G2/3 sample subset). However, the lack of a specific gene signature of HIF-2α activity in ccRCC limits the robustness of our analysis. In summary, in the context of previously published data, our results raise the possibility that cell-extrinsic signaling through cytokines (largely released by immune cells) are not solely responsible for PD-L1 expression in ccRCC cancer cells, which might require concomitant tumor cell-intrinsic pro-oncogenic signals. Importantly, however, some of the pathways found to be associated with TC PD-L1 expression and considered to be tumor cell-intrinsic in this study (e.g. glycolysis, mitotic spindle, fatty acid metabolism) have been recently reported to be upregulated in immune cells (CD8 positive T cells) in the RCC microenvironment (84). Therefore, in absence of single cell RNA seq data, it is not possible to determine whether these transcriptional programs are activated in the tumor cells and/or in the tumor-infiltrating immune cells. In conclusion, further analyses of ccRCC model systems are needed to formally test the hypothesis that tumor cell-intrinsic pathways play a central role in the is regulation of PD-L1 expression in ccRCC.
The anti-PD-1 antibody nivolumab is approved for the treatment of patients with metastatic ccRCC as monotherapy (after VEGFR inhibitor failure) and in combination with the anti-CTLA4 antibody ipilimumab or the multi-kinase inhibitor cabozantinib (as frontline therapies). PD-L1 expression in tumor cells is currently an FDA-approved biomarker for nivolumab-based therapies in other tumor types, including NSCLC (85) and Head and Neck Squamous Cell Carcinoma (86). Yet, in the context of ccRCC, its predictive value remains uncertain (3,87). Since accurate assessment of PD-L1 expression by immunohistochemistry is particularly challenging in ccRCC (4,88,89), the transcriptomic correlates of TC PD-L1 expression identified here not only provide insights into ccRCC biology the but might facilitate the development of more robust and clinically significant biomarkers for ICI response in this tumor type. Motzer and colleagues recently proposed a ccRCC classification system based on analysis of transcriptomic data and identified tumor subtypes with improved response to atezolizumab (anti PD-L1) in combination with bevacizumab (anti-VEGF) versus sunitinib (VEGFR inhibitor) in the IMmotion 151 trial (75). We noticed that the transcriptomic features of one of the subtypes associated with better outcome on atezolizumab plus bevacizumab (the T effector/proliferative subtype, cluster 4) largely overlapped with those of TC PD-L1 positive samples in our study. Indeed, cluster 4 displayed significant upregulation of both immune (T-effector, JAK/STAT, and INF-α and -ɣ) and cell cycle (G2M, E2F targets, MYC targets) transcriptional programs. As predicted, when we classified tumors from the CM-025 trial using the clustering algorithm from Motzer et al, we found that cluster 4 was enriched for tumors expressing PD-L1 on tumor cells. Of note, TC PD-L1 expression itself was not associated with clinical outcome in the CM-025 trial, either in the entire patient population (55) or in the subset of patients analyzed here. In contrast, our analysis showed that patients in cluster 4 displayed improved ORR and PFS on nivolumab relative to everolimus. These findings suggest that, compared to TC PD-L1 status, co-expression of immune and cell cycle gene signatures (i.e. transcriptomic correlates of PD-L1 expression) may provide better predictive value for response to anti-PD1-based immunotherapy in metastatic ccRCC. Additional investigations of clinical trial samples (especially in the frontline setting) are warranted to confirm our results. Interestingly, a recent analysis of RNA-seq data from the CheckMate 214 trial showed that overexpression of several gene sets associated with TC PD-L1 expression in our study (Apoptosis, Reactive Oxygen Species Pathway, IL-6/JAK/STAT3 Signaling, Allograft Rejection, and Inflammatory Response) was associated with longer PFS in patients treated with nivolumab plus ipilimumab but with shorter PFS in patients treated with sunitinib (90)
Through the analysis of paired samples, we have shown that ccRCC tumors can harbor PD-L1 positive and PD-L1 negative tumor regions that are characterized by distinct transcriptional states. It should be noted that this intratumoral heterogeneity represents a major challenge for the development of clinically meaningful biomarkers of response to immune checkpoint inhibitors in this tumor type.
