

Abstract
While sperm mosaicism has few consequences for men, the offspring and future generations are unwitting recipients of gonadal cell mutations, often yielding severe disease. Recent studies, fueled by emergent technologies, show that sperm mosaicism is a common source of de novo mutations that underlie severe pediatric disease as well as human genetic diversity. Sperm mosaicism can be divided into three types: Type I arises during sperm meiosis and is non-age dependent; Type II arises in spermatogonia and increases as men age; Type III arises during paternal embryogenesis, spreads throughout the body, and contributes stably to sperm throughout life. Where Types I and II confer little risk of recurrence, Type III may confer identifiable risk to future offspring. These mutations are likely the single largest contributor to human genetic diversity. New sequencing approaches may leverage this framework to evaluate and reduce disease risk for future generations.
Keywords: Advanced paternal age, sperm mosaicism, human variation, spermatogonial stem cells, primordial germ cells
Overview of sperm mosaicism and de novo mutations
As one of many types of genomic mosaicism, sperm mosaicism specifically refers to sperm cells carrying genetic variants that are not constitutively present in a man’s genome [1–3]. Because sperm carry all-male genetic information to the child, even a variant present in a single sperm can become part of the zygotic genome of the next generation. Thus, sperm mosaicism, unlike other forms of male mosaicism, has the potential to profoundly influence future generations.
Genetic variants present in sperm can arise anytime during the process of cell maturation, from as early as the 2-cell embryonic stage of the father to the moment sperm are ejaculated [4, 5]. Presumably any type of genetic perturbation can occur in sperm, including single nucleotide variants (SNVs), insertions/deletions (SNV/INDELs), copy number or structural variants (CNV/SVs), transposition of transposable elements (TE), repeat expansions or contractions, and (sub-) chromosomal aneuploidies or translocations [6–11] (Glossary Box). Any of these sperm mosaic variants can result in de novo mutations (DNMs) in the child if the sperm cell successfully fertilizes an egg and leads to birth. Across the genome, every child displays approximately 70 de novo SNVs and 6 de novo SVs when the average paternal age is 29 years [12]. While most of these DNMs are neutral, a small proportion can lead to congenital pediatric disorders in the offspring, either due to loss of one copy of a haploinsufficient gene or the generation of a toxic gain-of-function allele.
The observation that this subset of DNMs contributes significantly to human disease, especially congenital disorders, is now firmly established [13–15]. The scale of their contribution has only recently been appreciated following implementation of ‘trio sequencing’ (exome or genome) [16]. In non-consanguineous populations, DNMs are the leading identifiable cause of severe congenital disorders, often producing lethal outcomes or loss of fertility [17]. Because of this phenotypic severity, these mutations are unlikely to pass to subsequent generations, and are thus under purifying selection. A prime example is that of DNMs leading to intellectual disability, which greatly reduces reproductive fitness [18].
A surprising 80% of de novo SNVs (dSNVs) in an offspring arise on the paternal haplotype [19]. The number of dSNVs closely correlates with the age of the father at the time of conception [19, 20], supporting an age-dependent, linear accumulation of sperm mosaicism. On average, each decade of life increases the number of mutations in an offspring by a dozen or more [12, 19, 21]. As one of the main distinguishing features of oocyte development is the absence of mitotic activity, the post-pubescent proliferation of spermatogonial stem cells (SSCs, sometimes called spermatogonia) is the likely culprit of this stark asymmetry in paternal versus maternal contribution [22]. Massive spermatogonial proliferation [23], generating mature sperm at an impressive rate of 3-4 million per day [24], is a likely source of cell-cycle or age-dependent mutations as a result of DNA polymerase or other mitotic errors. Although the maternal age-dependent DNM accumulation is less pronounced than the paternal, some mutational types, such as aneuploidies, are in fact more frequent in females, attributed to prolonged oocyte meiotic arrest [25, 26].
While the existence of paternal age-dependent DNMs in offspring is indisputable, recent analysis of DNMs across species has cast doubt on the mechanisms [21, 27]. In one hypothesis, DNA damage incurred during meiosis or within the sperm cell may be repaired in a particularly inefficient way in the zygote. If borne out by more data, some DNMs might therefore be referred to as ‘primed sperm mosaicism’, in which defects in zygotic DNA repair serve to amplify their effect [9, 28, 29].
Four types of sperm mosaicism based upon the timing of origin
Mammalian testes are colonized by primordial germ cells (PGCs), which eventually give rise to SSCs [30]. SSCs are attached to the basement membrane and divide slowly throughout life, yielding transient amplifying stem cell subtypes, and eventually primary spermatocytes. These subsequently undergo meiosis to produce secondary spermatocytes, spermatids, and ultimately mature sperm [31, 32] (Fig. 1).
Figure 1.
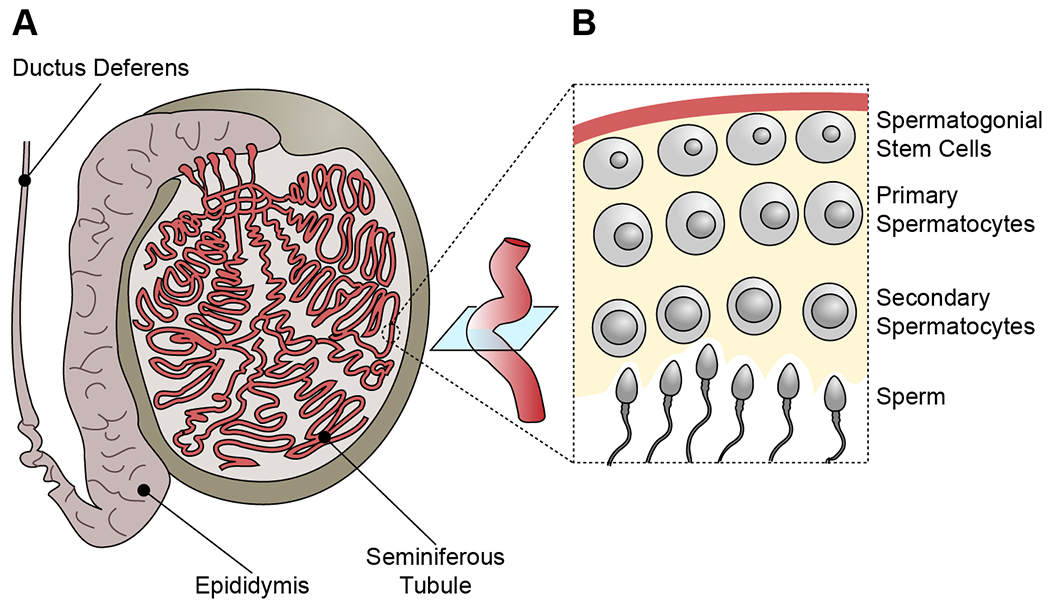
Anatomy of the spermatogonial stem cell (SSC) niche.
(A) Human testes contain a web of seminiferous tubules, which are the site of spermatogenesis. They connect to the epididymis and the ductus deferens (or vas deferens) as a portal to ejaculation.
(B) Cross-section of a seminiferous tubule. SSCs proliferate and self-renew, producing spermatocytes, which undergo meiosis and, depending on their progression, are distinguished as primary or secondary. Following secondary meiotic division, spermatids differentiate into mature sperm (or spermatozoa) that will shed into the lumen of the seminiferous tubule prior to ejaculation.
