

Abstract
Exosomes are cell-derived nanovesicles involved in regulating intercellular communications. In contrast to conventional nanomedicines, exosomes are characterized by unique advantages for therapeutic development. Despite their major successes in drug delivery, the full potential of exosomes for immunotherapy remains untapped. Herein we designed genetically engineered exosomes featured with surfaced-displayed antibody targeting groups and immunomodulatory proteins. Through genetic fusions with exosomal membrane proteins, Expi293F cell-derived exosomes were armed with monoclonal antibodies specific for human T-cell CD3 and epidermal growth factor receptor (EGFR) as well as immune checkpoint modulators, programmed death 1 (PD-1) and OX40 ligand (OX40L). The resulting genetically engineered multifunctional immune-modulating exosomes (GEMINI-Exos) can not only redirect and activate T cells toward killing EGFR-positive triple negative breast cancer (TNBC) cells but also elicit robust anti-cancer immunity, giving rise to highly potent inhibition against established TNBC tumors in mice. GEMINI-Exos represent candidate agents for immunotherapy and may offer a general strategy for generating exosome-based immunotherapeutics with desired functions and properties.
Keywords: immunotherapy, exosomes, protein engineering, synthetic biology, extracellular vesicles, triple negative breast cancer
Graphical abstract
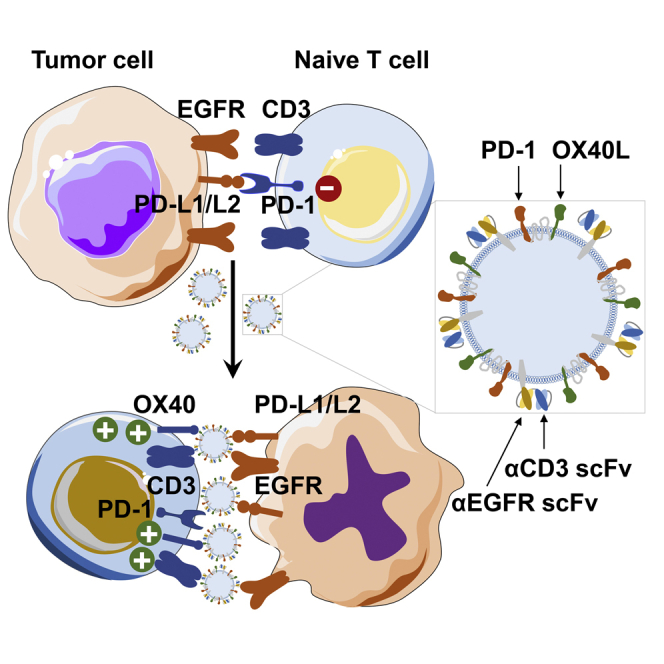
Cell-derived exosomes participate in intercellular communications. Through genetic engineering, exosomes could be armed with not only targeting antibodies but also immunomodulatory proteins. These genetically engineered multifunctional immune-modulating exosomes (GEMINI-Exos) are able to induce and sustain robust anti-tumor immunity, providing new strategies for developing therapeutic exosomes.
Introduction
Exosomes are naturally occurring membranous vesicles derived from a variety of types of cells. Featured with a diameter of 30 to 150 nm, exosomes carry a substantial amount of contents from parental cells.1,2 Through direct interactions with cell surface receptors and ligands and/or materials transfer via different modes, exosomes are capable of modulating physiology and pathophysiology of recipient cells.3,4 These nanoscale vesicles are known as key mediators for short- and long-range cell-to-cell communications.5,6
As a native form of nanocarriers, exosomes possess characteristic lipid bilayers and membrane proteins that constitute important functional components.7 By promoting directing membrane fusion to target cells and suppressing phagocytic clearance, proteins on exosome surfaces facilitate cytosolic delivery and increase half-lives in circulation, enhancing the pharmacological properties of exosomes.8,9 Together with excellent biocompatibility, these valuable features draw significant interests in developing exosomes as a class of nanomedicine.10, 11, 12, 13
To date, exosome-aided drug delivery shows broad utility in the treatment of various human diseases.13, 14, 15 But the potential of exosomes for immunotherapy has yet to be fully leveraged. Considering important roles and unique advantages of exosomes in intercellular communications, we envisioned that functionally reprogramming these cell-derived nanovesicles may result in an innovative form of agents with desired activities in eliciting disease-specific immune responses. In contrast to molecularly defined immunotherapeutics such as immune checkpoint inhibitors and bispecific antibodies, exosomes may enable multivalent expression of immunomodulatory proteins on spherical surface. This will increase their avidity and binding affinity to target receptors or ligands on immune and diseased cells and foster the formation of immunological synapses, resulting in enhanced activation of the immune system. Moreover, functional display of multiple immunomodulatory proteins on the same exosome vesicle, which target different signaling pathways, may promote synergistic actions, offering improved therapeutic efficacy in comparison with conventional combination therapies.
To explore this hypothesis, we managed to display not only distinct immune checkpoint modulators but also targeting moieties on exosome surfaces using genetic approaches. The resulting exosomes from Expi293F cells, named genetically engineered multifunctional immune-modulating exosomes (GEMINI-Exos) (Figure 1), are characterized by surface-displayed programmed death 1 (PD-1) and OX40 ligand (OX40L) as well as monoclonal antibodies specific for T-cell CD3 and epidermal growth factor receptor (EGFR), a receptor tyrosine kinase frequently overexpressed in many human cancers.16 The generated αCD3-αEGFR-PD-1-OX40L GEMINI-Exos display strong binding affinity to human CD3, EGFR, PD-1 ligands, and OX40. Preclinical studies using cellular and animal models of triple negative breast cancer (TNBC) indicated αCD3-αEGFR-PD-1-OX40L GEMINI-Exos can induce potent cellular immunity against EGFR-positive TNBC tumors by elevating infiltration of CD8+ T cells and alleviating immunosuppression of regulatory T cells (Tregs). This work demonstrates preclinical feasibility of GEMINI-Exos-based cancer immunotherapy and provides a genetic engineering-based strategy for developing immunotherapeutic exosomes.
Figure 1.

Schematic of αCD3-αEGFR-PD-1-OX40L GEMINI-Exos for targeted cancer immunotherapy
HA, hemagglutinin; PD-1, programmed death 1; OX40L, OX40 ligand; αCD3, anti-CD3; αEGFR, anti-EGFR; scFv, single-chain variable fragment; PDGFR TMD, transmembrane domain of human platelet-derived growth factor receptor; PD-L1/L2, programmed death-ligand 1/ligand 2.
Results
To determine whether endogenous exosomes could be reprogrammed as immunotherapeutics, we sought to genetically engineer exosomes with both targeting moieties and immunomodulatory agents and examine their activities in mounting immune responses using TNBC cellular and animal models in consideration of unmet medical needs for this disease. To this end, single-chain variable fragments (scFvs) targeting human T-cell CD3 and TNBC-associated EGFR were designed as targeting domains for GEMINI-Exos. Two immune checkpoint modulators, PD-1 and OX40L, were selected for display on GEMINI-Exos to augment cellular immunity by competing for PD-1 ligands’ binding and activating the OX40 signaling pathway, respectively. PD-1 and OX40 are expressed on activated T cells but play opposite roles in regulating T-cell activation. Engagement of PD-1 with its ligands, PD-L1 and PD-L2, produces inhibitory signals,17 whereas ligation of OX40 by its ligand OX40L generates stimulatory activities.18,19
To balance stability, biological activities, and expression yields of the four simultaneously displayed proteins on exosome surfaces, we chose to transfect and co-express two fusion proteins constructs in exosome-producing cells (Figure 1). By following our recently established strategy,20,21 αCD3 scFv and αEGFR scFv could be functionally anchored on exosomal surfaces through tandem fusion with the transmembrane domain (TMD) of human platelet-derived growth factor receptor (PDGFR). A hemagglutinin (HA) epitope tag was placed at the N-terminus of αCD3-αEGFR-PDGFR TMD fusion.
