

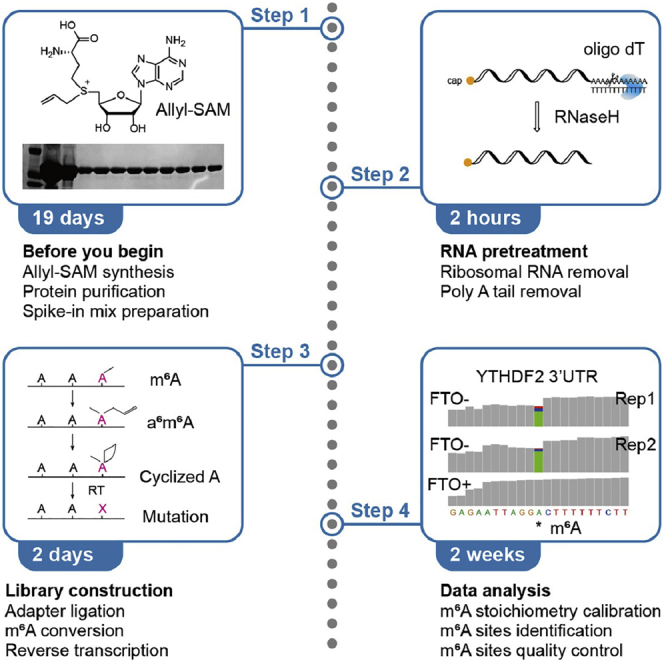
Summary
As the most abundant internal mRNA modification, N6-methyladenosine (m6A) was involved in almost all the aspects of RNA metabolism. Here, we introduce our protocol for m6A-SAC-seq, which enables the whole transcriptome-wide mapping of m6A RNA modification at single-nucleotide resolution with stoichiometry information. m6A-SAC-seq relies on selective allyl labeling of m6A by specific methyltransferase and chemical treatment that introduce mutation upon reverse transcription. The technique only requires ∼30 ng of input RNA.
For complete details on the use and execution of this protocol, please refer to Hu et al. (2022).
Subject areas: Bioinformatics, Biotechnology and bioengineering, Chemistry, Genomics, Molecular biology, Molecular/Chemical probes, Protein biochemistry, Protein expression and purification, Sequence analysis
Graphical abstract
Highlights
-
•
A protocol to label RNA m6A with allyl group
-
•
Incorporate mismatch and detect m6A at single-nucleotide resolution
-
•
Provide stoichiometric information of individual m6A sites with spike-in calibration
-
•
An optimized assay to detect and quantitate RNA m6A with only ∼ 30 ng input RNA
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
As the most abundant internal mRNA modification, N6-methyladenosine (m6A) was involved in almost all the aspects of RNA metabolism. Here, we introduce our protocol for m6A-SAC-seq, which enables the whole transcriptome-wide mapping of m6A RNA modification at single-nucleotide resolution with stoichiometry information. m6A-SAC-seq relies on selective allyl labeling of m6A by specific methyltransferase and chemical treatment that introduce mutation upon reverse transcription. The technique only requires ∼30 ng of input RNA.
Before you begin
Synthesis of Allyl-SAM
Timing: 3 days
Synthesize the allyl-SAM to replace SAM as cofactor of dimethyl transferase MjDim1. The allyl-SAM is used in “step-by-step method details” 4b and 4c.
((2S)-2-Amino-4-[(RS)-{[(2S,3S,4R,5R)-5-(4-amino-9H-purin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl} v(prop-2-enyl)sulfaniumyl]butanoate) (MW: 552).
-
1.
Add 100 mg of S-(5′-Adenosyl)-L-homocysteine in a 50 mL round bottom flask. Add 1 mL of acetic acid and 1 mL of formic acid. Add 52.0 mg of AgClO4. Add 2.125 mL of allyl bromide. Add a stir bar and stir at 25°C for 8 h. Quench the reaction by adding 20 mL of 0.1% TFA (Trifluoroacetic acid, CF3CO2H).
CRITICAL: Perform these steps in the hood with Personal Protective Equipment (PPE).
-
2.
Transfer the quenched reaction mixture into a 100 mL separation funnel. Wash the reaction mixture with 10 mL of diethyl ether. Keep the lower phase after separation. Repeat the washing step for three times, using 10 mL of diethyl ether for each time. Transfer the washed aqueous phase to a 50 mL syringe with the nozzle attached to a 0.22 μm filter. Carefully attach the plunger. Pass the solution through the filter by applying a steady pressure to the plunger. Transfer the solution into a 50 mL round-bottom flask. Evaporate residual ether with a rotatory evaporator. Chill the solution at 4°C by submerging the flask in ice-water bath.
-
3.
Add 0.1% of the filtrate’s volume of TFA (add 20 μL of TFA to 20 mL of filtrate, for example). Aliquot the acidified solution into 1.5 mL tubes. Snap-freeze and lyophilize the solution using a VirTis Sentry Lyophilizer.
-
4.
Purify the allyl-SAM by reconstituting the lyophilized crude in 0.1% TFA. Purify the solution on a Waters Alliance HPLC system with a Higgins Analytical 5 μm, 250 × 4.6 mm C18 reversed phase HPLC column. Set the gradient to 5% of Phase B in 30 min, where Phase A = 0.1% TFA in deionized water; Phase B = 0.1% TFA in acetonitrile. Troubleshooting 1.
-
5.
Determine the concentration of purified allyl-SAM by measuring A260 on NanoDrop. The molar extinction coefficient is 15.4 mM-1 (The molar extinction coefficient is calculated as follows: weigh the synthesized allyl-SAM powder, dissolve the powder in nuclease free H2O to get the accurate molar concentration. The molar extinction coefficient is obtained by dividing A260 with molar concentration.). Aliquot the eluate into 1.5 mL tubes each containing 0.8 μmol of ally-SAM. Lyophilize the eluate. Store the compound at –80°C. The lyophilized allyl-SAM is stable at –80°C for at least a year.
Preparation of spike-in probes
Synthesize the spike-in probes to generate the calibration curve of the m6Astoichiometry versus mutation generated by m6A-SAC-seq. The spike-in probes are used in “step-by-step method details” 3c.
-
6.
Design and synthesize the RNA probes (key resources table and Table S1) with modification using an EXPEDIT DNA Synthesizer 8909. Purchase the unmodified probes from Integrated DNA Technologies. Generate the 41bp-Probes for the calibration spike-in mix (used for the method validation part) by splint ligation of barcoded 12 mer purchased oligo with the 29 mer m6A containing synthesized probe (Table S1).
-
7.
Synthesize a6m6A-containing RNA oligo by incorporating N6-phenyl-adenosine phosphoramidite into the designed sequence containing a GGACU motif. Treat the beads with N-methyl-N-allylamine to convert O6-phenyl to the N6-methyl N6-allyl group. After the regular procedure to remove 2′-silyl protecting group, precipitate the crude RNA oligo with ethanol further purify the RNA oligo by RP HPLC.
-
8.Confirm the structure of the probe by rapifleX MALDI-TOF MS with THAP as the matrix.
-
a.Dissolve the HPLC purified probe in deionized water. Treat 1 nmol of N6-methyl N6-allyl containing probe in 200 μL water with 8 μL of 0.2 M iodine dissolved in 0.2 M KI. After incubation at 25°C for 1 h, add 8 μL 0.2 M Na2S2O3 to quench the reaction.
-
b.Filter the mixture and inject it into HPLC with the same gradient. It can be observed that the original peak disappeared at retention time (32.3 min), while two identical peaks eluted at 29.3 and 29.6 min, which are the two isomers of the cyclized products. Both peaks show the same MS at 3018 in the MALDI-TOF mass spectrum, consistent with the proposed structure (Figure S3).
-
a.
-
9.Order 8 RNA probes (spike-in probe 1–8) with the sequence listed in the “key resources table” to make the spike-in mix (used for library construction).
-
a.Probes 5–8 contain a N6-methyladenosine modification (m6A, code /iN6Me-rA/ in IDT) (key resources table). All probes contain random ribonucleotides designated as rN, which should be ordered as standard mixed bases with an equal ratio of rA/rU/rC/rG. The probes should be ordered with RNase-free HPLC purification. Dissolve each oligo-ribonucleotide in RNase-free water to produce a 100 ng/μL stock solution.
-
b.Prepare the spike-in mix by adding the stock solutions in the following ratio, so that we could get the spike-in mix probe having 0%, 25%, 50%, 75%, 100% m6A stoichiometry with specific barcode, respectively:
Spike-in probe (for library construction) ID 1 2 3 4 5 6 7 8 Volume of 100 ng/μL spike-in probe stock (μL) 20 15 10 5 5 10 15 20 -
c.Prepare 100 ng/μL spike-in mix (100 μL of volume) and dilute the mix 10 times with UltraPure™ DEPC-Treated water into 10 ng/μL. Aliquot the 10 ng/μL mix into 1.5 mL tubes per 50 μL volume. Store the aliquots at –80°C. The aliquoted spike-in mix is stable at −80°C for at least a year without frequent freeze-thaw cycles.
