

Summary
In this report, we provide evidence showing diminished expression of the mineral dust-induced gene (mdig), a previously identified oncogenic gene, in human triple negative breast cancer (TNBC). Using a mouse model of orthotopic xenograft of the TNBC MDA-MB-231 cells, we demonstrate that mdig promotes the growth of primary tumors but inhibits metastasis of these cells in vivo. Knockout of mdig resulted in an enhancement of H3K36me3 in the genome and upregulation of some X chromosome-linked genes for cell motility, invasion, and metastasis. Silencing MAGED2, one of the most upregulated and H3K36me3-enriched genes resulted from mdig depletion, can partially reverse the invasive migration of the mdig knockout cells. The anti-metastatic and inhibitory role of mdig on H3K36me3 was cross-validated in another cell line, A549 lung cancer cells. Together, our data suggest that mdig is antagonist against H3K36me3 that enforces expression of genes, such as MAGED2, for cell invasion and metastasis.
Subject areas: Molecular biology, Molecular mechanism of gene regulation, Cancer
Graphical abstract
Highlights
-
•
Loss of mdig expression in TNBC and metastatic breast cancer
-
•
Knockout of mdig enforces metastasis of the TNBC cells
-
•
Mdig antagonizes H3K36me3 that promotes expression of X-linked metastatic genes
-
•
Silencing MAGED2 reduces invasive migration of the mdig knockout cells
Molecular biology; Molecular mechanism of gene regulation; Cancer
Introduction
Breast cancer is the most common cancer among women and is the second leading cause of cancer related deaths after lung cancer. Advanced tumor growth and metastasis remain the major reason that approximately 70% of the breast cancer patients are poor candidates for surgical intervention (Tao et al., 2011). The metastatic dissemination of breast cancer cells is a complex cascade comprised of several components pertaining to genetics, epigenetics, and therapeutic responses that are involved in local invasion, intravasation, and extravasation of the tumor cells in circulation, embedding in the regional lymph nodes and/or distal organs, and interactions with the non-cancerous neighboring cells in the microenvironment of the primary and metastatic tumors (Polyak, 2011). Using molecular profiling approaches, some studies addressed distinct mutational spectra between the primary and metastatic breast cancers lesions (Yates et al., 2017; Zhao and Powers, 2017). However, challenges remain in the interpretation of the data from such studies because of the extensive genomic heterogeneity in both primary and metastatic tumors and patient-to-patient variability in genomic profiles, as well as the discordances introduced by the different subtypes of breast cancers, such as luminal A/B breast cancer, Her2-enriched breast cancer, and triple negative breast cancer (TNBC). Hence, additional measures are required to define the critical biomarkers and their implication in breast carcinogenesis and metastasis for the development of robust breast cancer therapies.
It has long been suspected that multiple driver mutations occur on the trajectory of metastatic evolution of the primary tumor cells, despite several studies having revealed a lack of metastasis-specific driver mutations in human cancer (Hu et al., 2020; Reiter et al., 2018; Turajlic and Swanton, 2016), which opens up the possibility that specific epigenetic landscapes or the unique pattern of chromatin remodeling may be critical for the acquisition of metastatic potential of the cancer cells. Emerging evidence suggests that changes in the epigenetic landscapes on chromatin not only affect the overall dynamics of gene expression but determine the cell lineage commitment, differentiation, or plasticity also. A recent report by Cai et al. (2020) using integrated epigenomic analysis uncovered a distinct open chromatin status in the breast cancer cells that metastasized to the lung or brain. It was believed that such a chromatin reconfiguration created a metastasis specific access of transcription factors (TFs) for the transcription of metastasis-related genes. However, the question of how metastasized cells achieve such a chromatin configuration remains to be characterized.
A mineral dust-induced gene (mdig) was identified from human glioblastoma cell line T98G engineered with c-Myc overexpression and from coal miners’ alveolar macrophages through differential display RT-PCR, respectively (Tsuneoka et al., 2002; Zhang et al., 2005, 2019a). The protein product of the mdig gene contains a conserved JmjC domain that was found among a number of histone demethylases. Several lines of evidence unraveled the properties of mdig in promoting cell proliferation, immune regulation, pulmonary inflammation, epigenetic regulation, and neoplasia (Thakur et al., 2015; Zhang et al., 2019a, 2020). Elevated expression of mdig was frequently observed in many different types of human cancers, suggesting an oncogenic-like role of mdig in malignancies. In human breast cancer, high expression of mdig is associated with poor overall survival of the patients. However, in patients with lymph node metastasis, a high level of mdig predicted better overall survival (Thakur et al., 2014). A further detailed study that stratified different disease stages found that mdig expression was lost from aggressive or late stage breast cancers and that mdig exhibited inhibitory activity on the invasion and metastasis of the cancer cells (Thakur et al., 2018). Likewise, a recent investigation of mdig in human lung cancer also revealed that mdig is strongly positive in earlier stage lung cancer, but the expression of mdig tends to decrease with an increase of tumor grades and staging (Shi et al., 2021). It is very likely, thus, that the role of mdig in human cancer may be dependent on the context of the tumor stages. In the present report, we tried to understand how mdig affects the metastatic potential of the TNBC MDA-MB-231 cells through approaches of orthotopic mouse breast cancer model, CRISPR-Cas9 gene editing, quantitative proteomics, and global ChIP-seq analysis. Our data suggest that knockout of mdig facilitates the acquisition of the metastatic potential of the cancer cells through an enhanced enrichment of histone H3 lysine 36 trimethylation (H3K36me3) on the genome and some specific genes, including IQSEC2, NMNAT1, MAGED2, HGFBP7, LAMP2, CTSD, and so on, that are involved in cell motility, invasion, and metastasis. Silencing of melanoma-associated antigen D2 (MAGED2), one of the most upregulated X-linked genes in the mdig knockout cells, can partially reverse the capability of invasive migration of the cells that have mdig depletion. Taken together, this study demonstrated a distinct biological property of mdig and H3K36me3 in tumor metastasis, a previously unrecognized mechanism of tumor progression, which may serve as actionable targets to treat patients with later stage breast cancer.
Results
Clinical features of the breast cancer with different expression status of mdig
Our earlier studies revealed a distinctive expression pattern of mdig among different histological subtypes of human breast cancer and noted the highest expression of mdig in ductal carcinoma in situ and the mucinous breast cancer (Thakur et al., 2018). To further characterize the clinical relevance of mdig in breast cancer, we evaluated the protein level of mdig through immunohistochemistry (IHC) for 98 human breast cancer samples classified by the hormone expression status. Among 50 hormone positive breast cancer tissues (ER+, PR+ and HER2+), the positivity rate of mdig is 54% (Figure 1A). The positivity rate of mdig, however, is much lower in 48 triple negative breast cancer (TNBC). Only 18% of the TNBC cases showed detectable expression of mdig, whereas 82% of these cancer samples are negative for mdig expression. These observations, accordingly, clearly indicate that loss of mdig expression may be a common feature of the more aggressive breast cancers, such as TNBC.
Figure 1.

Loss of mdig expression in triple negative breast cancer (TNBC)
(A) Immunohistochemistry (IHC) of mdig expression in hormone positive breast cancer and TNBC. Both groups of tumors contain mdig positive and mdig negative cancer tissue samples. In hormone positive group, 54% of tumor samples are mdig positive. In TNBC, only 18% of these samples are mdig positive. Scale bar = 50 μm, magnification is 40×.
(B) Kaplan-Meier overall survival (OS) analysis of 1,879 cases of breast cancer with high and low mdig expression. Right two panels show the prognostic outcomes of mdig expression in 201 TNBC cases. Both probes of mdig in the database are shown.
(C) High level of mdig predicts poor survival of the patients without lymph node metastasis (left two panels) but predicts better survival of the patients with lymph node metastasis (right two panels).
To additionally address the clinical relevance of mdig in human breast cancer, we also analyzed transcriptomics data linked to the overall survival of 1,879 breast cancer patients. When data from all breast cancer subtypes were pooled in a period of 180 months after diagnosis, a high expression of mdig correlated with poor survival outcome (Figure 1B). If we restricted our analysis on 201 breast cancer patients who were ER IHC negative, PR IHC negative, and HER2 array negative, both mdig probes in the database predicted that a high level of mdig is associated with a prolonged survival of these TNBC patients, whereas a low expression of mdig has a significantly worse prognosis (Figure 1B, right two panels). Because TNBC tends to be more metastatic and aggressive, we next stratified breast cancers based on the status of lymph node metastasis. Kaplan-Meier survival estimation showed that the lymph node negative patients with high mdig expression had a short overall survival. In contrast, the lymph node positive patients with high mdig expression exhibited a much improved overall survival (Figure 1C). These findings, thus, clearly suggested that a high mdig expression was a significantly unfavorable prognostic factor for hormone positive and non-metastatic breast cancer patients, and vice versa, a high mdig expression in TNBC and breast cancer with lymph node metastasis unexpectedly correlates with an increased probability of survival.
Loss of mdig impaired primary breast tumor formation in NSG mice
Knockout (KO) of mdig gene in MDA-MB-231 cells through CRISPR-Cas9 gene editing was reported previously (Zhang et al., 2020). To understand the role of mdig in tumor development and metastasis, we injected wide type (WT) or mdig KO MDA-MB-231 cells into the fourth and fifth mammary fat pads of the severely immunocompromised NSG mice. Tumors were measured once a week, beginning 15 days after implantation. Mice were sacrificed for analysis after 7 weeks and primary mammary tumors were dissected and measured. As expected, the injected KO cells generated smaller mammary tumors compared to their WT counterparts (Figure 2A, right panels). In comparison with WT tumors, tumors from the KO cells were significantly smaller in size at every measuring time point (Figure 2A, bottom left panel). This indicates that mdig is essential for breast tumorigenesis and loss of mdig is sufficient to impair tumor formation in vivo. This also suggests that mdig is important for the early stages of tumor development, a stage at which mdig is likely to serve as a pro-proliferative factor for the cancer cells.
Figure 2.

Breast tumor formation in NSG mice injected with WT and mdig KO MDA-MB-231 cells
(A) Photographs of the freshly resected mammary tumors from the mice injected with WT and KO cells, respectively. The WT cells form large tumors than the KO cells. Lower panels show quantification of the tumor size. Data are represented as mean ± SD. Tumors from the KO cells not only grow much slower (left panel) but are significantly smaller in size at the end of experiments (right panel). Only three tumors from each group of mice were shown.
(B) Photographs of the resected axillary and lumbar lymph nodes from the mice received with WT and KO cells. An overall enlargement of lymph nodes in the mice received with KO cells was observed. Bottom panel shows the summary of the tumor size, lymph node and the visually observed metastasis in the mice received with WT and KO cells.
Unexpectedly, however, examination of lymph nodes revealed a striking difference in lymph node size and the number between the mice injected with WT and KO MDA-MB-231 cells. In mice inoculated with KO cells, the axillary and lumbar lymph nodes are enlarged (Figure 2B). Also, metastasis was detected by visual exam in two of the five KO cell-inoculated mice where the pancreas and upper region of the abdominal cavity near the lung showed abnormal tissue mass (summarized in bottom panel of Figure 2B). These data indicate that mdig is important during the initial events of tumorigenesis but may negatively regulate the metastasis of breast cancer cells. This notion corroborates our previous findings showing that mdig serves as a pro-proliferative factor for lung (Yu et al., 2014) and breast cancer (Thakur et al., 2018) but acts as a negative regulator for the motility and invasion of the cancer cells (Yu et al., 2014). In other words, mdig may have different roles during the early and later phases of cancer development. In the early phase, mdig enforces the proliferation and survival of the cancer cells, and in the later phase, mdig may be inhibitory for the migration and invasion of the cancer cells.