It has been hypothesized that in ccRCC, aberrantly expressed ERVs might represent tumor antigens that are recognized by tumor infiltrating immune cells. Indeed, ERV expression levels have been shown to correlate with levels of intratumoral immune cells, cytolytic activity, and immune checkpoint molecules and have been associated with response to ICI (57,73,91,92). Here we attempted to identify ERVs upregulated in tumor samples expressing PD-L1 in the tumor cells. Only one ERV (ERVH-7) was overexpressed in PD-L1 positive samples of both cohorts. The function of ERVH-7 in tumor biology is unexplored and further investigation is necessary to determine if ERVH-7 plays a role in eliciting anti-tumor immunity in ccRCC. Please note that none of the ERVs overexpressed in either PD-L1 positive or PD-L1 negative samples of both cohorts were found to be associated with clinical outcome to anti-PD1 therapy in a prior study from our group (56).
Our study has some limitations. First, the discovery cohort included only a small number of PD-L1 positive and negative paired samples. Second, although we utilized samples from a prospective clinical trial, this was an ad hoc analysis that should be considered exploratory in nature. Third, tumor samples from the CM-025 trial were largely collected from surgeries (mostly nephrectomies) that took place before the patients received any systemic therapy. Since patients were enrolled in the trial after failing VEGFR TKIs, the tumor tissues that we analyzed might not entirely be representative of VEGFR TKI resistant metastatic lesions that were targeted by the treatment with nivolumab or everolimus. As a consequence, our findings need to be further investigated in the context of patient cohorts that have received front-line anti-PD-1-based therapy. Fourth, the ERV analysis that we performed is highly exploratory and the clinical significance of the findings is uncertain.
In conclusion, our results suggest that PD-L1 regulation in ccRCC cells is a complex process involving multiple tumor cell-intrinsic and -extrinsic pathways, which might act in an interconnected and synergistic fashion. The quantitation of these transcriptional programs might better predict benefit from anti-PD-1 based therapy compared to TC PD-L1 expression alone in metastatic ccRCC.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Statement of translation relevance.
The anti-PD-1 antibody nivolumab is approved, either alone or in combination with other agents, for the treatment of patients with metastatic clear cell renal cell carcinoma (ccRCC). While tumor cell (TC) PD-L1 expression by immunohistochemistry is a biomarker of response to nivolumab-based therapies in some tumor types, its clinical utility in ccRCC is uncertain. By analyzing paired samples from individual tumors as well as tissue specimens from a randomized phase 3 clinical trial of nivolumab versus everolimus (CheckMate 025), we demonstrated that in ccRCC, PD-L1 expression in TC is associated with increased activity of both immune- and cell-proliferation-related pathways and that a molecular subtype of RCC characterized by combined overexpression of these pathways shows better outcome to nivolumab relative to everolimus. Our findings indicate that the quantitation of transcriptional programs correlated with TC PD-L1 expression may be a clinically useful predictor of response to anti-PD-1-based therapy in metastatic ccRCC.
Acknowledgments of research support for the study:
This work was supported by Dana-Farber / Harvard Cancer Center Kidney Cancer SPORE (P50-CA101942-12), DOD CDMRP (W81XWH-18-1-0480 and W81XWH-18-1-0366), and Bristol-Myers Squibb. S.S.,T.K.C., and D.F.M. are supported in part by the Dana-Farber/Harvard Cancer Center Kidney Program (P30 CA06516). M.B.A. is supported in part by the Georgetown-Lombardi Comprehensive Cancer Center CCSG (P30 CA051008) and the William M. Scholl Foundation. T.K.C. is supported in part by the Kohlberg Chair at Harvard Medical School and the Trust Family, Michael Brigham, and Loker Pinard Funds for Kidney Cancer Research at Dana-Farber Cancer Institute. Patients treated at Memorial Sloan Kettering Cancer Center were supported in part by Memorial Sloan Kettering Cancer Center Support Grant/Core Grant (P30 CA008748)
Footnotes
Disclosures:
S.S. reports receiving commercial research grants from Bristol-Myers Squibb, AstraZeneca, Exelixis and Novartis; is a consultant/advisory board member for Merck, AstraZeneca, Bristol-Myers Squibb, CRISPR Therapeutics AG, AACR, and NCI; receives royalties from Biogenex; and mentored several non-US citizens on research projects with potential funding (in part) from non-US sources/Foreign Components.
