

ABSTRACT
After myocardial infarction, the massive death of cardiomyocytes leads to cardiac fibroblast proliferation and myofibroblast differentiation, which contributes to the extracellular matrix remodelling of the infarcted myocardium. We recently found that myofibroblasts further differentiate into matrifibrocytes, a newly identified cardiac fibroblast differentiation state. Cardiac fibroblasts of different states have distinct gene expression profiles closely related to their functions. However, the mechanism responsible for the gene expression changes during these activation and differentiation events is still not clear. In this study, the gene expression profiling and genome-wide accessible chromatin mapping of mouse cardiac fibroblasts isolated from the uninjured myocardium and the infarct at multiple time points corresponding to different differentiation states were performed by RNA sequencing (RNA-seq) and the assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq), respectively. ATAC-seq peaks were highly enriched in the promoter area and the distal area where the enhancers are located. A positive correlation was identified between the expression and promoter accessibility for many dynamically expressed genes, even though evidence showed that mechanisms independent of chromatin accessibility may also contribute to the gene expression changes in cardiac fibroblasts after MI. Moreover, motif enrichment analysis and gene regulatory network construction identified transcription factors that possibly contributed to the differential gene expression between cardiac fibroblasts of different states.
KEYWORDS: Myocardial infarction, cardiac fibroblast, chromatin accessibility, transcription factor
Introduction
In response to myocardial infarction (MI), cardiac fibroblasts proliferate and differentiate into myofibroblasts expressing elevated levels of contractile proteins and extracellular matrix (ECM) proteins which are important in maintaining the structural integrity of the infarcted myocardium [1–3] but may have a detrimental effect on the long-term cardiac function due to permanent replacement fibrosis [4]. Depletion of myofibroblasts after MI increased the early incidence of cardiac rupture [2] but also improved the long-term cardiac function [2,3], indicating the complicated role of cardiac fibroblasts during the post-MI tissue repair. Thus, a complete understanding of the post-MI cardiac fibroblast activities and underlying mechanisms is important, as it may allow the fine-tuning of cardiac fibroblast activities, which may ultimately lead to a novel treatment strategy. Using lineage-tracing, we recently found that the proliferative state and myofibroblast state of cardiac fibroblasts were rather transient and largely limited to the first week after MI [1]. Myofibroblasts then further differentiate into matrifibrocytes, a newly identified cardiac fibroblast differentiation state featured by the expression of select cartilage and bone genes and is likely an adaptation to the highly fibrotic environment. Cryoinjury-induced depletion of matrifibrocytes resulted in reduced scar stability and cardiac function, suggesting a critical role of matrifibrocytes. Using Affymetrix microarray, we also studied the gene expression profiles of cardiac fibroblasts of different differentiation states and identified state-specific gene expression in these cells that corresponded to their unique functions [1]. However, the mechanism responsible for the differential gene expression is still not known. Despite a large amount of effort that has been made to explore the mechanism responsible for cardiac fibroblast activation and myofibroblast differentiation, our understanding of these processes is still relatively limited. An increasing number of studies have reported the presence of multiple signalling pathways involved in the post-injury activation and myofibroblast differentiation of cardiac fibroblasts [5] besides the well-known TGFβ signalling [6–9], suggesting the complexity of these processes. Moreover, the molecular mechanism regulating the newly identified matrifibrocyte differentiation remains unknown. The accessibility of chromatin directly regulates gene transcription [10,11]. Thus, chromatin remodelling likely contributes to the dynamic gene expression during the sequential activation and differentiation events in cardiac fibroblasts after MI.
In this study, using a transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) protocol optimized for low cell numbers and a cardiac fibroblast lineage-tracing mouse line, we performed open chromatin profiling using cardiac fibroblasts isolated from the uninjured myocardium and the infarct at different time points after MI. The same population of cardiac fibroblasts was also subjected to RNA sequencing (RNA-seq) analysis. The integrated analysis of ATAC-seq and RNA-seq data revealed the important role of chromatin remodelling in post-MI gene expression regulation in cardiac fibroblasts. Transcription factor (TF) binding motif analysis and gene regulatory network construction also revealed motifs and TFs that are possibly responsible for the differential gene expression between cardiac fibroblasts of different differentiation states.
Materials and methods
Mice and animal procedures
Mouse lines used in this study are as follows: Tcf21-MerCreMer [12] and R26-EGFP [13]. These two mouse lines were crossed to obtain Tcf21MCM/+; R26eGFP mice. To induce the activity of the MerCreMer, mice were treated with tamoxifen (Sigma T5648) dissolved in corn oil through intraperitoneal (IP) injections at a dosage of 75 mg/kg body weight/day for 5 days. MI was induced in mice via permanent surgical ligation of the left coronary artery [14]. Briefly, mice were anaesthetized using isoflurane and a left lateral thoracotomy was performed. The left coronary artery was identified and ligated just below the left atrium. For pain management related to surgical procedures, mice were given Carprofen at 5 mg/kg before the procedure and 12 hours after the procedure. Mice used in this study include both males and females. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Louisiana State University.
Tissue processing and imaging
Mouse heart samples were fixed in 4% paraformaldehyde for 3.5 hours, rinsed in PBS for 30 min, and immersed in PBS containing 30% sucrose overnight at 4°C. Heart samples were embedded in OCT for cryosectioning. Cryosections were stained with a chicken anti-GFP primary antibody (Abcam, ab13970) and a goat anti-chicken Alexa Fluor 488 (Life Technologies, A-11039) as previously described [1], followed by imaging. For whole-mount imaging, the left ventricles of uninjured hearts and infarcts were carefully removed and trimmed before imaging. Imaging was performed using a Leica SP8 confocal microscope or a Nikon A1 confocal microscope. Images obtained from cryosections were used for cell size measurement using ImageJ. Only EGFP+ cells with Dapi signals were included in cell size measurement. In case that cell boundaries between adjacent EGFP+ cells could not be identified, the entire EGFP+ area divided by the number of EGFP+ cells in this area was used as the average EGFP+ cell size in this area. Three samples per group were included in the analysis. At least three random images were used to calculate the average EGFP+ cell size of each sample.
FACS
FACS. Heart tissue was digested as previously described with modification [15]. Briefly, the heart tissue samples were minced and digested in DMEM containing 0.75 U/ml Collagenase D (Roche 11,088,866,001), 1.0 U/ml Dispase II (Roche 10,165,859,001), and 1 mM CaCl2 at 37°C for 40 min. The slurry was then passed through a 100-μm cell strainer and then a 40-μm cell strainer. Cells were collected by centrifugation at 350 × g for 10 min. The cell pellet was then resuspended in PBS. Tcf21 lineage-traced eGFP+ cardiac fibroblasts were sorted in FACSaria II (BD Biosciences). Gates were made based on WT eGFP− control. Sorted eGFP+ cardiac fibroblasts were used for RNA-seq and ATAC-seq analysis.
RNA-seq library construction and sequencing
Total RNA was extracted from 10,000 FACS-sorted Tcf21 lineage-traced fibroblasts using the miRNeasy Micro Kit (217,084) from Qiagen. cDNA libraries were constructed using the NEBNext single cell/low input RNA library prep kit for Illumina (E6420) from New England BioLabs. The cDNA libraries were sequenced on the Illumina Hi-seq platform using 150 bp paired-end sequencing. Around 30 million read pairs were obtained for each sample. Two samples from each group were analysed.
