

Graphical abstract
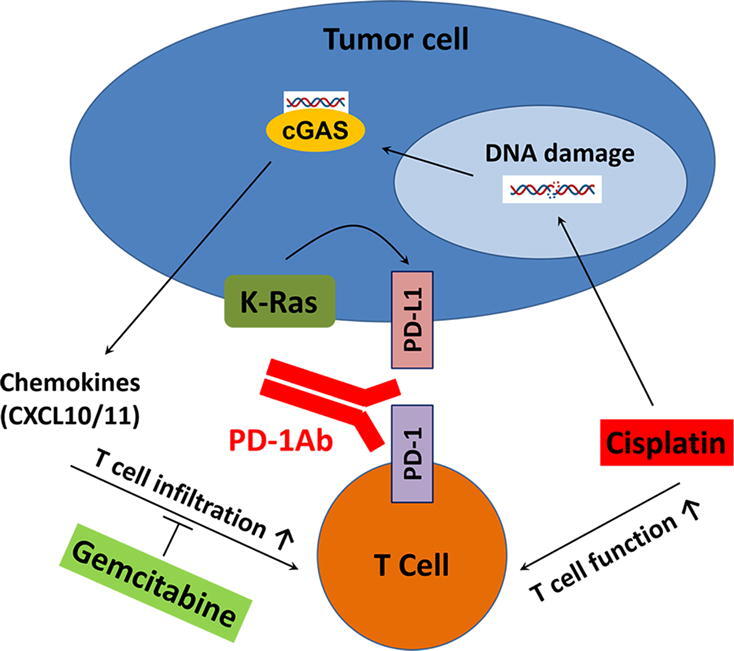
Keywords: K-ras, Cisplatin, Gemcitabine, PD-1/PD-L1, Immunochemotherapy
Highlights
-
•
Two common chemotherapeutic drugs, cisplatin and gemcitabine, exert opposite effect on the efficacy of PD-1 antibody in K-ras-driven cancers.
-
•
Gemcitabine antagonizes PD-1Ab due to its inhibition on T cell infiltration in tumor tissues.
-
•
Combination PD-1Ab and cisplatin leads to complete tumor eradication in vivo due to activation of the cGAS-mediated immune response.
-
•
The impact of drugs on T cell functions should be considered as a critical factor in selecting drugs for immunochemotherapy to achieve optimal therapeutic outcome.
Abstract
Introduction
Immunochemotherapy using PD-1/PD-L1 antibodies in combination with chemotherapeutic agents has become a mainstream treatment for cancer patients, but it remains unclear which drug combinations would produce best therapeutic outcome.
Objectives
The purpose of this study was to investigate two common chemotherapeutic drugs, gemcitabine and cisplatin, for their impacts on the therapeutic efficacy of PD-1 antibody in K-ras-driven cancers known to overexpress PD-L1.
Methods
Both in vitro assays and syngeneic mouse tumor models were used in this study. Biochemical and molecular assays were used to determine the effects of drugs on T cell functions in cell culture models and in mouse/human tumor tissues. Allograft tumor models with K-ras mutation were used to investigate the combination effect of gemcitabine or cisplatin with immunotherapy. Data of lung cancer patients with K-ras mutation treated with cisplatin and toripalimab were analyzed to evaluate the clinical relevance of the lab findings.
Results
Cisplatin and gemcitabine unexpectedly exert opposite effect on the therapeutic activity of PD-1 antibody in vivo. Gemcitabine antagonizes the therapeutic effect of PD-1 antibody due to its significant inhibition on CD8+ T cell infiltration, which was observed both in mouse tumor allografts and in human pancreatic cancer tissues. In contrast, cisplatin shows synergistic activity with PD-1 antibody by activation of CD8+ T cells through the DNA damage-mediated cGAS-STING sensing mechanism, leading to increase of T cell infiltration and secretion of antitumor cytokines. Clinical data show that a combination of cisplatin with PD-1 antibody toripalimab could be effective in advanced lung cancer patients with K-ras mutation who failed prior therapies.
Conclusions
Our study shows that a key factor in selecting chemotherapeutic agents for immunochemotherapy is the drug’s impact on T cell functions, and that cisplatin-based chemotherapy is an excellent choice for combination with immune checkpoint antibody to achieve favorable clinical outcome.
Introduction
K-ras mutations are often found in human tumors and associated with poor clinical outcome [1]. The vast majority of pancreatic cancer cells exhibit a constitutive activation of K-ras due to gene mutations and overexpression [2], and approximately 30–40% in lung and colon cancers also harbor K-ras mutation [3], [4]. Recent studies from multiple groups including our laboratory revealed a link between oncogenic K-ras signaling and the expressions of immune function-modulating molecules such as CD137 [5] and PD-L1 (programmed death-ligand 1) [6], [7], [8]. Among these new findings, a particularly interesting finding is that the abnormal activation of K-ras by mutations in pancreatic cancer cells promotes the expression of PD-L1 through reactive oxygen species (ROS)-mediated growth factor signaling [8]. Such K-ras-induced elevation of PD-L1 expression may provide the tumor cells an important mechanism to escape immune surveillance [9], and thus contribute to the more aggressive cancer progression and poor clinical outcome.
PD-L1 is localized at the surface of various cell types including those of the immune system and non-immune cells [10], [11], [12], [13], [14]. The PD-1 receptor (programmed death 1) is mainly expressed on T cells, and its physiologic interaction with PD-L1 leads to inhibition of T cell functions [10], [11], [15], [16]. Expression of PD-L1 has also been detected in various types of tumor cells and is considered as an important mechanism of tumor immune evasion [11], [15]. High PD-L1 expression is often associated with poor clinical outcome in patients with malignant diseases due to the inhibition of antitumor immune functions [17], [18], [19]. The disruption of PD-1/PD-L1 interaction thus becomes a logical strategy to unlock the suppressed immune functions, and constitutes the basis for using antibodies against PD-1 or PD-L1 in cancer immunotherapy. Indeed, targeting PD-1 or PD-L1 with monoclonal antibodies has demonstrated long-lasting therapeutic activity against cancer in multiple clinical trials [20], [21], [22]. A combination of the immune checkpoint blockade agents such as PD-1 antibody and standard chemotherapeutic drugs has been shown to further enhance the antitumor activity and improve the clinical outcome of the cancer patients [23]. In the context of immunochemotherapy in pancreatic ductal adenocarcinoma (PDAC) or colorectal cancer, immunotherapy as monotherapy offers no significant therapeutic activity. Combination of gemcitabine and nab-paclitaxel with PD-L1 antibody has recently showed no overall survival benefit for PDAC patients [24]. However, regimen comprising of PD-1 antibody and 5-fluorouracil/leucoverin/irinitocan together with BL-8040 (a CXCR4 antagonist) seems to achieve significant clinical responses in certain patients [25], providing new hope to treat this highly aggressive cancer. In resectable lung cancer, nivolumab (PD-1 antibody) monotherapy could achieve complete responses in almost half of the cases [26], and therapeutic effects of combination of various chemotherapies (i.e., carboplatin, paclitaxel and etoposide) with immunotherapy are currently evaluated in clinical trials [23].
In the context of using PD-1/PD-L1 blockade as an antitumor strategy, the expression of PD-L1 on the surface of tumor cells is an important factor that largely affects the sensitivity of tumor cells to treatment with PD-1 or PD-L1 antibodies [27]. However, it is important to recognize that the presence of functional T cells in the tumor tissue is another important determinant that affects the outcome of the immunotherapy. Without sufficient functional CD8+ T cells in the tumor tissues, the use of PD-1 antibody is unlikely to achieve satisfactory therapeutic effect. As such, the impact of chemotherapeutic drugs on T cell functions would be a critically important consideration in selecting drug for use in immunochemotherapy. Our recent study using a T cell-based screening assay showed that the chemotherapeutic agents commonly used in clinical treatment of cancer exhibited diverse effects on T cell functions, with some of the drugs exhibiting stimulatory or inhibitory effect on T cells [28]. These new findings underscore the importance of selecting proper drugs for combination with PD-1 antibody (PD-1Ab) to avoid drugs with antagonist effect on immune cells. In this study, we used the K-ras-driven tumor models, which express high level of PD-L1 [8], to evaluate the impact of cisplatin and gemcitabine, two chemotherapeutic agents commonly used in cancer treatment and often used in immunochemotherapy, on the in vivo therapeutic effect of PD-1 antibody. After observing the surprising opposite effect of the two drugs, we used both in vitro and animal experimental models to investigate the underlying mechanisms. We also further validated these findings in pancreatic cancer specimens and clinical data from advanced-stage lung cancer patients with K-ras mutation who failed prior therapies and subsequently treated with cisplatin plus PD-1 antibody.
Materials and methods
Cell lines and cell culture
Mouse CT26.WT colon cancer cells with K-ras G12D mutation (#CRL-2638), mouse LLC lung cancer cells with G12C K-ras mutation (#CRL-1642), human A549 lung cancer cells with K-ras G12S mutation (#CRM-CCL-185) and human CFPAC-1 pancreatic cancer cells with K-ras G12V mutation (#CRL-1918) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). CT26 and A549 cells were cultured in RPMI (Roswell Park Memorial Institute) and F12K (Kaighn's Modification of Ham's F-12 medium) medium complemented with 10% fetal bovine serum (FBS), respectively. LLC and CFPAC-1 cells were cultured in DMEM (Dubbelco’s modified Eagle’s medium) medium with 10% FBS. Mouse ovalbumin-specific B3Z T cell line was a kind gift of Dr. Nilabh Shastri (University of California, Berkeley, USA) [29] and maintained in RPMI medium with 10% FBS.
For generation of the stable doxycycline inducible HPNE/K-RasG12D cells (human pancreatic epithelial cell line), KRAS4BG12D sequence (Addgene, #83131) was cloned into pInduce20 plasmid (Addgene, #83131) via Gateway LR Clonase II Enzyme (Invitrogen, Waltham, MA, USA; #11791–020). Lentiviruses were collected from human 293 T embryonic kidney cells (ATCC, #CRL-3216) transfected with K-rasG12D–pInducer20 plasmid and viral package vectors (Addgene, #12260 and #12259), and were used to infect HPNE (ATCC, #CRL-4023) cells. The HPNE cells were screened for two weeks with 1.5 mg/mL of G418. The cell line was maintained in DMEM supplemented with 10% tetracycline-free FBS.
