

ABSTRACT
Fibronectin (Fn1) fibrils have long been viewed as continuous fibers composed of extended, periodically aligned Fn1 molecules. However, our live-imaging and single-molecule localization microscopy data are inconsistent with this traditional view and show that Fn1 fibrils are composed of roughly spherical nanodomains containing six to eleven Fn1 dimers. As they move toward the cell center, Fn1 nanodomains become organized into linear arrays, in which nanodomains are spaced with an average periodicity of 105±17 nm. Periodical Fn1 nanodomain arrays can be visualized between cells in culture and within tissues; they are resistant to deoxycholate treatment and retain nanodomain periodicity in the absence of cells. The nanodomain periodicity in fibrils remained constant when probed with antibodies recognizing distinct Fn1 epitopes or combinations of antibodies recognizing epitopes spanning the length of Fn1. Treatment with FUD, a peptide that binds the Fn1 N-terminus and disrupts Fn1 fibrillogenesis, blocked the organization of Fn1 nanodomains into periodical arrays. These studies establish a new paradigm of Fn1 fibrillogenesis.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Fibril, Fibrillogenesis, Fibronectin, Live imaging, Super-resolution microscopy
Summary: Fibronectin (Fn1) fibrils form when centripetally moving Fn1 nanodomains become organized into arrays. Fn1 fibrils are arrays of periodically spaced nanodomains containing multiple Fn1 dimers.
INTRODUCTION
Fibronectin (Fn1) is a requisite component of the extracellular matrix (ECM) and is necessary for embryogenesis and homeostasis (Schwarzbauer and DeSimone, 2011). In the absence of Fn1 fibrillogenesis, the binding of Fn1 to cells is not sufficient to mediate critical biological processes such as embryonic development, angiogenesis, vascular remodeling or cartilage condensation (Chiang et al., 2009; Rozario et al., 2009; Singh and Schwarzbauer, 2014; Zhou et al., 2008). Therefore, understanding the mechanisms by which Fn1 proteins assemble into fibrils is essential for gaining insights into the various functions of Fn1. Fn1 is secreted as a dimer in which two Fn1 molecules are held in an anti-parallel orientation by two disulfide bonds close to their C-termini (Skorstengaard et al., 1986; Wagner and Hynes, 1979). Fn1 fibrillogenesis is a cell-dependent process (McKeown-Longo and Mosher, 1983), which occurs following the binding of Fn1 dimers to cell-surface integrins (Schwarzbauer and DeSimone, 2011). Following integrin binding, intracellular cytoskeletal forces generate pulling forces on Fn1 dimers, exposing epitopes that promote Fn1 fibrillogenesis (Chernousov et al., 1987; Hocking et al., 1994; Smith et al., 2007; Zhang et al., 1994, 1997; Zhong et al., 1998). At the cellular level, the process of Fn1 fibrillogenesis is correlated with the formation of fibrillar adhesions, wherein complexes containing Fn1 and integrin α5β1 elongate while translocating toward the nucleus, giving rise to long filaments (≥1 µm) containing Fn1 and intracellular cytoplasmic effectors linking Fn1 and the actin cytoskeleton (Geiger et al., 2001; Geiger and Yamada, 2011; Lu et al., 2020; Pankov et al., 2000; Zamir et al., 1999, 2000). Fn1 fibrils generated in this process are incorporated into the ECM.
It has been thought that Fn1 fibrils resemble ropes, in which extended Fn1 dimers align such that regions containing overlapping N-termini alternate with regions containing C-termini (Chen et al., 1997; Dzamba and Peters, 1991; Früh et al., 2015; also illustrated in Fig. S5A). To understand how the process of fibrillogenesis occurs in real time and at the nanoscale level, we adopted a CRISPR/Cas9-mediated mutagenesis approach to generate fluorescently labeled Fn1, which was subjected to physiological regulation by expression and splicing. This approach enabled visualization of Fn1 fibrillogenesis over an extended period of time. Using live imaging and super-resolution microscopy, we uncovered an unexpected mechanism of Fn1 fibrillogenesis in which Fn1 fibrils formed as a result of centripetally moving Fn1 nanodomains. As Fn1 nanodomains moved toward the cell center, they assembled into arrays of periodically spaced nanodomains. The arrays became longer as the movement toward the cell center continued and as more nanodomains were added. We show that these nanodomain arrays have properties of bona fide Fn1 fibrils and that each Fn1 nanodomain contained multiple Fn1 dimers. Our live-imaging and single-molecule localization microscopy (SMLM) data revealed that the interactions mediated by the N-terminal assembly domain of Fn1 regulate the organization of Fn1 nanodomains into nanodomain arrays. These studies provide significant new insights into the mechanisms of Fn1 ECM formation, remodeling and signaling.
RESULTS
Diffraction-limited microscopy reveals the beaded structure of Fn1 fibrils
Staining wild-type mouse embryos with saturating amounts of anti-Fn1 antibodies revealed that Fn1 fibrils in the pharyngeal arches and the heart appeared dotted, with regularly spaced regions of high and low fluorescence intensity (Fig. 1; Movie 1). The dotted appearance of Fn1 fibrils in embryonic tissues suggests that the distribution of Fn1 molecules in Fn1 fibrils is not homogenous. This was puzzling, and therefore, we decided to investigate how Fn1 fibrils form. For this purpose, we employed a CRISPR/Cas9 knock-in strategy to modify the endogenous Fn1 locus by replacing the termination codon of Fn1 with a sequence encoding a short linker fused to a fluorescent protein. We used this strategy to generate cell lines and Fn1mEGFP/mEGFP homozygous knock-in mice expressing Fn1 fused to monomeric enhanced green fluorescent protein (mEGFP) or other monomeric fluorescent proteins (FPs) (Fig. S1A–D). Homozygous Fn1mEGFP/mEGFP mice were obtained at the correct Mendelian ratio (Fig. S1B, panels 4 and 5), and were viable and fertile, indicating that Fn1–mEGFP supports all functions of Fn1 necessary for embryonic development, fetal viability and adult homeostasis. Examination of Fn1 expression patterns in knock-in mice showed that Fn1–mEGFP was expressed in the same pattern as the unmodified Fn1 in embryos (Peters and Hynes, 1996), i.e. there were no regions that were GFP-positive but Fn1-negative and vice versa in Fn1mEGFP/+ embryos expressing one wild-type allele of Fn1 (Fig. S1H). In addition, we used CRISPR/Cas9 mutagenesis to generate five independent lines of mouse embryonic fibroblasts (MEFs) expressing Fn1–mEGFP, Fn1–mScarlet-I, Fn1–Neon Green, or Fn1–tdTomato. Western blots showed that fluorescent protein (FP) fusions to Fn1 were specific: FPs were only fused to Fn1 as no other FP fusion proteins were detected by either western blotting or immunofluorescence (IF) (Fig. S1C,D).
Fig. 1.
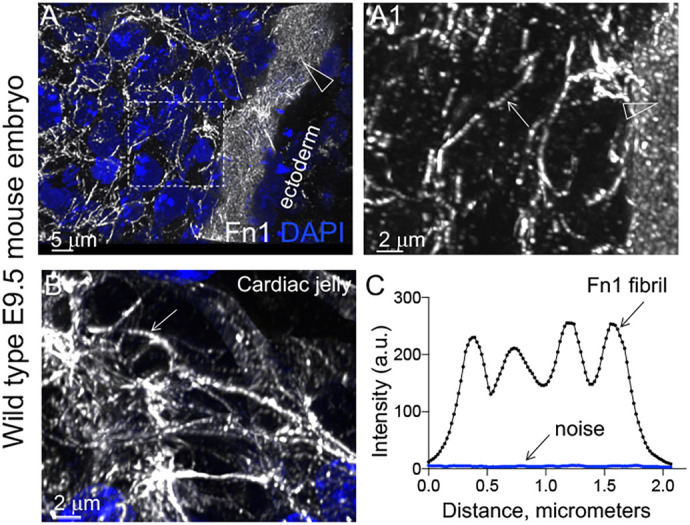
Beaded architecture of Fn1 fibrils in embryonic ECM. (A,A1,B) Wild-type E9.5 mouse embryos were fixed and stained with a 1:300 dilution of the Abcam monoclonal antibody to Fn1 (white) and DAPI (blue) and imaged using a 100× oil objective, with NA 1.49, pinhole 0.8 and a sampling rate of 40 nm/pixel. The sagittal optical section through the first pharyngeal arch (A,A1) and the cardiac jelly (B) are shown. Large arrowheads in A,A1 point to the ECM at the ectoderm-mesenchyme boundary of the first pharyngeal arch. The dotted box in A is magnified in A1 to show Fn1 microarchitecture. The arrow in A1 points to a beaded Fn1 fibril within the first arch mesenchyme. The arrow in B indicates the beaded architecture of Fn1 fibrils in cardiac jelly. (C) Intensity profile plot of an Fn1 fibril. a.u., arbitrary units. Note that at least 1:400 dilutions of this antibody are saturating (see below and Fig. 5F). Images are representative of three independent experiments. Scale bars: 5 μm (A); 2 μm (A1,B).
Measuring deoxycholate (DOC) insolubility of Fn1 ECM is a classical biochemical assay for stable incorporation of Fn1 proteins into the ECM (Choi and Hynes, 1979; McKeown-Longo and Mosher, 1983; Schwarzbauer, 1991; Singh et al., 2010; Wierzbicka-Patynowski et al., 2004). For these assays, we carefully controlled the number of cells plated, as cell density affects the extent of Fn1 fibrillogenesis (Hynes, 1990). We performed DOC insolubility assays using five independently generated cell lines expressing Fn1-FP fusions (Fn1–FPs). These experiments demonstrated that the incorporation of Fn1–FPs into the ECM was indistinguishable from that of wild-type, untagged Fn1 (Fig. S1E). In addition, DOC insolubility assays showed that Fn1–mEGFP proteins isolated from mouse embryos were incorporated into a deoxycholate-insoluble embryonic ECM, similarly to unmodified Fn1 (Fig. S1F,G). Taken together with the viability of Fn1mEGFP/mEGFP homozygous knock-in mice, these data demonstrate that Fn1–FPs are suitable reagents for investigating the mechanisms of Fn1 fibrillogenesis.
To visualize the process of fibrillogenesis, we plated Fn1mEGFP/+ MEFs on gelatin-coated coverslips and imaged cells 16 h after plating using total internal reflection fluorescence (TIRF) microscopy at the critical angle of incidence. These experiments showed that Fn1 fibrillogenesis was initiated at the cell periphery as distinct, brightly fluorescent Fn1 densities that moved centripetally in parallel with F-actin and became aligned into linear arrays of ‘beads’ (arrows in Movie 2). TIRF imaging also showed that domains of higher fluorescence intensity of Fn1 co-localized with areas of higher intensity of integrin α5β1 in both non-fibrillar and fibrillar adhesions (Fig. 2), and that the staining of Fn1 and α5β1 in fibrillar adhesions appeared ‘beaded’ (Fig. 2B–B2). Beaded architecture of Fn1 fibrils was also observed by an independent imaging method using Zeiss Airyscan technology (Fig. S2). To test whether the beaded appearance of Fn1 fibrils depended on antibody epitope availability, we stained wild-type MEFs or mouse endothelial cells using three different antibodies: the 297.1 polyclonal antibody raised against the rat plasma Fn1 protein and recognizing multiple Fn1 epitopes in mouse Fn1 (Fig. S3A–C); the 3E2 monoclonal antibody recognizing the alternatively spliced EIIIA exon of Fn1; and an Abcam monoclonal antibody recognizing an epitope within the central region of Fn1 (see Table S4). The beaded appearance of Fn1 fibrils did not depend on the type of antibody used (Fig. S4). Moreover, staining with the 297.1 polyclonal antibody, which recognizes multiple epitopes along the mouse Fn1 molecule, resulted in the beaded appearance of Fn1 fibrils (Fig. S4A,A1). Fn1 fibrils formed by cells plated on uncoated coverslips, as well as on coverslips coated with gelatin, laminin 111 or vitronectin, also appeared beaded (Fig. S2, Fig. S4E,F). Similarly, Fn1 fibrils in cell-free areas (Fig. S4A,A1) and between cells appeared beaded (Fig. S4B,B1,C,C1). Fn1 fibrils produced by cells plated on soft substrata, such as hydrogels of variable stiffness, also appeared beaded (Fig. S4G,G1,H,H1). In this latter experiment, Fn1 was detected by imaging the fluorescence of the Fn1–mEGFP protein, indicating that the beaded appearance of Fn1 fibrils was independent of antibody staining. To determine whether Fn1 fibrils in cell-free, fibrillar ECM were beaded, cultures were treated with 2% DOC (Lu et al., 2020). The 2% DOC treatment dissolved cell membranes and cytoplasmic components, leaving behind ECM devoid of cell contact (see Movie 3 for time-lapse of the dissolution of cellular components, F-actin and DNA). This experiment showed that Fn1 fibrils retained their beaded architecture in the absence of cell contact (Fig. S4D). Taken together, these studies indicated that the beaded appearance of Fn1 fibrils is a general feature of the Fn1 ECM, which was seen in multiple contexts: in the ECM devoid of cells, in DOC-resistant cell-free fibrils, in different embryonic tissues in vivo, and in fibrils produced by cells that were cultured on various substrata in vitro. Moreover, the beaded appearance of Fn1 fibrils was independent of the antibody and method of detection; it was observed by indirect detection, such as by immunofluorescence, and by direct detection of the fluorescent Fn1–mEGFP protein.
Fig. 2.

Integrin α5 and Fn1 co-localize in beaded adhesions. Wild-type MEFs were cultured for 16 h on glass coverslips without coating, fixed and stained with the Abcam monoclonal Fn1 antibody (cyan) and anti-integrin α5 (Itga5) antibody (magenta). Cells were imaged at the critical angle of incidence using a 100× oil objective, with NA 1.49. (A–A2) Representative images of the cell periphery. Arrows in A–A2 point to examples of non-fibrillar Fn1 adhesions. (B–B2) Representative images of the medial portion of a cell containing beaded fibrillar adhesions (arrows). Magnifications in all panels are the same. Images are representative of three experiments. Scale bar: 2 μm
Nanoarchitecture of Fn1 fibrils revealed by standardized single-molecule localization microscopy
To examine the structure of Fn1 fibrils at the nanoscale level, we adopted SMLM techniques by using direct stochastic optical reconstruction microscopy (dSTORM) (Heilemann et al., 2008; Rust et al., 2006). To optimize SMLM imaging conditions and to determine the effective labeling efficiency (ELE) of our reagents, we used the methodology and NUP96–mEGFP U2OS cells as a reference cell line (Thevathasan et al., 2019), as gold-standard tools to optimize image quality, measure ELE and quantify the number of mEGFP molecules in Fn1 nanodomains (Lelek et al., 2021; Thevathasan et al., 2019). In this reference cell line, both copies of the NUP96 gene are modified by CRISPR/Cas9 mutagenesis, thus generating the NUP96–mEGFP protein, a component of the nuclear pore complex (NPC) (Thevathasan et al., 2019). Thirty-two copies of NUP96 are evenly distributed among the eight corners of the NPC at known distances (Bui et al., 2013; Thevathasan et al., 2019; von Appen et al., 2015). To optimize SMLM imaging, we followed the current best SMLM practices (Lelek et al., 2021; Mund and Ries, 2020) and used the methodology and the super-resolution microscopy analysis platform (SMAP) developed by Diekmann et al. (2020), Ries (2020) and Thevathasan et al. (2019). Fourier ring correlation analysis (Nieuwenhuizen et al., 2013) implemented in the SMAP software indicated that the resolution of our images ranged between 14 and 28 nm. The localization precision was 9.1±1.9 nm on average. By using antibodies to detect NUP96–mEGFP and the SMAP software to analyze images from three independent experiments, eight nuclei and 4571 NPCs, we determined the apparent NPC radius to be 63.6±0.86 nm (Fig. 3A–C; Table S1). This radius is consistent with the reported NPC radius of 64.3±2.6 nm, measured using this cell line and a combination of commercial anti-GFP primary and Alexa Fluor 647 (AF647)-conjugated secondary antibodies (Thevathasan et al., 2019). We then used NUP96–mEGFP U2OS cells to optimize the ELE of our reagents. ELE summarily measures how well the reagents and methods used, including the SMLM imaging protocol, generate images that reflect the actual structure under study (Thevathasan et al., 2019). SMLM imaging of NUP96 in the xy plane should produce eightfold symmetrical ring-like structures, in which four NUP96 molecules are positioned at each of the eight vertices of the NPC at known intervals and with known dimensions (Thevathasan et al., 2019). Using NUP96–mEGFP U2OS as a reference cell line, anti-GFP primary antibodies, commercially labeled AF647-conjugated secondary antibodies and the SMAP software (Thevathasan et al., 2019), we determined the ELE to be 79.6±5.4% (Table S1), reflecting that the majority of NPCs in our images have eight NUP96-positive corners (Fig. 3A–C). This ELE and the standard deviation of our measurements (<10%) are comparable with the best ELE measured for this system (∼74%) (Thevathasan et al., 2019). Taken together, these experiments indicate that our reagents and SMLM imaging conditions are within the accepted SMLM standards (Lelek et al., 2021).