PD-1 and OX40L are type I and II membrane proteins, respectively. To display both on the exosome surface in a single-chain fusion format with correct orientations, we attempted to genetically link full-length PD-1 and OX40L to the N- and C-terminus of CD9, respectively (Figure 1). Considering the high abundance and intracellular termini for CD9 in exosome membranes, CD9-based fusions may facilitate display of transmembrane proteins on exosome surfaces. The designed PD-1-CD9-OX40L fusion contains an N-terminal HA tag, a C-terminal 6×His tag, and flexible (GGGGS)2 linkers before and after the fused CD9 domain.
The expression and function of the PD-1-CD9-OX40L fusion protein in exosomes were examined before generating the αCD3-αEGFR-PD-1-OX40L GEMINI-Exos. PD-1-OX40L-Exos were produced through transient transfection of Expi293F cells with the designed fusion construct, followed by purification from harvested media via differential centrifugations and ultracentrifugation.20, 21, 22, 23, 24, 25, 26, 27 Immunoblot results confirmed expression of the PD-1-CD9-OX40L fusion protein in exosomes (Figure 2A). Nanoparticle tracking analysis (NTA) revealed that the mean and mode size of PD-1-OX40L-Exos are around 115 nm and 105 nm, respectively, similar to that of native exosomes (Figure 2B). Sandwich ELISA indicated that unlike native exosomes, the PD-1-OX40L-Exos can simultaneously bind to both the human PD-L1 and OX40 (Figure 2C). The bindings of PD-1-OX40L-Exos to PD-L1/PD-L2 and OX40 were further analyzed by flow cytometry (Figure 2D) using BT-20 cells with constitutive and upregulated expression of PD-L1 and PD-L2 upon stimulation (Figures 2D and S1), activated human T cells (Figures 2E and S2), and negative MDA-MB-468 cells (Figures 2F and S1). PD-1-OX40L-Exos display tight binding to both the PD-L1+/PD-L2+ BT-20 cells and OX40+ T cells and no binding to PD-L1−/PD-L2−/OX40− MDA-MB-468 cells. These results support functional displays of PD-1 and OX40L on exosome surfaces.
Figure 2.

Generation and characterization of PD-1-OX40L-Exos
(A) Immunoblot analysis of purified exosomes. (B) Size distribution of native exosomes and PD-1-OX40L-Exos. (C) Sandwich ELISA analysis of the binding of PD-1-OX40L-Exos to human PD-L1 and OX40. Recombinant human PD-L1 and biotinylated OX40 were used as capture and detection reagents, respectively. Data are shown as mean ± SD of duplicates. (D–F) Flow cytometry of the binding of PD-1-OX40L-Exos to IFN-γ stimulated BT-20 cells (D), activated human T cells (E), and MDA-MB-468 cells (F). Anti-PD-L1, anti-PD-L2, and anti-OX40 antibodies were used as positive controls. Arrows show signal levels of indicated target proteins being detected after the incubation with antibodies, native Exos, or PD-1-OX40L-Exos. (G and H) Dose-dependent activation of human T cells by PD-1-OX40L-Exos. Human PBMCs were incubated with pre-coated anti-human CD3 monoclonal antibody in the presence of various concentrations of PD-1-OX40L-Exos or native exosomes for 48 h. The levels of secreted IFN-γ (G) and IL-2 (H) were measured by ELISA. Data are shown as mean ± SD of triplicates. (I) PD-1-OX40L-Exos restore T-cell activation from PD-L1-mediated inhibition. Human PBMCs were incubated with pre-coated anti-human CD3 monoclonal antibody without or with pre-coated human PD-L1 in the absence or presence of 10 μg mL−1 PD-1-OX40L-Exos or native exosomes for 48 h. The levels of secreted IL-2 were measured by ELISA. Data are shown as mean ± SD of triplicates. ∗p < 0.05 and ∗∗∗∗p < 0.0001 (two-tailed unpaired t test).
The stimulatory activity of PD-1-OX40L-Exos on activation of primary T cells was then evaluated. Incubation of PD-1-OX40L-Exos with human peripheral blood mononuclear cells (PBMCs) activated primarily by an anti-CD3 antibody result in dose-dependent increases of secreted interferon (IFN)-γ and interleukin (IL)-2 cytokines (Figures 2G and 2H). In contrast, native exosomes have little effects on stimulating T-cell activation. On the basis of secreted IL-2 levels, additions of PD-1-OX40L-Exos restore T-cell activation that is inhibited by immobilized PD-L1 introduced to anti-CD3 antibody-activated PBMCs (Figure 2I). In addition, αCD3-αEGFR-Exos were prepared as previously described20 and used to recruit and activate human T cells against EGFR+ BT-20 TNBC cells in the absence or presence of PD-1-OX40L-Exos. Significantly higher levels of IL-2 release were observed for PBMC:BT-20 mixtures with PD-1-OX40L-Exos (Figure S3). These results demonstrate in vitro immune stimulating activities for PD-1-OX40L-Exos.
Next, αCD3-αEGFR-PD-1-OX40L GEMINI-Exos were generated by co-transfecting exosome-producing cells with αCD3-αEGFR-PDGFR TMD and PD-1-CD9-OX40L fusion expression constructs. Immunoblot analysis indicated that both fusion proteins were successfully expressed in exosomes (Figure 3A) and the three genetically modified exosomes showed comparable yields (Figure S4). The size distribution for GEMINI-Exos is comparable to those of native exosomes and PD-1-OX40L-Exos, according to NTA analysis (Figure 3B). ELISA results showed that the αCD3-αEGFR-PD-1-OX40L GEMINI-Exos retain strong bindings to PD-L1, PD-L2, and OX40 targets, despite slightly decreased binding affinity in comparison with PD-1-OX40L-Exos (Figure 3C). Flow cytometry revealed that the GEMINI-Exos exhibit the tightest binding to BT-20 cells (EGFR+ PD-L1+ PD-L2+) compared with αCD3-αEGFR-Exos and PD-1-OX40L-Exos (Figures 3D, S1, and S5), possibly due to dual targeting capability to EGFR and PD-L1/L2. The binding affinity of GEMINI-Exos to Jurkat cells (CD3+ OX40−) is comparable to that of αCD3-αEGFR-Exos (Figure 3D). Moreover, αCD3-αEGFR-PD-1-OX40L GEMINI-Exos show strong binding to MDA-MB-468 cells (PD-L1− PD-L2− OX40− CD3− EGFR+) as detected by an anti-6×His antibody, indicating co-expression of the HA-αCD3-αEGFR-PDGFR TMD and HA-PD-1-CD9-OX40L-6×His fusion proteins on the surface of the same exosome (Figure S6). These results demonstrate that both targeting antibodies and immunoregulatory proteins are functionally displayed on exosomal surfaces and may allow exosomes to engage TNBC cells by T cells for eliciting cancer-specific cellular immunity. Notably, incubation of human PBMC:BT-20 cell mixtures with the αCD3-αEGFR-PD-1-OX40L GEMINI-Exos results in significantly higher and more sustainable levels of IL-2 release in contrast to ones with αCD3-αEGFR-Exos or a mixture (1:1) of αCD3-αEGFR-Exos and PD-1-OX40L-Exos (Figure 3E), suggesting the GEMINI-Exos may induce potent anti-cancer immune responses by modulating PD-1 and OX40-associated immune checkpoint pathways.
