

Abstract
The development of methyl transverse relaxation optimized spectroscopy has greatly facilitated the study of macromolecylar assemblies by solution NMR spectroscopy. However, limited sample solubility and stability has hindered application of this technique to ongoing studies of complexes formed on membranes by the neuronal SNAREs that mediate neurotransmitter release and synaptotagmin-1, the Ca2+ sensor that triggers release. Since the 1H NMR signal of a tBu group attached to a large protein or complex can be observed with high sensitivity if the group retains high mobility, we have explored the use of this strategy to analyze presynaptic complexes involved in neurotransmitter release. For this purpose, we attached tBu groups at single cysteines of fragments of synaptotagmin-1, complexin-1 and the neuronal SNAREs by reaction with 5-(tert-butyldisulfaneyl)-2-nitrobenzoic acid (BDSNB), tBu iodoacetamide or tBu acrylate. The tBu resonances of the tagged proteins were generally sharp and intense, although tBu groups attached with BDSNB had a tendency to exhibit somewhat broader resonances that likely result because of the shorter linkage between the tBu and the tagged cysteine. Incorporation of the tagged proteins into complexes on nanodiscs led to severe broadening of the tBu resonances in some cases. However, sharp tBu resonances could readily be observed for some complexes of more than 200 kDa at low micromolar concentrations. Our results show that tagging of proteins with tBu groups provides a powerful approach to study large biomolecular assemblies of limited stability and/or solubility that may be applicable even at nanomolar concentrations.
Keywords: Tert-butyl NMR, slow relaxation, macromolecular assemblies, synaptotagmin, SNARE complex, complexin, neurotransmitter release
Introduction
NMR spectroscopy in solution provides many avenues to study the biophysical basis underlying the functions of macromolecular assemblies. Although structure determination at atomic resolution becomes increasingly difficult as the molecular weight escalates beyond 30 kDa, a wealth of structural and dynamic information can still be obtained by a variety of approaches for complexes well above this molecular weight (Huang and Kalodimos, 2017; Rosenzweig and Kay, 2014). Particularly powerful among these strategies is the use of methyl-transverse relaxation optimized spectroscopy (TROSY) on samples specifically 13CH3-labeled on a perdeuterated background (Tugarinov et al., 2004), which has allowed studies of protein complexes in the 1 MDa range (Rosenzweig and Kay, 2014). However, this approach also has some limitations, as high temperatures and/or relatively high sample concentrations are required to overcome the strong resonance broadening that still occurs for large assemblies, and such conditions may not be applicable in many cases because of limited sample stability or solubility. Thus, whereas in our extensive studies of the machinery that controls neurotransmitter release using NMR spectroscopy (Rizo et al., 2012) methyl-TROSY experiments yielded crucial insights on soluble protein complexes of up to 100 kDa even at low micromolar concentrations (e.g. (Brewer et al., 2015; Ma et al., 2011)), it has been much more challenging to apply this methodology to analyze membrane-anchored protein complexes. Some of the difficulties that we encountered are illustrated by the studies of membrane-anchored complexes between the Ca2+ sensor synaptotagmin-1 (Syt1) and the neuronal soluble N-ethylmaleimide sensitive factor attachment protein receptors (SNAREs) summarized below.
An interesting strategy to tag proteins with probes that can be observed with high sensitivity by NMR spectroscopy entails the use of tert-butyl groups (tBu). Because they contain three chemically identical methyl groups, the tBu moiety exhibits exceptionally narrow and intense NMR signals even when attached to large proteins or complexes, as long as fast internal motions remain around the moiety. A dramatic demonstration of this notion was provided by the sharp 13C signal observed for a tBu group that was attached to a peptide covalently linked to 50 μM BioBeads (molecular weight in the petaDalton range) and that had a longitudinal relaxation time comparable to that of a tBu group attached to a single amino acid (Giralt et al., 1984). tBu groups attached to proteins by chemical methods or by site-specific introduction of non-native amino acids were also shown to yield sharp, readily identifiable signals in 1H NMR spectra of protein complexes in the 100-300 kDa range (Chen et al., 2015; Chen et al., 2016; Jabar et al., 2017; Loh et al., 2018). Trimethylsilyl groups have also been used as protein tags that yield sharp signals observable with high sensitivity in a region of protein 1H spectra with little overlap (Abdelkader et al., 2021; Becker et al., 2018; Hu et al., 2019; Jabar et al., 2017; Liu et al., 2020; Loh et al., 2018). Both approaches provide highly sensitive reporters to facilitate measurement of NMR parameters that can provide structural information such of nuclear Overhauser effects (NOEs), pseudocontact shifts (PCSs) and paramagnetic relaxation effects (PREs) (Abdelkader et al., 2021; Chen et al., 2016; Jabar et al., 2017), and can also be used to monitor ligand binding, conformational changes, enzyme inhibition and oligomer symmetry (Abdelkader et al., 2021; Becker et al., 2018; Chen et al., 2015; Hu et al., 2019; Liu et al., 2020; Loh et al., 2018). While the results obtained with these probes so far are highly promising, the signals of tBu or trimethylsilyl groups attached to the surface of proteins may be severely broadened if they have interactions with other groups that restrict their mobility, which would strongly hinder the observation of these signals for large complexes at low concentrations. Because there are only a few examples of the use of these groups as protein tags in the literature, it is currently unclear whether such broadening might occur frequently and to what extent it depends on the method used to attach a tBu or trimethylsilyl group to a protein.
The study described below was designed to investigate the generality of this overall approach by examining the 1H NMR signals of tBu groups placed in a variety of environments on protein surfaces and testing how these signals are affected by incorporation of the proteins into complexes of diverse molecular weights. For this purpose, we attached tBu groups to various components of the neurotransmitter release machinery through distinct chemical reactions that link the tBu group to a cysteine side chain through different moieties. Our results indicate that tBu groups attached to surface-exposed positions of proteins commonly yield sharp, intense tBu resonances. Binding of the tagged proteins to targets can lead to strong broadening of the tBu resonance in some cases, which likely arises because of immobilization at the binding interface, but in other cases we still observed relatively sharp tBu resonances that can be detected at low micromolar concentrations even upon formation of complexes of over 200 kDa. Our data support the notion that tBu tagging constitutes a versatile tool to study macromolecular assemblies by solution NMR methods with high sensitivity.
Materials and methods
Protein expression and purification
Constructs to express the following proteins or protein fragments were described previously: rat synaptobrevin-2 SNARE motif (residues 29–93), rat synaptobrevin-2 (residues 49–93), full-length rat synaptobrevin-2, human SNAP-25A fragments encoding its SNARE motifs (residues 11–82 and 141–203), full-length rat syntaxin-1A, rat syntaxin-1A (residues 183-288), rat syntaxin-1A (residues 191–253), rat syntaxin-1A (residues 2–253), rat synaptotagmin-1 C2B domain (residues 271–421), rat synaptotagmin-1 C2AB fragment (residues 140–421), rat complexin-1 (residues 26–83) and MSP1E3D1 (pMSP1E3D1 was a kind gift from Stephen Sligar; Addgene plasmid # 20066; http://n2t.net/addgene:20066; RRID:Addgene_20066) (Brewer et al., 2015; Chen et al., 2006; Chen et al., 2008; Chen et al., 2002; Denisov et al., 2007; Ma et al., 2013; Rizo et al., 2006; Xu et al., 2013; Zhou et al., 2013). All these proteins were expressed in E. coli BL21 (DE3) cells and purified as previously described in these references, with the exceptions noted below.
Expression vectors for mutant proteins were generated using a combination of the QuikChange site-directed mutagenesis kit (Stratagene) and standard PCR-based techniques with custom designed primers. These mutants included: Syt1 C2B (271–421) C277A/N319C, Syt1 C2B (271–421) C277A/E346C, Syt1 C2AB (140–421) C277A/E346C, complexin-1 (26–83) V61C, syntaxin-1 (191-253) D214C, and SNAP-25A (141–203) D166C. All mutant proteins were purified as the WT proteins, including 0.5 mM TCEP in the final purification step for cysteine containing proteins. The only exception was SNAP-25A (141–203) D166C which was purified into a buffer containing 1 mM DTT.
15N-labeled Syt1 C2B and C2AB mutants, Cpx-1 (26–83) mutant, and syntaxin-1 (191-253) mutant were expressed using M9 media containing 15NH4Cl as the sole nitrogen source (1 g/L). Uniformly perdeuterated, 15N-labeled proteins were expressed using M9 expression media in 99.9% D2O with D-glucose (1,2,3,4,5,6,6-D7, 97–98%) as the sole carbon source (3 g/L) and 15NH4Cl as the sole nitrogen source (1 g/L). Specific 13CH3-labeling at the Met and Ile δ1 methyl groups of the Syt1 C2AB fragment was achieved by adding [3,3–2H] 13C-methyl α-ketobutyric acid (80 mg/L) and 13C-methyl methionine (250 mg/L) (Cambridge Isotope Laboratories) to the cell cultures 30 min prior to Isopropyl β-D-1-thiogalactopyranoside (IPTG) induction.
Synthesis of (5-tert-butyldisulfanyl)-2-nitrobenzoic acid.