Fig. 3.

Standardized SMLM imaging results in high resolution and high effective labeling efficiency. NUP96mEGFP/mEGFP homozygous knock-in U2OS cells (A–C) and Fn1mEGFP/mEGFP homozygous knock-in MEFs (D–F) were analyzed using SMLM imaging protocol I (see Materials and Methods). The boxed regions in A,D are shown in B,E, respectively, and the boxed regions in B,E are shown in C,F, respectively. The resolution in C and F measured by the Fourier ring curve method are 18.3±6.7 nm and 14.5±1.1 nm, respectively. The nanodomain architecture of Fn1–mEGFP fibrils is shown in subsequent magnifications of the fibril boxed in D. Arrows in F point to Fn1 nanodomains. N1 is the number of grouped localizations in each panel. The vertical bar in F shows color coding according to localization density for all panels. Images are representative of three independent experiments. Scale bars: 1 μm (A,B,D,E); 100 nm (C,F).
To attain a comparable ELE with that measured with NUP96–mEGFP, we stained homozygous Fn1mEGFP/mEGFP MEFs at the same time as NUP96–mEGFP U2OS cells, using aliquots of the same mixtures of reagents. Furthermore, stained Fn1mEGFP/mEGFP MEFs and NUP96–mEGFP U2OS cells were imaged on the same day, using aliquots of the same imaging buffer, and the same off-switching and imaging protocols (see SMLM imaging protocol I). Similar to previously published results (Früh et al., 2015), we saw that Fn1 fibrils appeared as arrays of nanodomains situated along Fn1 fibrils with a regular periodicity (Fig. 3D–F, Fig. 4D–D2, Fig. 5D–D″). As shown in Früh et al. (2015), nanodomain periodicity was analyzed using autocorrelation, in which the position of the first autocorrelated peak marked the nanodomain periodicity (Früh et al., 2015). Autocorrelation analysis of 14 fibrils from seven cells and three independent experiments indicated that the nanodomain periodicity of GFP epitopes in Fn1–mEGFP fibrils was 102±29 nm (Fig. 4D–D2, Fig. 5D–D″,F; Table S2). This periodicity is comparable with the nanodomain periodicity measured by Früh et al. (2015), which was 99±17 nm, and the periodicity determined by immunoelectron microscopy using an anti-EIIIA antibody, which was on average 84 nm and ranged between 40 and 280 nm (Dzamba and Peters, 1991).
Fig. 4.

Nanodomain architecture of Fn1 fibrils detected with four different antibodies. Fn1mEGFP/mEGFP MEFs were plated on glass coverslips without coating, cultured overnight and stained with the following antibodies: (A) R457 (1:200 dilution); (B) R184 (1:100 dilution); (C) Abcam monoclonal antibody (1:200 dilution); and (D) anti-GFP antibody (1:100 dilution). Boxed regions in A–D are magnified to show fibrils in A1–D1, and bracketed regions in A1–D1 are magnified in A2-D2. Fourier ring correlation (FRC) was used to determine image resolution. The vertical bar in D shows color coding according to localization density for all panels in this figure. Cells were imaged using SMLM imaging protocol I. Arrowheads point to examples of sparse Fn1 localizations between nanodomains. Images are representative of three independent experiments. Scale bars: 10 μm (A–D); 1 μm (A1–D1); 100 nm (A2–D2).
Fig. 5.

Periodical labeling of Fn1 fibrils by multiple antibodies, their dilutions and combinations. (A–E) Fn1mEGFP/mEGFP MEFs were plated on glass coverslips without coating, cultured overnight and stained with the indicated antibodies. Cells were imaged using SMLM imaging protocol I. Bracketed regions in A–E are magnified in A′–E′. Arrowheads point to examples of sparse Fn1 localizations between nanodomains. Fourier ring correlation (FRC) was used to determine image resolution. (A″–E″) Autocorrelation traces for fibrils shown in A–E. Plots were truncated at 1 µm for clarity. Ljung–Box Q test for autocorrelation at the peak positions marked by the red arrows resulted in h=1 and p=0, where h is the null hypothesis that the first 4 autocorrelations are jointly zero. h=1 rejects this hypothesis and p is the probability that the null hypothesis is correct, indicating strong evidence of autocorrelation at least up to the fourth peak. (F) Summary of autocorrelation analyses, shown as violin plots of the distances between nanodomains in fibrils. Red lines indicate the median and black dotted lines indicate the quartiles. Plots of data from stainings with combinations of antibodies are highlighted in yellow. One-way ANOVA with Tukey's correction for multiple testing showed no significant differences among the means. *Anti-GFP and R457 antibodies were both used at 1:100 dilution. **The five antibodies used against Fn1–mEGFP and their dilutions were as follows: R457 (1:400), R184 (1:200), Abcam (1:400), anti-GFP (1:100) and 297.1 (1:400). (G–I) Clustering using DBSCAN. The fibril in G was analyzed by DBSCAN in H and I. The minimum number of points per neighborhood (k) was set to 4. (H) The radius of the neighborhood ε was set to 14 nm (Table S2). Adjacent clusters are labeled with different colors. (I) ε was estimated automatically by the DBSCAN algorithm. Images are representative of fibrils from at least three independent experiments. Scale bars: 1 μm (A–E, G–I); 0.1 μm (A′–E′).
Localization of Fn1 epitopes within periodical nanodomain arrays
Fn1 is a large, multi-domain, ∼250 kDa glycoprotein secreted as a dimer, in which two Fn1 subunits are linked in an anti-parallel orientation by two disulfide bonds at their C-termini (Fig. S3D) (Schwarzbauer and DeSimone, 2011; Skorstengaard et al., 1986; Wagner and Hynes, 1979). To investigate the relationship between the protein domains of Fn1 and the nanodomain architecture of Fn1 fibrils, we first used antibodies specific to different regions of the Fn1 protein (Fig. S3E, Table S4). For these experiments, Fn1mEGFP/mEGFP MEFs were plated on uncoated glass coverslips. Cells were then fixed and stained with the following antibodies recognizing different Fn1 epitopes: R457 rabbit polyclonal antibodies raised against the N-terminal 70 kDa domain of Fn1 (Aguirre et al., 1994; Sechler et al., 2001), R184 rabbit polyclonal antibodies raised to recognize the domain immediately following the 70 kDa N-terminal domain and containing the first six type III repeats of Fn1 (Fn1 III1-6) (Raitman et al., 2018), a rabbit monoclonal antibody (Abcam) recognizing an epitope within the central region of Fn1 (see Table S4 for further description of this antibody), and a polyclonal anti-GFP antibody recognizing the C-terminus of Fn1–mEGFP proteins. In our analyses, we focused on long fibrillar adhesions that were over 1 µm in length, characteristic of mature assembled Fn1 fibrils (Lu et al., 2020).
Irrespective of the antibodies used, Fn1 fibrils appeared as arrays of periodically spaced nanodomains (Figs 4 and 5A–F). To ensure that the beaded appearance of fibrils was not due to undersampling, we tested two to four dilutions of each antibody, followed by SMLM imaging and autocorrelation analysis, as outlined by Früh et al. (2015). Our analyses showed that the nanodomain periodicity remained constant at all antibody dilutions tested (Fig. 4, Fig. 5A–F). The periodicity of nanodomains detected with either R457, R184 or the Abcam monoclonal antibody was similar to that measured with the anti-GFP antibody, suggesting that these antibodies were used at an ELE comparable with that of GFP-labeling reagents. The finding that the nanodomain periodicity was independent of antibody specificity supports the conclusion that the beaded appearance of Fn1 fibrils is not a result of a particular Fn1 protein conformation, as staining using antibodies specific to three different regions of Fn1 as well as antibodies against GFP resulted in the same pattern. In all cases, nanodomain periodicity was independent of the antibody concentrations, arguing against the possibility of undersampling.
Analysis using density-based spatial clustering of applications with noise (DBSCAN) (Caetano et al., 2015) discovered clusters corresponding to nanodomains when the radius of the neighborhood (ε) was set to 14 nm, the average apparent radius of an Fn1 nanodomain (Table S2; compare Fig. 5G and Fig. 5H). Unbiased DBSCAN clustering using the neighborhood radius automatically estimated by the SMAP software discovered larger clusters of Fn1 nanodomains (indicated by brackets in Fig. 5I), suggesting that Fn1 nanodomains are organized in a higher-order structure within long fibrils, consistent with the observation of ‘beads’ in lower-resolution diffraction-limited images of Fn1 (Figs 1 and 2; Figs S2 and S4).
Fn1 nanodomains contain multiple full-length Fn1 molecules
It was previously thought that Fn1 nanodomains detected by immunoelectron microscopy or by immunofluorescence SMLM corresponded to particular regions in the Fn1 protein sequence (Dzamba and Peters, 1991; Früh et al., 2015), illustrated in Fig. S5A,B. Surprisingly, staining using the 297.1 polyclonal anti-Fn1 antibody raised against full-length rat plasma Fn1 and recognizing multiple epitopes along the Fn1 protein (Fig. S3A–C) resulted in the same periodicity of nanodomains as staining with antibodies against distinct regions of the Fn1 protein (Fig. 5E,F, Fig. 6A,D). Interestingly, studies using polyclonal antibodies raised against the full-length Fn1 from human plasma also showed an apparent periodical staining of Fn1 fibrils by immunoelectron microscopy (Furcht et al., 1980a,b,c).
Fig. 6.

Polyclonal antibody to full-length Fn1 and combinations of antibodies recognizing epitopes along the length of Fn1 reveal periodical nanodomain architecture of Fn1 fibrils. Fn1mEGFP/mEGFP MEFs were plated on glass coverslips without coating, cultured overnight and stained with the indicated antibodies. Cells were imaged using SMLM imaging protocol I. (A–C) Representative image of cells stained with the 297.1 antibody (1:200 dilution) (A); a combination of anti-GFP antibody (1:100) and R457 antibody (1:100) (B); and a combination of the five antibodies, R457 (1:400), R184 (1:200), Abcam (1:400), anti-GFP (1:100) and 297.1 (1:400) (C). These antibodies are depicted in the schematic above panel C, and red arrows mark their epitopes. Boxed fibrils in A–C are magnified in A′–C′, and bracketed regions in A′–C′ are magnified in A″–C″. The vertical bar in C shows color-coding according to localization density for A–C. C′-1 and C″-1 represent the same images in C′ and C″, respectively, but are color-coded according to Voronoi cluster density (horizontal bar below C″-1). Arrows in C″-1 indicate Voronoi tessellation clusters corresponding with nanodomains. Arrowheads in A″–C″ point to examples of sparse Fn1 localizations between nanodomains. (D–F) Autocorrelation analysis and Ljung–Box Q test for autocorrelation at the peak positions marked by the red arrows resulted in h=1 and P=0, indicating strong evidence for autocorrelation. h, the null hypothesis for this test, is that the first four autocorrelations are jointly zero. h=1 rejects this hypothesis. Images are representative of three independent experiments. Scale bars: 10 μm (A–C); 1 μm (A′–C′,C′-1); 0.1 μm (A″–C″,C″-1).
As the 297.1 polyclonal antibody recognizes multiple epitopes along Fn1, including an epitope in the 70 kDa N-terminal assembly region of Fn1 (Fig. S3C), our experiments suggested that each Fn1 nanodomain contained at least one entire Fn1 dimer. To determine the number of Fn1 molecules per nanodomain, we used the methodology developed by Thevathasan et al. (2019) as well as identical labeling reagents and imaging conditions for detecting mEGFP in NUP96–mEGFP U2OS cells and Fn1–mEGFP MEFs (Thevathasan et al., 2019). This analysis indicated that each Fn1 nanodomain contains 16.85±5.1 mEGFP molecules on average (Table S2). As Fn1 assembled in ECM fibrils is an obligate dimer of two disulfide-bonded Fn1 molecules (Schwarzbauer, 1991), these findings indicate that Fn1 nanodomains contain six to 11 Fn1–mEGFP dimers on average.
These results are not consistent with the canonical model of Fn1 fibrillogenesis, which stipulates that the thinnest Fn1 fibrils are made of extended single Fn1 dimers that are periodically aligned in a regular, end-to-end fashion, with regions containing N-termini alternating with regions containing C-termini (Chen et al., 1997; Dzamba and Peters, 1991; Früh et al., 2015; illustrated in Fig. S5A,B). This periodic alignment of Fn1 dimers necessitates that staining using antibodies recognizing multiple epitopes along the length of the Fn1 molecule would result in uniform, non-periodic labeling of Fn1 fibrils (illustrated in Fig. S5C). However, this was not what we observed; staining with the polyclonal 297.1 antibody showed periodically spaced nanodomains (Fig. 6A–A″,D). Autocorrelation analysis showed that the spacing was periodical at multiple concentrations of the 297.1 antibody (Fig. 5E–E″,F, Fig. 6D). Notably, the periodicity of nanodomains detected using the 297.1 antibody was statistically indistinguishable from periodicities seen with any region-specific antibodies tested, including anti-GFP antibodies (Fig. 5F). This suggested that the 297.1 polyclonal antibody was used at an ELE that was at least as high as that for the GFP-labeling reagents. Finally, the periodical architecture of Fn1 fibrils detected by the 297.1 antibody and the presence of multiple Fn1 dimers per nanodomain do not fit the model proposed by Dzamba and Peters (1991) and suggest a different model of Fn1 fibrillogenesis.
To further evaluate the hypothesis that Fn1 nanodomains contain full-length Fn1 dimers, we used combinations of antibodies and imaging parameters resulting in the high ELE (Fig. 5F). We first stained Fn1mEGFP/mEGFP MEFs utilizing a combination of R457 and anti-GFP antibodies detecting both the N- and C-termini of Fn1–mEGFP. Each of these antibodies was used at saturating levels (Fig. 5F, see the legend for dilution details). The periodicity of nanodomains detected with a mixture of R457 and anti-GFP antibodies was 101±45 nm. This periodicity is statistically indistinguishable from the periodicities observed when either of these antibodies was used alone (Fig. 5F, Fig. 6B–B″,E).