The mouse pancreatic cancer (KPC) cells harboring mutant K-ras (G12D) and mutant p53 (R172H), were obtained according to the procedures previously described, and maintained in DMEM medium with 10% FBS [8]. Mouse cGAS (cGAMP synthase) knockout PDAC cells were constructed as previously described [30]. The absence of mycoplasma in culture media of all cell lines was confirmed using the LookOut mycoplasma PCR detection kit (Sigma, Saint Louis, MO, USA). The authentication of cell lines was performed by short-tandem repeat (STR) genotyping (Microread Genetics, Beijing, China). All culture media were from Gibco (Invitrogen). Doxycycline (#D9891) was from Sigma-Aldrich (Saint Louis, MO, USA). Cisplatin was from Hospira (Lake Forest, IL, USA) and gemcitabine was from Eli Lilly and Company (Indianapolis, IN, USA).
Immunoblotting
The following antibodies were used for immunoblotting analyses using standard western blotting procedures: beta-actin (#ab6276) and PD-L1 (#ab174838) were purchased from Abcam (Cambridge, UK); K-ras (#sc-30) was from Santa-Cruz biotechnology (Santa Cruz, CA, USA); Chk1 (#2360S), P-Chk1 S345 (#2341S) and STING (#13647) were from Cell Signaling Technology (Beverly, MA, USA). The protein bands were detected by chemiluminescence, using an ECL detection kit (Pierce, Thermo Scientific, Rockford, IL, USA).
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Cellular RNA was isolated using Trizol (Invitrogen), and was reverse-transcribed into cDNA using the Primer Script RT reagent Kit with gDNA Eraser (Takara BIO INC, Kusatsu, Shiga, Japan). SYBR Premix Ex Taq RNAse H+ kit (Takara) was used to detect and quantify target mRNA in cells. Quantitative RT-PCR was analyzed using the Bio-Rad detection system (Bio-Rad, Hercules, CA, USA), and the results were calculated by the delta-delta CT method (formula: 2-(Ct target-Ct Reference)) and matched to the control samples. The specific oligonucleotide primers (Supplementary material Table S1) were purchased from Sangon Biotech (Shanghai, China).
T cell activation and ELISA (enzyme-linked immunosorbent assay)
T lymphocytes from healthy donors were isolated by Ficoll method (GE Healthcare, Little Chalfont, UK) followed by positive selection using magnetic beads coated with anti-CD8 antibody (Miltenyi Biotec, Bergisch Gladbach, Germany). Cytotoxic CD8+ T lymphocytes were stimulated with 500 ng/mL ionomycin (Cayman, Ann Arbor, MI, USA; #1932) and 20 ng/mL phorbol 12-myristate-13-acetate (PMA, Adipogen, San Diego, CA, USA; #AG-CN2-0010) for 24 h. Secretions of mouse and human IFN-γ (interferon gamma) were measured by the respective ELISA kits according to the protocols from the manufacturer (eBioscience, San Diego, CA, USA; #88-7314-88 and #88-7316-88).
B16-OVA tumor cells were exposed to cisplatin or oxaliplatin (TargetMol, Boston, MA, USA) for 24 h and then co-cultured with immune cells (B3Z and bone marrow-derived dendritic cells) as previously described [28]. Platins were not removed from the culture media in order to expose immune cells to the drugs. Interleukin-2 (IL-2) levels in the culture supernatants were measured by ELISA (eBioscience, San Diego, CA, USA; #88-7024-88).
Mouse CXCL10 (C-X-C Motif Chemokine Ligand 10) protein levels in culture medium were quantified by ELISA according to the protocol provided by the manufacturer (R&D systems, #DY466-05).
Cell transfection
The dsDNA fragments (60-mer of HSV-1 sequence) was synthesized and annealed as previously described [31]. X-tremeGENE HP transfection reagent (Roche, Basel, Switzerland) was used to deliver dsDNA (4 μg/mL), a substrate of cGAS enzyme, into cancer cells (Supplementary material Table S1).
Flow cytometry
For the analysis of tumor-infiltrating lymphocytes, mice were injected subcutaneously with 2x106 pancreatic cancer cells driven by mutant K-ras. For drug treatment, anti-mouse PD-1 antibody, cisplatin, and gemcitabine were administrated twice a week for 3 times as indicated. Tumors were collected on day 15, and dissociated by gentleMACS Dissociator (MiltenyiBiotec) and filtered through 70-μm cell strainers to generate single-cell suspensions. Cells were stained with fluorochrome-labeled antibodies against CD45 (Biolegend, San Diego, CA, USA; #103125) in order to identify TIL (tumor-infiltrating lymphocytes) populations, and then CD3 (T cell marker, Biolegend, #100305), CD4 (CD4+ T cell marker, Biolegend, #100549), CD8 (CD8+ T cell marker, BD Biosciences, #564297), CD19 (B cell marker, BD Biosciences, San Jose, CA, USA; #561736), CD11b (myeloid cells marker, Biolegend, #101207), CD11c (dendritic cells marker, Biolegend, #117322), F4/80 (macrophage marker, Biolegend, #123107) to identify the respective subpopulations. Fluorescence data were acquired on a BD LSR Fortessa Flow Cytometer (BD Biosciences). FlowJo software (https://www.flowjo.com) was used to analyze immunostaining, with at least 10,000 cells per sample counted.
Cell migration assay
B3Z cells (density 1x106/ mL) were incubated with gemcitabine or cisplatin for 4 h in serum-free RPMI medium. The cells were then transferred in the upper chemotaxis chamber (QCM Chemotaxis 5 μm Cell migration assay; Millipore, Darmstadt, Germany). The lower chamber contained RPMI medium with FBS as chemoattractants. Lymphocytes that migrated into the lower chamber were measured according to the manufacturer’s protocol.
TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay
TUNEL assay was performed with paraffin-embedded tissues according to manufacturer’s instructions (Biotool TUNEL Apo-Green Detection kit; Selleck chemicals, Houston, TX, USA). DAPI was used as counterstaining to reveal cellular nuclei.
Immunohistochemistry (IHC)
IHC staining of mouse and human tumor tissue sections was processed and analyzed as previously described [8]. Primary antibodies were rabbit anti-mouse CD8 (Bioss, Woburn, MA, USA; #bs-0648R), rabbit anti-human CD8 (ZSGB-Bio, Beijing, China; #ZA-0508), rabbit anti-human CD4 (Abcam; #ab133616), and rabbit anti-mouse FoxP3 (Cell Signaling Technology; #12653). The number of intratumoral CD4+, CD8+ or FoxP3+ lymphocytes and tissue surface areas were quantified using ImageJ software on five randomly chosen 20 × fields per section.
IHC procedures for measuring intratumoral T cells were performed at MD Anderson Cancer Center (Houston, Texas, USA) as described previously [32]. Primary antibodies used were mouse anti-human CD8 (Thermoscientific; #MA5-13473) and rabbit anti-human CD4 (Cell Marque, Rocklin, CA, USA; #104R14). The number of CD4+ and CD8+ T lymphocytes in human PDAC tumors was calculated as the percentage of positive staining area versus the total tumor area as previously described [32].
MTT assay
Cell metabolic status was assessed by following the reduction of MTT (3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide) to blue formazan. Cancer cells were seeded into 96-well plates at a density of 2000 cells per well for 24 h, and then incubated with the test compounds (3 wells were used for each condition) for 48 h. Cells were then washed with PBS and incubated with MTT (0.5 mg/mL) for 2 h at 37 °C. Blue formazan crystals were solubilized by adding 100 µL DMSO per well, and the colored solution was subsequently read at 550 nm. Results are expressed as % of MTT reduction compared to untreated control conditions.
Syngeneic tumor mouse models
Cohorts of 6–7 weeks old female black C57BL/6, white BALB/c, or nude BALB/c mice were purchased from Vital River Laboratory Animal Technology (Beijing, China). Mice were first kept for an acclimation period of 7–14 days in a controlled pathogen-free environment before study began. Mouse KPC pancreatic cancer cells (1-2x106) or CT26 colon cancer cells (0.3x106) were injected into the right flanks of the 7–8 weeks old mice. Before drug administration, mice were randomly divided into the indicated groups without blinding. For drug treatment, anti-mouse PD-1 antibody (BioXcell, West Lebanon, NH, USA; #BE0146), cisplatin (Hospira, Lake Forest, IL, USA; CAS 15663-27-1), and gemcitabine (Eli Lilly and Company, Indianapolis, IN, USA; CAS 122111-03-9) were administrated twice a week for 3 weeks as indicated. Body weights and tumor volumes were measured twice per week. Tumor volume was calculated using the following formula: tumor volume (mm3) = (length (mm) × width2 (mm))/2. Moribund mice or animals with tumors of greater than 15 mm in length or with ulcerated tumors were promptly sacrificed to avoid unnecessary pain and suffering.
Human subjects and clinical samples
Clinical data of seven advanced non-small cell lung cancer (NSCLC) patients with wild-type or mutated K-ras who failed prior therapies and subsequently treated with a combination of cisplatin and PD-1 antibody were analyzed and used to show the clinical relevance of this study. The patient clinical characteristics, K-ras and EGFR (epidermal growth factor receptor) mutation status and other relevant information are shown in Fig. 9G. All seven patients were at advanced disease stage and had failed prior chemotherapy. The patients received immunochemotherapy using a combination of toripalimab (3 mg/kg, i.v. drip, q3w) and cisplatin (35 mg/m2, i.v. drip, q3w) as a second-line treatment. The clinical study using toripalimab and various chemotherapeutic agents in lung cancer patients was a separate clinical research project at Hubei Cancer Hospital (Wuhan, China).
Fig. 9.