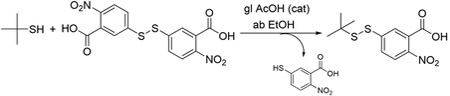
Reagents: 2-Methyl-2-propanethiol; 2,2'-dinitro-5,5'-dithiodibenzoic acid (Ellman’s reagent); absolute ethanol (ab EtOH) and glacial acetic acid (gl AcOH) as catalyst were supplied by Sigma-Aldrich and used without further treatment.
2,2'-Dinitro-5,5'-dithiodibenzoic acid (Ellman’s Reagent) (725 mg, 1.83 mmol) was added into a round bottom flask, previously dried in the oven at 120 °C, and equipped with a stirring magnetic bar and a septum. Then, it was purged several times with a stream of nitrogen, and next ab EtOH (5 mL), gl AcOH (140 μL) and 2-methyl-2-propanethiol (190 μL, 1.66 mmol) were added through a syringe. The reaction was left to stir overnight at room temperature. After this time, the HPLC showed that the reaction was completed. The solvent was removed on a rotary evaporator and 415 mg of the crude product (approximately 50% of total crude), which was a mixture of the target product and the thiol side product, were purified directly on Waters semi-preparative HPLC-MS, separation module 2545, UV detector 2487 with dual absorbance, Waters ZQ micromass mass detector. Waters 2767 injector-manifold. Column, XBridge BEH130 semi-preparative C18 (5 μm, 19x100 mm). Eluent solvent system: A (H2O with 0.1% HCOOH), B (ACN with 0.7% HCOOH), flow rate 16 mL/min. Detection wavelength 220 nm. Gradient 50 to 100% of B in 10 minutes. We obtained 32 mg of the final product (25% overall yield), which was a yellow solid with a purity of 99% as assessed by HPLC-PDA analysis (retention time 5.289 min., Supplementary Figure 1) performed on a Waters Alliance 2695 with a photodiode array detector Waters 2998, Column XBridge BEH130 C18 (3.5 μm, 4.5mm x 100mm). Elution solvent system: A (H2O with 0.045% trifluoroacetic acid (TFA)), B (ACN with 0.036% TFA), flow rate of 1.0 mL/min, temperature 25 °C. Detection wavelength (λ=220nm). Gradient 40 to 70 % of B in 8 minutes. The identity of the product was confirmed by High resolution mass spectrometry (HR-MS) performed in an LC / MSD-TOF Agilent Technologies G1969A instrument, ESI-TOF at 150V and using H2O-acetonitrile (ACN) (1: 1) as eluent (Supplementary Figure 2), and by 1H and 13C NMR spectroscopy performed on a Varian VNMRS500 500 MHz spectrometer (Supplementary Figures 3, 4). HR-MS: calculated mass [M-H]−1= 286.0276; found mass [M-H]−1 286.0209; [M-COOH]−1 = 242.0313). 1H NMR (500 MHz, DMSO): δ1.31 (9H, s); δ7.94 (2H, m); δ8.01 (1H, m); δ13.99 (1H, s). 13C NMR (500 MH, DMSO): 29.31 ppm; 50.32 ppm; 124.79 ppm; 125.52 ppm; 127.93; 128.94 ppm; 145.28 ppm; 145.46 ppm; 165.74 ppm. 1H and 13C chemical shifts were measured relative to the deuterated DMSO signal at 2.5 ppm and 39.5 ppm, respectively.
Labeling proteins with tBu groups and 13CH3 iodide
Single cysteine mutants of proteins were first buffer exchanged into 20 mM HEPES pH 7.4 125 mM KCl 0.5 mM TCEP using concentration and dilution. For labeling with N-tBu-iodoacetamide (CAS number 103615-48-1), tBu-acrylate (CAS number 1663-39-4), (5-tert-butyldisulfanyl)-2-nitrobenzoic acid or 13CH3 iodide (CAS Number: 4227-95-6), the proteins (25- 200 μM concentration) were incubated with 1- 2 mM reagent at room temperature. The reactions were monitored by following the perturbations observed in the 1H-15N heteronuclear single quantum coherence (HSQC) spectra of the proteins. The incubation times varied between 1- 16 hours, and 10 mM DTT was added at the end to quench the reactions except for reactions with (5-tert-butyldisulfanyl)-2-nitrobenzoic acid. Unreacted reagents were separated from the labeled proteins by size exclusion chromatography on either a Superdex 75 column (GE 10/300) or a Superdex 200 column (GE 10/300) in 20 mM HEPES pH 7.4 125 mM KCl buffer.
Labeling proteins with Dy3+-C2
SNAP-25A (141–203) D166C was first treated with 10 mM DTT which was subsequently removed by size exclusion chromatography on a Superdex 75 column (GE 16/60). The fresh protein was pooled and immediately incubated with 3- fold molar excess of Dy3+-C2 (Brewer et al., 2015; Graham et al., 2011). Reaction progress was measured by monitoring the absorbance of the reaction byproduct at 345 nm. The tagged protein was directly used for SNARE complex assembly.
Preparation of soluble SNARE complex
The SNARE motifs were mixed in equimolar ratios in the following order: synaptobrevin-2 (29–93), SNAP-25A(141–203), SNAP-25A(11–82) and syntaxin-1A (191–253), in the presence of 1 M NaCl. The mixture contained the following protease inhibitors (protease inhibitor cocktail A): Antipain Dihydrochloride 0.016 mg/ml (Thermo Fischer Scientific: 50488492); Leupeptin 0.33 mg/ml (Gold Bio: L01025); Aprotinin 0.08 mg/ml (Gold Bio: A655100). The assembly reaction was incubated at room temperature overnight while rotating. The SNARE motifs that did not incorporate into complex were removed by concentration-dilution at room temperature using 30 kDa molecular weight cutoff (MWCO) Amicon centrifugation filters. The almost quantitative formation of SDS-resistant SNARE complex was verified by SDS-PAGE and Coomassie blue staining.
Preparation of nanodiscs, cisSC-nanodiscs and transSC-nanodiscs
Appropriate lipid mixtures (specific to each experiment as indicated in the text) were prepared by mixing chloroform stocks of 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt) (DOPS), L-α-phosphatidylinositol-4,5-bisphosphate (Brain, Porcine) (ammonium salt) (PIP2), 1,2-dipalmitoyl-d62-sn-glycero-3-phosphocholine (deuterated PC) and/or 1-palmitoyl-d31-2-oleoyl-sn-glycero-3-[phospho-L-serine] (sodium salt) (partially deuterated POPS) in glass test tubes. These mixtures were dried under a stream of nitrogen and stored overnight in a vacuum desiccator. The lipids were solubilized in a 20 mM HEPES pH 7.4 125 mM KCl 1% β-OG (octyl-beta-glucoside) buffer by vortexing for 5 min. To form nanodiscs, MSP1E3D1 was incubated with solubilized lipids at a ratio of 1:110 in the presence of 1% β-OG (final concentration) at 4°C for 30 min. The mixture was passed over a 4 cm-high Thermo Scientific Pierce Detergent Removal Resin (87780) column (approximately 3 mL of the slurry; the final volume of the mixture was always between 3–4 mL). The nanodiscs were purified by size exclusion chromatography using a Superdex 200 column (GE 16/60). Appropriate fractions as assessed by SDS-PAGE followed by Coomassie blue staining were pooled and concentrated to a desired concentration using a 30 kDa MWCO Amicon centrigufation filter.
To prepare cisSNARE complex nanodiscs (cisSC-NDs), detergent solubilized cisSNARE-complex was formed by incubating either 10 μM full-length rat syntaxin-1A with 15 μM of synaptobrevin-2 (29–93), SNAP-25A (11-82) and SNAP-25A (141-203) or 10 μM full-length rat synaptobrevin with 15 μM of rat syntaxin-1A (191-253), SNAP-25A (11-82) and SNAP-25A (141-203) in the presence of 1% β–OG, 0.5 M NaCl and protease inhibitor cocktail A overnight at 4°C. For incorporation into nanodiscs, cisSNARE-complexes were mixed with MSP1E3D1 and solubilized lipids at a ratio of 1:3:300 and incubated at 4 °C for 30 minutes. The detergent was removed using Thermo Scientific Pierce® Detergent Removal Resin, as described for the isolated nanodiscs, and size exclusion chromatography using a Superdex 200 column (GE 16/60). Fractions from size exclusion chromatography were assessed by SDS-PAGE and Coomassie blue staining. Fractions that contained approximately one cisSNARE-complex per two MSP1E3D1 molecules were pooled together, mixed with protease inhibitor cocktail A and concentrated. To prepare transSNARE complex nanodiscs (trSC-NDs), separate nanodiscs containing 15N-labeled full-length synaptobrevin-2 or syntaxin-1A (183-288) were prepared as described above for cisSC-NDs but replacing the solubilized cisSNARE complex with full-length synaptobrevin-2 or syntaxin-1A (183-288). Syntaxin-1A (183-288) NDs were incubated with 15N-synaptobrevin-2 NDs, SNAP-25 (11–82) and SNAP-25 (141–203) fragments overnight at 4 °C in the presence of protease inhibitor cocktail A. Formation of the trans-SNARE complex was verified from the disappearance of the cross-peaks in the 1H-15N HSQC spectrum of 15N-synaptobrevin-2. For final NMR sample preparation, samples were buffer exchanged into the appropriate buffer by using either concentration dilution, PD-10 Desalting Column (GE Healthcare) or Zeba Spin Desalting Columns.
