

ABSTRACT
Coordinated migration of the mesoderm is essential for accurate organization of the body plan during embryogenesis. However, little is known about how mesoderm migration influences posterior neural tube closure in mammals. Here, we show that spinal neural tube closure and lateral migration of the caudal paraxial mesoderm depend on transmembrane protein 132A (TMEM132A), a single-pass type I transmembrane protein, the function of which is not fully understood. Our study in Tmem132a-null mice and cell models demonstrates that TMEM132A regulates several integrins and downstream integrin pathway activation as well as cell migration behaviors. Our data also implicates mesoderm migration in elevation of the caudal neural folds and successful closure of the caudal neural tube. These results suggest a requirement for paraxial mesodermal cell migration during spinal neural tube closure, disruption of which may lead to spina bifida.
KEY WORDS: Transmembrane protein 132A, Neural tube closure, Spina bifida, Mesodermal cell migration, Integrin pathway, Cytoskeletal remodeling
Summary: Loss of TMEM132A severely impairs lateral migration of the paraxial mesoderm in the posterior trunk, leading to failure of caudal neural tube closure and contributing to spina bifida.
INTRODUCTION
Neural tube closure is a fundamental process of morphogenesis that culminates in the elevation, apposition and fusion of the neural folds in the dorsal midline (Wilde et al., 2014). The neural tube is the embryonic precursor of the brain and spinal cord, and when the neural tube does not close properly, neural tube defects (NTDs) occur. NTDs are a common malformation worldwide affecting approximately 1 in 1000 live births and causing severe childhood morbidity and mortality (Greene and Copp, 2014). Maternal prenatal folic acid (FA) supplementation can reduce the incidence of NTDs, which is now implemented in many countries (Wald et al., 2018; Wilde et al., 2014). However, FA supplementation in humans and animal models have not approached 100% NTD prevention, and at least one-third of NTDs are not folate responsive (Greene and Copp, 2014). Hence, NTDs remain of high clinical significance. Thus, elucidating the molecular mechanisms that control neural tube closure is of great relevance.
The Knockout Mouse Project (KOMP) under the International Mouse Phenotyping Consortium (IMPC) created transmembrane protein 132a (Tmem132a)-null mice, and homozygous mutant embryos show spina bifida and limb defects. TMEM132A is a single-pass type I transmembrane protein which was first identified as an interacting protein with the endoplasmic reticulum (ER)-resident chaperone 78-kDa glucose-regulated protein GRP78, also called BIP or HSPA5, and was previously named GRP78 binding protein (GBP) or HSPA5 binding protein 1 (HSPA5BP1) (Oh-hashi et al., 2003, 2010). The TMEM132 family is composed of five members (TMEM132A to E), the structure and molecular function of which are underinvestigated. TMEM132B is linked to intracranial aneurysm and excessive daytime sleepiness (Farlow et al., 2015; Lane et al., 2017). TMEM132D variants are reported in lifetime prevalence of panic disorder (Erhardt et al., 2012; Haaker et al., 2014; Quast et al., 2012; Shimada-Sugimoto et al., 2016). TMEM132E is associated with autosomal-recessive nonsyndromic hearing impairment (Li et al., 2015; Liaqat et al., 2020). To date, TMEM132A or C have not been associated with human diseases. Knockdown of TMEM132A increased resistance to cell death induced by serum starvation in cultured cells (Oh-hashi et al., 2010), Our previous study identified TMEM132A as a regulator of Wnt signaling by interacting with the Wnt ligand trafficking protein Wntless (WLS) (Li and Niswander, 2020). Wnt signaling is important in neuromesodermal progenitor (NMP) self-renewal as well as lineage choice to neural and mesodermal tissue of the trunk (Garriock et al., 2015; Gouti et al., 2014, 2017). However, our data suggest the spina bifida phenotype is unrelated to NMP differentiation, indicating a different function of TMEM132A during mammalian caudal neural tube closure.
Here, we systematically investigated the role of TMEM132A in NTD pathogenesis in the mouse. Tmem132a−/− embryos die prenatally and show ∼85% penetrance of spina bifida. Mutant embryos exhibit severely impaired lateral migration of the paraxial mesoderm in the posterior trunk. Molecularly, TMEM132A regulates the levels of several integrin proteins and activates the integrin pathway, with consequent effects on cytoskeletal remodeling. Together, our studies indicate that TMEM132A ensures successful paraxial mesoderm migration, failure of which contributes to spina bifida.
RESULTS AND DISCUSSION
Tmem132a deletion causes embryonic lethality and caudal neural tube defects
The Tmem132atm1b(KOMP)Wtsi mouse was generated by Baylor College of Medicine (TX, USA) and obtained from KOMP. This allele removes exons 2 and 3 of Tmem132a and introduces En2 splice acceptor (SA), β-galactosidase (lacZ) and a polyadenylation signal (pA) sequence to create a presumed null allele (Fig. 1A-C). Homozygous deletion of Tmem132a in mice results in defects in multiple tissues including the neural tube and limbs (Fig. 1D; Fig. S1A). Tmem132a homozygous mutant pups were never observed alive, but embryos survived at least to embryonic day (E) 18.5. The cause of lethality was not explored. Over 85% of mutant embryos at E10.5 showed severe caudal NTD of spina bifida, extending posteriorly from the hindlimb level, with curly or missing tail (Fig. 1E). Almost every embryo showed digit abnormalities in both the forelimbs and hindlimbs (Fig. S1A), and occasionally one or both hindlimbs were missing. Homozygous mutants were developmentally delayed, as judged by fewer somites compared with littermates (E8.5-E10.5; Fig. S1B) but were morphologically normal for any given somite age beyond NTD and limb defects. At E18.5, mutants remained growth retarded based on body and brain weight (Fig. S1C). To determine whether NTD could be prevented by FA, female and male mice were fed control (2 ppm) or FA-supplemented (10 ppm) diets recapitulating serum levels in humans (Marean et al., 2011) over multiple generations and F3 embryos examined. NTD severity or penetrance was not affected (Fig. S1D), indicating that loss of TMEM132A cannot be rescued by FA supplementation. These results indicate that TMEM132A plays crucial roles in mammalian embryogenesis, particularly caudal neural tube closure and limb development.
Fig. 1.

Tmem132a deletion causes embryonic lethality and spina bifida. (A) Schematic of wild-type (WT) and Tmem132atm1b(KOMP)Wtsi mutant allele. (B) Genotyping with primers targeting either WT or substituted regions as shown in A, and with Prdm14 primers as internal control. (C) TMEM132A (T132A) protein expression in WT, heterozygous or homozygous mutant embryos with relative band intensity normalized to GAPDH internal control listed below. (D) Representative whole-mount images of E8.5, E9.5 and E10.5 WT and mutant embryos. Green arrowheads mark posterior neuropore (PNP) in WT and red arrowheads indicate enlarged PNP in mutants. (E) All genotypes are observed at expected frequency but ∼85% of homozygous mutant embryos exhibit spina bifida at E10.5. Numbers of embryos examined are shown in each column. Scale bars: 1 mm.
TMEM132A is required for lateral migration of caudal paraxial mesodermal cells
To determine the temporal and spatial expression pattern of TMEM132A during neural tube closure, we examined E8.5-E10.5 embryos for β-galactosidase expression (Fig. 2A) or RNA in-situ hybridization (ISH; Fig. S2A). Expression of TMEM132A is widespread, with stronger expression in neuroepithelium, limb buds, mesoderm and endoderm.