Therapeutic effect of toripalimab in combination with cisplatin in advanced stage NSCLC patients with mutant or wild-type K-ras. (A) Thoracic CT image of a NSCLC patient (Pt No. 1) with K-rasG12C mutation at advanced stage (T2N3M1). The red arrow indicates a large tumor before treatment. (B) Thoracic CT image of the tumor after 6 cycles of chemotherapy with docetaxel plus cisplatin. (C) Thoracic CT image of the tumor after additional radiotherapy (SBRT). (D) Thoracic CT image showing tumor regrowth at disease progression. (E) Thoracic CT image showing partial regression (PR) of the tumor after 2 cycles of immunochemotherapy with toripalimab (3 mg/kg, i.v. q3w) and cisplatin (35 mg/m2, i.v., q3w). (F) Thoracic CT image of the tumor in partial regression after 4 cycles of immunochemotherapy. (G) Clinical characteristics and therapeutic outcome of 7 advanced NSCLC patients with mutant or wild-type K-ras receiving immunochemotherapy with a combination of toripalimab and cisplatin as a second-line treatment. Abbreviations: Cis, cisplatin; Tor, toripalimab; SD, stable disease; PD, progressive disease; PR, partial remission; wt, wild-type.
T lymphocytes were obtained from blood samples of anonymous healthy donors. Tumor tissues from untreated or gemcitabine-treated patients were de-identified.
Statistics
All experiments were performed and repeated under the same conditions for at least three times (3 or more independent experiments). Q-Q plots were used to check the assumption that the experimental data were normally distributed, either on the original scale or on the log scale, and if the points were aligned on the diagonal, data were considered as normally distributed. Results are expressed as mean ± SD (standard deviation) or mean ± SEM (standard error of the mean) as indicated. Student T-tests were used to compare the means between two groups when data were normally distributed. ANOVA with F tests were used to compare the means of more than two independent groups when data where normally distributed. When ANOVA was significant, Tukey or Dunnett post hoc tests were performed to find two-by-two significant differences. The tumor volumes obtained from the animal experiments were first log-transformed before performing a generalized linear model (GLM) analysis, with treatment as a 6-level between group factor, with time as a within group factor, and with their interaction. For the F-test, F was reported with its 2 degrees of freedom and the corresponding P-value. Post-hoc comparisons between the drug-treated groups and the PBS (phosphate-buffered saline)-treated group were performed using Dunnett’s contrasts. P-values for two-by-two comparisons were corrected for multiple comparisons using the Bonferroni method. GraphPad Prism software (San Diego, CA, USA) and SAS 9.4 software (SAS Institute Inc; Chicago, IL, USA) were used to perform statistical analyses. No data were excluded and no statistical method was used to calculate sample sizes, which were determined empirically in this study. All statistical analyses were two-tailed, and a P-value equal or <0.05 was considered statistically significant.
Data availability
The experimental data and materials, generated and analyzed during this study, are available from the corresponding authors on reasonable request.
Ethics statement
The experiments involving animals were conducted according to the ethical policies and procedures approved by the Animal Care and Use Committee of Sun Yat-Sen University Cancer Center, Guangzhou, China (Approval no. L102012016010E).
Analysis of clinical data was performed retrospectively using data from a separate study of lung cancer patients treated with toripalimab and various chemotherapeutic agents with proper informed consent, which was reviewed and approved by the Committee for Ethical Review of Research Involving Human Subjects of Hubei Cancer Hospital Affiliated to Tongji Medical College, Wuhan, China (Approval no. LLHBCH2020LW024).
Results
Gemcitabine and cisplatin exhibit opposite effects on the therapeutic activity of PD-1 antibody in K-ras-driven cancer
Our recent study showed that mutant K-ras signaling could induce high expression of PD-L1 through a novel mechanism involving ROS-mediated growth factor signaling [8]. The ability of mutant K-ras to promote PD-L1 expression was consistently observed in human pancreatic normal epithelial cells (HPNE) harboring a doxycycline-inducible K-rasG12D expression system, identified as HPNE/K-rasG12D cells (Fig. 1A). This observation prompted us to test the potential in vivo therapeutic effect of PD-1 antibody alone or in combination with chemotherapeutic drugs in mice bearing syngeneic pancreatic cancer allografts. Gemcitabine and cisplatin were used in combination with immunotherapy. Gemcitabine is the standard first-line treatment for PDAC. Cisplatin demonstrated immunostimulant activity in a T cell-based screening assay [28] and is used in combination with gemcitabine for treatment of cancers harboring BRCA mutations (5–10% of PDAC). Cisplatin is also commonly used in regimens to treat colon and lung cancers, which often contain K-ras mutation. As shown in Fig. 1B, pancreatic cancer cells derived from the K-rasG12D transgenic KPC (Pdx1-Cre/LSL-K-rasG12D/LSL-p53R172H) mouse model were injected into the right flank of C57BL/6 mice. When the tumor size reached approximately 100 mm3, the mice were divided into 6 groups for treatment with PD-1Ab, gemcitabine (Gem), cisplatin (Cis), or their combination as indicated. The time trends of tumor growth were significantly different among the 6 groups of mice over the period of drug treatment between day 7 to day 28 (Interaction F(5,36) = 12.4; P < 0.001) and the post-treatment period between day 28 to day 45 (Interaction F(5,36) = 10.2; P < 0.001), indicating various impacts of the different therapeutic agents (Fig. 1C and D). Specifically, the mean tumor volume growth was + 3.7% per day (F(1,6) = 20.7; P = 0.004) in the control mice treated with PBS. Gemcitabine appeared to have little therapeutic effect since the tumor growth in this drug treatment group was similar to that of the control mice. Treatment with PD-1Ab exhibited significant therapeutic activity, evidenced by an obvious delay of tumor growth (+0.7%/day, F(1,6) = 0.10) during the treatment period. The tumors in 4 of the 7 mice in this treatment group disappeared. However, the remaining tumors in 3 mice started to grow significantly a week after the treatment stopped (P < 0.001, Dunnett’s test). Surprisingly, a combination of PD-1Ab and gemcitabine did not show any therapeutic benefit, as evidenced by significant tumor growth in mice treated with both drugs (+3.9%/day, F(1,6) = 10.8, P = 0.02). This tumor growth rate (+3.9%/day) was similar to that of the control mice (+3.7%/day) and significantly worse than that of the mice treated with PD-1Ab alone (+0.7%/day, Fig. 1C), indicating an antagonistic effect between gemcitabine and PD-1Ab.
Fig. 1.

In vivo therapeutic activity of PD-1 antibody in combination with cisplatin or gemcitabine. (A) HPNE/K-RasG12D cell line was incubated without (OFF) or with doxycycline for 24–72 h to induce mutant K-ras expression. K-ras (21 kDa), PD-L1 (45 kDa) and beta-actin (43 kDa) proteins were detected by immunoblotting. (B) Mouse pancreatic cancer (KPC) cells harboring K-rasG12D mutation (2x106 cells per injection) were inoculated in C57BL/6 mice. The mice were randomly divided into 6 groups (7 mice for each group). Drug treatment started on day 7. PD-1 antibody (PD-1Ab, 100 μg, i.p.), gemcitabine (Gem, 30 mg/kg; i.p.), and cisplatin (Cis, 3 mg/kg; i.p) were injected as indicated. The control mice were treated with PBS. (C-D) Tumor volumes are means ± SD, and were log-transformed before performing GLM analysis. The number on the right side of each curve indicates the number of tumor-free mice at the end of the study. Groups treated with PBS or PD-1Ab are same in (C) and (D). (E) Mice were inoculated with syngeneic PDAC, and divided into 6 groups for treatment with PBS, gemcitabine, cisplatin, and PD-1 antibody as indicated (3 mice/group). (F-G) Tumor tissues were processed on day 15 for TUNEL staining (green). DAPI was used to stain nuclei (blue). Representative images are shown in (G), and quantitative data are means ± SEM and shown in (F). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (one-way ANOVA and Tukey post hoc test).
In contrast, combination of PD-1Ab with another chemotherapeutic agent cisplatin exhibited striking synergistic effect (Fig. 1D), resulting in a significant decrease of tumor size (growth rate −5.9%/day, F(1,6) = 33.4, P < 0.001) and eventually a complete disappearance of tumors in 6 out of the 7 mice. The remaining tumor did not show any significant growth after treatment stopped (Fig. 1D). Treatment with cisplatin alone showed a detectable therapeutic effect with a substantial delay in tumor growth (-0.6%/day, F(1,6) = 0.2) during drug treatment. There was no complete tumor regression in the mice treated with cisplatin alone. After the stop of cisplatin treatment for a week, tumors started to grow significantly (+4.3%/day, F(1,6) = 35.3, P < 0.001).
In a separate set of animal experiments, mice were treated with the individual drugs or their combination using the dose-schedule indicated in Fig. 1E. After treatment for two weeks, tumor tissues were isolated 24 h after the last drug treatment and examined for drug-induced cell death in vivo using TUNEL assay. Cisplatin and PD-1Ab were able to induce significant cell death in vivo, whereas gemcitabine showed minimum effect (Fig. 1F and G).
To test if the mice that became tumor-free after treatment could retain their immunity against the same tumor, the tumor-free mice (the 4 mice from the PD-1Ab treated group and 6 mice from the PD-1Ab + cisplatin combination group) were re-inoculated with the same mouse pancreatic cancer cells (Pdx1-Cre/LSL-K-rasG12D/LSL-p53R172H) at higher cell density (1x107 cells per injection site) at 100 days after the end of the first run of drug treatment. None of the 10 mice developed any tumor (Fig. 2A). In contrast, inoculation of same number of cells to control mice without prior exposure to cancer cells (naïve mice, comparable age) resulted in tumor development in all three mice, which grew rapidly to 400 mm3 within two weeks after inoculation (Fig. 2A). These data together suggest that the mice that became tumor-free after the first run of drug treatment likely retained long-term memory T cells against the previously inoculated cancer cells.
Fig. 2.