NMR spectroscopy
All NMR spectra except for syntaxin-1A (191-253) 214C were acquired at 25°C on Agilent DD2 spectrometers operating at 600 or 800 MHz and equipped with cold probes. Spectra of syntaxin-1A (191-253) 214C were collected at 20°C. All 1D 1H NMR spectra listed in Table 1 were acquired on the same Agilent DD2 600 MHz spectrometer. The 1H-13C HMQC spectra of Fig. 1 were collected with 100% D2O as the solvent. All other 1Ds, 1H-15N HSQC, 1H-15N TROSY-HSQC and 1H-13C HMQC spectra were acquired on samples with 10% D2O as the solvent. The buffer used for all the NMR experiments was 20 mM HEPES pH 7.4, 125 mM KCl with protease inhibitor cocktail A added. Some experiments were performed in the presence of 1 mM Ca2+ or 1 mM EGTA as indicated in the figures. All 1D 1H NMR spectra were processed and analyzed with Agilent VNMRJ. Two-dimensional spectra were processed with NMRPipe (Delaglio et al., 1995) and analyzed with NMRView (Johnson and Blevins, 1994).
Table 1.
Key acquisition conditions and NMR parameters measured in 1D 1H NMR spectra of tBu-tagging reagents and tBu-tagged proteins alone or bound to various ligandsa.
| Sample | Conc. (μM) | mw (kDa) | ntb | δ (ppm) | Δν1/2 (Hz) | S:N (x:1)c | S:N10μMd | Figuree |
|---|---|---|---|---|---|---|---|---|
| tBu-iodoacetamide | 1,000 | 0.24 | 4 | 1.12 | 2.0 | 2,200 | 124 | |
| tBu-acrylate | 1,000 | 0.13 | 4 | 1.31 | 2.1 | 1,400 | 79 | |
| BDSNB | 1,000 | 0.29 | 4 | 1.14 | 2.3 | 2,000 | 113 | |
| Cpx1(26-83)61I | 16 | 6.9 | 128 | 1.18 | 1.8 | 200 | 125 | 4a |
| Cpx1(26-83)61A | 13 | 6.9 | 128 | 1.31 | 1.8 | 250 | 192 | 4a |
| Cpx1(26-83)61B | 16 | 6.9 | 128 | 1.19 | 2.9 | 140 | 88 | 4a |
| C2B319I | 100 | 17.2 | 128 | 1.09 | 4.6 | 208 | 42 | 4b |
| C2B319A | 65 | 17.2 | 128 | 1.24 | 3.2 | 470 | 72 | 4b |
| C2B319B | 6 | 17.2 | 512 | 1.12 | 2.0 | 65 | 54 | 4b |
| C2B346I | 92 | 17.2 | 512 | 1.21 | 3.3 | 1,400 | 76 | 4c |
| C2B346A | 100 | 17.2 | 512 | 1.32 | 2.7 | 1,400 | 70 | 4c |
| C2B346B | 23 | 17.2 | 512 | 1.22 | 5.5 | 300 | 65 | 4c |
| C2AB346I | 100 | 32.2 | 128 | 1.21 | 3.8 | 730 | 73 | 4d |
| C2AB346A | 50 | 32.2 | 128 | 1.32 | 5.2 | 430 | 86 | 4d |
| C2AB346B | 50 | 32.2 | 128 | 1.22 | 9.2 | 200 | 40 | 4d |
| Syx241I | 25 | 7.5 | 128 | 1.18 | 2.7 | 160 | 64 | 5a |
| Syx214I-SC | 30 | 31.4 | 128 | 1.18 | 9.0 | 78 | 26 | 5a |
| Cpx1(26-83)61I + SC | 18 | 38.3 | 128 | 1.19 | 3.7 | 130 | 72 | 5b |
| Cpx1(26-83)61A + SC | 18 | 38.3 | 128 | 1.33 | 3.7 | 140 | 78 | 5b |
| Cpx1(26-83)61A | 8 | 6.9 | 128 | 1.32 | 2.3 | 140 | 175 | |
| Cpx1(26-83)61A + cisSC-ND | 8 | ~280 | 128 | 1.33 | n.d. | n.d. | n.d. | 5b |
| dC2B319I | 7 | 17.3 | 768 | 1.10 | 4.5 | 140 | 82 | 6c |
| dC2B319A | 7 | 17.3 | 768 | 1.24 | 4.5 | 110 | 64 | 6b |
| dC2B346I | 7 | 17.3 | 768 | 1.21 | 2.9 | 200 | 117 | 6e |
| dC2B346A | 7 | 17.3 | 768 | 1.33 | 2.7 | 200 | 117 | 6d |
| dC2B319A + PND + EGTA | 7 | ~260 | 768 | 1.24 | 5.7 | 65f | 38 | 6b |
| dC2B319A + PND + Ca2+ | 7 | ~260 | 768 | 1.24 | 5.3 | 84f | 49 | 6b |
| dC2B319I + PND + EGTA | 7 | ~260 | 768 | 1.09 | 5.5 | 65f | 38 | 6c |
| dC2B319I + PND + Ca2+ | 7 | ~260 | 768 | 1.09 | 5.5 | 74f | 43 | 6c |
| Cpx1(26-83)61A | 1 | 6.9 | 2048 | 1.31 | 2.4 | 25 | 65 | S7a |
| Cpx1(26-83)61A | 0.3 | 6.9 | 6900 | 1.31 | 3.1 | 8 | 36 | S7b |
All spectra were acquired with spectral width 8,103 Hz, acquisition time 0.6 s, zero filling to 8,192 complex points (digital resolution 1.0 Hz) and relaxation delay 1 s for a total recycling time of 1.6 s. n.d. not determined.
Number of transients (scans).
Signal to noise ratio for the tBu resonance calculated with Agilent VNMRJ.
Signal to noise ratio normalized to 10 μM nt=128 assuming a linear dependence with the concentration and with the square root of nt.
Figure where the corresponding spectrum is shown.
Corrected to account for the fact that in difference spectrum the noise increases by a factor of 20.5.
Figure 1.

Analysis presynaptic complexes on nanodiscs by methyl-TROSY. a-h Expansions corresponding to the methionine ε (a-d) or isoleucine δ1 (e-h) methyl regions of 1H-13C HMQC spectra of 50 μM 2H,13CH3-IM-C2AB in the presence of: i) 50 μM NDs composed of POPC:POPS 85:15 and EDTA (black contours, a-h) or Ca2+ (red contours, a,b,e,f); ii) 50 μM cisSC-NDs (anchored through the synaptobrevin TM region) and EDTA (blue contours, a,c,e,g); or iii) 50 μM trSC-NDs and Ca2+ (orange contours, d,h). All spectra were plotted at the same contour levels except for the red contours in b,f and the blue contours in c,g, which were plotted at five-fold lower levels. The C2A domain spans residues 140-265 and the C2B domain residues 270-421.
Results
1H-13C HMQC spectra of Syt1 complexes bound to nanodiscs and nanodisc-anchored SNARE complex
The release of neurotransmitters by Ca2+-triggered synaptic vesicle exocytosis is a crucial event for interneuronal communication and is exquisitely controlled by a sophisticated protein machinery (Rizo, 2018). Central components of this machinery are the SNARE proteins syntaxin-1, SNAP-25 and synaptobrevin, which form a tight four-helix bundle called the SNARE complex that brings the synaptic vesicle and plasma membranes together and is critical for membrane fusion (Hanson et al., 1997; Poirier et al., 1998; Sollner et al., 1993; Sutton et al., 1998). Syt1 acts as the major Ca2+ sensor that triggers neurotransmitter release (Fernandez-Chacon et al., 2001) through the two C2 domains that form its cytoplasmic region (the C2A and C2B domain), which bind three and two Ca2+ ions, respectively through loops at the tip of β-sandwich structures (Fernandez et al., 2001; Sutton et al., 1995; Ubach et al., 1998). The function of Syt1 depends on Ca2+-dependent binding of these loops to membranes (Fernandez-Chacon et al., 2001) and on interactions with the SNARE complex (Brewer et al., 2015; Zhou et al., 2015; Zhou et al., 2017) in a tight interplay with the SNAREs and complexins (Tang et al., 2006), which are small soluble proteins that bind tightly to the SNARE complex (Chen et al., 2002; McMahon et al., 1995). Thus, elucidating how synaptotagmin-1 binds to the SNARE complex is critical to understand how Ca2+ sensing is coupled to membrane fusion, but structural studies of these interactions using soluble fragments yielded three distinct binding modes involving the Syt1 C2B domain (Brewer et al., 2015; Zhou et al., 2015; Zhou et al., 2017) and some of these binding modes involved Syt1 sequences that had also been implicated in binding to membranes. These and other findings (Voleti et al., 2020) emphasized the importance of studying Syt1-SNARE complex interactions on a membrane environment. For this purpose, we designed a strategy that takes advantage of the high sensitivity of methyl-TROSY experiments and is based on measuring PCSs caused by lanthanide tags placed at strategic positions of the SNARE complex (Brewer et al., 2015; Pan et al., 2016) on 1H-13C HMQC cross-peaks of the Syt1 C2 domains. Here we just outline some of our initial efforts to start this project that led us to explore alternatives for high-sensitivity detection of NMR signals of these proteins.