Fig. 2.

TMEM132A deficiency leads to defective lateral migration of caudal mesoderm. (A) Whole-mount β-galactosidase staining of Tmem132a expression during neural tube closure from E8.5 to E10.5. (B) Mutant embryos have an elongated PNP at E9.5 (22-24 somites; n=4). (C) Mutant embryos have convex everted neuroepithelium (NE) by bright field and by H&E staining of transverse sections around the neuropore (sections in panels 1-4 refer to whole-mount picture, reproduced from Fig. 1D). Green arrowheads mark posterior neuropore (PNP) in WT and red arrowheads indicate enlarged PNP in mutants. (D) H&E staining shows all tissue types in mutants, but NE shows reverse bending at the medial hinge point (MHP)/notochord (NC), exaggerated dorsolateral hinge points (DLHP) and abnormal positioning of mesoderm near the midline. SE, surface ectoderm. Panels D2 and D2′ are labeled copies of C2 and C2′, respectively. (E) Paraxial somite marker Uncx4.1 and presomitic mesoderm marker Meox1 expression is grossly normal but transverse sections highlight the abnormal mesoderm mass that is Meox1 positive. (F) Schematic (adapted from de Goederen et al., 2022) indicating measurements to quantify mesoderm area flanking NE and curvatures of the DLHP and MHP (n=3 for WT and mutant). Data are mean±s.d. **P<0.01 (two-tailed Student's t-test). Scale bars: 1 mm for embryos; 100 μm for sections.
Focusing on E9.5 embryos during caudal neural tube closure, the length of the posterior neuropore (PNP) is significantly increased in mutants (Fig. 2B), with a markedly convex and everted appearance of the neuroepithelium (NE) (Fig. 2C). Transverse sections around the PNP zippering point showed the presence of all tissues – NE, non-neural ectoderm or surface ectoderm (SE), notochord (NC), mesoderm and endoderm in mutants (Fig. 2C,D). However, the medial hinge point (MHP) is severely disrupted, exhibiting reversed bending of the NE overlaying the NC. Anteriorly where the neural tube has closed, the pair of dorsal-lateral hinge points (DLHPs) form in the proper direction but the degree of bending is greatly exaggerated. In regions where the neural folds remain open, the NE is everted in mutant embryos, compared with wild type in which the neural folds are elevated to ultimately bring them into apposition to fuse. Strikingly, mutants have a mass of mesoderm-like cells underneath the medial NE and NC, whereas normally the mesoderm migrates anteriorly and laterally to form the paraxial mesoderm/somites. In wild-type embryos there is little to no mesodermal tissue left between NE/NC and endothelial tissues at the midline.
To assess the lineage of the abnormally localized mesoderm-like cells, we used various molecular markers. Trunk NE and adjacent paraxial mesoderm arise from shared, dual-fated NMPs marked by co-expression of neural progenitor cell regulator Sox2 and pan-mesodermal transcription factor brachyury (T) (Henrique et al., 2015; Rodrigo Albors and Storey, 2016). The antagonistic activities of Sox2 and T control NMP fate choice into neural or mesodermal lineage (Gouti et al., 2017; Gouti et al., 2014; Koch et al., 2017). An imbalance in NMP differentiation, for example mutations that impact pathways regulating NMP fate choice like Wnt signaling, can cause caudal NTDs (Garriock et al., 2015; Nowotschin et al., 2012; Takemoto et al., 2011). Evaluation of E9.5 mouse embryos with Sox2 and T, neural lineage markers Sox1 and Sox3, and mesodermal lineage makers Tbx6 and Msgn1 (Fig. S2B,C), showed no significant change due to TMEM132A loss, suggesting NMP differentiation was not disrupted in mutant embryos.
Paraxial/presomitic mesoderm (PSM) originates from and then migrates anteriorly and laterally out of caudal-lateral epiblast (CLE) (Koch et al., 2017; Steventon and Martinez Arias, 2017). The gross expression of the paraxial somite marker Uncx4.1 (also known as Uncx) and the PSM marker Meox1 appeared to be normal in E9.5 Tmem132a mutants (Fig. 2E). However, Meox1 positive mesoderm cells were abnormally located underneath the medial NE/NC of mutants, corresponding with the histology in Fig. 2C,D. These data indicate aberrant positioning of the paraxial mesoderm in Tmem132a mutant embryos.
Recent mathematical modeling and quantitative image analysis suggests that mesoderm expansion laterally is important for caudal neural tube closure (de Goederen et al., 2022). This modeling highlights extrinsic forces, including lateral expansion of the paraxial mesoderm, as being sufficient for MHP formation and neural fold elevation. Fig. 2C-E histology shows abnormal paraxial mesoderm expansion in Tmem132a mutants, whereas another parameter modeled of notochord adhesion to the neural plate is not disrupted. Furthermore, the modeling by de Goederen et al. (2022) shows that low mesoderm expansion results in exaggerated DLHPs. Indeed, at E9.5 during mode 2 closure, the mesoderm area flanking the NE is sharply decreased in mutant embryos, whereas the DLHPs are greatly exaggerated and MHP bending is severely reduced (Fig. 2F). Thus, our data provide in vivo evidence to support the computational modeling by de Goederen et al. (2022) that increased mesoderm expansion is a necessary parameter in neural fold elevation and closure. We suggest that the abnormal mass of paraxial mesoderm underlying the NE forces the NE to bend in the reverse direction and/or the mesoderm is not present laterally to help to elevate the neural folds. In summary, our data indicate an important role for TMEM132A in appropriate positioning of the paraxial mesoderm.
TMEM132A does not regulate tissue patterning
Based on the Tmem132a mutant histology, several additional possible deficits were examined. We found two other rodent models in the literature with similar histology to Tmem132a mutants. The first is pharmacological disruption of heparan sulphate by chlorate treatment of cultured E8.5 mouse embryos (Yip et al., 2002). Treated embryos had a flat or convex NE and accentuated DLHP bending. However, the authors noted chlorate treatment accelerates PNP closure, whereas the Tmem132a mutant PNP is enlarged and does not close. The second model is excess all-trans-retinoic acid (RA) treatment of E9.5 rat embryos in culture, which caused NE eversion at the caudal neuropore (Seegmiller et al., 1991). In mammals, anterior-posterior patterning is controlled by the antagonistic activities of RA versus fibroblast growth factor (FGF) and Wnt signaling pathways (Gouti et al., 2015; Kumar and Duester, 2014). Comparison of wild-type and Tmem132a mutant E9.5 embryos showed no major changes in RNA expression of RA synthetic enzyme gene Aldh1a2, RA degrading-enzyme gene Cyp26a1 and Fgf8, except that the most-caudal epiblast expression of Aldh1a2 is largely missing in mutants (Fig. S2D). These data suggest a different mechanism for TMEM132A function.
We examined additional tissue patterning markers to determine whether their expression was impacted in mutants. Cdx2 transcription factor is required for axial elongation and hindgut endoderm (Chawengsaksophak et al., 2004; Savory et al., 2009, 2011). In Tmem132a mutants, Cdx2 was present in the tail bud, with perhaps a change in the anterior extent of expression, but was significantly downregulated in the hindgut (Fig. S2D). Dorsal-ventral patterning is controlled by sonic hedgehog (Shh), Wnt and BMP signaling in chicken and mouse embryos (Cai and Liu, 2017; Dessaud et al., 2008; Eom et al., 2011). Expression was similar between E9.5 wild type and mutants for the roof plate marker Msx1, the dorsal neural fold, neural crest cell and somite marker Pax7, the ventral neural plate marker Foxa2 and the floor plate and NC marker Shh (Fig. S2E). Foxa2 expression depends on high Shh signaling (Cai and Liu, 2017). The apparently normal Foxa2 expression suggests normal Shh signaling from the NC and floor plate/MHP.