ATAC-seq library construction and sequencing
The ATAC-seq was performed following a protocol as previously described [16]. Briefly, 10,000 FACS-sorted Tcf21 lineage-traced fibroblasts were lysed to isolate the nuclei. Isolated nuclei were then incubated with the Tn5 transposase (TDE1, Illumina) and tagmentation buffer at 37°C for 30 minutes with shaking on a thermomixer at 500 g. Tagmentated DNA was purified using MinElute Reaction Cleanup Kit (Qiagen). PCR was performed to amplify the ATAC-seq libraries using Illumina TrueSeq primers and multiplex by indexes primers. After the PCR reaction, libraries were purified with the 1.1X AMPure beads (Beckman). The concentration of the sequencing libraries was determined using the Qubit dsDNA HS Assay Kit (Life Technologies). The size of the sequencing libraries was determined using a High-Sensitivity D5000 Assay kit with a Tapestation 4200 system (Agilent). ATAC-seq libraries were sequenced on the Illumina Hi-seq platform using 150 bp paired-end sequencing. Around 50 million read pairs were obtained for each sample. Two samples of each group were analysed.
RNA-seq data processing
RNA-seq reads that passed filters were trimmed to remove low-quality reads and adapters by TrimGalore-0.6. The quality of reads after filtering was assessed by fastQC, followed by alignment to the mouse genome (MM10) by STAR (2.5.3a) with default parameters. Individual mapped reads were adjusted to provide TPM (transcripts per million) values with mouse genome as the reference. Differential gene expression analysis was done using DESeq2 [17] and OneStopRNAseq [18]. Genes expressed in at least one sample (TPM > 0.1) are considered expressed genes. Genes not expressed in any of the samples were deemed as unexpressed genes. The differential expression of a gene is considered to be significant when the absolute value of estimated shrunken Log2 fold change (LFC) in TPM is over 0.585 and the adjusted P value is less than 0.05. To classify genes by expression level, a Z-score for each gene in each sample was first obtained by standardizing the TPM values of all genes in the sample to have a mean of 0 and a variance of 1. Then, genes with a z-score below (above) zero in all sample groups were deemed to have consistently low (high) expression, with the remaining genes classified as having dynamic expression.
ATAC-seq data processing
Sequencing reads of all samples underwent adapter removal using TrimGalore-0.6, followed by quality assessment using FastQC. Reads were then aligned to the mouse reference genome MM10 using Bowtie 2.3 with the following options: very-sensitive -X 2000 – no-mixed – no-discordant. Only unique alignments within each sample were retained in subsequent analysis. Moreover, alignments resulting from PCR duplicates or located in mitochondria were excluded. The mouse genome was tiled with consecutive non-overlapping 300bp bins. The accessibility of each of the 300bp bins was assessed by the number of fragments per million mapped (FPM) that was aligned to the bin. There was a total of 8,466,536 bins with at least one aligned fragment in at least one sample. Pairwise Poisson distance between samples was calculated using package PoiClaClu in R. The Principal Component Analysis (PCA) was also done in R.
Peak calling and enrichment of genomic features in peaks
ATAC-seq peaks were called separately for each sample by MACS2 [19] with the following options: keep-dup all – nolambda – nomodel. Peaks in individual samples from each group were subsequently merged using bedtools (https://bedtools.readthedocs.io/en/latest). The annotations of genomic features, including transcription start sites (TSS), transcription end sites (TES), exons, introns, and CpG islands and repeat-elements: long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), long terminal repeats (LTRs) and simple sequence repeats (Simple) were downloaded from UCSC genome browser. Promoters were defined as 500bp up- and downstream from the TSS of each annotated gene (TSS ± 500bp). Intergenic regions were defined as genomic regions before the TSS of the first gene and after the TES of the last gene in each chromosome, and in-between the TES and TSS of two consecutive genes. Peaks that did not overlap with annotated promoters were deemed as distal peaks. To evaluate the enrichment of the above genomic features with identified ATAC-seq peaks, a set of random peaks was first generated by matching the length of ATAC-seq peaks. The enrichment was then assessed by the ratio (or the log of the ratio) between the numbers of ATAC-seq and random peaks that overlap with the corresponding genetic feature. The enrichment of transcriptional factor motifs in peaks was evaluated using HOMER (http://homer.ucsd.edu/homer/motif).
Assessment of promoter accessibility
Promoter accessibility in each sample of cardiac fibroblasts at distinct stages was assessed by the number of ATAC-seq fragments (FPM) mapped to the defined promoter region (i.e., TSS ± 500bp). To classify genes by promoter accessibility, a Z-score for each promoter in each sample was first obtained by standardizing the FPM values in the sample to have a mean of 0 and a variance of 1 across all promoters. Promoters with Z-scores below zero in all sample groups were deemed to have consistently low accessibility, while those with z-scores above zero in all sample groups were deemed to have consistently high accessibility. The remaining promoters were deemed to have dynamic accessibility.
Assessment of the correlation between promoter/distal region accessibility and gene expression
The promoter/distal region accessibility and gene expression at each stage was determined by the average over samples from the stage. Pearson correlation (cor) was calculated between promoter/distal region accessibility and corresponding gene expression across all stages. cor> 0.5 was considered to have a strong positive correlation. Raw correlation data are included in Online Supplemental Data (ATACseq-RNAseq correlation-distal.csv and ATACseq-RNAseq correlation-promoter.csv). When making heat maps, to help visualize the expression and accessibility changes of individual genes/promoters/distal regions among sample groups, the data of accessibility and expression were standardized such that each row (i.e., gene/promoter/distal region) has a mean of 0 and a standard deviation of 1 (Z-score).
Assessment of promoter CpG density
CpG density of annotated promoters (TSS ± 500bp) was assessed using the function CpGDensityByRegion in the R package BSgenome. In our analysis, when categorization of the promoter CpG density was needed, it was done as follows: low (<25), medium (≥25 and ≤75) and high (>75).
Gene ontology and pathway analysis
Gene ontology (GO) enrichment analysis was performed using OneStopRNAseq [18] and on geneontology.org.
Gene regulatory network reconstruction
For each annotated gene, we first identified TFs with a binding motif that appears in ATAC-seq peaks overlapping with the regulatory region of the gene, which is defined as 100kb up- and down-stream of its TSS. A set of trans-regulators was then created for each gene to include all the identified TFs, and subsequently refined by retaining only TFs that were co-expressed with the gene (the absolute value of cor ≥ 0.6). The network topological analysis and visualization were done in Cytoscape [20] with additional plugins: NetAnalyzer [21] and yFiles [22].
Other statistical analyses
All data in bar graphs are expressed as mean ± SEM. Data were analysed using GraphPad Prism 9 (GraphPad Software, Inc.). One-way ANOVA with multiple comparisons was used to determine the significance of differences between each post-MI group and the uninjured group. A two-tailed test was used to determine the significance of differences between means when only two groups were compared. P < 0.05 was considered significant.
Data availability
All raw sequencing data have been deposited into the public database at NCBI (GEO #GSE186079).