Next, we stained Fn1mEGFP/mEGFP MEFs with a cocktail of five antibodies recognizing the following epitopes distributed along the length of the Fn1–mEGFP molecule (Table S4; Fig. S3E): the N-terminal 70 kDa assembly domain of Fn1 (R457 antibody), the sequence immediately following the 70 kDa domain (R184 antibody), an epitope in the middle of Fn1 (Abcam monoclonal antibody), multiple epitopes along the Fn1 protein (297.1 antibody) and the C-terminus of Fn1–mEGFP (anti-GFP antibody) (Fig. 6C–C″,F). Each antibody was used at a dilution resulting in a periodic staining (Fig. 5F, see the legend for dilution details). If Fn1 fibrils were composed of continuous, linear arrays of extended and periodically aligned Fn1 molecules, this cocktail of antibodies recognizing epitopes from the N- to the C-terminus of Fn1 would uniformly label Fn1 fibrils (e.g. Fig. S5C). Even in the scenario in which the epitopes recognized by R457 did not extend to the N-terminal-most sequence of Fn1, the staining using this mixture of five antibodies would not be expected to produce periodical staining. However, we observed that the cocktail of five antibodies labeled nanodomains with an average periodicity of 94±47 nm at a spatial resolution of 23 nm in thin Fn1 fibrils (Fig. 6C–C″,F). This periodicity was statistically indistinguishable from the periodicities detected by individual antibodies at all tested dilutions (Fig. 5F).
Unbiased clustering using the Voronoi tessellation method (Andronov et al., 2016b) implemented in SMAP showed that Fn1 fibrils stained with the five-antibody cocktail could be segmented into clusters (Fig. 6C″-1). This analysis indicates that the nanodomain structure of Fn1 fibrils can be discovered using an unbiased computational approach. The sizes of the nanodomains detected with the five-antibody cocktail were larger than those of nanodomains detected using any single antibody (e.g. compare Fig. 6A″ with Fig. 6C″). This behavior is expected in SMLM when molecules are clustered (Baumgart et al., 2016).
In agreement with the results described above, periodically spaced nanodomains were detected in Fn1 fibrils using two additional cell lines, heterozygous Fn1mEGFP/+ MEFs and wild-type MEFs (Fig. S6). The presence of periodically organized nanodomains in fibrils produced by wild-type cells indicated that the periodical structure of Fn1 fibrils was not an artifact of Fn1–mEGFP fusion. It is interesting to note that in all fibrils imaged, Fn1 nanodomains containing on average 159±78 localizations (Table S2) were separated by regions containing a low number of Fn1 localizations, which could be detected by any antibody used in this study (e.g. arrowheads in Fig. 4A2–D2, Fig. 5A′–E′, Fig. 6A″–C″). These data suggest that at least one entire Fn1 dimer or subunit is located between Fn1 nanodomains in fibrils.
Nanodomain spacing in Fn1 fibrils treated with 2% DOC, which removes cells and cellular components, was similar to that of untreated fibrils (Fig. S6), indicating that Fn1 fibrils retained their periodical architecture in the absence of cells. Three-dimensional (3D)-STORM imaging showed that Fn1 nanodomains were roughly globular (Movie 4). Finally, bovine Fn1 present in the MEF culture medium is recognized by the 297.1 polyclonal antibody (Fig. S8B) and would be detected if it were incorporated into Fn1 fibrils. Therefore, the sparsity of Fn1 localizations between nanodomains was not due to the inability of antibodies to recognize bovine plasma Fn1. Together, these data showed that Fn1 fibrils are arrays of periodically spaced nanodomains containing multiple Fn1 dimers. This periodical nanodomain architecture of Fn1 fibrils was independent of the antibodies used for staining, was seen in cells expressing wild-type Fn1, and remained intact in the absence of cell contact.
To further test the hypothesis that Fn1 nanodomains contain full-length Fn1 molecules, we performed double-color dSTORM experiments, in which the N-terminus of Fn1 was labeled with the R457 primary antibody and anti-rabbit CF680-conjugated secondary immunoglobulins, and the C-terminus of Fn1–mEGFP was labeled with anti-GFP primary antibody and AF647-conjugated anti-chicken secondary antibodies. As a control, we stained Fn1mEGFP/mEGFP MEFs using two different polyclonal anti-GFP antibodies, one made in rabbit (detected using CF680-conjugated secondary antibodies) and the other in chicken (detected with AF647-conjugated anti-chicken secondary antibodies). Most nanodomains detected using the two anti-GFP antibodies contained overlapping CF680 and AF647 signals, suggesting a high ELE for both reagents (Fig. 7A–C,A′–C′; gray lines in Fig. 7A′,B′ mark overlapping staining in nanodomains, and nanodomains in which the two labels did not overlap are marked by pink lines). To determine the extent of co-localization between the two labels, we performed coordinate-based co-localization (CBC) analysis, a widely accepted method for detecting co-localization in SMLM data (Malkusch et al., 2012). The CBC coefficients were calculated using algorithms implemented by the Abbelight software or the publicly available ThunderSTORM software (Ovesný et al., 2014). The coefficient ‘+1’ indicates a high probability of co-localization, whereas CBC≤0 indicates a low likelihood of co-localization. CBC analysis using the Abbelight software and color coding according to the CBC coefficient showed that the parameters chosen for these analyses, r=30 nm and Rmax=300 nm, reflected the overlap (e.g. yellow color in Fig. 7D,E and gray lines in Fig. 7A′,B′) and the lack of overlap (pink color and arrows in Fig. 7D,E and pink lines and arrows in Fig. 7A′,B′) seen in SMLM images. These figures also show that we could detect non-overlapping signals in nanodomains located next to one another (Fig. 7D,E). CBC analyses performed on 22 regions containing long (≥1 µm) Fn1 fibrils from Fn1mEGFP/mEGFP MEFs showed extensive overlap among localizations arising from labeling with rabbit and chicken anti-GFP antibodies (Fig. 7F). Taken together, these control experiments show that, as expected, staining of Fn1–mEGFP proteins with different antibodies against GFP results in overlapping localizations.
Fig. 7.

Double-color dSTORM shows that N- and C-termini of Fn1 overlap within Fn1 nanodomains. (A–C) Fn1 fibrils were detected in Fn1mEGFP/mEGFP cells plated on glass coverslips without coating using polyclonal rabbit and chicken anti-GFP antibodies. A′,B′ show the same images as in A,B with gray and pink lines marking overlapping and non-overlapping nanodomains, respectively. Pink dots under the pink arrows indicate nanodomains in which the signal in one channel is higher than in the other. C and C′ show the merged signals in color and grayscale, respectively. <100% overlap is expected as ELE<100%. (D,E) Co-distribution of localizations in one channel with those in another channel (e.g. Rab αGFP with chick αGFP in D) is color-coded according to the coordinate-based co-localization (CBC) coefficient; (Rmax=300, r=30). +1 indicates complete overlap; 0 indicates no overlap. Pink arrows in D,E correspond to those in A′,B′. Arrowheads in A′,B′,D,E indicate overlapping signals, and the arrow next to the arrowhead in B′ and E indicates adjacent non-overlapping signals. (F) CBC coefficients for 22 regions containing long fibrils (gray traces) and their average (blue trace). (G–I) Fn1 fibrils were detected in Fn1mEGFP/mEGFP cells plated on glass coverslips without coating using rabbit polyclonal R457 antibody and chicken anti-GFP antibody. G′–I′ show the same images as in G–I with overlapping (gray lines) and non-overlapping (pink lines and arrows) staining. I and I′ show the merged signals in color and grayscale, respectively. White arrows in I,I′ point to nanodomains. (J,K) Co-distribution of localizations in one channel with those in another channel (e.g. R457 with chick αGFP) is color-coded according to the CBC coefficient; (Rmax=300, r=30). Pink arrows shown in J correspond to those in G′. Pink arrowheads in G correspond to pink arrowheads in I and J, and point to non-overlapping nanodomains. White arrowheads in G, H correspond to black arrowheads in J, K and point to overlapping nanodomains. Note that settings for CBC analysis distinguish overlapping and non-overlapping localizations in adjacent nanodomains. (L) CBC coefficients for 20 long fibrils (gray traces) and their average (blue trace). (M,M′,N,N′) CBC using ThunderSTORM (Rmax=50 nm, r=5 nm) for 22 fibrils, four cells (M,M′) and 17 fibrils, four cells (N,N′). Images are representative of fibrils in six cells. Scale bars: 1 μm.
Next, we analyzed Fn1 fibrils stained with antibodies recognizing the N- and the C-termini of Fn1–mEGFP. CBC analyses of 20 regions containing long fibrils from Fn1mEGFP/mEGFP MEFs stained with R457 and anti-GFP antibodies showed extensive overlap between R457 and anti-GFP antibody localizations, which detect the N- and the C- termini of Fn1–mEGFP, respectively (Fig. 7G–L). Using ThunderSTORM software, we varied the parameters of CBC analysis, e.g. by setting the radius r from 2 or 5 nm and Rmax from 20 or 50 nm, respectively. These analyses had the same outcome as above, i.e. CBC coefficients were close to +1, indicating a high probability of overlap among localizations resulting from R457 and anti-GFP antibody binding (compare Fig. 7M,M′ with Fig. 7N,N′). Taken together, these studies show that both the N- and C-termini of Fn1 are contained within each Fn1 nanodomain and support the model in which Fn1 nanodomains in Fn1 fibrils contain multiple full-length Fn1 molecules.
Nanoarchitecture of Fn1 fibrils formed by ectopically added Fn1
It is known that ectopic Fn1 added to cells assembles into fibrils (McKeown-Longo and Mosher, 1983). To determine whether ectopically added Fn1 was assembled into nanodomains in fibrils, we first used live imaging to film the assembly of ectopic Fn1. In these experiments, MEFs producing Fn1–mEGFP were cultured on an uncoated glass dish inside an insert, whereas a confluent monolayer of MEFs producing Fn1–tdTomato was grown in the space surrounding the insert. Before imaging, the insert was lifted and Fn1–mEGFP-positive MEFs were imaged at ∼17 min intervals for 16 h. These movies show that Fn1–tdTomato proteins became clearly visible as strands of fibrils on the surface of Fn1–mEGFP cells by ∼3 h of co-culture without first accumulating inside cells, suggesting that ectopic Fn1 is assembled at the cell surface (Movie 5). To determine the nanoarchitecture of Fn1 fibrils formed by ectopically added Fn1, we cultured Fn1mEGFP/mEGFP MEFs in the presence of 10 µg of Fn1–tdTomato for 24 h. The ECM in the regions between cells was imaged using TIRF microscopy to detect Fn1–mEGFP and Fn1–tdTomato fluorescence, and dSTORM to detect Fn1–tdTomato due to the AF647 label. Fn1–tdTomato extensively co-assembled with Fn1–mEGFP (Fig. S7A,B), and high-resolution SMLM reconstructions of thin and thick fibrils showed that these fibrils contained nanodomain arrays composed of Fn1–tdTomato proteins (Fig. S7C–F). Taken together, these experiments demonstrated that Fn1 fibrils deposited into the intercellular ECM space are composed of nanodomain arrays and that Fn1 ectopically added to cells assembled into arrays of periodically spaced nanodomains.
Fibrillogenesis inhibitor FUD disrupts the organization of Fn1 nanodomains into arrays
To understand the relationship between the nanodomain architecture of Fn1 fibrils and the process of fibrillogenesis, we adopted a live-imaging approach using Fn1mEGFP/+ MEFs and inhibitors of fibrillogenesis. One such inhibitor is a 49-amino-acid peptide derived from Streptococcus pyogenes adhesin F1, termed the functional upstream domain (FUD), a highly potent inhibitor of Fn1 fibrillogenesis (Tomasini-Johansson et al., 2001). Fn1 fibrillogenesis critically depends on the interactions mediated by the N-terminal 70 kDa assembly domain of Fn1 (marked in blue in Fig. S3D), and FUD is one of the inhibitors that interferes with these interactions (Filla et al., 2017; Morla et al., 1994; Schwarzbauer, 1991; Sechler et al., 2001, 1996; Tomasini-Johansson et al., 2001).
To further investigate the mechanism of Fn1 fibrillogenesis and the role of the N-terminal domain of Fn1 in this process, Fn1mEGFP/+ MEFs were plated on uncoated coverslips for 4 h, and then imaged for 15–18 h either in the imaging medium alone or in the medium containing 225 nM FUD. We also imaged cells incubated with 274 nM III-11C peptide, a 68-amino-acid control peptide that does not interfere with Fn1 fibrillogenesis (Morla et al., 1994; Sottile and Chandler, 2005). Untreated cells or cells treated with the control peptide developed and accumulated long Fn1 fibrils (Movie 6). In contrast, treatment with FUD led to the disassembly of the pre-existing Fn1 fibrils and inhibition of the formation of new Fn1 fibrils (Movie 7). Cells cultured in the presence of FUD contained centripetally moving Fn1–mEGFP fluorescent ‘beads’, which rarely assembled into fibrils (Movie 7). These experiments suggested that FUD inhibits fibrillogenesis by interfering with the process by which Fn1 ‘beads’ are organized into linear arrays. To test this hypothesis, Fn1mEGFP/+ MEFs were cultured for 16 h in the presence of either 225 nM FUD or 274 nM III-11C control peptides, or were left untreated. Cells were then washed with PBS, fixed, and stained without permeabilization using monoclonal anti-Fn1 antibodies and AF647-conjugated secondary antibodies, and imaged at the critical angle of incidence. This approach maximizes the detection of cell-surface Fn1. These experiments demonstrated that the organization of Fn1 nanodomains into linear arrays was lost upon incubation with FUD (compare Fig. 8A,A1,B,B1 with Fig. 8C,C1). Non-fibrillar Fn1 nanodomains in cells treated with FUD had a similar number of Fn1 localizations per nanodomain and were of similar sizes compared with nanodomains in fibrillar or non-fibrillar Fn1 adhesions in untreated cells or cells incubated with the control peptide (Fig. 8A2,B2,C2,D). These data suggest that FUD does not interfere with the formation of Fn1 nanodomains, but inhibits the organization of Fn1 nanodomains into linear arrays (Fig. 8E).
Fig. 8.

The N-terminal Fn1 assembly domain regulates the organization of Fn1 nanodomains into linear fibrillar arrays. Fn1mEGFP/+ MEFs were plated on glass and were either left untreated (A–A2) or incubated with the control III-11C peptide (B–B2) or the FUD peptide (C–C2). Cells were fixed, stained without permeabilization and imaged using SMLM protocol II. Boxes marked 1 and 2 in A,B are magnified in A1,B1 and in A2,B2, respectively. The box in C is magnified in C1 and C2. Arrows in A1,B1 point to Fn1 nanodomains (NDs) in fibrils. Arrows in C1 point to non-fibrillar (NF) nanodomains, which are magnified in C2. Images are representative of three independent experiments. Scale bars: 5 μm (A–C); 500 nm (A1–C1); 100 nm (A2–C2). (D) Quantification of grouped Fn1 localizations in nanodomains. Red lines mark medians, black lines indicate the quartiles. Differences are not statistically significant, determined by Kruskal–Wallis test with Dunn's correction for multiple testing. (E) Models of Fn1 fibril formation. (E1,E2) Nanodomain periodicity in fibrils might be due to an extended Fn1 dimer (E1) or Fn1 subunits (E2). Fn1 molecules are colored according to the scheme shown in Fig. S3D, mEGFP is marked in green, and C-terminal disulfide bonds in red.
DISCUSSION
Our SMLM and live-imaging data show that Fn1 fibrillogenesis entails the formation of mobile nanodomains containing six to 11 Fn1 dimers. The joining of Fn1 nanodomains generates long linear arrays of nanodomains with a periodicity of ∼100 nm. The periodical Fn1 nanodomain arrays can be visualized between cells in culture and within tissues, and have properties of bona fide Fn1 fibrils. Fn1 fibrils have a punctate appearance at the 20 nm resolution by SMLM and 120–200 nm resolution by diffraction-limited microscopy. These observations and unbiased DBSCAN clustering analysis indicate that Fn1 nanodomains in Fn1 fibrils are organized in higher-order periodical structures. Taken together, these data support a novel mechanism of Fn1 fibrillogenesis through the organization of Fn1 nanodomains into linear arrays. In this model, nanodomains co-localize with α5β1 integrins and become aligned as integrins move toward the cell center with the actin flow.