In initial experiments, we prepared samples of a fragment spanning the two C2 domains of Syt1 (C2AB) that was specifically 13CH3-labeled at Ile δ1, Val and Leu methyl groups on a perdeuterated background (2H,13CH3-ILV-labeled), but we observed complete disappearance of almost all 1H-13C HMQC cross-peaks corresponding to the C2B domain upon addition of SNARE complex anchored through the transmembrane (TM) region of synaptobrevin on nanodiscs composed of POPC:POPS 85:15 (cisSC-NDs). To decrease the density of protons and thus further limit the rate of transverse relaxation, we prepared C2AB that was specifically 13CH3-labeled at Ile δ1 and Met methyl groups on a perdeuterated background (2H,13CH3-IM-labeled). As expected, 1H-13C HMQC spectra of 50 μM 2H,13CH3-IM-C2AB acquired in 1.5 hr in the presence of nanodiscs (NDs; also composed of POPC:POPS 85:15) and EDTA (Fig. 1, black contours) exhibited very high sensitivity that was comparable to that observed for 2H,13CH3-IM-C2AB alone because there is no substantial binding under these conditions. In contrast, analogous experiments performed in the presence of Ca2+ led to strong broadening and hence dramatic decreases in 1H-13C HMQC cross-peak intensities (Fig. 1A,E, red contours) due to Ca2+-induced binding of 2H,13CH3-IM-C2AB to the NDs (molecular weight ca. 240 kDa). Nevertheless, most cross-peaks of 2H,13CH3-IM-C2AB were still observable at low contour levels (Fig. 1B,F, red contours). 1H-13C HMQC spectra of 2H,13CH3-IM-C2AB in the presence of cisSC-NDs and EDTA also exhibited strong broadening, particularly for cross-peaks corresponding to the C2B domain (Fig. 1A,E, blue contours), which mediates SNARE complex binding (Brewer et al., 2015; Voleti et al., 2020; Zhou et al., 2015; Zhou et al., 2017). Indeed, a few cross-peaks from the C2B domain were not observable even at low contours levels (Fig. 1C,G, blue contours).
Naturally, longer acquisition times increased the sensitivity, which could be further enhanced by increasing the sample concentrations, but such efforts were hindered by sample precipitation that increases over time, particularly in the presence of Ca2+, which caused further cross-peak broadening. In the most challenging experiments, we used trans-SNARE complexes with synaptobrevin anchored on one nanodisc and syntaxin-1 anchored on another nanodisc (trSC-NDs; ca. 520 kDa). Ca2+-dependent binding of 2H,13CH3-IM-C2AB to trSC-NDs led to very strong broadening and we could observe a few weak cross-peaks from the C2B domain only after long acquisitions (e.g. 53 hr; Fig. 1D,H). However, we observed substantial sample degradation during the experiments and the observation of weak cross-peaks was hindered by noise caused by the signals from the nanodiscs. Although the overall data indicate that methyl-TROSY experiments in combination with PCS measurements could still be used to study how 2H,13CH3-IM-C2AB binds to cisSC-NDs in the absence of Ca2+, the considerable expense expected for such studies led us to explore whether strategies based on protein-tagging with tBu groups could provide a viable alternative.
tBu groups attached to neuronal exocytotic proteins exhibit sharp 1H NMR signals
The exceptionally intense signal that is normally observed in 1H NMR spectra for tBu groups attached to small organic compounds arises in part because tBu groups contain nine identical protons with no scalar couplings and in part because of the narrow linewidths resulting from the fast motions undergone by small compounds. Since attachment of a tBu group to a large protein results in much longer rotational correlation times, the 1H tBu resonance is expected to remain sharp only if it remains sufficiently mobile. Thus, in order to exhibit a sharp resonance, the tBu group should ideally be attached to the surface of a protein, minimizing the possibility of long-lasting interactions with other parts of the protein. Internal rotations around the bonds that link the methyl groups of the tBu moiety to the quaternary carbon and around the bond linking the quaternary carbon to the protein (Fig. 2A) should be very fast (in the ps time scale) in the absence of contacts and thus should help to maintain tBu resonances sharp. However, additional flexibility in one or a few bonds linking the tBu to rigid parts of the protein is required for the tBu protons to experience the fast, isotropic overall motion necessary for narrow linewidths. Thus, longer linkers between the tBu group and the protein should increase the probability of observing narrow tBu resonances. However, longer linkers lead to larger uncertainty in the location of the tBu group and hence in the structural information that can be obtained from observed perturbations (e.g. PCSs) of the tBu resonance. These arguments led us to test three different reactions to attach a tBu group to a cysteine, each yielding a different moiety between the tBu group and the cysteine (Fig. 2B): i) reaction with N-tBu-iodoacetamide, which was described previously (Jabar et al., 2017) and leaves a CH2-CO-NH linker; ii) Michael addition to tBu-acrylate, which leaves a CH2-CH2-CO-O linker; and iii) disulfide bond formation with 5-(tert-butyldisulfaneyl)-2-nitrobenzoic acid (BDSNB), which leaves only a S atom between the tBu group and the cysteine S.
Figure 2.

Chemical reactions and positions selected for tBu tagging in presynaptic proteins. a Diagram of the tBu group, with arrows illustrating the fast rotations that are expected to occur in the ps time scale around the bonds that link the methyl groups to the quaternary carbon and around the bond linking the quaternary carbon to the protein (P) if the tBu group does not pack against other atoms of the protein. b Diagrams of the chemical reactions used to attach tBu groups to protein cysteine residues. c Close up views of ribbon diagrams of the three structures of complexes between the Syt1 C2B domain (blue) and the SNARE four-helix bundle (green, red and yellow) that have been elucidated (Brewer et al., 2015; Zhou et al., 2015; Zhou et al., 2017) (PDB accession codes 2N1T, 5KJ7 and 5W5D, respectively). The structures were superimposed using the Syt1 C2B domain and only the C2B domain molecule corresponding to 5KJ7 is shown. The residues corresponding to the positions that were selected for mutation to cysteine for tagging with a tBu group (N319 and E346) are shown as orange spheres. Other side chains are shown by sticks. The Ca2+-binding sites of the C2B domain are located on the opposite end of the C2B domain β-sandwich, behind the plane of view, and are not observable in this diagram. d Ribbon diagram of the crystal structure of Cpx1(26-83) (pink) bound to the SNARE complex (synaptobrevin red, syntaxin-1 yellow, SNAP-25 green) (Chen et al., 2002) (PDB accession code 1KIL). The residues of syntaxin-1 and Cpx1(26-83) selected for mutation to cysteine and tagging with a tBu group (D214 and V61, respectively) are shown as purple and cyan spheres, respectively. Other side chains are shown by sticks.
We used these three reactions separately to attach tBu groups to the following fragments of Syt1 and complexin-1, all of which contained a single cysteine: i) two fragments spanning the Syt1 C2B domain (residues 270-421) that had the native cysteine mutated to alanine and contained either a N319C or a E346C mutation; ii) a fragment spanning both Syt1 C2 domains (C2AB) that had the native cysteine mutated to alanine and contained a E346C mutation; and iii) a fragment spanning residues 26-83 of complexin-1 and containing a V61C mutation. The N319C or a E346C mutations in the Syt1 C2B domain were chosen because they involve residues that are exposed on the surface and are not expected to interfere with binding to the SNARE complex in any of the three binding modes that have been described (Brewer et al., 2015; Zhou et al., 2015; Zhou et al., 2017) (Fig. 2C). Moreover, these residues are in regions that have not been implicated in membrane binding (Fernandez et al., 2001; Rufener et al., 2005) or membrane-membrane bridging (Arac et al., 2006). We also used the larger C2AB fragment with one of these two mutations (E346C) as a model to examine whether an increase in molecular weight affects the signal observed for the attached tBu group. The Cpx1(26-83) fragment is flexible in solution, with a mixture of helical and random coil conformation, and was previously co-crystallized with the SNARE complex (Chen et al., 2002). Cpx1(26-83) forms a long α-helix upon binding to the SNARE complex (Fig. 2D) and therefore offers the opportunity to examine how the expected restriction in the overall mobility of the fragment upon binding affects the tBu resonance.