-
a.
Preparation of ULP1
Purify the ULP1 protein to cleave the SUMO tag and release MjDim1 protein during MjDim1 purification. The ULP1 protein is used during MjDim1preparation (step 15, d).
-
10.Day 1. Prepare liquid medium and recombinant ULP1 bacteria.
-
a.Prepare 1 L of 2 × YT liquid medium, adjust the pH to be 7.0 and autoclave.
-
b.Transform the ULP1 plasmid and spread the recombinant bacteria competent cell on the LB-agar plate in a Petri dish (in the morning, at around 7:00 AM).
-
i.The ULP1 gene was cloned into a pET28a vector and is a generous gift from Professor Yanhui Xu’s lab at Fudan University in China. Transform the T7 Express Competent Escherichia coli (NEB) with the plasmid and culture at 37°C for 1 h.
-
ii.Harvest the recombinant bacteria competent cells at 2,500 × g for 3 min and spread the bacteria on the LB-agar plate with 50 μg/mL kanamycin. Put the plate in 37°C constant-temperature incubator for 30 min, turn the plate upside down and culture it at 37°C for 12–16 h.
-
i.
-
c.Culture the recombinant bacteria in LB medium for 12 h (in the evening, at around 8:00 PM).
-
a.
Pick up colonies from the plate and culture for 12 h at 37°C in LB medium with 50 μg/mL kanamycin for 12–16 h.
-
11.Day 2. Culture recombinant bacteria in flasks.
-
a.At around 9:00 AM, collect 1 mL of culture and mix with an equal volume of glycerol. Store at −80°C. The glycerol stock can be stored for at least a year if caution is taken to minimize freeze-saw cycles.
-
b.At around 10:00 AM, inoculate the remaining recombinant bacteria into 1 L of liquid medium in the flask. Prepare 6 L of liquid medium in total. Culture the recombinant bacteria at 37°C in shaking incubator for 3–4 h.
-
c.At around 2:00 PM, cool the cells to 16°C when the optical density at 600 nm (OD600) reached 1.0. After ∼2 h, add IPTG to a final concentration of 0.1 mM for inducible expression, and culture the cells at 16°C for additional 18 h.
-
d.Load nickel columns with 3 mL of Ni-NTA resin for each. Wash Ni-NTA resin using 50 mL of deionized H2O for three times. Then wash the resin with 50 mL of 0.1 M imidazole, pH 8.0 (4°C, 3 months for storage), followed by thorough deionized H2O washing (more than 200 mL) to get rid of the imidazole. Then wash the resin with 50 mL of ULP1 lysis buffer (25 mM Tris, pH 8.0, 150 mM NaCl, 4°C, 3 months for storage) for three times. Store at 4°C.
-
a.
-
12.Day 3. Purify ULP1 protein.
-
a.Collect the cells by centrifuging at 2,500 × g for 10 min. Resuspend the cells in 300 mL of ULP1 lysis buffer (50 mL/1 L culture). Add 100 μL of 10 mg/mL DNase I. Keep the resuspension on ice.
-
b.Rinse the interior of EmulsiFlex C3 with deionized water and ULP1 lysis buffer. Crush the cells by passing the resuspension through the machine while applying 1000–1500 bars of high pressure. Keep the lysate flow-through on ice.CRITICAL: The whole crushing process should be chilled by ice-water mixture.
-
c.Centrifugate the lysate at 14,000 × g for 30 min. Load the supernatant onto the prepared nickel column. After the loaded volume completely passes through, wash the column with 300 mL of ULP1 washing buffer (25 mM Tris, pH 8.0, 150 mM NaCl, 20 mM Imidazole, 4°C, 3 months for storage).
-
d.Elute the ULP1 protein with ULP1 elution buffer (25 mM Tris, pH 8.0, 150 mM NaCl, 250 mM Imidazole, 4°C, 3 months for storage), dilute the eluate with ten times the volume of ULP1 lysis buffer. Aliquot the ULP1 enzyme into 15 mL tubes per 10 mL volume. The aliquoted ULP1 enzyme is stable at −80°C for at least a year without frequent freeze-thaw cycles.CRITICAL: The dilution of ULP1 eluate with ULP1 lysis buffer is necessary to reduce the concentration of imidazole to less than 20 mM. Otherwise, the ULP1 enzyme will precipitate and lose activity after freeze-and-thaw.
-
a.
Preparation of methyltransferase MjDim1
Purify the MjDim1 protein to convert m6A to allyl6m6A. MjDim1 protein is used in “step-by-step method details” 4b and 4c.
-
13.Day 1. Prepare liquid medium and recombinant MjDim1 bacteria.
-
a.Prepare 6 L of 2 × YT liquid medium, adjust the pH to be 7.0 and autoclave.
-
b.Transform the Mjdim1 plasmid and spread the recombinant bacteria competent cell on the LB-agar plate in a Petri dish (in the morning, at around 7:00 AM).
-
i.The MjDim1 gene was codon optimized for BL21 (DE3) expression and synthesized by Thermo Fisher Scientific and cloned into a pET-His-SUMO vector. Transform the T7 Express Competent Escherichia coli (NEB) with the plasmid and culture at 37°C for 1 h.
-
ii.Harvest the recombinant bacteria competent cells at 2,500 × g for 3 min and spread the bacteria on the LB-agar plate with 50 μg/mL kanamycin. Put the plate in 37°C constant-temperature incubator for 30 min, turn the plate upside down and culture it at 37°C for 12–16 h.
-
i.
-
c.Culture the recombinant bacteria in LB medium for 12 h (in the evening, at around 8:00 PM).
-
a.
Pick up colonies from the plate and culture for 12 h at 37°C in LB medium with 50 μg/mL kanamycin for 12–16 h.
-
14.Day 2. Culture recombinant bacteria in flasks.
-
a.At around 9:00 AM, collect 1 mL of culture and mix with an equal volume of glycerol. Store at −80°C. The glycerol stock can be stored for at least a year if caution is taken to minimize freeze-saw cycles.
-
b.At around 10:00 AM Inoculate the remaining recombinant bacteria into 1 L liquid medium in the flask. Prepare 6 L of liquid medium in total. Culture the recombinant bacteria at 37°C in shaking incubator for 3–4 h.
-
c.At around 2:00 PM, cool the cells to 16°C when the optical density at 600 nm (OD600) reached 1. After ∼2 h, add IPTG to the final concentration of 0.1 mM for inducible expression, and culture the cells at 16°C for an additional 18 h.
-
d.Load four nickel columns with 3 mL of Ni-NTA resin for each. Was Ni-NTA resin using 50 mL of deionized H2O for three times. Then wash the resin with 50 mL of 0.1 M imidazole, pH 8.0, followed by thorough deionized H2O washing to get rid of the imidazole. Then wash the resin with 50 mL of MjDim1 lysis buffer (1 × PBS, pH 7.4 150 mM NaCl, 4°C, 3 months for storage) for three times. Store at 4°C.
-
a.
-
15.Day 3. Collect the bacteria cells and purify MjDim1 protein.
-
a.Collect the cells by centrifuging at 2,500 × g for 10 min. Resuspend the cells in 300 mL of lysis buffer (50 mL/1 L culture). Add 100 μL of 10 mg/mL DNase I. Keep the resuspension on ice.
-
b.Rinse the interior of EmulsiFlex C3 with deionized water and MjDim1 lysis buffer. Crush the cells by passing the resuspension through the machine while applying 1000–1500 bars of high pressure. Keep the lysate flow-through on ice.CRITICAL: Although the Mjdim1 enzyme is thermostable, the whole crushing process should be chilled by ice-water mixture.
-
c.Centrifugate the lysate at 14,000 × g for 30 min. Load the supernatant onto the prepared nickel column. After the loaded volume completely passes through, wash the column with 300 mL of MjDim1 washing buffer (1 × PBS, pH 7.4, 150 mM NaCl, 20 mM Imidazole, 4°C, 3 months for storage).
-
d.Add 15 mL of 0.5 mg/mL ULP1 to the column. Let the buffer pass through the column and save ∼ 2 mL buffer above the nickel resin. Then seal the outlet nozzle with cap and put the column in an upright position. Resuspend the resin thoroughly and incubate at 4°C for 12 h to cleave the SUMO tag and release MjDim1 protein.CRITICAL: As the ULP1 has 6 × his-tag and it binds with the nickel resin, resuspend the resin with added ULP1 thoroughly is important for efficient cleavage.
-
e.Equilibrate the SOURCE 15Q-column and a 15S-column by washing both sequentially in 60 mL of 300 mM NaOH, 60 mL of H2O, 60 mL of buffer B (20 mM Bis-tris, pH 6.0, 1 M NaCl, 4°C, 3 months for storage), and 60 mL of buffer A (20 mM Bis-tris, pH 6.0, 4°C, 3 months for storage). Store at 4 °.
-
a.
-
16.Day 4. Purify the MjDim1 protein to high purity with High Performance Liquid Chromatography (HPLC).