Deficiency in mdig enhanced the metastatic propensity of breast cancer cells
After having determined that the loss of mdig can impair primary tumor formation, we next evaluated the influence of mdig on the metastasis of the MDA-MB-231 cells. For this, paraffin embedded lung, lymph node, and liver tissues from the mice that received WT or KO cells were sectioned and stained for H&E and Cytokeratin 18 (Figure S1). H&E staining of the primary tumors and the secondary organs revealed the presence of tumor cells with no distinct difference between the WT and KO categories (Figure 3, upper panel). Lesions harboring abnormal cell morphology in comparison to the resident cells of the host tissue were observed in the lymph nodes, lung, and liver, which is suggestive of the presence of tumor cells. When these tissues were stained with Cytokeratin 18, all of them were found to be positive in some regions for the marker indicating the presence of metastatic lesions. We identified several micro-metastases in the lung, liver, and lymph nodes of both groups of mice injected with WT and KO cells, respectively (Figure S1). Of interest, compared with the livers from mice inoculated with WT cells, the livers from the KO category showed a significant increase in the number of metastatic lesions. However, no difference was observed in the lung tissues between these two groups of mice (Figure 3, lower panel). These results are suggestive of an increased ongoing micro metastasis in the livers of the KO cells.
Figure 3.

Histological evaluation of the primary tumors and distant organs for metastasis
Paraffin embedded tissue sections were stained for H&E and microscopic examination of the tumors formed by WT and KO MDA-MB-231 cells. Primary tumors as well as tumor cells in the lymph nodes, lung, and liver tissues were shown. Dashed line circles show metastasized tumors in lymph node, lung and liver. Images are representatives from 5 mice received WT cells and 5 mice received KO cells. T: Tumor. Scale bar = 50 μm, magnification is 40× except for lymph nodes which is 20×. Bottom panels show the quantification of the cytokeratin 18 positive lesions in the liver (left) and the lung (right). Data are represented as mean ± SD.
Distinct proteomic profiles between WT and KO cells
The above data clearly indicate an enhanced metastatic potential of the cells in which mdig was depleted through genetic gene editing. To gain insight into how mdig is involved in the metastasis of the cancer cells, we next performed quantitative proteomics analysis through LC-MS/MS of 5 WT clones and 12 KO clones of the MDA-MB-231 cells. The raw data were analyzed to determine the fold change (FC) of proteins as a normalized ratio for KO compared to WT cells. In addition to considering the pvalue, we arbitrarily set a 2-fold t-Statistic cutoff of the differentially expressed proteins between two groups. Relative to the WT cells, the KO cells exhibited an increased expression of 886 proteins and decreased expression of 980 proteins (Figure 4A). The top 25 down- and upregulated proteins, all of which showed a more than 5-fold differences between two groups of the cells, are listed in the lower panel of Figure 4A. A strong enrichment of cell motility- or metastasis-related genes was noted among those top upregulated genes in the KO cells. Ontology analysis for molecular function suggested that downregulated proteins in KO cells are mostly involved in rRNA processing, rRNA metabolism, fatty acid oxidation, and biogenesis of ribosome (Figure 4B, upper panel). This result is consistent with the notion that mdig is normally localized in nucleoli and associated with the ribosomal dynamics. The upregulated proteins in the KO cells, on the other hand, are mostly in the pathways of binding to cadherin, actin, ubiquitin, GTPase, etc. (Figure 4B, lower panel), suggesting that the proteins contributing to the cell motility or invasion were increased in the KO cells. Among the top-25 most upregulated proteins in the KO cells, 9 proteins are encoded by the genes located in chromosome X, including WASH6P, IQSEC2, MAGED2, HUWE1, PLAS3, TCEAL3, UPF3B, LAMP2, and CUL4B (marked with X in Figure 4A), which possibly indicated a previously unidentified role of chromosome X in cancer cell metastasis. In addition, 4 of the top-25 upregulated proteins in KO cells are known lysosomal proteins that potentially contribute to autophagy and the remodeling of extracellular matrix during cancer cell invasion and migration (marked with ∗ in Figure 4A).
Figure 4.

Quantitative proteomics analysis for protein expression between WT and KO MDA-MB-231 cells
(A) Volcano plot shows increased and decreased proteins in the mdig KO cells (upper panel). Bottom panel shows top-25 the most downregulated (left) and upregulated proteins (right) in the KO cells. Gene names with red color are those genes with decreased enrichment of H3K36me3. Gene names with green color are the genes with an enhanced enrichment of H3K36me3 in the mdig KO cells. Asterisks indicate proteins located in lysosomes. X denotes the proteins that are encoded by genes located in chromosome X.
(B) Ontology GO molecular function assay of the downregulated proteins (upper panel) and the upregulated proteins (bottom panel) in the KO cells.
The mdig KO cells are more invasive with perplexing expression of epithelial-mesenchymal transition (EMT) proteins
EMT is generally viewed as one of the key features of metastatic cells. Despite proteomics data suggested an increased expression of proteins supporting cell motility, it is unclear whether the KO cells acquired properties of EMT and an enhanced metastasis resulted from mdig depletion. One of our earlier studies suggested a significant morphological change of the epithelial cells following overexpression of the JmjC deletion-mutated mdig, such as elongated fibroblast-like shape of the cells, increased formation of membrane ruffles and protrusions (Yu et al., 2014). This notion was supported by the mesenchymal-like features of the mdig KO cells (Figure 5A). All WT cells have flat and cuboidal-like shape, whereas majority of the KO cells showed long and thin filopodia-like structures, indicative of active movement of the cells. To investigate whether depletion of mdig alters the invasive migration capacity, we first performed cell invasion assay using trans-well Boyden chamber with a PET membrane pre-coated with extracellular matrix at medium density in vitro. All four KO clones showed a substantial enhancement of invasive migration relative to four WT clones (Figure 5B). This observation not only correlates with the increased expression of proteins in actin and cadherin bindings as determined by quantitative proteomics (Figure 4B) but also indicates possible acquirement of EMT of the KO cells. To validate the proteomics data and have a focus on EMT, we conducted immunoblotting for the selected proteins that showed differential expression between WT and KO cells. The mdig protein was detected in all five WT clones but not in any of the 12 KO clones (clone number 10 of the KO cells was not included because of the limited quantity of cell lysate) (Figure 5C). The KO cells also showed a reduced expression of MYC, and a minor degree of reduction of EGFR. The levels of epithelial cell marker, E-cadherin (CDH1) in WT and KO cells were inconclusive. The KO cells showed reduced expression of the mesenchymal marker Snail1, but the expression of other mesenchymal markers, including VIM and TWIST1, was increased. We also checked the abundances of other proteins that showed more than 2-fold increase in the mass spectrometry (MS) analysis in KO cells and had been previously reported as potential regulators for cancer cell metastasis, including MAGED2, CTSD and STMN1. In agreement with the proteomic data, all KO clones showed higher levels of MAGED2, CTSD, and STMN1 relative to the WT clones. Although there were no significant differences in MS data for the known metastasis suppressors and tumor suppressor KISS1R, KISS1, and IRX1, we also checked the levels of these proteins in WT and KO clones in immunoblotting. Corroborating with the proteomic data, roughly similar levels of these proteins were detected among the WT clones and KO clones (Figure 5C).
Figure 5.

Knockout of mdig enhances invasive migration of the MDA-MB-231 cells and affects the expression of genes associated with invasion and EMT
(A) Cell morphology of the WT and KO cells.
(B)Trans-well invasion assay of the WT and KO cells. WT clones 2, 3 and 4, and KO clones 6, 7 and 8 were shown. Bottom panel shows relative numbers of invasive cells of the WT and KO cells. Data are represented as mean ± SD.
(C) Immunoblotting of the indicated proteins between 5 WT clones and 12 KO clones.
Enhanced repressive histone trimethylation and compromised MYC signaling in the mdig KO cells
We had previously shown a forward feedback regulation between mdig and MYC (Wu et al., 2016; Zhang et al., 2019a). The reduced MYC expression in the mdig KO cells (Figure 5B) suggests that the decreased tumor growth of the KO cells in mice (Figure 2A) is likely a result of MYC depletion following mdig KO. To further elucidate how mdig regulates MYC, we reanalyzed our ChIP-seq data of the repressive histone trimethylation markers, H3K9me3 and H4K20me3, in the WT and mdig KO MDA-MB-231 cells. By arbitrarily focusing on the genomic region encompassing -2 kb upstream to 2 kb downstream of the gene loci, the average plot of ChIP-seq for the merged peaks showed a substantial enhanced enrichment of H3K9me3 and H4K20me3 in the mdig KO cells (Figure 6A), which confirmed our previous findings showing the antagonistic property of mdig on these repressive histone methylation markers (Zhang et al., 2020). Further analysis of the more than 2-fold downregulated 980 proteins in the mdig KO cells by an analysis of TF perturbations followed by expression demonstrated that the top three pathways of these downregulated proteins are covered by the cells with MYC knockout (KO), MYC knockdown (KD), and MYCN shRNA silencing (Figure 6B). Most of these downregulated MYC pathway genes, including MYC itself, in the mdig KO cells exhibited an elevated enrichment of the repressive histone trimethylation marker, H3K9me3 or H4K20me3, in the gene body, proximal upstream, and proximal downstream (Figure 6C). Glutamate-ammonia ligases (GLUL), a well-documented MYC target gene participating in MYC-mediated cell proliferation and tumorigenesis (Bott et al., 2015), showed a significant diminishment of the active histone marker H3K4me3 and H3K36me3, in addition to the elevated enrichment of the repressive histone trimethylation marker, H4K20me3, in the mdig KO cells. Another potential MYC target gene encoding a lipid metabolic enzyme DGAT2 (Monroe et al., 2022), also exhibited loss of both H3K4me3 and H3K36me3 in the mdig KO cells (Figure 6C). Thus, the growth promoting activity of mdig for the tumors may achieved, at least partially, through the MYC-mediated oncogenic pathway. Knockout of mdig, thus, will impair the oncogenic activity of MYC and repress tumor growth.
Figure 6.

Enhanced repressive histone trimethylation and down-regulation of MYC signaling in the mdig KO cells
(A) Average plots of the merged peaks of ChIP-seq for H3K9me3, H3K36me3 and H4K20me3 in the regions from -2 kb to 2 kb of gene loci in WT and mdig KO cells. Right panel shows heatmaps of the merged peak distribution of H3K9me3 and H4K20me3 between WT and mdig KO cells.
(B) Transcription factor pathway analysis using Enrichr TF Perturbations Followed by Expression for the 980 downregulated proteins in the mdig KO MDA-MB-231 cells.
(C) Histone trimethylation profiles of the selected MYC pathway genes in both WT and mdig KO cells. Block arrowheads depict the enhanced enrichment of the repressive histone trimethylation markers on the indicated genes in KO cells. Empty arrowheads indicate diminished active histone trimethylation markers in the indicated gene in WT and KO cells.