All reactions were performed with 1–2 mM concentration of the corresponding reagent and using 15N-labeled proteins so that we could monitor the reactions through perturbations observed in 1H-15N HSQC spectra. In some cases, the reactions were complete when we acquired the first spectrum (ca. 1 hr) after adding the reagent, while other reactions were complete in several hours, as judged from the complete disappearance of selected cross-peaks that shift due to the presence of the tBu tag, and the appearance of new cross-peaks nearby. Fig. 3 shows examples of 1H-15N HSQC spectra acquired before adding the reagent and after the reaction was completed for selected cases, and spectra for almost all of the proteins are shown in Supplementary Figure 5. The spectra illustrate the cross-peak shifts caused by the reactions. Multiple cross-peaks of Cpx1(26-83) shifted (e.g. Fig. 3A), which can be attributed to the flexible nature of this fragment and suggests that the tBu tag alters the conformational ensemble to some extent. For the C2B domain and the C2AB fragment, the shifts caused by the reactions were more limited and involved only cross-peaks from residues that are very close to the single cysteine (e.g. Fig. 3B,C, Supplementary Figure 5). Below we refer to the purified tBu-tagged proteins using the abbreviated protein fragment name, the residue number corresponding to the single cysteine, and the suffix I, A or B for the reactions with the tBu iodoacetamide, acrylate or BDSNB, respectively [e.g. Cpx1(26–83)61I, Cpx1(26–83)61A or Cpx1(26–83)61B]. Fig. 4 shows 1D 1H NMR spectra of the four protein fragments tagged with the three different reactions and purified by gel filtration to remove the excess reagent. In each spectra we observed a sharp resonance (marked with a *) that is not present in the spectra of the untagged protein (Supplementary Figure 6) and can be attributed to the attached tBu group. This assignment was unambiguous for all the proteins except for C2B319B, as the sharp resonance attributed to the tBu group could overlap with a spurious sharp signal at 1.11 pm that was observed in some of the spectra. Table 1 lists the tBu signal positions, linewidths and signal-to-noise (N:S) ratios corresponding to these signals for the 12 protein fragments, for 1H NMR spectra of the three reagents and for other samples described below. These data need to be interpreted with caution, as the spectra were acquired over a period of two years as this project developed, and were not designed to perform a systematic analysis. Thus, spectra were acquired with different number of transients for signal averaging and different protein concentrations. Moreover, although we optimized the shims for each sample as we normally do for protein samples, we did not make any additional efforts to optimize the field homogeneity, which would be required for accurate measurement of linedwidths of resonances as sharp as those that we observed for the tBu groups in some of the samples. Nevertheless, a few firm conclusions can be drawn from the overall results.
Figure 3.

1H-15N HSQC spectra illustrating cross-peak shifts caused by tBu tagging. a-c 1H-15N HSQC spectra of Cpx1(26-83) V61C (a), Syt1 C2B domain E346C (b) and Syt1 C2AB E346C (c) before (black contours) and after (red contours) tagging with a tBu group via the acrylate (a), BDSNB (b) or iodoacetamide (c) reaction. Selected cross-peaks that shifted due to tagging with the tBu group are labeled. Although resonance assignments are available for WT Cpx1(26-83) (Trimbuch et al., 2014), assignment of cross-peaks in the middle of the spectrum of the V61C mutant is hindered by the multiple shifts caused by the mutation in this flexible, partially helical fragment (Chen et al., 2002; Pabst et al., 2000). The cross-peak of R67, which can be unambiguously assigned, is indicated in (a). Assignments for the Syt1 C2A and C2B domains are also available (Fernandez et al., 2001; Shao et al., 1998; Voleti et al., 2020). Assignments of selected cross-peaks that shifted because of attachment of the tBu group to residue 346 of the Syt1 C2B domain are indicated in (b, c).
Figure 4.

Gallery of 1D 1H NMR spectra of tBu-labeled presynaptic proteins. a-d Expansions corresponding to the methyl region of 1D 1H NMR spectra of Cpx1(26-83), Syt1 C2B domain and Syt1 C2AB tagged with a tBu group at different positions as indicated by the labels at the top right corner of each box. The protein concentrations and parameters of each spectrum are listed in Table 1. The resonance corresponding to the tBu group in each spectrum is indicated by a *.
It is clear that the tBu resonance was sharp for almost all the protein fragments, with line widths below 5 Hz and in some cases similar to those observed for the reagents (ca. 2 Hz), which have a much smaller molecular weight. The tBu signal was particularly prominent for the tagged Cpx1(26-83) fragment, which has a paucity of residues with methyl groups in its sequence. Even for the tagged C2B or C2AB fragments, which have a normal abundance of methyl-containing side chains, the tBu resonance had generally a higher intensity than the strongest side chain methyl group resonances at around 0.7-0.8 ppm. These observations show that the tBu group retained high mobility in all these proteins. However, the mobility appeared to be somewhat more limited for Cpx1(26-83)61, C2B346 and C2AB346 labeled with BDSNB compared to the same fragments labeled with tBu iodoacetamide or acrylate, as the tBu resonance of Cpx1(26-83)61B, C2B346B and C2AB346B was somewhat broader and its relative intensity with respect to the side chain methyl signals at 0.7-0.8 ppm was smaller. These observations suggest that labeling with BDSNB is more likely to lead to broader signals because of the closer proximity of the tBu group to the main chain, but the tBu group attached through a disulfide bond can still exhibit very high mobility in some positions, depending on the nearby environment on the protein surface.
To facilitate comparisons of the S:N ratios for the tBu resonances observed in the different spectra, we converted each measured S:N ratio to a normalized value calculated for a 10 μM concentration and 128 transients (S:N10μM), assuming that the S:N ratio is proportional to the concentration and to the square root of the number of transients. Although there was a natural variability in the calculated S:N10μM values (Table 1), it is clear that the values observed for the tBu-tagged proteins were generally comparable. The S:N10μM values calculated for Cpx1(26-83) were similar to those observed for the reagents and somewhat higher than those observed for the C2B and C2AB fragments, most likely because Cpx1(26-83) is smaller and flexible. Nevertheless, the overall data show that the 1H resonances of the tBu groups attached to two different surface exposed positions of the C2B or C2AB fragments by the three different reactions can be observed with high sensitivity at 10 μM concentrations in 3.4 minutes (corresponding to 128 transients with a 1.6 s recycling delay). These results suggest that tBu resonances should be observable even at nanomolar concentrations. To test this prediction, we acquired 1D 1H NMR spectra of 1 μM and 300 nM samples of Cpx1(26-83)61A in 1 and 3 hr, respectively. The sharp tBu resonance was readily observable in both samples, with S:N ratios of 25:1 and 8:1 for the 1 μM and 300 nM samples, respectively (Supplementary Figure 7a,b; Table 1). These ratios correspond to S:N10μM values of 65:1 and 36:1, respectively, which are lower than those calculated for 8 and 13 μM samples of Cpx1(26-83)61A (175:1 and 192:1; Table 1). This finding likely arises because proteins have a tendency to bind to glass and the percentage of protein bound to the NMR tube becomes substantial at low micromolar concentrations (Arac et al., 2003). Hence, it is likely that the actual concentrations of Cpx1(26-83)61A that remained in solution were considerably smaller than 1 μM and 300 nM, and that use of agents that prevent glass binding would yield even higher sensitivity. We note however that the intensity of the tBu resonance in the 300 nM sample was comparable to those of impurities that are typically present in buffers or NMR tubes, as shown by a 1D 1H NMR spectra of the buffer used for these experiments (Supplementary Figure 7c). Hence, studies at such low concentrations may benefit from the use of 13C-labeled tBu groups and acquisition of 1D 13C-edited 1H NMR spectra.
Analysis of tBu resonances in large presynaptic complexes
We next examined whether tBu resonances could still be observed for tBu groups attached to presynaptic proteins incorporated into complexes of increasing molecular weight (Figs. 5 and 6). First with attached a tBu probe to the SNARE motif of syntaxin-1 (residues 191-253) bearing a cysteine mutation at residue 214 using the iodoacetamide reaction (Syx214I). This syntaxin-1 fragment is unstructured in isolation (Dulubova et al., 1999) but becomes helical upon binding to the SNARE motifs of synaptobrevin and SNAP-25 to form the SNARE complex (Chen et al., 2002; Poirier et al., 1998; Sutton et al., 1998). The tBu signal of isolated Syx214I was sharp (2.7 Hz, Table 1) and became broader (9 Hz) but still readily observable upon incorporation into the SNARE complex (Fig. 5A, left and middle panels), indicating that the tBu group was partially immobilized but still retained substantial flexibility. We also acquired a 1D 1H NMR spectrum of an analogous sample in which the SNARE complex was additionally labeled with a DOTA-based tag called C2 (no relation to the term C2 domain) loaded with Dy3+ and did not observe the tBu signal (Fig. 5A, right panel), likely because the negative PCSs that are expected to be induced by this tag around residue 214 of syntaxin-1 (Pan et al., 2016) moved the tBu signal below the SNARE complex resonances at 0.6-0.8 ppm. It is also plausible that the tBu resonance was broadened by paramagnetic broadening, but the broadening is not expected to be strong because the distance between the tBu group and the center of the tensor is predicted to be more than 25 Å. Overall, these results illustrate that tBu groups can provide useful probes to measure PCSs at defined positions but at the same time emphasize the need to use methods that can distinguish the tBu resonance from other signals (e.g. if the tBu group is 13C-labeled).
Figure 5.