SMLM analyses of Fn1 fibrils visualized with a number of different reagents showed a periodical architecture with regions containing a high number of Fn1 dimers alternating with regions containing low levels of Fn1. This periodical pattern was detected with multiple different antibodies and their combinations. Importantly, anti-GFP antibody staining of Fn1 fibrils assembled by MEFs expressing Fn1–mEGFP showed a periodical architecture similar to that of wild-type MEF fibrils detected with anti-Fn1 antibodies. Others have reported fibrillar structures similar to nanodomain arrays, such as the bulbous appearance of Fn1 fibrils in cryo-scanning transmission electron tomography images (Lansky et al., 2019) and the beaded appearance of Fn1 fibrils in atomic force microscopy (Gudzenko and Franz, 2015). These two methods, which did not rely on antibodies to detect Fn1, validated that the beaded architecture of Fn1 fibrils is independent of antibody labeling and supported the notion that a periodical nanodomain architecture is a general feature of Fn1 fibrils.
Two pieces of evidence suggest that Fn1 nanodomains in arrays are linked: (1) the finding that the periodical nanoarchitecture of Fn1 fibrils is preserved in the absence of cell contact, such as in fibrils deposited into the extracellular space or after the treatment of cells with DOC which dissolves cell membranes; and (2) that nanodomain arrays are lost in cultures treated with the FUD peptide, which blocks interactions involving the N-terminal domain of Fn1. Two important questions arising from our work are how the nanodomains are organized into the arrays and what determines nanodomain periodicity. Because the FUD peptide disrupts the organization of Fn1 nanodomains into arrays, the simplest explanation for forming nanodomain arrays is through Fn1–Fn1 interactions. Indeed, the N-terminal 27 kDa assembly domain is known to be required for Fn1 fibrillogenesis (Schwarzbauer, 1991). This domain mediates intermolecular Fn1–Fn1 interactions, and an N-terminal 70 kDa fragment of Fn1 and antibodies against that fragment have been shown to inhibit fibrillogenesis (McDonald et al., 1987; McKeown-Longo and Mosher, 1985; Sechler et al., 2001). The FUD peptide acts as a potent inhibitor of fibril formation by binding tightly to the N-terminal assembly domain of Fn1 (Ma et al., 2015; Maurer et al., 2010; Tomasini-Johansson et al., 2001) and fluorescent FUD co-localizes with Fn1 fibrils in cell culture (Filla et al., 2017). The packing of Fn1 dimers within nanodomains could leave protruding subunits, which could then interact through their N-termini, explaining the ∼100 nm periodicity of Fn1 nanodomains within the arrays (Fig. 8E1,E2). This model with nanodomains of 11 Fn1 dimers linked by an extended Fn1 molecule would result in a tenfold difference in the number of epitopes in nanodomains versus the intervening regions, and would explain the observed sparsity of antibody localization between nanodomains in arrays (arrowheads in Fig. 4A2–D2, Fig. 5A′–E′, Fig. 6A″–C″).
In vitro studies have suggested that Fn1 fibrils can self-assemble in the absence of cells, obviating the need for additional factors during Fn1 fibril assembly (Nelea and Kaartinen, 2010; Salmerón-Sánchez et al., 2011). However, whether the architecture of self-assembled Fn1 is the same as that of Fn1 fibrils produced by cells is unknown. At this time, we cannot exclude the interesting possibility that molecules other than Fn1 can participate in the organization of Fn1 nanodomains into arrays. For example, prior studies suggested that Fn1 fibrillogenesis can be initiated through the interactions of the N-terminal assembly domain of Fn1 with large apparent molecular mass cell-surface complexes (LAMMs) (Tomasini-Johansson et al., 2006; Zhang and Mosher, 1996). FUD would be able to disrupt LAMM-mediated fibrillogenesis by blocking assembly domain interactions with a yet unknown component of LAMMs. In these studies, biochemical evidence obtained using the 70 kDa Fn1 fragment and Fn1-null cells suggests a role for additional molecules besides Fn1 in fibril assembly. Determining the mechanisms of Fn1 nanodomain linkage and periodicity in nanodomain arrays are areas for future investigation.
The beaded architecture of the Fn1 ECM has important implications for the mechanisms of ECM formation, remodeling and signal transduction. The movement of Fn1 nanodomains in our movies resembles mobile fibrillar adhesions in which integrin α5β1 and tensin are linked to the actin cytoskeleton and move centripetally with the rearward actin flow (Pankov et al., 2000; Zamir et al., 1999, 2000). The relationship between mobile and stationary adhesions and Fn1 nanodomain formation into arrays remains to be determined. It has been shown that the tensile strength of knotted strings is significantly lower than that of strings with uniformly aligned fibers (Arai et al., 1999; Saitta et al., 1999); thus, the beaded architecture of Fn1 fibrils might facilitate their rupture under strain (Ohashi et al., 1999). The non-uniform nanodomain architecture of Fn1 might facilitate the accessibility of Fn1 fibrils to matrix metalloproteases, allowing the degradation of Fn1 fibrils by metalloprotease cleavage of Fn1 nanodomain linkages and thus facilitating dynamic ECM remodeling. Finally, Fn1 binds growth factors (Martino and Hubbell, 2010; Saunders and Schwarzbauer, 2019; Wijelath et al., 2002) and cell adhesion to the ECM is known to orchestrate growth factor signaling (Hynes, 2009). Thus, Fn1 nanodomains could serve as platforms for the binding and presentation of concentrated packets of growth factors to cells. The organization of Fn1 nanodomains into arrays could further facilitate clustering and signaling by growth-factor receptors.
MATERIALS AND METHODS
Generation of Fn1-fluorescent protein-expressing cell lines
Sequences of monomeric (m)EGFP, mNeonGreen, mScarlet-I and tdTomato were obtained from FPbase (https://www.fpbase.org). The sequences encoding the above FPs were knocked into the Fn1 locus following the last coding exon of mouse Fn1, and separated from the last coding amino acid by a flexible, proline-rich linker, PPPELLGGP (Snitkovsky and Young, 1998). Targeting was achieved by CRISPR/Cas9 (Ran et al., 2013). The guide RNA (gRNA) sequence was chosen and off-target sites were identified using GuideScan and Off-Spotter software (Perez et al., 2017; Pliatsika and Rigoutsos, 2015). The gRNA sequence 5′-AGCGGCATGAAGCACTCAAT-3′ targeting the last coding exon of Fn1 was subcloned downstream of the U6 promoter into the PX459 vector (Addgene, #62988) encoding the Cas9-2A-puromycin cassette (Ran et al., 2013). The homology-directed repair (HDR) template was constructed using the pBS-KS (+) vector (Agilent, cat. #212205) (Fig. S1A). The sequence of the last coding exon of Fn1 5′-AACGTAAATTGCCCCATTGAGTGCTTCATGCCGCTAGATGTGCAAGCTGACAGAGACGATTCTCGAGAG-3′ was modified to 5′-AACGTAAATTGCCCCATcGAaTGCTTCATGCCGCTAGATGTGCAAGCTGACAGAGACGATTCTCGAGAG-3′ in the HDR template by introducing silent mutations (lowercase underlined) to prevent targeting of the template by the gRNA. Homology arm 1 contained 677 bp encoding exon 45, an intron and a portion of the last exon (exon 46) of the transcript ENSMUST00000055226.12. Homology arm 2 encoded a 1739 bp sequence immediately downstream of the Fn1 termination codon and included the unmodified 3′-UTR of Fn1. Knockin Fn1mEGFP/+ mice were generated by Biocytogen using the same HDR construct and a longer gRNA, 5′-TAGCGGCATGAAGCACTCAATGG-3′, targeting the same sequence in the last coding exon (the differences between the two gRNAs are underlined). Targeting was confirmed by sequencing and Southern blotting (Fig. S1B). About 500 bp of the top ten predicted off-target sites were sequenced, and no mutations were found in the founder mice. Mice containing correctly targeted Fn1 locus were used to establish colonies of Fn1mEGFP/mEGFP animals. Wild-type, Fn1mEGFP/+ and Fn1mEGFP/mEGFP mice were genotyped using the following primers: Fn1-WT-Fwd, 5′-TCCCCGAAACACACACACTTTTGGT-3′; Fn1-WT-Rev, 5′-GTCACCCTGTTCTGCTTCAGGGTTT-3′; and Fn1GFP-Rev, 5′-GACCCGCGCCGAGGTGAAG-3′. A 372 bp band was detected using the Fn1-WT-Fwd and Fn1-WT-Rev primers for the wild-type allele, whereas the Fn1-WT-Fwd primer and the Fn1GFP-Rev primer located in the GFP sequence gave rise to a 512 bp if the targeted allele was present. Mice were housed in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-approved barrier facility. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Rutgers University and conducted in accordance with the federal guidelines for the humane care of animals.
Cell lines and cell culture
Wild-type mouse embryonic fibroblasts (MEFs) were isolated from embryonic day (E) 13.5 embryos derived from the C57BL/6J strain (Jackson Labs, stock #664), according to established protocols (Behringer et al., 2014), and cultured in complete medium (see Table S3 for culture medium details). For propagation, MEFs were plated in flasks pre-coated with 0.1% gelatin solution made in water, and grown in a complete medium at 37°C with 5% CO2.
Fn1flox/+;RosamTmG/+ MEFs were isolated from E13.5 embryos, as above, and expressed membrane-bound tdTomato and wild-type Fn1 in the absence of Cre recombinase (Muzumdar et al., 2007). Fn1-null MEFs were generated by treating Fn1flox/−;RosamTmG/+ cells with the Ad-Cre-IRES-GFP adenovirus (Vector Biolabs, #1710) according to the manufacturer's recommendations. In this experiment, Cre recombinase is expressed transiently and mediates site-specific recombination between a pair of loxP sites flanking the first exon of Fn1 (Sakai et al., 2001) and another pair of loxP sites flanking the STOP cassette in the mTmG reporter (Muzumdar et al., 2007). Three days following infection with Ad-Cre-IRED-GFP, GFP-positive cells were sorted, resulting in a pure population of Fn1-null MEFs (confirmed by immunofluorescence, Fig. S8). Fn1-null MEFs were cultured on gelatin-coated dishes in complete medium.
Fn1–FP-expressing cells (except Fn1–mEGFP cells) were generated by CRISPR/Cas9 mutagenesis of wild-type MEFs. CRISPR/Cas9 targeting was performed by transfecting wild-type MEFs using the PX459 plasmid encoding Fn1 gRNA and the HDR template using Lipofectamine 3000 (Thermo Fisher Scientific, cat. #L3000015), as described (Ran et al., 2013). MEFs expressing Fn1–mEGFP proteins that were used for live imaging and STORM were generated from homozygous E13.5 Fn1mEGFP/mEGFP embryos or, when noted, from Fn1mEGFP/+ embryos. Fn1mEGFP/mEGFP MEFs were used in experiments to quantify Fn1 molecule number in nanodomains and to measure nanodomain spacing and nanodomain diameter, as well as in experiments to label both the N- and the C-termini of Fn1. For live imaging, Fn1mEGFP/+ MEFs were used. Fibronectins from Fn1mEGFP/mEGFP and Fn1mEGFP/+ cells behaved equivalently in Fn1 matrix assembly assays (Fig. S1F).
NUP96–mEGFP-expressing U2OS cells (Thevathasan et al., 2019) (clone #195, Cell Line Services, #300174) were cultured according to the vendor's specifications in McCoy's 5A medium (VWR, cat. #MSPP-302007) containing GlutaMax (Thermo Fisher Scientific, #35050061), 10% v/v fetal bovine serum (Gemini Biosciences, #100-106) and penicillin/streptomycin (GE Healthcare, #SV30010).
Reagents and buffers
FUD and III-11C peptides were generated as described previously (Sottile and Chandler, 2005; Tomasini-Johansson et al., 2001) and stored in PBS at −80°C. A 4% DOC solution was prepared by dissolving 0.4 g deoxycholate salt (Sigma-Aldrich, #D6750) in 10 ml of imaging medium (see Table S3 for imaging medium details); the solution was then vortexed and filter sterilized. The pH of the final solution was 8.01. A 16% paraformaldehyde (PFA) solution (Electron Microscopy Sciences; #50-980-487) was diluted in PBS to prepare 4% PFA. The 4% PFA solution was aliquoted into 1 ml microfuge tubes, stored at −80°C, and thawed at 37°C immediately before use.
Phosphate buffered saline (PBS, pH 7.5) was prepared from 10× PBS (VWR, #76180-740). PBST was prepared using 0.1% Triton X-100 (Sigma-Aldrich, #T-8787) and used for all staining protocols except those involving NUP96–mEGFP U2OS cells, as detailed below. Blocking buffer was prepared by adding 10% donkey serum (Sigma-Aldrich, #D9663) to PBST. A 5 mg/ml stock of DAPI (Thermo Fisher Scientific, #D3571) was prepared in water and used at 1:300 dilution. Stain Buffer (BD Pharmingen, #554656) was used for antibody dilutions and washing of cells that were stained without permeabilization. Hoechst 33342 Trihydrochloride (Thermo Fisher Scientific, #H1399, 10 mg/ml) was used for labeling live MEFs at 1:300 dilution. In live MEFs, F-actin was labeled using SiR actin (Cytoskeleton, #CY-SC001, used at 1 µM final concentration). mCardinal-Lifeact-7 was acquired from Addgene (plasmid #54663; deposited by Michael Davidson). Vectashield antifade mounting medium (Vector Laboratories, #H-1000) was used for coverslipping.
For STORM imaging, we used 25 mm high-precision glass coverslips #1.5H (Marienfeld, #0117650; obtained from Azer Scientific, PA, #ES0117650) without coating. Before their use, glass coverslips were cleaned using concentrated nitric acid, washed in water, air dried and autoclaved, as described in Kaech and Banker (2006). Clean coverslips were stored in sealed six-well plates for no longer than a week before their use.
GLOX/BME STORM buffer contained 50 mM Tris-HCl, pH 8.0 (Thermo Fisher Scientific, #T-395-1), 10 mM NaCl (Sigma-Aldrich, #S-7653), 10% glucose (Sigma-Aldrich, #G8270), 0.5 mg/ml glucose oxidase (Sigma-Aldrich, #G2133), 40 µg/ml catalase (Sigma-Aldrich, #C40) and 143 mM β-mercaptoethanol (BME, Sigma-Aldrich, #444203) (Thevathasan et al., 2019). As described in Jimenez et al. (2020), stocks of enzyme solutions were prepared and stored at −20°C. GLOX/BME buffer was used for STORM imaging of cells plated on coverslips.
GLOX/MEA STORM buffer was used for STORM imaging of cells plated in ibidi glass-bottomed eight-well chambers (cat. #80827). This buffer was prepared as above, but instead of BME, it contained 50 mM mercaptoethylamine (MEA, Sigma-Aldrich, #30070) (Jimenez et al., 2020). For double-color STORM imaging, we used the SMART Kit buffer (Abbelight).
Antibodies
All primary antibodies were checked for specificity on cells that were genetically null for the antigen (e.g. Fig. S8A) and tissues: Fn1-null tissue sections obtained from Fn1-null embryos were used to assay the specificity of each of the anti-Fn1 antibodies; tissues isolated from Itga5-null embryos (derived from the cross between Itga5+/− mice, Jackson Labs, cat. #002274), and GFP-null and mCherry-null embryos (derived from wild-type C57BL/6J mice (Jackson Labs, cat. #664) were used to authenticate the specificity of the anti-Itga5, anti-GFP, and anti-mCherry antibodies. For each of the antibodies, the staining of control tissues resulted in no more fluorescent signal than the background fluorescence produced by the use of secondary antibodies only. Rabbit polyclonal antibody R457 was raised against the 70 kDa N-terminal domain of Fn1 (Aguirre et al., 1994; Sechler et al., 2001) and rabbit polyclonal R184 was raised against the first six type III repeats of Fn1 (Raitman et al., 2018). The specificity of these antibodies was verified by enzyme-linked immunosorbent assay (ELISA) and western blotting, reported in the references in Table S4. The details of all antibodies used are given in Table S4.