We recently proposed several types of sperm mosaicism based upon results from sequencing sperm directly (Fig. 2, Table 1). Every male is expected to exhibit all of these types if assessed with similar sequencing approaches [9]. Type I (sperm) mutations occur in terminally- or near terminally-postmitotic spermatocytes and sperm cells, are likely stable in number throughout paternal aging, and may account for a significant portion of DNMs, especially in young men. Type II (SSC) mutations occur in SSCs and accumulate during aging as a result of environmental exposure and mitotic errors. These are further divided into IIa and IIb, the latter conferring selective growth advantage, whereas the former displays no evidence of selective advantage. Neither Type I nor II associate with intrafamilial DNM or disease recurrence (i.e. across siblings), but Type IIb can lead to increased population occurrence (i.e. interfamilial recurrence) as a result of selective growth advantage and their increased frequency in older men.
Figure 2.
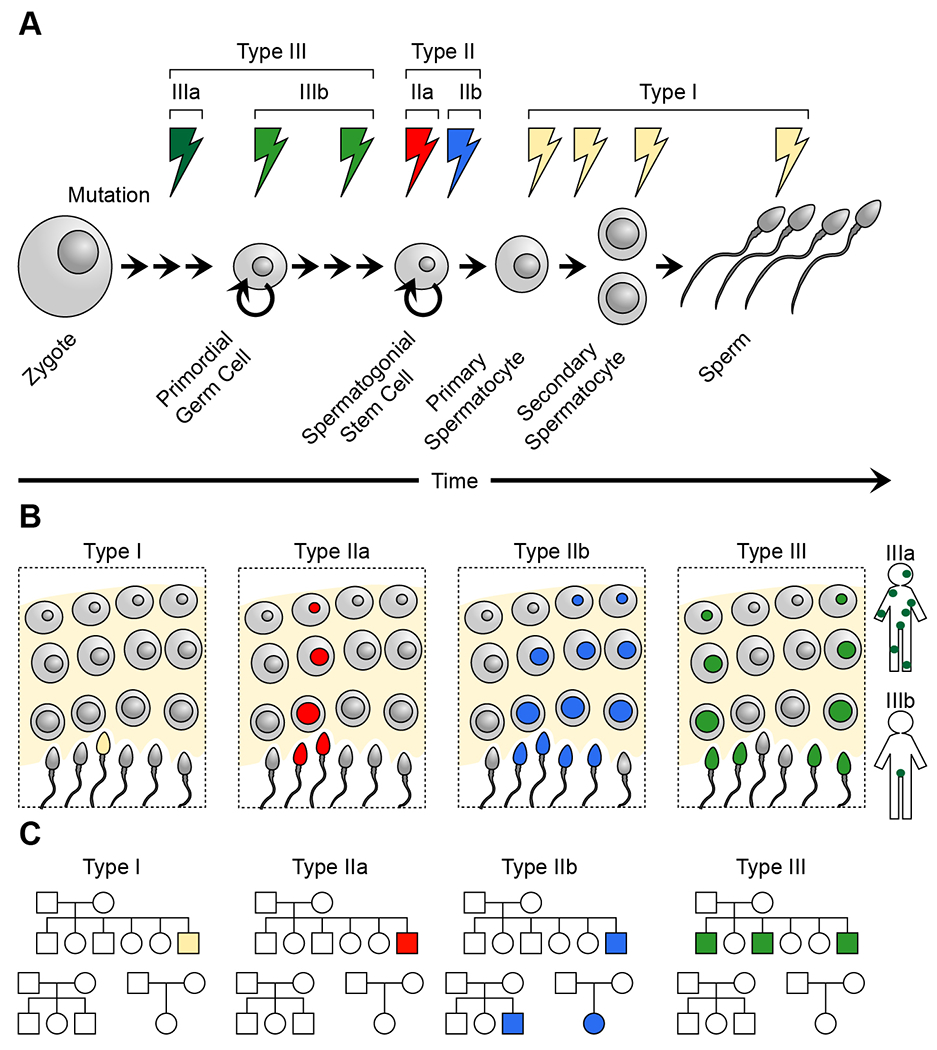
Temporal resolution of sperm mosaicism.
(A) Sperm mosaic mutations can arise at any point during sperm lineage, starting from the zygote, before or after the establishment of the primordial germ cells (PGCs), which are the embryonic progenitors of spermatogonial stem cells (SSCs), or during spermatogenesis. We broadly distinguish three types of sperm mosaicism, Types I (sperm), II (SSC), and III (embryonic), and further subdivide Types II and III into IIa, IIb, IIIa, and IIIb. Type III mutations arise during early embryogenesis prior to (IIIa) or after (IIIb) the establishment of the PGC population. Type IIa and IIb mutations arise within the SSCs and are distinguished by their impact on cellular proliferation. Type I mutations arise during the last stages of spermatogenesis and by definition are only ever-present in a few sperm of the same meiotic division.
(B) In the testicular stem cell niche, Type I mutations are the only type absent from SSCs. Type IIa includes mutations that do not impact the fitness of SSCs and stay contained within their lineage. Type IIb, also referred to as ‘selfish sperm’ mutations, provides a selective advantage to an SSC within the niche; however, this is at the cost of offspring, as these mutations typically result in severe congenital disorders (e.g., Apert, Noonan, and Costello syndromes). Finally, Type III is present in several SSCs due to their developmental origin. They are typically found throughout the seminiferous tubules and across the two testes, resulting in measurable allelic fractions in bulk sperm samples.
(C) The different types of sperm mosaicism result in distinct recurrence risk patterns. Type I and Type IIa mutations result in no or infinitesimally small recurrence risk within a family; as they are random, their recurrence across the population is as expected by chance. Type IIb mutations result in overproliferation within the stem cell niche, however, this results in little recurrence risk within a family. Like cancer mutations, however, due to their selection advantage, the same FGFR2/3, RET, HRAS, or KRAS mutations are found more frequently across the population than expected by chance. Finally, Type III mutations can exhibit high recurrence risk within a family (up to 25%), but their recurrence across the population is as expected by chance.
Table 1.
Types of sperm detectable mutations
| TYPE I | TYPE II | TYPE III | |||
|---|---|---|---|---|---|
| Type IIa | Type IIb | Type IIIa | Type IIIb | ||
| Stage of occurrence | Postnatal | Embryonic | |||
| Age Dependence | No? | Yes | Yes | No | |
| Abundance | Single sperm | Single sperm lineage | Sperm collections | Sperm collections | Sperm and soma |
| Recurrence risk | No | Effectively No | Yes(Population) | Yes(Family) | |
| Clinical relevance | Sporadic de novo genetic disorders | Sporadic common disorders/cancer | Familial de novo genetic disorders | ||
Type III (embryonic) mutations occur during embryogenesis of the male, later seeding multiple SSCs and contributing to a stable proportion of sperm throughout life. Type III mosaic mutations are not age-dependent, typically show no evidence of selective advantage, and are often measurable in sperm with bulk sequencing. Type III is further divided into IIIa and IIIb, the former is also associated with evidence of mosaicism in sampled somatic tissues like blood or saliva, whereas the latter shows mosaicism limited to sperm. Unlike other types, Type III can lead to intrafamilial recurrence. Type III can also occur in females, and based upon epidemiological evidence, is of likely similar magnitude in men and women [2, 33]. The proportional increase in Type III mosaicism in females is due to an apparent absence or reduction of Type I or II mosaicism and is consistent with the higher relative rates of intrafamilial recurrence for mutations arising on the maternal haplotype [8, 34, 35]. The combined burden of these four mutational types likely represents the total of sperm mosaicism.