TMEM132A regulates cell migration
The striking defect in paraxial mesoderm positioning led us to explore whether cell migration and cell behaviors are affected by loss of TMEM132A function. Using a scratch assay to visualize cells as they migrate into the free space, siRNA knockdown of TMEM132A in HeLa cells (Fig. 3A,B) and HEK293 cells (stably transfected with turbo GFP; Fig. S3A,B) showed a significant defect in cell migration (Fig. 3C-E; Fig. S3C; Movies 1-9,10-18 showing fluorescent and brightfield videos, respectively, collected concurrently in HeLa cells, and Movies 19-21 showing fluorescent videos in HEK293 cells). In control siRNA experiments, the cells moved directionally to close the wound (Fig. 3E) and most cells at the wound front displayed lamellipodium (Fig. 3F; Movies 10-18). In contrast, TMEM132A knockdown caused the cells to move randomly with erratic trajectories and the cells at the wound front had significantly thinner protrusions that resembled filopodium.
Fig. 3.

TMEM132A regulates cell migration in wound healing assay in HeLa cells. (A) Knockdown of Tmem132A in HeLa cells by siRNAs #1 and #2 relative to negative control siRNA, assayed 48 h post transfection by RT qPCR with GAPDH as internal control (n≥3, two-tailed Student's t-test, **P≤0.01). Data are mean±s.d. (B) Immunoblotting assay demonstrates successful knockdown of TMEM132A (48 h post siRNA transfection), repression of RhoA activation and reduced phosphorylation of cofilin and MLC2. Relative band intensity given below each corresponding panel (TMEM132A normalized to GADPH; activated RhoA and phosphorylated proteins normalized to corresponding total protein). Relative protein levels <0.80 shown in red. (C) Wound healing assay in HeLa cells shows defective cell migration into the wound upon TMEM132A siRNA knockdown (still images at indicated time points). (D) Percentage of wound area closed calculated with initial cell-free space set as 100% in each group. (E) Paths of individual cells in wound healing assay tracked over time. Upper panel: black dots indicate initial position, colored dots indicate final position. Middle panel: cell paths tracked normalized to an initial position. Lower panel: radial histograms calculated from degree of displacement of each cell, with 90 degrees indicating a direct horizontal path from the starting point. Degrees were normalized to account for which side of the wound the cell originated. (F) Visualization of cells at the leading edge of the wound 12 h post insert removal and quantification of percentage of lamellipodia and filopodia in control siRNA and TMEM132A knockdown cells. Green arrowheads mark lamellipodia and red arrowheads indicate filopodia. **P<0.01 (Fisher's exact test). Scale bars: 250 μm (C); 25 μm (F).
Efficient generation of lamellipodia underlies directional cell motility (Zhang et al., 2012). Lamellipodia formation is regulated by cytoskeletal remodeling through the small GTPases RhoA and Rac. Downstream targets of small GTPases are the actin-severing protein cofilin and LIM kinase, which phosphorylates cofilin at Ser3 to inhibit its severing activity, thus regulating actin depolymerization. Small GTPases also initiate phosphorylation of myosin light chain 2 (MLC2) at Ser19 to control Myosin ATPase-dependent contractile activity (Lawson and Ridley, 2018). Immunoblotting analysis showed that TMEM132A knockdown significantly impaired RhoA activation, and cofilin and MLC2 phosphorylation (Fig. 3B), indicating a defect in cytoskeleton remodeling due to alterations in actomyosin polymerization.
TMEM132A regulates several integrins
The WAVE regulatory complex (WRC) interacts with diverse membrane proteins such as PCDH10 containing the WRC interacting receptor sequence (WIRS) cytoplasmic motif to control actin dynamics by transmitting information from Rac GTPase to the actin nucleator Arp2/3 complex (Chen et al., 2014; Ismail et al., 2009). A conserved WIRS motif exists in the cytoplasmic domain of TMEM132A, C, D and E proteins (Sanchez-Pulido and Ponting, 2018) (Fig. S4A). The Caenorhabditis elegans homolog of the human panic–disorder risk gene TMEM132D orchestrates neuronal morphogenesis through the WRC (Wang et al., 2021). However, endogenous WAVE1, 2 and 3 (WASF1, 2 and 3), failed to be co-immunoprecipitated by overexpressed C-terminus of mouse TMEM132A. Parallel experiments using human PCDH10-positive control showed pull down of WAVE proteins, and PCDH10 interaction was abolished by mutating two conserved amino acids in the WIRS motif (Fig. S4B). This suggests that TMEM132A may not act via the WRC.
Efficient migration of a population of cells is regulated by cell-cell interactions, which are determined by tight junction and adhesion junction proteins (Friedl and Mayor, 2017). However, in TMEM132A knockdown HeLa cells, there was no obvious change in protein levels of apical tight junction protein ZO-1 (TJP1), or calcium-dependent adhesion junction proteins cadherin(s), P120 (CTNND1), β-catenin, α-catenin and IQGAP1, or calcium-independent adhesion junction proteins afadin and Nectin1 (Fig. S4C). Directional cell migration depends on cell-matrix adhesion through focal adhesions involving integrin-matrix interactions (Petrie et al., 2009; Shafaq-Zadah et al., 2016). Integrin synthesis, trafficking or stability affects cell attachment to the basement matrix, as well as downstream cytoskeletal remodeling inside cells. These extracellular and intracellular activities underlie cell behavior such as lamellipodia formation and directional migration (Nader et al., 2016; Paul et al., 2015; Shafaq-Zadah et al., 2016). Strikingly, TMEM132A knockdown in HeLa cells downregulated β1 integrin, but not β3 and β5 integrins (Fig. 4A,B; Fig. S4C).
Fig. 4.