Embryo staining
E9.5 embryos were isolated either from matings of wild-type C57BL/6J mice or from mating Fn1mEGFP/mEGFP mice with wild-type mice to obtain Fn1mEGFP/+ embryos. Embryos were fixed using cold 4% PFA overnight at 4°C, washed three times for 5 min in PBS and blocked overnight at 4°C in blocking buffer containing PBS, 0.1% Triton-X and 10% donkey serum. Embryos were stained either with Abcam monoclonal anti-Fn1 antibodies at 1:300 dilution in blocking buffer or with both Abcam monoclonal anti-Fn1 antibodies and anti-GFP antibodies at 1:300 dilution. The staining and imaging were performed exactly as described in Ramirez and Astrof (2020).
Treatment of cells with DOC
Around 104 Fn1mEGFP/+ MEFs were plated for 48 h in eight-well glass-bottomed ibidi dishes in complete medium and incubated at 37°C with 5% CO2. Two hours before imaging, SiR-actin was added at a final concentration of 1 µM. SiR-actin contains a far-red dye, silicon rhodamine, conjugated to jasplakinolide, which labels F-actin in live and fixed cells (Lukinavičius et al., 2014). Just before imaging, complete medium was replaced by 150 µl imaging medium containing 33 µg/ml of Hoechst 33342 (Thermo Fisher Scientific, #H1399, 10 mg/ml). Positions were marked in each well and live imaging was initiated in a humidified chamber at 37°C and 5% CO2. After 15 min, 150 µl of 4% DOC solution prepared in imaging medium containing 33 µg/ml Hoechst 33342 was added to the experimental well (final pH 8.01) and 150 µl imaging medium containing 33 µg/ml Hoechst 33342 but without DOC was added to the control well. Cells were imaged at 50 s intervals until F-actin and DNA disappeared (see Movie 3). The medium was then removed, cells were rinsed for 1 min with PBS that had been pre-warmed to 37°C, and fixed with 4% PFA that had been pre-warmed to 37°C. For staining, cells were permeabilized and blocked as described above, and incubated with the Abcam monoclonal anti-Fn1 antibody diluted to 1:300 in the blocking solution at 4°C overnight. Primary antibodies were detected with anti-rabbit secondary antibodies conjugated to AF647. Enhanced-resolution imaging was used to image the DOC-treated fibrils, as described below.
Confocal settings for enhanced resolution imaging
Confocal images of fixed samples were recorded using a Nikon A1-HD25 inverted confocal microscope equipped with CFI Apochromat TIRF 100XC Oil objective with the pinhole set to 0.8 Airy units, and imaged through 2–4 μm with a step size of 0.125–0.15 µm at a sampling of 40 nm per pixel and 180 nm optical resolution. Deconvolution was done using Nikon 3D deconvolution software (v5.11.01). Airyscan imaging was performed using a Zeiss LSM 880 microscope fitted with a 32 array Airyscan GaAsP-photomultiplier tube (PMT) detector and the Plan Apochromat 63× Oil (NA 1.4) objective. Deconvolution and pixel reassignment were done using Zeiss LSM software.
Live imaging using TIRF
Fn1mEGFP/+ MEFs were plated on 35 mm round glass-bottomed MatTek dishes (#P35G-1.5-14-C), and the complete medium was switched to the imaging medium prior to filming. Live TIRF microscopy was performed using the Nikon A1-HD25 inverted confocal microscope equipped with four laser lines of 100 mW per line at 405, 488 and 561 nm, and 125 mW at 640 nm, and motorized TIRF illumination. CFI Apochromat TIRF 100XC Oil objective and EMCCD camera were used. Before imaging, the lasers were aligned and the critical angle of incidence for imaging was determined by the software. The exposure time was 20 ms and readout speed was set at 10 MHz.
Live imaging using confocal point-scanning microscopy
Live-cell imaging was performed using ibidi glass-bottomed eight-well chambers (#80827). Approximately 0.6×104 wild-type or Fn1mEGFP/+ MEFs were plated in each well of the ibidi glass-bottomed eight-well chambers, and allowed to grow overnight prior to staining and imaging by dSTORM (see SMLM imaging protocol II below). For FUD and III-11C treatment, Fn1mEGFP/+ MEFs were plated in eight-well glass-bottomed ibidi dishes (1 cm2 growth area) without coating at a density of 0.6×104 cells/well in complete medium. After 5 h, the complete medium was removed and cells were rinsed once with PBS. Subsequently, the medium was changed to imaging medium. For FUD experiments, the imaging medium was supplemented with either 225 nM FUD or 274 nM of the control III-11C peptide. Untreated wells contained cells incubated with the imaging medium. Following the addition of the imaging medium (with or without the peptides), the chamber was immediately set up for imaging in the humidified Tokai Hit stage-top incubator at 37°C and 5% CO2. Live imaging was performed using the Nikon A1-HD25 inverted confocal microscope with the DUG four-channel detector, two GaAsP, two high-sensitivity PMTs, and a motorized xyz stage with Nikon's Perfect Focus 4 system, and Plan Fluor 40× Oil (numerical aperture 1.3, #MRH01401). mEGFP was excited using a 488 nm laser at 1% power and pinhole set to 1 Airy unit. An optical zoom of 2 and z-step size of 0.5 µm were used, and the stack size was set to 10–15 μm, allowing imaging of the entire cell. For overnight movies, each position was filmed every 1.5–4 min, as noted in the Movie legends. Movies in the MP4 format were generated using Imaris 9.5.1 (Bitplane), and titles and arrows were added using Adobe Premiere Elements Editor 2020.
Preparation of coverslips for SMLM imaging
Coverslips with plated cells were rinsed with GLOX/BME buffer and mounted onto single-cavity glass slides (VWR, #10118-600) and pre-filled with 80 µl GLOX/BME buffer immediately prior to placement of the coverslip. Excess buffer was fully adsorbed from the sides and the top of the coverslip using Whatman paper, taking care to keep the coverslip centered on top of the cavity. Coverslips were sealed onto the cavity slide by pipetting Acid-Free Elmer's No-Wrinkle Rubber Cement around the edge, and allowing the rubber cement to cure for ∼15 min. After imaging, the rubber cement was gently peeled off, and the slides were soaked in PBST for 5 min at room temperature (RT) to remove coverslips which were then stored in six-well plates filled with PBST containing 0.02% NaN3 at 4°C until further use. In this set up, the GLOX/BME buffer remained at pH 8 for at least 12 h.
Quantification of ELE and the number of Fn1 molecules in Fn1 nanodomains
To quantify the number of Fn1 molecules per nanodomain and to measure the ELE, we used NUP96–mEGFP U2OS cells as a reference cell line and SMAP software (Ries, 2020; Thevathasan et al., 2019). The stoichiometry of NUP96–mEGFP in nucleopores is well characterized, and imaging methodology and software have been developed for careful measurements of the stoichiometry (Diekmann et al., 2020; Ries, 2020; Thevathasan et al., 2019). Together, these tools allow the use of the NUP96–mEGFP cell line as a reference to assess the quality of SMLM imaging, measure ELE and determine the number of Fn1–mEGFP molecules in Fn1 nanodomains.
NUP96–mEGFP U2OS cells and Fn1–mEGFP MEFs were cultured as described above. Approximately 3×105 NUP96–mEGFP cells and 5×104 Fn1–mEGFP cells were plated in their respective culture mediums on 25 mm high-precision glass coverslips #1.5H (Marienfeld, #0117650, obtained from Azer Scientific, #ES0117650) positioned in six-well plates (Corning, #353046). Prior to their use, glass coverslips were cleaned using concentrated nitric acid, washed in water and autoclaved, as described in Kaech and Banker (2006). Coverslips were used without any coating. 24 h after plating, cells were fixed and stained as described in Thevathasan et al. (2019) with minor modifications. Cover slips with NUP96–mEGFP cells and Fn1–mEGFP cells were handled simultaneously in pairs at each step. For fixation, permeabilization and washing, coverslips were kept in six-well plates. Cells were fixed in PBS containing 2.4% PFA for 20 min at RT. PFA was aspirated and cells were incubated with 100 mM NH4Cl in PBS for 5 min at RT, and then washed three times for 5 min in PBS with agitation. Cells were permeabilized for 20 min at RT using PBS containing either 0.2% Triton-X for NUP96–mEGFP cells or 0.1% Triton-X for Fn1–mEGFP cells. Cells were then washed in PBS three times for 5 min at RT, washed once more for 5 min in PBS containing 0.1% Triton-X, and then either used immediately for staining or stored at 4°C in PBST until further use. For each experiment, sufficient amounts of each solution were prepared such that NUP96–mEGFP and Fn1–mEGFP cells were incubated with the same mixtures. Solutions containing blocking reagents and antibodies were spun for 5 min at 16,000 g using tabletop centrifuges to get rid of particulates. Humidified chambers were prepared from empty pipette-tip boxes with water placed in the lower chamber and parafilm partially covering the surface of the tip rack. Drops of solutions were placed on the parafilm, and cells were incubated with various solutions by inverting coverslips on top of the droplets. To block non-specific antibody binding, cells were first incubated with a blocking solution (10% donkey serum in PBST) for 30 min at RT. Coverslips were then gently lifted, drained and incubated with anti-GFP antibody (Aves Labs, #GFP-1010) diluted 1:100 in the blocking solution overnight at 4°C. Coverslips were then placed into six-well plates and washed three times for 5 min in PBST with agitation. Cells were then incubated with AF647-conjugated anti-chicken F(ab)2 (Jackson ImmunoResearch, 703-606-155) diluted 1:300 in the blocking solution for 4 h at RT, and then washed three times for 5 min in PBST with agitation. Stained coverslips were stored at 4°C until imaging.
SMLM imaging
Protocol I: Quantification of the ELE and the number of mEGFP molecules in Fn1–mEGFP nanodomains
For the following experiments, two-dimensional (2D) imaging was used to maximize the resolution in the plane of imaging. To minimize fluorophore bleaching and maximize the number of collected photons, we followed the protocol developed by Diekmann et al. (2020). Stained NUP96–mEGFP and Fn1–mEGFP cells were imaged in pairs using the same preparation of the GLOX/BME buffer and the same imaging conditions (described below). For each independent experiment (n=3) and for each round of measurements, images of NUP96–mEGFP and Fn1–mEGFP cells taken on the same day were analyzed. SMLM was performed using a Nikon A1-HD25 Ti2E microscope equipped with motorized TIRF illumination, 125 mW 640 nm solid-state laser, Perfect Focus and CFI Apochromat TIRF 100XC Oil objective with numerical aperture 1.49 (#MRD01905). All images were acquired at the critical angle of incidence (57°) and recorded using a 512×512 EMCCD camera (Princeton Instruments), using 128×128 central region on the camera. Gain was set to 3 and multiplication gain amplifier was set to 20 MHz. Prior to acquisition, the centered region of interest (ROI) (128×128) was bleached at a 57° angle and 10% laser power for 500 frames, followed by an additional 500 frames at 20% laser power, with the exposure of 60 ms per frame. Images were then acquired at a 57° angle and 20% laser power for 60,000 frames at 60 ms per frame. Images were processed, rendered and quantified using SMAP and MATLAB version 2020a (Ries, 2020), as described in Diekmann et al. (2020) and the documentation found on GitHub (https://github.com/jries/SMAP). Settings for peak finding in the SMAP software were set according to our camera manufacturer’s specifications and were as follows: EM was set to ‘on’, camera pixel size was set to 0.162 µm, EM gain was set to 300 and e-/ADU conversion factor was set to 2.35. Data was fitted using a Gaussian point spread function (PSF) model, the cutoff parameter for peak finding was set to 2 and ROI size was set to 7 pixels. Localizations were then grouped and rendered using the default parameters in SMAP. Drift correction was performed on rendered localizations in timepoint blocks of 10 or 20, as recommended (Thevathasan et al., 2019; https://github.com/jries/SMAP). Following satisfactory drift correction judged by the overlapping cross-correlations, localizations were filtered according to the recommended settings for AF647 (Thevathasan et al., 2019). In brief, localizations with poor precision were filtered out by limiting localization precision to 0–15 nm, out-of-focus localizations were excluded by setting the PSF range to 0–150 nm in the xy plane, poorly fitted localizations were further filtered out by setting the LLrel parameter (relative log likelihood) in SMAP to a negative cut-off value leaving the majority of the peak intact, and the first 1000 frames were excluded from the analyses. All the remaining grouped localizations were rendered according to 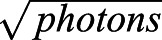 , and images were color-coded using look-up tables set according to localization density.
, and images were color-coded using look-up tables set according to localization density.
Image resolution was measured using the Fourier ring correlation (FRC) method described by Nieuwenhuizen et al. (2013) and implemented in SMAP. Voronoi cluster discovery was performed according to Andronov et al. (2016a) and their algorithm implemented in SMAP. DBSCAN cluster analysis was performed according to Caetano et al. (2015) and Ester et al. (1996) implemented in SMAP. For DBSCAN, the minimum number of points in the neighborhood (k) was set to 4, as recommended for all 2D data (Ester et al., 1996). The neighborhood radius ε was either set to 14 nm (the average apparent radius of Fn1 nanodomains was determined as described below) or automatically estimated by the DBSCAN algorithm in SMAP (Ries, 2020).
NPC radius, ELE and the number of grouped localizations per fluorophore were determined using the algorithm in SMAP following the published automated workflow to segment and analyze NPCs, using the parameters recommended by Thevathasan et al. (2019) without modifications. In brief, NPCs in focus (mean fitted PFS size of each NPC was less than 145 nm) were automatically segmented using circular ROIs of 220 nm diameter. The number of grouped localizations per NPC, NPC radius and ELE were determined using published algorithms implemented in SMAP without modifications, as described in Diekmann et al. (2020), Thevathasan et al. (2019) and the SMAP user manual ‘Analysis of NPCs using SMAP’ (https://www.embl.de/download/ries/Documentation/SMAP_manual_NPC.pdf). As we used identical staining, imaging and processing parameters for NUP96–mEGFP and Fn1–mEGFP cells, the number of Fn1–mEGFP molecules per nanodomain was calibrated to the number of grouped localizations per NUP96–mEGFP protein, as described previously (Thevathasan et al., 2019). In brief, we first determined the number of merged localizations per NPC in the ROI (Lref). Each NPC contains 32 NUP96–mEGFP proteins; therefore, the number of merged localizations per NUP96–mEGFP (Nref) is Nref=Lref/32. To determine the number of Fn1–mEGFP proteins per Fn1 nanodomain, we segmented Fn1 nanodomains manually using a circular ROI of 60 nm diameter, and the number of merged localizations per ROI (Lt) was determined by using the countingStatistics plugin in SMAP, as outlined in the SMAP manual. The number of Fn1–mEGFP proteins per nanodomains (Nt) is Lt/Nref. As Fn1 incorporated into fibrils is an obligate dimer, the number of Fn1 dimers per nanodomain is Nt/2.