Antagonization of mdig on histone H3 lysine 36 trimethylation (H3K36me3)
In addition to the repressive histone trimethylation markers, we also checked the status of H3K36me3, an active transcriptional elongation marker of the genes, in both WT and mdig KO cells. A pronounced magnification of H3K36me3 on the gene bodies in the KO cells relative to the WT cells was noted, indicating that mdig is also antagonistic toward H3K36me3 (Figure 7A). Visualization of the ChIP-seq data using the UCSC Genome Browser revealed a prominent increase in the level of H3K36me3 in some chromosome regions of the KO cells, as shown for chromosome 9 and chromosome X (Figure 7B). Indeed, there is a closer correlation between the level of H3K36me3 enrichment and gene expression, such as IQSEC2, HUWE1, MAGED2, UPF3B, LAMP2, and CUL4B whose genes showed elevated enrichment of H3K36me3 and are among the top-25 most upregulated proteins in the KO cells (Figure 4A). The regulatory role of mdig on H3K36me3 had been reported in the latent state of the HIV-1 proviruses and in demethylase assay in vitro of the recombinant mdig protein (Bi et al., 2021; Huang et al., 2019). Although some inconsistencies presence in different type of cancers, genetic manipulation of H3K36me3 had been associated with EMT and tumor cells migration (Lien et al., 2018; Yuan et al., 2020). In global ChIP-seq assay, we identified 566 genes with a significantly increase of H3K36me3 and 184 genes exhibited a reduced enrichment of H3K36me3 in the KO cells. Pathway analysis for molecular function of those genes with an increased enrichment of H3K36me3 in KO cells revealed that the top-ranked pathways are centered on β1 integrin interaction, DNA base excision repair and Gα signaling (Figure 7C). Integrins are superior and well-characterized cell surface receptors of various extracellular matrix (ECM) proteins and are critical regulators of metastasis (Ganguly et al., 2013; Gui et al., 1995) and angiogenesis (Yousefi et al., 2021). In fact, β1 integrin had been implicated in breast cancer progression and metastasis (Gui et al., 1995; Jonjić et al., 1993). Because histone markers control many cellular processes, these results reveal that with mdig disruption in breast cancer, H3K36me3 is enriched which further exerts its positive regulation on the genes implicated in angiogenesis, DNA damage repair and metastasis. It also suggests that the genes crucial for malignant characteristics are more likely to acquire an overall active epigenetic status in the mdig-depleted breast cancer cells. The genes with a reduced enrichment of H3K36me3 in KO cells are largely involved in IL-1-mediated extracellular matrix synthesis, cytokine signaling and autophagy (Figure 7D).
Figure 7.

Enhanced enrichment of H3K36me3 in the mdig KO cells
(A) ChIP-seq shows global enrichment levels of H3K36me3 on the gene bodies between WT and KO cells (top panel) and Pearson correlation of H3K36me3 between WT and KO cells (bottom panel).
(B) Genome browser screenshots of H3K36me3 between WT and KO cells using examples of chromosomes 9 (left panel) and X (right panel). Red arrows and dashed red boxes show regions in the chromosomes with an enhanced enrichment of H3K36me3 in the KO cells. Typical genes with enhanced enrichment of H3K36me3 are listed at the bottom of the panels.
(C) Ontology molecular function assay of the genes with a more than 2-fold increase of H3K36me3 in the KO cells.
(D) Ontology molecular function assay of the genes with a more than 2-fold decrease of H3K36me3 in the KO cells.
Gain of H3K36me3 of the top expressed genes involved in EMT and metastasis
To understand how H3K36me3 contributes to the higher metastatic feature of the KO cells further, we next examined the enrichment status of H3K36me3 on these genes that showed the most significant upregulation as determined by quantitative proteomics indicated in Figure 4A. Among the top 25 most increased genes, only 7 genes did not show amplified enrichment of H3K36me3, whereas 18 genes showed remarkable enhancement of H3K36me3 in their entire gene bodies (Figure 4A, marked in green). Figure 8A depicted ChIP-seq signals for H3K36me3 as well as H3K4me3, H3K9me3 and H4K20me3 of the randomly selected genes from the top 25 upregulated genes in the KO cells. The IQSEC2 and its family members are Arf-nucleotide exchange factors and have been implicated in angiogenesis and tumor metastasis (D'Souza and Casanova, 2016). Several other genes among them, including NMNAT1 (Yaku et al., 2018), MAGED2 (Jia et al., 2019), SLC12A7 (Brown et al., 2018), IGFBP7 (Paulitschke et al., 2019), HUWE1 (Vaughan et al., 2015), PLS3 (Ueo et al., 2015), PRNP (Cha et al., 2021), HMGB2 (Cui et al., 2019), LAMP2 (Damaghi et al., 2015), CTSD (Ketterer et al., 2020), etc., have also been linked to the glycolysis, EMT, stemness, and metastasis of the cancer cells. Thus, enhancement of H3K36me3 may preferentially favor the expression of genes contributing to EMT and cancer cell metastasis.
Figure 8.

Correlation between the most increased expression of genes in metastasis and enhanced enrichment of H3K36me3 in the KO cells
(A) Genome browser screenshots of the histone methylation pattern on the selected most upregulated genes in mdig KO cells.
(B) Different degrees of correlation between the upregulated genes and the enhanced enrichment of H3K36me3 in KO cells. The top 50 most upregulated genes in KO cells were analyzed for an increase of H3K36me3. In the top 1 to 15 range, 80% of these genes showed an elevated enrichment of H3K36me3, whereas from top 16 to 50 range, 43% of the genes showed an increase in H3K36me3.
(C) Genome browser screenshots depicts representative downregulated genes with a pronounced diminishment of H3K36me3 in the KO cells.
H3K36me3 has been found to be tightly associated with the active transcription marker, H3K4me3, for transcriptional elongation. It is generally believed that intensified enrichment of H3K36me3 on the individual genes is indicative of elevated expression of genes (Sims and Reinberg, 2009). Indeed, we found that genes that have a dramatic increase in expression are marked with an augmented H3K36me3 in their gene bodies. Among the top 15 expressed genes in KO cells as indicated in Figures 4A, 80% of them showed correlation between the levels of expression and H3K36me3 enrichment. Among the genes ranked at top 16 to 50, the correlation is about 43% (Figure 8B). However, there appears to be no necessary correlation between the increased expression of many genes that showed 2- to 4-fold increases and the enrichment of H3K36me3 on their gene bodies in the KO cells. For those genes that are downregulated in the KO cells as indicated in Figure 4A, only two genes, CKB and PLEKHH3, showed notable enhancement of H3K36me3; six genes, including DOCK6, PPM1D, ITGB2, RAD50, TFPI2, and PPP2R1B, exhibited substantial decrease of H3K36me3; and the remaining 17 genes showed no change of H3K36me3 on their gene bodies (Figure 8C and data not shown).
Stathmin1 (STMN1) is a negative regulator for the invasion of KO cells
Stathmin1 (STMN1) is an important regulator of microtubule filament system by destabilizing microtubules. Depending on the types of cancer, STMN1 had been shown to be able to either foster or impede metastasis of the tumor cells (Han et al., 2017; Williams et al., 2012). A few earlier reports revealed that high STMNI correlates with poor prognosis of the breast cancer patients (Baquero et al., 2012; Saal et al., 2007). In both proteomics study and immunoblotting analysis (Figure 5), we found that knockout of mdig resulted in a pronounced elevation of STMN1, which is correlated with an amplified enrichment of H3K36me3 on the STMN1 gene in KO cells as determined by ChIP-seq (Figure S2A). Kaplan-Meier relapse-free survival (RFS) assay showed that high STMN1 correlated with the poor survival of breast cancer patients with either lymph node positive or negative metastasis (Figure S2B). The increased expression of STMN1 in KO cells was additionally validated by immunofluorescent staining of the STMN1 protein (Figure S2C). To determine whether increased metastasis of the KO cells is a result of high STMN1 expression, we first silenced the STMN1 expression using siRNAs targeting the STMN1 mRNA in three KO cell lines (Figure S2D). After successful knockdown of STMN1 in the KO cells, we proceeded for the trans-well invasion assays to assess the invasion potential of the cells with or without STMN1 silencing. It is unanticipated that cells with STMN1 knockdown even showed an enhanced invasion relative to the cells treated with control siRNA (Figure S2E). Therefore, the increased level of STMN1 is not responsible for the enhanced metastatic potential of the KO cells. It is very likely that STMN1, in fact, is an anti-metastatic factor in the breast cancer cells with mdig deficiency.
Melanoma-associated antigen D2 (MAGED2) positively regulates the metastasis of breast cancer cells
Because the STMN1 appears to be not a pro-metastatic factor in the KO cells (Figure S2), we next put our attention on MAGED2, which has greater than a 7-fold increase in expression (Figures 4A and 5) and a strong enrichment of H3K36me3 on this gene in the KO cells as determined by ChIP-seq (Figure 8). MAGED2 had been previously shown to be capable of promoting cell adhesion and metastasis of colorectal and gastric cancer cells (Kanda et al., 2016). As MAGED2 was upregulated in the KO cells, we silenced the MAGED2 expression using siRNAs targeting the MAGED2 gene in the KO cells (Figure 9A). Invasive migration assay in vitro suggested that knockdown of MAGED2 abrogated the invasive potential of the KO cells substantially (Figure 9B), indicating that MAGED2 is required for the strengthened invasion or metastasis capabilities of the KO cells. This observation is highly relevant to the clinical data of breast cancer patient survival. Overall survival (OS) analysis of the TNBC revealed that high expression of MAGED2 is a poor prognostic factor for patient survival (Figure 9C). Additionally, we found that high MAGED2 expression is a common feature of invasive breast cancers and is expressed in conjunction with other genes that are implicated in the malignant phenotypes of breast cancer (Figure 9D). Both TCGA and Curtis breast cancer datasets suggested a more than 2-fold increased expression of MAGED2 in invasive lobular breast cancer relative to the normal breast tissues (only Curtis database was shown in Figure 9D). Because the molecular function of MAGED2 is largely unknown, we used TNMplot RNA-seq database to analyze correlation genes of MAGED2 in invasive breast carcinoma with a range of Spearman’s Rho correlation value from 0.3 to 0.61. Within this range, a total of 2,021 genes were included for GO Biological Pathway assay. The results showed that the top 3 ranked pathways are clustered on the organization, assembly and epithelial movement of the cilium (Figure 9E), suggesting that MAGED2 is an essential regulator for the motility of the breast cancer cells. Figure 9F depicted a few of the top-correlated genes of MAGED2 in breast cancer, including KCNK15, THSD4, TBC1D9, and FAM174A that are functioning as potassium channel, matrix cleavage, GTPase activator, and membrane protein. It is somehow unexpected to note a strong correlation of MAGED2 with a group of genes regulating the organization, assembly and function of cilium, a cell surface organelle involved in cell locomotion. Furthermore, cilium is also important in mediating growth signals from pathways of Hedgehog, Wnt, Notch, Hippo, GPCR, and others (Wheway et al., 2018). These data clearly indicated the pro-oncogenic and pro-metastatic roles of MAGED2.
Figure 9.

MAGED2 positively regulates the invasive migration of the breast cancer cells
(A) Three clones of mdig KO MDA-MB-231 cells were silenced for MAGED2 gene using siRNA targeting MAGED2. GAPDH was used as loading control for the Western blotting. MAGED2 was completely silenced by the MAGED2 siRNA, but not the control siRNA.
(B) Loss of MAGED2 through siRNA silencing resulted in lower invasion capability of the KO cells, indicating MAGED2 is a positive regulator for the invasive migration. Data are represented as mean ± SD.