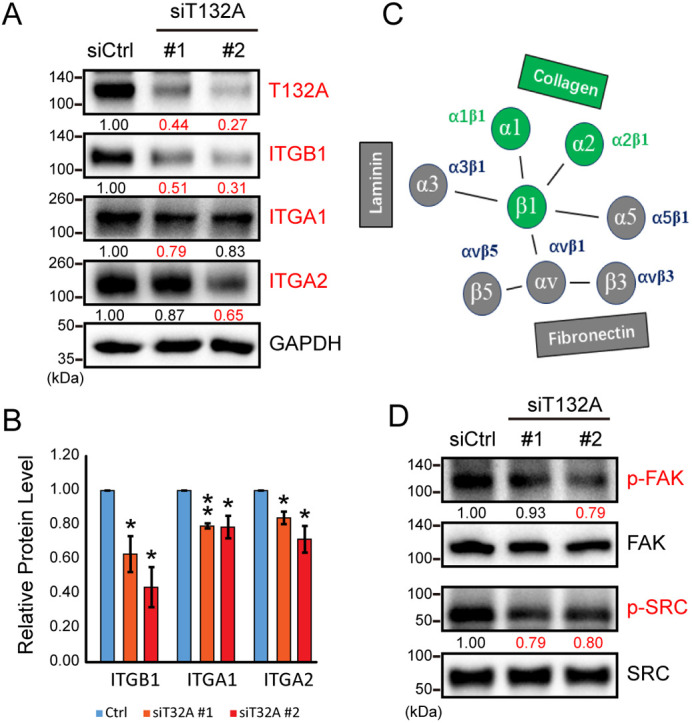
TMEM132A regulates integrin protein levels. (A) Integrin proteins α1, α2 and β1, which form the collagen binding receptors, were repressed by TMEM132A knockdown in HeLa cells (48 h post-transfection; GAPDH internal control). (B) Quantification of β1, α1 and α2 integrin protein levels from western blot band intensity (n=3). Data are mean±s.d. *P<0.05, **P<0.01 (two-tailed Student's t-test). (C) Representative integrin heterodimers that bind different matrix proteins. Collagen-binding integrin heterodimers that are repressed by TMEM132A loss are highlighted in green. (D) TMEM132A knockdown shows decreased phosphorylation of FAK and SRC in HeLa cells 48 h post-transfection. Relative band intensity of phosphorylated proteins normalized to corresponding total proteins is given below corresponding panel. Relative protein levels <0.80 are shown in red.
In mammals, heterodimerization of the 18 α integrin and 8 β integrin subunits occurs within the ER, leading to assembly of 24 distinct integrin receptors. Further post-translational modifications occur in the Golgi, followed by trafficking of integrin receptors to the cell surface where they perform a myriad of functions (Paul et al., 2015). Integrins are broadly categorized into RGD receptors, collagen receptors, laminin receptors and leukocyte-specific receptors (Fig. 4C). An RGD receptor of α5β1 integrin mediates basal anchorage to drive epithelial zippering during mouse spinal neural tube closure (Mole et al., 2020). However, we did not observe obvious changes in protein expression of α5 or αv, which assemble into RGD receptors, or α3, which forms laminin receptors. Instead, there was specific downregulation of α1 and α2 integrin proteins that form the collagen receptors along with β1, which was also downregulated (Fig. 4A,B). This indicates that TMEM132A functions in controlling the protein levels of collagen-binding integrin heterodimers.
Integrins and other proteins form a focal adhesion complex to establish a mechanical link between the extracellular matrix and intracellular cytoskeleton, and this is crucial for cell migration. However, there was no significant change in protein levels of other focal adhesion complex components such as talin, paxillin, vinculin and ACTN1 in TMEM132A-knockdown cells (Fig. S4C). Phosphorylation of FAK and SRC kinases is an indication of integrin pathway activation (Nader et al., 2016). As would be predicted by the decreased levels of several integrins, TMEM132A knockdown resulted in reduction of both FAK and SRC phosphorylation (Fig. 4D). These results of decreased integrin protein levels and reduced integrin pathway activation are consistent with disrupted cytoskeletal remodeling and cell migration observed upon loss of TMEM132A.
In summary, our studies of Tmem132a−/− mouse model highlight the role of TMEM132A in regulating mesoderm migration, which contributes to neural fold elevation and successful caudal neural tube closure. Loss of TMEM132A disrupts migration of paraxial mesodermal cells in the caudal trunk region. Our data suggests that lack of TMEM132A function in the mesoderm is the predominant cause of caudal NTD but tissue-specific knockout experiments are needed to confirm a specific role in the paraxial mesoderm. TMEM132A regulates the level of integrin proteins that preferentially bind collagen. This serves to activate the integrin signaling pathway to regulate actomyosin polymerization and cytoskeletal remodeling, which is required for lamellipodium formation, cell motility and PSM migration during mammalian caudal neural tube closure.
MATERIALS AND METHODS
Mouse model
Tmem132atm1b(KOMP)Wtsi mice (Colony Tmem132a EPD0111_5_B06 in C57BL/6N genetic background) was generated by Baylor College of Medicine and obtained from KOMP. This study and animal handling were reviewed and approved by Institutional Animal Care and Use Committee (IACUC) at the University of Colorado Boulder.
Phenotypic analysis of embryos was either performed by us or by microCT (Fig. S1A) by Baylor College of Medicine at indicated embryonic stages. Three-dimensional reconstructions of microCT data are available through https://www.mousephenotype.org/embryoviewer/?mgi=MGI:2147810. DNA isolated from the embryonic yolk sac was used for genotyping with primers targeting wild-type allele (W1 F/R: 5′-CCGATCCCTGACTCTCCAAC-3′/5′-ATGCTCAGGGGCATCTAGGA-3′; W2 F/R: 5′-AATCCCACCTTCCCAGACCT-3′/5′-AGCCCTCTCCTAGGGCAATC-3′), mutant allele (M1 F/R: 5′-TGCTGCATGGACCTATTTGG-3′/5′-GGGAAAGGGTTCGAAGTTCC-3′; M2 F/R: 5′-TCTGGATCCGGAATAACTTCG-3′/5′-ACGGCTCTTTCCAGTGGGTA-3′) and Prdm14 as internal control (Int. Ctrl F/R: 5′-CTAGGCCACAGAATTGAAAGATCT-3′/5′-GTAGGTGGAAATTCTAGCATCATCC-3′).
The FA supplementation experiment was performed as previously described (Marean et al., 2011). Briefly, males and females were bred and maintained on two diets that only differed in the concentration of FA (2 ppm for the control FA diet or 10 ppm for the high FA diet) and F1 and F2 animals were kept on one of these diets. The F2 heterozygous males and females from the same diet were timed-mated (with noon of the day a seminal plug was detected considered as E0.5). F3 embryos were dissected at E10.5 and scored for phenotype and genotype.
Histology of mouse embryos
Timed-mated pregnant female mice were euthanized and embryos dissected in PBS at various stages, the number of somites counted, and yolk sac used for genotyping. For all wild type to mutant comparisons, we used somite-matched embryos (two somite or less difference). For cryosectioning, embryos were cryoprotected in 30% sucrose (in PBS) overnight at 4°C, then rinsed sequentially in 2:1, 1:1 and 1:2 (v/v) of 30% sucrose:O.C.T (Tissue-Tek). They were then embedded in O.C.T, sealed and stored at −80°C until use. Cryosections (12 µm) were cut using a Leica CM3050 S research cryostat and mounted on Fisherbrand Superfrost Plus Microscope Slides and stored at −80°C before use.
For whole-mount β-galactosidase staining, embryos were fixed at room temperature in Fix solution (2% formaldehyde, 0.2% glutaraldehyde in PBS) for 15 min, 30 min or 60 min for embryos at E8.5, E9.5 or E10.5, respectively. After a wash and 30 min incubation in PBS, embryos were transferred into Staining solution [PBS with 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, 2 mM magnesium chloride and 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) in dimethylformamide (DMF)] overnight in the dark at room temperature with gentle agitation. Embryos were rinsed quickly and then incubated for 30 min in PBS, fixed again overnight and then photographed.