Figure 3.

Generation and characterization of αCD3-αEGFR-PD-1-OX40L GEMINI-Exos
(A) Immunoblot analysis of purified exosomes. (B) Size distribution of αCD3-αEGFR-PD-1-OX40L GEMINI-Exos. (C) ELISA analysis of the binding of αCD3-αEGFR-PD-1-OX40L GEMINI-Exos to human PD-L1, PD-L2, and OX40. PD-1-OX40L-Exos, αCD3-αEGFR-PD-1-OX40L GEMINI-Exos, and native exosomes at various concentrations were coated on 96-well ELISA plates overnight, followed by incubation with recombinant PD-L1-Fc, PD-L2-Fc, or OX40-Fc and detection with an anti-human IgG-HRP. Data are shown as mean ± SD of duplicates. (D) Flow cytometry of the binding of αCD3-αEGFR-PD-1-OX40L GEMINI-Exos to BT-20 cells (EGFR+ PD-L1+) and Jurkat cells (CD3+). Arrows show signal levels of indicated target proteins being detected after the incubation with various types of exosomes. (E) Time-dependent activation of human T cells by αCD3-αEGFR-PD-1-OX40L GEMINI-Exos. Human PBMCs were incubated with BT-20 cells at a ratio of 2:1 for 24 to 96 h in the presence of native exosomes, PD-1-OX40L-Exos, αCD3-αEGFR-Exos, a mixture (1:1) of PD-1-OX40L- and αCD3-αEGFR-Exos, or αCD3-αEGFR-PD-1-OX40L GEMINI-Exos. The levels of secreted IL-2 were measured by ELISA. Data are shown as mean ± SD of triplicates. ns = not significant, ∗p < 0.05, and ∗∗∗∗p < 0.0001 (ordinary one-way ANOVA test).
In vivo efficacy of the αCD3-αEGFR-PD-1-OX40L GEMINI-Exos was then evaluated using BT-20 xenograft mouse models with engrafted human PBMCs. As shown in Figures 4A and 4I, mice treated with PD-1-OX40L-Exos, αCD3-αEGFR-Exos, a mixture (1:1) of PD-1-OX40L-Exos and αCD3-αEGFR-Exos, or GEMINI-Exos display significant growth inhibition against established tumors in comparison to mice treated with PBS or native exosomes. Combination of PD-1-OX40L-Exos and αCD3-αEGFR-Exos shows enhanced anti-tumor efficacy compared with PD-1-OX40L-Exos. Importantly, mice treated with αCD3-αEGFR-PD-1-OX40L GEMINI-Exos exhibit the most pronounced tumor growth inhibition. The weights and photographs of collected tumors at the endpoint are consistent with these results (Figures 4B and S7). No overt toxicity or loss in body weight was observed for mice in all groups (Figure 4C). In addition, no significant changes were found for alanine aminotransferase (ALT) activities (a liver injury marker) and creatinine concentrations (a kidney injury marker) in plasma across all groups at the end of studies (Figures 4D and 4E). These results indicate the excellent anti-tumor activity and safety for αCD3-αEGFR-PD-1-OX40L GEMINI-Exos.
Figure 4.

In vivo evaluation of αCD3-αEGFR-PD-1-OX40L GEMINI-Exos
(A) Anti-tumor activity of GEMINI-Exos. BT-20 cells were subcutaneously implanted into the flank of female NSG mice (n = 5). In vitro expanded human PBMCs from the same healthy donor were intraperitoneally injected into mice on days 12 and 18 post tumor implantation. One day post the first PBMC administration, mice were treated with PBS or different types of exosomes (10 mg/kg for monotherapy and 20 mg/kg for combination therapy) every other day for a total of six times via intravenous injections. Data are shown as mean ± SD (n = 5). ns = not significant, ∗p < 0.05, and ∗∗p < 0.01(one-way repeated measures ANOVA test with the Geisser-Greenhouse correction). (B) Tumor weights at the end of study. (C) Body weights of mice during the study. (D) ALT activities in plasma at the end of study. (E) Creatinine concentrations in plasma at the end of study. (F) Percentage CD8+ T cells in CD45+ cells in tumors. (G) Percentages of CD4+ CD25+ FoxP3+ Tregs in CD45+ cells in tumors. (H) CD8+ T cell/Treg ratios in tumors. At the end of the study, tumors were harvested and disaggregated into single-cell suspensions. After immunostaining, cells were analyzed by flow cytometry for the expression of CD45, CD4, CD8, CD25, and FoxP3. (I) Tumor growth curves for individual mice during the study. Data in (B, D–H) are shown as mean ± SD (n = 5). ns = not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001 (ordinary one-way ANOVA test).
Tumor-infiltrating lymphocytes were harvested and analyzed at the endpoint (Figures 4F–4H and S8–S11). Compared with PBS- or native exosomes-treated groups, mice administered with PD-1-OX40L-Exos, αCD3-αEGFR-Exos, the mixture (1:1) of PD-1-OX40L-Exos and αCD3-αEGFR-Exos, or GEMINI-Exos show significantly increased intratumoral CD8+ T cells (Figure 4F). Tregs reduction in tumors were also seen for mice treated with exosomes expressing the PD-1-CD9-OX40L fusion (Figures 4G and S10B), supporting immunostimulatory roles for exosomal surface-displayed PD-1 and OX40L in the tumor microenvironment. Consistent with anti-tumor efficacy results, GEMINI-Exos-treated mice displayed the most significant changes in tumor-infiltrating CD8+ T cells and CD8+ T cell/Tregs ratios among all the treatment groups (Figures 4F–4H and S10B). Mice treated with GEMINI-Exos showed slight but not significant increases of CD4+ T cells in tumors (Figure S10A). Moreover, immunohistofluorescence imaging showed marked infiltration of T cells for tumors implanted in mice receiving αCD3-αEGFR-Exos, the mixture (1:1) of PD-1-OX40L-Exos and αCD3-αEGFR-Exos, or GEMINI-Exos (Figure S11). In addition, moderate increases of CD8+ T cells were found in the spleen of mice receiving GEMINI-Exos and no significant differences for T cell subsets in blood were seen across all groups (Figure S12). These results suggest that the αCD3-αEGFR-PD-1-OX40L GEMINI-Exos induce potent anti-tumor immune responses through promoting CD8+ T cell infiltration and depleting immunosuppression of Tregs.