Nanodomain periodicity in Fn1 fibrils stained by a variety of different antibodies was assayed and quantified according to Früh et al. (2015). In brief, MEFs were stained using a variety of antibodies at different dilutions or using combinations of antibodies (noted in Figs 5, 6) according to the staining protocol described above. MEFs were then imaged using GLOX/BME buffer and the exact imaging settings as described above for imaging NUP96–mEGFP U2OS cells and Fn1–mEGFP MEFs stained with anti-GFP antibodies. Images were processed and rendered in SMAP according to the parameters described above. Gaussian rendering of the localizations (min σ Gaussian was set to 3 nm) were saved as uncompressed TIFF files and opened in Fiji version 2.1.0/1.53c, and intensity line profiles along each fibril were generated and imported into MATLAB 2021a. To assay periodicity, we used the autocorrelation function implemented in the MATLAB's 2021a Econometrics toolbox and the criteria outlined in Früh et al. (2015). In brief, the presence of at least four regularly spaced peaks in the autocorrelation profile was considered to reflect the periodical nature of Fn1 nanodomains, and the position of the first autocorrelation maximum was taken as a quantitative measure of nanodomain periodicity, as extensively discussed and computationally modelled in the supplementary information of Früh et al. (2015). Ljung–Box Q test for residual autocorrelation was performed using MATLAB's 2021a Econometrics toolbox to assess statistical significance of autocorrelation.
To determine the apparent diameter of Fn1 nanodomains, we imported fibril intensity line profiles obtained from SMLM images of Fn1mEGFP/mEGFP MEFs stained for GFP into MATLAB 2021a. To automate the analyses, we used the Signal Processing toolbox in MATLAB 2021a to fit each intensity peak in the line profile with a Gaussian curve and calculate full width at half height (FWHT) for each peak. Altogether, 1292 nanodomains were assessed in 27 long (>1 µm in length) fibrils from six cells and three independent experiments. We also performed this analysis manually on a subset of Fn1 nanodomains (n=248) in long fibrils by fitting a Gaussian to an intensity profile of each nanodomain and calculating FWHT. The results were the same.
Protocol II: Imaging cells plated on ibidi glass-bottomed dishes
This protocol was used for images shown in Fig. S6, and for SMLM that followed live imaging in ibidi plates (Fig. 8). Following fixation with 4% PFA, cells were washed with PBS, incubated with blocking buffer containing PBS, 0.1% Triton-X and 10% donkey serum for 30 min at RT. Cells were then incubated with the Abcam monoclonal anti-Fn1 antibody diluted at 1:300 in blocking buffer overnight at 4°C. Following three washes at 5 min each in PBST, cells were incubated with AF647-conjugated anti-rabbit secondary antibodies diluted 1:300 in blocking buffer for 4 h at RT and washed three times for 5 min each in PBST. Before imaging, freshly prepared GLOX/MEA buffer was added, and the chamber was immediately sealed using parafilm. STORM was performed using a Nikon A1-HD25 Ti2E microscope equipped with motorized TIRF illumination, 125 mW 640 nm solid-state laser, Perfect Focus, and a 100×/1.49 NA objective. Images were acquired at the critical angle of incidence and recorded using a 512×512 EMCCD camera (Princeton Instruments). Calibration was obtained by imaging 100 nm Tetraspeck beads (Life Technologies, #T-7279) using the same glass surface and buffer conditions. To drive AF647 into the dark state, samples were pre-bleached by the illumination at 640 nm for 10 s at 100% laser power. 40,000 frames were acquired at 8.4 ms exposure. Blinking events were fitted using the Nikon N-STORM localization software. Images were analyzed using Nikon Elements AR Software (v5.11.01). Localization events with fewer than 800 or more than 50,000 photons were filtered out to remove blinking events that were either too faint or too bright (Jimenez et al., 2020). In addition, blinking events were filtered out if they occurred in more than five consecutive frames or were outside the z-range determined by the calibration using 100 nm Tetraspeck beads. Images in which z-rejection was below 50% were used for further analyses.
Analysis of localization numbers in fibrillar and non-fibrillar nanodomains
To enrich for non-fibrillar nanodomains, Fn1mEGFP/+ MEFs were plated in eight-well glass-bottomed ibidi dishes (1 cm2 growth area) without coating. Cells were plated at a density of 0.6×104 cells/well in the imaging medium with or without FUD (225 nM) or III-11C (274 nM) and incubated at 37°C with 5% CO2 for 1 h. Subsequently, MEFs were rinsed once in warm PBS and fixed using pre-warmed 4% PFA for 20 min. After fixation, wells were rinsed three times for 5 min each with Stain Buffer (BD Pharmingen, #554656), blocked for 30 min at room using 5% donkey serum prepared in Stain Buffer, and incubated with the monoclonal anti-Fn1 (Abcam, #199056) overnight at 4°C. Cells were then washed with Stain Buffer three times for 10 min each, and incubated with anti-rabbit antibodies conjugated with AF647 for 1 h at RT. Cells were then rinsed again with Stain Buffer three times for 10 min each, and stored at 4°C in PBS. STORM imaging was performed, as described in SMLM imaging protocol II. To quantify the number of grouped localizations per nanodomain, we used the free-hand ROI tool in the STORM window (Nikon Elements AR Software v5.11.01) to determine the number of localizations within non-fibrillar and fibrillar Fn1 nanodomains. Fn1 nanodomains were analyzed in five random regions from three independently acquired images (a total of 15 fields) for each sample. To determine the number of localizations in Fn1 nanodomains within fibrils, we analyzed more than 20 nanodomains from three or more independently acquired images. The counts were plotted in Prism 8.2.1 (GraphPad Software) and compared using either a one-way ANOVA test with Tukey's correction or the Kruskal–Wallis test with Dunn's correction for multiple testing.
Double-color dSTORM acquisition
Samples were mounted with the SMART Kit buffer (Abbelight). 2D or 3D STORM images were acquired using a SAFe360 module (Abbelight) coupled to an inverted bright-field Olympus IX83 microscope, equipped with a 100× oil-immersion objective (1.49 NA). This dual-camera system (sCMOS cameras, Orcaflashv4, Hamamatsu) allows for performing multicolor STORM by the spectral demixing strategy coupled with the use of far-red dyes. Briefly, a dichroic filter at 700 nm separates the fluorescence emission from AF647 and CF680 dyes. The PSF of each detection can be retrieved on both cameras, and the measured photon numbers are related to the spectral separation of the fluorophore (Fig. S8C,D). We acquired 60,000 frames at 20 ms exposure time on a camera sensor size of 1024×1024 pixels to collect single-molecule detections. The irradiation at the sample was tuned according to the ASTER technology (Mau et al., 2021) implemented on the SAFe360 Abbelight module. The resulting coordinate tables and images were processed and analyzed using NEO software (Abbelight). As the PSF is captured on both cameras, transmitted and reflected, a ratiometric analysis was applied. A ratio for each detection was calculated, and the final ratio distribution used for lambda assignment:
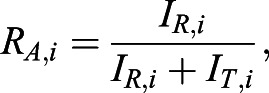 |
in which the suffix A is the fluorophore, i is the localization, and IR and IT are the intensities measured (i.e. the number of photons emitted per molecule) on camera R and camera T, respectively, as shown in Fig. S8C.
Average ratio distributions obtained from measurements are shown in Fig. S8E. Following the ratio distributions measured in the samples, the following parameters were used for separation: detections with ratios between 0–0.45 were assigned to CF680, and detections with ratios between 0.5–1 were assigned to AF647. On average, 3% of detections were rejected while keeping the crosstalk below 1%. After de-mixing, co-localization analysis was performed using NEO software according to the CBC algorithm (Malkusch et al., 2012). Parameters set for CBC analysis were Rmax at 300 nm and the number of steps equal to 10. In addition, the CBC algorithm implemented in ThunderSTORM, with Rmax=50 nm and the number of steps equal to 10 (Ovesný et al., 2014) was used to perform CBC analyses (Malkusch et al., 2012).
Epitope mapping of the rabbit 297.1 polyclonal antibody
Epitopes recognized by the 297.1 polyclonal antibody were mapped by generating custom overlapping peptide arrays (PEPperPRINT, Heidelberg, Germany). The Fn1 protein sequence including alternatively spliced exons was encoded by 15-amino-acid peptides with a peptide-peptide overlap of 13 amino acids. The resulting Fn1 peptide microarrays contained 1239 different peptides printed in duplicate (2478 peptide spots) and were framed by an additional HA-tag (YPYDVPDYAG, 106 spots) as a control peptide. The HA-tag peptide was used to monitor array quality and served as a positive control for clean antibody binding detected with the anti-HA-tag antibody (mouse monoclonal anti-HA DyLight800, Thermo Fisher Scientific, cat. #26183-D800; used at 0.5 µg/ml concentration). Prior to staining, arrays were incubated with blocking buffer (Rockland, #MB-070) for 30 min at RT. To measure background antibody binding, arrays were first incubated with secondary goat anti-rabbit IgG (Fc) DyLight680 (Thermo Fisher Scientific, cat. #35568) at 0.2 µg/ml concentration diluted in PBS, pH 7.4, containing 0.05% Tween 20 and 10% blocking buffer, and imaged. To determine specific 297.1 antibody binding, arrays were incubated for 16 h with shaking at 4°C with two dilutions of the 297.1 antibody (1:300 and 1:1000). Antibody dilutions were made in PBS, pH 7.4 containing 0.05% Tween 20 and 10% blocking buffer. Arrays were then washed with PBS at pH 7.4 containing 0.05% Tween 20, and incubated with goat anti-rabbit IgG (Fc) DyLight680 (0.2 µg/ml) for 45 min at RT. Arrays were then washed and imaged using LI-COR Odyssey Imaging System, with a scanning offset of 0.65 mm, resolution of 21 µm and scanning intensities of 7/7 (red=700 nm/green=800 nm). Quantification of spot intensities and peptide annotations were done with PepSlide Analyzer (PEPperPRINT, Heidelberg, Germany).
Analysis of Fn1 matrix assembly and western blotting using MEFs
Matrix assembly was performed according to established protocols (Wierzbicka-Patynowski et al., 2004). MEFs were plated in six-well dishes (9 cm2 growth area) at a density of 2×105 cells per well for 48 h in complete medium and incubated under sterile conditions at 37°C with 5% CO2. Cells were washed twice with ice-cold PBS (supplemented with Mg2+ and Ca2+), scraped with a cell scraper and lysed with either 500 µl RIPA lysis buffer, pH 8.0 [50 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, 1% v/v NP-40, 0.5% w/v sodium deoxycholate, 0.1% w/v SDS and 1× protease inhibitor cocktail (Cell Signaling Technology, #5871)], or DOC lysis buffer, pH 8.8 (20 mM Tris-HCl, 2 mM EDTA, 2% w/v sodium deoxycholate, 1× protease inhibitor cocktail). Extracts were carefully transferred to Eppendorf tubes containing 1 µl (250 units) benzonase nuclease (Sigma-Aldrich, #E1014), mixed by inverting a few times and incubated at 37°C for 15 min. The samples were then centrifuged at 16,000 g for 15 min at 4°C. For cells lysed with DOC lysis buffer, the supernatant containing DOC-soluble material was carefully removed and the pellet containing the DOC-insoluble material was resuspended in 100 µl SDS solubilization buffer, pH 8.8 (20 mM Tris-HCl, 2 mM EDTA, 1% w/v SDS, 1× protease inhibitor cocktail). The DOC-insoluble pellet was thoroughly dissolved by heating the sample to 95°C and vortexing. All samples were aliquoted and stored at −80°C until further use. Prior to quantification of Fn1 in the samples, the total protein concentration of the RIPA and DOC lysates was determined using the Pierce BCA Protein Assay Kit (23225). Lysates containing Fn1 and Fn1–FP fusion proteins were reduced and resolved using 66–440 kDa Wes Separation Module (ProteinSimple, #SM-W007). Primary antibodies were used at the following dilutions: anti-Fn1, 1:1000 (Abcam, ab199056); anti-GFP, 1:1000 (Roche, 11814460001); and anti-mCherry, 1:1000 (Abcam, ab167453). Primary antibodies were detected using horseradish peroxidase-conjugated secondary antibodies (anti-Rabbit Detection Module, ProteinSimple, DM-001), and chemiluminescence was quantified using the Compass for SW software (v3.1.8). Prior to running experimental samples, care was taken to optimize the dilutions of lysates to be within the linear range of the detection.
Analysis of Fn1 matrix assembly and western blotting using E9.5 embryos
To analyze Fn1–mEGFP matrix assembly in vivo, we mated Fn1mEGFP/+ mice to obtain wild-type, Fn1mEGFP/+ and Fn1mEGFP/mEGFP littermate embryos. At the time of dissection, each embryo was frozen on dry ice in individual Eppendorf tubes and stored at −80°C until the genotyping was complete. Yolk sacs were used for genotyping. 300 µl of DOC ice-cold lysis buffer at pH 8.8 containing protease inhibitors were added to each embryo, and the embryos were dissociated by passing through a 27-gauge syringe needle five times, keeping the tubes on ice. Lysates were centrifuged at 16,000 g for 15 min at 4°C. The supernatant containing DOC-soluble material was transferred to another tube and was supplemented with 100 µl of 4× SDS loading buffer and BME at a final concentration of 350 mM BME. The DOC-insoluble pellet was washed twice with 300 µl DOC lysis buffer on ice, and the pellet was resuspended with 200 µl SDS solubilization buffer with protease inhibitors. The pellet was dissolved by heating at 95°C and vortexing, and supplemented with 66.6 µl of 4× SDS loading buffer and BME, at a final concentration of 350 mM BME. DOC-soluble and insoluble samples were heated at 95°C for 5 min and 40 µl was loaded on 4–12% acrylamide gels (Invitrogen, #XP04120BOX). Fn1 was detected using rabbit anti-Fn1 primary antibody (Abcam, ab199056) and IRDye 680RD donkey anti-rabbit secondary antibody (LiCor, #926-68073). Membranes were imaged using LiCor Odyssey 9120 Gel Imaging System (#ODY-2425) and quantified using Fiji software.
Testing the reactivity of the 297.1 polyclonal antibody to bovine Fn1 present in fetal bovine serum
To test whether the 297.1 antibody could bind bovine Fn1 present in fetal bovine serum, we used a complete medium (CM, see Table S3) and, as a control, a conditioned completed medium (CCM). To prepare CCM, 2×105 cells plated in a single well of six-well plates were incubated for 48 h with 2 ml of CM, and the medium was then collected and centrifuged at 300 g for 3 min to discard dead cells. 100 µl of trichloroacetic acid (TCA) was added to 900 µl of CM or CCM and incubated on ice for 30 min to precipitate proteins. Samples were centrifuged at 16,000 g at 4°C for 15 min. Pellets were washed with 700 µl of 100% acetone and resuspended in a gel loading buffer containing 100 µl 0.1N NaOH, 75 µl 4× SDS-PAGE loading buffer, 0.35 M BME and 125 µl H2O. Samples were heated at 95°C for 5 min and 10 µl of each sample was resolved using Novex WedgeWell 4 to 12%, Tris-Glycine, 1.0 mm, Mini Protein Gels (Invitrogen, #XP04120BOX) and Tris-Gly SDS Running Buffer (Invitrogen, #LC2675). Following the transfer to nitrocellulose membranes (Bio-Rad, #1620122), Fn1 was detected by immunoblotting using 1:1000 dilution of 297.1 polyclonal antibodies and IRDye 680RD conjugated to donkey anti-rabbit secondary antibody. Protein standards were from Invitrogen (#LC5925). Membranes were imaged using LiCor Odyssey 9120 Gel Imaging System.
Coating of coverslips with different ECM proteins
#1.5 round glass coverslips (Electron Microscopy Sciences, #72230-01) were coated with the following ECM proteins as described below: gelatin [Sigma-Aldrich, #G2500; 0.1% (w/v) of gelatin was prepared in Milli-Q water and autoclaved to dissolve], vitronectin [Sigma-Aldrich, #SRP3186; stock solution was prepared as 200 µg/ml in 0.1% bovine serum albumin (BSA) and water] and laminin (R&D Systems, #3400-010-02; stock solution of 1 mg/ml was pipetted into 10 μl aliquots and stored at −80°C). To coat with gelatin, glass surfaces were incubated with 0.1% gelatin solution for 5 min at RT. To coat with vitronectin or laminin, glass coverslips were incubated at 37°C for 1 h in 20 µg/ml of either vitronectin or laminin, excess liquid was removed, and the coverslips were rinsed once with PBS and blocked with 10 µg/ml heat-denatured BSA for 30 min before plating cells (Lu et al., 2020).