Whole-mount in situ hybridization (WISH) was performed according to published protocols (Biris and Yamaguchi, 2014). Mouse riboprobe constructs for Uncx4.1 (SalI-T7) was kindly provided by Dr Terry Yamaguchi (Chalamalasetty et al., 2014) and Msgn1 (XbaI-Sp6) was a gift from Dr Olivier Pourquié (Chal et al., 2015). Sox2 (AccI-T3), Brachyury (T) (BamHI-T7), Tbx6 (HindIII-T3), Fgf8 (PstI-T7), Msx1(BamHl-T7), Foxa2 (BamHI-T3) and Shh (HindIII-T3) are Niswander lab mouse ISH stock #53, #25, #27, #10, #133, #42 and #41, respectively. The following mouse riboprobes were amplified by the following primer pairs and subcloned into pCR-Blunt II-TOPO (Thermo Fisher Scientific, #450245). Meox1 (EcoRV-Sp6) 646 bp amplicon generated by primer pair 5′-TGTGGGAGCCTTGAATTTCC-3′/5′-CACAGGGGTCTCTTCCAAGG-3. Tmem132a (EcoRV-Sp6) 336 bp amplicon with primer pair 5′-CGCTCAGACACCAGGATCT-3′/5′-TGCGAGCCTTCTTACTCTGC-3′. Sox1 (EcoRV-Sp6) 549 bp amplicon with primer pair 5′-TGTATGACTGCCGGCTCTGT-3′/5′-ACTCTTGGGCCCTGCAGATA-3′. Sox3 (SpeI-T7) 468 bp amplicon with primer pair 5′-AGCGGAAATGGGACTTGCTA-3′/5′-AAATAACCCCTTCCCCACCA-3′. Aldh1a2 (SpeI-T7) 439 bp amplicon with primer pair 5′-CCCCTTGTCCATTCACCACT-3′/5′-TGGGTGCACTGTAGGAGGAA-3. Cyp26a1 (SpeI-T7) 526 bp amplicon with primer pair 5′-AGTAACCTGGGCGGCCTTAT-3′/5′-GGCTGAAGGCCTGCATAATC-3′. Cdx2 (SpeI-T7) 644 bp amplicon with primer pair 5′-TCCTCTCCTCCTACCCACGA-3′/5′-GGGACAGGAAGTCCAGGTTG-3′. Pax7 (SpeI-T7) 555 bp amplicon with primer pair 5′-TGGATCACCCTCATCCAGTG-3′/5′-TGTGGAGGAGGATGCATTTG-3′.
Hematoxylin and Eosin (H&E) staining protocol was performed on cryosections according to procedure posted on IHCWORLD website (https://www.ihcworld.com/_protocols/special_stains/h&e_ellis.htm). Sections were then mounted and stored at 4°C before imaging.
Whole-mount imaging was performed using a Nikon SMZ18 stereomicroscope. Section images were captured on Nikon TiU wide field with color brightfield camera under 10×0.45NA Plan Apo DIC N1 WD 4 (mm) objective. Images were processed and analyzed using Fiji (NIH) software (Schindelin et al., 2012).
Cell culture and transfection
HEK293 cells [American Type Culture Collection (ATCC), #CRL-1573] stably transfected with pSCDTG(P) expressing turbo GFP (Li and Niswander, 2020), HEK293 T cells (ATCC, #CRL-3216) and HeLa cells (ATCC, #CCL-2) were cultured in high-glucose Dulbecco's Modified Eagle Medium (DMEM, Thermo Fisher Scientific, #11995065) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, #A3840101) at 37°C with 5% CO2.
Cells were seeded and maintained to reach ∼50% confluency at the time of transfection. PEI MAX 40 K (Polysciences, #24765-1, 3-5 μg per μg plasmid) or Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific, #L3000015, 3 μl per µg plasmid) were used for plasmid transfection. Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific, #13778-150, 3 µl per 10 pmol siRNA) was used for siRNA transfection, following the manufacturer's protocol. The following Silencer Select siRNAs from Thermo Fisher Scientific were used at 10 mM final concentration: Negative control No. 1 siRNA (siCtrl, #4390843), human TMEM132A No.1 siRNA (siT132A #1, #s29890) and human TMEM132A No. 2 siRNA (siT132A #2, #s29892).
Real-time quantitative PCR
Total RNA was extracted from cultured cells 48 h post siRNA transfection using TRIzol Reagent (Thermo Fisher Scientific, #15596018) and cDNA generated by reverse transcription using SuperScript IV First-Strand Synthesis System (Thermo Fisher Scientific, #18091050) according to the manufacturer's instructions. Real-time quantitative PCR was performed on a Roche LC480 thermocycler using Luna Universal Probe qPCR Master Mix (New England Biolabs, #M3004X). Relative fold change in gene expression was calculated as described previously (Li et al., 2013a,b). Primers and Universal Probe Library (UPL, Roche Diagnostics) used were: human TMEM132A F/R (+ UPL #1), 5′-CCCTGGACGTCGTGAGAG-3′/5′-GAAGTGTTCAGGGGCGTCTA-3′; human GAPDH F/R (+ UPL #45), 5′-TCCACTGGCGTCTTCACC-3′/5′-GGCAGAGATGATGACCCTTTT-3′.
Cell lysis, immunoprecipitation and immunoblotting assay
Medium was removed from cultures and cells washed once in PBS. Cytoplasmic proteins were extracted for immunoprecipitation (IP)/co-IP experiments using n-Dodecyl-beta-Maltoside Detergent (DDM, Thermo Fisher Scientific, #89903) or NP-40 lysis buffer [50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% glycerol (v/v), 1 mM EDTA, 1% DDM or 0.5% NP-40 (v/v)], supplemented with proteinase and phosphatase inhibitor cocktail [1 µg/ml Aprotinin (Calbiochem, #616370), 1 µg/ml Leupeptin (Calbiochem, #108975), 1 µg/ml Pepstatin (Calbiochem, #516481), 1 mM Na3VO4, 10 mM NaF] and 10 units/ml Benzonase Nuclease (MilliporeSigma, #E1014). Lysate was clarified by centrifugation to remove insoluble precipitates (∼20,000 g, 15 min at 44°C) and protein concentration was measured using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, #23225). Anti-Flag M2 magnetic beads (MilliporeSigma, #M8823) were used for the IP/co-IP experiment. Beads were then washed three times using lysis buffer and boiled at 70°C for 10 min in LDS Sample Buffer (Thermo Fisher Scientific, #84788) supplemented with 50 mM dithiothreitol (DTT).
For whole cell lysis, RIPA buffer [50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 10% glycerol (v/v), 1 mM EDTA, 1% NP-40 (v/v), 0.5% sodium deoxycholate (w/v), 0.1% sodium dodecyl sulfate (SDS) (v/v)], supplemented with proteinase and phosphatase inhibitor cocktail and 10 units/ml Benzonase Nuclease was used. Lysate was clarified by centrifugation (∼20,000 g, 15 min at 4°C), supernatant collected, and protein concentration was measured by BCA assay. Whole cell lysate was then denatured by the addition of LDS sample buffer supplemented with 50 mM DTT, followed by boiling at 70°C for 10 min.