M.B.A reports an advisory role for Bristol-Myers Squibb, Merck, Novartis, Eisai, Aveo, Pfizer, Werewolf, Fathom, Pneuma, Leads, Pyxis Oncology, PACT, Elpis, X4Pharma, ValoHealth, ScholarRock, Surface, Takeda, Simcha, Roche, SAB Bio and GSK and a consultant role: Bristol-Myers Squibb, Merck, Novartis, Pfizer, Roche, Exelixis, Iovance, COTA, Idera, Agenus, Apexigen, Asher Bio, Neoleukin, AstraZeneca, Calithera, SeaGen, and Sanofi. He reports research support to his institution from Bristol-Myers Squibb, Merck and Pfizer. He holds stock/stock options in Pyxis Oncology, Werewolf and Elpis.
G.J.F. has patents/pending royalties on the PD-1/PD-L1 pathway from Roche, Merck MSD, Bristol-Myers-Squibb, Merck KGA, Boehringer-Ingelheim, AstraZeneca, Dako, Leica, Mayo Clinic, and Novartis. GJF has served on advisory boards for Roche, Bristol-Myers-Squibb, Xios, Origimed, Triursus, iTeos, NextPoint, IgM, Jubilant, Trillium, GV20, IOME, and Geode. GJF has equity in Nextpoint, Triursus, Xios, iTeos, IgM, Trillium, Invaria, GV20, and Geode.
D.A.B. reports personal fees from LM Education and Exchange, Adnovate Strategies, MDedge, Cancer Network, Cancer Expert Now, OncLive, Catenion, AVEO, and grants and personal fees from Exelixis, outside the submitted work.
R.J.M. reports grants and personal fees from Pfizer, grants and personal fees from Eisai, grants and personal fees from Aveo Pharmaceuticals, grants and personal fees from Exelixis, grants and fees from Merck, grants and personal fees from Genentech, personal fees from Incyte, grants and personal fees from Roche, grants from Bristol Myers Squibb, personal fees from Astra Zeneca
J-C.P. is employed by Jounce Therapeutics.
T.K.C. reports institutional and personal, for research, advisory boards, consultancy, and honoraria (paid and unpaid support ) from: AstraZeneca, Aravive, Aveo, Bayer, Bristol Myers-Squibb, Calithera, Circle Pharma, Eisai, EMD Serono, Exelixis, GlaxoSmithKline, IQVA, Infinity, Ipsen, Jansen, Kanaph, Lilly, Merck, Nikang, Nuscan, Novartis, Pfizer, Roche, Sanofi/Aventis, Surface Oncology, Takeda, Tempest, Up-To-Date, CME events (Peerview, OncLive, MJH and others), outside the submitted work. Institutional patents filed on molecular mutations and immunotherapy response, and ctDNA. Equity: Tempest, Pionyr, Osel, NuscanDx. Committees: NCCN, GU Steering Committee, ASCO/ESMO, ACCRU, KidneyCan. Medical writing and editorial assistance support may have been funded by Communications companies in part. No speaker’s bureau. Mentored several non-US citizens on research projects with potential funding (in part) from non-US sources/Foreign Components. The institution (Dana-Farber Cancer Institute) may have received additional independent funding of drug companies or/and royalties potentially involved in research around the subject matter.
D.F.M. received honoraria for consulting from BMS, Pfizer, Merck, Alkermes, Inc., EMD Serono, Eli Lilly and Company, Iovance, Eisai Inc., Werewolf Therapeutics, Calithera Biosciences, Synthekine, Inc., Johnson & Johnson, Aveo. D.F.M. received research support from BMS, Merck, Genentech, Pfizer, Exelixis, X4 Pharma, Alkermes, Inc., Checkmate Pharmaceuticals, CRISPR Therapeutics.