-
a.Elute the SUMO-cleaved protein from the nickel column with 80 mL of MjDim1 elution buffer (20 mM Tris, pH 8.0, 50 mM NaCl, 4°C, 3 months for storage). Add 20 μg of HeLa total RNA into the eluate. Mix well and incubate at 37°C for 30 min.CRITICAL: This step is essential to exhaust the endogenous SAM-cofactors binding in the catalytic core of the MjDim1 methyltransferase. It’s important to reduce non-specific labeling introduced by the methyltransferase.
-
b.Load the eluate with RNA onto the SOURCE 15Q-column. Collect the flowthrough. Then apply 100% Buffer A and collect another 50 mL of flowthrough. If using AKTA Pure FPLC system, a significant reduction in UV-280 nm (UV280) signal should be observed around this time point.CRITICAL: This step is important to remove all the DNA and RNA binding with the MjDim1 enzyme. Negatively charged nucleic acid will be retained on the SOURCE 15Q-column but the positively charged MjDim1 enzyme will escape from the Q-column. Save the flowthrough.
-
c.Load the flowthrough from the Q-column onto S-column. Equilibrate the S-column with 60 mL of buffer A. Then apply a gradient of 50% buffer B for 20 min. If using AKTA Pure FPLC system, a significant peak in UV280 signal should be observed peaking at 35 mS/cm conductivity, and it should be the only peak.
-
d.Concentrate the MjDim1 enzyme with 10 kD MWCO spin filters to 1 mL. Spin at 3,000 × g for 30 min each time. Add sterile glycerol, making its final concentration 30%, v/v. Measure the A280 with NanoDrop with the ten times diluted MjDim1 protein. Troubleshooting 2 The concentration should be 1.2–2 mM (Molar extinction coefficient 23.38 mM-1). The molar extinction coefficient is calculated by “ProtParam tool” (https://web.expasy.org/protparam/). We obtain the molar concentration by dividing A280 with molar extinction coefficient. Aliquot the MjDim1 enzyme into 1.5 mL tubes per 25 μL volume. The aliquoted MjDim1 enzyme is stable at −80°C for at least a year without frequent freeze-thaw cycles.CRITICAL: Protein of too high concentration tends to form emulsion and should be diluted before being loaded on Nanodrop detector. Otherwise, the A280 value is not accurate.
-
a.
Quality control of MjDim1 activity
Validate the activity of the MjDim1 protein. MjDim1 protein is used in “step-by-step method details” 4b and 4c.
-
17.Measure the reactivity of purified Mjdim1 by performing the labeling reaction on 50 ng of Mjdim1 QC probe (key resources table).
-
a.Resuspend 40 μL of Dynabeads MyOne Streptavidin C1 beads in 40 μL of 0.1 M NaOH. Wash again in 40 μL of 10 mM Tris HCl, pH 7.5. Resuspend the beads in 40 μL of 1 × Binding/Wash buffer. Add 2 μL of 50 ng/μL Mjdim1 QC probe. Mix well and incubate at 25°C for 15 min.
-
b.Place the beads on a magnetic rack. Decant the beads. Wash the beads once in 50 μL of 1 × Binding/Wash buffer. Then twice in 50 μL of 10 mM Tris HCl, pH 7.5 (using 50 μL each time). Resuspend the beads in 12 μL of RNase-free H2O. Divide into two halves. Use one half for labeling and the other as control. Keep the control on ice.
-
c.Prepare neutralized allyl-SAM. Dilute 3 μL of 20 mM Allyl-SAM with 3 μL of Nuclease free H2O and neutralize it with 0.6 μL of 1 M Tris HCl, pH 8.3.CRITICAL: The Allyl-SAM is dissolved in 0.1% TFA and it needs to be neutralized right before usage. Do not re-freeze neutralized allyl-SAM.
-
d.Add the following components to the beads (resuspended in 6 μL of RNase-free H2O).
Reagent Final concentration Amount Neutralized Allyl SAM (∼9 mM) 2.7 mM 6 μL SUPERase In RNase Inhibitor (20 U/μL) 2 U/μL 2 μL 10 × MjDim1 reaction buffer (400 mM HEPES, pH 8.0, 400 mM NH4Cl and 40 mM MgCl2) 1 × 2 μL Purified Mjdim1 (1.2–2 mM) 0.4 mM 4 μL Total N/A 14 μL Mix well and incubate at 50°C for 1 h. Wash the beads sequentially with 50 μL of 0.1% PBST (DPBS + 0.1% Tween™ 20, 4°C, 3 months for storage) (v/v), 50 μL of 1× Binding/Wash buffer (5 mM Tris HCl, pH 7.5, 0.5 mM EDTA and 1 M RNase free NaCl, 4°C, 3 months for storage) and twice with 50 μL of 10 mM Tris HCl, pH 7.5 (using 50 μL each time). Decant and resuspend in 6 μL of RNase-free H2O (UltraPure™ DEPC-Treated Water). -
e.For both the labeled probe and the control group (resuspended in 6 μL of RNase-free H2O), add 0.3 μL of 1 M NH4Ac, pH 5.3 and 1 μL of Nuclease P1 (100 U/μL). Mix well and incubate at 37°C for 12 h. Then add 3 μL of 10 × Fast AP Reaction Buffer and 1 μL of Fast AP (1 U/μL). Mix well and incubate at 37°C for 2 h.
-
f.Load the supernatant onto 0.22 μM PVDF spin filter. Centrifuge at 10,000 × g. Transfer the filtrate into 250 μL 11-mm cap LC vials. Measure the two samples using a SCIEX Triple Quad LC-MS/MS System with Agilent Eclipse XDB-C18 reversed phase HPLC column. Use the following LC program.
Time (min) A (%) B (%) 0.00 98.00 2.00 3.00 82.00 18.00 4.00 50.00 50.00 5.00 10.00 90.00 6.00 10.00 90.00 6.10 2.00 98.00 7.00 2.00 98.00 7.10 98.00 2.00 9.00 98.00 2.00 -
g.The Phase A is 0.1% formic acid in water, and Phase B is 0.1% formic acid in Methanol. Set the scan type to MRM. Polarity = Positive. Duration = 9 min, Cycle = 1270 × 0.425 s per cycle. Use the following parameters:
Base m/z transition Time (ms) CE (V) A 268->136 100.0 47.000 m6A 282->150 100.0 25.000 G 284->152 50.0 17.000 C 245->113 50.0 19.000 U 244->112 100.0 17.000 -
h.The conversion rate is calculated as follows:Conversion Rate %= (1-((Area m6A, treated/Area G, treated)/ (Area m6A, control/Area G, control))) × 100%Area stands for integrated peak area. The conversion rate should not be lower than 50% for one-round enzyme labeling and more than 90% for four-round enzyme labeling.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| S-(5′-Adenosyl)-L-homocysteine, C14H20N6O5S | Sigma-Aldrich | Cat#A9384 |
| Silver perchlorate, AgClO4 | Sigma-Aldrich | Cat#674583 |
| Allyl bromide, CH2=CHCH2Br | Sigma-Aldrich | Cat#A29585 |
| Acetic acid, CH3CO2H | Sigma-Aldrich | Cat#695092 |
| Formic acid, HCOOH | Sigma-Aldrich | Cat#695076 |
| TFA (Trifluoroacetic acid, CF3CO2H) | Sigma-Aldrich | CAS#T6508 |
| N-methyl-N-allylamine | Sigma-Aldrich | Cat#255130 |
| THAP(2′,4′,6′-Trihydroxyacetophenone) | Sigma-Aldrich | Cat#T64602 |
| UltraPure™ DEPC-Treated Water | Thermo Fisher Scientific | Cat#750023 |
| IPTG | Sigma-Aldrich | Cat#I6758 |
| Ni-NTA resin | GE Healthcare | Cat#17-5318-02 |
| DNase I, grade II, from bovine pancreas | Roche | Cat#10104159001 |
| SUPERase In RNase Inhibitor (20 U/μL) | Thermo Fisher Scientific | AM2696 |
| Nuclease P1 | New England Biolabs | Cat# M0660S |
| FastAP Thermosensitive Alkaline Phosphatase (1 U/μL) and 10 × FastAP Buffer | Thermo Fisher Scientific | Cat# EF0651 |