Common clinical encounters of sperm mosaicism
There are dozens of reports of sperm mosaicism detected in fathers where two or more offspring share the same DNM-related disease, and where the mutation traces to the father [36–44]. In fact, depending on the disorder, up to 10% of clinically relevant DNMs are detectable as mosaic in father’s sperm, most likely representing Type III mosaicism [42, 45–58]. This rate was much higher than previously assumed from disease recurrence estimates, suggesting that such disease-causing mutations may be hiding in male gonads. These studies mostly focused on individual genes, but were supported by our recent assessment of fathers of children with autism attributed to DNMs, where 4 out of 20 families showed evidence of a disease-causing mutation representing Type III mosaicism in father’s sperm [9].
While most sperm mosaic mutations are probably neutral to sperm progenitors (i.e. do not confer positive or negative selection), Type IIb mutations yield a selective growth advantage, best evidenced in the ‘selfish sperm’ model [59]. Incidence of certain conditions like ‘RASopathies’ in children is known to increase with advanced paternal age (APA) [60], leading to the hypothesis that mutations in a subset of genes could yield a selective growth advantage in SSCs. Specialized PCR methods called ‘peptide nucleic-acid amplification’ (PNA) demonstrated the presence of such mutations in an average of 4 per 1,000,000 sperm, with a 5-fold increase in incidence in 70 to 80-year-old compared with 10 to 30-year-old males [61].
Most men over 50 years of age demonstrate testicular clonal microfoci containing these specific gain of function mutations in a handful of genes including FGFR2, FGFR3, HRAS, and NRAS [59, 60, 62–65]. Rarely, these microfoci, when combined with other mutations like trisomy 9 or 20, monosomy 7, or amplification of DMRT1, can progress to testicular tumors [66]. For the most part, these microfoci remain stable and are of little health consequence to the man. Instead, they produce dominant RASopathy phenotypes in offspring, leading to the concept of ‘selfish sperm’ gaining a growth advantage at the expense of causing disease in the offspring [65, 67]. Due to their cancer-like selection, the incidence of these mutations is higher than expected by chance, resulting in interfamilial recurrence. It would be interesting to evaluate pathways beyond ‘RASopathies’ for similar effects.
Sperm mosaicism has also been described in other disease contexts, although the origin and disease mechanism are still under investigation. For instance, the trinucleotide repeat length in the Huntington gene can show dramatic differences in sperm in mutation or pre-mutation carriers [11]. This may be a result of repeated Type I or II mutations, but we have previously demonstrated that repeat expansions and contractions can also manifest as Type III mosaicism [9]. Male infertility has also been connected to mosaic aneuploidies and epigenetic mosaicism [68, 69]. However, it is unclear whether these observations are a cause or a consequence of infertility, and more studies are required to understand these interactions.
Identifying sperm mosaicism from bulk or single-cell sequencing
Sperm mosaicism can be assessed either from ‘bulk’ or ‘single sperm sequencing. The abundance of a mutation and the ability to detect mosaicism from bulk samples are related to the allelic fraction (AF) and thus the developmental timing of mutations (Fig. 3). SSCs are generated from a founder pool of perhaps a few dozen PGCs that arise in the epiblast at early embryonic stages [70]. SSCs likely eventually number in the millions in humans [71], and thus mutations in any one cell will contribute only a tiny fraction of sperm to a given ejaculate. Given the current technical detection sensitivities, assessing sperm mosaicism using bulk sequencing without prior knowledge of the mutation can limit detection to mutations that are present in >1% of sperm (i.e. AF>0.01). Thus, the same mutation would need to be present in tens of thousands of individual SSCs, and therefore only Type III mutations are readily detected from these approaches. Sequencing single sperm offers an alternative, but suffers from ‘undersampling’ (i.e. sequencing of only a few cells) given the throughput of current methods and their uneven coverage. Additionally, this approach has a high false-discovery rate (FDR) due to whole-genome amplification steps prior to sequencing, though some of these shortcomings are likely to be solved in the near future with new technologies.
Figure 3.
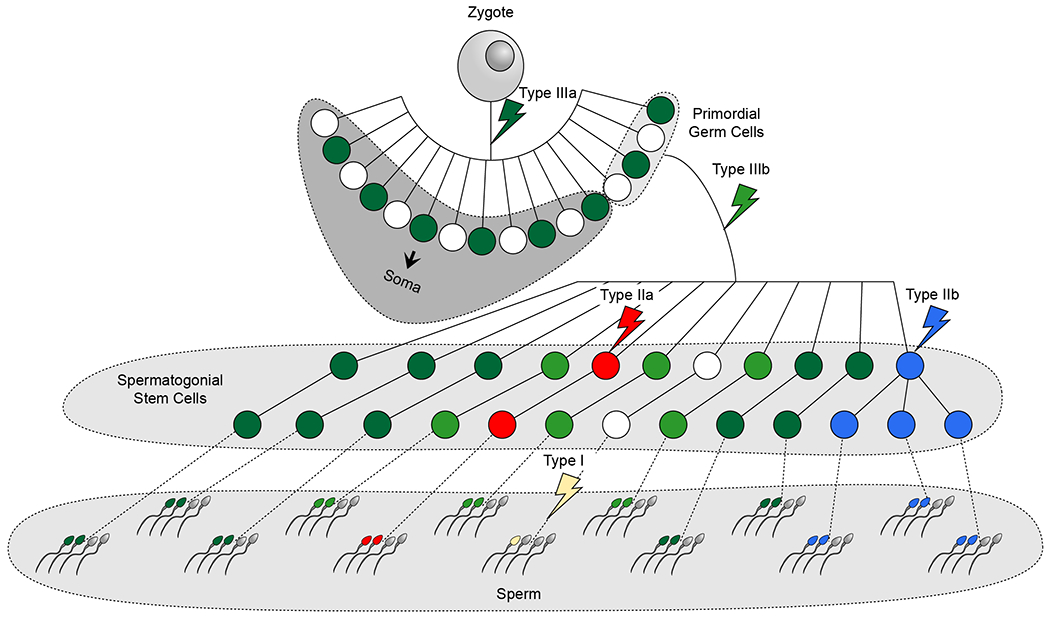
The timing of a mutation and its selective potential determine abundance in sperm.
Schematic of the developing lineage of germ cells and the soma. The different types of sperm mosaicism are labeled and occur prior to or following Primordial Germ Cell (PGC) specification (Types IIIa and IIIb, respectively), within the SSCs (Type II), or are present only within one or two sperm cells (Type I). Only Type IIb mutations exhibit positive selection. The abundance of a mutation among sperm cells is a function of its timing during development and the presence or absence of positive selection. Note that for simplicity all mutations are shown to occur in separate sperm lineages. However, in reality, individual sperm lineages will show a combination of all types of sperm mosaicism.