(C) High MAGED2 predicts poorer relapse free survival (RFS) of breast cancer patients, concomitant with p53 mutation.
(D) High MAGED2 expression is a common feature of the invasive breast cancer. Data are derived from Curtis Breast cancer that showed a more than 2-fold elevation of MAGED2 gene in invasive lobular breast cancer compared to normal breast tissue.
(E) Volcano plot shows strong correlation of MAGED2 expression with the genes involved in the organization, assembly and regulation of cilium for cell locomotion.
(F) Selected examples of top-correlated genes of MAGED2 in breast cancer.
Negative correlation of mdig with MAGED2 and other metastatic genes in breast cancer
Data from quantitative proteomics and ChIP-seq of the MDA-MB-231 cells revealed a negative correlation of mdig with MAGED2 and several other genes linked to EMT and metastasis of the breast cancer cells (Figures 4, 5, 7, and 8). However, there is no clinical evidence indicating such negative correlations in human breast cancer. To explore the relationship between mdig and MAGED2 in human breast cancer, we examined genes negatively co-expressed with mdig in 242 normal breast tissues and 7,569 breast cancer samples using TNMplot database. There is a clear negative correlation between mdig and MAGED2 in both normal breast tissues and breast cancer samples (Figure 10A). Such a negative correlation was also detected between mdig and IQSEC2 (bottom panel of Figure 10A) and others that showed the most upregulated expression in mdig KO cells among these clinical breast cancer tissues. We did a WikiPathways analysis for 3,299 genes that showed a more than 20% negative correlation with mdig in human breast cancers and found the top four most enriched clusters are G protein-coupled receptor pathways that are critically involved in cell adhesion, migration, invasion, and metastasis (Khalil et al., 2016) (Figure 10B). Further analysis using the Epigenomics Roadmap of histone modification ChIP-seq unraveled that majority of these negatively correlated genes with mdig is clustered in the histone modification pathway of H3K27me3, H3K9me3 and H3K36me3 (Figure 10B, bottom panel), which corroborates to the findings of the enhanced enrichment of H3K9me3 and H3K36me3 in the genome of the mdig KO cells (Figures 6 and 7). Lastly, we investigated the expression trends of mdig, MAGED2 and IQSEC2 as determined by RNA-seq among normal breast tissues, primary breast tumors and metastatic breast tumors using the TNMplot dataset. Resembling the expression pattern of mdig between hormone positive breast cancer and TNBC (Figure 1A), a substantial decrease of mdig expression was noted in the metastatic tumors relative to the primary tumors and normal breast tissues (Figure 10C, top panel). This result, therefore, supports the notion that metastatic breast cancers tend to lose expression of mdig. On the other hand, there is a tendency of increased expression of MAGED2 and IQSEC2 in primary tumors relative to the normal tissues, which were further enforced in the metastatic tumors (Figure 10C, bottom panels).
Figure 10.

Negative correlation of mdig with MAGED2 and other metastasis-related genes
(A) Negative correlation of mdig with MAGED2 in both normal breast tissues and breast cancer tissues. Bottom panel shows negative correlation of mdig with IQSEC2.
(B) WikiPathways analysis of 3,299 genes that showed more than 20% of negative correlation with mdig. The genes enriched on the major GPCRs pathways are marked in red. Bottom panel shows epigenomics clustering of the negatively correlated genes with mdig in breast cancer samples.
(C) Inverse correlation of mdig with MAGED2 and IGSEC2 in metastatic breast cancers.
Cross-validation of the roles of mdig on H3K36me3 and invasion in A549 cells
To investigate whether the observed effect of mdig on H3K36me3 and cell motility or metastasis can be recapitulated in other cancer cell lines, we tested mdig overexpression in a widely used human lung cancer cell line, A549 cells, followed by measuring the level of H3K36me3 and the invasion of the cells. In several tests, the level of H3K36me3 in the cells with expression of exogenous mdig is measurably lower than the cells transfected with an empty expression vector (EV) (Figure 11A), whichsupports the observation showing demethylation of mdig on H3K36me3 in demethylation assay as reported by Huang et al. (2019). The EV expressing cells are highly invasive in in vitro invasion assay. Enforced expression of mdig, on the other hand, abolished the invasive capability of the cells substantially (Figure 11B). Quantification of the invaded cells showed that overexpression of mdig resulted in a 3- to 5-fold reduction of cell invasion (Figure 11C). Although to a lesser degree, expression of the exogenous mdig reduced cell migration relative to the EV expression cells (Figure 11D), which agrees with our earlier report showing negative regulation of mdig on cell migration and invasion (Yu et al., 2014). Accordingly, it is probably common that mdig is anti-metastatic through antagonizing H3K36me3 in some different types of human cancers.
Figure 11.

Overexpression of mdig represses H3K36me3 in and invasion of the A549 lung cancer cell
(A) Western blotting for mdig, H3K36me3 and histone H3 (His H3) in A549 cells transfected with an empty expression vector (EV) or mdig expressing vector. Ex-mdig: exogenous mdig; En-mdig: endogenous mdig.
(B) Invasive migration assay of the A549 cells expressing empty vector or mdig.
(C) Quantification of invasive cells in B. Data are represented as mean ± SD.
(D) Migration assay of the A549 cells transfected with empty vector and mdig. Lower panel shows quantification of the migrated cells. Data are represented as mean ± SD.
Discussion
Triple negative breast cancers (TNBC) comprising 10 to 20% of all breast cancers, are biologically more aggressive, highly metastasizing and have a worse five-year prognosis (Carey et al., 2010; Desmedt et al., 2008). These cancers are resistant to conventional therapies such as hormone treatment, chemotherapy, and radiotherapy. Surgical interventions are the only effective approach to treat these highly aggressive cancers. Therefore, there is a dire need to understand the biology of TNBCs and the biological components implicated in their malignant behavior. We have identified an environment factor-associated gene, mdig whose expression status influences the overall survival of breast cancer patients (Thakur et al., 2014, 2018). High expression of mdig predicts poor overall survival, whereas predicts better survival in the subset of patients who are positive for lymph node metastasis. Of interest, mdig appears to be an oncogenic factor that promotes tumor growth of the early stage breast cancer, whereas at the later stages, it acts as a tumor suppressor by inhibiting the tumor cell motility and invasion (Thakur et al., 2018). In human lung cancer cell line A549 cells using approaches of overexpression and siRNA knockdown, we also noted that mdig contributes to cell proliferation but restrains migration and invasion of the cells (Yu et al., 2014). Although the metastasis suppressor-like activity of mdig has been elucidated in breast cancer and lung cancer cells, a comprehensive knowledge regarding a detailed repertoire of genes and cellular processes affected by mdig in the pathways of EMT and metastasis is lacking. Therefore, to better understand mdig’s impact on breast cancer, we tested human TNBC cell line MDA-MB-231 cells with and without mdig and subjected them for orthotopic tumorigenesis analysis in NSG mice along with quantitative proteomics and global histone methylation profiling by ChIP-seq. Our data provided the first evidence showing the anti-metastatic role of mdig in an animal model.
The absence of biomarkers facilitating early detection, development of resistance toward chemotherapeutics and rapid onset of metastasis is a major factor contributing to increased breast cancer mortality rates. Therefore, understanding the biological, cellular, and molecular processes is essential for gaining a profound insight into the genomic and epigenetic contributions to the pathobiology of breast cancer. This further widens our understanding in terms of un-discovering the biological entities that can be exploited for therapeutic interest. The mdig gene was originally identified from coal miners’ alveolar macrophages, human glioblastoma cell line T98G cells with c-myc overexpression and Xenopus laevis, respectively. The encoded protein contains a conversed JmjC domain that can be found in most members of the histone demethylases (Zhang et al., 2019a). Enzymatic activity assay suggested that mdig, also named mina53, NO52 and Riox2, has histidine hydroxylase activity toward 60S ribosomal protein RPL27a (Ge et al., 2012). In addition to RPL27a, our most recent mass spectrometry analysis indicated possible hydroxylation of XRCC2, TPX2 and OASL by mdig in the MDA-MB-231 cells (Qiu Y et al., unpublished). The involvement of mdig in histone demethylation was noted in some cells with mdig overexpression or deletion (Zhang et al., 2019a, 2020). We had reported recently that depletion of mdig by CRISPR-Cas9-based gene editing strengthens the overall repressive histone trimethylation markers on the genome, including H3K9me3, H3K27me3 and H4K20me3 in bronchial epithelial cells and MDA-MB-231 cells (Zhang et al., 2020). Most recent study by Liu et al. suggested that mdig is recruited by H3K36me2/3 binding protein HDGFL2 (HRP2) for the demethylation H3K27me3 in multiple myeloma cells (Wang et al., 2022). It is interesting that a notable increment of H3K36me3, an epigenetic marker for transcriptional elongation, is observed in the mdig KO cells, suggesting an antagonistic role of mdig on H3K36me3. The demethylase-like activity of mdig had also been reported by Huang et al. who demonstrated that recombinant mdig (mina53) preferentially demethylates H3K36me3 in an in vitro demethylation assay (Huang et al., 2019).
H3K36me3 has long been associated with elongating Pol II and is enriched within the body of transcriptionally active genes. Meanwhile, H3K36me3 has also been implicated in DNA repairing and pre-mRNA splicing (Luco et al., 2011). The methyltransferase catalyzing the formation of H3K36me3 in histone H3, SETD2, has long been viewed as a tumor suppressor in some types of cancer (Duns et al., 2010). However, recent study unraveled indirect tumor promoting property of SETD2 in negatively regulating the tumor suppressive-like activity of the NK cells and/or altering the subpopulation of the group 3 innate lymphoid cells (ILC3s) (Chang et al., 2022). Knockout of SETD2 reduced the expression of several key inflammatory cytokines in ILC3, including IL-17A, IL-17F, NRP1, CCR6, etc, which will also weaken the pro-tumorigenic environment of the cancer cells. Of interest, our previous and recent studies also revealed important roles of mdig on the infiltration of Th17 cells into the lung and the expression of NRP1 in lung epithelial cells (Thakur et al., 2015; Zhang et al., 2021). Thus, there must be some degrees of coordination between mdig and SETD2 in the pathogenesis of cancer. It appears to be inconclusive, nevertheless, whether the level of H3K36me3 affects the pathogenesis of cancer. In human hepatocellular carcinoma (HCC), immunohistochemistry analysis revealed strong staining of H3K36me3 in HCC, especially in the peripheries and invasive fronts of the tumors (Lien et al., 2018). Moreover, there was a strong association between the positivity of H3K36me3 and adverse HCC outcomes, including high level of serum α-fetoprotein, high tumor grade, high tumor stage, and poor prognosis. In prostate cancer cells, NSD2, another methyltransferase for the formation of H3K36me2 and H3K36me3, is believed to be able to recruit NF-κB transcription factor for the transcription of those genes, such as IL-6, IL-8 and VEGF, involved in inflammation, tumor cell survival and metastasis (Yang et al., 2012). Indeed, in a study using genetically engineered mouse prostate cancer models, NSD2 was found robustly expressed in the metastasized but not the primary tumors and considered as a key driving factor for metastasis (Aytes et al., 2018). The pro-oncogenic or metastatic role of NSD2 was also noted in some human breast cancer tissues or cell lines (Gao et al., 2020). These observations, thus, provided unequivocal evidence linking H3K36me3 with cancer cell EMT and metastasis.