| UltraPure™ 1 M Tris-HCI Buffer, pH 7.5 | Thermo Fisher Scientific | Cat#15567027 |
| UltraPure™ 0.5 M EDTA, pH 8.0 | Thermo Fisher Scientific | Cat#15575020 |
| NaCl (5 M), RNase-free | Thermo Fisher Scientific | Cat#AM9760G |
| UltraPure™ DEPC-Treated Water | Thermo Fisher Scientific | Cat#750023 |
| Ethyl alcohol, Pure, CH3CH2OH | Sigma-Aldrich | Cat#E7023 |
| Methanol, CH3OH | Sigma-Aldrich | Cat#34860 |
| Acetonitrile, CH3CN | Sigma-Aldrich | Cat#34851 |
| Nuclease Decontamination Solution | IDT | Cat#11-05-01-01 |
| HEPES | Fisher Scientific | Cat# BP310-100 |
| Sodium hydroxide, NaOH | Sigma-Aldrich | Cat#221465 |
| Ammonium chloride, NH4Cl | Sigma-Aldrich | Cat#1.01142 |
| Magnesium chloride, MgCl2 | Sigma-Aldrich | Cat# M4880 |
| Iodine, I2 | Sigma-Aldrich | Cat#376558 |
| Potassium iodide, KI | Sigma-Aldrich | Cat#207969 |
| Tween™ 20 | Fisher Scientific | Cat#BP337-100 |
| DPBS, no calcium, no magnesium | Thermo Fisher Scientific | Cat#14190144 |
| Dynabeads™ mRNA DIRECT™ Purification Kit | Thermo Fisher Scientific | Cat#61012 |
| RiboMinus™ Eukaryote System v2 | Thermo Fisher Scientific | Cat#A15026 |
| RNA Clean & Concentrator-5 | Zymo Research | Cat#R1013 |
| Qubit™ RNA HS Assay Kit | Thermo Fisher Scientific | Cat#Q32852 |
| NEBNext® Magnesium RNA Fragmentation Module | New England Biolabs | Cat#E6150S |
| Oligo(dT)18 Primer | Thermo Fisher Scientific | Cat#SO132 |
| RNase H and 10 × RNase H Reaction Buffer | New England Biolabs | Cat#M0297L |
| RNaseOUT™ Recombinant Ribonuclease Inhibitor | Thermo Fisher Scientific | Cat#10777019 |
| T4 Polynucleotide Kinase and T4 Polynucleotide Kinase Reaction Buffer | New England Biolabs | Cat# M0201L |
| T4 RNA Ligase 2, truncated KQ and T4 RNA Ligase Reaction Buffer, PEG 8000 | New England Biolabs | Cat#M0373L |
| 5′ Deadenylase | New England Biolabs | Cat#M0331S |
| RecJf | New England Biolabs | Cat#M0264L |
| Dynabeads™ MyOne™ Streptavidin C1 | Thermo Fisher Scientific | Cat#65002 |
| Reverse Transcriptase, Recombinant HIV | Worthington Biochemical | Cat#LS05006 |
| SuperScript III first strand synthesis system | Thermo Fisher Scientific | Cat#18080051 |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | Cat#N0447L |
| T4 RNA Ligase 1 (ssRNA Ligase), High Concentration | New England Biolabs | Cat#M0437M |
| Adenosine-5′-Triphosphate (ATP) | New England Biolabs | Cat#M0437M |
| HIV-RT enzyme | Worthington Biochemical Corporation | Cat# LS05003 |
| DNA Clean & Concentrator-5 | Zymo Research | Cat# D4013 |
| NEBNext® Ultra™ II Q5® Master Mix | New England Biolabs | Cat#M0544L |
| NEBNext® Multiplex Oligos for Illumina (Index Primers Set 1–4) | New England Biolabs | Cat#E7500L, E7710L, E7730L respectively |
| SYBR™ Green I Nucleic Acid Gel Stain | Thermo Fisher Scientific | Cat#S7563 |
| Qubit™ dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat#Q32854 |
| AMPure XP beads (SPRI beads) | Beckman Coulter | Cat#A63880 |
| Oligonucleotides | ||
| Mjdim1 QC probe: (rCrUrCrUrCrGrArCrGrUrGrG/iN6Me-rA/rCrUrGrGrCrArUrUrGrCrGrCrUrCrUrC/3Bio/) | (Hu et al., 2022) | Home-synthesized |
| 3′ Adaptor (/5rApp/AGATCGGAAGAGCGTCGTG/3Bio/) | (Hu et al., 2022) | IDT |
| RT Primer (ACACGACGCTCTTCCGATCT) | (Hu et al., 2022) | IDT |
| cDNA Adaptor (/5Phos/NNNNNNAGATCGGAAGAGCACAC GTCTG/3SpC3/) |
(Hu et al., 2022) | IDT |
| Spike-in Probe 1: rUrArUrCrUrGrUrCr UrCrGrArCrGrUrNrNrArNrNrGrGrCr CrUrUrUrGrCrArArCrUrArGrArArUrUr ArCrArCrCrArUrArArUrUrGrCrU |
(Hu et al., 2022) | IDT |
| Spike-in Probe 2: rUrArUrCrUrGrUr CrUrCrGrArCrGrUrNrNrArNrNrGr GrCrArUrUrCrArArGrCrCrUrArGrAr ArUrUrArCrArCrCrArUrArArUrUrGrCrU |
(Hu et al., 2022) | IDT |
| Spike-in Probe 3: rUrArUrCrUr GrUrCrUrCrGrArCrGrUrNrNrArNr NrGrGrCrGrArGrGrUrGrArUrCrUr ArGrArArUrUrArCrArCrCrArUrAr ArUrUrGrCrU |
(Hu et al., 2022) | IDT |
| Spike-in Probe 4: rUrArUrCrUrGr UrCrUrCrGrArCrGrUrNrNrArNr NrGrGrCrUrUrCrArArCrArArCr UrArGrArArUrUrArCrArCrCrArUr ArArUrUrGrCrU |
(Hu et al., 2022) | IDT |
| Spike-in Probe 5: rUrArUrCrUrGr UrCrUrCrGrArCrGrUrNrN/iN6Me-rA/rNrNrGrGrCrArUrUrCrArArGrCr CrUrArGrArArUrUrArCrArCrCr ArUrArArUrUrGrCrU |
(Hu et al., 2022) | IDT |
| Spike-in Probe 6: rUrArUrCrUr GrUrCrUrCrGrArCrGrUrNrN/iN6Me-rA/rNrNrGrGrCrGrArGrGrUrGrArUrCr UrArGrArArUrUrArCrArCrCrArUrAr ArUrUrGrCrU |
(Hu et al., 2022) | IDT |
| Spike-in Probe 7: rUrArUrCrUrGr UrCrUrCrGrArCrGrUrNrN/iN6Me-rA/rNrNrGrGrCrUrUrCrArArCrArArCr UrArGrArArUrUrArCrArCrCrArUr ArArUrUrGrCrU |
(Hu et al., 2022) | IDT |
| Spike-in Probe 8: rUrArUrCrUrGrUr CrUrCrGrArCrGrUrNrN/iN6Me-rA/rNrNrGrGrCrGrArUrGrGrUrUrUrCr UrArGrArArUrUrArCrArCrCrArUr ArArUrUrGrCrU) |
(Hu et al., 2022) | IDT |
| Biological samples | ||
| Recombinant ULP1 bacteria | Gift from Professor Yanhui Xu’s lab | NA |
| T7 Express Competent Escherichia coli | NEB | Cat#C2566H |
| Recombinant MjDim1 bacteria | Prepared by transforming the Mjdim1 plasmid into bacteria competent cell | NA |
| The Mjdim1 plasmid | Synthesized by Thermo Fisher Scientific | NA |
| pET-His-SUMO vector | Gift from Professor Yanhui Xu’s lab | NA |
| Deposited data | ||
| Method validation of m6A-SAC-seq and application during Hematopoiesis | (Hu et al., 2022) | GSE162357 |
| Software and algorithms | ||
| Cutadapt v1.15 | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/ |
| FASTX-Toolkit | Hannon Lab | http://hannonlab.cshl.edu/fastx_toolkit/ |
| HISAT2 v2.1.0 | (Kim et al., 2015) | http://daehwankimlab.github.io/hisat2/ |
| Samtools v1.7 | (Li et al., 2009) | http://www.htslib.org/ |
| STAR v2.5.3a | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| VarScan v2.3 | (Koboldt et al., 2012) | http://varscan.sourceforge.net/ |
| R 3.5.1 | The R Project | https://www.r-project.org/ |
| MACS2 v2.1.1 | (Zhang et al., 2008) | https://github.com/macs3-project/MACS |
| Subread v1.6.4 | (Liao et al., 2013) | http://subread.sourceforge.net/ |
| Cluster 3.0 (v1.59) | (de Hoon et al., 2004) | http://bonsai.hgc.jp/∼mdehoon/software/cluster/software.htm |
| Java TreeView (v1.1.6r4) | (Saldanha, 2004) | http://jtreeview.sourceforge.net/ |
| Mfuzz | (Kumar and Futschik, 2007) | https://www.bioconductor.org/packages/release/bioc/html/Mfuzz.html |
| Metascape | (Zhou et al., 2019) | https://metascape.org/ |
| g:Profiler | (Raudvere et al., 2019) | https://biit.cs.ut.ee/gprofiler/gost |
| edgeR | (Robinson et al., 2010) | http://bioconductor.org/packages/release/bioc/html/edgeR.html |
| TRRUST v2 | (Han et al., 2018) | https://www.grnpedia.org/trrust/ |
| rMATS v4.0.2 | (Shen et al., 2014) | http://rnaseq-mats.sourceforge.net/index.html |
| Trimmomatic v0.39 | (Bolger et al., 2014) | https://github.com/usadellab/Trimmomatic |
| Other | ||