Generally, 15-30 µg of each lysate was loaded onto 4-12% NuPAGE Bis-Tris gels (Thermo Fisher Scientific, #NP0321BOX), followed by SDS-PAGE with NuPAGE MOPS SDS running buffer (Thermo Fisher Scientific, NP0001) or MES SDS running buffer (Thermo Fisher Scientific, #NP0002) supplemented with NuPAGE Antioxidant (Thermo Fisher Scientific, #NP0005). The proteins were electro-transferred to immobilon-FL PVDF, 0.45 µm (MilliporeSigma, #IPFL85R) or Nitrocellulose Membrane, 0.2 µm (Bio-Rad, #1620112), and subjected to immunoblotting assay against the following antibodies (at 1:1000 dilution unless otherwise stated): TMEM132A (Proteintech, #25301-1-AP), GAPDH (MilliporeSigma, #G9545, 1:5000), RhoA (provided in Rho Activation Assay Biochem Kit, Cytoskeleton, #BK036), Cofilin [Cell Signaling Technology (CST), #5175S], phospho-Cofilin (S3) (CST, #3313S), MLC2 (CST, #8505S), phosphor-MLC2 (S19) (CST, #3671S), ZO-1 (Thermo Fisher Scientific, #61-7300), pan-Cadherin (CST, 4068S), P120 (BD Biosciences, #610133), β-Catenin (ZYMED, #18-0266), α-Catenin (CST, #3236S), IQGAP1 (CST, #2901S), Afadin (R&D Systems, MAB78921), Nectin1 (Thermo Fisher Scientific, #37-5900), ITGB1 (BD Biosciences, #610467), ITGB3 (CST, #13166T), ITGB5 (CST, #3629T), ITGA1a (Proteintech, #29042-1-AP), ITGA2 (Thermo Fisher Scientific, #14-0498-80), ITGA3 (Proteintech, #66070-1-Ig), ITGA5 (CST, #4705T), ITGAv (CST, 4711T), Talin (CST, 9021S), Paxillin (CST, #12065S), Vinculin (MilliporeSigma, #V9131), ACTN1 (CST, #6487S), FAK (CST, #3285S), phosphor-FAK (Y397) (CST, 3283S), SRC (CST, #2109S), phosphor-SRC (Y416) (CST, 2101S), WAVE1 (MilliporeSigma, #MABN503), WAVE2 (CST, #3659T), WAVE3 (CST, #2806) and Flag (MilliporeSigma, #F1804). Goat anti-mouse or rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, HRP (Thermo Fisher Scientific, #A16078 or A16110, 1:1000) was used. Immunoblot signal was developed using SuperSignal West Pico Chemiluminescent or Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, #34578 or 34096) and captured on Bio-Rad ChemiDoc MP Imaging system and quantification was calculated by measuring band grayscale using Fiji (NIH) software (Schindelin et al., 2012).
Wound healing assay
HEK293 cells stably expressing turbo GFP or HeLa cells were transfected with control or TMEM132A-specific siRNAs at ∼50% confluency. Cells were trypsinized and counted 48 h later, and ∼0.44×106 cells were seeded into each well of Culture-Insert 2 Well (Ibidi, #80209) mounted on a 35 mm glass-bottom dish (cell culture treated, Collagen1-coated for HEK293 cells to increase the attachment). The following day, when cells reached full confluency and appropriate attachment, inserts were removed to create a cell-free gap of ∼500 µm. Time-lapse live cell images spanning 36 h with 10 min intervals were captured on Yokogawa & Olympus CellVoyager CV1000 confocal scanner under a 10×0.4NA UPLSAPO WD 3.1 (mm) objective. To visualize nuclei of HeLa cells, SPY505-DNA (Cytoskeleton, #CY-SC101) was added to the medium at the recommended concentration 6 h before insert removal. Cell images were captured by phase contrast using bright field or by z-stack (10 slices) maximum projection using the green fluorescence channel (emission filter: 500-550 nm).
Movies of image stacks were processed using CV1000 confocal scanner operating software. All wound healing assay analysis was performed in Fiji (NIH) software (Schindelin et al., 2012). To calculate wound area over time, the brightness was adjusted to make every cell clearly visible. For HeLa cells, the videos were bandpass filtered to remove floating cells from the middle of the wound area. For HEK293 cells, each image was made into a binary and the binary image was dilated to close gaps between cells not in the wound area. The wound area was then defined in each video by calculating the ‘Measure Stack’ function.
Cell tracking analysis was performed using the TrackMate macro (version 7.5.0) (https://www.biorxiv.org/content/10.1101/2021.09.03.458852v1) in Fiji. For all analyses, cells were detected using a Laplacian of a Gaussian (LoG) detector. For HeLa cell analysis, the estimated object diameter was set to 25 px and the threshold was set to 1. For HEK293 cell analysis, the estimated object diameter was set to 35 px and the threshold was set to 0.1. Tracks were created using the LAP tracker. For HeLa cells, the maximum linking distance and track segment gap closing were set to 15 px, with a maximum frame gap of 2. For analysis, tracks analyzed were limited to those where the cell could be tracked for more than 103/207 video frames.
GTPase activity assay
HeLa cells were transfected with 140 pmol (∼10 mM final concentration) control or TMEM132A-specific siRNAs at ∼50% confluency in 10 cm dishes. Cells were trypsinized and counted 48 h post transfection, and re-plated into the rectangular cell culture plates provided in the Cell Comb Scratch Assay kit (MilliporeSigma, #17-10191) at ∼50% confluency. The following day, when cells reached full confluency and appropriate attachment, scratches were evenly applied extensively to the mono-cell layer using the combs provided in the kit. Detached cell debris was removed by replacing the medium. Cells were cultured further for 12 h to initiate migration into free space and then subjected to RhoA activity assay using the Rho Activation Assay Biochem Kit according to the manufacturer's instruction. Briefly, cells were lysed in 1 ml lysis buffer per plate provided in kit and clarified by centrifugation (∼20,000 g, 15 min at 4°C). A small amount of supernatant was used for protein concentration determination using the Pierce BCA Protein Assay Kit, and the rest was flash frozen and stored at −80°C before use. An equal amount of lysate was subjected to 50 µg amino acids 7-89 of rhotekin Rho binding domain (RBD)-coupled beads and incubated at 4°C on a rotator for 1 h to enrich the active form of RhoA (GTP-bound state). Beads were then washed once and boiled at 70°C for 10 min in LDS Sample Buffer supplemented with 50 mM DTT. Total RhoA protein level in cell lysate serves as internal control.
Data collection and statistical analysis
All experiments were performed at least twice, and representative results are shown. For real-time quantitative PCR at least three biological replicates were performed. Data are presented as the mean±s.d. P-value was calculated by Fisher's exact test or two-tailed Student's t-test as needed and considered significant when less than 0.05.
Supplementary Material
Acknowledgements
We are grateful to Lori Bulwith for her continuous support in mouse husbandry. We thank the Molecular, Cellular and Developmental Biology (MCDB) Light Microscopy Core Facility (LMCF) at University of Colorado Boulder and Dr James Orth and Joaquin Quintana for their help and advice with microscopy.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Methodology: B.L., L.B.; Software: B.L., L.B.; Investigation: B.L., J.D.; Writing - original draft: B.L.; Writing - review & editing: L.A.N.; Supervision: L.A.N.; Funding acquisition: L.A.N.
Funding
This work was supported by the National Institutes of Health (R01-HD081562 and P01-HD104436). Deposited in PMC for release after 12 months.