Effect of cisplatin, gemcitabine and their combination with PD-1Ab in KPC and CT26 allograft mouse models. (A) The tumor-free mice from PD-1Ab (n = 4) or cisplatin + PD-1Ab (n = 6) groups in (Fig. 1D) were re-inoculated with 1x107 syngeneic PDAC (KPC) one hundred days after the first inoculation. Age-matched mice without prior exposure to cancer cells (naïve, n = 3) were inoculated with identical cancer cells as a control. The number above each group indicates the number of mice that developed tumors on day 14, and tumor volumes are shown as means ± SEM. (B) Mouse CT26 colon cancer cells (0.3x106 cells per injection) were inoculated in BALB/c mice. The mice were randomly divided into 6 groups (8 mice for each group). Cisplatin (Cis, 3 mg/kg; i.p.), gemcitabine (Gem, 30 mg/kg; i.p.) and/or PD-1 antibody (PD-1Ab, 100 μg, i.p.) were injected as indicated; the control group was treated with phosphate-buffered saline (PBS). (C-D) Tumor sizes were measured twice a week. Tumor volumes are shown as means ± SD. Groups treated with PBS or PD-1Ab are same in (C) and (D). Data were log-transformed for GLM analysis (between days 24 and 35).
We also used another mouse tumor model, the CT26 colon cancer with homozygous K-rasG12D mutation known to be insensitive to anti-PD-1 treatment [33], [34], to test if combination of cisplatin with anti-PD-1 could overcome the tumor’s resistance to anti-PD-1. As shown in Fig. 2B, mice bearing CT26 tumor were treated with cisplatin, gemcitabine, anti-PD-1 antibody, or their combination as indicated. Mice treated with anti-PD-1 alone (100 μg, i.p, twice a week) did not exhibit any therapeutic activity compared to PBS-treated control group (P = 0.99, Bonferroni corrected P-value, Fig. 2C and D). Cisplatin alone showed a significant therapeutic activity against this colon cancer allograft model (P = 0.003; Bonferroni corrected P-value, compared with PBS-treated control). Importantly, combination of cisplatin with PD-1Ab resulted in a further increase of in vivo anticancer activity, compared with mice treated with cisplatin alone (P = 0.009, Bonferroni corrected P-value, between days 24–35, Fig. 2C). In contrast, gemcitabine showed better therapeutic effect than cisplatin but no synergy in combination with PD-1Ab (P = 0.49, Bonferroni corrected P-value, between days 24–35, Fig. 2D).
PD-1 antibody and cisplatin enhance T cell functions whereas gemcitabine inhibits intratumoral T cell infiltration
The results of TUNEL assay (Fig. 1E–G) seemed to provide a reasonable explanation for the superior in vivo therapeutic activity of cisplatin and PD-1Ab combination, but could not explain why gemcitabine and PD-1Ab had antagonist effect. To explore the potential underlying mechanisms, we first tested whether gemcitabine and cisplatin might have different impacts on T cell functions such as their ability to infiltrate the tumor tissues and to secrete cytokines. As shown in Fig. 3A, administration of PD-1 antibody alone significantly increased the number of tumor-infiltrating CD8+ T lymphocytes compared to the control samples from mice treated with PBS. Gemcitabine treatment significantly reduced the number of CD8+ T cells in the tumor tissues, whereas cisplatin did not show such a suppressive effect (Fig. 3A and B). Importantly, combination of gemcitabine with PD-1Ab almost completely abolished the ability of PD-1Ab to promote CD8+ T cell infiltration, while cisplatin did not compromise the CD8+ T cell infiltration induced by PD-1Ab (Fig. 3A and B). To gain further insight into the role of the immune system in affecting tumor growth in vivo, we inoculated treated immunocompetent mice (C57BL/6) and immunodeficient mice (nude BALB/c athymic mice that lack T cells) with the same number of pancreatic cancer cells (1x106 cells per injection), and treated with gemcitabine or cisplatin as shown in Fig. 3C. We observed that PDAC tumors grew much faster in nude mice than in B6 mice, suggesting a significant role of the immune system in suppressing tumor growth. In the immune-deficient mice, gemcitabine and cisplatin showed a slight inhibitory effect on tumor growth. In immune-competent mice, gemcitabine seemed to promote tumor growth instead of showing therapeutic effect (Fig. 3D–F), likely reflecting its immunosuppressive effect. The tumor CD8+/CD4+ T cell ratio decreased 6-fold in PDAC-bearing C57BL/6 mice treated with gemcitabine (Fig. 3G).
Fig. 3.

Impact of PD-1 antibody, cisplatin, and gemcitabine on CD8+ T cell tumor infiltration. (A) PDAC tumor-bearing C57BL/6 mice were treated as described in Fig. 1E and tumor tissues were immunostained to detect and quantify CD8+ T lymphocytes. (B) Immunostaining of CD8+ T lymphocytes in PDAC tumor-bearing C57BL/6 mice treated as described in Fig. 1E. Scale bars: 100 μm. (C) Mouse pancreatic cancer (KPC) cells (1x106 cells per injection) expressing K-rasG12D (8–10 mice/ group) were inoculated in C57BL/6 mice and nude BALB/c mice. Gemcitabine (Gem, 30 mg/kg; i.p.) was given twice a week (at days 7, 11, 14, 18, 21, 25 and 28), and cisplatin (Cis, 3 mg/kg; i.p) was given once a week (at days 7, 14, 21 and 28). (D) Tumor growth in immune-competent C57BL/6 mice treated with cisplatin or gemcitabine as indicated. (E) Tumor growth in immune-deficient BALB/c nude mice treated with cisplatin or gemcitabine as indicated. (F) Mice were sacrificed at day 33 and tumor weights were measured. (G) PDAC tumor-bearing mice were treated as Fig. 1E. Comparison of CD8/CD4 ratio in tumor tissues from PBS and gemcitabine treated groups (n = 3). (H) Comparison of CD8/CD4 ratio in pancreatic tumor tissues from patients treated with gemcitabine (n = 6) or not (Untreated; n = 10). (I) Immunostaining of CD4+ and CD8+ T lymphocytes in human PDAC tumor tissues. Representative images are shown of tumors from one untreated PDAC patient, treated with gemcitabine or with combination gemcitabine + paclitaxel. Scale bars: 50 μm. Statistical analyses: data are means ± SEM of at least three separate biological replicates (for A, G) except for F and H (means ± SD); one-way ANOVA and Tukey post hoc test for A, F; two-tailed unpaired t-test for G-H. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To evaluate the clinical relevance of the change in CD8+/CD4+ T cell ratio induced by gemcitabine observed in the mouse model, we further analyze clinical specimens from pancreatic cancer patients with or without gemcitabine treatment. As shown in Fig. 3H and I, a significant decrease of CD8+/CD4+ T cell ratio was also observed in the clinical samples from pancreatic cancer patients treated with gemcitabine. In a tumor specimen from a patient treated with gemcitabine and paclitaxel, the CD8+/CD4+ T cell ratio was 2.47, compared to 3.53 ± 1.60 in the untreated tissues and 1.13 ± 0.49 in the gemcitabine-treated samples. The number of CD4+ T cells in the gemcitabine + paclitaxel treated sample was 91 cells/mm2, lower than that of the gemcitabine-treated tumor sample (185 ± 50 cells/mm2) and the untreated samples (106 ± 42 cells/mm2). The number of CD8+ T cells were not significantly different among the three groups (376 ± 217, 204 ± 97, 226 cells/mm2 respectively for the untreated, gemcitabine and gemcitabine + paclitaxel-treated tumors) (Fig. 3H and I, Supplementary material Fig. S1). In a separate set of pancreatic cancer tissues from patients of another cancer hospital, we also observed a significant decrease in intratumoral CD8+ T cells in the samples from gemcitabine-treated patients, with a concurrent increase of CD4+ T cells, leading to a substantial decrease of CD8+/CD4+ ratio (Table 1).
Table 1.
Intensity of staining for CD8 and CD4 in pancreatic tumor tissues from patients treated with gemcitabine or not (Untreated).
| Score CD8 | Mean | SD | n | P-value |
|---|---|---|---|---|
| Untreated | 1.8772 | 2.0120 | 47 | |
| Gemcitabine | 1.2472 | 1.2605 | 88 | 0.0273 |
| Score CD4 | Mean | SD | n | P-value |
| Untreated | 0.1689 | 0.2866 | 46 | |
| Gemcitabine | 0.6748 | 0.7474 | 88 | <0.001 |
Statistical analysis: unpaired T-test.
We then performed immuno-phenotyping using a panel of cell surface markers to further analyze the detail changes of immune cells in mice treated with PD-1 antibody and/or chemotherapeutic drugs. The results showed that the combination of cisplatin and PD-1Ab increased mainly the proportion of CD8+ T cells and gemcitabine increased the proportion of CD4+ T cells within the TIL population (Fig. 4A and B). The number of CD19+ B cells and CD11c+ dendritic cells remained unchanged in different treatment groups (Fig. 4C and D). However, the proportion of intratumoral CD11b+ myeloid cells and F4/80+ macrophages increased in the PD-1Ab treatment group, whereas the number of these cell types in the PD-1Ab + Cis combination group was similar to the control group (Fig. 4E and F). Consistent with clinical specimen (Table 1), an increase in CD4+ cell populations was also observed in tumors from gemcitabine-treated mice (Fig. 4B). We thus explored the possibility that gemcitabine might trigger the infiltration of immunosuppressive CD4+/FoxP3+ T regulator cells but the number of FoxP3+ cells remained low and similar among the six groups (Fig. 4G, Supplementary material Fig. S2). Consistently, in the CT26 subcutaneous tumor model, the number of CD8+ T cells increased about 3-fold in the mice treated with anti-PD-1 in combination with cisplatin. Conversely, the number of intratumoral CD8+ T cells was unchanged in mice treated with gemcitabine or the combination gemcitabine and PD-1Ab (Fig. 4H, Supplementary material Fig. S3).
Fig. 4.