We had detected a limited degree of increase of H3K36me3 in the mdig KO MDA-MB-231 cells through immunoblotting of total cellular proteins previously (Zhang et al., 2020). Global ChIP-seq in the present report, however, unraveled a pronounced elevation of H3K36me3 in the KO MDA-MB-231 cells relative to the WT cells, indicating the demethylase-like or antagonistic role of mdig on H3K36me3. In A549 cells, enforced expression of the exogenous mdig reduced the level of H3K36me3 and the cell invasion. These notions are complementarily supported by the CRISPR-Cas9 functional genomic screens that revealed demethylation of H3K36me3 by mdig during the maintenance of HIV-1 provirus latency (Huang et al., 2019). In conjunction with quantitative proteomics analysis, it further demonstrated that elevated H3K36me3 is particularly important for the expression of those top-ranked proteins contributing to invasive migration and/or metastasis of the KO cells. Except for WASH6P that participated in actin polymerization, and a few others, most genes of the top-25 most upregulated proteins in KO cells have exhibited substantially enhanced enrichment of H3K36me3 and H3K4me3. Although none of the top-25 increased proteins in KO cells had been established as an EMT identity previously, most of them had been associated with the regulation of cytoskeleton, glycolysis, stemness, and cell motility. For example, IQSEC2, an ARF family guanine nucleotide exchange factor (ARFGEF) regulating cytoskeletal assembly, is frequently mutated in human lung adenocarcinoma (Zhang et al., 2019b). IQSEC1, the same family member of IQSEC2, has been linked to the metastasis of MDA-MB-231 breast cancer cells (Morishige et al., 2008). Similarly, high level of NMNAT1, a nuclear form rate-limiting enzyme catalyzing the biosynthesis of nicotinamide adenine dinucleotide (NAD) in the salvage pathway, has been noted in tumors with higher grade, metastasis, and poor survival (Santidrian et al., 2014). It is worth noting that four lysosome proteins, PRNP, LAMP2, NPC2 and CTSD, all of which featured with an enhanced enrichment of H3K36me3, are also among the top 25 most upregulated proteins in the mdig KO cells. Emerging evidence suggests that lysosomal proteases are critically involved in the remodeling of extracellular matrix for the invasion of human breast cancer cells (Glunde et al., 2003). Both LAMP2 and CTSD had been shown to be highly capable of fostering metastasis of the breast cancer cells (Damaghi et al., 2015; Ketterer et al., 2020), whichsupports the findings that invasive and metastatic cells have large numbers of lysosomes located at the periphery of the cells for the degradation of focal adhesion proteins and extracellular matrix (Hamalisto and Jaattela, 2016).
MAGED2, a functionally elusive protein, has been designated as a molecular biomarker predictive of some human cancers, including colorectal cancer (Chung et al., 2010), liver cancer (Hashimoto et al., 2015), gastric cancer (Kanda et al., 2016), lung cancer (Tsai et al., 2007), and metastasis of small intestinal carcinoid (Kidd et al., 2006). In breast cancer, a higher level of MAGED2 predicts poorer survival of the patients (Jia et al., 2019). Based on the fact of nucleolar localization, MAGED2 was considered as a possible regulator for pre-rRNA processing (Pirlot et al., 2016). Under stress conditions, MAGED2 can be redistributed to the cytoplasm. There is evidence showing that genetic mutations in MAGED2 cause X-linked antenatal Bartter’s syndrome featured with an impaired expression of the sodium chloride cotransporters SLC12A1 and SLC12A3 in the distal renal tubule. Although SLC12A1 and SLC12A3 were not detected in both WT and mdig KO MDA-MB-231 cells, SLC12A7, a trans-membrane potassium chloride cotransporter, was found to be highly increased in the KO cells, indicating possible regulation of MAGED2 on this ion transporters. It has been well-established that ion imbalance can directly affect the attachment kinetics of cell membrane, organization of cytoskeleton, and cell motility (Stock and Schwab, 2015). Indeed, analysis of the MAGED2-correlating genes in TNMplot RN-seq database revealed that the most correlated genes of MAGED2 in invasive breast cancer are clustered in the organization, assembly and epithelial movement of the cilium, a primary organelle important for cell locomotion (Figure 9E). Thus, it is perceivable that MAGED2 may serve as one of the key regulators in certain steps of EMT and metastasis of the cancer cells. This assumption was supported by the findings in this report that mdig KO cells showed elevated expression of MAGED2 and metastasis, and siRNA silencing of MAGED2 diminishes invasive migration of the mdig KO cells.
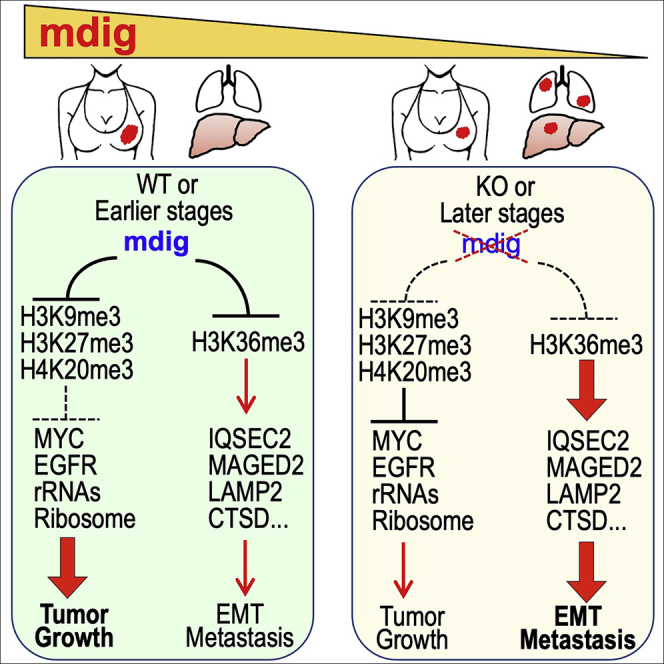
Tumor cell EMT, invasive migration and metastatic dissemination rely on a plethora of molecules that regulate the reorganization of cytoskeleton and adhesion molecules, remodeling of extracellular matrix, release of cytokines and extracellular vesicles, extravasation of the cancer cells, and communication with neighboring non-cancerous cells within the primary tumor and the remote embedding sites (Birkbak and McGranahan, 2020). In addition to the driving factors for metastasis, there are some negative regulators that limit the motility and colonization of the cancer cells in distal organs. In our previous report using siRNA to silence mdig, we observed an increased expression of several genes associated with EMT, including CXCL12, UPA (PLAU), PAI-1 (SERPINE1), VIM, and MMP9. This notion was supported by the increased expression of UPA, VIM, MMP9, MMP14, and MMP19 in the mdig KO cells in current study (data not shown). Knockout of mdig and the consequent enhancement of H3K36me3, which reprograms the epigenetic landscape on the genome, thus, may tip the balance between pro- and anti-metastasis factors to alter the metastatic evolutionary trajectories and increase the likelihood of a subset of cancer cells acquiring the traits of metastatic potential. Given the fact of a reduced expression of mdig in later stage breast cancer (Thakur et al., 2018) and lung cancer (Shi et al., 2021), the data presented in this report suggest the different roles of mdig played during the earlier and later phases of tumorigenesis. Through its antagonistic properties on the repressive histone trimethylation markers, including H3K9me3, H3K27me3 and H4K20me3, mdig promotes the expression of oncogenes, such as c-Myc, EGFR, and others, to promote tumor initiation and growth in the early phase of tumorigenesis (Zhang et al., 2020). When tumors are well-developed in the later phase, loss of mdig will result in an elevation of H3K36me3 that mostly promotes the transcription and expression of genes contributing to EMT, invasive migration, and metastasis of the cancer cells (Figure 12). It will be interesting to know how the expression of mdig is regulated in the different stages of tumors. Understanding the expression and functional dynamics of mdig, histone methylation, oncogenes, and major regulators for cancer cell EMT, invasive migration and metastasis, accordingly, will yield insights on crafting new strategies for targeted therapies in breast cancer and possibly, other malignant diseases.
Figure 12.

Schematic diagram shows the regulatory roles of mdig on histone protein methylations, tumor cells growth and metastasis
In earlier stage breast cancer or WT breast cancer cells, increased expression of mdig antagonizes the repressive trimethylation of histone proteins, including H3K9me3, H3K27me3, and H4K20me3, leading to derepression of the major oncogenes, such as MYC, EGFR, and others, that promote tumor growth. Mdig is also antagonistic toward H3K36me3 that preferentially enforces expression of the genes in cancer cell motility, EMT and metastasis. In later stage breast cancer or the mdig KO breast cancer cells, because of the loss of mdig expression, the repressive trimethylations of histone proteins compromise the expression of the growth promoting oncogenes. Meanwhile, loss of mdig causes enhanced enrichment of H3K36me3 that boosts transcriptional elongation of the genes involved in cell motility, EMT and metastasis. Thus, metastasis or uplifted metastatic potential is dominant in the later stage breast cancer or the breast cancer cells with mdig knockout.
Limitations of the study
Most cancer deaths can be attributed to metastatic spread of the cancer cells from the primary site to distal organs. We provided evidence showing that mdig is oncogenic in early stages of breast cancer but anti-metastatic for the metastatic cancer through antagonizing H3K36me3 that linked to the expression of genes involved in cancer cell motility and invasive migration. However, this study has some limitations. First, although the cellular models used in the present report have advantages for detailed mechanistic studies, a well-known caveat of such models is that they only partially recapitulate the clinical features of breast cancer. Thus, the current findings will be validated through the patient-derived xenograft (PDX) mouse models and clinic studies in the future. Second, knockout of mdig resulted in upregulation of several metastasis-related genes that are located in chromosome X. Because the question whether chromosome X is an important contributor to breast cancer metastasis has never been addressed or discussed currently, it will be imperative to distinguish whether the increased expression of X-linked metastatic genes following mdig deletion is resulted from sporadic overactivation of the genes in the active X chromosome or the reactivation of the inactivated X chromosome (Barr body). Finally, the sample size of metastasis for the comparison of gene expression among normal tissue, primary tumor and metastasis is too small to be significant. To make the data more significantly, future studies should include more cases of metastatic breast cancer. Accordingly, this study represents a first step in understanding how an oncogenic gene, mdig, can have tumor suppressor-like activity in later stages of breast cancer through dampening metastasis. More integrated experimental and clinical studies are needed to fully investigate the effect of mdig on the acquisition of metastatic potential of the cancer cells, circulation of the metastatic cells and embedding and growth of the metastatic cells in distal organs. It will also be critical to delineate how MAGED2 can enforce the metastasis of the breast cancer cells and find targeting strategies to this newly identified pro-metastatic factor to reduce metastasis and improve the survival of the patients.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mdig (MINA53) | Invitrogen | 39-7300 |
| EGFR | Invitrogen | MA5-13070 |
| KISS1R | Invitrogen | 15505-1-AP |
| Sox2 | Cell Signaling Technology | 3579 |
| c-Myc | Cell Signaling Technology | 13987 |
| Vimentin | Cell Signaling Technology | 5741 |
| Slug | Cell Signaling Technology | 9585 |
| b-Catenin | Cell Signaling Technology | 9562 |
| Cathepsin D | Cell Signaling Technology | 2284 |
| Snail | Cell Signaling Technology | 3879 |
| KLF4 | Cell Signaling Technology | 4038 |
| GAPDH | Cell Signaling Technology | 5174 |
| E-Cadherin | Novus | NBP-16258 |
| PCDH7 | Santa Cruz Biotechnology | sc-517042 |
| Twist1 | Santa Cruz Biotechnology | sc-81417 |
| MAGED2 | Santa Cruz Biotechnology | sc-130443 |
| KISS1 | Santa Cruz Biotechnology | sc-101246 |
| IRX1 | Novus Biologicals | NBP2-85107 |
| HRP-Conjugated IgG | Santa Cruz Biotechnology | sc-516087 |
| RACK1 | Santa Cruz Biotechnology | sc-17754 |
| Deposited data | ||
| ChIP-seq of MDA-MB-231 for H3K4me3, H3K9me3, H3K36me3, H4K20me3 | NCBI GEO | GSE207505 |
| Experimental models: Cell lines | ||
| MDA-MB-231 | ATCC | CRM-HTB-26 |
| A549 | ATCC | CRM-CCL-185 |
| Experimental Models: Mice | ||
| NSGTM variants | The Jackson Lab | 005557 |
| Recombinant DNA | ||
| MINA53 (mdig)-GFP | OriGENE | RG214829 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Fei Chen at Fei.Chen.1@stonybrook.edu.