Peer review history
The peer review history is available online at https://journals.biologists.com/dev/article-lookup/doi/10.1242/dev.200442
References
- Biris, K. K. and Yamaguchi, T. P. (2014). Two-color in situ hybridization of whole-mount mouse embryos. Methods Mol. Biol. 1092, 17-30. 10.1007/978-1-60327-292-6_2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, H. and Liu, A. (2017). Spop regulates Gli3 activity and Shh signaling in dorsoventral patterning of the mouse spinal cord. Dev. Biol. 432, 72-85. 10.1016/j.ydbio.2017.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chal, J., Oginuma, M., Al Tanoury, Z., Gobert, B., Sumara, O., Hick, A., Bousson, F., Zidouni, Y., Mursch, C., Moncuquet, P.et al. (2015). Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol. 33, 962-969. 10.1038/nbt.3297 [DOI] [PubMed] [Google Scholar]
- Chalamalasetty, R. B., Garriock, R. J., Dunty, W. C., Jr, Kennedy, M. W., Jailwala, P., Si, H. and Yamaguchi, T. P. (2014). Mesogenin 1 is a master regulator of paraxial presomitic mesoderm differentiation. Development 141, 4285-4297. 10.1242/dev.110908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawengsaksophak, K., de Graaff, W., Rossant, J., Deschamps, J. and Beck, F. (2004). Cdx2 is essential for axial elongation in mouse development. Proc. Natl. Acad. Sci. USA 101, 7641-7645. 10.1073/pnas.0401654101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, B., Brinkmann, K., Chen, Z., Pak, C. W., Liao, Y., Shi, S., Henry, L., Grishin, N. V., Bogdan, S. and Rosen, M. K. (2014). The WAVE regulatory complex links diverse receptors to the actin cytoskeleton. Cell 156, 195-207. 10.1016/j.cell.2013.11.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Goederen, V., Vetter, R., McDole, K. and Iber, D. (2022). Hinge point emergence in mammalian spinal neurulation. Proc. Natl. Acad. Sci. USA 119, e2117075119. 10.1073/pnas.2117075119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessaud, E., McMahon, A. P. and Briscoe, J. (2008). Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development 135, 2489-2503. 10.1242/dev.009324 [DOI] [PubMed] [Google Scholar]
- Eom, D. S., Amarnath, S., Fogel, J. L. and Agarwala, S. (2011). Bone morphogenetic proteins regulate neural tube closure by interacting with the apicobasal polarity pathway. Development 138, 3179-3188. 10.1242/dev.058602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt, A., Akula, N., Schumacher, J., Czamara, D., Karbalai, N., Muller-Myhsok, B., Mors, O., Borglum, A., Kristensen, A. S., Woldbye, D. P.et al. (2012). Replication and meta-analysis of TMEM132D gene variants in panic disorder. Transl. Psychiatry 2, e156. 10.1038/tp.2012.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlow, J. L., Lin, H., Sauerbeck, L., Lai, D., Koller, D. L., Pugh, E., Hetrick, K., Ling, H., Kleinloog, R., van der Vlies, P.et al. (2015). Lessons learned from whole exome sequencing in multiplex families affected by a complex genetic disorder, intracranial aneurysm. PLoS One 10, e0121104. 10.1371/journal.pone.0121104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedl, P. and Mayor, R. (2017). Tuning collective cell migration by cell-cell junction regulation. Cold Spring Harb. Perspect. Biol. 9, a029199. 10.1101/cshperspect.a029199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriock, R. J., Chalamalasetty, R. B., Kennedy, M. W., Canizales, L. C., Lewandoski, M. and Yamaguchi, T. P. (2015). Lineage tracing of neuromesodermal progenitors reveals novel Wnt-dependent roles in trunk progenitor cell maintenance and differentiation. Development 142, 1628-1638. 10.1242/dev.111922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouti, M., Tsakiridis, A., Wymeersch, F. J., Huang, Y., Kleinjung, J., Wilson, V. and Briscoe, J. (2014). In vitro generation of neuromesodermal progenitors reveals distinct roles for wnt signalling in the specification of spinal cord and paraxial mesoderm identity. PLoS Biol. 12, e1001937. 10.1371/journal.pbio.1001937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouti, M., Metzis, V. and Briscoe, J. (2015). The route to spinal cord cell types: a tale of signals and switches. Trends Genet. 31, 282-289. 10.1016/j.tig.2015.03.001 [DOI] [PubMed] [Google Scholar]
- Gouti, M., Delile, J., Stamataki, D., Wymeersch, F. J., Huang, Y., Kleinjung, J., Wilson, V. and Briscoe, J. (2017). A Gene Regulatory Network Balances Neural and Mesoderm Specification during Vertebrate Trunk Development. Dev. Cell 41, 243-261.e247. 10.1016/j.devcel.2017.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene, N. D. and Copp, A. J. (2014). Neural tube defects. Annu. Rev. Neurosci. 37, 221-242. 10.1146/annurev-neuro-062012-170354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaker, J., Lonsdorf, T. B., Raczka, K. A., Mechias, M. L., Gartmann, N. and Kalisch, R. (2014). Higher anxiety and larger amygdala volumes in carriers of a TMEM132D risk variant for panic disorder. Transl. Psychiatry 4, e357. 10.1038/tp.2014.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrique, D., Abranches, E., Verrier, L. and Storey, K. G. (2015). Neuromesodermal progenitors and the making of the spinal cord. Development 142, 2864-2875. 10.1242/dev.119768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail, A. M., Padrick, S. B., Chen, B., Umetani, J. and Rosen, M. K. (2009). The WAVE regulatory complex is inhibited. Nat. Struct. Mol. Biol. 16, 561-563. 10.1038/nsmb.1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, F., Scholze, M., Wittler, L., Schifferl, D., Sudheer, S., Grote, P., Timmermann, B., Macura, K. and Herrmann, B. G. (2017). Antagonistic activities of Sox2 and brachyury control the fate choice of neuro-mesodermal progenitors. Dev. Cell 42, 514-526.e517. 10.1016/j.devcel.2017.07.021 [DOI] [PubMed] [Google Scholar]
- Kumar, S. and Duester, G. (2014). Retinoic acid controls body axis extension by directly repressing Fgf8 transcription. Development 141, 2972-2977. 10.1242/dev.112367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane, J. M., Liang, J., Vlasac, I., Anderson, S. G., Bechtold, D. A., Bowden, J., Emsley, R., Gill, S., Little, M. A., Luik, A. I.et al. (2017). Genome-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits. Nat. Genet. 49, 274-281. 10.1038/ng.3749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson, C. D. and Ridley, A. J. (2018). Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 217, 447-457. 10.1083/jcb.201612069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B. and Niswander, L. A. (2020). TMEM132A, a novel Wnt signaling pathway regulator through wntless (WLS) interaction. Front. Cell Dev. Biol. 8, 599890. 10.3389/fcell.2020.599890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B., Li, S., Yin, J. and Zhong, J. (2013a). Identification and characterization of the Spodoptera Su(var) 3–9 histone H3K9 trimethyltransferase and its effect in AcMNPV infection. PLoS One 8, e69442. 10.1371/journal.pone.0069442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, B., Yu, C., Yin, J. and Zhong, J. (2013b). Enhanced transgene expression in mammalian cells by recombinant baculovirus vector containing bovine papillomavirus type 1 replication elements. J. Gene Med. 15, 282-290. 10.1038/gim.2012.129 [DOI] [PubMed] [Google Scholar]