Analyses of tumor cell populations in KPC and CT26 tumor-bearing mice. (A–F) PDAC (KPC) tumor-bearing C57BL/6 mice were treated as described in Fig. 1E (3 mice/ group). Mice were sacrificed on day 15 and proportion of (A) CD3+ CD8+, (B) CD3+ CD4+, (C) CD3+ CD19+, (D) CD11c+, (E) CD11b+ and (F) F4/80+ cells within CD45+ population were analyzed by flow cytometry. (G) KPC tumor tissues (from Fig. 1E) were immunostained to detect and quantify FoxP3+ Treg lymphocytes. Data are mean ± SD of three to four biological replicates. (H) CT26 tumors were harvested at end-points (from Fig. 2C-D) and were processed for immunostaining of CD8+ T cells. The number of CD8+ lymphocytes was quantified in each group. Data are mean ± SD of five to six biological replicates. Statistical analysis: data are mean ± SEM of three biological replicates (A-F); one-way ANOVA and Tukey post hoc test for A-H. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We then sought to quantify the expressions of CD4+ T cell population markers. Interestingly, the mRNA levels of Tbx21 (T-box transcription factor 21) and IFNγ decreased while the mRNA levels of GATA3 increased in the whole tumor tissues from immunocompetent mice treated with gemcitabine (Fig. 5A and B). These findings suggested that there was a decrease in Th1 in favor of a Th2 response, consistent with the immunosuppressive effect of gemcitabine. Quantitative PCR results confirmed that the populations of Treg did not increase (Fig. 5C), and most of the markers for Th9 (interleukin-9) and Th17 (interleukin 17–22-23) populations were not detectable (Fig. 5D).
Fig. 5.

CD4+ T cell population markers (Th1, Th2, Treg and others) in mouse pancreatic tumor tissues. (A-D) PDAC (KPC) tumor-bearing C57BL/6 mice were treated as described in Fig. 1E. The expressions of CD4+ T cell markers for (A) Th1, (B) Th2, (C) Treg, and (D) other T cell populations were measured by qRT-PCR. Statistical analyses: data are means ± SEM of three biological replicates; two-tailed unpaired T-test for A-D. *, P < 0.05.
The impact of gemcitabine and cisplatin on T cell migration was further tested in vitro, using a mouse CD8+ T cell line (B3Z). As shown in Fig. 6A, serum (FBS)-induced T cell migration in vitro was significantly suppressed by a relatively low concentration of gemcitabine (5 nmol/L), whereas no significant inhibition of T cell migration was observed in B3Z cells treated with 250 nmol/L cisplatin.
Fig. 6.

Impact of PD-1 antibody, gemcitabine, and platinum drugs on T cell function and cytokine secretion. (A) B3Z cells (mouse CD8+ T cells, 1x106/mL) were incubated with 5 nmol/L gemcitabine or 250 nmol/L cisplatin for 4 h in medium without FBS. The cells were then transferred to the chemotaxis chamber (upper chamber). The lower chamber contains RPMI medium with FBS. After 16 h incubation, lymphocytes migrated to the lower chamber was quantified by fluorescent assay. “RPMI” indicates the background fluorescent of the medium. (B) Human CD8+ T cells (1x106) were first purified and pre-treated with gemcitabine or cisplatin for 24 h, before stimulation with PMA/Ionomycin for 24 h. IFN-γ levels were measured by ELISA (n = 4). (C) B3Z cells (2.5x105) were incubated with gemcitabine or cisplatin for 24 h, and then stimulated with PMA/ionomycin for 24 h. IFN-γ levels were measured by ELISA. (D-F) The mRNA levels of IFNγ, GZMB (granzyme B) and PRF1B (perforin) were analyzed by qRT-PCR in KPC tumors from mice treated with PD-1Ab, cisplatin, gemcitabine, or their combination as indicated (3 mice/ group; from Fig. 1E). (G-I) Quantitative RT-PCR analysis of CXCL9, CXCL10 and CXCL11 mRNA expression in KPC tumor tissues from mice treated with PD-1Ab, cisplatin, gemcitabine, or their combination as indicated (3 mice/ group; from Fig. 1E). (J) B16-OVA cells and immune cells (B3Z T cells + BMDC) were co-cultured and exposed to DMSO (0.3% v/v) or the indicated platinum drugs at their respective IC25 concentrations. IL-2 levels in the culture medium were then measured using ELISA assay. Statistical analyses: data are means ± SEM of at least three separate experiments or biological replicates; data were log-transformed before analysis for B; one-way ANOVA and Tukey post hoc test for A, D-I; one-way ANOVA and Dunnett post hoc test for B-C (to PBS), J (to DMSO). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We then further tested if gemcitabine and cisplatin might have different impacts on the ability of CD8+ T cells to secrete IFN-γ as another indicator of functional changes. Quantitative analysis by ELISA showed that gemcitabine had no significant effect on IFN-γ secretion, whereas cisplatin treatment could significantly stimulate IFN-γ secretion in both mouse and human CD8+ T cells (Fig. 6B and C). Consistent with these findings, the expression of T cell activation markers (IFN-γ, granzyme B and perforin) was enhanced in the tumor tissues (containing T cells) from mice treated with the combination of PD1 antibody plus cisplatin, whereas gemcitabine did not affect their expression (Fig. 6D–F).
To further explore the mechanisms for different effects of gemcitabine and cisplatin on T cell infiltration, we measured the expression of chemokines in tumor tissues. As shown in Fig. 6G–I, the expression of chemoattractant molecules (CXCL9, CXCL10 and CXCL11) decreased in tumors from mice treated with gemcitabine, whereas cisplatin increased their expression. Anti-PD-1 administration alone also augmented the expression of these chemokines, which was inhibited by gemcitabine (P < 0.05, unpaired T-test versus PBS). In contrast, cisplatin in combination with PD-1Ab further enhanced the expression of these chemoattractant molecules (Fig. 6G–I).
We also tested the effect of cisplatin on T cell secretion of IL-2 as another indicator of the drug impact on T cell functions. Ovalbumin (OVA)-specific B3Z T cells and bone marrow-derived dendritic cells were co-cultured with B16-OVA cells to induce IL-2 secretion, and the co-culture samples were exposed to DMSO (0.3% v/v) or cisplatin. Oxaliplatin, a platinum derivative used in clinical treatment of colon and pancreatic cancers, was also included for comparison at their respective IC25 concentrations. As shown in Fig. 6J, cisplatin and oxaliplatin significantly enhanced T cell secretion of IL-2.
Cisplatin and gemcitabine induce chemokine expression in a cGAS-dependent manner
In an attempt to investigate the mechanisms for the differential impact of gemcitabine and cisplatin on the expression of chemoattractant chemokines, we first treated the mouse pancreatic cancer cells with gemcitabine or cisplatin in vitro and then assay for type I interferon molecule (IFNB1) known to regulate the expression of these chemokines [35]. As shown in Fig. 7A–C, cisplatin (but not gemcitabine) induced a high expression of IFN-β and its downstream chemokines CXCL10/11 (Supplementary material Fig. S4A). To further explore the mechanism responsible for the differential effect of cisplatin and gemcitabine on IFN-β, we then tested the involvement of cGAMP synthase, which is known to regulate type I IFN signaling in response to certain DNA damage such as double-strand breaks through the cGAS/STING (stimulator of interferon genes) signaling pathway [36]. We used sgRNA guided CRISPR/cas9 technology to disrupt cGAS gene in mouse pancreatic cancer cells, and then compare them with the wild-type cells for their expression of IFN-β, and CXCL10 in response to double-strand DNA fragments (dsDNA 60-mers) transfection or to treatment with cisplatin or gemcitabine (Fig. 7D). As shown in Fig. 7E-G (and Supplementary material Fig. S4B), the wild-type cells exhibited increased expression of the cytokines after transfection with double-stranded DNA fragments, but such cytokine induction was not observed in the cGAS-deficient cells. Cisplatin, which is known to induce double-strand DNA breaks, was able to induce a significant increase of the cytokine expression in the wild-type cells but not in the cGAS-deficient cells (Fig. 7H–J, Supplementary material Fig. S4C). Interestingly, although transfection of wild-type cells with dsDNA fragments induces the expression of IFN-β1 and CXCL10/11, gemcitabine did not cause any further changes in the expression of these cytokines (Fig. 7K and M, Supplementary material Fig. S4D). Similar results were observed in the human CFPAC-1 pancreatic cancer cell line with K-rasG12V mutation. Indeed, CXCL10 and CXCL11 mRNA levels were augmented when this cell line was incubated for 48 h with cisplatin, but not with gemcitabine (Fig. 7N and O). These data together suggest an important role of cGAS in recruiting T cells to the tumor sites through the secretion of chemoattractant chemokines in response to DNA damages caused by cisplatin.
Fig. 7.

Cisplatin induces the expression of chemokines in a cGAS-dependent manner. (A-C) Mouse pancreatic (KPC) cancer cells were incubated with gemcitabine (Gem) or cisplatin (Cis) for 48 h. The expressions of IFN-β and CXCL10 were measured by qRT-PCR and ELISA. (D) Experimental design: cGAS-wild-type and cGAS-depleted mouse PDAC cells were exposed to drugs or dsDNA (cGAS inducer) for 48 h. The expression of chemokines was then quantified by qRT-PCR and ELISA. (E-G) Mouse pancreatic wild-type (WT) or the sgRNA-mediated cGAS-disrupted cells (cGAS-sgRNA) were transfected with dsDNA fragments for 48 h. The expressions of IFN-β and CXCL10 were measured by qRT-PCR and ELISA. (H-J) Mouse pancreatic wild-type (WT) or cGAS-disrupted cells were incubated with cisplatin for 48 h. The expressions of IFN-β and CXCL10 were measured by qRT-PCR and ELISA. (K-M) Wild-type mouse pancreatic cancer cells were incubated with gemcitabine. Eight hours post-treatment, cells were transfected with dsDNA fragments for 40 h. The expressions of IFN-β and CXCL10 were measured by qRT-PCR and ELISA. (N-O) Human pancreatic CFPAC-1 cancer cells were incubated with gemcitabine or cisplatin for 48 h. The expressions of CXCL10 and CXCL11 were quantified by qRT-PCR. Statistical analyses: data are means ± SEM of three separate experiments; some values (i.e., untreated cells) are similar for C, G, J and M; one-way ANOVA and Tukey post hoc test for A-C, E-O. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We then sought to determine whether gemcitabine and its combination with cisplatin could alter chemokine expression in K-ras-driven cancer cells. We found that combination of 10 µmol/L cisplatin and gemcitabine exerted very high cytotoxicity (Fig. 8A, D and G). As shown in Fig. 8B, a low concentration gemcitabine (30 nmol/L) did not induce significant DNA damage in KPC cells, while cisplatin (3 µmol/L) induced substantial DNA damage as indicated by the phosphorylation of Chk1 (checkpoint kinase 1). As previously described, only cisplatin could upregulate CXCL10 in KPC cells and gemcitabine-cisplatin combination did not influence the levels of CXCL10 expression (Fig. 8C). Mouse LLC lung cancer cells (K-rasG12C) were more sensitive to gemcitabine compared to KPC cells (Fig. 8D). Gemcitabine (30 nmol/L) induced DNA damage in LLC cells (Fig. 8E), and was able to induce CXCL10 expression (Fig. 8F). These data suggest that gemcitabine might not inhibit cGAS-STING pathway and could activate it at certain concentrations. However, combination of various concentrations of gemcitabine (10–30 nmol/L) and cisplatin (3 µmol/L) slightly decreased CXCL10 expression compared to cisplatin alone (Fig. 8F), likely due to high cytotoxicity. At subtoxic concentrations, gemcitabine did not induce CXCL10 expression in CT26 cancer cells (Fig. 8G–I). Unlike in LLC cells, combination of gemcitabine and cisplatin did not alter chemokine expression in CT26 cells (Fig. 8I). Finally, we tested the drug combination in human A549 (K-rasG12S) lung cancer cells. This cell line was more resistant to the drugs (Fig. 8J), and cisplatin (10 µmol/L) and gemcitabine (100 nmol/L) were able to induce DNA damage (Fig. 8K). However, CXCL10 mRNA was not detected in any of the tested samples, likely due to lack of STING expression in A549 cells (Fig. 8K). This result confirmed the important role of cGAS-STING pathway in the drugs-mediated chemokine upregulation.