Discussion
Cell-derived exosomes have been widely used for the delivery of various types of cargos including chemotherapeutics,28 interfering RNAs,10 peptides,12 and proteins.29 Meanwhile, genetically modified exosomes have been emerging as an increasingly important class of therapeutic modality, such as SIRPα-exosomes to block CD47 and increase cancer cell phagocytosis,30 exoIL-12 to stimulate local and systemic anti-tumor activity,31 and Exo-PH20 to penetrate deeply into tumor foci via hyaluronan degradation.11 In this study, native exosomes were genetically modified to express four distinct proteins on surfaces. By simultaneously targeting tumor-associated EGFR and immunomodulatory molecules, the rationally designed αCD3-αEGFR-PD-1-OX40L GEMINI-Exos exhibit excellent activity in directing, activating, and modulating T cell-mediated immunity against EGFR-positive TNBC tumors. To the best of our knowledge, this work represents the first report of successful generation of multifunctional exosomes via genetic engineering approaches for targeted cancer immunotherapy.
In comparison with molecular immunotherapeutics such as bispecific antibodies and immune checkpoint inhibitors, GEMINI-Exos with integrated immunoregulatory proteins are likely to augment therapeutic efficacy by engaging and modulating multiple immune checkpoint pathways. Despite improved efficacy for combination therapies that can target different immunomodulators, physically restricting these therapeutic agents on the same vesicle may facilitate their synergistic actions on individual target cells, resulting in increased potency. The GEMINI-Exos feature full-length transmembrane proteins displayed via CD9-fusion. Unlike physical and chemical methods, this genetic approach for incorporating functional proteins into exosome membranes may enable them to retain their native folding, generating engineered exosomes with desired functions and properties. And these intact membrane proteins on GEMINI-Exos may possess higher stability and activities than those on synthetic nanoparticles. Furthermore, the functions of GEMINI-Exos can be further expanded through packing with small-molecule and nucleic acid agents to improve therapeutic efficacy by leveraging their potential for intracellular drug delivery. In addition, GEMINI-Exos-based therapeutics may show higher biocompatibility than synthetic and viral nanomedicines.
To more effectively fight tumors, immunotherapeutic candidates are required to not only recruit immune effector cells but also sustain anti-tumor immunity in response to dynamic immunosuppressive tumor microenvironments. The GEMINI-Exos were designed to meet these requirements. Surfaced-displayed αCD3 and αEGFR antibodies can redirect cytotoxic T cells toward attacking EGFR-positive TNBC tumors. PD-1 expressed on GEMINI-Exos is expected to block immune checkpoint inhibitory pathway activated by upregulated PD-L1/L2 on tumor surfaces. Multivalent OX40L on GEMINI-Exos is anticipated to engage with OX40 expressed on activated T cells to turn on immune checkpoint stimulatory signals. Furthermore, co-expressed PD-1-OX40L fusion and αCD3-αEGFR antibodies on the surface of the same GEMINI-Exos may increase targeting capabilities toward EGFR-, PD-L1-, PD-L2-positive tumor cells and CD3- and OX40-positive T cells as well as maximize cellular immunity against tumors. Collectively, these GEMINI-Exos-enabled molecular interactions establish robust and sustainable immune responses specific for TNBC tumors.
Exosomes are defined as extracellular vesicles (EVs) that originate as intraluminal vesicles within multivesicular bodies (MVBs) and are secreted upon fusion of the MVBs with plasma membranes.2,32 As the most commonly used method for exosome isolation currently,33 differential ultracentrifugation was employed in this study. The purified GEMINI-Exos are likely to carry other types of EVs with similar morphology, size, and protein expression.
Depending on the cells of origin, exosomes may possess immunomodulatory potentials. Expi293F cells, a suspension-adapted HEK293 cell line, were used here to produce GEMINI-Exos. Exosomes from HEK293 cells were shown to have minimal toxicity and immunogenicity,34 representing an excellent source of exosomes for the additions of new functions. Moreover, the use of Expi293F cells may facilitate the bioreactor-based, large-scale production of clinical grade exosomes.
In comparison with PDGFR TMD-based fusions, CD9 tetraspanin allows for displaying both type I and II transmembrane proteins on the exosome surface in native orientations, expanding choices of functional proteins for exosome expression. But the extracellular loops of CD9 could impact binding affinity and specificity of the fused protein. Further engineering and optimization may be required for improving biological activities of proteins displayed by CD9. Moreover, the generated GEMINI-Exos can be loaded with therapeutic agents targeting other immune checkpoint pathways for augmented efficacy. Further in vivo studies with different animal models are also needed to evaluate biodistribution, therapeutic efficacy, systemic toxicity, and animal survival for PD-1-OX40L-Exos, αCD3-αEGFR-Exos, the combination of PD-1-OX40L-Exos and αCD3-αEGFR-Exos, and GEMINI-Exos. Improvement of pharmacokinetics and exploration of different administration methods could also be carried out to optimize pharmacological activities of GEMINI-Exos for clinical translation. Notably, development of stable cell lines expressing optimized fusion constructs can further facilitate studies of biological and pharmacological activities of GEMINI-Exos and streamline production of pharmaceutical-grade exosomes. In addition, GEMINI-Exos-based immunotherapeutics for other human cancers and diseases can be developed by extending to different disease-associated antigens and immunoregulatory molecules.
In summary, we designed and generated αCD3-αEGFR-PD-1-OX40L GEMINI-Exos by genetically displaying both the monoclonal antibodies and immunomodulatory proteins on exosome surfaces. The generated GEMINI-Exos can not only recruit and activate human T cells against EGFR-positive tumors but also induce robust cancer-specific immune responses, leading to remarkable in vivo anti-tumor efficacy. This work demonstrates the potential for GEMINI-Exos in cancer immunotherapy and may provide a general approach for the development of immunotherapeutic exosomes with desired pharmacological activities.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM), Roswell Park Memorial Institute (RPMI) 1640 medium, and Dulbecco’s phosphate-buffered saline (DPBS) were purchased from Corning Inc. (Corning, NY). BalanCD HEK293 medium and L-glutamine (200 mM) were purchased from FUJIFILM Irvine Scientific. Fetal bovine serum (FBS), Opti-modified Eagle’s medium (Opti-MEM), QuantaBlu fluorogenic peroxidase substrate, and Coomassie Plus (Bradford) assay kit were purchased from Thermo Fisher Scientific (Waltham, MA). Collagenase (Type II) was purchased from Worthington Biochemical Corporation. DNase I, Triton X-100, sodium pyruvate, alanine, sodium 2-oxoglutarate monobasic, 2, 4-dinitrophenylhydrazine (2,4-DNPH), picric acid solution (1.3% in H2O), trichloroacetic acid (TCA), and creatinine were purchased from Sigma-Aldrich (St. Louis, MO).
Cell lines
Human PBMCs were purchased from HemaCare (Van Nuys, CA). Human embryonic kidney 293 cells (HEK293) were purchased from the American Type Culture Collection (ATCC) (Manassas, VA) and maintained in DMEM medium supplemented with 10% FBS at 37°C in 5% CO2. Breast cancer cell lines MDA-MB-468 and MDA-MB-231 were obtained from ATCC and maintained in RPMI 1640 medium supplemented with 10% FBS at 37°C in 5% CO2. BT-20 cell line was purchased from ATCC and cultured in MEM with 10% FBS at 37°C in 5% CO2. Expi293F cells were purchased from Thermo Fisher Scientific, and maintained in BalanCD HEK293 medium with 4 mM L-glutamine with shaking at a speed of 125 rpm min−1 at 37°C in 8% CO2. All the cell lines were tested for mycoplasma every half-year.
Molecular cloning and expression of engineered exosomes in mammalian cells
All DNA fragments and PCR primers used in this study are listed in Table S1.