An alternative to identify sperm mosaicism a priori is to first identify DNMs in a child and then search for these mutations in sperm from the father, with the knowledge that 80% of DNMs arise on the paternal haplotype. Using this method, coupled with 200× whole genome sequencing (WGS), we found that 2.1% of DNMs detectable in the child could be detected in the father’s sperm [9]. Note that this genome-wide rate of mosaicism is lower than that detected for disease-relevant and exonic variants [53, 57, 70], possibly as a result of distinct mutation AFs, effects on sperm health, or differences in the method of detection. By limiting the variants to just those on the paternal haplotype, this increased to 4.0%. Thus ~2-4% of DNMs in a child are detectable in father’s sperm as type III mutations. One criticism of this work was that the 200× WGS likely limited detection to AFs >1.5%, given that detection required at least three mutant reads per allele. We partially overcame this limitation using Multiplex Accurate Sensitive Quantitation (MASQ) on sperm DNA, which is capable of detecting AFs as low as 10−4 to 10−6 (i.e. 1:10,000 to 1:1,000,000) on a subset of variants. However, we detected no additional mosaic variants [9]. These data suggest that the vast majority of the paternally phased DNMs in a child represent Type I and II, rather than III, and are either not present in repeated sampling, or are present at extremely low levels.
Type III mutations are predicted to demonstrate three key properties: 1) Because they arise during embryogenesis of the father, the mutations have the potential to be spread more widely and should be of a relatively higher AF than other types. 2) Since widely spread SSCs are thought to contribute relatively equally to ejaculates, these AFs should remain stable over repeated sampling. Thus, Type III mutations should represent a lifelong risk to DNMs in offspring, and not change as a function of the father’s age. Indeed, we found that over a 1-year course of repeated sperm sampling, these mutations remained remarkably stable, and were similar in number in young men compared to aged men [70]. This is also consistent with prior family-based observations that assessed transmitted variants across multiple generations [12, 33]. 3) A subset of Type III mutations should be shared with other tissues in the body of the father, assuming each arose in a cell that contributed to both SSCs and other cell types. We found that about one-third to one-half of Type III mutations detected in the father’s sperm were evident in the father’s blood, saliva, or both [70], distinguishing Type IIIa (sperm and other samples) mutations from Type IIIb (sperm-specific).
Could Type I and Type IIa mutations be distinguished by either sequencing sperm at higher read depth or by single-cell sperm sequencing? Prior work suggests the presence of ‘mutational signatures’ that become more pronounced with increasing parental age [72, 73], pointing to different mutational mechanisms for Type III (embryonic) compared with Type I or II (sperm or SSC). These differences alone though are unlikely to distinguish Type I from II. Moreover, as Type I and IIa mutations are likely to present in less than 1:1,000,000 cells, bulk sequencing has little ability to reliably distinguish these mutations over background noise, given its intrinsic error rate. Single-cell sperm sequencing, on the other hand, could detect individual mutations at great sensitivity, especially as sperm are haploid. Thus single sperm WGS could likely distinguish these mutation types, given the expectation that IIa mutations should increase in older men [19]. Single-cell sperm sequencing should identify an excess of mutations in older men which, as a population, should be bona fide Type IIa. So far, single sperm sequencing has been applied with low read depth to determine factors regulating meiotic recombination and rates of structural variants, CNVs, and chromosomal events [7, 74]. Analogous approaches geared towards SNVs could potentially reveal the origins of Type I and II sperm mosaicism mutations before their transmission to a child. However, as mentioned, single sperm sequencing is currently still limited by current technologies.
Allelic fractions can reveal the time-of-origin of sperm mosaicism
Lineage origins of cells and cell populations can be deconvolved by tracing individual mosaic mutations that act as neutral barcodes, revealing shared lineages. Because modern next-generation sequencing (NGS) approaches reveal base changes at the individual strand level rather than an average of all bases in the sample, identifying these barcodes has become increasingly possible for both diploid and haploid cells [75]. Using the last-common ancestor as a reference point, cells accumulating mutations at earlier time points will be spread more widely (Fig. 3). Assuming that mutations do not confer purifying selection, earlier mutations will also be present at higher AFs if sampling from a cellular pool. At the single-cell level, the concept of AF breaks down and is instead replaced by the commonality of the mutation among individually sampled cells. While the AFs are expected to follow a stepwise function that decreases by half for every cell division, in actuality because of the number of cells being sampled and the binomial sampling distributions, exponential decay is most often observed in a rank-plot of AFs.
Assessing risk of recurrence of DNMs for paternally phased variants
Empiric population data suggests that the risk of recurrence of a paternally-phased DNM is ~2%, attributed to sperm mosaicism [76, 77]. Now that sperm mosaicism has been directly quantitated, we can infer that this empiric risk represents an average from separate and quantifiable effects, dependent on the type and AF of the mutation. Type I mutations have little to no chance to recur, having occurred in a terminally postmitotic cell. Type IIa mutations have an infinitesimal chance to recur, considering they likely exist in a single SSC lineage out of the millions that contribute to sperm. Type III mutations, on the other hand, account for a minority of DNMs in a child but have a potentially much higher and directly quantifiable risk of recurrence. Thus, the vast majority of DNMs have a near-zero recurrence risk but a small minority have a substantially higher risk, which could be assessed by sperm sequencing to assess AF, directly predicting risk [78]. The AFs for Type III mutations can range to as high as 20%, thus imparting a quantifiable risk to offspring, which might be useful to couples and their practitioners prior to conception. While unable to provide a family-specific recurrence risk, some of these concepts—as observed from family-based studies—have yielded models and ‘recurrence risk calculators’ that take parental age into account [2, 33].
Deconstruction of the cumulative risk of DNMs to offspring
To understand the parental origin of DNMs, it is necessary to turn back the clock to embryogenesis of the parents to understand their individual contribution to germ cells (Fig. 4). Mutations arising during embryogenesis of the father have the potential to contribute to germ cells in the form of Type III mosaicism. Similarly, women very likely have a counterpart to Type III mosaicism that originates during embryogenesis, probably very comparable in number and AFs (Box 1). Men additionally contribute mutations arising from Type I and II mosaicism, and likely make up the majority of DNMs in an offspring. While women undoubtedly have a counterpart to Type I and II, these are less numerous and nearly impossible to quantify due to the limitation in sampling female germ cells. Finally, there are likely to be mutations that arise in the fertilized, one-cell stage zygote which were not present in the germ cells of either parent. As these would necessarily occur in a single cell, prior to the first cell division, these are probably few in number and difficult to measure directly [21, 28, 29].
Figure 4.