Hydrogels
Methacrylated alginate synthesis
Methacrylated alginate (MeAlg) was synthesized according to a previously established protocol (Khetan et al., 2013). In brief, alginic acid sodium salt from brown algae (Sigma-Aldrich) (3% w/v) was fully dissolved in Dulbecco's phosphate buffered saline (DPBS, Sigma-Aldrich). Then, methacrylic anhydride (Sigma-Aldrich) (8% v/v) was added dropwise to the alginate solution and stirred for 12 h at 4°C, using 2 M NaOH (Sigma-Aldrich) to ensure that the pH remained between 8 and 9 for the duration of the reaction. The resulting solution was passed through filter paper (GE Whatman) and poured into Spectra/Por dialysis membrane with a 6–8 kDa molecular weight cutoff (Thermo Fisher Scientific) and kept in deionized water with stirring for 7 days to eliminate the unreacted methacrylic anhydride and salts. The dialyzed solution was then freeze dried for 4 days to obtain MeAlg foam.
Fabrication of the hydrogel substrates
MeAlg substrates were fabricated using a previously established protocol (Guvendiren and Burdick, 2012). Briefly, Petri dishes with glass bottoms were treated with ultraviolet light or ozone for 30 min, immediately followed by a coating of 3-(trimethoxysilyl)propyl methacrylate (TMS) (Sigma-Aldrich) to methacrylate the glass surfaces (Guvendiren et al., 2009). The dishes were left in a desiccator overnight. The hydrogels were fabricated using Michael-type addition polymerization. First, 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (I2959; 0.5% w/v; Sigma-Aldrich), a photoinitiator, was completely dissolved in DPBS, followed by the lyophilized MeAlg (3% w/v) synthesized previously. This was kept at room temperature until a clear solution was achieved. Crosslinking occurred with the introduction of DL-dithiothreitol (DTT) (Sigma-Aldrich) to the solution, along with 0.2 M triethanolamine (Sigma-Aldrich) at pH 10. To form 3 kPa and 12 kPa gels, 20% and 30% (w/v) DTT were used, respectively. To promote cell adhesion, GRGDSPC peptide (1% w/v) (GenScript) was added to the solution. After all contents were thoroughly mixed, 5 μl of MeAlg solution was pipetted onto the surface of the dish before being covered with a glass coverslip in order to create gels less than 30 μm thick. These were left at room temperature for 1 hour to crosslink before being submerged in DPBS to remove the coverslip.
Atomic force microscopy
To determine hydrogel stiffness, hydrogel samples were submerged in PBS and placed in a Dimension Icon atomic force microscope with ScanAsyst (Bruker). Using the PeakForce-QNM mode, hydrogel samples were indented using an MLCT-Bio probe tip with pyramidal geometry (Bruker) and a nominal spring constant of 0.03 N/m, checked by thermal calibration.
Assembly of exogenously added Fn1
Live imaging
A three-well culture insert (ibidi, #80366) was placed in the middle of the 35 mm glass-bottomed dish (ibidi, #81158). Then, 0.8×106 Fn1–tdTomato-expressing MEFs were plated surrounding the inserts and cultured for 24 h to reach confluency and to establish the Fn1–tdTomato matrix. At a 24 h time point, Fn1–mEGFP-expressing MEFs were plated inside the inserts on glass without coating for 5 h. Prior to imaging, the culture medium was removed and replaced with the imaging medium. Live imaging was performed using Plan Fluor 40× Oil objective (NA 1.3, Nikon). Positions containing Fn1–mEGFP-expressing MEFs were imaged at ∼17–18 min intervals for ∼16 h in a humidified Tokai Hit stage-top incubator at 5% CO2. Each position was imaged by acquiring 40–43 confocal slices at 0.5 µm thickness, and the pinhole was set to 1 Airy unit.
SMLM
The medium containing secreted Fn1–tdTomato fusion proteins was collected after a 72 h culture of confluent Fn1–tdTomato MEFs. The medium was centrifuged at 300 g for 5 min to pellet debris. The concentration of Fn1–tdTomato in the supernatant was quantified using an ELISA kit specific to mouse Fn1, which does not cross react with bovine Fn1 (Abcam, ab210967). Fn1–tdTomato-containing supernatant was diluted 1:4 with fresh MEF culture medium to the final concentration of 5 µg/ml of Fn1–tdTomato, and 2 ml of this supernatant was added to Fn1–mEGFP MEFs that had been plated on 25 mm #1.5H glass coverslips the day before at 105 cells per well in a six-well plate. Following 24 h of incubation at 37°C with 5% CO2, cells were washed with PBS and fixed in 4% PFA in PBS for 20 min at RT. PFA was quenched with 100 mM NH4Cl in PBS, cells were washed three times for 5 min in PBS and permeabilized with PBST. Cells were then incubated with a blocking solution containing 10% donkey serum in PBST for 30 min at RT. To detect Fn1–tdTomato, cells were incubated at 4°C overnight with rabbit anti-mCherry antibody (Abcam, ab167453) diluted 1:100 in the blocking solution. Primary antibodies were detected with AF647-conjugated donkey anti-rabbit F(ab)2 secondary antibodies (Jackson Labs, #711-606-152) diluted 1:300 in blocking solution and incubated for 4 h at RT. Cells were then washed three times for 5 min in PBST and stored at 4°C until imaging. dSTORM imaging was performed using GLOX/BME buffer. Cells were imaged at a 57° angle using the SMLM imaging protocol I. Images were processed and reconstructed using SMAP.
Supplementary Material
Acknowledgements
We thank Richard Hynes and Nathan Astrof for insightful discussions and careful reading of the manuscript, and Rae Astrof for love, encouragement and help with data entry. We also thank Patrick Murphy for the gift of wild-type endothelial cells, Richard Hynes for the 297.1 antibody, Tung Chan for help with western blotting using ProteinSimple, and Jonas Ries for the advice to improve our SMLM imaging and adopt the SMAP SMLM analysis software.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: D.T., J.E.S., S.A.; Methodology: D.T., V.C., S.A.; Validation: D.T., V.C., S.A.; Formal analysis: D.T., V.C., S.A.; Investigation: D.T., C.A., S.M., B.E.A., B.F., P.V., V.C., A.H., S.A.; Resources: M.G., A.K., J.E.S., S.A.; Data curation: D.T., S.A.; Writing - original draft: S.A.; Writing - review & editing: S.A., J.E.S.; Visualization: D.T., P.V., V.C., S.A.; Supervision: S.A.; Project administration: S.A.; Funding acquisition: M.G., A.K., J.E.S., S.A.
Funding
This work was supported by funding from the National Heart, Lung, and Blood Institute of the National Institutes of Health (R01 HL103920, R01 HL134935 and R01 HL158049), a Transformational Project Award from the American Heart Association (20TPA35490074) to S.A., the NIH Office of the Director (R21 OD025323-01) to S.A., by a American Heart Association postdoctoral fellowship (#836254) to C.A.; a pre-doctoral fellowship from the National Heart, Lung, and Blood Institute (F31HL151046) to B.E.A., the National Institute of General Medical Sciences (R35GM122505) to A.K., the National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01 AR073236) to J.E.S., the Faculty Seed Grant from the Center for Engineering MechanoBiology (CEMB), a National Science Foundation Science and Technology Center, under the grant agreement CMMI: 15-48571. Any opinions, findings, conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. Desposited in PMC for release after 12 months.
Peer review history
The peer review history is available online at https://journals.biologists.com/jcs/lookup/doi/10.1242/jcs.260120.reviewer-comments.pdf.
References
- Aguirre, K. M., McCormick, R. J. and Schwarzbauer, J. E. (1994). Fibronectin self-association is mediated by complementary sites within the amino-terminal one-third of the molecule. J. Biol. Chem. 269, 27863-27868. 10.1016/S0021-9258(18)46866-4 [DOI] [PubMed] [Google Scholar]
- Andronov, L., Lutz, Y., Vonesch, J.-L. and Klaholz, B. P. (2016a). SharpViSu: integrated analysis and segmentation of super-resolution microscopy data. Bioinformatics 32, 2239-2241. 10.1093/bioinformatics/btw123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronov, L., Orlov, I., Lutz, Y., Vonesch, J.-L. and Klaholz, B. P. (2016b). ClusterViSu, a method for clustering of protein complexes by Voronoi tessellation in super-resolution microscopy. Sci. Rep. 6, 24084. 10.1038/srep24084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai, Y., Yasuda, R., Akashi, K.-I., Harada, Y., Miyata, H., Kinosita, K., Jr and Itoh, H. (1999). Tying a molecular knot with optical tweezers. Nature 399, 446-448. 10.1038/20894 [DOI] [PubMed] [Google Scholar]
- Baumgart, F., Arnold, A. M., Leskovar, K., Staszek, K., Fölser, M., Weghuber, J., Stockinger, H. and Schütz, G. J. (2016). Varying label density allows artifact-free analysis of membrane-protein nanoclusters. Nat. Methods 13, 661-664. 10.1038/nmeth.3897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer, R., Gertsenstein, M., Vintersten, K. and Nagy, A. (2014). Manipulating the Mouse Embryo: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press, 814pp. [Google Scholar]
- Bui, K. H., von Appen, A., DiGuilio, A. L., Ori, A., Sparks, L., Mackmull, M.-T., Bock, T., Hagen, W., Andrés-Pons, A., Glavy, J. S.et al. (2013). Integrated structural analysis of the human nuclear pore complex scaffold. Cell 155, 1233-1243. 10.1016/j.cell.2013.10.055 [DOI] [PubMed] [Google Scholar]
- Caetano, F. A., Dirk, B. S., Tam, J. H. K., Cavanagh, P. C., Goiko, M., Ferguson, S. S. G., Pasternak, S. H., Dikeakos, J. D., de Bruyn, J. R. and Heit, B. (2015). MIiSR: molecular interactions in super-resolution imaging enables the analysis of protein interactions, dynamics and formation of multi-protein structures. PLoS Comput. Biol. 11, e1004634. 10.1371/journal.pcbi.1004634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., Zardi, L. and Peters, D. M. P. (1997). High-resolution cryo-scanning electron microscopy study of the macromolecular structure of fibronectin fibrils. Scanning 19, 349-355. 10.1002/sca.4950190505 [DOI] [PubMed] [Google Scholar]
- Chernousov, M. A., Faerman, A. I., Frid, M. G., Printseva, O. Y. and Koteliansky, V. E. (1987). Monoclonal antibody to fibronectin which inhibits extracellular matrix assembly. FEBS Lett. 217, 124-128. 10.1016/0014-5793(87)81255-3 [DOI] [PubMed] [Google Scholar]
- Chiang, H.-Y., Korshunov, V. A., Serour, A., Shi, F. and Sottile, J. (2009). Fibronectin is an important regulator of flow-induced vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 29, 1074-1079. 10.1161/ATVBAHA.108.181081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, M. G. and Hynes, R. O. (1979). Biosynthesis and processing of fibronectin in NIL.8 hamster cells. J. Biol. Chem. 254, 12050-12055. 10.1016/S0021-9258(19)86426-8 [DOI] [PubMed] [Google Scholar]
- Diekmann, R., Kahnwald, M., Schoenit, A., Deschamps, J., Matti, U. and Ries, J. (2020). Optimizing imaging speed and excitation intensity for single-molecule localization microscopy. Nat. Methods 17, 909-912. 10.1038/s41592-020-0918-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamba, B. J. and Peters, D. M. (1991). Arrangement of cellular fibronectin in noncollagenous fibrils in human fibroblast cultures. J. Cell Sci. 100, 605-612. 10.1242/jcs.100.3.605 [DOI] [PubMed] [Google Scholar]
- Ester, M., Krigel, H.-P., Sander, J. and Xu, X. (1996). A density-based algorithm for discovering clusters in large spatial databases with noise. Proc. Second Int. Conf. Knowl. Discov. Data Min. 96, 226-231. [Google Scholar]
- Filla, M. S., Dimeo, K. D., Tong, T. and Peters, D. M. (2017). Disruption of fibronectin matrix affects type IV collagen, fibrillin and laminin deposition into extracellular matrix of human trabecular meshwork (HTM) cells. Exp. Eye Res. 165, 7-19. 10.1016/j.exer.2017.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Früh, S. M., Schoen, I., Ries, J. and Vogel, V. (2015). Molecular architecture of native fibronectin fibrils. Nat. Commun. 6, 7275. 10.1038/ncomms8275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furcht, L. T., Smith, D., Wendelschafer-Crabb, G., Mosher, D. F. and Foidart, J. M. (1980a). Fibronectin presence in native collagen fibrils of human fibroblasts: immunoperoxidase and immunoferritin localization. J. Histochem. Cytochem. 28, 1319-1333. 10.1177/28.12.7014712 [DOI] [PubMed] [Google Scholar]
- Furcht, L. T., Wendelschafer-Crabb, G., Mosher, D. F. and Foidart, J. M. (1980b). Ascorbate-induced fibroblast cell matrix: reaction of antibodies to procollagen I and III and fibronectin in an axial periodic fashion. Prog. Clin. Biol. Res. 41, 829-843. [PubMed] [Google Scholar]
- Furcht, L. T., Wendelschafer-Crabb, G., Mosher, D. F. and Foidart, J. M. (1980c). An axial periodic fibrillar arrangement of antigenic determinants for fibronectin and procollagen on ascorbate treated human fibroblasts. J. Supramol. Struct. 13, 15-33. 10.1002/jss.400130103 [DOI] [PubMed] [Google Scholar]
- Geiger, B. and Yamada, K. M. (2011). Molecular architecture and function of matrix adhesions. Cold Spring Harbor Perspect. Biol. 3, a005033. 10.1101/cshperspect.a005033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger, B., Bershadsky, A., Pankov, R. and Yamada, K. M. (2001). Transmembrane crosstalk between the extracellular matrix and the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2, 793-805. 10.1038/35099066 [DOI] [PubMed] [Google Scholar]
- Gudzenko, T. and Franz, C. M. (2015). Studying early stages of fibronectin fibrillogenesis in living cells by atomic force microscopy. Mol. Biol. Cell 26, 3190-3204. 10.1091/mbc.e14-05-1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvendiren, M. and Burdick, J. A. (2012). Stiffening hydrogels to probe short- and long-term cellular responses to dynamic mechanics. Nat. Commun. 3, 792. 10.1038/ncomms1792 [DOI] [PubMed] [Google Scholar]
- Guvendiren, M., Yang, S. and Burdick, J. A. (2009). Swelling-induced surface patterns in hydrogels with gradient crosslinking density. Adv. Funct. Mater. 19, 3038-3045. 10.1002/adfm.200900622 [DOI] [Google Scholar]
- Heilemann, M., van de Linde, S., Schuttpelz, M., Kasper, R., Seefeldt, B., Mukherjee, A., Tinnefeld, P. and Sauer, M. (2008). Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew. Chem. Int. Ed. Engl. 47, 6172-6176. 10.1002/anie.200802376 [DOI] [PubMed] [Google Scholar]
- Hocking, D. C., Sottile, J. and McKeown-Longo, P. J. (1994). Fibronectin's III-1 module contains a conformation-dependent binding site for the amino-terminal region of fibronectin. J. Biol. Chem. 269, 19183-19187. 10.1016/S0021-9258(17)32292-5 [DOI] [PubMed] [Google Scholar]
- Hynes, R. O. (1990). Fibronectins. New York: Springer-Verlag. [Google Scholar]
- Hynes, R. O. (2009). The extracellular matrix: not just pretty fibrils. Science 326, 1216-1219. 10.1126/science.1176009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez, A., Friedl, K. and Leterrier, C. (2020). About samples, giving examples: optimized single molecule localization microscopy. Methods 174, 100-114. 10.1016/j.ymeth.2019.05.008 [DOI] [PubMed] [Google Scholar]
- Kaech, S. and Banker, G. (2006). Culturing hippocampal neurons. Nat. Protoc. 1, 2406-2415. 10.1038/nprot.2006.356 [DOI] [PubMed] [Google Scholar]