Fig. 8.

Effects of gemcitabine and cisplatin combination in upregulation of CXCL10 expression in multiple types of cancer cells. (A) Mouse KPC pancreatic cancer cells were incubated with various concentrations of gemcitabine, cisplatin or in combination for 48 h. Cell survival was quantified by MTT assay. (B) KPC cells were treated with drugs for 24 h. P-Chk1 (S345), Chk1, STING and beta-actin proteins were detected by immunoblotting. (C) KPC cells were treated with drugs for 48 h. The expression of CXCL10 was then quantified by qRT-PCR. (D) Mouse LLC lung cancer cells were incubated with various concentrations of gemcitabine, cisplatin or in combination for 48 h. Cell survival was quantified by MTT assay. (E) LLC cells were treated with drugs for 24 h. P-Chk1 (S345), Chk1, STING and beta-actin proteins were detected by immunoblotting. (F) LLC cells were treated with drugs for 48 h. The expression of CXCL10 was then quantified by qRT-PCR. (G) Mouse CT26 colon cancer cells were incubated with various concentrations of gemcitabine, cisplatin or in combination for 48 h. Cell survival was quantified by MTT assay. (H) CT26 cells were treated with drugs for 24 h. P-Chk1 (S345), Chk1, STING and beta-actin proteins were detected by immunoblotting. (I) CT26 cells were treated with drugs for 48 h. The expression of CXCL10 was then quantified by qRT-PCR. (J) Human A549 lung cancer cells were incubated with various concentrations of gemcitabine, cisplatin or in combination for 48 h. Cell survival was quantified by MTT assay. (K) A549 cells were treated with drugs for 24 h. P-Chk1 (S345), Chk1, STING and beta-actin proteins were detected by immunoblotting. Statistical analyses: data are means ± SEM of three separate experiments; one-way ANOVA and Tukey post hoc test for C, F and I. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Combination of toripalimab with cisplatin in lung cancer patients with K-ras mutation shows promising therapeutic effect
The observation that PD-1 antibody in combination with cisplatin or gemcitabine produced opposite therapeutic effect in mice bearing K-ras-driven cancer models prompted us to evaluate its clinical relevance by analyzing clinical data from a separate clinical study in Hubei Cancer Hospital where toripalimab (a PD-1 antibody) and various chemotherapeutic agents were used for treatment of lung cancer patients. A retrospective review of the NSCLC patients who had been treated with toripalimab in combination with gemcitabine and cisplatin revealed that this immunochemotherapy produced variable results, and some of the patients exhibited rapid disease progression. For instance, a patient (male, age 57) with K-rasG12C mutation (EGFR wild-type) at advanced stage (T4N3M0) was initially treated with a combination of docetaxel (75 mg/m2, i.v. q3w) and cisplatin (75 mg/m2, i.v. q3w) for 3 cycles, followed by intensity-modulated radiotherapy (IMRT). The disease progressed in approximately one month, and the patient then received a combination of cisplatin (35 mg/m2, i.v. q3w), gemcitabine (1000 mg/m2, i.v on day 1 and day 8, q3w), and toripalimab (3 mg/kg, i.v. q3w) for 2 cycles (6 weeks). This immunochemotherapy did not induce any therapeutic response, and the patient succumbed to rapid disease progression.
In contrast, data from four advanced NSCLC patients with the same K-ras mutation (G12C) treated with toripalimab in combination with cisplatin without gemcitabine showed encouraging therapeutic results. Fig. 9A–F illustrates the results in one of these patients. This patient (male, age 45) was in advanced stage with metastasis (T2N3M1) with a large tumor in the left lung (Fig. 9A), and had K-rasG12C mutation with wild-type EGFR (Fig. 9G, Pt. No. 1). He was initially treated with a combination of docetaxel (75 mg/m2, i.v. q3w) and cisplatin (75 mg/m2, i.v. q3w) for 6 cycles (18 weeks) and stereotactic body radiotherapy (SBRT). This treatment induced partial remission (PR) for approximately one year (Fig. 9B and C) until the disease progressed (Fig. 9D). Immunochemotherapy was then initiated, using a combination of low-dose cisplatin (35 mg/m2, i.v. q3w) and toripalimab (3 mg/kg, i.v. q3w). After two cycles of treatment with toripalimab and cisplatin, the tumor shrunk (Fig. 9E). This partial remission was maintained following four cycles of treatment (Fig. 9F), consistent with the durable effect of immunotherapy. The patient is still in partial remission at the time of this article preparation (23 months after toripalimab + cisplatin treatment). Fig. 9G shows all four NSCLC patients with K-rasG12C mutation at advanced stage treated with a combination of toripalimab and cisplatin as a second-line therapy. Two out of the four patients had partial remission, one patient had stable disease and one patient exhibited disease progression. Among three NSCLC patients with wild-type K-ras treated with cisplatin plus toripalimab, two patients had stable disease and one patient deceased. These data appear promising, since the therapeutic effect seemed superior to that of PD-1 antibody alone as a second-line treatment in advanced NSCLC patients with K-ras mutation.
Discussion
Targeting immune checkpoint molecules is a promising antitumor strategy [37], and therapeutic antibodies against PD-1 and PD-L1 are effective for certain patients with melanomas, lung and renal cancers [20], [21], [22]. However, PD-1 antibody monotherapy showed limited therapeutic activity for certain cancers such as pancreatic cancer, and the results of clinical trials of immune checkpoint therapy in combination with chemotherapeutic agents in pancreatic cancer patients remain to be seen [38]. Since the vast majority of pancreatic cancer cells harbor oncogenic K-ras mutations which promote PD-L1 expression [8], it is possible that PD-1/PD-L1 antibodies could still be effective against pancreatic cancer if other major immune suppressive factors could also be identified and alleviated. Our study showed that administration of PD-1 antibody was effective in immunocompetent mice bearing syngeneic pancreatic cancer allografts, but its combination with gemcitabine surprisingly yielded antagonist results due to the drug inhibition of T-cell infiltration in the tumor tissues. While gemcitabine seems able to eliminate myeloid-derived suppressive cells (MDSC) and therefore activate the T cell response [39], this chemotherapy was also known to induce the release of IL-1β by MDSC and the stimulation of T helper 17 cells which in turn enhance tumor growth [40], [41]. Our study suggests that gemcitabine could have inhibitory effects on the immune system due to its suppression of T cell infiltration into the tumor tissues. It would be interesting to test such drug-induced immunosuppression in KPC murine model of spontaneous PDAC. The precise molecular mechanisms by which gemcitabine inhibits T cell infiltration remains unclear and requires further study. Our experimental findings suggest that it is unlikely that gemcitabine could inhibit cGAS-STING pathway, nor could it induce Treg-mediated immunosuppression. Since gemcitabine is often used in pancreatic cancer treatment and might also be included in PD-1 antibody clinical trials [38], it is possible that this drug at high concentration could have inhibited T cell functions in tumors and thus might have contributed to the poor clinical outcome of PD-1 antibody treatment in pancreatic cancer. In our animal study, mice treated with PD-1 antibody alone showed significant therapeutic response, whereas addition of gemcitabine abolished the therapeutic effect of PD-1 antibody, suggesting that it was gemcitabine that produced the negative effect in the animal model. Importantly, since the decrease of intratumoral CD8+ T cells was observed both in tumor samples from gemcitabine-treated mice and in two sets of clinical samples from pancreatic cancer patients treated with gemcitabine, the inhibition of cytotoxic T cells by gemcitabine seems significant and clinically relevant. While immunotherapy using immune checkpoint inhibitors has not produced significant therapeutic effect in PDAC patients so far, efforts are still ongoing to evaluate various integrated strategies to improve the outcome of immunotherapies in pancreatic cancer, based on the beneficial effects of combination therapies observed in other cancer types [42]. Gemcitabine did not enhance the tumor immunogenicity in combination with other immunotherapy such as anti-CD40 in PDAC, and showed disparate effects on patient survival [43]. Recent clinical trials showed that the combination of gemcitabine with PD-1 antibodies seemed to exhibit inferior efficiency (i.e., ORR, PFS, OS) for NSCLC patients compared to other chemotherapeutic agents (paclitaxel, pemetrexed, platinum) in combination with PD-1 antibodies [44], [45]. As such, caution should be exercised in considering using gemcitabine in combination with PD-1 antibody, although some study suggests that gemcitabine could target MDSC and enhance immune function [39].