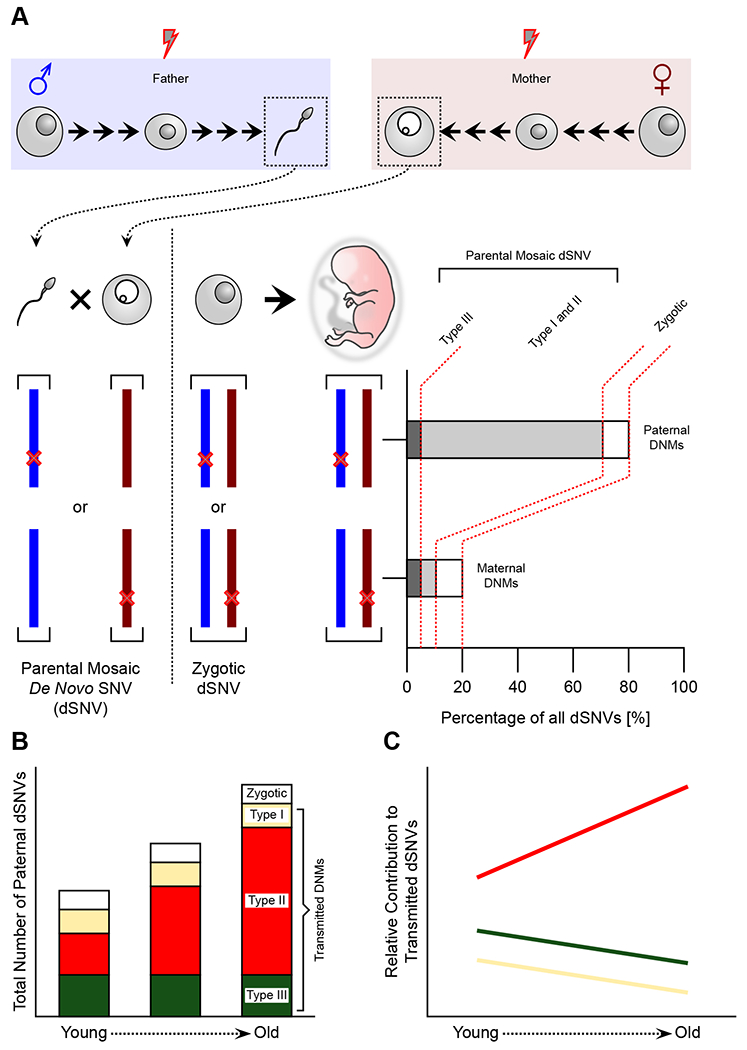
Paternal mutations in offspring are largely due to age-dependent sperm mosaicism.
(A) Single nucleotide variants (SNVs) occurring during spermatogenesis or oogenesis of the parents are present in the respective germ cells prior to conception (parental mosaic de novo SNVs, i.e. dSNVs) and will reside on the paternal or maternal haplotype of the embryo, respectively. dSNVs that occur following conception are defined as zygotic, and should be stochastically distributed across both haplotypes. Currently, we further assume that Type III mutations appear at similar rates in the male and female germ cell lineage. Thus the imbalance (80:20%) of dSNVs in favor of the paternal haplotype must derive from Type I and II sperm mosaicism.
(B) Type III mutations are determined during embryonic development and should remain constant with age. Similarly, the stochastic nature of Type I is likely independent of age effects. Thus, the observed increase of dSNVs with paternal age should derive mostly from Type II mutations that accumulate with each cell cycle.
(C) As a consequence of the increase of Type II mutations with age, the relative contribution of Types I and III decreases. Likewise, the average intrafamilial recurrence risk of a given dSNV decreases with age.
Box 1. Types of oocyte mosaicism.
While there are clear differences in oocytes compared to sperm, the developmental processes are similar. For instance, there appears little to no sex difference at the time of PGC establishment and early amplification, supported by transcriptional signatures and similar mosaicism rates of Type IIIa mutations [8, 77]. PGCs in females differentiate into oogonial stem cells, then meiotically arrested oocytes, which, like sperm, can accumulate Type I mutations, whereas Type II will be significantly reduced due to the smaller number of cell divisions in oocytes. In contrast, the lifelong meiotic arrest results in Type I mosaicism that can be distinguished from that found in sperm. For instance, analysis of maternal mutations revealed the occurrence of clustered mutations in oocytes [33, 82]. In addition, meiotically derived CNVs and aneuploidies are more likely to occur in oocytes, and increase with maternal age [83].
The DNM source should also be considered in the context of paternal aging, when the number of Type II mutations increases dramatically. Thus, in young men, the contribution of Type II mutations is relatively small, compared with the sum of Type I and III and zygotic mutations, but these increase with the age of the father, and, on average, a 60-year-old father transmits 60 more dSNVs than a 20-year-old. At the same time, the number of Type I, III, and zygotic mutations likely remains stable. Thus the relative contribution of Type II mutations increases dramatically while the relative contribution of Type I and III and zygotic mutations decrease as men age. The larger contribution of non-type III mutations in men is likely the reason that a larger percent of maternally-phased DNMs from the child demonstrate parental mosaicism than the corresponding paternally-phased DNMs [9].
The majority of human genetic diversity likely has origins in sperm mosaicism
It seems likely that the majority of rare and possibly common variants in humans originated as sperm mosaicism, as they are the major mutagenic source passed to the next generation (Box 2). Assuming, each child harbors ~120 DNMs (considering all types of mutations), and the paternal contribution is ~80%, i.e. 100 DNMs, the vast majority of these paternal haplotype mutations will be present in sperm prior to conception. Given a stable population size, in addition to 100 paternal variants from each generation, an additional half of the mutations from the prior generation are also transmitted (Fig. 5). However, assuming a population doubling, following the same 3 generations, genomic diversity increases by over 1000 new variants [12, 79]. This difference results because a greater portion of DNMs from the prior generation are perpetuated to subsequent generations. Within 10 generations, the offspring from a single founder genome would harbor over 100,000 new variants. The contribution from the other two identifiable sources of genetic diversity (maternal mosaic and zygotic mutations), collectively likely represent less than 25% of this diversity. These estimates do not take into account variants lost due to purifying selection. With paternal age increasing over the past century, these numbers may underestimate sperm’s contribution to human genetic diversity.
Box 2. Somatic mosaicism: an evolutionary dead end.
Most somatic mutations in the body are inconsequential, although a fraction can lead to cancer or other conditions like focal cortical dysplasia [84–86]. Because these mutations are largely not shared with gonadal cells, there is little to no risk to transmit to future generations, and are thus considered ‘dead end’ mutations. The only somatic mutations capable of transmission to future offspring are those that are found in gonadal germ cells. The design of the mammalian germ cell niche is separated developmentally from the rest of the soma, likely in part to safeguard against excessive mutational accumulation [87].
Figure 5.
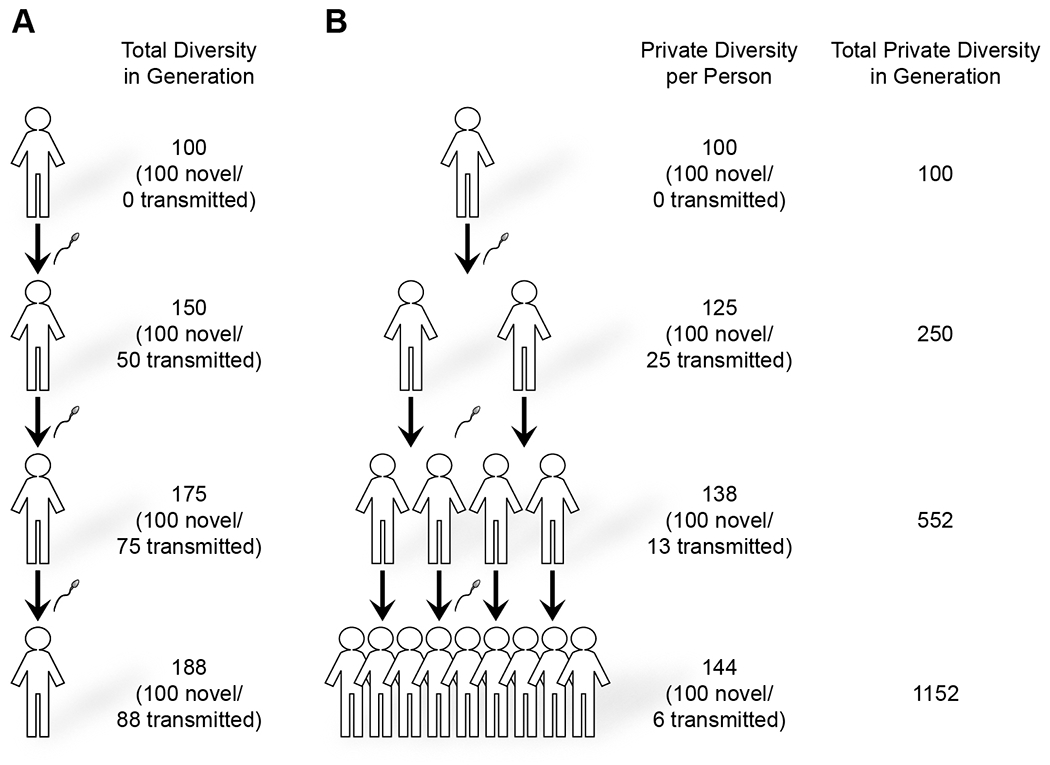
Sperm mosaicism contributes to human genomic diversity.