C.J.W. holds equity from BioNTech, Inc. and receives research funding from Pharmacyclics., S.A.S. reports nonfinancial support from Bristol-Myers Squibb, and equity in Agenus Inc., Agios Pharmaceuticals, Breakbio Corp., Bristol-Myers Squibb and Lumos Pharma
T.D., Y.H., E.W., D.J.W. declare no conflict of interest
References
- 1.Choueiri TK, Atkins MB, Bakouny Z, Carlo MI, Drake CG, Jonasch E, et al. Summary from the First Kidney Cancer Research Summit, September 12–13, 2019: A Focus on Translational Research. J Natl Cancer Inst. 2021;113:234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choueiri TK, Albiges L, Atkins MB, Bakouny Z, Bratslavsky G, Braun DA, et al. From Basic Science to Clinical Translation in Kidney Cancer: A Report from the Second Kidney Cancer Research Summit. Clin Cancer Res. 2021;1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mori K, Abufaraj M, Mostafaei H, Quhal F, Fajkovic H, Remzi M, et al. The Predictive Value of Programmed Death Ligand 1 in Patients with Metastatic Renal Cell Carcinoma Treated with Immune-checkpoint Inhibitors: A Systematic Review and Meta-analysis. Eur Urol. 2021;79:783–92. [DOI] [PubMed] [Google Scholar]
- 4.Callea M, Albiges L, Gupta M, Cheng SC, Genega EM, Fay AP, et al. Differential expression of PD-L1 between primary and metastatic sites in clear-cell renal cell carcinoma. Cancer Immunol Res. 2015;3:1158–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakazawa A, Dotan I, Brimnes J, Allez M, Shao L, Tsushima F, et al. The Expression and Function of Costimulatory Molecules B7h and B7-H1 on Colonic Epithelial Cells. Gastroenterology. 2004;126:1347–57. [DOI] [PubMed] [Google Scholar]
- 6.Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, Hersey P. Inducible but Not Constitutive Expression of PD-L1 in Human Melanoma Cells Is Dependent on Activation of NF-κB. Cheriyath V, editor. PLoS One. 2015;10:e0123410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8:793–800. [DOI] [PubMed] [Google Scholar]
- 8.Lamano JB, Lamano JB, Li YD, DiDomenico JD, Choy W, Veliceasa D, et al. Glioblastoma-derived IL6 induces immunosuppressive peripheral myeloid cell PD-L1 and promotes tumor growth. Clin Cancer Res. 2019;25:3643–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun L, Wang Q, Chen B, Zhao Y, Shen B, Wang H, et al. Gastric cancer mesenchymal stem cells derived IL-8 induces PD-L1 expression in gastric cancer cells via STAT3/mTOR-c-Myc signal axis. Cell Death Dis. 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Yang L, Huang F, Zhang Q, Liu S, Ma L, et al. Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol Lett. 2017;184:7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carbotti G, Barisione G, Airoldi I, Mezzanzanica D, Bagnoli M, Ferrero S, et al. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget. 2015;6:43267–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen S, Crabill GA, Pritchard TS, McMiller TL, Wei P, Pardoll DM, et al. Mechanisms regulating PD-L1 expression on tumor and immune cells. J Immunother Cancer. 2019;7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The Activation of MAPK in Melanoma Cells Resistant to BRAF Inhibition Promotes PD-L1 Expression That Is Reversible by MEK and PI3K Inhibition. Clin Cancer Res. 2013;19:598–609. [DOI] [PubMed] [Google Scholar]
- 14.Bu LL, Yu GT, Wu L, Mao L, Deng WW, Liu JF, et al. STAT3 Induces Immunosuppression by Upregulating PD-1/PD-L1 in HNSCC. J Dent Res. 2017;96:1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bouillez A, Rajabi H, Jin C, Samur M, Tagde A, Alam M, et al. MUC1-C integrates PD-L1 induction with repression of immune effectors in non-small-cell lung cancer. Oncogene. 2017;36:4037–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue J, Chen C, Qi M, Huang Y, Wang L, Gao Y, et al. Type Iγ phosphatidylinositol phosphate kinase regulates PD-L1 expression by activating NF-κB. Oncotarget. 2017;8:42414–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O’Donnell E, et al. Constitutive AP-1 Activity and EBV Infection Induce PD-L1 in Hodgkin Lymphomas and Posttransplant Lymphoproliferative Disorders: Implications for Targeted Therapy. Clin Cancer Res. 2012;18:1611–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.David JM, Dominguez C, McCampbell KK, Gulley JL, Schlom J, Palena C. A novel bifunctional anti-PD-L1/TGF-β Trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. Oncoimmunology. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science (80- ). 2016;352:227–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atsaves V, Tsesmetzis N, Chioureas D, Kis L, Leventaki V, Drakos E, et al. PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia. 2017;31:1633–7. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Jia Y, Zhao S, Zhang X, Wang X, Han X, et al. BIN1 reverses PD-L1-mediated immune escape by inactivating the c-MYC and EGFR/MAPK signaling pathways in non-small cell lung cancer. Oncogene. 2017;36:6235–43. [DOI] [PubMed] [Google Scholar]