Results
Transcriptome profiling of cardiac fibroblasts of different differentiation state
To study the gene expression profiles of cardiac fibroblasts in different differentiation states, we employed the same cardiac fibroblast lineage-tracing mouse line (Tcf21MCM/+; R26eGFP) that we used in our previous publication (Figure 1(a)) [1]. Tcf21MCM/+; R26eGFP mice were treated with tamoxifen and subjected to surgical induction of MI (Figure 1(b)). Whole-mount confocal imaging of the uninjured myocardium and the infarct showed a significant expansion of the cardiac fibroblast population after MI (Figure 1(c), Online Figure 1). Cardiac fibroblasts were also enlarged after MI (Figure 1(c,d)), likely due to the highly abundant smooth muscle alpha-actin (SMαA) stress fibres. Moreover, cardiac fibroblasts appeared to be more organized as the infarct became more stabilized, likely due to progressed infarct remodelling (Figure 1(c)). Freshly sorted Tcf21 lineage-traced cardiac fibroblasts from the uninjured myocardium (quiescent cardiac fibroblast) and the infarct on day 3 (proliferative early myofibroblast), day 7 (mature myofibroblast), week 2 (transitioning matrifibrocyte), and week 4 (matrifibrocyte) after MI were subjected to RNA-seq. RNA-seq data were validated by principal component analysis (PCA) and Poisson distance analysis (Figure 2(a,b)). Distinct transcriptomes were identified in cardiac fibroblasts in different states. The expression of a total of ~20,000 genes was detected. Around 5,000 genes were differentially expressed between groups, out of which 3,829 showed differential expression in cardiac fibroblasts after MI as compared to cardiac fibroblasts from the uninjured myocardium including both upregulated and downregulated genes (Figure 2(c,d)). Figure 2(e) shows the expression of some representative genes which are classified into different groups based on their functions. The expression of proliferation genes peaked at day 3 after MI (Figure 2(e)), which is in line with the peak proliferation rate of cardiac fibroblasts at this time point [1]. Stress fibre genes, myocardium ECM genes, and cell migration genes were most enriched on both days 3 and 7 post-MI (Figure 2(e)), corresponding to the myofibroblast state of cardiac fibroblasts. Moreover, the expression of some cartilage genes was most enriched at week 4 post-MI (Figure 2(e)), indicating the matrifibrocyte identity of cardiac fibroblasts in more stabilized infarcts [1]. Gene ontology (GO) enrichment analysis of genes that were upregulated in cardiac fibroblasts isolated from infarcts at different time points after MI as compared to cardiac fibroblasts isolated from the uninjured myocardium showed the enrichment of multiple biological processes. Some representative GO terms are listed in Online Table 1. The data are largely consistent with our previous microarray analysis [1].
Figure 1.
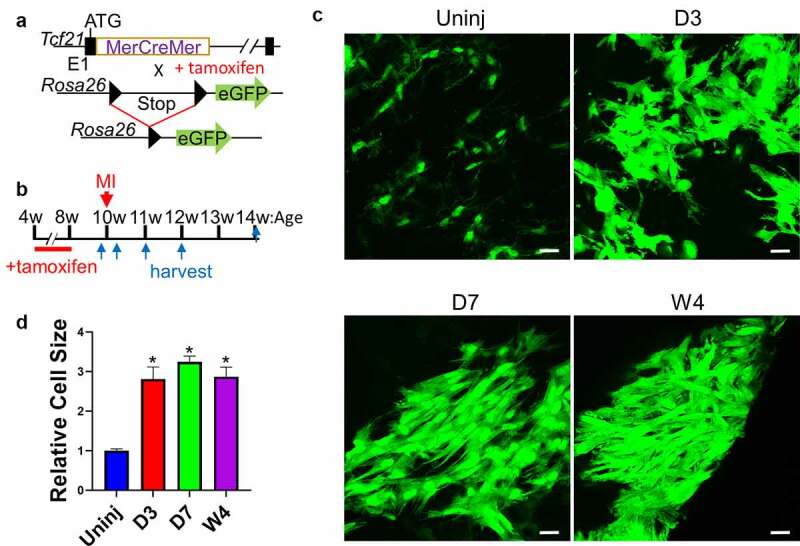
Lineage-tracing of cardiac fibroblast after MI.
(a) Schematic of the Tcf21MCM/+; R26eGFP mouse line. (b) Experimental scheme whereby Tcf21MCM/+; R26eGFP mice were treated with TM subjected to MI surgery, and then euthanized for heart sample collection at different time points after MI. (c-d) Whole-mount confocal microscopy images showing Tcf21 lineage-traced cardiac fibroblasts (GFP+) in the uninjured myocardium and the infarct at indicated time points after MI (C). d shows the relative sizes of cardiac fibroblasts at indicated time points. Data are shown as mean ± SEM (n = 3). *P < 0.05 vs uninjured, one-way ANOVA with multiple comparisons. Scale bar = 10 µm. Uninj, uninjured; D3, day 3; D7, day 7; W4, week 4.
Figure 2.
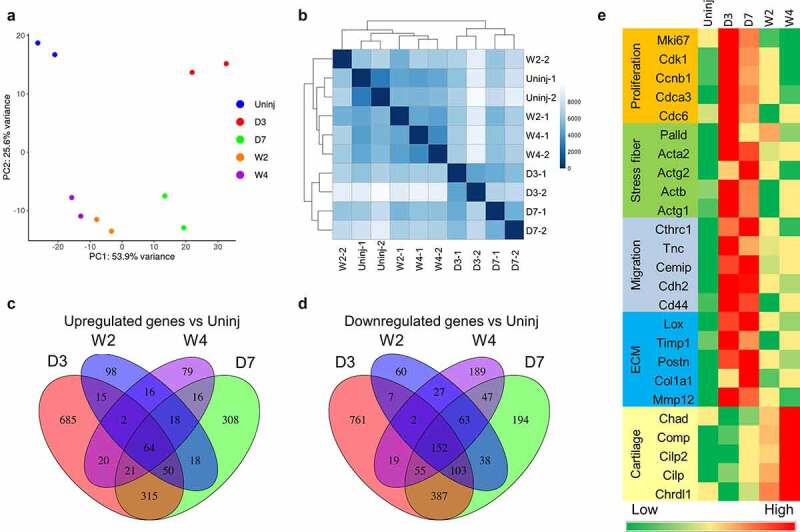
RNA-seq analysis of cardiac fibroblasts of different differentiation states.
Tcf21 lineage-traced cardiac fibroblasts were sorted from the uninjured myocardium and the infarct at different time points after MI for RNA-seq analysis. (a-b) PCA analysis (a) and Poisson distance analysis (b) of RNA-seq data showing tight clustering of repeats of each group. (c-d) Venn diagram showing the overlaps between genes that were differentially expressed between Tcf21 lineage–traced fibroblasts isolated from the uninjured myocardium and those from the infarct at different time points after MI. Genes that were upregulated after MI compared to cardiac fibroblasts isolated from the uninjured myocardium are shown in C. Downregulated genes are shown in D. (e) Heat maps showing the normalized gene expression of some representative genes in different categories. Uninj, uninjured; D3, day 3; D7, day 7; W2, week 2; W4, week 4.
Accessible chromatin landscape in cardiac fibroblasts of different differentiation state
To investigate the chromatin accessibility in cardiac fibroblasts during their post-MI activation and differentiation, we performed ATAC-seq analysis on Tcf21 lineage-traced cardiac fibroblasts isolated from the uninjured myocardium and the infarct on day 3, day 7, week 2, and week 4 after MI. Using our recently improved ATAC-seq protocol that is suitable for low cell numbers, we could obtain high-quality ATAC-seq libraries and data using 10,000 cells per sample. ATAC-seq peaks were highly enriched in the regions around the transcription start site (TSS) and the transcription end site (TES), and in exons (Figure 3(a,b)). In addition, ATAC-seq peaks were especially highly enriched at CpG islands that likely included many promoters [23], suggesting that DNA methylation may play a role in chromatin remodelling during the activation and differentiation of cardiac fibroblasts (Figure 3(b)). An increasing number of studies have shown that repetitive elements such as interspersed elements (LINEs), short interspersed elements (SINEs), long terminal repeats (LTR), and simple sequence repeats (SSRs) are prevalent in mammalian genomes and play some important roles in the regulation of chromatin accessibility and gene expression [24–29]. However, we did not identify significant enrichment of ATAC peaks in these repetitive elements or observe any difference in the enrichment of ATAC peaks in these repetitive elements among different sample groups, which suggests that repetitive elements are not the major regulator responsible for the changes in chromatin accessibility and gene expression during the post-MI activation and differentiation of cardiac fibroblasts (Figure 3(b)). PCA and Poisson distance analysis showed high consistency within each sample group and distinct chromatin accessibility profiles among different groups (Figure 3(c,d)). A total of ~200,000 peaks were identified. Around 100,000 peaks showed differential accessibility between cardiac fibroblasts of different groups, out of which 36,348 were differentially accessible in cardiac fibroblasts after MI as compared to cardiac fibroblasts from the uninjured myocardium (Figure 3(e,f)).