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Ethics statement and animal experiments
The animal experiments described in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at the Wayne State University (protocol number 19-08-1218). Female NSG mice (NOD.CgPrkdcscid Il2rgtm1Wjl/SzJ) were purchased from Jackson Laboratories (Bar Harbor, ME). All animals were maintained in accordance with IACUC guidelines. MDA-MB-231 WT and KO cells (1 × 106) were collected by trypsinization and were mixed on ice with PBS and phenolred free Cultrex basement membrane extract (BME, Trevigen) to a concentration of 10 mg/mL of BME. Two hundred μl of the cocktail was injected near the area of fourth and fifth mammary fat pad of the 10-week-old female NSG mice. Tumors were measured once a week, beginning 15 days after transplantation. Mice were sacrificed for analysis 48 days after initial transplantation. Primary mammary tumors were dissected and measured. Additionally, lungs, livers, axillary, and lumbar lymph nodes were harvested for histology.
Method details
Cell culture
The human triple negative breast cancer cell (TNBC) line MDA-MB-231 was purchased from the American Type Culture Collection (Manassas, VA). Cells were cultured in DMEM F-12 medium and were supplemented with 10% FBS and 1% penicillin-streptomycin (Thermo Fisher, MA) and grown in 37°C-humidified incubators in the presence of 5% CO2.
Invasive migration assay
Four individual WT and KO cell clones in duplicates, were utilized for the assay. Twenty-four well plates, 8.0-μm pore membranes, Trans-well and Matrigel invasion chambers (Corning USA) were used according to the manufacturer’s protocol. First, chambers were rehydrated with serum-free medium for 2 h at 37°C. Each upper chamber was supplied with 700 μL of serum-free medium containing 5 × 104 cells for migration and 1 × 105 cells/well for the invasion assay. Simultaneously, 250 μL of cell culture medium with 5% FBS (without antibiotics) was added to the lower chamber as a chemoattractant, and cells were incubated for 24 h at 37°C. The inserts (upper chambers) were removed, and the non-migrated and non-invaded cells remaining on the upper surface of the membrane were scrapped off using cotton swabs. The inserts were then stained with Diff-Quik stain kit (Dade Behring Inc., Newark, DE) according to the manufacturer’s instructions. Inserts were dried and imaged under a bright field microscope. Images were captured at 10× magnification for five different fields and were counted using ImageJ software (National Institute of Health, Bethesda, USA) (https://imagej.nih.gov/ij/).
Quantitative mass spectrometry
Cell harvesting for proteomics analysis protocol has been adopted from (Rogers et al., 2010). Thereafter, the samples in duplicates were subjected to proteomics analysis. In total, 34 cell pellets were weighed and volumes matched with the addition of HPLC-grade water. 1% LiDS final was added to the samples and heated at 95°C for 5 min, followed by filtering through Pierce Handee Spin Columns (Thermo Scientific) to remove non-soluble material. Protein amount was determined by BCA Protein Assay (range from 0.278 mg to 1.064 mg). Fifty μg aliquots of each were buffered with 100 mM ammonium bicarbonate (AMBIC), reduced with 5 mM dithiothreitol (DTT), and alkylated with 15 mM iodoacetamide (IAA) under standard conditions. Excess IAA was quenched with an additional 5 mM DTT. Samples were diluted to decrease LiDS to 0.1% then an overnight digestion was performed with sequencing-grade trypsin (Promega, Madison, WI) in 100 mM AMBIC, 0.3 M urea, and 15% acetonitrile. The next day, detergent was removed from the samples using Pierce Detergent Removal Columns. Samples were speed vac’ed to dryness and solubilized in 0.1% FA for analysis. The peptides, 4 μg per analysis, were separated by reversed-phase chromatography (Easy Spray PepMap RSLC C18 50 cm column, Thermo Scientific), followed by ionization with the Easy Spray Ion Source (Thermo Scientific), and introduced into a Fusion Orbitrap mass spectrometer (Thermo Scientific). Abundant species were fragmented with collision-induced dissociation (CID).
Mass spectrometry data analysis
For protein quantification and pathway analysis, mass spectrometry raw files were searched against the Uniprot human complete database downloaded 2017.07.14 (20,201 entries) using MaxQuant v1.6.2.10 with the default version of the Andromeda search engine. Match between runs was enabled and just one peptide was required for protein quantification. All other parameters were left at their default values, including: tryptic cleavage with at most 1 missed cleavage was the protease, methionine oxidation and protein N-terminus acetylation were variable modifications, cysteine carbamidomethylation was a fixed modification, fragment ion tolerance was 0.5 Da, precursor tolerance was 20 ppm for the first search and 4.5 ppm for the second, peptide identifications were allowed at a 1% false discovery rate as determined by a reversed database. For all analyses, peptide spectra matches were accepted at a 1% false discovery rate as determined by a reversed database search. Peptide area under the curve was used to generate quantitative values.
Chromatin immunoprecipitation and global sequencing (ChIP-seq) for H3K36me3
The wild type (WT) and mdig knockout (KO) MDA-MB-231 cells were established as reported previously (Zhang et al., 2020). For ChIP-seq, ten million WT and KO MDA-MB-231 cells were fixed and subjected to immunoprecipitation using ChIP-grade antibody from Active Motif (Carlsbad, CA) against H3K36me3. ChIP and input/control DNA were prepared for sequencing using Illumina NextSeq 500, and the 75-nt sequence reads (tags) were aligned to the reference genome hg19 using the Burrows-Wheeler Aligner (BWA) algorithm with default settings. Reads were extended at their 3′ends to a length of 200 base pair (bp) fragments in a size-selected library. The densities of fragments (signal map) were determined by the number of fragments in each 32-nt bins along genome. The tag number of samples within a comparison group was normalized by random sampling to the number of tags in the smallest sample. Data was visualized by the University of California, Santa Cruz (UCSC) genome browser.
Western blotting
Total cellular proteins were prepared by lysing cells via sonication in 1 × RIPA buffer (Millipore, Billerica, MA) supplemented with phosphatase/protease inhibitor cocktail and 1 mM PMSF. Lysed cells were then centrifuged and supernatant isolated as protein, which was quantified using the Micro BCA Protein Assay Reagent Kit (Thermo Scientific, Pittsburgh, PA). Before loading onto SDS–PAGE gels, samples were boiled in 4 × NuPage LDS sample buffer (Invitrogen) containing 1 mM dithiothreitol (DTT). Samples were run on 7.5%, 10% or 15% SDS-PAGE gels, and separated proteins were then transferred to methanol-wetted PVDF membranes (Invitrogen). Membranes were subsequently blocked in 5% nonfat milk in TBST and probed with the indicated primary antibodies at dilutions of 1:1000, or 1:5000 overnight at 4°C. The next day, membranes were washed with TBST and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at dilutions of 1:2000 or 1:5000 at room temperature for 1 h. Immunoreactive bands were visualized through SuperSignal™ West Pico Chemiluminescent Substrate detection system (Thermo Scientific, Rockford, IL). Mdig (mouse) antibody, EGFR and KISS1R was purchased from Invitrogen. Antibodies for Sox-2, Nanog, c-myc, Vimentin, Slug, beta-catenin, Cathepsin D, RACK1, STMN1, SNAIL, KLF4, and GAPDH were purchased from Cell Signaling Technology (Danvers, MA, USA). Oct4 and E-Cadherin were from Novus Biologicals, PCDH7, Twist, MAGED2, KISS1R, KISS1, and IRX1 were from Santa Cruz Biotechnology (Dallas, TX, USA). All presented data are representative of at least three independent experiments.
Histology
Mice were sacrificed and perfused with PBS (PBS). Lungs, axillary and lumbar lymph nodes, and liver were dissected and fixed with 4% buffered paraformaldehyde and paraffin embedded. Tissue blocks were sectioned to 6 μm sections by using a manual rotary microtome (Leica). For histological analysis of paraffin-embedded tissues, sections were deparaffinized in xylene, rehydrated in graded series of alcohol and stained with hematoxylin and eosin (H&E).
Immunohistochemistry and immunofluorescence staining
Paraffin-embedded tissue sections were deparaffinized with xylene and hydrated in a series of alcohol gradients. To quench endogenous peroxidase activity, slides were incubated with 1.5–3% H2O2 in PBS for 20 min at room temperature. Heat-mediated antigen retrieval was performed by boiling tissue sections in citrate buffer with pH6.0 for 20 min in a microwave. To block nonspecific binding of immunoglobulin, slides were incubated with a solution containing 5% goat serum, 0.2% Triton X-100 in PBS for 2 h at room temperature, followed by incubation with primary antibodies against mouse Cytokeratin 18 (from Abcam with 1:50 dilution) overnight at 4°C. Goat anti-mouse biotinylated secondary antibody was subsequently applied at 1:200 dilution and incubated for 2 h at room temperature. Slides were then incubated with ABC reagent (Vectastatin Elite ABC kit) for 45 min at room temperature, and the chromogen was developed with diaminobenzidine (DAB). Slides were counterstained with hematoxylin (Sigma-Aldrich, St. Louis, MO) and mounted with Entellan® (Electron Microscopy Sciences, Hatfield, PA). All incubation steps were carried out in a humidified chamber, and all washing steps were performed with 1 × PBS. Images as showing in Supplementary material (Figure S1) were captured under bright field of a Nikon Eclipse Ti-S Inverted microscope (Mager Scientific, Dexter, MI, USA) and analyzed using Nikon’s NIS Elements BR 3.2 software.
For stathmin (STMN1) immunofluorescence, ten thousand WT and KO cells per well in 400 μL of medium were seeded and were incubated overnight. Next day, cells were blocked with 1%BSA in PBS for 1 h, then subsequently incubated with Stathmin antibody (Rabbit mAb from Cell Signaling) in 1:1000 dilution, overnight at 4°C. Next day, cells were washed with tween 20-PBS solution, and secondary antibody Goat anti-Rabbit Alexa Fluor 488 (Invitrogen, 1:1000) was applied for 1 h at room temperature. Next the cells were washed and mounting medium (Fluoroshield with DAPI, Sigma) was applied. Slides were observed under the blue and green channels of the fluorescent microscope. The data of STMN1 immunofluorescence can be found in Supplementary material (Figure S2).