(A) In a simplified assumption that each sperm contributes 100 mutations before conception and a stable paternal age at conception, each man will transmit 50 of these private variants to the next generation. In the fourth generation, if each man has one son, this will amount to a total of 188 private variants that are only found in any one individual within this generation. Ultimately, this will increase to double the number of DNMs, with ~100 arising from prior generations and ~100 arising from the most recent generation.
(B) Assuming that each man has two sons, using a similar rate of ~100 sperm mosaic mutations, sperm mosaicism will contribute 144 private variants (not shared with any sibling or cousin) per person, or 1152 across all 16 offspring in the fourth generation. The total contribution in the population now numbers 1152 novel variants.
Concluding Remarks
Our understanding of human genetics has been proven a powerful asset. Genetic literacy and cascade screening in the population has reduced the incidence of congenital disorders like Tay-Sachs disease and cystic fibrosis [80, 81]. However, the likely largest monogenic burden on pediatric health in outbred populations remains due to DNMs. To address this, sperm mosaicism assessment will shed light on the landscape of possible genetic disease at an individual level, serving as an important tool for family planning.
We suggest at least two different applications to clinical practice. The first would be an assessment of the father’s sperm after a child with disease is born in order to evaluate the risk of recurrence. The second would be an assessment of sperm before conception in order to identify clinically relevant sperm mosaicism and provide parental consultation. These practices have great potential to reduce disease burden, although their implementation would be challenging before advances in mosaicism detection and variant classification technology are made. This framework promises a robust approach to preventing crippling genetic diseases prior to conception.
Highlights.
Sperm is the only cell type transmitting male genetic information to offspring, with each cell containing dozens of mutations unique to this cell or shared with only a subset of others.
The three types of sperm mosaicism are distinguished based on their cell and time of origin and their potential of familial or population recurrence.
Genome-wide risk assessment of individual mutations in spermcan characterize the transmission risk for individuals to their offspring.
Sperm mosaicism contributes to human genetic diversity derived from de novo mutation and increases as the population expands and paternal age increases.
Outstanding questions.
Can genes with mutations in a father’s sperm be predicted or identified prior to the conception of a child?
Are there gene networks beyond the RASopathies seen in the selfish sperm model that – if overactivated – provide a spermatogonial stem cell with a growth advantage?
Can we use ‘sperm sequencing’ as a general tool to identify men at risk for sperm mosaicism, to reduce the DNM disease burden in the population?
What is the effect of paternal age or environment on the three types of sperm mosaicism?
Can we experimentally differentiate between mosaicism arising in sperm vs spermatogonia?
Are the same selective pressures exerted on sperm as on oocytes at the individual or population level?
Acknowledgments
The authors wish to acknowledge the Simons Foundation Autism Research Initiative (SFARI, grant 571583) and the NIH (U01MH108898). We also want to thank Drs. Miles Wilkinson, Sangmoon Lee, and Guoliang Chai (UC, San Diego), as well as Jennifer Silhavy (Illumina, Inc), Swapnil Mittal, and Isaac Tang (UC, San Diego) for useful comments and language edits.
Glossary
- Alleles
alternative genetic versions of the same genomic region.
- Allelic fraction (AF)
the fraction of an allele represented in sequencing results, equal to (haploid cells; i.e., sperm) or half (diploid cells, most somatic cells) the fraction of cells carrying the allele.
- Copy-number variant (CNV)
a variant that has multiplied (gain) or lost (loss) a specific region of the genome. De novo mutations (DNMs): new mutations that arise in the child as heterozygous but are not constitutively heterozygous in either of the parents. Primary spermatocytes: diploid cells (2n) that are derived from SSCs and replicate before meiosis I.
- Primordial germcell (PGC)
germ cell progenitors that still have to reach the gonads and divide repeatedly on their migratory route through the gut and into the developing gonads. Secondary spermatocytes: diploid cells (2n) after meiosis I that contain only one duplicated haplotype (other than crossover regions). They will further divide during meiosis II into spermatids that differentiate into mature sperm.
- Single-nucleotide variant (SNV)
a single-base change in the genome.
- Small insertion or deletion (INDEL)
the insertion or deletion of a small (≤50bp) DNA sequence compared with the reference genome.
- Spermatogonial stem cell (SSC)
undifferentiated self-renewal (Type Ad) or differentiating (Type Ap and B) germinal stem cells that line the seminiferous epithelium in the testis; also known as spermatogonia.
- Structural variant (SV)
variation in the structure of the chromosome; can include large (>50 bp) DNA fragment deletion, insertion, inversion, translocation, and complex combinations of these.
- Transposable element (TE)
also called a ‘jumping gene’; a DNA sequence that can change its position in a genome or duplicate itself to be integrated at a different location.
Glossary box
- Primordial Germ Cell (PGC)
Germ cell progenitors that still have to reach the gonads and divide repeatedly on their migratory route through the gut and into the developing gonads.
- Spermatogonial Stem Cell (SSC)
Undifferentiated self-renewal (Type Ad) or differentiating (Type Ap and B) germinal stem cells that line the seminiferous epithelium in the testis. Also known as spermatogonia.
- Primary Spermatocytes
Diploid cells (2n) that are derived from SSCs and replicate before Meiosis I.
- Secondary Spermatocytes
Diploid cells (2n) after Meiosis I that only contain one duplicated haplotype (other than cross-over regions). They will further divide during Meiosis II into spermatids that differentiate into mature sperm.
- De Novo Mutation (DNM)
New mutations that arise in the child as heterozygous but are not constitutively heterozygous in either of the parents.
- Alleles
Alternate genetic versions of the same genomic region.
- Allelic Fraction (AF)
The fraction of an allele represented in the sequencing results, equal to (haploid cells, i. e. sperms) or half (diploid cells, most somatic cells) the fraction of cells carrying the allele.
- Single Nucleotide Variant (SNV)
A single base change in the genome.
- Small Insertion or Deletion (INDEL)
The insertion or deletion of a small (<=50 bp) DNA sequence compared with the reference genome.
- Copy Number Variant (CNV)
A variant that has multiplied (gain) or lost (loss) a specific region of the genome.
- Structural Variant (SV)
The variation in the structure of the chromosome can include large (>50 bp) DNA fragment deletion, insertion, inversion, translocation, and complex combinations of the above.
- Transposable Element (TE)
Also called a ‘jumping gene’, is a DNA sequence that can change its position within a genome or duplicate itself to be integrated at a different location.