Figure 3.
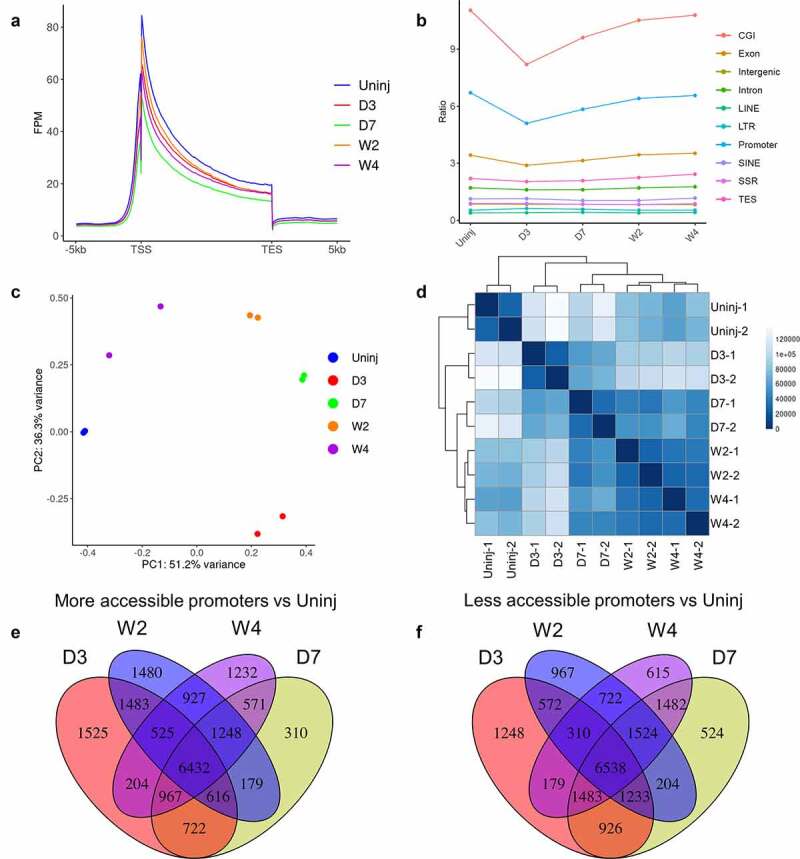
ATAC-seq analysis of cardiac fibroblasts of different differentiation states.
Tcf21 lineage-traced cardiac fibroblasts were sorted from the uninjured myocardium and the infarct at different time points after MI for ATAC-seq analysis. (a) Enrichment of ATAC-seq peaks around TSS and TES of genes. (b) Relative abundance of ATAC-seq peaks in different genomic features. (c-d) PCA analysis (c) and Poisson distance analysis (d) of ATAC-seq data show tight clustering of repeats of each group. (e-f) Venn diagram showing the overlaps between ATAC-seq peaks that had different abundances between Tcf21 lineage–traced fibroblasts isolated from the uninjured myocardium and those from the infarct at different time points after MI. Peaks that were upregulated after MI compared to cardiac fibroblasts isolated from the uninjured are shown in E. Downregulated peaks are shown in F. Uninj, uninjured; D3, day 3; D7, day 7; W2, week 2; W4, week 4.
Differential gene expression between cardiac fibroblasts of different differentiation states is associated with dynamic chromatin accessibility
Chromatin accessibility of genes especially in the promoter regions may directly affect the transcription activity of genes. To study if promoter accessibility is correlated with gene expression in cardiac fibroblasts, RNA-seq and ATAC-seq data were combined for an integrated analysis. It was found that the promoter accessibilities of genes expressed by at least one state of cardiac fibroblasts were higher than that of genes not expressed by any states of cardiac fibroblasts (average Log10(FPM), 1.76 vs 0.87, p< 2.2e-16) (Figure 4(a)). An analysis of expressed genes and their promoter accessibilities showed that a strong positive correlation (cor > 0.5) between promoter accessibility and transcription were identified in 30.77% of the expressed genes (Figure 4(b)), such as Tcf21 (a quiescent cardiac fibroblast marker), Ki67 (a proliferation marker), Acta2 (a myofibroblast marker), and Comp (a matrifibrocyte marker) (Figure 4(c-f)).
Figure 4.
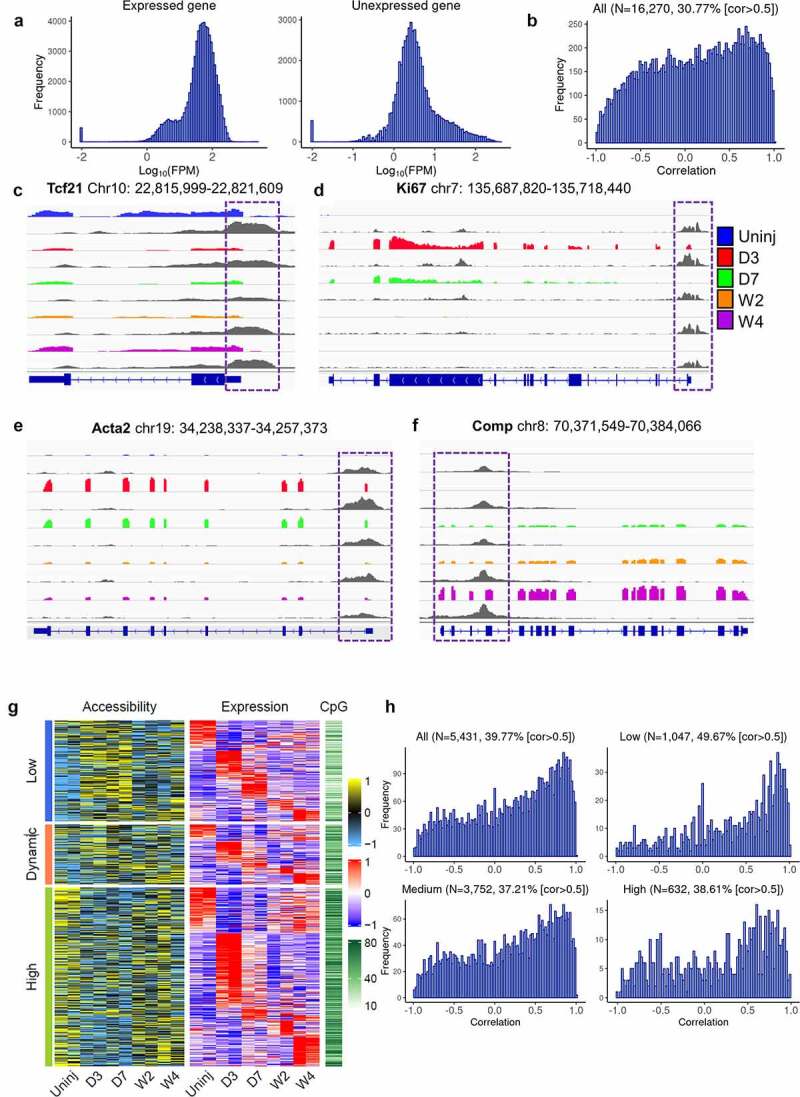
Differential gene expression and corresponding changes in promoter accessibility among cardiac fibroblasts of different differentiation states.