| 50 mL round bottom flask | Synthware | Cat#F300050V |
| 250 mL Erlenmeyer flask | Synthware | Cat#F663825 |
| 100 mL separation funnel | Synthware | Cat#F479100A |
| Fisherbrand™ Octagon Spinbar™ Magnetic Stirring Bars | Fisher Scientific | Cat#14-513-51 |
| IKA 3622001 RET basic MAG Stirring Hot Plate | IKA | Cat#3622001 |
| Rotavapor® R-215 | BUCHI | Cat#R-215 |
| Millex-GS Syringe Filter Unit, 0.22 μm | Millipore | Cat#SLGSV255F |
| VirTis Sentry™ 12SL Freeze Dryer | VirTis | Cat#12SL |
| Waters Alliance HPLC System | Waters | Cat# Alliance HPLC |
| 5 μm C18 HPLC Column 250 × 4.6 mm | Higgins Analytical, S/N 289460 | Cat# CS-2546-C185 |
| NanoDrop™ 8000 Spectrophotometer | Thermo Fisher Scientific | Cat# ND8000 |
| EXPEDITE DNA Synthesizer 8909 | Applied Biosystems | Cat#EXPEDITE 8909 |
| rapifleX MALDI-TOF | Brucker | Cat#rapifleX |
| Avestin EmulsiFlex C3 | AVESTIN | Cat#EmulsiFlex-C3 |
| Ni Sepharose® High Performance | Cytiva | Cat#GE17-5268-01 |
| ÄKTA pure protein purification system | Cytiva | Cat#GE29-0182-24 |
| SOURCE™ 15Q 4.6/100 PE | Cytiva | Cat#GE17-5181-01 |
| SOURCE™ 15S 4.6/100 PE | Cytiva | Cat#GE17-5182-01 |
| Vivaspin® 6, 10 kDa MWCO Polyethersulfone | Cytiva | Cat#GE28-9322-96 |
| Millex Syringe Filter, Durapore® (PVDF), Non-sterile | Millipore | Cat#SLGVR04NL |
| SCIEX Triple Quad™ 6500+ LC-MS/MS Systems | SCIEX | Cat#Triple Quad 6500+ |
| Agilent Eclipse XDB-C18 reversed phase HPLC column 4.6 mm ID × 250 mm (5 μm) 80 Ǻ | Agilent | Cat#990967-902 |
| 11-mm cap LC vials | Agilent | Cat#5182-0548 |
| Bioruptor® Plus sonication device | Diagenode | Cat#B01020001 |
| 1.5 mL Low Adhesion Microcentrifuge Tubes | USA Scientific | Cat#1415-2600 |
| Tubes and Domed Caps, strips of 8 | Thermo Fisher Scientific | Cat#AB0266 |
| Reach Olympus Premium Barrier Tips | Genesee Scientific | 10 μL: Cat#23-401; 200 μL: Cat#23-412; 1000 μL: Cat#23-430 |
| ART™ Barrier Speciality Pipette tips | Thermo Fisher Scientific | Cat#2149 |
| Pipetman L single channel pipette | Gilson | P2L, P10L, P20L; P100L, P200L, P1000L: Cat#FA10001M-10006M, respectively. |
| Fisherbrand™ Elite™ Multichannel Pipettes | Fisher Scientific | 1–10 μL: Cat#FBE1200010 30–300 μL: Cat#FBE1200030 |
| 12 -Tube Magnetic Separation Rack | New England Biolabs | Cat#S1509S |
| DiaMag 0.2 mL - magnetic rack |
Diagenode | Cat#B04000001 |
| Qubit™ 2.0 Fluorometer | Thermo Fisher Scientific | Cat#Q33216 |
| Qubit™ Assay Tubes | Thermo Fisher Scientific | Cat#Q32856 |
| QuantStudio™ 6 Pro Real-Time PCR System, 96-well, 0.2 mL, desktop | Thermo Fisher Scientific | Cat#A43180 |
| MicroAmp™ Optical 96-Well Reaction Plate | Thermo Fisher Scientific | Cat#4316813 |
| 2100 Bioanalyzer Instrument | Agilent | Cat#G2939BA |
| NovaSeq 6000 System | Illumina | Cat#NovaSeq 6000 |
Materials and equipment
| Home-made reagent and equipment | ||
|---|---|---|
| Home-made reagent | Final concentration | Amount |
| TFA | 0.1% (v/v) | 10 mL |
| N6-phenyl-adenosine phosphoramidite | NA | 2 mg |
| Imidazole, pH 8.0 | 5 M | 500 mL |
| ULP1 lysis buffer | 25 mM Tris, pH 8.0, 150 mM NaCl | 1 L |
| ULP1 washing buffer | 25 mM Tris, pH 8.0, 150 mM NaCl,20 mM Imidazole | 1 L |
| ULP1 elution buffer | 25 mM Tris, pH 8.0, 150 mM NaCl,250 mM Imidazole | 1 L |
| MjDim1 lysis buffer | 1 × PBS, pH 7.4, 150 mM NaCl | 1 L |
| MjDim1 washing buffer | 1 × PBS, pH 7.4, 150 mM NaCl, 20 mM Imidazole | 1 L |
| MjDim1 elution buffer | 20 mM Tris, pH 8.0, 50 mM NaCl | 1 L |
| Buffer A for Q-column and S-column | 20 mM Bis-tris, pH 6.0 | 1 L |
| Buffer B for Q-column and S-column | 20 mM Bis-tris, pH 6.0, 1 M NaCl | 1 L |
| HEPES, PH 8.0 | 1 M | 1 L |
| NH4Cl | 1 M | 1 L |
| MgCl2 | 1 M | 1 L |
| KI | 1 M | 10 mL |
| I2 dissolved in KI | 125 mM I2 (in 200 mM KI solution) | 1 mL |
| Na2S2O3 | 200 mM | 1 mL |
| PBST | 0.1% | 50 mL |
| 1 × Binding/Wash Buffer | 5 mM Tris HCl, pH 7.5, 0.5 mM EDTA and 1 M NaCl | 50 mL |
| 2 × Binding/Wash Buffer | 10 mM Tris HCl, pH 7.5, 1 mM EDTA and 2 M NaCl | 50 mL |
| 10 × Mjdim1 reaction buffer | 400 mM HEPES, pH 8.0, 400 mM NH4Cl and 40 mM MgCl2 | 10 mL |
| MjDim1 activity test (quality control) system | 2.7 mM neutralized Allyl SAM, 2 U/μL SUPERase In RNase Inhibitor, 1 × Mjdim1 Reaction Buffer, 0.4 mM Purified Mjdim1 | 20 μL |
Step-by-step method details
RNA ribodepletion and sample concentration
In this section, we remove the ribosomal RNA in the total RNA sample.
-
1.
For cell line and fresh-frozen tissue samples, purify total RNA with Trizol or spin-column based extraction kits. Use tissue homogenizer when necessary.
-
2.
To detect mRNA m6A modification, extract mRNA with Dynabeads mRNA DIRECT Kit. To detect whole-transcriptome wide m6A modification, deplete ribosomal RNA with RiboMinus Eukaryote System v2. Then purify the longer (>200 nt) RNA using RNA Clean & Concentrator-5. The concentration of RNA was measured by Qubit™ RNA HS Assay Kit.
Library preparation
In this section, we convert m6A into a6m6A, perform the cyclization, reverse transcription, and construct the library.
-
3.
Remove poly A and ligate adapters.
The library construction strategy was modified from m1A-MAP approach (Li et al., 2017).-
a.Anneal 30–100 ng poly A+ RNA or ribo- RNA (300 ng to 1 μg total RNA) with oligo-dT, digest the hybrid with RNase H (NEB), followed by DNase I (NEB) to remove oligo-dT and purified by RNA Clean & Concentrator (RCC) Kits (Zymo Research).
-
b.Purify the RNA and fragment the RNA by sonication using Bioruptor® Plus sonication device (Diagenode) or NEBNext® Magnesium RNA Fragmentation Module, and the program is 30 s on/off, 30 cycles to ∼ 150 nt. End-repair the RNA with PNK enzyme (NEB) at 37°C for 30 min to expose the 3′ hydroxyl group.
-
c.Add 0.6% calibration spike-in mix in the reaction. Ligate the RNA fragment with 3′ adapter (key resources table) using T4 RNA ligase2, truncated KQ (NEB) at 25°C for 2 h.
-
d.Digest the excessive RNA adaptor by adding 1 μL of 5′ Deadenylase (NEB) into the ligation mix followed by incubation at 30°C for 1 h. Then add 1 μL of RecJf (NEB), incubating at 37°C for another 1 h. Add 1 μL of RT primer (50 μM), anneal at 75°C for 5 min, 37°C for 15 min, and 25°C for 15 min.
 Pause point: The ligated RNA could be stored at −80°C for at least one month.CRITICAL: Poly A tail acts as competitor of m6A sites for MjDim1 Methyltransferase and Allyl-SAM cofactor. We add poly A elimination step with RNase H.
Pause point: The ligated RNA could be stored at −80°C for at least one month.CRITICAL: Poly A tail acts as competitor of m6A sites for MjDim1 Methyltransferase and Allyl-SAM cofactor. We add poly A elimination step with RNase H.