In contrast with the immunosuppressive effect of gemcitabine, we showed that cisplatin and PD-1 antibody produced synergistic effect against pancreatic and colon cancer harboring mutant K-ras in mice, likely due to activation of certain T cell functions such as increased secretion of IFN-γ. Unlike gemcitabine, cisplatin did not suppress T cell infiltration into tumor tissues, and was able to increase the expression of chemoattractant molecules as IFN-β1 and CXCL9/10/11. Several studies have already showed a promising synergistic effect between cisplatin and immunotherapies in animal models [46], [47], [48] and in breast cancer patients [49], but the mechanisms by which cisplatin enhances an immune response still remained poorly understood. Cisplatin was previously classified as a poor ICD (immune cell death) inducer [50], whereas other studies suggested that cisplatin could function as a potential immune-modulator capable of upregulating major histocompatibility complex (MHC) class I expression and promoting the activity of human cytotoxic T cells [51], [52]. In our study, we demonstrated that cisplatin enhanced the expression of the chemokines likely through induction of DNA damage to stimulate the cGAS/STING signaling pathway. A recent study suggests an important role of cGAS in affecting the antitumor effect of monoclonal antibodies targeting PD-L1 and CTLA4 (cytotoxic T-lymphocyte associated protein 4) [53]. The presence of intratumoral CD8+ lymphocytes is considered as a strong prognostic factor of responsiveness to immunotherapies [54]. Our study showed that cisplatin was effective in promoting CD8+ T cell infiltration in tumor tissues and enhancing the in vivo therapeutic effect of PD-1 blockade in both syngeneic pancreatic and colon cancer models harboring mutant K-ras, suggesting a possibility that this drug could potentially improve the therapeutic activity of anti-PD-1 in patients with K-ras-driven cancer. Another explanation for the superior effect of cisplatin could be the release of damage-associated molecular patterns (DAMPs) from dying cancer cells. These molecules would then stimulate antigen-presenting cells to activate T cells.
Our analysis of clinical data from clinical study suggests that the combination of cisplatin and toripalimab has promising therapeutic effect in advanced NSCLC patients with who failed prior chemotherapy and radiotherapy, especially in patients with K-rasG12C mutation. Two of the four patients with K-rasG12C mutation treated with cisplatin and toripalimab showed partial response and one patient maintained stable disease without progression at the time of this manuscript preparation. Our data analysis also suggests that inclusion of gemcitabine in this immunochemotherapy might not bring addition therapeutic benefits in patients with K-ras mutation, and could potentially compromise the clinical outcome. However, it is important to note that the above observations were from data of a very small number of patients. Further clinical studies with large number of patients with tumors harboring wild-type and mutant K-ras are needed to test the therapeutic effect of cisplatin in combination with immune checkpoint blockade therapy. It is of significant interest to note that recent studies demonstrated that gemcitabine-cisplatin (GP) chemotherapy in combination with PD-1 antibody toripalimab or camrelizumab as a treatment for patients with recurrent or metastatic nasopharyngeal carcinoma is superior compared to GP alone [55], [56]. However, there was no direct comparison of the therapeutic outcome of GP plus toripalimab regimen with that of cisplatin alone plus toripalimab.
Although currently cisplatin is not a standard drug for pancreatic cancer, this compound is commonly used in clinical treatment of lung cancers, which also have relatively high frequency of K-ras mutations. Since our study showed that a combination of cisplatin with anti-PD-1 antibody produced promising therapeutic effect even in the mouse colon cancer model (CT26) known to be resistant to anti-PD-1 and in lung cancer patients with K-ras mutation, these new findings merit further evaluation of this drug combination in a clinically relevant setting such as randomized clinical trials. The results of such clinical studies will provide a basis to consider the feasibility to use cisplatin in combination with PD-1 antibodies for treatment of pancreatic cancer, for which limited treatment options are currently available.
Immunotherapy using antibodies against PD-1/PD-L1 have shown objective clinical responses in several cancer types including melanoma, NSCLC, renal cell carcinoma, bladder cancer and Hodgkin lymphoma [57]. Based on the findings from our study, it would be interesting to investigate whether immunochemotherapy regimens that include cisplatin could provide added benefits in multiple tumor types, in addition to K-ras-driven tumors. Since most cancers are driven by activation of oncogenes or/and loss of tumor suppressor function due to various mutations, PD-L1 expression is often dysregulated due in part to the changes in these oncogenic signals (i.e., K-ras, c-myc, EGFR, etc.). The effectiveness of immunotherapy using PD-1/PD-L1 antibodies for treatment of cancers driven by oncogenic mutations appears limited [9]. As such, the use of cisplatin or other DNA-damaging drugs that stimulate immune response in combination with PD-1/PD-L1 antibodies would seem a plausible strategy to enhance the therapeutic efficacy for treatment of diverse cancer types, and merits further preclinical and clinical studies.
In the context of immunochemotherapy, a recent study using an in vitro assay to screen a panel of anticancer drugs revealed the diverse effects of chemotherapeutic agents on T cell functions [28]. DNA-damaging agents (i.e., melphalan and doxorubicin) and cisplatin were shown to have stimulatory effects on T cell functions, whereas arsenic trioxide and gemcitabine exhibited inhibitory effect. It is also important to note that drug concentrations seem to be a critical factor in the drug impact on the immune system. For instance, Paclitaxel at IC25 showed a moderate inhibitory effect on T cells function, but exhibited strong stimulatory effect at a clinically relevant concentration [28]. Thus, the selection of chemotherapeutic agents for use in immunochemotherapy should consider not only their mechanisms of action but also the drug dosage. As such, it is possible that gemcitabine at low or high concentrations might not suppress T cell functions or could even exert stimulatory effect due to its impact on MDSC [39]. Further studies are needed to test such possibilities.
Conclusion
Our study shows that cisplatin and gemcitabine exert opposite effects on immunochemotherapy with PD-1 antibody in K-ras-driven cancer due to their different impacts on CD8+ T cell functions, and suggests that it is extremely important to select appropriate chemotherapeutic drugs for combination with immunotherapy such as PD-1 antibody in order to achieve optimal anticancer activity. The impact of chemotherapeutic agents on T cell functions including their tumor infiltration seems to be the key determinant. Cisplatin-based chemotherapy seems to be an excellent choice for combination with immune checkpoint antibody to achieve favorable clinical outcome due to the ability of cisplatin to activate T cells, whereas drugs such as gemcitabine that inhibit T cells functions should be considered with caution.
Compliance with ethics requirements
All experiments involving animals were conducted according to the ethical policies and procedures approved by the Animal Care and Use Committee of Sun Yat-Sen University Cancer Center (Guangzhou, China).
Analysis of clinical data was performed retrospectively using data from a separate study of lung cancer patients treated with toripalimab and various chemotherapeutic agents with proper informed consent, which was reviewed and approved by the Committee for Ethical Review of Research Involving Human Subjects of Hubei Cancer Hospital Affiliated to Tongji Medical College (Wuhan, China).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors are grateful to Dr. Nilabh Shastri (University of California Berkeley, USA) for the B3Z cells. The authors also thank Yunhua Guo and Jingyu Tian for their excellent assistance. We acknowledge the research funding from the National Key R&D Program of China (2020YFA0803302 and 2018YFC0910203).
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2021.12.005.
Contributor Information
Xiaobing Li, Email: lixiaobing0629@126.com.