PD-1 fragment was amplified from human PD-1 cDNA purchased from Horizon Discovery Ltd. (clone ID: 6,147,966) with a hemagglutinin (HA)-tag fused at N-terminus. Synthetic genes encoding CD9-OX40L-6×His was purchased from Integrated DNA Technologies, Inc. (Skokie, IL) with a (GGGGS)2 linker and a SalI restriction enzyme site inserted between CD9 and OX40L. To generate HA-PD-1-CD9-OX40L-6×His fusion gene fragment, overlap extension PCR was performed and a GGGGS linker and a NheI restriction enzyme site were inserted between PD-1 and CD9 fragments. The amplified HA-PD-1-CD9-OX40L-6×His fragment was ligated in-frame using T4 DNA ligase (New England Biolabs, Ipswich, MA) between the EcoRI and NotI restriction enzyme sites in a modified pDisplay vector in which the N-terminal signal peptide and the TMD of human PDGFR were deleted. The generated expression vector pDisplay-PD-1-CD9-OX40L was confirmed by DNA sequencing provided by GENEWIZ (South Plainfield, NJ). The construct pDisplay-αCD3-αEGFR-PDGFR TMD was generated from our previous work.20,21
Transfection-level plasmids for the sequence-verified expression constructs were purified using ZymoPURE II plasmid kits (ZYMO Research, Irvine, CA) and transiently transfected into Expi293F cells using PEI MAX 40K (Polysciences, PA) by following the manufacturer’s instructions. Cell culture supernatants were collected on day 3 and day 6 post transfection through centrifugation and stored at −80°C.
Exosome purification
Exosomes were isolated from cell culture supernatants of Expi293F cells through differential centrifugation and ultracentrifugation as previously described with modifications.20,21,27,35 Briefly, cell cultures were first centrifuged at 100 × g for 10 min to remove Expi293F cells and then centrifuged at 4,000 × g for 30 min to remove dead cells and cell debris using a Heraeus Megafuge 40R refrigerated centrifuge with a TX-750 swinging bucket rotor (Thermo Fisher Scientific). The collected supernatant was then centrifuged at 14,000 × g for 40 min by J2-21 floor model centrifuge with a JA-17 fixed-angle aluminum rotor (Beckman Coulter, Indianapolis, IN) to remove large vesicles. Clarified supernatants were then centrifuged in a Type 70 Ti rotor by Optima L-80 XP ultracentrifuge (Beckman Instruments) at 60,000 rpm (371,000 × g) for 1.5 h to pellet exosomes. All the centrifuge processes were performed at 4°C. The resulting exosome pellets were washed twice with PBS, resuspended in PBS, and followed by filtration using 0.2-μm syringe filters. Protein concentrations of the purified exosomes were determined by Bradford assays by following manufacturer’s instructions.
Nanoparticle tracking analysis
The size distribution and concentration of the purified exosomes were determined through NTA using a Nanosight LM10 (Malvern Instruments, UK) by following the manufacturer’s instructions. Ten replicates of analysis with 60 s for each were performed.
Immunoblot analysis
Western blots were performed as previously described.20,21 Briefly, 4 μg of purified exosomes (whole protein amount) was lysed in NuPAGE LDS sample buffer (Thermo Fisher Scientific) with or without 10 mM dithiothreitol and boiled at 95°C for 5 min. The lysates were then resolved by 4% to 20% ExpressPlus-PAGE gels (GeneScript, Piscataway, NJ), transferred to Immun-Blot PVDF membranes (Bio-Rad Laboratories, Inc, Hercules, CA), blocked with 5% BSA (Thermo Fisher Scientific) in PBS with 0.5% Tween 20 (PBST) and probed with appropriate primary antibodies (anti-HA [2–2.2.14] from Thermo Fisher Scientific, anti-CD9 [D8O1A] from Cell Signaling Technology, anti-CD81 [5A6] and anti-CD63 [H5C6] from BioLegend) and secondary antibodies (anti-mouse immunoglobulin [Ig]G-horseradish peroxidase [HRP] [62-6520] and anti-rabbit IgG-HRP [65-6120] from Thermo Fisher Scientific). HA-tagged fusion proteins and CD9 were resolved under fully denaturing and reducing conditions, while CD81 and CD63 were resolved under non-reducing conditions. The immunoblots were developed by additions of SuperSignal West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific) and imaged with a ChemiDoc Touch Imaging System (Bio-Rad Laboratories, Inc).
ELISA analysis of binding of engineered exosomes to ligands and receptors
The bindings of PD-1-OX40L-Exos and αCD3-αEGFR-PD-1-OX40L GEMINI-Exos to PD-L1, PD-L2, and OX40 were determined by immunocapture-based ELISA. High-binding 96-well plates (Greiner Bio-One, Monroe, NC) were coated with various concentrations of exosomes overnight at room temperature. Following extensive washing with PBST, wells were blocked with PBS containing 1% BSA for 2 h at room temperature, followed by extensive washing with PBST. Corresponding ligands or receptors (0.4 μg mL−1; PD-L1-hFc and PD-L2-hFc from PeproTech, Inc. and OX40-hFc from BioLegend) were added and incubated for 2 h at room temperature, followed by extensive washing with PBST. Goat anti-human IgG-HRP was subsequently added for 1-h incubation at room temperature, followed by extensive washing. QuantaBlu fluorogenic peroxidase substrate (Thermo Fisher Scientific) was then added. Fluorescence intensities (Ex: 325 nm; Em: 420 nm) were measured using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader (BioTek, VT).
To detect the binding of PD-1-OX40L-Exos to both PD-L1 and OX40, ELISA was performed as described above with PD-L1-hFc (0.4 μg mL−1) as the capture protein and biotinylated OX40-hFc as the detection reagent.
Flow cytometric analysis of binding of engineered exosomes to target-expressing cells
BT-20 cells were treated with 100 U/mL IFN-γ (BioLegend) for 2 days to induce expression of PD-L1. Purified human PBMCs from HemaCare were treated with immobilized anti-CD3 antibody (5 μg mL−1; clone: OKT3 from BioLegend) and soluble anti-CD28 antibody (2 μg mL−1; clone: CD28.2, from BioLegend) for 2 days to induce expression of OX40. PD-L1/PD-L2/EGFR-expressing BT-20 cells, OX40-expressing PBMCs, and CD3-expressing Jurkat cells were then used to verify the binding of PD-1-OX40L-Exos or αCD3-αEGFR-PD-1-OX40L GEMINI-Exos to their targets. PD-L1/PD-L2/OX40-negtive MDA-MB-468 cells were used as negative controls for assessing the binding of PD-1-OX40L-Exos to their targets. Briefly, cells (300,000 cells per tube) were stained with 100 μg mL−1 of exosomes for 30 min at 4°C. Cells were washed three times with PBS containing 2% FBS and stained with an anti-HA antibody (2–2.2.14, from Thermo Fisher Scientific) for 30 min at 4°C. After washing three times, cells were stained with an Alexa Fluor 488-labeled goat anti-mouse IgG H&L antibody (Catalog #A28175 from Thermo Fisher Scientific) for 30 min at 4°C. Positive controls were performed by staining the cells with respective antibodies (PE anti-PD-L1, clone: 29E.2A3; APC anti-PD-L2, clone: MIH18; PE/Cyanine7 anti-OX40, clone: Ber-ACT35; BioLegend) for 30 min at 4°C. Thereafter, cells were washed and resuspended in PBS containing 2% FBS, followed by analysis using a BD Fortessa X20 flow cytometer. Data were processed by FlowJo_V10 software (Tree Star Inc., Ashland, OR).