Gallery of 1D 1H NMR spectra of tBu-labeled presynaptic proteins and complexes. a Expansions corresponding to the methyl region of 1D 1H NMR spectra of Syx214I alone (25 μM, left) or incorporated into the SNARE complex (30 μM) without (middle, Syx214I-SC) or with a Dy3-C2 tag on residue 166 of SNAP-25 (Brewer et al., 2015) (right, Syx214I-SC-DY). b Expansions corresponding to the methyl region of 1D 1H NMR spectra of 18 μM Cpx1(26-83)61I in the presence of 21 μM SNARE complex (left), of 18 μM Cpx1(26-83)A in the presence of 21 μM SNARE complex (middle), and of 8 μM Cpx1(26-83)A in the presence of 12 Cpx1(26-83)A cis-SNARE complex anchored on nanodiscs through the synaptobrevin TM region (right). The inset on the right panel shows an expansion of the region corresponding to the tBu resonance plotted at a much larger vertical scale and with baseline correction to account for the slope of the large lipid resonances that overlap with the tBu signal. The parameters of each spectrum are listed in Table 1. The nanodiscs contained 85% POPC and 15% DOPS. The resonance corresponding to the tBu group in each spectrum is indicated by a *.
Figure 6.

Gallery of 1D spectra of tBu-tagged Syt1 C2B domain bound to nanodiscs and cis-SNARE complex nanodiscs. a Expansions corresponding to the methyl region of 1D 1H NMR spectra of PIP2-containing nanodiscs (PNDs), cis-SC-PNDs and dC2B319A or DC2B319I bound to PNDs in the presence of Ca2+. The * on the two right panels indicates the tBu signal overlapped with the strong resonances of the partially deuterated nanodiscs. b-e Expansions corresponding to the methyl region of 1D 1H NMR spectra of dC2B319A (b), dC2B319I (c), dC2B346A (d) or DC2B346I (e) alone or bound to PNDs in the presence of EGTA or Ca2+, or bound to cisSC-PNDs in the presence of EGTA. 1D 1H NMR spectra of PNDs or cisSC-PNDs (left panels in a) were subtracted from the spectra of the tBu-tagged dC2B domains acquired in the presence of PNDs or cisSC-PNDs, respectively. All spectra in each row were plotted at the same vertical scale so that signal intensities can be compared. The resonance corresponding to the chemical shift of the tBu group in each spectrum is indicated by a *. The concentrations of all tBu-tagged proteins were 7 μM, and those of PNDs or cisSC-PNDs were 14 μM. The lipid compositions of PNDs and cisSC-PNDs were 82% perdeuterated PC, 15% partially deuterated POPS and 3% PIP2. D2O was used as the solvent for each sample. The parameters of the spectra where the tBu resonance could be identified unambiguously are listed in Table 1.
We next tested how binding to the SNARE complex affects the tBu resonance attached via the iodoacetamide or acrylate reactions to residue 61 of Cpx1(26-83). We observed a slight broadening of the tBu signal in both cases, but they still remained intense and sharp [3.7 Hz line width, compared to 1.8 Hz in isolated Cpx1(26-83)61I or Cpx1(26-83)61A] (Table 1; Fig. 5B, left and middle panels). Importantly, when we analyzed a sample of 8 μM Cpx1(26-83)61A bound to SNARE complex that was anchored on nanodiscs through the synaptobrevin TM region (cisSC-NDs) (ca. 270 kDa), its tBu signal was still distinguishable within the envelop of the strong resonances of the nanodiscs around 1.2 ppm. Reliable measurement of the linewidth of the tBu signal in this sample was hindered by the overlap, but the tBu signal intensity above the baseline formed by the nanodisc resonances was about one fifth of the tBu intensity observed in the 1H NMR spectrum of a sample of 8 μM isolated Cpx1(26-83)61A ran side-by-side, which had a linewidth of 2.3 Hz (Table 1). Hence, although the immobilization of Cpx1(26-83)61A caused by binding to SC-NDs broadened the tBu resonance substantially, sufficient mobility remained to yield a signal that can be observed in 3.4 min for a ca. 280 kDa complex at 8 μM concentration.
Although the Syt1 C2B domain does not bind substantially to nanodiscs containing POPC:POPS 85:15 in the absence of Ca2+, inclusion of phosphatidylinositol 4,5-bisphosphate (PIP2) in the membrane induces Ca2+-independent binding (Bai et al., 2004). Interestingly, our recent studies suggested that PIP2 enhances Ca2+-independent interactions of the C2B domain with membrane-anchored SNARE complexes that underlie the primed state of synaptic vesicles, and Ca2+-dependent binding of the C2B domain to PIP2-containing membranes leads to dissociation from the SNARE complex as a key step to trigger neurotransmitter release (Voleti et al., 2020). These findings led us to examine whether the tBu resonances of the Syt1 C2B domain tagged at residue 319 or 346 with a tBu group through the iodoacetamide or acrylate reactions can still be observed upon Ca2+-independent and Ca2+-dependent binding to PIP2-containing nanodiscs (PNDs, ca. 240 kDa), or Ca2+-independent binding to analogous nanodiscs containing membrane-anchored SNARE complexes (cisSC-PNDs; ca. 270 kDa). To decrease the density of protons that might be near the tBu group and facilitate the observation of its resonance in 1H NMR spectra, these experiments were performed in D2O with uniformly perdeuterated, 15N-labeled C2B domain and some of the lipids used to prepare the nanodiscs were also deuterated.
1H NMR spectra of control samples containing isolated PNDs or cisSC-PNDs still exhibited strong resonances in the methyl region (Fig. 6A, two left panels), but sharp tBu resonances of dC2B319A or dC2B319I bound to PNDs were still observable on the shoulders of the nanodisc resonances near 1.2 or 1.1 ppm, respectively (e.g. Fig. 6A, two right panels). To facilitate analysis of these resonances, we subtracted the spectra of the isolated PNDs or cisSC-PNDs from those of the samples containing in addition tBu-tagged dC2B domain. These difference spectra are compared with the spectrum of the corresponding isolated dC2B domain in Fig. 6B-E, where the four spectra of each panel were plotted at the same vertical scale to allow direct comparison of the tBu resonance intensities. Relatively sharp tBu signals were observed for dC2B319A and dC2B319I bound to PNDs in the absence and presence of Ca2+ (Fig. 6B,C), and the signals exhibited S:N ratios in the 65-85 range for spectra acquired during 20 minutes (768 transients) despite the low protein concentration used in these experiment (7 μM protein). Much weaker and broader signals were observed at the corresponding tBu positions in 1H NMR spectra of dC2B319A or dC2B319I bound to cisSC-PNDs in the absence of Ca2+ (Fig. 6B,C), but we cannot be certain that the resonances indeed correspond to the tBu group. Similarly, very weak and broad signals were observed at the corresponding tBu positions for dC2B346A or dC2B346I bound to PNDs or cisSC-PNDs (Fig. 6D,E), but it is unclear whether they correspond to the tBu group. These results show that the tBu group on residue 319 of the C2B domain remains flexible upon binding to nanodiscs and hence does not contact the lipids, but the tBu group becomes substantially more immobilized when the C2B domain binds to nanodiscs containing anchored SNARE complex, suggesting that residue 319 contacts the SNAREs or that SNARE complex binding leads to rearrangements that induce contact of this residue with the lipids. In contrast residue 346 appears to contact the lipids even in the absence of the SNARE complex, perhaps because of the proximity of this residue to a polybasic region that binds to PIP2 (Bai et al., 2004).
The decrease in the intensity of the tBu signal of dC2B319A upon binding to PNDs was only about 50% (Fig. 6B), which shows that incorporation of the C2B domain into a 260 kDa complex can result in very limited broadening when the tBu group remains sufficiently mobile and contrasts with the stronger decreases in intensities that we observed for the methyl cross-peaks of 2H,13CH3-IM-C2AB upon Ca2+-dependent binding to nanodiscs (Fig. 1A,B). However, it is difficult to compare these effects because the broadening induced by nanodisc binding depends on the position of the methyl group. To allow a direct comparison of the benefits of methyl-TROSY effects to observe resonances in large complexes with those afforded by the tBu tagging approach, we labeled the cysteine sulfur atoms of perdeuterated, 15N-labeled N319C and E346C mutants with 13CH3 by reaction with 1 mM 13CH3 iodide. The reactions were complete after overnight incubation, as judged from perturbations observed in 1H-15N HSQC spectra. 1H-13C HMQC spectra of the purified proteins (dC2B319-Me and dC2B346-Me) revealed single cross-peaks that appeared near the cross-peaks of cysteine tagged with 13CH3 by the same procedure (Fig. 7A). The methyl cross-peak could be observed with reasonably good S:N in 1H-13C HMQC spectra of 7 μM dC2B319-Me and dC2B346-Me acquired in 6.1 and 14.6 hr, respectively, as shown by the 1H traces taken along the corresponding 13C chemical shift (top traces in Fig. 7B,C). However, the sensitivity is clearly much lower than that observed the tBu resonances of the tBu-tagged dC2B319 or dC2B346 fragments at the same concentration in 1D 1H NMR spectra acquired in just 20 min (Fig. 6B,E). Moreover, Ca2+-dependent binding to PNDs led to strong broadening of the 1H-13C HMQC cross-peak of dC2B319-Me and dC2B346-Me, which was barely detectable for the latter under these conditions (Fig. 7B,C, middle traces), and the cross-peak was broadened beyond detection for both proteins upon Ca2+-independent binding to cisSC-PNDs. Overall, these results show that the tBu tagging approach may be advantageous over methyl-TROSY-based methods for analyses of large macromolecular assemblies with limited solubility and/or stability.