Peng Huang, Email: huangpeng@sysucc.org.cn.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Papke B., Der C.J. Drugging RAS: Know the enemy. Science. 2017;355(6330):1158–1163. doi: 10.1126/science.aam7622. [DOI] [PubMed] [Google Scholar]
- 2.Bardeesy N., DePinho R.A. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2(12):897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7. [DOI] [PMC free article] [PubMed]
- 4.Herbst R.S., Heymach J.V., Lippman S.M. Lung cancer. N Engl J Med. 2008;359(13):1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glorieux C., Huang P. Regulation of CD137 expression through K-Ras signaling in pancreatic cancer cells. Cancer Commun (Lond) 2019;39(1):41. doi: 10.1186/s40880-019-0386-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen N., Fang W., Lin Z., Peng P., Wang J., Zhan J., et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol Immunother. 2017;66(9):1175–1187. doi: 10.1007/s00262-017-2005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coelho M.A., de Carné Trécesson S., Rana S., Zecchin D., Moore C., Molina-Arcas M., et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity. 2017;47(6):1083–1099.e6. doi: 10.1016/j.immuni.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glorieux C., Xia X., He Y.-Q., Hu Y., Cremer K., Robert A., et al. Regulation of PD-L1 expression in K-ras-driven cancers through ROS-mediated FGFR1 signaling. Redox Biol. 2021;38:101780. doi: 10.1016/j.redox.2020.101780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glorieux C., Xia X., Huang P. The Role of Oncogenes and Redox Signaling in the Regulation of PD-L1 in Cancer. Cancers (Basel) 2021;13(17):4426. doi: 10.3390/cancers13174426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong H., Zhu G., Tamada K., Chen L. B7–H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5(12):1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 11.Freeman G.J., Long A.J., Iwai Y., Bourque K., Chernova T., Nishimura H., et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keir M.E., Liang S.C., Guleria I., Latchman Y.E., Qipo A., Albacker L.A., et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203(4):883–895. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazanet M.M., Hughes C.C.W. B7–H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J Immunol. 2002;169(7):3581–3588. doi: 10.4049/jimmunol.169.7.3581. [DOI] [PubMed] [Google Scholar]
- 14.Nakazawa A., Dotan I., Brimnes J., Allez M., Shao L., Tsushima F., et al. The expression and function of costimulatory molecules B7H and B7–H1 on colonic epithelial cells. Gastroenterology. 2004;126(5):1347–1357. doi: 10.1053/j.gastro.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Dong H., Strome S.E., Salomao D.R., Tamura H., Hirano F., Flies D.B., et al. Tumor-associated B7–H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 16.Dong H., Zhu G., Tamada K., Flies D.B., van Deursen J.M.A., Chen L. B7–H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity. 2004;20(3):327–336. doi: 10.1016/s1074-7613(04)00050-0. [DOI] [PubMed] [Google Scholar]
- 17.David R. PD-L1 expression by circulating breast cancer cells. Lancet Oncol. 2015;16(7):e321. doi: 10.1016/S1470-2045(15)00074-1. [DOI] [PubMed] [Google Scholar]
- 18.Gao Q., Wang X.-Y., Qiu S.-J., Yamato I., Sho M., Nakajima Y., et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15(3):971–979. doi: 10.1158/1078-0432.CCR-08-1608. [DOI] [PubMed] [Google Scholar]
- 19.Kingwell K. Neuro-oncology: Glioblastoma prognosis linked to neuronal PD-L1 expression in tumour-adjacent tissue. Nat Rev Neurol. 2013;9(11):602–603. doi: 10.1038/nrneurol.2013.197. [DOI] [PubMed] [Google Scholar]
- 20.Borghaei H., Paz-Ares L., Horn L., Spigel D.R., Steins M., Ready N.E., et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(17):1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brahmer J.R., Tykodi S.S., Chow L.Q.M., Hwu W.-J., Topalian S.L., Hwu P., et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herbst R.S., Soria J.-C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salas-Benito D., Pérez-Gracia J.L., Ponz-Sarvisé M., Rodriguez-Ruiz M.E., Martínez-Forero I., Castañón E., et al. Paradigms on Immunotherapy Combinations with Chemotherapy. Cancer Discov. 2021;11(6):1353–1367. doi: 10.1158/2159-8290.CD-20-1312. [DOI] [PubMed] [Google Scholar]
- 24.Renouf D.J., Knox J.J., Kavan P., Jonker D., Welch S., Couture F., et al. LBA65 The Canadian Cancer Trials Group PA.7 trial: Results of a randomized phase II study of gemcitabine (GEM) and nab-paclitaxel (Nab-P) vs GEM, nab-P, durvalumab (D) and tremelimumab (T) as first line therapy in metastatic pancreatic ductal adenocarcinoma (mPDAC) Ann Oncol. 2020;31:S1195. doi: 10.1016/j.annonc.2020.08.2300. [DOI] [Google Scholar]
- 25.Bockorny B., Semenisty V., Macarulla T., Borazanci E., Wolpin B.M., Stemmer S.M., et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. 2020;26(6):878–885. doi: 10.1038/s41591-020-0880-x. [DOI] [PubMed] [Google Scholar]
- 26.Forde P.M., Chaft J.E., Smith K.N., Anagnostou V., Cottrell T.R., Hellmann M.D., et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N Engl J Med. 2018;378(21):1976–1986. doi: 10.1056/NEJMoa1716078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z., Wu X. Study and analysis of antitumor resistance mechanism of PD1/PD-L1 immune checkpoint blocker. Cancer Med. 2020;9(21):8086–8121. doi: 10.1002/cam4.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glorieux C., Cui L., Zeng P., Xia X., Huang P. Diverse effects of chemotherapeutic agents on immune cell function and implications in immunochemotherapy. Cancer Commun (Lond) 2021;41(5):432–435. doi: 10.1002/cac2.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shastri N., Gonzalez F. Endogenous generation and presentation of the ovalbumin peptide/Kb complex to T cells. J Immunol. 1993;150(7):2724–2736. [PubMed] [Google Scholar]
- 30.Wang Z., Chen J., Hu J., Zhang H., Xu F., He W., et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J Clin Invest. 2019;129(11):4850–4862. doi: 10.1172/JCI127471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jønsson K.L., Laustsen A., Krapp C., Skipper K.A., Thavachelvam K., Hotter D., et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun. 2017;8(1):14391. doi: 10.1038/ncomms14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nejati R., Goldstein J.B., Halperin D.M., Wang H., Hejazi N., Rashid A., et al. Prognostic significance of tumor-infiltrating lymphocytes in patients with pancreatic ductal adenocarcinoma treated with neoadjuvant chemotherapy. Pancreas. 2017;46(9):1180–1187. doi: 10.1097/MPA.0000000000000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dyck L., Wilk M.M., Raverdeau M., Misiak A., Boon L., Mills K.H.G. Anti-PD-1 inhibits Foxp3(+) Treg cell conversion and unleashes intratumoural effector T cells thereby enhancing the efficacy of a cancer vaccine in a mouse model. Cancer Immunol Immunother. 2016;65(12):1491–1498. doi: 10.1007/s00262-016-1906-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Germano G., Lamba S., Rospo G., Barault L., Magrì A., Maione F., et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature. 2017;552(7683):116–120. doi: 10.1038/nature24673. [DOI] [PubMed] [Google Scholar]
- 35.Groom J.R., Luster A.D. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89(2):207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun L., Wu J., Du F., Chen X., Chen Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J.M., Robert L., et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johansson H., Andersson R., Bauden M., Hammes S., Holdenrieder S., Ansari D. Immune checkpoint therapy for pancreatic cancer. World J Gastroenterol. 2016;22(43):9457–9476. doi: 10.3748/wjg.v22.i43.9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin F., Apetoh L., Ghiringhelli F. Role of myeloid-derived suppressor cells in tumor immunotherapy. Immunotherapy. 2012;4(1):43–57. doi: 10.2217/imt.11.154. [DOI] [PubMed] [Google Scholar]
- 40.Bruchard M., Mignot G., Derangère V., Chalmin F., Chevriaux A., Végran F., et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19(1):57–64. doi: 10.1038/nm.2999. [DOI] [PubMed] [Google Scholar]
- 41.Ghiringhelli F., Apetoh L. The interplay between the immune system and chemotherapy: emerging methods for optimizing therapy. Expert Rev Clin Immunol. 2014;10(1):19–30. doi: 10.1586/1744666X.2014.865520. [DOI] [PubMed] [Google Scholar]
- 42.Das S., Berlin J., Cardin D. Harnessing the Immune System in Pancreatic Cancer. Curr Treat Options Oncol. 2018;19(10):48. doi: 10.1007/s11864-018-0566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beatty G.L., Torigian D.A., Chiorean E.G., Saboury B., Brothers A., Alavi A., et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19(22):6286–6295. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rizvi N.A., Hellmann M.D., Brahmer J.R., Juergens R.A., Borghaei H., Gettinger S., et al. Nivolumab in Combination With Platinum-Based Doublet Chemotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2016;34(25):2969–2979. doi: 10.1200/JCO.2016.66.9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Y., Lin Z., Zhang X., Chen C., Zhao H., Hong S., et al. First-line treatment for patients with advanced non-small cell lung carcinoma and high PD-L1 expression: pembrolizumab or pembrolizumab plus chemotherapy. J Immunother Cancer. 2019;7(1):120. doi: 10.1186/s40425-019-0600-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fournel L., Wu Z., Stadler N., Damotte D., Lococo F., Boulle G., et al. Cisplatin increases PD-L1 expression and optimizes immune check-point blockade in non-small cell lung cancer. Cancer Lett. 2019;464:5–14. doi: 10.1016/j.canlet.2019.08.005. [DOI] [PubMed] [Google Scholar]
- 47.Grimaldi A., Cammarata I., Martire C., Focaccetti C., Piconese S., Buccilli M., et al. Combination of chemotherapy and PD-1 blockade induces T cell responses to tumor non-mutated neoantigens. Commun Biol. 2020;3(1):85. doi: 10.1038/s42003-020-0811-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran L., Allen C.T., Xiao R., Moore E., Davis R., Park S.-J., et al. Cisplatin Alters Antitumor Immunity and Synergizes with PD-1/PD-L1 Inhibition in Head and Neck Squamous Cell Carcinoma. Cancer Immunol Res. 2017;5(12):1141–1151. doi: 10.1158/2326-6066.CIR-17-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Voorwerk L., Slagter M., Horlings H.M., Sikorska K., van de Vijver K.K., de Maaker M., et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25(6):920–928. doi: 10.1038/s41591-019-0432-4. [DOI] [PubMed] [Google Scholar]
- 50.Michaud M., Sukkurwala A.Q., Di Sano F., Zitvogel L., Kepp O., Kroemer G. Synthetic induction of immunogenic cell death by genetic stimulation of endoplasmic reticulum stress. Oncoimmunology. 2014;3:e28276. doi: 10.4161/onci.28276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Biasi A.R., Villena-Vargas J., Adusumilli P.S. Cisplatin-induced antitumor immunomodulation: a review of preclinical and clinical evidence. Clin Cancer Res. 2014;20(21):5384–5391. doi: 10.1158/1078-0432.CCR-14-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Markasz L., Skribek H., Uhlin M., Otvos R., Flaberg E., Eksborg S., et al. Effect of frequently used chemotherapeutic drugs on cytotoxic activity of human cytotoxic T-lymphocytes. J Immunother. 2008;31(3):283–293. doi: 10.1097/CJI.0b013e3181628b76. [DOI] [PubMed] [Google Scholar]
- 53.Wang H., Hu S., Chen X., Shi H., Chen C., Sun L., et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci USA. 2017;114(7):1637–1642. doi: 10.1073/pnas.1621363114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tang H., Wang Y., Chlewicki L., Zhang Y., Guo J., Liang W., et al. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell. 2016;30(3):500. doi: 10.1016/j.ccell.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 55.Fang W., Yang Y., Ma Y., Hong S., Lin L., He X., et al. Camrelizumab (SHR-1210) alone or in combination with gemcitabine plus cisplatin for nasopharyngeal carcinoma: results from two single-arm, phase 1 trials. Lancet Oncol. 2018;19(10):1338–1350. doi: 10.1016/S1470-2045(18)30495-9. [DOI] [PubMed] [Google Scholar]
- 56.Mai H.-Q., Chen Q.-Y., Chen D., Hu C., Yang K., Wen J., et al. Toripalimab or placebo plus chemotherapy as first-line treatment in advanced nasopharyngeal carcinoma: a multicenter randomized phase 3 trial. Nat Med. 2021;27(9):1536–1543. doi: 10.1038/s41591-021-01444-0. [DOI] [PubMed] [Google Scholar]
- 57.Voena C., Chiarle R. Advances in cancer immunology and cancer immunotherapy. Discov Med. 2016;21(114):125–133. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The experimental data and materials, generated and analyzed during this study, are available from the corresponding authors on reasonable request.