- Li, J., Zhao, X., Xin, Q., Shan, S., Jiang, B., Jin, Y., Yuan, H., Dai, P., Xiao, R., Zhang, Q.et al. (2015). Whole-exome sequencing identifies a variant in TMEM132E causing autosomal-recessive nonsyndromic hearing loss DFNB99. Hum. Mutat. 36, 98-105. 10.1002/humu.22712 [DOI] [PubMed] [Google Scholar]
- Liaqat, K., Hussain, S., Bilal, M., Nasir, A., Acharya, A., Ali, R. H., Nawaz, S., Umair, M., Schrauwen, I., Ahmad, W.et al. (2020). Further evidence of involvement of TMEM132E in autosomal recessive nonsyndromic hearing impairment. J. Hum. Genet. 65, 187-192. 10.1038/s10038-019-0691-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marean, A., Graf, A., Zhang, Y. and Niswander, L. (2011). Folic acid supplementation can adversely affect murine neural tube closure and embryonic survival. Hum. Mol. Genet. 20, 3678-3683. 10.1093/hmg/ddr289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mole, M. A., Galea, G. L., Rolo, A., Weberling, A., Nychyk, O., De Castro, S. C., Savery, D., Fassler, R., Ybot-Gonzalez, P., Greene, N. D. E.et al. (2020). Integrin-mediated focal anchorage drives epithelial zippering during mouse neural tube closure. Dev. Cell 52, 321-334.e326. 10.1016/j.devcel.2020.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nader, G. P., Ezratty, E. J. and Gundersen, G. G. (2016). FAK, talin and PIPKIgamma regulate endocytosed integrin activation to polarize focal adhesion assembly. Nat. Cell Biol. 18, 491-503. 10.1038/ncb3333 [DOI] [PubMed] [Google Scholar]
- Nowotschin, S., Ferrer-Vaquer, A., Concepcion, D., Papaioannou, V. E. and Hadjantonakis, A. K. (2012). Interaction of Wnt3a, Msgn1 and Tbx6 in neural versus paraxial mesoderm lineage commitment and paraxial mesoderm differentiation in the mouse embryo. Dev. Biol. 367, 1-14. 10.1016/j.ydbio.2012.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-hashi, K., Naruse, Y., Amaya, F., Shimosato, G. and Tanaka, M. (2003). Cloning and characterization of a novel GRP78-binding protein in the rat brain. J. Biol. Chem. 278, 10531-10537. 10.1074/jbc.M212083200 [DOI] [PubMed] [Google Scholar]
- Oh-hashi, K., Imai, K., Koga, H., Hirata, Y. and Kiuchi, K. (2010). Knockdown of transmembrane protein 132A by RNA interference facilitates serum starvation-induced cell death in Neuro2a cells. Mol. Cell. Biochem. 342, 117-123. 10.1007/s11010-010-0475-9 [DOI] [PubMed] [Google Scholar]
- Paul, N. R., Jacquemet, G. and Caswell, P. T. (2015). Endocytic trafficking of integrins in cell migration. Curr. Biol. 25, R1092-R1105. 10.1016/j.cub.2015.09.049 [DOI] [PubMed] [Google Scholar]
- Petrie, R. J., Doyle, A. D. and Yamada, K. M. (2009). Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 10, 538-549. 10.1038/nrm2729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast, C., Altmann, A., Weber, P., Arloth, J., Bader, D., Heck, A., Pfister, H., Muller-Myhsok, B., Erhardt, A. and Binder, E. B. (2012). Rare variants in TMEM132D in a case-control sample for panic disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159B, 896-907. 10.1002/ajmg.b.32096 [DOI] [PubMed] [Google Scholar]
- Rodrigo Albors, A. and Storey, K. G. (2016). Mapping body-building potential. Elife 5, e14830. 10.7554/eLife.14830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Pulido, L. and Ponting, C. P. (2018). TMEM132: an ancient architecture of cohesin and immunoglobulin domains define a new family of neural adhesion molecules. Bioinformatics 34, 721-724. 10.1093/bioinformatics/btx689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savory, J. G., Bouchard, N., Pierre, V., Rijli, F. M., De Repentigny, Y., Kothary, R. and Lohnes, D. (2009). Cdx2 regulation of posterior development through non-Hox targets. Development 136, 4099-4110. 10.1242/dev.041582 [DOI] [PubMed] [Google Scholar]
- Savory, J. G., Mansfield, M., Rijli, F. M. and Lohnes, D. (2011). Cdx mediates neural tube closure through transcriptional regulation of the planar cell polarity gene Ptk7. Development 138, 1361-1370. 10.1242/dev.056622 [DOI] [PubMed] [Google Scholar]
- Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., Preibisch, S., Rueden, C., Saalfeld, S., Schmid, B.et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seegmiller, R. E., Harris, C., Luchtel, D. L. and Juchau, M. R. (1991). Morphological differences elicited by two weak acids, retinoic and valproic, in rat embryos grown in vitro. Teratology 43, 133-150. 10.1002/tera.1420430206 [DOI] [PubMed] [Google Scholar]
- Shafaq-Zadah, M., Gomes-Santos, C. S., Bardin, S., Maiuri, P., Maurin, M., Iranzo, J., Gautreau, A., Lamaze, C., Caswell, P., Goud, B.et al. (2016). Persistent cell migration and adhesion rely on retrograde transport of beta(1) integrin. Nat. Cell Biol. 18, 54-64. 10.1038/ncb3287 [DOI] [PubMed] [Google Scholar]
- Shimada-Sugimoto, M., Otowa, T., Miyagawa, T., Khor, S. S., Omae, Y., Toyo-Oka, L., Sugaya, N., Kawamura, Y., Umekage, T., Miyashita, A.et al. (2016). Polymorphisms in the TMEM132D region are associated with panic disorder in HLA-DRB1*13:02-negative individuals of a Japanese population. Hum. Genome Var. 3, 16001. 10.1038/hgv.2016.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steventon, B. and Martinez Arias, A. (2017). Evo-engineering and the cellular and molecular origins of the vertebrate spinal cord. Dev. Biol. 432, 3-13. 10.1016/j.ydbio.2017.01.021 [DOI] [PubMed] [Google Scholar]
- Takemoto, T., Uchikawa, M., Yoshida, M., Bell, D. M., Lovell-Badge, R., Papaioannou, V. E. and Kondoh, H. (2011). Tbx6-dependent Sox2 regulation determines neural or mesodermal fate in axial stem cells. Nature 470, 394-398. 10.1038/nature09729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wald, N. J., Morris, J. K. and Blakemore, C. (2018). Public health failure in the prevention of neural tube defects: time to abandon the tolerable upper intake level of folate. Public Health Rev. 39, 2. 10.1186/s40985-018-0079-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X., Jiang, W., Luo, S., Yang, X., Wang, C., Wang, B., Dang, Y., Shen, Y. and Ma, D. K. (2021). The C. elegans homolog of human panic-disorder risk gene TMEM132D orchestrates neuronal morphogenesis through the WAVE-regulatory complex. Mol. Brain 14, 54. 10.1186/s13041-021-00767-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde, J. J., Petersen, J. R. and Niswander, L. (2014). Genetic, epigenetic, and environmental contributions to neural tube closure. Annu. Rev. Genet. 48, 583-611. 10.1146/annurev-genet-120213-092208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip, G. W., Ferretti, P. and Copp, A. J. (2002). Heparan sulphate proteoglycans and spinal neurulation in the mouse embryo. Development 129, 2109-2119. 10.1242/dev.129.9.2109 [DOI] [PubMed] [Google Scholar]
- Zhang, Y., Kim, T. H. and Niswander, L. (2012). Phactr4 regulates directional migration of enteric neural crest through PP1, integrin signaling, and cofilin activity. Genes Dev. 26, 69-81. 10.1101/gad.179283.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.