Figure 7.

Methyl-TROSY of cysteine-tagged Syt1 C2B domain bound to nanodiscs and cis-SNARE complex nanodiscs. a Superposition of 1H-13C HMQC spectra of cysteine tagged at the S atom with 13CH3 (Cys-Me) (black contours), dC2B319-Me (cyan contours) and dC2B346-Me (red contours). b,c One dimensional traces taken along the 1H dimension at the 13C chemical shift corresponding to the 13CH3 of 7 μM dC2B319-Me (b) or dC2B346-Me (c) alone or in the presence of 14 μM PNDs and 1 mM Ca2+, or 14 μM cisSC-PNDs and 1 mM EGTA. The lipid compositions of PNDs and cisSC-PNDs were 82% perdeuterated PC, 15% partially deuterated POPS and 3% PIP2. SNARE complexes were anchored through the synaptobrevin TM region. All spectra were acquired in 6.1 hr (b) or 14.6 hr (c) using D2O was used as the solvent, and the three traces in each panel were plotted at the same vertical scale.
Discussion
Analysis of weak protein interactions on membranes or between two membranes is very challenging, as illustrated by ongoing research on the mechanisms underlying intracellular membrane fusion in general and neurotransmitter release in particular (Rizo, 2018). NMR spectroscopy provides a powerful tool to study such systems, particularly through the use of methyl-TROSY based approaches that offer high sensitivity even for large macromolecular assemblies (Rosenzweig and Kay, 2014). However, application of such approaches is hindered when sample stability and/or solubility is limited. Strategies based on attaching tBu groups to proteins constitute promising alternative tools to obtain structural information on large complexes using solution NMR spectroscopy [e.g. (Chen et al., 2015; Jabar et al., 2017)]. Here we have further explored this methodology using proteins involved in neurotransmitter release and their complexes as benchmarks. Our results show that tBu groups can be observed with very high sensitivity even when attached to complexes of over 200 kDa. With some technical improvements that we discuss below, this methodology might be applicable to study biomolecular complexes of any molecular weight at low micromolar and even nanomolar concentrations.
It is noteworthy that our 1D 1H spectra were acquired on a 600 MHz Agilent spectrometer equipped with a 14-year old cold probe (S:N 4,500:1 in the standard ethylbenzene assay). Thus, tBu signals should be observable with even higher sensitivity using higher fields and more modern cryoprobes. The sensitivity of this approach is much higher than that offered by methyl-TROSY experiments or by approaches involving the introduction of specific 19F-labeled tags on proteins, which have been successfully used in multiple applications to large biomolecular systems, including membrane proteins (Rose-Sperling et al., 2019). Clearly, methyl TROSY-based approaches have a big advantage over the use of tBu tags in that they can provide information on multiple methyl groups of a protein in the same spectrum and hence they do not require preparation of one mutant protein for each probe position. However, the high sensitivity of the tBu-based approach is advantageous for studies of samples with limited solubility and/or stability, and requires very small amounts of protein, which dramatically reduces the cost of preparing each mutant. It is also worth noting that streamlined production of multiple mutants of a protein has been extensively used in electron paramagnetic resonance studies involving site-directed spin labeling (Garcia-Rubio, 2020).
Retaining high mobility is critical for tBu groups attached to large proteins or complexes to exhibit sharp signals and hence to be observable with high sensitivity. The use of different chemical linkages to attach the tBu group, as described here, offers different options to retain such high mobility. The attachment through a disulfide bond using BDSNB leads to the shortest chemical link to the protein and hence would be advantageous for the purpose of deriving structural information because of the lower uncertainty regarding the location of the tBu. However, the shorter link is also expected to lead to a lower probability for high mobility, as the tBu is more likely to pack with nearby atoms of the protein, even if it is only transiently. Our results confirm that, although it is possible to observe very sharp resonances for tBu groups attached with BDSNB (e.g. for C2B319E), the resonances of tBu groups attached through the iodoacetamide or acrylate reactions are more like to remain sharp (Table 1), most likely because the longer chemical linkages to the protein favor higher mobility. In general, it is expected that protein perdeuteration should help to keep tBu resonances sharp, but perdeuteration may not be necessary when very high mobility is retained (e.g. for Cpx1(26-83)61A bound to cisSC-NDs; Fig. 5B). A priori, it may be difficult to predict which surface residues may be more appropriate for mutation to cysteine and tagging with a tBu group that could retain high mobility. While visual inspection might be helpful, analyses by molecular dynamics simulations are likely to be very useful to predict mobility and the likelihood of packing against nearby groups in the surface.
Identification of the resonances of tBu groups attached to proteins is somewhat hindered by overlap with protein resonances, but in general we were able to readily distinguish the tBu resonances in the examples shown in this study. However, the tBu resonance of Syx241C-SC-DY could not be distinguished (Fig. 5A), either because it overlapped with protein resonances due to the pseudocontact shift induced by the lanthanide or because of paramagnetic broadening. Moreover, the overlap with lipid signals strongly hinders the identification of tBu resonances. In samples with even lower concentrations than those used in our studies, tBu resonances might also be difficult to distinguish from those of sharp spurious signals likely arising from sample or buffer impurities (e.g. see spectra of Figs. 4-6). From this perspective, the trimethylsilyl groups could offer a clear advantage over the tBu group because it appears in a much less crowded region of 1D 1H NMR spectra, near 0 ppm (Becker et al., 2018; Jabar et al., 2017). Alternatively, the tBu group can be 13C-labeled to facilitate identification of its resonance using one dimensional 1H-13C HMQC or HSQC spectra.
In summary, the use of tBu (or potentially trimethylsilyl) tags offers a promising avenue for structural studies of protein complexes that are difficult to study by other methods because of limited stability and/or solubility. The observation of resonances from these tags attached to different positions should allow the measurement of parameters that provide structural information such as PCSs induced by lanthanides attached to other positions of the complex. Conversely, disappearance of the resonance of a tBu group attached to a protein upon binding to a target can inform on the surface involved in binding. This approach can also be readily adapted for structural studies of DNA, RNA and their complexes, thus providing a versatile tool to study biomolecular assemblies.
Supplementary Material
Acknowledgments
We thank Ad Bax for the suggestion of exploring the use tBu groups as probes for structural studies of the neurotransmitter release machinery, and Ad Bax, Lewis Kay and Charampalos Kalodimos for fruitful discussions on this subject. The Agilent DD2 console of the 800 MHz spectromenter used for the research presented here was purchased with a shared instrumentation grant from the NIH (S10OD018027 to JR). Rashmi Voleti was supported by a fellowship from the Howard Hughes Medical Institute. The preparation of BDSNB was performed at the NANBIOSIS –CIBER BBN Peptide Synthesis Unit (U3). This work was supported by grant I-1304 from the Welch Foundation (to JR) and by NIH Research Project Award R35 NS097333 (to JR).
Data availability statement
All the data associated with this manuscript are available upon reasonable request.