Transfection of siRNAs
Reverse transfections were performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocol as described previously (Thakur et al., 2018). Stathmin 1 and MAGED2 Human siRNA Oligo Duplex was purchased from Origene. Universal scrambled negative control siRNA duplex was used as Control siRNA.
Quantification and statistical analysis
Statistics
The quality control of ChIP-seq was made by Active Motif (Carlsbad, CA). At least three independent experiments were performed for western blotting. A Student’s ttest was used for statistics analysis of other quantitative data. A p-value of <0.05 was considered to be statistically significant.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R01 ES031822, R01 ES028335, R01 ES028263, and Research Start-up fund of the Stony Brook University to FC. PMS is supported by NIH grants P30 ES020957, P30 CA022453 and S10 OD030484.
Author contributions
Conceptualization, F.C.; Experiments, C.T., Y.Q., Q.Z., M.Y., Z.B., Y.F., P.W., B.A., and A.S.; Proteomics and data analysis, N.J.C. and P.M.S.; Writing – Initial Draft, C.T.; Writing – Review and Editing, F.C.
Declaration of interests
The authors declare no competing interests.
Published: October 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105057.
Supplemental information
Data and code availability
This article does not report original code. Any additional information required to reanalyze the data reported in this article is available from the lead contacton request. Raw and processed ChIP-seq data have been deposited at GEO and publicly available at the date of publication. Accession numbers are listed in the key resources table.
References
- Aytes A., Giacobbe A., Mitrofanova A., Ruggero K., Cyrta J., Arriaga J., Palomero L., Farran-Matas S., Rubin M.A., Shen M.M., et al. NSD2 is a conserved driver of metastatic prostate cancer progression. Nat. Commun. 2018;9:5201. doi: 10.1038/s41467-018-07511-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero M.T., Hanna J.A., Neumeister V., Cheng H., Molinaro A.M., Harris L.N., Rimm D.L. Stathmin expression and its relationship to microtubule-associated protein tau and outcome in breast cancer. Cancer. 2012;118:4660–4669. doi: 10.1002/cncr.27453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi Z., Zhang Q., Fu Y., Seno A., Wadgaonkar P., Qiu Y., Almutairy B., Xu L., Zhang W., Thakur C., Chen F. Cooperation between NRF2-mediated transcription and MDIG-dependent epigenetic modifications in arsenic-induced carcinogenesis and cancer stem cells. Semin. Cancer Biol. 2021;76:310–318. doi: 10.1016/j.semcancer.2021.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkbak N.J., McGranahan N. Cancer genome evolutionary trajectories in metastasis. Cancer Cell. 2020;37:8–19. doi: 10.1016/j.ccell.2019.12.004. [DOI] [PubMed] [Google Scholar]
- Bott A.J., Peng I.C., Fan Y., Faubert B., Zhao L., Li J., Neidler S., Sun Y., Jaber N., Krokowski D., et al. Oncogenic Myc induces expression of glutamine synthetase through promoter demethylation. Cell Metab. 2015;22:1068–1077. doi: 10.1016/j.cmet.2015.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown T.C., Murtha T.D., Rubinstein J.C., Korah R., Carling T. SLC12A7 alters adrenocortical carcinoma cell adhesion properties to promote an aggressive invasive behavior. Cell Commun. Signal. 2018;16:27. doi: 10.1186/s12964-018-0243-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W.L., Greer C.B., Chen J.F., Arnal-Estape A., Cao J., Yan Q., Nguyen D.X. Specific chromatin landscapes and transcription factors couple breast cancer subtype with metastatic relapse to lung or brain. BMC Med. Genomics. 2020;13:33. doi: 10.1186/s12920-020-0695-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey L., Winer E., Viale G., Cameron D., Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010;7:683–692. doi: 10.1038/nrclinonc.2010.154. [DOI] [PubMed] [Google Scholar]
- Cha S., Sin M.J., Kim M.J., Kim H.J., Kim Y.S., Choi E.K., Kim M.Y. Involvement of cellular prion protein in invasion and metastasis of lung cancer by inducing Treg cell development. Biomolecules. 2021;11:285. doi: 10.3390/biom11020285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J., Ji X., Deng T., Qiu J., Ding Z., Li Z., Ma Y., Hu X., Li L., Qiu J. Setd2 determines distinct properties of intestinal ILC3 subsets to regulate intestinal immunity. Cell Rep. 2022;38:110530. doi: 10.1016/j.celrep.2022.110530. [DOI] [PubMed] [Google Scholar]
- Chung F.Y., Cheng T.L., Chang H.J., Chiu H.H., Huang M.Y., Chang M.S., Chen C.C., Yang M.J., Wang J.Y., Lin S.R. Differential gene expression profile of MAGE family in taiwanese patients with colorectal cancer. J. Surg. Oncol. 2010;102:148–153. doi: 10.1002/jso.21580. [DOI] [PubMed] [Google Scholar]
- Cui G., Cai F., Ding Z., Gao L. HMGB2 promotes the malignancy of human gastric cancer and indicates poor survival outcome. Hum. Pathol. 2019;84:133–141. doi: 10.1016/j.humpath.2018.09.017. [DOI] [PubMed] [Google Scholar]
- D'Souza R.S., Casanova J.E. The BRAG/IQSec family of Arf GEFs. Small GTPases. 2016;7:257–264. doi: 10.1080/21541248.2016.1219442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damaghi M., Tafreshi N.K., Lloyd M.C., Sprung R., Estrella V., Wojtkowiak J.W., Morse D.L., Koomen J.M., Bui M.M., Gatenby R.A., Gillies R.J. Chronic acidosis in the tumour microenvironment selects for overexpression of LAMP2 in the plasma membrane. Nat. Commun. 2015;6:8752. doi: 10.1038/ncomms9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmedt C., Haibe-Kains B., Wirapati P., Buyse M., Larsimont D., Bontempi G., Delorenzi M., Piccart M., Sotiriou C. Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin. Cancer Res. 2008;14:5158–5165. doi: 10.1158/1078-0432.CCR-07-4756. [DOI] [PubMed] [Google Scholar]
- Duns G., van den Berg E., van Duivenbode I., Osinga J., Hollema H., Hofstra R.M., Kok K. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res. 2010;70:4287–4291. doi: 10.1158/0008-5472.CAN-10-0120. [DOI] [PubMed] [Google Scholar]
- Ganguly K.K., Pal S., Moulik S., Chatterjee A. Integrins and metastasis. Cell Adh. Migr. 2013;7:251–261. doi: 10.4161/cam.23840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B., Liu X., Li Z., Zhao L., Pan Y. Overexpression of EZH2/NSD2 histone methyltransferase Axis predicts poor prognosis and accelerates tumor progression in triple-negative breast cancer. Front. Oncol. 2020;10:600514. doi: 10.3389/fonc.2020.600514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge W., Wolf A., Feng T., Ho C.H., Sekirnik R., Zayer A., Granatino N., Cockman M.E., Loenarz C., Loik N.D., et al. Oxygenase-catalyzed ribosome hydroxylation occurs in prokaryotes and humans. Nat. Chem. Biol. 2012;8:960–962. doi: 10.1038/nchembio.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glunde K., Guggino S.E., Solaiyappan M., Pathak A.P., Ichikawa Y., Bhujwalla Z.M. Extracellular acidification alters lysosomal trafficking in human breast cancer cells. Neoplasia. 2003;5:533–545. doi: 10.1016/s1476-5586(03)80037-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui G.P., Wells C.A., Browne P.D., Yeomans P., Jordan S., Puddefoot J.R., Vinson G.P., Carpenter R. Integrin expression in primary breast cancer and its relation to axillary nodal status. Surgery. 1995;117:102–108. doi: 10.1016/s0039-6060(05)80236-3. [DOI] [PubMed] [Google Scholar]
- Hamalisto S., Jaattela M. Lysosomes in cancer-living on the edge (of the cell) Curr. Opin. Cell Biol. 2016;39:69–76. doi: 10.1016/j.ceb.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han G., Wu Z., Zhao N., Zhou L., Liu F., Niu F., Xu Y., Zhao X. Overexpression of stathmin plays a pivotal role in the metastasis of esophageal squamous cell carcinoma. Oncotarget. 2017;8:61742–61760. doi: 10.18632/oncotarget.18687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R., Kanda M., Takami H., Shimizu D., Oya H., Hibino S., Okamura Y., Yamada S., Fujii T., Nakayama G., et al. Aberrant expression of melanoma-associated antigen-D2 serves as a prognostic indicator of hepatocellular carcinoma outcome following curative hepatectomy. Oncol. Lett. 2015;9:1201–1206. doi: 10.3892/ol.2014.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z., Li Z., Ma Z., Curtis C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 2020;52:701–708. doi: 10.1038/s41588-020-0628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Kong W., Jean M., Fiches G., Zhou D., Hayashi T., Que J., Santoso N., Zhu J. A CRISPR/Cas9 screen identifies the histone demethylase MINA53 as a novel HIV-1 latency-promoting gene (LPG) Nucleic Acids Res. 2019;47:7333–7347. doi: 10.1093/nar/gkz493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia B., Zhao X., Wang Y., Wang J., Wang Y., Yang Y. Prognostic roles of MAGE family members in breast cancer based on KM-Plotter Data. Oncol. Lett. 2019;18:3501–3516. doi: 10.3892/ol.2019.10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonjić N., Lucin K., Krstulja M., Iternicka Z., Mustać E. Expression of beta-1 integrins on tumor cells of invasive ductal breast carcinoma. Pathol. Res. Pract. 1993;189:979–984. doi: 10.1016/S0344-0338(11)80668-0. [DOI] [PubMed] [Google Scholar]
- Kanda M., Nomoto S., Oya H., Takami H., Shimizu D., Hibino S., Hashimoto R., Kobayashi D., Tanaka C., Yamada S., et al. The expression of melanoma-associated antigen D2 both in surgically resected and serum samples serves as clinically relevant biomarker of gastric cancer progression. Ann. Surg Oncol. 2016;23:S214–S221. doi: 10.1245/s10434-015-4457-8. [DOI] [PubMed] [Google Scholar]
- Ketterer S., Mitschke J., Ketscher A., Schlimpert M., Reichardt W., Baeuerle N., Hess M.E., Metzger P., Boerries M., Peters C., et al. Cathepsin D deficiency in mammary epithelium transiently stalls breast cancer by interference with mTORC1 signaling. Nat. Commun. 2020;11:5133. doi: 10.1038/s41467-020-18935-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil B.D., Hsueh C., Cao Y., Abi Saab W.F., Wang Y., Condeelis J.S., Bresnick A.R., Backer J.M. GPCR signaling mediates tumor metastasis via PI3Kbeta. Cancer Res. 2016;76:2944–2953. doi: 10.1158/0008-5472.CAN-15-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd M., Modlin I.M., Mane S.M., Camp R.L., Eick G., Latich I. The role of genetic markers--NAP1L1, MAGE-D2, and MTA1--in defining small-intestinal carcinoid neoplasia. Ann. Surg Oncol. 2006;13:253–262. doi: 10.1245/ASO.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Lien H.C., Jeng Y.M., Jhuang Y.L., Yuan R.H. Increased Trimethylation of histone H3K36 associates with biliary differentiation and predicts poor prognosis in resectable hepatocellular carcinoma. PLoS One. 2018;13:e0206261. doi: 10.1371/journal.pone.0206261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco R.F., Allo M., Schor I.E., Kornblihtt A.R., Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011;144:16–26. doi: 10.1016/j.cell.2010.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe J.D., Fraher D., Huang X., Mellett N.A., Meikle P.J., Sinclair A.J., Lirette S.T., Maihle N.J., Gong Z., Gibert Y. Identification of novel lipid biomarkers in xmrk- and Myc-induced models of hepatocellular carcinoma in zebrafish. Cancer Metab. 2022;10:7. doi: 10.1186/s40170-022-00283-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishige M., Hashimoto S., Ogawa E., Toda Y., Kotani H., Hirose M., Wei S., Hashimoto A., Yamada A., Yano H., et al. GEP100 links epidermal growth factor receptor signalling to Arf6 activation to induce breast cancer invasion. Nat. Cell Biol. 2008;10:85–92. doi: 10.1038/ncb1672. [DOI] [PubMed] [Google Scholar]