References
- 1.Hall JG (1988) Review and hypotheses: somatic mosaicism: observations related to clinical genetics. Am J Hum Genet 43 (4), 355–63. [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell IM et al. (2014) Parent of origin, mosaicism, and recurrence risk: probabilistic modeling explains the broken symmetry of transmission genetics. Am J Hum Genet 95 (4), 345–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biesecker LG and Spinner NB (2013) A genomic view of mosaicism and human disease. Nat Rev Genet 14 (5), 307–20. [DOI] [PubMed] [Google Scholar]
- 4.Morrow EM (2020) Paternal sperm DNA mosaicism and recurrence risk of autism in families. Nat Med 26 (1), 26–28. [DOI] [PubMed] [Google Scholar]
- 5.Samuels ME and Friedman JM (2015) Genetic mosaics and the germ line lineage. Genes (Basel) 6 (2), 216–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu S et al. (2012) Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing. Science 338 (6114), 1627–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell AD et al. (2020) Insights into variation in meiosis from 31,228 human sperm genomes. Nature 583 (7815), 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell IM et al. (2014) Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet 95 (2), 173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breuss MW et al. (2020) Autism risk in offspring can be assessed through quantification of male sperm mosaicism. Nat Med 26 (1), 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W et al. (2019) Association of sperm methylation at LINE-1, four candidate genes, and nicotine/alcohol exposure with the risk of infertility. Front Genet 10, 1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Telenius H et al. (1995) Somatic mosaicism in sperm is associated with intergenerational (CAG)n changes in Huntington disease. Hum Mol Genet 4 (2), 189–95. [DOI] [PubMed] [Google Scholar]
- 12.Sasani TA et al. (2019) Large, three-generation human families reveal post-zygotic mosaicism and variability in germline mutation accumulation. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iossifov I et al. (2012) De novo gene disruptions in children on the autistic spectrum. Neuron 74 (2), 285–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Epi KC et al. (2013) De novo mutations in epileptic encephalopathies. Nature 501 (7466), 217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Homsy J et al. (2015) De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350 (6265), 1262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veltman JA and Brunner HG (2012) De novo mutations in human genetic disease. Nat Rev Genet 13 (8), 565–75. [DOI] [PubMed] [Google Scholar]
- 17.Deciphering Developmental Disorders S (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature 542 (7642), 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vissers LE et al. (2010) A de novo paradigm for mental retardation. Nat Genet 42 (12), 1109–12. [DOI] [PubMed] [Google Scholar]
- 19.Kong A et al. (2012) Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488 (7412), 471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Francioli LC et al. (2015) Genome-wide patterns and properties of de novo mutations in humans. Nat Genet 47 (7), 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao Z et al. (2019) Overlooked roles of DNA damage and maternal age in generating human germline mutations. Proc Natl Acad Sci U S A 116 (19), 9491–9500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner M et al. (2020) Single-cell analysis of human ovarian cortex identifies distinct cell populations but no oogonial stem cells. Nat Commun 11 (1), 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sohni A et al. (2019) The Neonatal and Adult Human Testis Defined at the Single-Cell Level. Cell Rep 26 (6), 1501–1517 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulze W and Rehder U (1984) Organization and morphogenesis of the human seminiferous epithelium. Cell Tissue Res 237 (3), 395–407. [DOI] [PubMed] [Google Scholar]
- 25.Oliver TR et al. (2008) New insights into human nondisjunction of chromosome 21 in oocytes. PLoS Genet 4 (3), e1000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.MacLennan M et al. (2015) Oocyte development, meiosis and aneuploidy. Semin Cell Dev Biol 45, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu FL et al. (2020) A comparison of humans and baboons suggests germline mutation rates do not track cell divisions. PLoS Biol 18 (8), e3000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang AY et al. (2018) Distinctive types of postzygotic single-nucleotide mosaicisms in healthy individuals revealed by genome-wide profiling of multiple organs. PLoS Genet 14 (5), e1007395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye AY et al. (2018) A model for postzygotic mosaicisms quantifies the allele fraction drift, mutation rate, and contribution to de novo mutations. Genome Res 28 (7), 943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.M. DF (2013) Origin, migration, and proliferation of human primordial germ cells. In Oogenesis (G. C et al. eds), Springer. [Google Scholar]
- 31.de Kretser DM et al. (1998) Spermatogenesis. Hum Reprod 13 Suppl 1, 1–8. [DOI] [PubMed] [Google Scholar]
- 32.Fayomi AP and Orwig KE (2018) Spermatogonial stem cells and spermatogenesis in mice, monkeys and men. Stem Cell Res 29, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jonsson H et al. (2018) Multiple transmissions of de novo mutations in families. Nat Genet. [DOI] [PubMed] [Google Scholar]
- 34.Miller TE et al. (2004) Recurrent third-trimester fetal loss and maternal mosaicism for long-QT syndrome. Circulation 109 (24), 3029–34. [DOI] [PubMed] [Google Scholar]
- 35.Riviere JG et al. (2020) Uncovering low-level maternal gonosomal mosaicism in X-linked agammaglobulinemia: implications for genetic counseling. Front Immunol 11, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edwards MJ et al. (1992) Recurrence of lethal osteogenesis imperfecta due to parental mosaicism for a mutation in the COL1A2 gene of type I collagen. The mosaic parent exhibits phenotypic features of a mild form of the disease. Hum Mutat 1 (1), 47–54. [DOI] [PubMed] [Google Scholar]
- 37.Namikawa C et al. (1995) Recurrence of osteogenesis imperfecta because of paternal mosaicism: Gly862-->Ser substitution in a type I collagen gene (COL1A1). Hum Genet 95 (6), 666–70. [DOI] [PubMed] [Google Scholar]
- 38.Dobrovolny R et al. (2005) Recurrence of Fabry disease as a result of paternal germline mosaicism for alpha-galactosidase a gene mutation. Am J Med Genet A 134A (1), 84–7. [DOI] [PubMed] [Google Scholar]
- 39.Choi HJ et al. (2008) Familial focal segmental glomerulosclerosis associated with an ACTN4 mutation and paternal germline mosaicism. Am J Kidney Dis 51 (5), 834–8. [DOI] [PubMed] [Google Scholar]
- 40.Pauli S et al. (2009) Proven germline mosaicism in a father of two children with CHARGE syndrome. Clin Genet 75 (5), 473–9. [DOI] [PubMed] [Google Scholar]
- 41.Mensa-Vilaro A et al. (2016) Brief Report: First Identification of Intrafamilial Recurrence of Blau Syndrome due to Gonosomal NOD2 Mosaicism. Arthritis Rheumatol 68 (4), 1039–44. [DOI] [PubMed] [Google Scholar]
- 42.Rowczenio DM et al. (2016) Brief report: Association of tumor necrosis factor receptor-associated periodic syndrome with gonosomal mosaicism of a novel 24-nucleotide TNFRSF1A deletion. Arthritis Rheumatol 68 (8), 2044–9. [DOI] [PubMed] [Google Scholar]
- 43.Hancarova M et al. (2019) Parental gonadal but not somatic mosaicism leading to de novo NFIX variants shared by two brothers with Malan syndrome. Am J Med Genet A 179 (10), 2119–2123. [DOI] [PubMed] [Google Scholar]
- 44.Sihombing NRB et al. (2020) Pathogenic variant in NFIX gene affecting three sisters due to paternal mosaicism. Am J Med Genet A 182 (11), 2731–2736. [DOI] [PubMed] [Google Scholar]