(a) Histograms of promoter accessibility of genes that were expressed (left) and unexpressed (right) in cardiac fibroblasts measured by ATAC-seq. (b) Histogram of correlation between promoter accessibility and expression of all genes that have ATAC-seq reads mapped to their promoters and mapped RNA-seq reads. The percentage of genes with cor > 0.5 is indicated. (c-f) Integrated Genome Viewer (IGV) views of the RNA-seq data and ATAC-seq data of representative genes showing differential gene expression and promoter accessibility among Tcf21 lineage-traced cardiac fibroblasts isolated from the uninjured myocardium and the infarct at different time points after injury. Promoter regions are highlighted in purple boxes. Coloured tracks are RNA-seq data, followed by ATAC-seq tracks (grey) of the same sample groups. (g) Heat map showing the promoter accessibility (Z-score), expression level (Z-score), and promoter CpG density of genes that were differentially expressed among cardiac fibroblasts isolated at different time points. Genes were divided into three groups based on their promoter accessibility (low, dynamic, and high). The data for accessibility and expression were standardized such that each row (i.e., promoter/gene) has a mean of 0 and a standard deviation of 1 (Z-score). (h) Histograms of correlations between promoter accessibility and expression level of all differentially expressed genes and their subgroups are classified according to average promoter CpG density, low, medium, and high. Percentages of genes with cor > 0.5 are indicated. Uninj, uninjured; D3, day 3; D7, day 7; W2, week 2; W4, week 4.
We then classify the detected promoters based on their accessibility. Accessibility data for each sample were standardized to have mean 0 and standard deviation 1 across promoters of genes included. Promoters received a standardized value that was no more than 0 in any of the samples and were deemed to have consistently low accessibility. Those with values above 0 in all samples were deemed to have consistently high accessibility. The rest were deemed to have dynamic accessibility. The 5,431 differentially expressed genes were then assigned to these 3 promoter accessibility groups. Interestingly, even though a strong promoter accessibility-transcription correlation was identified in many genes of all three promoter accessibility groups, a weaker correlation, or lack of correlation was detected in some genes especially those with high promoter accessibilities and were upregulated at day 3 post-MI, suggesting that after MI some other mechanisms besides chromatin accessibility in the promoter region may be responsible for the activation of the expression of genes that have generally accessible promoters (Figure 4(g)). DNA methylation also regulates gene transcription activity. Increased DNA methylation may result in the recruitment of histone modification enzymes, which induces chromatin accessibility changes [30,31]. DNA methylation may also regulate gene expression by inhibiting TF binding [32,33]. We then investigated the CpG density in the promoters of different accessibility groups. Interestingly, genes with high promoter accessibility had the highest CpG density, while genes with low promoter accessibility had the lowest CpG density. These data suggest that DNA methylation may be a mechanism contributing to the low promoter accessibility-transcription correlation of some genes with constitutively high promoter accessibility (Figure 4(g)). Indeed, when the 5,431 differentially expressed genes were divided into 3 groups based on the CpG densities in their promoter, a higher percentage of genes (49.67%) with low CpG density had a strong positive correlation (cor > 0.5) between promoter accessibility and expression compared to the other 2 groups (37.21% and 38.61% for genes with medium and high CpG densities, respectively), further suggesting the presence of gene transcription regulation by DNA methylation that is independent of promoter chromatin accessibility (Figure 4(h)).
Differential chromatin accessibilities in promoters and distal regions are correlated with differential gene expression
The analysis performed above focused on the identification of corresponding changes in chromatin accessibility for genes that were differentially expressed between cardiac fibroblasts of different states. We then selected genes with differential promoter accessibility and those with differential accessibility in distal regions and study the changes in their expression levels between cardiac fibroblasts of different states. Three thousand five hundred and eighteen promoters with differential accessibilities were identified, out of which 30.96% had a strong positive correlation (cor > 0.5) between promoter accessibility and gene expression (Figure 5(a,b)). This suggests that a large portion of promoters (69.04%) with differential accessibilities lack corresponding changes in the expression of their associated genes. Moreover, a higher percentage of promoters with low CpG density (38.45%) showed a strong positive correlation (cor > 0.5) as compared to promoters with medium or high CpG density (both ~27.5%), further suggesting that the expression of genes with higher CpG density in the promoters may be more actively regulated by DNA methylation rather than promoter accessibility (Figure 5(b)).
Figure 5.
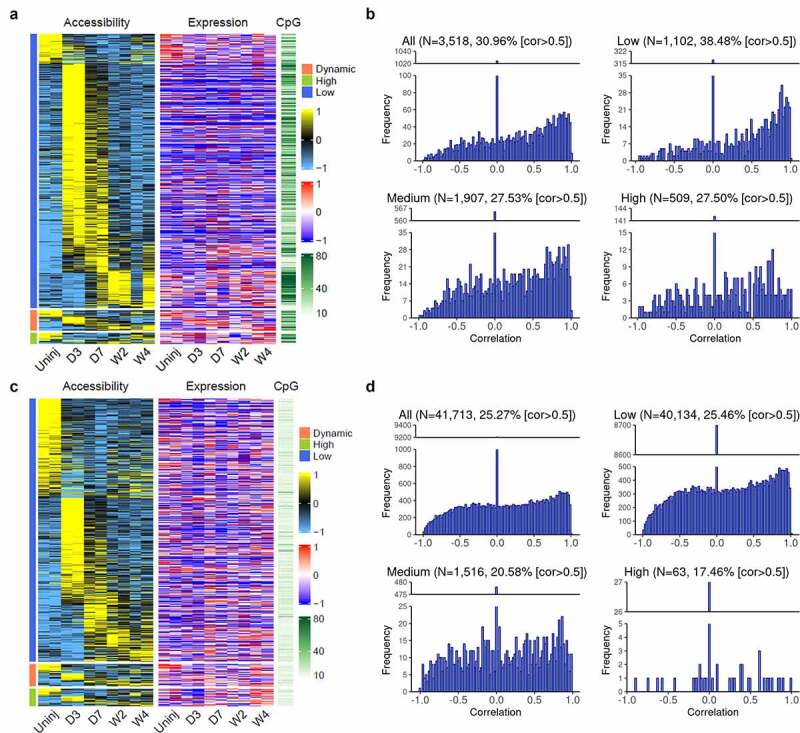
Differential accessibility in the promoter and distal regions and corresponding gene expression changes among cardiac fibroblasts of different differentiation states.
(a) Heat map showing the accessibility (Z-score) and CpG density of promoters that had differential accessibility among Tcf21 lineage-traced cardiac fibroblasts isolated from the uninjured myocardium and those isolated from the infarct at different time points after MI, together with the expression level (Z-score) of corresponding genes. Promoters were divided into three groups based on the expression of their corresponding genes (low, dynamic, and high). The data of accessibility and expression were standardized such that each row has a mean of 0 and a standard deviation of 1 (Z-score). (b) Histogram of correlation between accessibility and expression level of corresponding genes of all differentially accessible promoters and those with low, medium, and high CpG densities. Percentages of genes with cor > 0.5 are indicated. (c) Heat maps showing the accessibilities and CpG density of distal regions that had differential accessibility among Tcf21 lineage-traced cardiac fibroblasts isolated from the uninjured myocardium and those isolated from the infarct at different time points after MI, together with the expression level of proximal genes. Distal peaks were divided into three groups based on the expression of their proximal genes (low, dynamic, and high). The data of accessibility and expression were standardized such that each row has a mean of 0 and a standard deviation of 1 (Z-score). (d) Histogram of correlation between accessibility and expression level of proximal genes of all differentially accessible distal regions and those with low, medium, or high CpG densities. Percentages of genes with cor > 0.5 are indicated. Uninj, uninjured; D3, day 3; D7, day 7; W2, week 2; W4, week 4.