- Khetan, S., Guvendiren, M., Legant, W. R., Cohen, D. M., Chen, C. S. and Burdick, J. A. (2013). Degradation-mediated cellular traction directs stem cell fate in covalently crosslinked three-dimensional hydrogels. Nat. Mater. 12, 458-465. 10.1038/nmat3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansky, Z., Mutsafi, Y., Houben, L., Ilani, T., Armony, G., Wolf, S. G. and Fass, D. (2019). 3D mapping of native extracellular matrix reveals cellular responses to the microenvironment. J. Struct. Biol. X 1, 100002. 10.1016/j.yjsbx.2018.100002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelek, M., Gyparaki, M. T., Beliu, G., Schueder, F., Griffié, J., Manley, S., Jungmann, R., Sauer, M., Lakadamyali, M. and Zimmer, C. (2021). Single-molecule localization microscopy. Nat. Rev. Methods Primers 1, 39. 10.1038/s43586-021-00038-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J., Doyle, A. D., Shinsato, Y., Wang, S., Bodendorfer, M. A., Zheng, M. and Yamada, K. M. (2020). Basement membrane regulates fibronectin organization using sliding focal adhesions driven by a contractile winch. Dev. Cell 52, 631-646.e34. 10.1016/j.devcel.2020.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavičius, G., Reymond, L., D'Este, E., Masharina, A., Göttfert, F., Ta, H., Güther, A., Fournier, M., Rizzo, S., Waldmann, H.et al. (2014). Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 11, 731-733. 10.1038/nmeth.2972 [DOI] [PubMed] [Google Scholar]
- Ma, W., Ma, H. and Mosher, D. F. (2015). On-off kinetics of engagement of FNI modules of soluble fibronectin by beta-strand addition. PLoS ONE 10, e0124941. 10.1371/journal.pone.0124941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkusch, S., Endesfelder, U., Mondry, J., Gelléri, M., Verveer, P. J. and Heilemann, M. (2012). Coordinate-based colocalization analysis of single-molecule localization microscopy data. Histochem. Cell Biol. 137, 1-10. 10.1007/s00418-011-0880-5 [DOI] [PubMed] [Google Scholar]
- Martino, M. M. and Hubbell, J. A. (2010). The 12th-14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 24, 4711-4721. 10.1096/fj.09.151282 [DOI] [PubMed] [Google Scholar]
- Mau, A., Friedl, K., Leterrier, C., Bourg, N. and Lévêque-Fort, S. (2021). Fast widefield scan provides tunable and uniform illumination optimizing super-resolution microscopy on large fields. Nat. Commun. 12, 3077. 10.1038/s41467-021-23405-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer, L. M., Tomasini-Johansson, B. R., Ma, W., Annis, D. S., Eickstaedt, N. L., Ensenberger, M. G., Satyshur, K. A. and Mosher, D. F. (2010). Extended binding site on fibronectin for the functional upstream domain of protein F1 of Streptococcus pyogenes. J. Biol. Chem. 285, 41087-41099. 10.1074/jbc.M110.153692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, J. A., Quade, B. J., Broekelmann, T. J., LaChance, R., Forsman, K., Hasegawa, E. and Akiyama, S. (1987). Fibronectin's cell-adhesive domain and an amino-terminal matrix assembly domain participate in its assembly into fibroblast pericellular matrix. J. Biol. Chem. 262, 2957-2967. 10.1016/S0021-9258(18)61453-X [DOI] [PubMed] [Google Scholar]
- McKeown-Longo, P. J. and Mosher, D. F. (1983). Binding of plasma fibronectin to cell layers of human skin fibroblasts. J. Cell Biol. 97, 466-472. 10.1083/jcb.97.2.466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown-Longo, P. J. and Mosher, D. F. (1985). Interaction of the 70,000-mol-wt amino-terminal fragment of fibronectin with the matrix-assembly receptor of fibroblasts. J. Cell Biol. 100, 364-374. 10.1083/jcb.100.2.364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morla, A., Zhang, Z. and Ruoslahti, E. (1994). Superfibronectin is a functionally distinct form of fibronectin. Nature 367, 193-196. 10.1038/367193a0 [DOI] [PubMed] [Google Scholar]
- Mund, M. and Ries, J. (2020). How good are my data? Reference standards in superresolution microscopy. Mol. Biol. Cell 31, 2093-2096. 10.1091/mbc.E19-04-0189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar, M. D., Tasic, B., Miyamichi, K., Li, L. and Luo, L. (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593-605. 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- Nelea, V. and Kaartinen, M. T. (2010). Periodic beaded-filament assembly of fibronectin on negatively charged surface. J. Struct. Biol. 170, 50-59. 10.1016/j.jsb.2010.01.009 [DOI] [PubMed] [Google Scholar]
- Nieuwenhuizen, R. P. J., Lidke, K. A., Bates, M., Puig, D. L., Grünwald, D., Stallinga, S. and Rieger, B. (2013). Measuring image resolution in optical nanoscopy. Nat. Methods 10, 557-562. 10.1038/nmeth.2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi, T., Kiehart, D. P. and Erickson, H. P. (1999). Dynamics and elasticity of the fibronectin matrix in living cell culture visualized by fibronectin-green fluorescent protein. Proc. Natl. Acad. Sci. USA 96, 2153-2158. 10.1073/pnas.96.5.2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovesný, M., Křížek, P., Borkovec, J., Švindrych, Z. and Hagen, G. M. (2014). ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 30, 2389-2390. 10.1093/bioinformatics/btu202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankov, R., Cukierman, E., Katz, B.-Z., Matsumoto, K., Lin, D. C., Lin, S., Hahn, C. and Yamada, K. M. (2000). Integrin dynamics and matrix assembly: tensin-dependent translocation of alpha(5)beta(1) integrins promotes early fibronectin fibrillogenesis. J. Cell Biol. 148, 1075-1090. 10.1083/jcb.148.5.1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, A. R., Pritykin, Y., Vidigal, J. A., Chhangawala, S., Zamparo, L., Leslie, C. S. and Ventura, A. (2017). GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol. 35, 347-349. 10.1038/nbt.3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, J. H. and Hynes, R. O. (1996). Fibronectin isoform distribution in the mouse. I. The alternatively spliced EIIIB, EIIIA, and V segments show widespread codistribution in the developing mouse embryo. Cell Adhes. Commun. 4, 103-125. 10.3109/15419069609010766 [DOI] [PubMed] [Google Scholar]
- Pliatsika, V. and Rigoutsos, I. (2015). “Off-Spotter”: very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAs. Biol. Direct 10, 4. 10.1186/s13062-015-0035-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raitman, I., Huang, M. L., Williams, S. A., Friedman, B., Godula, K. and Schwarzbauer, J. E. (2018). Heparin-fibronectin interactions in the development of extracellular matrix insolubility. Matrix Biol. 67, 107-122. 10.1016/j.matbio.2017.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, A. and Astrof, S. (2020). Visualization and Analysis of Pharyngeal Arch Arteries using Whole-mount Immunohistochemistry and 3D Reconstruction. J. Vis. Exp. 157, e60797. 10.3791/60797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A. and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281-2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries, J. (2020). SMAP: a modular super-resolution microscopy analysis platform for SMLM data. Nat. Methods 17, 870-872. 10.1038/s41592-020-0938-1 [DOI] [PubMed] [Google Scholar]
- Rozario, T., Dzamba, B., Weber, G. F., Davidson, L. A. and DeSimone, D. W. (2009). The physical state of fibronectin matrix differentially regulates morphogenetic movements in vivo. Dev. Biol. 327, 386-398. 10.1016/j.ydbio.2008.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust, M. J., Bates, M. and Zhuang, X. (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793-796. 10.1038/nmeth929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitta, A. M., Soper, P. D., Wasserman, E. and Klein, M. L. (1999). Influence of a knot on the strength of a polymer strand. Nature 399, 46-48. 10.1038/19935 [DOI] [PubMed] [Google Scholar]
- Sakai, T., Johnson, K. J., Murozono, M., Sakai, K., Magnuson, M. A., Wieloch, T., Cronberg, T., Isshiki, A., Erickson, H. P. and Fässler, R. (2001). Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat. Med. 7, 324-330. 10.1038/85471 [DOI] [PubMed] [Google Scholar]
- Salmerón-Sánchez, M., Rico, P., Moratal, D., Lee, T. T., Schwarzbauer, J. E. and García, A. J. (2011). Role of material-driven fibronectin fibrillogenesis in cell differentiation. Biomaterials 32, 2099-2105. 10.1016/j.biomaterials.2010.11.057 [DOI] [PubMed] [Google Scholar]
- Saunders, J. T. and Schwarzbauer, J. E. (2019). Fibronectin matrix as a scaffold for procollagen proteinase binding and collagen processing. Mol. Biol. Cell 30, 2218-2226. 10.1091/mbc.E19-03-0140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzbauer, J. E. (1991). Identification of the fibronectin sequences required for assembly of a fibrillar matrix. J. Cell Biol. 113, 1463-1473. 10.1083/jcb.113.6.1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzbauer, J. E. and DeSimone, D. W. (2011). Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harbor Perspect. Biol. 3, a005041. 10.1101/cshperspect.a005041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechler, J. L., Takada, Y. and Schwarzbauer, J. E. (1996). Altered rate of fibronectin matrix assembly by deletion of the first type III repeats. J. Cell Biol. 134, 573-583. 10.1083/jcb.134.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechler, J. L., Rao, H., Cumiskey, A. M., Vega-Colón, I., Smith, M. S., Murata, T. and Schwarzbauer, J. E. (2001). A novel fibronectin binding site required for fibronectin fibril growth during matrix assembly. J. Cell Biol. 154, 1081-1088. 10.1083/jcb.200102034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, P. and Schwarzbauer, J. E. (2014). Fibronectin matrix assembly is essential for cell condensation during chondrogenesis. J. Cell Sci. 127, 4420-4428. 10.1242/jcs.150276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, P., Carraher, C. and Schwarzbauer, J. E. (2010). Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 26, 397-419. 10.1146/annurev-cellbio-100109-104020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorstengaard, K., Jensen, M. S., Sahl, P., Petersen, T. E. and Magnusson, S. (1986). Complete primary structure of bovine plasma fibronectin. Eur. J. Biochem. 161, 441-453. 10.1111/j.1432-1033.1986.tb10464.x [DOI] [PubMed] [Google Scholar]
- Smith, M. L., Gourdon, D., Little, W. C., Kubow, K. E., Eguiluz, R. A., Luna-Morris, S. and Vogel, V. (2007). Force-induced unfolding of fibronectin in the extracellular matrix of living cells. PLoS Biol. 5, e268. 10.1371/journal.pbio.0050268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snitkovsky, S. and Young, J. A. T. (1998). Cell-specific viral targeting mediated by a soluble retroviral receptor-ligand fusion protein. Proc. Natl. Acad. Sci. USA 95, 7063-7068. 10.1073/pnas.95.12.7063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile, J. and Chandler, J. (2005). Fibronectin matrix turnover occurs through a caveolin-1-dependent process. Mol. Biol. Cell 16, 757-768. 10.1091/mbc.e04-08-0672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevathasan, J. V., Kahnwald, M., Cieśliński, K., Hoess, P., Peneti, S. K., Reitberger, M., Heid, D., Kasuba, K. C., Hoerner, S. J., Li, Y.et al. (2019). Nuclear pores as versatile reference standards for quantitative superresolution microscopy. Nat. Methods 16, 1045-1053. 10.1038/s41592-019-0574-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasini-Johansson, B. R., Kaufman, N. R., Ensenberger, M. G., Ozeri, V., Hanski, E. and Mosher, D. F. (2001). A 49-residue peptide from adhesin F1 of Streptococcus pyogenes inhibits fibronectin matrix assembly. J. Biol. Chem. 276, 23430-23439. 10.1074/jbc.M103467200 [DOI] [PubMed] [Google Scholar]
- Tomasini-Johansson, B. R., Annis, D. S. and Mosher, D. F. (2006). The N-terminal 70-kDa fragment of fibronectin binds to cell surface fibronectin assembly sites in the absence of intact fibronectin. Matrix Biol. 25, 282-293. 10.1016/j.matbio.2006.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Appen, A., Kosinski, J., Sparks, L., Ori, A., DiGuilio, A. L., Vollmer, B., Mackmull, M.-T., Banterle, N., Parca, L., Kastritis, P.et al. (2015). In situ structural analysis of the human nuclear pore complex. Nature 526, 140-143. 10.1038/nature15381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner, D. D. and Hynes, R. O. (1979). Domain structure of fibronectin and its relation to function. Disulfides and sulfhydryl groups. J. Biol. Chem. 254, 6746-6754. 10.1016/S0021-9258(18)50432-4 [DOI] [PubMed] [Google Scholar]
- Wierzbicka-Patynowski, I., Mao, Y. and Schwarzbauer, J. E. (2004). Analysis of fibronectin matrix assembly. Curr. Protoc. Cell Biol. Chapter 10, Unit 10.12. 10.1002/0471143030.cb1012s25 [DOI] [PubMed] [Google Scholar]
- Wijelath, E. S., Murray, J., Rahman, S., Patel, Y., Ishida, A., Strand, K., Aziz, S., Cardona, C., Hammond, W. P., Savidge, G. F.et al. (2002). Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ. Res. 91, 25-31. 10.1161/01.RES.0000026420.22406.79 [DOI] [PubMed] [Google Scholar]
- Zamir, E., Katz, B. Z., Aota, S., Yamada, K. M., Geiger, B. and Kam, Z. (1999). Molecular diversity of cell-matrix adhesions. J. Cell Sci. 112, 1655-1669. 10.1242/jcs.112.11.1655 [DOI] [PubMed] [Google Scholar]
- Zamir, E., Katz, M., Posen, Y., Erez, N., Yamada, K. M., Katz, B.-Z., Lin, S., Lin, D. C., Bershadsky, A., Kam, Z.et al. (2000). Dynamics and segregation of cell-matrix adhesions in cultured fibroblasts. Nat. Cell Biol. 2, 191-196. 10.1038/35008607 [DOI] [PubMed] [Google Scholar]
- Zhang, Q. and Mosher, D. F. (1996). Cross-linking of the NH2-terminal region of fibronectin to molecules of large apparent molecular mass. Characterization of fibronectin assembly sites induced by the treatment of fibroblasts with lysophosphatidic acid. J. Biol. Chem. 271, 33284-33292. 10.1074/jbc.271.52.33284 [DOI] [PubMed] [Google Scholar]
- Zhang, Q., Checovich, W. J., Peters, D. M., Albrecht, R. M. and Mosher, D. F. (1994). Modulation of cell surface fibronectin assembly sites by lysophosphatidic acid. J. Cell Biol. 127, 1447-1459. 10.1083/jcb.127.5.1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q., Magnusson, M. K. and Mosher, D. F. (1997). Lysophosphatidic acid and microtubule-destabilizing agents stimulate fibronectin matrix assembly through Rho-dependent actin stress fiber formation and cell contraction. Mol. Biol. Cell 8, 1415-1425. 10.1091/mbc.8.8.1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, C., Chrzanowska-Wodnicka, M., Brown, J., Shaub, A., Belkin, A. M. and Burridge, K. (1998). Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J. Cell Biol. 141, 539-551. 10.1083/jcb.141.2.539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, X., Rowe, R. G., Hiraoka, N., George, J. P., Wirtz, D., Mosher, D. F., Virtanen, I., Chernousov, M. A. and Weiss, S. J. (2008). Fibronectin fibrillogenesis regulates three-dimensional neovessel formation. Genes Dev. 22, 1231-1243. 10.1101/gad.1643308 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.