- Paulitschke V., Eichhoff O., Gerner C., Paulitschke P., Bileck A., Mohr T., Cheng P.F., Leitner A., Guenova E., Saulite I., et al. Proteomic identification of a marker signature for MAPKi resistance in melanoma. EMBO J. 2019;38:e95874. doi: 10.15252/embj.201695874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirlot C., Thiry M., Trussart C., Di Valentin E., Piette J., Habraken Y. Melanoma antigen-D2: a nucleolar protein undergoing delocalization during cell cycle and after cellular stress. Biochim. Biophys. Acta. 2016;1863:581–595. doi: 10.1016/j.bbamcr.2015.12.010. [DOI] [PubMed] [Google Scholar]
- Polyak K. Heterogeneity in breast cancer. J. Clin. Invest. 2011;121:3786–3788. doi: 10.1172/JCI60534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter J.G., Makohon-Moore A.P., Gerold J.M., Heyde A., Attiyeh M.A., Kohutek Z.A., Tokheim C.J., Brown A., DeBlasio R.M., Niyazov J., et al. Minimal functional driver gene heterogeneity among untreated metastases. Science. 2018;361:1033–1037. doi: 10.1126/science.aat7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers L.D., Fang Y., Foster L.J. An integrated global strategy for cell lysis, fractionation, enrichment and mass spectrometric analysis of phosphorylated peptides. Mol. Biosyst. 2010;6:822–829. doi: 10.1039/b915986j. [DOI] [PubMed] [Google Scholar]
- Saal L.H., Johansson P., Holm K., Gruvberger-Saal S.K., She Q.B., Maurer M., Koujak S., Ferrando A.A., Malmström P., Memeo L., et al. Poor prognosis in carcinoma is associated with a gene expression signature of aberrant PTEN tumor suppressor pathway activity. Proc. Natl. Acad. Sci. USA. 2007;104:7564–7569. doi: 10.1073/pnas.0702507104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santidrian A.F., LeBoeuf S.E., Wold E.D., Ritland M., Forsyth J.S., Felding B.H. Nicotinamide phosphoribosyltransferase can affect metastatic activity and cell adhesive functions by regulating integrins in breast cancer. DNA Repair. 2014;23:79–87. doi: 10.1016/j.dnarep.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Thakur C., Zhao Y., Li Y., Nie L., Zhang Q., Bi Z., Fu Y., Wadgaonkar P., Almutairy B., et al. Pathological and prognostic indications of the mdig gene in human lung cancer. Cell. Physiol. Biochem. 2021;55:13–28. doi: 10.33594/000000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims R.J., 3rd, Reinberg D. Processing the H3K36me3 signature. Nat. Genet. 2009;41:270–271. doi: 10.1038/ng0309-270. [DOI] [PubMed] [Google Scholar]
- Stock C., Schwab A. Ion channels and transporters in metastasis. Biochim. Biophys. Acta. 2015;1848:2638–2646. doi: 10.1016/j.bbamem.2014.11.012. [DOI] [PubMed] [Google Scholar]
- Tao M., Ma D., Li Y., Zhou C., Li Y., Zhang Y., Duan W., Xu X., Wang R., Wu L., Liu H. Clinical significance of circulating tumor cells in breast cancer patients. Breast Cancer Res. Treat. 2011;129:247–254. doi: 10.1007/s10549-011-1512-4. [DOI] [PubMed] [Google Scholar]
- Thakur C., Lu Y., Sun J., Yu M., Chen B., Chen F. Increased expression of mdig predicts poorer survival of the breast cancer patients. Gene. 2014;535:218–224. doi: 10.1016/j.gene.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur C., Wolfarth M., Sun J., Zhang Y., Lu Y., Battelli L., Porter D.W., Chen F. Oncoprotein mdig contributes to silica-induced pulmonary fibrosis by altering balance between Th17 and Treg T cells. Oncotarget. 2015;6:3722–3736. doi: 10.18632/oncotarget.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur C., Chen B., Li L., Zhang Q., Yang Z.-Q., Chen F. Loss of mdig expression enhances DNA and histone methylation and metastasis of aggressive breast cancer. Signal Transduct. Target Ther. 2018;3:25. doi: 10.1038/s41392-018-0027-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai J.R., Chong I.W., Chen Y.H., Yang M.J., Sheu C.C., Chang H.C., Hwang J.J., Hung J.Y., Lin S.R. Differential expression profile of MAGE family in non-small-cell lung cancer. Lung Cancer. 2007;56:185–192. doi: 10.1016/j.lungcan.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Tsuneoka M., Koda Y., Soejima M., Teye K., Kimura H. A novel myc target gene, mina53, that is involved in cell proliferation. J. Biol. Chem. 2002;277:35450–35459. doi: 10.1074/jbc.M204458200. [DOI] [PubMed] [Google Scholar]
- Turajlic S., Swanton C. Metastasis as an evolutionary process. Science. 2016;352:169–175. doi: 10.1126/science.aaf2784. [DOI] [PubMed] [Google Scholar]
- Ueo H., Sugimachi K., Gorges T.M., Bartkowiak K., Yokobori T., Muller V., Shinden Y., Ueda M., Ueo H., Mori M., et al. Circulating tumour cell-derived plastin3 is a novel marker for predicting long-term prognosis in patients with breast cancer. Br. J. Cancer. 2015;112:1519–1526. doi: 10.1038/bjc.2015.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan L., Tan C.T., Chapman A., Nonaka D., Mack N.A., Smith D., Booton R., Hurlstone A.F., Malliri A. HUWE1 ubiquitylates and degrades the RAC activator TIAM1 promoting cell-cell adhesion disassembly, migration, and invasion. Cell Rep. 2015;10:88–102. doi: 10.1016/j.celrep.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Zhu X., Dang L., Jiang H., Xie Y., Li X., Guo J., Wang Y., Peng Z., Wang M., et al. Epigenomic reprogramming via HRP2-MINA dictates response to proteasome inhibitors in multiple myeloma with t(4;14) translocation. J. Clin. Invest. 2022;132:e149526. doi: 10.1172/JCI149526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheway G., Nazlamova L., Hancock J.T. Signaling through the primary cilium. Front. Cell Dev. Biol. 2018;6:8. doi: 10.3389/fcell.2018.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K., Ghosh R., Giridhar P.V., Gu G., Case T., Belcher S.M., Kasper S. Inhibition of stathmin1 accelerates the metastatic process. Cancer Res. 2012;72:5407–5417. doi: 10.1158/0008-5472.CAN-12-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K., Li L., Thakur C., Lu Y., Zhang X., Yi Z., Chen F. Proteomic Characterization of the World Trade Center dust-activated mdig and c-myc signaling circuit linked to multiple myeloma. Sci. Rep. 2016;6:36305. doi: 10.1038/srep36305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaku K., Okabe K., Hikosaka K., Nakagawa T. NAD metabolism in cancer therapeutics. Front. Oncol. 2018;8:622. doi: 10.3389/fonc.2018.00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P., Guo L., Duan Z.J., Tepper C.G., Xue L., Chen X., Kung H.J., Gao A.C., Zou J.X., Chen H.W. Histone methyltransferase NSD2/MMSET mediates constitutive NF-kappaB signaling for cancer cell proliferation, survival, and tumor growth via a feed-forward loop. Mol. Cell Biol. 2012;32:3121–3131. doi: 10.1128/MCB.00204-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates L.R., Knappskog S., Wedge D., Farmery J.H.R., Gonzalez S., Martincorena I., Alexandrov L.B., Van Loo P., Haugland H.K., Lilleng P.K., et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. 2017;32:169–184.e7. doi: 10.1016/j.ccell.2017.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi H., Vatanmakanian M., Mahdiannasser M., Mashouri L., Alahari N.V., Monjezi M.R., Ilbeigi S., Alahari S.K. Understanding the role of integrins in breast cancer invasion, metastasis, angiogenesis, and drug resistance. Oncogene. 2021;40:1043–1063. doi: 10.1038/s41388-020-01588-2. [DOI] [PubMed] [Google Scholar]
- Yu M., Sun J., Thakur C., Chen B., Lu Y., Zhao H., Chen F. Paradoxical roles of mineral dust induced gene on cell proliferation and migration/invasion. PLoS One. 2014;9:e87998. doi: 10.1371/journal.pone.0087998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S., Natesan R., Sanchez-Rivera F.J., Li J., Bhanu N.V., Yamazoe T., Lin J.H., Merrell A.J., Sela Y., Thomas S.K., et al. Global regulation of the histone mark H3K36me2 underlies epithelial plasticity and metastatic progression. Cancer Discov. 2020;10:854–871. doi: 10.1158/2159-8290.CD-19-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Lu Y., Yuan B.Z., Castranova V., Shi X., Stauffer J.L., Demers L.M., Chen F. The Human mineral dust-induced gene, mdig, is a cell growth regulating gene associated with lung cancer. Oncogene. 2005;24:4873–4882. doi: 10.1038/sj.onc.1208668. [DOI] [PubMed] [Google Scholar]
- Zhang Q., Thakur C., Shi J., Sun J., Fu Y., Stemmer P., Chen F. New discoveries of mdig in the epigenetic regulation of cancers. Semin. Cancer Biol. 2019;57:27–35. doi: 10.1016/j.semcancer.2019.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.C., Wang J., Shao G.G., Wang Q., Qu X., Wang B., Moy C., Fan Y., Albertyn Z., Huang X., et al. Comprehensive genomic and immunological characterization of Chinese non-small cell lung cancer patients. Nat. Commun. 2019;10:1772. doi: 10.1038/s41467-019-09762-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Thakur C., Fu Y., Bi Z., Wadgaonkar P., Xu L., Liu Z., Liu W., Wang J., Kidder B.L., Chen F. Mdig promotes oncogenic gene expression through antagonizing repressive histone methylation markers. Theranostics. 2020;10:602–614. doi: 10.7150/thno.36220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Wadgaonkar P., Xu L., Thakur C., Fu Y., Bi Z., Qiu Y., Almutairy B., Zhang W., Stemmer P., Chen F. Environmentally-induced mdig contributes to the severity of COVID-19 through fostering expression of SARS-CoV-2 receptor NRPs and glycan metabolism. Theranostics. 2021;11:7970–7983. doi: 10.7150/thno.62138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Powers S. New views into the genetic landscape of metastatic breast cancer. Cancer Cell. 2017;32:131–133. doi: 10.1016/j.ccell.2017.07.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This article does not report original code. Any additional information required to reanalyze the data reported in this article is available from the lead contacton request. Raw and processed ChIP-seq data have been deposited at GEO and publicly available at the date of publication. Accession numbers are listed in the key resources table.