Enhancers located in distal regulatory regions also play an important role in gene expression regulation [34]. A total of 41,713 distal ATAC-seq peaks that showed differential accessibility were identified. We then assumed that genes proximal to the distal regulatory regions are their target genes and analysed the correlation between the accessibility of these distal ATAC-seq peaks and the expression of their assigned (proximal) genes. Even though strong positive correlations (cor > 0.5) were identified for some distal ATAC-seq peaks (25.27%), this number is lower than the percentage of promoters with positive correlations between accessibility and gene expression (30.96%), which may be partially due to the oversimplified method used to assign distal ATAC-seq peaks to their target genes (Figure 5(c,d)). Moreover, the majority of distal ATAC-seq peaks had low CpG density (Figure 5(d)), suggesting that DNA methylation may not play a major role in the regulation of the interaction between TFs and enhancers.
Prediction of major enhancers and targeting TFs regulating cardiac fibroblast activation and differentiation after MI
Gene expression is directly regulated by TFs. Another application of ATAC-seq other than measuring chromatin accessibility is to predict TF binding in open chromatin regions. Unlike Chromatin Immunoprecipitation Sequencing (ChIP-seq) which is highly specific and accurate but can only locate the binding motifs of a single TF per assay, ATAC-seq is able to locate all known TF binding motifs in accessible genomic sites [35]. Studies have found that TFs mainly target enhancers in the distal regions, which play an important role in gene expression regulation [36]. Using the findMotifsGenome.pl from HOMER, 440 known motifs were identified in the distal ATAC-seq peaks, out of which 263 corresponding TFs had detectable expression in cardiac fibroblasts. The most enriched motifs including those for zinc finger proteins (e.g., Ctcf and Egr2), bZIP TFs (e.g., AP-1 family, C/EBP family, Nfil3), Nf1, Tead family (e.g., Tead2), EBF family (e.g., Ebf2), Stat family (e.g., Stat1), ETS family (e.g., ETS1), NF-κB family (e.g., Rela), MEF2 family (e.g., Mef2d), Runx family (e.g., Runx1), bHLH proteins (e.g., Tcf21), and ROR family (e.g., Rora). To identify motifs and corresponding TFs that are most likely responsible for the post-MI differentiations of cardiac fibroblasts, these motifs were classified into several groups based on their enrichment in different states of cardiac fibroblasts (Figure 6(a-e)). The enrichment of Ctcf motif was found to be relatively unchanged across different sample groups, suggesting a constitutively active function of Ctcf in cardiac fibroblasts of all states (Figure 6(a)). Interestingly, the enrichment of Rora motif significantly dropped in myofibroblasts, and a prolonged and progressive reduction in the enrichment of Tcf21 motifs was identified in both myofibroblasts and matrifibrocytes (Figure 6(b)). The enrichment of some motifs (Nf1, Nfil3, Rela, Ets1, and AP-1 TFs) was found to be temporarily increased in myofibroblasts and then decreased in matrifibrocytes to a level around or below their levels in quiescent cardiac fibroblasts (Figure 6(c,d)). Several other motifs with similar changes in enrichment but lack statistical significance include Ebf2, Cebpg, and Stat1 (Figure 6(c)). In matrifibrocytes (week 2 post-MI and beyond), a sustained elevation in the enrichment was identified for 4 motifs which are Tead2, Mef2s, Egr2, and Runx1 (Figure 6(e)). Online Figure 2 shows the prevalence of these TF motifs in target sequences (ATAC-seq peaks) and background sequences (sequences other than ATAC-seq peaks), which suggests that the changes in their prevalence in target peaks and background peaks together mediated the differential enrichment of these motifs in cardiac fibroblasts after MI. The change in the expression level of a TF can be a major factor regulating the enrichment of its targeting motif in ATAC-seq peaks since its expression level may directly influence its protein abundance. TFs whose motif enrichment change and gene expression change roughly match to each other in cardiac fibroblasts after MI include Tcf21, Rora, Nf1, Stat1, Fosl1, Tead2, and Runx1 (Figure 6(a-e)). Interestingly, despite the progressive reduction in the enrichment of Tcf21 motif after MI, its expression level returned to the pre-injury level in matrifibrocytes. Surprisingly, the expression of most AP-1 TFs, which represent the largest TF group whose motifs were significantly enriched in myofibroblasts, dropped in myofibroblasts (Figure 6(d)). One exception was Fosl1 whose expression was upregulated in myofibroblasts (Figure 6(d)). Given the fact that neither the motif enrichment nor expression of Ctcf was different among cardiac fibroblasts of different states, it is likely not responsible for the gene expression change during myofibroblast and matrifibrocyte differentiation. We then assigned distal ATAC-seq peaks to their proximal genes. An analysis was performed to study the prevalence of these 17 TF motifs in all distal ATAC-seq peaks for each sample group and in those assigned to genes that were upregulated in cardiac fibroblasts in each post-MI sample group as compared to the uninjured group. Such an analysis revealed that for most of these TF motifs in most of the post-MI sample groups their enrichment in distal ATAC-seq peaks assigned to upregulated genes was greater compared to all identified distal ATAC-seq peaks except for Ctcf (Figure 7(a-e)), further suggesting that Ctcf may not contribute to the gene expression alteration in cardiac fibroblasts after MI. Fosl1, Rora, Stat1, Nf1, Tead2, Runx1, and Tcf21 were then selected to study their potential contributions to the post-MI activation and differentiations of cardiac fibroblast. Genes that were differentially expressed between cardiac fibroblasts of different states and possibly targeted by each of these TFs (corresponding motifs were identified in associated distal peaks) were identified. A comparison of differentially expressed genes targeted by individual TFs revealed that the largest overlapping was among Tcf21, Runx1, Tead2, Nf1, and Fosl1 (Figure 8), suggesting that many genes that are associated with the post-MI activation and differentiation of cardiac fibroblasts are co-regulated by multiple key TFs.
Figure 6.