References
- Abdelkader EH, Qianzhu H, Tan YJ, Adams LA, Huber T, Otting G (2021) Genetic Encoding of N(6)-(((Trimethylsilyl)methoxy)carbonyl)-l-lysine for NMR Studies of Protein-Protein and Protein-Ligand Interactions. J Am Chem Soc 143:1133–1143 [DOI] [PubMed] [Google Scholar]
- Arac D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Sudhof TC, Rizo J (2006) Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol 13:209–217 [DOI] [PubMed] [Google Scholar]
- Arac D, Murphy T, Rizo J (2003) Facile detection of protein-protein interactions by one-dimensional NMR spectroscopy. Biochemistry 42:2774–2780 [DOI] [PubMed] [Google Scholar]
- Bai J, Tucker WC, Chapman ER (2004) PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol 11:36–44 [DOI] [PubMed] [Google Scholar]
- Becker W, Adams LA, Graham B, Wagner GE, Zangger K, Otting G, Nitsche C (2018) Trimethylsilyl tag for probing protein-ligand interactions by NMR. J Biomol NMR 70:211–218 [DOI] [PubMed] [Google Scholar]
- Brewer KD, Bacaj T, Cavalli A, Camilloni C, Swarbrick JD, Liu J, Zhou A, Zhou P, Barlow N, Xu J, Seven AB, Prinslow EA, Voleti R, Haussinger D, Bonvin AM, Tomchick DR, Vendruscolo M, Graham B, Sudhof TC, Rizo J (2015) Dynamic binding mode of a Synaptotagmin-1-SNARE complex in solution. Nat. Struct. Mol. Biol 22:555–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WN, Kuppan KV, Lee MD, Jaudzems K, Huber T, Otting G (2015) O-tert-Butyltyrosine, an NMR tag for high-molecular-weight systems and measurements of submicromolar ligand binding affinities. J Am Chem Soc 137:4581–4586 [DOI] [PubMed] [Google Scholar]
- Chen WN, Nitsche C, Pilla KB, Graham B, Huber T, Klein CD, Otting G (2016) Sensitive NMR Approach for Determining the Binding Mode of Tightly Binding Ligand Molecules to Protein Targets. J Am Chem Soc 138:4539–4546 [DOI] [PubMed] [Google Scholar]
- Chen X, Arac D, Wang TM, Gilpin CJ, Zimmerberg J, Rizo J (2006) SNARE-Mediated Lipid Mixing Depends on the Physical State of the Vesicles. Biophys. J 90:2062–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Lu J, Dulubova I, Rizo J (2008) NMR analysis of the closed conformation of syntaxin-1. J. Biomol. NMR 41:43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Tomchick DR, Kovrigin E, Arac D, Machius M, Sudhof TC, Rizo J (2002) Three-dimensional structure of the complexin/SNARE complex. Neuron 33:397–409 [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) Nmrpipe - A Multidimensional Spectral Processing System Based on Unix Pipes. Journal of Biomolecular Nmr 6:277–293 [DOI] [PubMed] [Google Scholar]
- Denisov IG, Baas BJ, Grinkova YV, Sligar SG (2007) Cooperativity in cytochrome P450 3A4: linkages in substrate binding, spin state, uncoupling, and product formation. J Biol Chem 282:7066–7076 [DOI] [PubMed] [Google Scholar]
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Sudhof TC, Rizo J (1999) A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J 18:4372–4382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Sudhof TC (2001) Synaptotagmin I functions as a calcium regulator of release probability. Nature 410:41–49 [DOI] [PubMed] [Google Scholar]
- Fernandez I, Arac D, Ubach J, Gerber SH, Shin O, Gao Y, Anderson RG, Sudhof TC, Rizo J (2001) Three-dimensional structure of the synaptotagmin 1 c(2)b-domain. Synaptotagmin 1 as a phospholipid binding machine. Neuron 32:1057–1069 [DOI] [PubMed] [Google Scholar]
- Garcia-Rubio I (2020) EPR of site-directed spin-labeled proteins: A powerful tool to study structural flexibility. Arch Biochem Biophys 684:108323. [DOI] [PubMed] [Google Scholar]
- Giralt E, Rizo J, and Pedroso E. 1984. Application of Gel-Phase 13C-NMR to monitor Solid Phase Peptide Synthesis. Tetrahedron 40: 4141–4152. [Google Scholar]
- Graham B, Loh CT, Swarbrick JD, Ung P, Shin J, Yagi H, Jia X, Chhabra S, Barlow N, Pintacuda G, Huber T, Otting G (2011) DOTA-amide lanthanide tag for reliable generation of pseudocontact shifts in protein NMR spectra. Bioconjug. Chem 22:2118–2125 [DOI] [PubMed] [Google Scholar]
- Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE (1997) Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell 90:523–535 [DOI] [PubMed] [Google Scholar]
- Hu W, Wang H, Hou Y, Hao Y, Liu D (2019) Trimethylsilyl reporter groups for NMR studies of conformational changes in G protein-coupled receptors. FEBS Lett 593:1113–1121 [DOI] [PubMed] [Google Scholar]
- Huang C, Kalodimos CG (2017) Structures of Large Protein Complexes Determined by Nuclear Magnetic Resonance Spectroscopy. Annu Rev Biophys 46:317–336 [DOI] [PubMed] [Google Scholar]
- Jabar S, Adams LA, Wang Y, Aurelio L, Graham B, Otting G (2017) Chemical Tagging with tert-Butyl and Trimethylsilyl Groups for Measuring Intermolecular Nuclear Overhauser Effects in a Large Protein-Ligand Complex. Chemistry 23:13033–13036 [DOI] [PubMed] [Google Scholar]
- Johnson BA, Blevins RA (1994) Nmr View - A Computer-Program for the Visualization and Analysis of Nmr Data. Journal of Biomolecular Nmr 4:603–614 [DOI] [PubMed] [Google Scholar]
- Liu Q, He QT, Lyu X, Yang F, Zhu ZL, Xiao P, Yang Z, Zhang F, Yang ZY, Wang XY, Sun P, Wang QW, Qu CX, Gong Z, Lin JY, Xu Z, Song SL, Huang SM, Guo SC, Han MJ, Zhu KK, Chen X, Kahsai AW, Xiao KH, Kong W, Li FH, Ruan K, Li ZJ, Yu X, Niu XG, Jin CW, Wang J, Sun JP (2020) DeSiphering receptor core-induced and ligand-dependent conformational changes in arrestin via genetic encoded trimethylsilyl (1)H-NMR probe. Nat Commun 11:4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh CT, Adams LA, Graham B, Otting G (2018) Genetically encoded amino acids with tert-butyl and trimethylsilyl groups for site-selective studies of proteins by NMR spectroscopy. J Biomol NMR 71:287–293 [DOI] [PubMed] [Google Scholar]
- Ma C, Li W, Xu Y, Rizo J (2011) Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat. Struct. Mol. Biol 18:542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Su L, Seven AB, Xu Y, Rizo J (2013) Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339:421–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Missler M, Li C, Sudhof TC (1995) Complexins: cytosolic proteins that regulate SNAP receptor function. Cell 83:111–119 [DOI] [PubMed] [Google Scholar]
- Pabst S, Hazzard JW, Antonin W, Sudhof TC, Jahn R, Rizo J, Fasshauer D (2000) Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J. Biol. Chem 275:19808–19818 [DOI] [PubMed] [Google Scholar]
- Pan YZ, Quade B, Brewer KD, Szabo M, Swarbrick JD, Graham B, Rizo J (2016) Sequence-specific assignment of methyl groups from the neuronal SNARE complex using lanthanide-induced pseudocontact shifts. J Biomol NMR 66:281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier MA, Xiao W, Macosko JC, Chan C, Shin YK, Bennett MK (1998) The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat. Struct. Biol 5:765–769 [DOI] [PubMed] [Google Scholar]
- Rizo J (2018) Mechanism of neurotransmitter release coming into focus. Protein Sci 27:1364–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Chen X, Arac D (2006) Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol 16:339–350 [DOI] [PubMed] [Google Scholar]
- Rizo J, Rosen MK, Gardner KH (2012) Enlightening molecular mechanisms through study of protein interactions. J. Mol. Cell Biol 4:270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose-Sperling D, Tran MA, Lauth LM, Goretzki B, Hellmich UA (2019) 19F NMR as a versatile tool to study membrane protein structure and dynamics. Biol Chem 400:1277–1288 [DOI] [PubMed] [Google Scholar]
- Rosenzweig R, Kay LE (2014) Bringing dynamic molecular machines into focus by methyl-TROSY NMR. Annu. Rev. Biochem 83:291–315 [DOI] [PubMed] [Google Scholar]
- Rufener E, Frazier AA, Wieser CM, Hinderliter A, Cafiso DS (2005) Membrane-bound orientation and position of the synaptotagmin C2B domain determined by site-directed spin labeling. Biochemistry 44:18–28 [DOI] [PubMed] [Google Scholar]
- Shao X, Fernandez I, Sudhof TC, Rizo J (1998) Solution structures of the Ca2+-free and Ca2+-bound C2A domain of synaptotagmin I: does Ca2+ induce a conformational change? Biochemistry 37:16106–16115 [DOI] [PubMed] [Google Scholar]
- Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE (1993) A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75:409–418 [DOI] [PubMed] [Google Scholar]
- Sutton RB, Davletov BA, Berghuis AM, Sudhof TC, Sprang SR (1995) Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell 80:929–938 [DOI] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395:347–353 [DOI] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Sudhof TC (2006) A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126:1175–1187 [DOI] [PubMed] [Google Scholar]
- Trimbuch T, Xu J, Flaherty D, Tomchick DR, Rizo J, Rosenmund C (2014) Re-examining how complexin inhibits neurotransmitter release. elife 3:e02391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugarinov V, Sprangers R, Kay LE (2004) Line narrowing in methyl-TROSY using zero-quantum 1H-13C NMR spectroscopy. J. Am. Chem. Soc 126:4921–4925 [DOI] [PubMed] [Google Scholar]
- Ubach J, Zhang X, Shao X, Sudhof TC, Rizo J (1998) Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J 17:3921–3930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voleti R, Jaczynska K, Rizo J (2020) Ca(2+)-dependent release of Synaptotagmin-1 from the SNARE complex on phosphatidylinositol 4,5-bisphosphate-containing membranes. Elife 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Brewer KD, Perez-Castillejos R, Rizo J (2013) Subtle Interplay between Synaptotagmin and Complexin Binding to the SNARE Complex. J. Mol. Biol 425:3461–3475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A, Brewer KD, Rizo J (2013) Analysis of SNARE complex/synaptotagmin-1 interactions by one-dimensional NMR spectroscopy. Biochemistry 52:3446–3456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB, Brewster AS, Sauter NK, Cohen AE, Soltis SM, onso-Mori R, Chollet M, Lemke HT, Pfuetzner RA, Choi UB, Weis WI, Diao J, Sudhof TC, Brunger AT (2015) Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature 525:62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Zhou P, Wang AL, Wu D, Zhao M, Sudhof TC, Brunger AT (2017) The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 548:420–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data associated with this manuscript are available upon reasonable request.