- 22.Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A Mechanism of Hypoxia-Mediated Escape from Adaptive Immunity in Cancer Cells. Cancer Res. 2014;74:665–74. [DOI] [PubMed] [Google Scholar]
- 23.Guo R, Li Y, Bai H, Wang J. KRAS mutants to regulate PD-L1 expression through NF-ƘB and HIF-1α pathways in non-small cell lung cancer cells. J Clin Oncol. 2017;35:e20049–e20049. [Google Scholar]
- 24.Kim MH, Kim CG, Kim SK, Shin SJ, Shin EC, Park SH, et al. YAP-induced PD-L1 expression drives immune evasion in BRAFi-resistant melanoma. Cancer Immunol Res. 2018;6:255–66. [DOI] [PubMed] [Google Scholar]
- 25.Song M, Chen D, Lu B, Wang C, Zhang J, Huang L, et al. PTEN Loss Increases PD-L1 Protein Expression and Affects the Correlation between PD-L1 Expression and Clinical Parameters in Colorectal Cancer. Sun J, editor. PLoS One. 2013;8:e65821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Atefi M, Avramis E, Lassen A, Wong DJL, Robert L, Foulad D, et al. Effects of MAPK and PI3K Pathways on PD-L1 Expression in Melanoma. Clin Cancer Res. 2014;20:3446–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lastwika KJ, Wilson W, Li QK, Norris J, Xu H, Ghazarian SR, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016;76:227–38. [DOI] [PubMed] [Google Scholar]
- 28.Papalexi E, Mimitou EP, Butler AW, Foster S, Bracken B, Mauck WM, et al. Characterizing the molecular regulation of inhibitory immune checkpoints with multimodal single-cell screens. Nat Genet. 2021;53:322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sumimoto H, Takano A, Teramoto K, Daigo Y. RAS–Mitogen-Activated Protein Kinase Signal Is Required for Enhanced PD-L1 Expression in Human Lung Cancers. Chellappan S, editor. PLoS One. 2016;11:e0166626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Coelho MA, de Carné Trécesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity. 2017;47:1083–1099.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen N, Fang W, Lin Z, Peng P, Wang J, Zhan J, et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol Immunother. 2017;66:1175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 Pathway Contributes to Immune Escape in EGFR-Driven Lung Tumors. Cancer Discov. 2013;3:1355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J Thorac Oncol. 2015;10:910–23. [DOI] [PubMed] [Google Scholar]
- 34.Zhang N, Zeng Y, Du W, Zhu J, Shen D, Liu Z, et al. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol. 2016;49:1360–8. [DOI] [PubMed] [Google Scholar]
- 35.Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A. 2008;105:20852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamamoto R, Nishikori M, Tashima M, Sakai T, Ichinohe T, Takaori-Kondo A, et al. B7-H1 expression is regulated by MEK/ERK signaling pathway in anaplastic large cell lymphoma and Hodgkin lymphoma. Cancer Sci. 2009;100:2093–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, et al. Induction of PD-L1 Expression by the EML4–ALK Oncoprotein and Downstream Signaling Pathways in Non–Small Cell Lung Cancer. Clin Cancer Res. 2015;21:4014–21. [DOI] [PubMed] [Google Scholar]
- 38.Koh J, Jang J, Keam B, Kim S, Kim M-Y, Go H, et al. EML4-ALK enhances programmed cell death-ligand 1 expression in pulmonary adenocarcinoma via hypoxia-inducible factor (HIF)-1α and STAT3. Oncoimmunology. 2016;5:e1108514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun C, Mezzadra R, Schumacher TN. Regulation and Function of the PD-L1 Checkpoint. Immunity. 2018;48:434–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lucibello G, Mograbi B, Milano G, Hofman P, Brest P. PD-L1 regulation revisited: impact on immunotherapeutic strategies. Trends Mol Med. 2021;27:868–81. [DOI] [PubMed] [Google Scholar]
- 41.Farrukh H, El-Sayes N, Mossman K. Mechanisms of PD-L1 regulation in malignant and virus-infected cells. Int J Mol Sci. 2021;22:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Twa DDW, Chan FC, Ben-Neriah S, Woolcock BW, Mottok A, Tan KL, et al. Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood. 2014;123:2062–5. [DOI] [PubMed] [Google Scholar]