-
a.
-
4.Label m6A sites and perform reverse transcription. Troubleshooting 3.
-
a.Add 15 μL of dynabeads C1 (Thermo Fisher Scientific) in the reaction to purify the 3′ adapter-ligated RNA. Wash the beads, resuspend the beads in 6 μL of H2O, and denature the RNA at 70°C for 30 s and cooled in ice to eliminate secondary structure.
-
b.Perform the m6A enzymatic labeling on beads. Add 2 μL of 10 × MjDim1 reaction buffer (400 mM HEPES, pH 8.0, 400 mM NH4Cl, 40 mM MgCl2,), 2 μL of SUPERase In RNase Inhibitor (Thermo Fisher Scientific), 6 μL of Allylic SAM, and 4 μL of MjDim1 enzyme (1.6 mM) in the reaction and incubate at 50°C for 1 h.
-
c.Remove the supernatant, then add 4 μL of H2O, 1 μL of 10 × MjDim1 reaction buffer, 1 μL of RNase inhibitor, 2 μL of allylic SAM, and 2 μL of MjDim1 enzyme in the reaction and incubate at 50°C for 20 min.
-
d.Repeat step c for 6 times to thoroughly label the most m6A sites.
-
e.Wash the beads and resuspend the beads in 25 μL of H2O. Add 1 μL of 125 mM I2 in the reaction and mix thoroughly. Keep the reaction in dark at 25°C for 1 h, then add 1 μL of 40 mM Na2S2SO3 to quench I2.CRITICAL: I2 should be kept in darkness. The dynabeads C1 would reduce some of the I2, do not reduce the concentration of I2.
-
f.Wash the beads and resuspend the beads in 9 μL of H2O. Add 10 × RT buffer (SuperScript™ III First-Strand Synthesis SuperMix from Thermo Fisher Scientific) 2 μL, 10 mM dNTP 2 μL, 25 mM MgCl2 2 μL, 0.1 M DTT 1.25 μL, RNaseOUT 2 μL, and HIV-RT enzyme (Worthington Biochemical Corporation) 2 μL in the tube to perform reverse transcription (RT) at 37°C for 3 h. (For Input RT 1 h with 1 μL of enzyme is sufficient). Then wash the beads and resuspend the beads with 8 μL of H2O.CRITICAL: Do not replace HIV-RT enzyme (Worthington Biochemical Corporation) with other reverse transcriptase, as they might stop at the N1, N6-ethanoadenine and N1, N6-propanoadenine (the derivates of a6m6A after treatment with I2) sites.
-
a.
-
5.Ligate cDNA 3′ adapter, construct library.
-
a.Add 1 μL of 10 × RNase H buffer, 1 μL of RNase H into the resuspended RT product, put the reaction into thermocycler (Bio-Rad) at 37°C for 30 min. Wash the beads and resuspend the beads in 50 μL of H2O. Boil the beads at 95°C for 10 min to elute the cDNA.
-
b.Purify the cDNA by DNA Clean & Concentrator-5 (Zymo Research) to remove short adapters and elute the RNA with 10 μL H2O.Pause point: The purified cDNA could be stored at −20°C for at least one month.
-
c.Add 2 μL of 10 × T4 RNA ligase buffer, 2 μL of 10 mM ATP, 10 μL of 50% PEG8000, 1 μL of cDNA 3′adapter (50 μM) (key resources table) and 1 μL of T4 RNA ligase 1 was added into the eluted cDNA and the ligation was performed at 25°C for 12 h. The reaction was purified by DNA Clean & Concentrator-5 (Zymo Research) and eluted with 21 μL of H2O.Pause point: The ligated cDNA could be stored at −20°C for at least one month.
-
d.Use 1 μL of supernatant for qPCR test and the remaining 15 μL for library construction. Use NEBNext® Ultra™ II Q5® Master Mix and NEBNext adaptors for the library amplification. Set up the qPCR reaction and parameters as follows:
Reagent Amount 10 μM Universal PCR Primer (NEB) 1.25 μL 10 μM PCR Index Primer (NEB, pre-indexed) 12.5 μL Purified supernatant 1 μL 25 × SYBR Green I 1 μL 2 × NEB Next Ultra II Q5 Master Mix 12.5 μL Nuclease free H2O 8 μL Total 25 μL Note: Set the PCR program as follows:Steps Temperature Time Cycles Initial Denaturation 98°C 30 s 1 Denaturation 98°C 10 s 25–35 cycles Annealing 65°C 15 s Extension 65°C 60 s Final extension 65°C 5 min 1 Hold 4°C Forever Calculate the ΔCt value of amplification and determine the optimal cycle number for library construction (Choose the cycle that is in the middle of the s-shape curve and before the exponential amplification curve reaching the plateau). -
e.Construct libraries. Set up the PCR reaction and parameters as follows:
Reagent Amount 10 μM Universal PCR Primer (NEB) 5 μL 10 μM PCR Index Primer (NEB, pre-indexed) 5 μL Purified supernatant 15 μL 2 × NEB Next Ultra II Q5 Master Mix 25 μL Total 50 μL Note: Set the PCR program as follows:Steps Temperature Time Cycles Initial Denaturation 98°C 30 s 1 Denaturation 98°C 10 s 25–35 cycles Annealing 65°C 15 s Extension 65°C 60 s Final extension 65°C 5 min 1 Hold 4°C Forever -
f.Purify the amplified libraries with 0.8 × Ampure beads.
-
i.Equilibrate the AMPure XP beads to 25°C for 30 min. Add 50 μL nuclease free H2O into the PCR tube to make a final volume of 100 μL. Add 80 μL (0.8 ×) AMPure XP beads to each PCR mixture. Mix well by pipetting up and down 10 times with the pipette set at 90 μL. Incubate at 25°C for 5 min.
-
ii.Place the beads on a magnetic rack for 5 min. Decant the beads.
-
iii.Add 200 μL freshly prepared 80% EtOH to the beads without mixing them. Incubate at 25°C for at least 30 s. Decant the beads. Repeat the wash for another time.CRITICAL: Use freshly prepared 80% EtOH. Do not disturb the beads.
-
iv.Aspirate the remaining EtOH and air dry for 2–5 min.CRITICAL: Do not over-dry the beads. If the beads crack, the recovery rate would be much lower.
-
v.Add 23 μL of RNase-free H2O. Mix well by pipetting up and down 10 times and incubate at 25°C for 5 min. Place the beads on a magnetic rack for 5 min.
-
vi.Transfer 21–22 μL of the eluate (there might be 1–2 μL dead volume) to a new 1.5 mL tube. Use 1 μL of supernatant for concentration measurement.
-
i.
-
g.Measure the concentration of the libraries by Qubit Fluorometer and Qubit dsDNA HS kit.
-
h.Check the quality of the purified libraries by 2100 Bioanalyzer Instrument.Pause point: The purified libraries could be stored at −20°C for at least one month.
-
i.Send the libraries for NGS deep sequencing. Sequence the libraries on NovaSeq 6000 System Ten with paired-end 2 × 150 bp read length.
-
a.
Bioinformatic analysis of the sequencing data
In this section, we analyze the data and identify bona fide RNA m6A sites.
-
6.Preparing and quality control of raw sequence data.
-
a.Firstly, we suggest the available memory of computer is more than 128 GB, and number of threads is more than 16. Download our perl, R, and Bash Shell scripts used below from GitHub https://github.com/CTLife/m6A-SAC-seq. Download the Human hg38 reference genome and transcriptome files from the UCSC Genome Browser (https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/) and the ncbiRefSeq GTF file from https://genome.ucsc.edu/cgi-bin/hgTables. Use these files to build index for the aligner STAR:> STAR --runMode genomeGenerate --genomeFastaFils hg38.genome.fa --genomeDir hg38 --sjdbGTFfile hg38.ncbiRefSeq.gtf
-
b.The scripts recognize paired-end FASTQ files by their suffixes automatically. Therefore, the two FASTQ files of each paired-end sequencing sample must be tagged by “R1″ and “R2″ respectively. Put all the raw FASTQ files into a same folder “1-rawFASTQ” and rename them, let their suffixes be “. R1.fastq.gz” or “. R2.fastq.gz”. For more details, please see “perl m6A-SAC-seq_1.pl -help”.
-
c.Before preprocessing, we should check adapter content, Troubleshooting 4 duplication level, Troubleshooting 5 and genome contamination Troubleshooting 6 to decide whether it is necessary to generate more reads or redo the experiment. Then check the quality of the raw FASTQ files by FASTQC, fastp, and FASTQ_screen, all of them were integrated as a script:> perl m6A-SAC-seq_1.pl -in 1-rawFASTQCRITICAL: ‘--sjdbGTFfile hg38. ncbiRefSeq.gtf’ should be indicated for better mapping reads with lots of mutation.
-
a.
-
7.Linear regression for m6A fraction and mutation rate.