- 45.Forissier JF et al. (2000) First description of germline mosaicism in familial hypertrophic cardiomyopathy. J Med Genet 37 (2), 132–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Youssoufian H and Pyeritz RE (2002) Mechanisms and consequences of somatic mosaicism in humans. Nat Rev Genet 3 (10), 748–58. [DOI] [PubMed] [Google Scholar]
- 47.Niu DM et al. (2006) Paternal gonadal mosaicism of NIPBL mutation in a father of siblings with Cornelia de Lange syndrome. Prenat Diagn 26 (11), 1054–7. [DOI] [PubMed] [Google Scholar]
- 48.Wilson M et al. (2008) The clinical phenotype of mosaicism for genome-wide paternal uniparental disomy: two new reports. Am J Med Genet A 146A (2), 137–48. [DOI] [PubMed] [Google Scholar]
- 49.Chiang PW et al. (2009) Somatic and germ-line mosaicism in Rubinstein-Taybi syndrome. Am J Med Genet A 149A (7), 1463–7. [DOI] [PubMed] [Google Scholar]
- 50.Chen CP et al. (2013) Prenatal diagnosis of recurrent autosomal dominant osteogenesis imperfecta associated with unaffected parents and paternal gonadal mosaicism. Taiwan J Obstet Gynecol 52 (1), 106–9. [DOI] [PubMed] [Google Scholar]
- 51.Bernkopf M et al. (2017) Quantification of transmission risk in a male patient with a FLNB mosaic mutation causing Larsen syndrome: Implications for genetic counseling in postzygotic mosaicism cases. Hum Mutat 38 (10), 1360–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fusco F et al. (2017) Unusual father-to-daughter transmission of incontinentia pigmenti due to mosaicism in IP males. Pediatrics 140 (3). [DOI] [PubMed] [Google Scholar]
- 53.Yang X et al. (2017) Genomic mosaicism in paternal sperm and multiple parental tissues in a Dravet syndrome cohort. Sci Rep 7 (1), 15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu A et al. (2018) Mosaicism and incomplete penetrance of PCDH19 mutations. J Med Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang X et al. (2019) ATP1A3 mosaicism in families with alternating hemiplegia of childhood. Clin Genet 96 (1), 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen L et al. (2020) Fetal cardiac rhabdomyoma due to paternal mosaicism of TSC2: A case report. Medicine (Baltimore) 99 (35), e21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Q et al. (2018) Genomic mosaicism in the pathogenesis and inheritance of a Rett syndrome cohort. Genet Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang X et al. (2020) MosaicBase: a knowledgebase of postzygotic mosaic variants in noncancer disease-related and healthy human individuals. Genomics Proteomics Bioinformatics 18 (2), 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Giannoulatou E et al. (2013) Contributions of intrinsic mutation rate and selfish selection to levels of de novo HRAS mutations in the paternal germline. Proc Natl Acad Sci U S A 110 (50), 20152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goriely A and Wilkie AO (2012) Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet 90 (2), 175–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glaser RL et al. (2003) The paternal-age effect in Apert syndrome is due, in part, to the increased frequency of mutations in sperm. Am J Hum Genet 73 (4), 939–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Henderson S et al. (2000) Germline and somatic mosaicism in achondroplasia. J Med Genet 37 (12), 956–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Altmuller F et al. (2017) Genotype and phenotype spectrum of NRAS germline variants. Eur J Hum Genet 25 (7), 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maher GJ et al. (2018) Selfish mutations dysregulating RAS-MAPK signaling are pervasive in aged human testes. Genome Res 28 (12), 1779–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goriely A et al. (2009) Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat Genet 41 (11), 1247–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giannoulatou E et al. (2017) Whole-genome sequencing of spermatocytic tumors provides insights into the mutational processes operating in the male germline. PLoS One 12 (5), e0178169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Goriely A et al. (2003) Evidence for selective advantage of pathogenic FGFR2 mutations in the male germ line. Science 301 (5633), 643–6. [DOI] [PubMed] [Google Scholar]
- 68.Kahraman S et al. (2020) High rates of aneuploidy, mosaicism and abnormal morphokinetic development in cases with low sperm concentration. J Assist Reprod Genet 37 (3), 629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Laurentino S et al. (2015) Epigenetic germline mosaicism in infertile men. Hum Mol Genet 24 (5), 1295–304. [DOI] [PubMed] [Google Scholar]
- 70.Yang X et al. (2020) Temporal stability of human sperm mosaic mutations results in lifelong threat of transmission to offspring. bioRxiv, 2020.10.14.339796. [Google Scholar]
- 71.Phillips BT et al. (2010) Spermatogonial stem cell regulation and spermatogenesis. Philos Trans R Soc Lond B Biol Sci 365 (1546), 1663–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goldmann JM et al. (2016) Parent-of-origin-specific signatures of de novo mutations. Nat Genet 48 (8), 935–9. [DOI] [PubMed] [Google Scholar]
- 73.Jonsson H et al. (2017) Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature 549 (7673), 519–522. [DOI] [PubMed] [Google Scholar]
- 74.Hinch AG et al. (2019) Factors influencing meiotic recombination revealed by whole-genome sequencing of single sperm. Science 363 (6433). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Adey AC (2017) Haplotype resolution at the single-cell level. Proc Natl Acad Sci U S A 114 (47), 12362–12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Acuna-Hidalgo R et al. (2016) New insights into the generation and role of de novo mutations in health and disease. Genome Biol 17 (1), 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rahbari R et al. (2016) Timing, rates and spectra of human germline mutation. Nat Genet 48 (2), 126–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pasmant E and Pacot L (2020) Should we genotype the sperm of fathers from patients with ‘de novo’ mutations? Eur J Endocrinol 182 (1), C1–C3. [DOI] [PubMed] [Google Scholar]
- 79.Simard M et al. (2019) Impact of paternal age at conception on human health. Clin Chem 65 (1), 146–152. [DOI] [PubMed] [Google Scholar]
- 80.Kaback M et al. (1993) Tay-Sachs disease--carrier screening, prenatal diagnosis, and the molecular era. An international perspective, 1970 to 1993. The International TSD Data Collection Network. JAMA 270 (19), 2307–15. [PubMed] [Google Scholar]
- 81.Moskowitz SM et al. (2008) Clinical practice and genetic counseling for cystic fibrosis and CFTR-related disorders. Genet Med 10 (12), 851–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goldmann JM et al. (2018) Germline de novo mutation clusters arise during oocyte aging in genomic regions with high double-strand-break incidence. Nat Genet 50 (4), 487–492. [DOI] [PubMed] [Google Scholar]
- 83.Snijders RJ et al. (1998) UK multicentre project on assessment of risk of trisomy 21 by maternal age and fetal nuchal-translucency thickness at 10-14 weeks of gestation. Fetal Medicine Foundation First Trimester Screening Group. Lancet 352 (9125), 343–6. [DOI] [PubMed] [Google Scholar]
- 84.Watson IR et al. (2013) Emerging patterns of somatic mutations in cancer. Nat Rev Genet 14 (10), 703–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marin-Valencia I et al. (2014) Pathogenetic mechanisms of focal cortical dysplasia. Epilepsia 55 (7), 970–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Blumcke I et al. (2017) Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med 377 (17), 1648–1656. [DOI] [PubMed] [Google Scholar]
- 87.Johnson AD and Alberio R (2015) Primordial germ cells: the first cell lineage or the last cells standing? Development 142 (16), 2730–9. [DOI] [PMC free article] [PubMed] [Google Scholar]