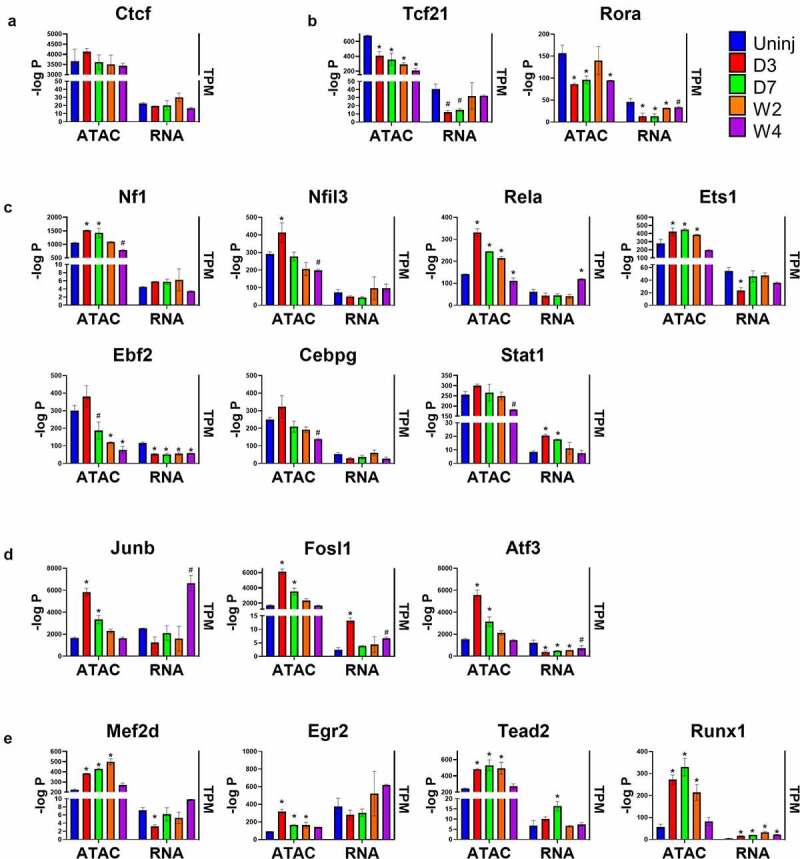
Identification of TF motifs in distal ATAC-seq peaks.
(a-d) The enrichment-log P values of some most-enriched motifs in ATAC-seq peaks and the mRNA expression levels (TPM) of the corresponding TFs in Tcf21 lineage-traced cardiac fibroblasts isolated from the uninjured myocardium and infarct regions at different time points after MI. Motifs were divided into uniformly enriched across all groups (a), more enriched in quiescent cardiac fibroblasts (b), more enriched in myofibroblasts (c-d), and more enriched in both myofibroblasts and matrifibrocytes (e). Data are shown as mean ± SEM (n = 2). #P < 0.1, *P < 0.05 vs uninjured, one-way ANOVA with multiple comparisons. Uninj, uninjured; D3, day 3; D7, day 7; W2, week 2; W4, week 4.
Figure 7.
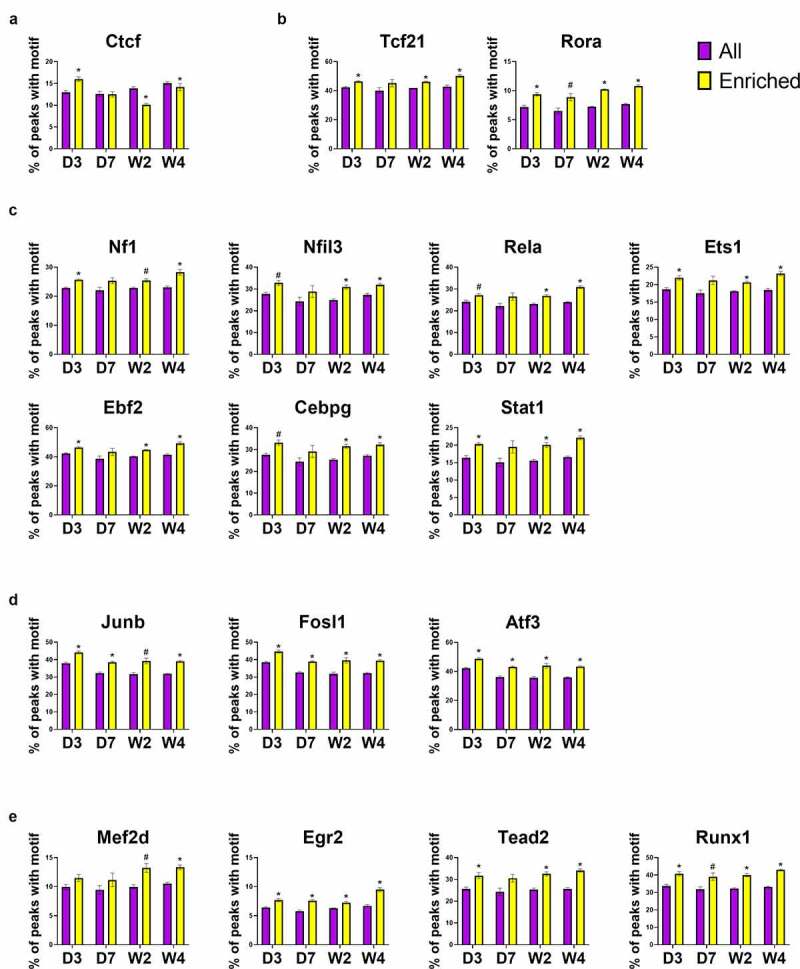
Increased enrichment of TF motifs in distal ATAC-seq peaks that are proximal to genes upregulated in cardiac fibroblasts after MI.
(a-e) The prevalence of selected TF motifs in all distal ATAC-seq peaks (All) and in distal ATAC-seq peaks assigned to genes that were upregulated (enriched) in cardiac fibroblasts at different time points after MI compared to cardiac fibroblasts isolated from the uninjured myocardium. Motifs were divided into uniformly enriched across all groups (a), more enriched in quiescent cardiac fibroblasts (b), more enriched in myofibroblasts (c-d), and more enriched in both myofibroblasts and matrifibrocytes (e) as shown in Figure 6. Data are shown as mean ± SEM (n = 2). #P < 0.1, *P < 0.05 enriched vs. all two-tailed t test. Uninj, uninjured; D3, day 3; D7, day 7; W2, week 2; W4, week 4.
Figure 8.
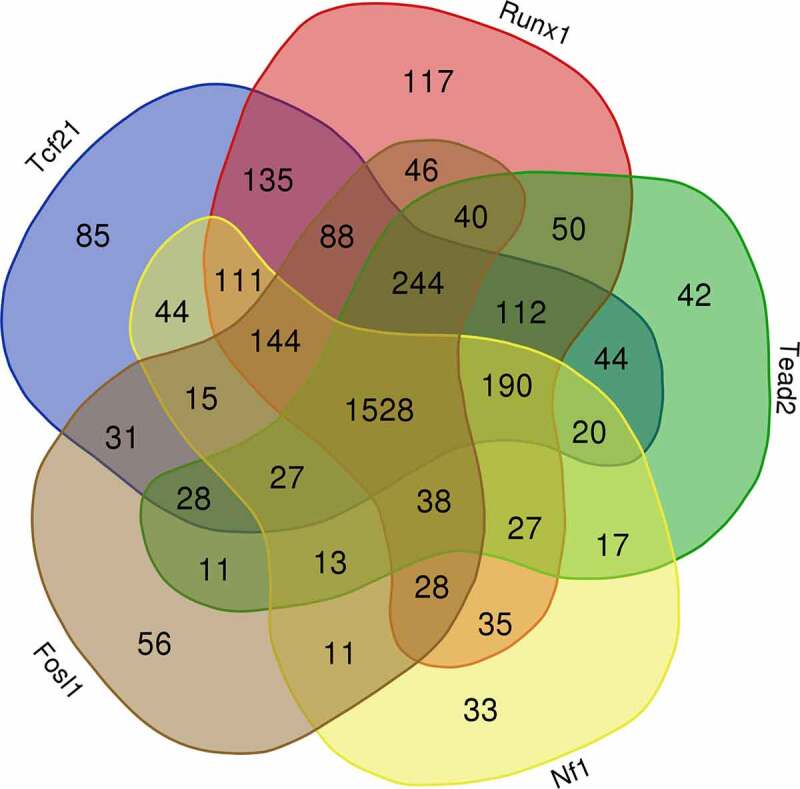
Overlaps between genes possibly targeted by individual TFs in the distal regions.