- 44.Ikeda S, Okamoto T, Okano S, Umemoto Y, Tagawa T, Morodomi Y, et al. PD-L1 Is Upregulated by Simultaneous Amplification of the PD-L1 and JAK2 Genes in Non–Small Cell Lung Cancer. J Thorac Oncol. 2016;11:62–71. [DOI] [PubMed] [Google Scholar]
- 45.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta S, Cheville JC, Jungbluth AA, Zhang Y, Zhang L, Chen YB, et al. JAK2/PD-L1/PD-L2 (9p24.1) amplifications in renal cell carcinomas with sarcomatoid transformation: implications for clinical management. Mod Pathol. 2019;32:1344–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qian Y, Deng J, Geng L, Xie H, Jiang G, Zhou L, et al. TLR4 signaling induces B7-H1 expression through MAPK pathways in bladder cancer cells. Cancer Invest. 2008;26:816–21. [DOI] [PubMed] [Google Scholar]
- 48.Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C, Savas P, et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin Cancer Res. 2016;22:1499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ju X, Zhang H, Zhou Z, Wang Q. Regulation of PD-L1 expression in cancer and clinical implications in immunotherapy. Am J Cancer Res. 2020;10:1–11. [PMC free article] [PubMed] [Google Scholar]
- 50.Yarchoan M, Albacker LA, Hopkins AC, Montesion M, Murugesan K, Vithayathil TT, et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quandt D, Jasinski-Bergner S, Müller U, Schulze B, Seliger B. Synergistic effects of IL-4 and TNFα on the induction of B7-H1 in renal cell carcinoma cells inhibiting allogeneic T cell proliferation. J Transl Med. 2014;12:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Messai Y, Gad S, Noman MZ, Le Teuff G, Couve S, Janji B, et al. Renal Cell Carcinoma Programmed Death-ligand 1, a New Direct Target of Hypoxia-inducible Factor-2 Alpha, is Regulated by von Hippel–Lindau Gene Mutation Status. Eur Urol. 2016;70:623–32. [DOI] [PubMed] [Google Scholar]
- 53.Ruf M, Moch H, Schraml P. PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int J Cancer. 2016;139:396–403. [DOI] [PubMed] [Google Scholar]
- 54.Banchereau R, Leng N, Zill O, Sokol E, Liu G, Pavlick D, et al. Molecular determinants of response to PD-L1 blockade across tumor types. Nat Commun. 2021;12:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Braun DA, Hou Y, Bakouny Z, Ficial M, Sant’ Angelo M, Forman J, et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat Med. 2020;26:909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ficial M, Jegede OA, Angelo MS, Hou Y, Flaifel A, Pignon JC, et al. Expression of T-Cell exhaustion molecules and human endogenous retroviruses as predictive biomarkers for response to nivolumab in metastatic clear cell renal cell carcinoma. Clin Cancer Res. 2021;27:1371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moch H, Humphrey PA, Ulbright TM, Reuter VE. WHO classification of tumours of the urinary system and male genital organs. 4th ed. Vol. 70, World Health Organization classifcation of tumours. 2016. 106–119 p. [DOI] [PubMed] [Google Scholar]
- 59.Mahoney KM, Sun H, Liao X, Hua P, Callea M, Greenfield EA, et al. PD-L1 antibodies to its cytoplasmic domain most clearly delineate cell membranes in immunohistochemical staining of tumor cells. Cancer Immunol Res. 2015;3:1308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Albers P, Albrecht W, Algaba F, Choueiri TK, Fay AP, Gray KP, et al. PD-L1 expression in nonclear-cell renal cell carcinoma. 2014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bellmunt J, Mullane SA, Werner L, Fay AP, Callea M, Leow JJ, et al. Association of PD-L1 expression on tumor-infiltrating mononuclear cells and overall survival in patients with urothelial carcinoma. Ann Oncol. 2015;26:812–7. [DOI] [PubMed] [Google Scholar]
- 62.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li B, Dewey CN. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deluca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research. 2016;4:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov M, Sergushichev A. Fast gene set enrichment analysis. 2016; [Google Scholar]