-
a.To correlate observed mutation rates and m6A fractions of the spike-in sequences in each motif, extract the spike-ins from raw FASTQ files:> perl spikeins.pl -in 1-rawFASTQ -out Spikeins> perl spikeins_RC.pl -in 1-rawFASTQ -out Spikeins_RC
-
b.Then, we can pool the results from same group together or detect mutation rates of spike-ins for each sample. Mutation rates and linear correlations of adenines (A) for each motif can be generated by:> Rscript linear_correlations.shLinear regression models were used: y=ax + b. Where y is the observed mutation rate and x is the m6A fraction. In the file “spikeins.fit.txt” from results, the first column is one of 256 5-mer motifs, the second column is the value of a, and the third column is the value of b.CRITICAL: Pool the spike-ins from same group together to increase the reliability of linear regression.
-
a.
-
8.Preprocessing and mapping.
-
a.Remove adaptors and bases with low quality by using Trimmomatic. This step with quality statistics was implemented by one script:> perl m6A-SAC-seq_2.pl -in 1-rawFASTQ -out 2-removedAdapters
-
b.Then adapter-free reads with barcodes will be removed PCR duplicates by using the clumpify.sh in BBMap:> clumpify.sh in=end1 in2=end2 out=output_end1 out2=output_end2 -Xmx60g reorder=f dedupes=t subs=0CRITICAL:‘-Xmx60g’ indicates that this step is memory consumption.CRITICAL: ‘dedupes=t subs=0’ parameters will keep only one of the duplicates with same barcode and sequence.This step with quality statistics was implemented by one script:> perl m6A-SAC-seq_3.pl -in 2-removedAdapters -out 3-removedDups
-
c.For using the variant detection tool VarScan, we need to separate stranded reads into plus-strand and minus-strand reads in further analysis. Therefore, we convert paired-end reads into single-end reads by reformat.sh in BBMap before mapping. If we use other tools to call mutation, this step is not required. One script with quality statistics was implemented (Optional):> perl m6A-SAC-seq_4.pl -in 3-removedDups -out 4-finalFASTQ
-
d.Remove barcodes and map reads to the reference genome using STAR:> STAR --runMode alignReads --clip3pNbases 6 --alignSJDBoverhangMin 1 --outSAMmultNmax 1 --outMultimapperOrder Random --outFilterMismatchNoverReadLmax 0.1 --outFilterMismatchNmax 999 --outFilterMultimapNmax 20 –outFileNamePrefix ouput --genomeDir STAR_index --readFilesIn input_end1 input_end2This step with quality statistics, and detecting gene expression with Salmon or Kallisto were implemented by one script:> perl m6A-SAC-seq_5.pl -genome hg38 -mis 0.1 -in 4-finalFASTQ-out 5-rawBAMCRITICAL: ‘--outFilterMismatchNoverReadLmax 0.1’ allows more mismatches of the reads with mutation.
-
e.Remove reads with low MAPQ, or on unplaced and unlocalized contigs (Optional):> perl m6A-SAC-seq_6.pl -genome hg38 -in 5-rawBAM -out 6-finalBAM
-
a.
-
9.Mutation calling and m6A sites identification.
-
a.Split each BAM file into two files, plus strand and minus strand (Optional):> perl split_strand.pl -in 6-rawBAM -out callVariants
-
b.Call mutations for each BAM file by SAMtools and VarScan:> samtools mpileup --fasta-ref hg38.genome. fa --output name.pileup name.bam> java -jar VarScan.jar somatic SAC-seq.pileup backfround.pileup outDir
-
c.Identify m6A sites from the outputs from VarScan. Most m6A sites were found in conserved motif DRACH (D = G/A/U, R = G/A, H = A/U/C), only a few sites located at non-DRACH motifs. So, we classified all the m6A sites into two categories: DRACH and nonDRACH. These several steps can be done by:> perl identify.pl -in inputDir -backgound 0.05 -p 0.05 -diff 0.1 -cov 10CRITICAL: ‘-backgound 0.05 -p 0.05 -diff 0.1 -cov 10’ indicates the thresholds of maximum background mutation rate, p-value, minimum mutation rate (background was removed), and coverage. These values should be changed based on your sequencing depth and species of samples.
-
a.
-
10.
Quality control of the identified m6A sites.
Some figures can be used to assess the quality, reliability, and features of the identified m6A sites:
> perl figures.pl -in inputDir -out outDir
Expected outcomes
We select the Methanocaldococcus jannaschii (a thermostable Archaea) homolog MjDim1, which shows highly processive kinetics of converting m6A into m62A (O'Farrell et al., 2006), and employed a chemically modified allyl-SAM as the co-factor (Shu et al., 2017) (Figures 1 and S1). Subsequent I2 treatment converts a6m6A and a6A into homologs of N1, N6-ethanoadenine and N1, N6-propanoadenine (Figure S2), respectively (Shu et al., 2017) (Figure 1).
Figure 1.

m6A-SAC-seq strategy
m6A-SAC-seq utilizes MjDim1 and allylic-SAM as a co-factor to convert m6A to allyl6m6A, followed by cyclization upon I2 treatment.
We can obtain 10 mg MjDim1 per 1 L of E. coli with the > 95% purity (Figure 2A). After 6 months in −80°C freezer, it kept the similar activity as shown below (Figures 2B and 2C). We have varied the concentration of DjDim1 from 2 nmol to 8 nmol and observed consistent m6A transfer using MALDI (Figure 2D).
Figure 2.

Validation of the consistency of the Mjdim1 enzyme activity
(A) MjDim1 was purified using Source 15S resin (GE Healthcare). 1: Protein marker; 2: enzyme on resin; 3: eluted enzyme; 4–12 purified fractions.
(B) UHPLC-QQQ-MS/MS quantitation of the m6A probe after allyl labeling using the fresh Mjdim1 enzyme (4 nmol) in 20 μL reaction solution. The retention time of am6A is around 5.5 min.
(C) Activity of MjDim1 remained the same after storage in −80°C freezer for 6–12 months. m6A/G ratio of the enzyme-treated reaction with MjDim1 that was stored in −80°C freezer for different time periods was normalized to fresh MjDim1 enzyme group. Error bars, mean ± s.e.m. n=3.
(D) 2, 3, 4, 5 nmol Mjdim1 showed consistent m6A transfer efficiency in 20 μL reaction solution. m6A/G ratio of the enzyme-treated reaction was normalized to 4 nmol MjDim1 enzyme group. Error bars, mean ± s.e.m. n=3.
After treatment, HIV-1 RT generate mutations at m6A sites but not unmodified sites nearby (Figure 3A). We could identify m6A sites by specific A -> U/C > G mutation locations and the stoichiometry could be quantified by addition of spike-in calibration probes (Figures 3B and S3). A background control in which RNA is treated with the m6A demethylase FTO before MjDim1 labeling helps to more reliably identify genuine m6A sites (Figure 3C). The average size of the libraries is about 300 bp, verified by bioanalyzer (Figure S4).
Figure 3.

Calibration of the m6A stoichiometries and identified m6A sites using m6A-SAC-seq
(A) Examples of mutation pattern of an m6A-modified 41-bp spike-in probe mix with 0%, 25%, 50%, 100% of m6A, respectively. After m6A -SAC-seq treatment, mutations are generated at m6A sites but not unmodified sites nearby.
(B) Calibration curve for each GGACU motif is generated by linear regression. P-values and goodness of fit (R2) of linear regression were also shown.
(C) The FTO (m6A demethylase) treatment could serve as a control to identify bona fide m6A sites more reliably. FTO- and FTO+ signal tracks for representative RNA (MALAT1) were displayed for visualization of the data.
We could identify more than 10,000 high-confidence m6A sites with stoichiometrical information in Hela cell lines with two biological replicates (Figure 4A). Most m6A sites are enriched around the stop codon and located in the 3′ UTR and CDS regions (Figure 4B), with identified m6A sites in the frequent m6A methylated GGACU and AGACU motifs displaying the highest frequency (Figure 4C). m6A-SAC-seq can determine the modification stoichiometry of m6A in almost all sequence contexts (Figure 4C). m6A stoichiometry is reproducible within technical replicates and the overlapping ratio between technical replicates could be 60% (Figure 4D). By comparing m6A-SAC-seq with MeRIP-seq, the overlapping ratio between m6A-SAC-seq sites and MeRIP-seq peaks could be ∼60% (Figure 4E).
Figure 4.

Characteristics of m6A sites in cell lines identified using m6A-SAC-seq
(A) Bar plots and boxplots showed the number and m6A fraction distribution of m6A sites in HeLa cell line, respectively. RNA regions were classified into 3′ UTR, CDS, intron, 5′ UTR, intergenic and promoter. In box plots, lower and upper hinges represent first and third quartiles, the center line represents the median, and whiskers represent ± 1.5 × the interquartile range.
(B) Average m6A fraction across all genes were showed by mRNA metagene profiles of m6A sites which were identified by m6A-SAC-seq compared with MeRIP-SAC-seq in HeLa cells.
(C) Number of m6A sites (upper), mutation frequencies (medium), and m6A fractions (bottom) in different m6A consensus motifs in transcripts from HeLa cells. The method could recover m6A sites in almost all DRACH canonical motifs with the allyl transfer reaction showing a preference for the GA sequence. In box plots, lower and upper hinges represent first and third quartiles, the center line represents the median, and whiskers represent ± 1.5 × the interquartile range.