MDA-MB-468 cells (PD-L1− PD-L2− OX40− CD3− EGFR+) were used to examine co-expression of the HA-αCD3-αEGFR-PDGFR TMD and HA-PD-1-CD9-OX40L-6×His fusion proteins on the surface of the same exosome by flow cytometry. Briefly, cells (300,000 cells per tube) were stained with 100 μg mL−1 of Native Exos, PD-1-OX40L-Exos, αCD3-αEGFR-Exos, or αCD3-αEGFR-PD-1-OX40L GEMINI-Exos for 30 min at 4°C. Cells were washed three times with PBS containing 2% FBS and stained with an anti-HA antibody (2–2.2.14, from Thermo Fisher Scientific) or an anti-6×His antibody (HIS.H8, from Thermo Fisher Scientific) for 30 min at 4°C. After washing three times, cells were stained with an Alexa Fluor 488-labeled goat anti-mouse IgG H&L antibody (Catalog #A28175 from Thermo Fisher Scientific) for 30 min at 4°C. Thereafter, cells were washed and resuspended in PBS containing 2% FBS, followed by analysis using a BD Fortessa X20 flow cytometer. Data were processed by FlowJo_V10 software (Tree Star Inc.).
Flow cytometry analysis of expression of EGFR, PD-L1, and PD-L2
HEK293 cells or three TNBC cells (MDA-MB-231, BT-20, MDA-MB-468) were treated with 100 U/mL IFN-γ or human PBMCs (PBMC:TNBC/HEK293 = 2:1) in the absence or presence of 20 ng/mL αCD3-αEGFR-Exos for 48 h at 37°C. Treated and non-treated cells were stained with PE anti-PD-L1 (clone: 29E.2A3, BioLegend), APC anti-PD-L2 (clone: MIH18, BioLegend), or anti-EGFR (clone: AY13, BioLegend), followed by staining with the Alexa Fluor 488-labeled goat anti-mouse IgG H&L antibody (Catalog # A28175 from Thermo Fisher Scientific). Thereafter, cells were washed and resuspended in PBS containing 2% FBS, followed by analysis with a BD Fortessa X20 flow cytometer (BD Biosciences, CA). Data were processed by FlowJo_V10 software (Tree Star Inc.).
T cell co-stimulation assays
Anti-human CD3 monoclonal Ab (clone: OKT3, BioLegend) was coated on surface-treated 96-well plates in 50 μL volume at a concentration of 10 μg mL−1 at 37°C for 3 h, followed by washing three times with DPBS and additions of human PBMCs (1 ×105 per well) in complete RPMI 1640 medium in the presence of various concentrations of PD-1-OX40L-Exos or native exosomes. After 48-h incubation, cell culture supernatants were collected and assayed for the levels of IFN-γ and IL-2 by ELISA kits (R&D Systems, Minneapolis, MN). Results are expressed as a mean ± SD from one of at least three separate experiments.
To examine inhibitory effects of PD-L1 on PD-1-OX40L-Exos-induced T cell co-stimulation, the anti-CD3 monoclonal antibody (clone: OKT3, 10 μg mL−1) together with PD-L1-6×His (10 μg mL−1; GenScript, NJ) were coated on surface-treated 96-well plates at 37°C for 3 h, followed by washing three times with DPBS and additions of human PBMCs (1 ×105 per well) in the presence of 10 μg mL−1 PD-1-OX40L-Exos or native exosomes for 48 h at 37°C. Cell culture supernatants were then collected and assayed for the levels of IL-2 by ELISA. Results are expressed as a mean ± SD from one of at least three separate experiments.
To analyze stimulatory effects of PD-1-OX40L-Exos on αCD3-αEGFR-Exos-mediated T cell activation, BT-20 cells and PBMCs were mixed at a ratio of 1:2 and incubated with αCD3-αEGFR-Exos (20 ng mL−1) in the absence or presence of PD-1-OX40L-Exos (10 μg mL−1) for 48 h at 37°C. Cell culture supernatants were then collected and assayed for the levels of IL-2 by ELISA. Results are expressed as a mean ± SD from one of at least three separate experiments.
To evaluate time-dependent T cell activation, BT-20 and PBMCs were incubated at a ratio of 1:2 in the presence of native exosomes (10 μg mL−1), PD-1-OX40L-Exos (10 μg mL−1), αCD3-αEGFR-Exos (10 μg mL−1), the combination of PD-1-OX40L-Exos (10 μg mL−1) and αCD3-αEGFR-Exos (10 μg mL−1), or αCD3-αEGFR-PD-1-OX40L GEMINI-Exos (10 μg mL−1) for 24, 48, 72, and 96 h. At each time point, cell culture supernatants were collected and assayed for the levels of IL-2 by ELISA. Results are expressed as a mean ± SD from one of at least three separate experiments.
In vivo efficacy studies
Six- to eight-week-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Southern California.
Mice (five per group) were injected subcutaneously on the right hind limbs on day 0 with 5 × 106 BT-20 cells in 0.1 mL of 50% matrigel (BD Biosciences). Human PBMCs were incubated in RPMI 1640 complete medium at a density of 2 × 106 cells/mL and stimulated in flasks with immobilized anti-human CD3 antibody (clone: OKT3, BioLegend), soluble anti-CD28 antibody (2 μg mL−1, clone: 28.2, BioLegend), and recombinant human IL-2 (rhIL-2) (40 IU mL−1, BioLegend) for 3 days at 37°C with 5% CO2. Cells were then expanded in RPMI 1640 complete medium with 40 IU mL−1 rhIL-2. All human PBMCs purchased from HemaCare Corporation were from the same healthy donor. When tumor sizes reached 80 to 100 mm3, 12 days post tumor implantation, mice received two intraperitoneal injections of expanded human PBMCs (20 × 106 cells per mouse) with a 6-day interval. One day following the first human PBMC injection, mice were treated intravenously every other day for a total of six times with vehicle (PBS), native exosomes (10 mg/kg), PD-1-OX40L-Exos (10 mg/kg), αCD3-αEGFR-Exos (10 mg/kg), the combination of PD-1-OX40L-Exos (10 mg/kg) and αCD3-αEGFR-Exos (10 mg/kg), and αCD3-αEGFR-PD-1-OX40L GEMINI-Exos (10 mg/kg). The loading amounts for exosomes were determined based on our previous studies.20,22 For the combination group, the total injection amount of 20 mg/kg exosomes per mouse per dose could serve as a strong competitor of 10 mg/kg GEMINI-Exos for comparison. Tumor volumes were measured three times weekly with a caliper and calculated as mm3 = 0.5 × (length) × (width).2 At the end of the study, mice were euthanized, and tumors, spleen, and blood were collected for lymphocyte isolation and analysis.
Lymphocyte isolation and analysis
The harvested blood samples were treated with red blood cell lysis buffer (BioLegend) by following the manufacturer’s instructions. Tumors and spleens were cut into small pieces and subjected to mechanical disruption and separation, followed by passing through 40-μm strainers and treatment with the red blood cell lysis buffer.