- 69.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018;24:749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Motzer RJ, Robbins PB, Powles T, Albiges L, Haanen JB, Larkin J, et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat Med. 2020;26:1733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Motzer RJ, Banchereau R, Hamidi H, Powles T, McDermott D, Atkins MB, et al. Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade. Cancer Cell. 2020;1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brunet JP, Tamayo P, Golub TR, Mesirov JP. Metagenes and molecular pattern discovery using matrix factorization. Proc Natl Acad Sci U S A. 2004;101:4164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vargiu L, Rodriguez-Tomé P, Sperber GO, Cadeddu M, Grandi N, Blikstad V, et al. Classification and characterization of human endogenous retroviruses mosaic forms are common. Retrovirology. 2016;13:1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anders S, Pyl PT, Huber W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Takahashi Y, Harashima N, Kajigaya S, Yokoyama H, Cherkasova E, McCoy JP, et al. Regression of human kidney cancer following allogeneic stem cell transplantation is associated with recognition of an HERV-E antigen by T cells (Journal of Clinical Investigation (2008) 118, (1099–1109) DOI: 10.1172/JCI34409). J Clin Invest. 2008;118:1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cherkasova E, Scrivani C, Doh S, Weisman Q, Takahashi Y, Harashima N, et al. Detection of an immunogenic HERV-E envelope with selective expression in clear cell kidney cancer. Cancer Res. 2016;76:2177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Webster WS, Lohse CM, Thompson RH, Dong H, Frigola X, Dicks DL, et al. Mononuclear cell infiltration in clear-cell renal cell carcinoma independently predicts patient survival. Cancer. 2006;107:46–53. [DOI] [PubMed] [Google Scholar]
- 83.Xie Y, Chen L, Ma X, Li H, Gu L, Gao Y, et al. Prognostic and clinicopathological role of high Ki-67 expression in patients with renal cell carcinoma: A systematic review and meta-analysis. Sci Rep. 2017;7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beckermann KE, Hongo R, Ye X, Young K, Carbonell K, Contreras Healey DC, et al. CD28 costimulation drives tumor-infiltrating T cell glycolysis to promote inflammation. JCI Insight. 2020;5:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sholl LM. Biomarkers of response to checkpoint inhibitors beyond PD-L1 in lung cancer. Mod Pathol. 2022;35:66–74. [DOI] [PubMed] [Google Scholar]
- 86.Cohen EEW, Bell RB, Bifulco CB, Burtness B, Gillison ML, Harrington KJ, et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J Immunother Cancer. 2019;7:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Albiges L, Flippot R, Escudier B. Immune Checkpoint Inhibitors in Metastatic Clear-cell Renal Cell Carcinoma: Is PD-L1 Expression Useful? Eur Urol. 2021;79:793–5. [DOI] [PubMed] [Google Scholar]
- 88.Jilaveanu LB, Shuch B, Zito CR, Parisi F, Barr M, Kluger Y, et al. PD-L1 expression in clear cell renal cell carcinoma: An analysis of nephrectomy and sites of metastases. J Cancer. 2014;5:166–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14:847–56. [DOI] [PubMed] [Google Scholar]
- 90.Motzer RJ, Choueiri TK, McDermott DF, Powles T, Vano YA, Gupta S, et al. Biomarker analysis from CheckMate 214: Nivolumab plus ipilimumab versus sunitinib in renal cell carcinoma. J Immunother Cancer. 2022;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Panda A, de Cubas AA, Stein M, Riedlinger G, Kra J, Mayer T, et al. Endogenous retrovirus expression is associated with response to immune checkpoint blockade in clear cell renal cell carcinoma. JCI insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Smith CC, Beckermann KE, Bortone DS, Cubas AA, Bixby LM, Lee SJ, et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J Clin Invest. 2018;128:4804–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Relevant data are available from the corresponding authors and/or are included in the manuscript. Gene signature scores are available in Supplementary Table 1 for the 8 pairs and in Supplementary Table 4 for the CheckMate 025 cohort. Clinical data for the CheckMate 025 cohort are available in Supplementary Table 4. Differentially expressed genes between PD-L1 positive and negative samples, gene set enrichment scores, and immune deconvolution data (by CIBERSORTx), are available in Supplementary Tables 2, 3, 5, respectively, for both the 8 pairs and the CheckMate 025 cohort. Normalized RNA-seq expression data are available in Supplementary Table 6 for the 8 pairs and in Supplementary Table 7 for the CheckMate 025 cohort. ERV expression (inferred from RNA-seq data) are available in Supplementary Table 8 for the 8 pirs and in Supplementary Table 9 for the CheckMate 025 cohort.