(D) The overlapping ratio of m6A sites between technical replicates identified by m6A-SAC-seq using the same RNA samples from HeLa cells.
(E) Upset plot displaying the overlap between m6A-SAC-seq sites and MeRIP-seq peaks identified in HeLa cells.
Limitations
m6A-SAC-seq shows a significant motif preference of GAC over AAC. Previous studies using chromatography (Schibler et al., 1977; Wei and Moss, 1977) as well as antibody-dependent approaches (e.g., miCLIP (Linder et al., 2015) (Kortel et al., 2021)) have reported that ∼70–75% of m6A sites occur in the GAC motif. It suggests that even though m6A-SAC-seq has a disadvantage in detecting AAC sites, it could still recover ∼ 80% of overall m6A sites. m6A-SAC-seq can reveal highly modified m6A sites in the AAC motif but miss out those with low stoichiometry. To break through the limitation, we intend to engineer the current version of MjDim1 methyltransferase, making it exhibits less sequence bias.
Troubleshooting
Problem 1
The yield of Allyl-SAM product is low and it shows multiples peaks in HPLC.
Potential solution
Reason: The crude Allyl-SAM has degraded. Refer to “synthesis of Allyl-SAM”.
Solution: Lyophilize the Allyl-SAM immediately after synthesis and determine the correct product peak via LC-MS.
Problem 2
The yield of MjDim1 is low. Refer to “preparation of methyltransferase MjDim1”.
Potential solution
Reason: The flowthrough after the Q-column purification is discarded, inoculation and culture of bacteria is non-standard, or the pH of purification buffer is wrong.
Solution: Save the flowthrough after Q-column purification. Take care to use a sanitizer to kill the recombinant bacteria before throwing them out to avoid phage infection. Make sure the OD600 and temperature is optimal before induction. The pH of the MjDim1 buffer should exactly follow the correct protocol.
Problem 3
The conversion of m6A is low.
Potential solution
Reason: The enzymatic activity of MjDim1 methyltransferase is impaired during purification or the misincorporation of HIV-RT enzyme during reverse transcription is not ideal.
Solution: Although MjdIM1 is thermostable, it should stay in ice whenever possible to make the activity stable and sustainable. During recombinant bacteria crushing step, keep the Emulsiflex chilled using ice bag. When concentrating MjDim1 enzyme, use the appropriate centrifuge speed (1,800 × g) and shorten the centrifuge time (30 min for each round until the appropriate protein concentration is obtained). During reverse transcription, the RNA amount should be less than 100 ng, as excessive RNA and insufficient HIV-RT enzyme could reduce the misincorporation efficiency. Avoid freeze-saw cycles of MjDim1, Allyl-SAM, and HIV-RT enzyme.
Problem 4
Library contains significant peaks under 150 bp.
Potential solution
Reason: Adaptor dimer, primer dimer, or primers are not removed completely.
RecJ treatment is necessary to digest the excessive 3′ adapter.
Solution: Purify cDNA with DNA Clean & Concentrator-5 (Zymo Research) to remove excessive 3′ adapter. The library should be purified with 0.8 × Ampure beads to get rid of primer dimers.
Problem 5
The reads contain high level of PCR duplication.
Potential solution
Reason: Over-amplification during library construction.
Solution: Reduce cycle number for NGS library construct. Under the premise of ensuring sufficient library concentration for NGS sequencing, reduce the PCR amplification cycles as many as possible.
Problem 6
Too many reads (more than 20%) mapped to rRNA or tRNA.
Potential solution
Reason: Incomplete removal of rRNA or tRNA.
Solution: Exactly follow the manufacturer’s instructions to extract mRNA with Dynabeads mRNA DIRECT Kit and remove rRNA with RiboMinus Eukaryote System v2. The purification step could be repeated twice to completely remove rRNA or tRNA.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lulu Hu (luluhu@fudan.edu.cn).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
The authors thank Dr. Pieter Faber, the University of Chicago Genomics Facility for sequencing support, Dr. Qin Jin for helping with UHPLC-QQQ-MS/MS, and Dr. Qing Dai for synthesizing RNA probes. The authors also thank T. Wu, J.B. Wei, and L.S. Zhang for discussions. L.H. is supported by National Key R&D Program of China, 2021YFA1100400. C.H. is supported by National Institutes of Health (NIH) Grant RM1 HG008935 and M.C. is supported by National Institutes of Health (NIH) Grant R01 GM126553, a Sloan Foundation Research Fellowship and a Human Cell Atlas Seed Network grant from Chan Zuckerberg Initiative. C.H. is an investigator of the Howard Hughes Medical Institute.
Author contributions
L.H. and C.H. conceived the study. M.C. supervised the bioinformatic analysis. L.H. and R.G. developed and optimized the experimental part of the protocol. Y.P. and S.L. developed the computational part of the protocol. H.M. prepared all the protein and optimized the protein purification protocol. L.H. and Y.P. wrote the manuscript with inputs from all authors. L.H. and C.H. supervised the study. All authors reviewed the manuscript.
Declaration of interests
A patent application for m6A-SAC-seq has been filed by the University of Chicago. C.H. is a scientific founder and a scientific advisory board member of Accent Therapeutics, Inc., and Inferna Green, Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2022.101677.
Contributor Information
Chuan He, Email: chuanhe@uchicago.edu.
Lulu Hu, Email: luluhu@fudan.edu.cn.
Supplemental information
Data and code availability
The accession number for the data reported in this paper is GEO: GSE162357.
The codes generated during this study are publicly available on GitHub (https://doi.org/10.5281/zenodo.6961497) (https://github.com/shunliubio/m6A-SAC-seq and https://github.com/CTLife/m6A-SAC-seq).
We have uploaded the supplemental information to Mendeley Data. The Mendeley Data DOI is: 10.17632/7nn9czh4zt.1.
References
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon M.J.L., Imoto S., Nolan J., Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H., Cho J.W., Lee S., Yun A., Kim H., Bae D., Yang S., Kim C.Y., Lee M., Kim E., et al. Trrust v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018;46:D380–D386. doi: 10.1093/nar/gkx1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L., Liu S., Peng Y., Ge R., Su R., Senevirathne C., Harada B.T., Dai Q., Wei J., Zhang L., et al. m(6)A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat. Biotechnol. 2022;40:1210–1219. doi: 10.1038/s41587-022-01243-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortel N., Ruckle C., Zhou Y., Busch A., Hoch-Kraft P., Sutandy F.X.R., Haase J., Pradhan M., Musheev M., Ostareck D., et al. Deep and accurate detection of m6A RNA modifications using miCLIP2 and m6Aboost machine learning. Nucleic Acids Res. 2021;49:e92. doi: 10.1093/nar/gkab485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar L., Futschik M.E. Mfuzz: a software package for soft clustering of microarray data. Bioinformation. 2007;2:5–7. doi: 10.6026/97320630002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Xiong X., Zhang M., Wang K., Chen Y., Zhou J., Mao Y., Lv J., Yi D., Chen X.W., et al. Base-resolution mapping reveals distinct m1A methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell. 2017;68:993–1005.e9. doi: 10.1016/j.molcel.2017.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y., Smyth G.K., Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B., Grozhik A.V., Olarerin-George A.O., Meydan C., Mason C.E., Jaffrey S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods. 2015;12:767–772. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10. [Google Scholar]
- O'Farrell H.C., Pulicherla N., Desai P.M., Rife J.P. Recognition of a complex substrate by the KsgA/Dim1 family of enzymes has been conserved throughout evolution. RNA. 2006;12:725–733. doi: 10.1261/rna.2310406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha A.J. Java Treeview–extensible visualization of microarray data. Bioinformatics. 2004;20:3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- Schibler U., Kelley D.E., Perry R.P. Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J. Mol. Biol. 1977;115:695–714. doi: 10.1016/0022-2836(77)90110-3. [DOI] [PubMed] [Google Scholar]
- Shen S., Park J.W., Lu Z.X., Lin L., Henry M.D., Wu Y.N., Zhou Q., Xing Y. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. USA. 2014;111:E5593–E5601. doi: 10.1073/pnas.1419161111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu X., Dai Q., Wu T., Bothwell I.R., Yue Y., Zhang Z., Cao J., Fei Q., Luo M., He C., Liu J. N6-allyladenosine: a new small molecule for RNA labeling identified by mutation assay. J. Am. Chem. Soc. 2017;139:17213–17216. doi: 10.1021/jacs.7b06837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.M., Moss B. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry. 1977;16:1672–1676. doi: 10.1021/bi00627a023. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the data reported in this paper is GEO: GSE162357.
The codes generated during this study are publicly available on GitHub (https://doi.org/10.5281/zenodo.6961497) (https://github.com/shunliubio/m6A-SAC-seq and https://github.com/CTLife/m6A-SAC-seq).
We have uploaded the supplemental information to Mendeley Data. The Mendeley Data DOI is: 10.17632/7nn9czh4zt.1.