The resulting single-cell suspensions were stained for live and dead cells with live/dead-fixable Zombie Aqua (BioLegend), followed by cell surface marker staining with PerCP/Cyanine5.5 anti-human CD45 antibody (clone: 2D1, BioLegend), FITC anti-human CD3 antibody (clone: UCHT1, BioLegend), APC/Cyanine7 anti-human CD4 antibody (clone: OKT4, BioLegend), Pacific blue anti-human CD8 antibody (clone: RPA-T8, BioLegend), PE anti-human CD25 antibody (clone: M-A251, BioLegend), and PE/Dazzle 594 anti-human CD127 antibody (clone: A019D5, BioLegend). Prior to the intracellular staining for FoxP3 with PE/Dazzle 594 anti-human FoxP3 antibody (clone: 206D, BioLegend), cells were fixed with 4% paraformaldehyde (PFA) solution (Thermo Fisher Scientific) and permeabilized with 0.1% Triton X-100. Data acquisition was performed on a BD Fortessa X20 flow cytometer and results were analyzed with FlowJo. Total numbers of CD4+, CD8+, CD4+ CD25+ CD127−, and CD4+ CD25+ FoxP3+ cells were analyzed within CD45+ hematopoietic cell populations.
Immunohistofluorescence analysis
Immunostaining of collected tumors was performed on 7-mm cryosections by following standard protocols. Tumor tissues were fixed with 4% paraformaldehyde (PFA) for 10 min and then blocked with PBS containing 5% goat serum for 1 h at room temperature. The tissue sections were then incubated with an anti-human CD3 antibody (clone: UCHT1, BioLegend) for 1 h, stained with Alexa Fluor 488-conjugated anti-mouse IgG (H + L) secondary antibody (catalog# A28175 from Thermo Fisher Scientific) for 1 h, followed by nuclei counterstaining with DAPI. Images were captured with a Leica SP8 confocal laser scanning microscope (Leica Microsystems Inc.) and processed using LAS X software (Leica Microsystems Inc.). For quantification, three random, non-overlapping regions along the margin and interior of each tumor (n = 3 mice/group) were imaged.
Alanine aminotransferase activity assay
At the end of in vivo efficacy study, alanine aminotransferase (ALT) activities in plasma samples were assayed. Collected mouse plasma samples (5 μL) or a series of dilutions of standard solution (sodium pyruvate) were added to wells of clear 96-well plates, followed by additions of 25 μL of ALT substrate solution (0.2 M alanine, 2 mM 2-oxoglutarate, pH 7.4) and incubation at 37°C for 20 min. Next, 50 μL of 2,4-DNPH (1 mM solution in 1 M HCl) was added and incubated at room temperature for 20 min. Then, 0.5 M sodium hydroxide was added, and absorbance was measured at 510 nm using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader (BioTek, VT). Amounts of generated pyruvate were calculated based on determined standard curves. ALT activity is reported as nmole/min/mL = unit/L, where 1 milliunit (mU) of ALT is defined as the amount of enzyme that generates 1.0 nmole of pyruvate per minute at 37°C.
Creatinine colorimetric assay
At the end of in vivo efficacy study, creatinine concentrations in plasma were determined by a colorimetric assay. Working solutions were prepared by mixing picric acid (38 mM) with sodium hydroxide (1.2 M) at 1:1 ratio. Mouse plasma samples were mixed with equal volume of TCA (10%) and centrifuged at 5,000 × g for 10 min. Collected supernatants and creatinine standards were added onto 96-well plates, followed by additions of working solution and incubation at room temperature for 45 min. Absorbance was measured at 500 nm using a BioTek Synergy H1 Hybrid Multi-Mode Microplate reader (BioTek, VT). Creatinine concentrations in mouse plasma samples were determined based on standard curves.
Statistical analysis
Raw data without pre-processing were used for statistical analysis. Matched, repeated measures one-way ANOVA with the Geisser-Greenhouse correction and Dunnett’s multiple comparisons test was used for comparing tumor growth curves in Figure 4A. Ordinary one-way ANOVA with Tukey’s multiple comparisons test was carried out for comparing other multiple groups. Two-tailed unpaired t test was performed for comparison between two groups. The statistical analyses were performed on independent biological replicates; n = 5 for in vivo animal studies and n ≥ 2 for in vitro assays. A p < 0.05 was considered statistically significant. Significance of finding was defined as follows: ns = not significant, p > 0.05; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001. Data are shown as mean ± SD. All statistical analyses were calculated using GraphPad Prism (GraphPad Software, La Jolla, CA).
Data availability
The raw/processed data required to reproduce these findings are available from the authors upon request.
Acknowledgments
This work was supported in part by STOP CANCER Research Career Development Award (to Y.Z.), Department of Defense CDMRP Career Development Award W81XWH-19-1-0272 (to Y.Z.), California Breast Cancer Research Program Innovative Development and Exploratory Award 25IB-0080 (to Y.Z.), Tobacco-Related Disease Research Program New Investigator Award T30KT1021 (to Y.Z.), National Institute of Biomedical Imaging and Bioengineering (NIBIB) of the National Institutes of Health (NIH) grant R01EB031830 (to Y. Z.), and NIH grant P30CA014089 to the USC Norris Comprehensive Cancer Center.
Author contributions
Q.C. and Y.Z. designed the research. Q.C. and Z.D. performed the research. G.S., A.L.E., and H.-J.L. provided resources and critical insights. Q.C. and Y.Z. analyzed data. Q.C. and Y.Z. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.06.013.
Supplemental information
References
- 1.Doyle L.M., Wang M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells. 2019;8:727. doi: 10.3390/cells8070727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalluri R., LeBleu V.S. The biology, function, and biomedical applications of exosomes. Science. 2020;367:eaau6977. doi: 10.1126/science.aau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ståhl A.L., Johansson K., Mossberg M., Kahn R., Karpman D. Exosomes and microvesicles in normal physiology, pathophysiology, and renal diseases. Pediatr. Nephrol. 2019;34:11–30. doi: 10.1007/s00467-017-3816-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boulanger C.M., Loyer X., Rautou P.-E., Amabile N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017;14:259–272. doi: 10.1038/nrcardio.2017.7. [DOI] [PubMed] [Google Scholar]
- 5.Maia J., Caja S., Strano Moraes M.C., Couto N., Costa-Silva B. Exosome-based cell-cell communication in the tumor microenvironment. Front. Cell Dev. Biol. 2018;6:18. doi: 10.3389/fcell.2018.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang H.-G., Grizzle W.E. Exosomes: a novel pathway of local and distant intercellular communication that facilitates the growth and metastasis of neoplastic lesions. Am. J. Pathol. 2014;184:28–41. doi: 10.1016/j.ajpath.2013.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haraszti R.A., Didiot M.-C., Sapp E., Leszyk J., Shaffer S.A., Rockwell H.E., Gao F., Narain N.R., DiFiglia M., Kiebish M.A., et al. High-resolution proteomic and lipidomic analysis of exosomes and microvesicles from different cell sources. J. Extracell. Vesicles. 2016;5:32570. doi: 10.3402/jev.v5.32570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van den Boorn J.G., Schlee M., Coch C., Hartmann G. SiRNA delivery with exosome nanoparticles. Nat. Biotechnol. 2011;29:325–326. doi: 10.1038/nbt.1830. [DOI] [PubMed] [Google Scholar]
- 9.Kamerkar S., LeBleu V.S., Sugimoto H., Yang S., Ruivo C.F., Melo S.A., Lee J.J., Kalluri R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature. 2017;546:498–503. doi: 10.1038/nature22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 11.Hong Y., Nam G.-H., Koh E., Jeon S., Kim G.B., Jeong C., Kim D.-H., Yang Y., Kim I.-S. Exosome as a vehicle for delivery of membrane protein therapeutics, PH20, for enhanced tumor penetration and antitumor efficacy. Adv. Funct. Mater. 2018;28:1703074. doi: 10.1002/adfm.201703074. [DOI] [Google Scholar]
- 12.Gao X., Ran N., Dong X., Zuo B., Yang R., Zhou Q., Moulton H.M., Seow Y., Yin H. Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy. Sci. Transl. Med. 2018;10:eaat0195. doi: 10.1126/scitranslmed.aat0195. [DOI] [PubMed] [Google Scholar]
- 13.Kojima R., Bojar D., Rizzi G., Hamri G.C.-E., El-Baba M.D., Saxena P., Ausländer S., Tan K.R., Fussenegger M. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat. Commun. 2018;9:1305. doi: 10.1038/s41467-018-03733-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu Q., Ling X., Yang Y., Zhang J., Li Q., Niu X., Hu G., Chen B., Li H., Wang Y., Deng Z. Embryonic stem cells-derived exosomes endowed with targeting properties as chemotherapeutics delivery vehicles for glioblastoma therapy. Adv. Sci. 2019;6:1801899. doi: 10.1002/advs.201801899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhuang X., Xiang X., Grizzle W., Sun D., Zhang S., Axtell R.C., Ju S., Mu J., Zhang L., Steinman L., et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011;19:1769–1779. doi: 10.1038/mt.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wieduwilt M.J., Moasser M.M. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008;65:1566–1584. doi: 10.1007/s00018-008-7440-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin H.T., Ahmed R., Okazaki T. Role of PD-1 in regulating T-cell immunity. Curr. Top. Microbiol. Immunol. 2011;350:17–37. doi: 10.1007/82_2010_116. [DOI] [PubMed] [Google Scholar]
- 18.Ishii N., Takahashi T., Soroosh P., Sugamura K. OX40-OX40 ligand interaction in T-cell-mediated immunity and immunopathology. Adv. Immunol. 2010;105:63–98. doi: 10.1016/S0065-2776(10)05003-0. [DOI] [PubMed] [Google Scholar]
- 19.Pauken K.E., Dougan M., Rose N.R., Lichtman A.H., Sharpe A.H. Adverse events following cancer immunotherapy: obstacles and opportunities. Trends Immunol. 2019;40:511–523. doi: 10.1016/j.it.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng Q., Shi X., Han M., Smbatyan G., Lenz H.J., Zhang Y. Reprogramming exosomes as nanoscale controllers of cellular immunity. J. Am. Chem. Soc. 2018;140:16413–16417. doi: 10.1021/jacs.8b10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng Q., Shi X., Zhang Y. Reprogramming exosomes for immunotherapy. Methods Mol. Biol. 2020;2097:197–209. doi: 10.1007/978-1-0716-0203-4_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi X., Cheng Q., Hou T., Han M., Smbatyan G., Lang J.E., Epstein A.L., Lenz H.-J., Zhang Y. Genetically engineered cell-derived nanoparticles for targeted breast cancer immunotherapy. Mol. Ther. 2020;28:536–547. doi: 10.1016/j.ymthe.2019.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeppesen D.K., Hvam M.L., Primdahl-Bengtson B., Boysen A.T., Whitehead B., Dyrskjøt L., Orntoft T.F., Howard K.A., Ostenfeld M.S. Comparative analysis of discrete exosome fractions obtained by differential centrifugation. J. Extracell Vesicles. 2014;3:25011. doi: 10.3402/jev.v3.25011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livshits M.A., Khomyakova E., Evtushenko E.G., Lazarev V.N., Kulemin N.A., Semina S.E., Generozov E.V., Govorun V.M. Isolation of exosomes by differential centrifugation: theoretical analysis of a commonly used protocol. Sci. Rep. 2015;5:17319. doi: 10.1038/srep17319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bond V.C., Powell M., Huang M.B., Ali S. World Intellectual Property Organization; 2013. Compositions and methods for exosome targeted expression. U.S. Patent WO 2013089738A1. [Google Scholar]
- 26.Schroit A.J., Thorpe P.E., Fussey S.P. U.S. Patent and Trademark Office; 2017. Methods and Compositions for Isolating Exosomes.https://patents.google.com/patent/US9835626B2/en U.S. Patent No. 9,835,626 B2. [Google Scholar]
- 27.Cheng Q., Dai Z., Shi X., Duan X., Wang Y., Hou T., Zhang Y. Expanding the toolbox of exosome-based modulators of cell functions. Biomaterials. 2021;277:121129. doi: 10.1016/j.biomaterials.2021.121129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian Y., Li S., Song J., Ji T., Zhu M., Anderson G.J., Wei J., Nie G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383–2390. doi: 10.1016/j.biomaterials.2013.11.083. [DOI] [PubMed] [Google Scholar]
- 29.Haney M.J., Klyachko N.L., Zhao Y., Gupta R., Plotnikova E.G., He Z., Patel T., Piroyan A., Sokolsky M., Kabanov A.V., Batrakova E.V. Exosomes as drug delivery vehicles for Parkinson's disease therapy. J. Control. Release. 2015;207:18–30. doi: 10.1016/j.jconrel.2015.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koh E., Lee E.J., Nam G.H., Hong Y., Cho E., Yang Y., Kim I.S. Exosome-SIRPα, a CD47 blockade increases cancer cell phagocytosis. Biomaterials. 2017;121:121–129. doi: 10.1016/j.biomaterials.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Lewis N.D., Sia C.L., Kirwin K., Haupt S., Mahimkar G., Zi T., Xu K., Dooley K., Jang S.C., Choi B., et al. Exosome surface display of IL12 results in tumor-retained pharmacology with superior potency and limited systemic exposure compared with recombinant IL12. Mol. Cancer Ther. 2021;20:523. doi: 10.1158/1535-7163.mct-20-0484. [DOI] [PubMed] [Google Scholar]
- 32.Witwer K.W., Théry C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J. Extracell. Vesicles. 2019;8:1648167. doi: 10.1080/20013078.2019.1648167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou M., Weber S.R., Zhao Y., Chen H., Sundstrom J.M. In: Chapter 2 - Methods for Exosome Isolation and Characterization. Exosomes Edelstein L., Smythies J., Quesenberry P., Noble D., editors. Academic Press; 2020. pp. 23–38. [Google Scholar]
- 34.Zhu X., Badawi M., Pomeroy S., Sutaria D.S., Xie Z., Baek A., Jiang J., Elgamal O.A., Mo X., Perle K.L., et al. Comprehensive toxicity and immunogenicity studies reveal minimal effects in mice following sustained dosing of extracellular vesicles derived from HEK293T cells. J. Extracell. Vesicles. 2017;6:1324730. doi: 10.1080/20013078.2017.1324730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi X., Cheng Q., Zhang Y. Reprogramming extracellular vesicles with engineered proteins. Methods. 2020;177:95–102. doi: 10.1016/j.ymeth.2019.09.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw/processed data required to reproduce these findings are available from the authors upon request.