Distal ATAC-seq peaks proximal to genes that were differentially expressed between cardiac fibroblasts of different states were analysed for the prevalence of TF motifs. The Venn diagram shows the overlaps between genes that are possibly targeted by individual TFs in the distal regions.
Construction of gene regulatory network in cardiac fibroblasts
To better understand the possible regulatory roles of these motifs and their corresponding TFs in the gene expression changes in cardiac fibroblasts after MI, we then identified genes for which at least one of the seven selected motifs (Fosl1, Rora, Stat1, Nf1, Tead2, Runx1, and Tcf21) are located in ATAC-seq peaks ± 100 kb of their TSSs. Among these genes, the top 500 most differentially expressed genes between cardiac fibroblasts of different states and the genes encoding the 7 selected TFs (506 total genes) were included in gene regulatory network (GRN) construction (Figure 9(a)). Four hundred and sixty-seven out of the 506 genes are possibly regulated by at least 1 of the 7 TFs, suggesting that these TFs contribute to the gene expression changes in cardiac fibroblasts. Again, most of these genes are possibly regulated by more than 1 TF. Interestingly, most genes connected to Tcf21 and Rora appeared to be negatively regulated by these two TFs. Surprisingly, even though both the expression and motif enrichment of Nf1 increased in cardiac fibroblasts after MI, Nf1 seems to function as a repressor during the post-MI activation and differentiation of cardiac fibroblasts. Both positive and negative regulation of gene expression by Fosl2, Stat1, Tead2, and Runx1 were identified.
Figure 9.
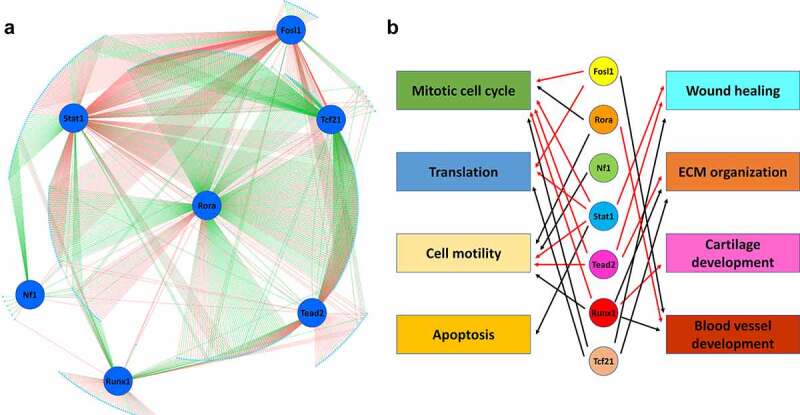
Gene regulatory network in cardiac fibroblasts.
(a) Top 500 most differentially expressed genes (cyan dots) among cardiac fibroblasts in different states with at least one of the 7 motifs (Fosl1, Rora, Stat1, Nf1, Tead2, Runx1, and Tcf21; blue circles) identified in the ATAC-seq peaks ± 100 kb of their TSSs were selected for gene regulatory network (GRN) construction. Red lines indicate positive regulation. Green lines indicate negative regulation. (b) Possible functions of TFs based on GO enrichment analysis of genes positively or negatively regulated by individual TFs. Red arrows indicate positive regulation. Black arrows indicate negative regulation.
To identify the possible contributions of these TFs to the post-MI activation and differentiation of cardiac fibroblasts, we performed GO enrichment analysis of genes that were possibly positively or negatively regulated by each TFs based on the constructed GRN. Significant enrichment of some GO biological process terms that are related to the physiological and functional changes of cardiac fibroblasts after MI were identified (Online Table 2) and used to predict the possible regulatory roles of individual TFs in the post-MI activation and differentiation of cardiac fibroblasts (Figure 9(b)). It was found that Fosl1, Stat1, Tead2, and Runx1 likely stimulate cardiac fibroblast proliferation through promoting pro-proliferative gene expression or inhibiting anti-proliferative gene expression (GO terms, mitotic cell cycle, negative regulation of cell population proliferation, and positive regulation of cell population proliferation) in newly activated cardiac fibroblasts, while Rora and Tcf21 possibly inhibit cardiac fibroblast proliferation. The protein synthesis (GO term, translation) in activated and differentiating cardiac fibroblasts seems to be promoted by Fosl1 and Stat1 but inhibited by Tcf21. The increased cell motility of activated cardiac fibroblasts is likely due to the inhibitory effect of Stat1 and Tead2 on the expression of genes that negatively regulate cell motility (GO term, negative regulation of cell motility), while Rora, Nf11, and Runx1 likely downregulate cell motility through inhibiting the expression of genes that promote cell motility (GO terms, regulation of cell motility, positive regulation of cell motility, and positive regulation of cell migration). Stat1, Runx1, and Tead2 are possibly the major TFs that enhance the myofibroblast-mediated ECM remodelling (GO terms, wound healing and ECM organization) after MI, which may be inhibited by Tcf21. The specific role of Stat1 in preventing the hypoxia-induced apoptosis in cardiac fibroblasts after MI was also identified (GO term, apoptosis). In addition, a possibly unique role of Runx1 in promoting cartilage development (GO term, cartilage development) was also identified, which may contribute to the differentiation of myofibroblasts into matrifibrocytes that share some gene expression signatures with chondrocytes. Moreover, a possible inhibitory role of Fosl1 and Runx1 in angiogenesis was identified (GO term, blood vessel development), which possibly contributes to the insufficient revascularization of the infarct scar after MI [37]
Discussion
We recently identified and described a series of events during the post-MI activation and differentiation of cardiac fibroblasts, which was associated with some drastic changes in gene expression [1]. In this study, we employed RNA-seq and ATAC-seq to study the correlation between chromatin accessibility and gene expression during these events, which is still poorly understood. The identification of a positive correlation between promoter accessibility and expression level in many genes that are differentially expressed between cardiac fibroblasts of different states indicates that ATAC-seq is a powerful tool to study promoter accessibility and the change of which may directly contribute to the gene expression in cardiac fibroblasts after MI. Interestingly, we found that a considerable number of differentially expressed genes between different states of cardiac fibroblasts lacked the corresponding changes in promoter accessibility especially in genes with high CpG density, suggesting that DNA methylation may regulate promoter activity without affecting their accessibility, possibly through preventing TFs from binding to target motifs in gene promoters [32,33]. However, future DNA methylome studies are required to reveal the collaborative and independent roles of DNA methylation and chromatin remodelling in the regulation of cardiac fibroblast gene expression during the post-MI activation and differentiation of cardiac fibroblasts. In addition, the preloading of promoters with RNA polymerase II and other transcription activation proteins have been described before, which is believed to enable the rapid activation of the expression of certain genes [38,39] and may contribute to the low correlation between promoter accessibility and expression of some genes that are rapidly activated in cardiac fibroblasts after MI. Moreover, many studies have identified that the relative abundance of mRNA is also influenced by the stability of mRNA [40,41]. Thus, the lack of correlation between promoter accessibility and expression of certain genes might be attributable to the changes in mRNA stability.
The binding of TFs to DNA mainly happens in the nucleosome-depleted regions which are abundant in promoter regions and even more in distal regions in the genome or enhancers that are away from TSSs [36]. Our GRN analysis identified several TFs that very likely regulate the post-MI activation, myofibroblast differentiation, and matrifibrocyte differentiation of cardiac fibroblasts by targeting promoters and enhancers proximal to genes that are differentially expressed between cardiac fibroblasts of different states. Some of these TFs/TF families, such as Tead, Tcf, and Runx1 were also reported to be cardiac fibroblast-specific in a recent study comparing TFs and their targeted enhancers between rat cardiac fibroblasts and cardiomyocytes [35], which, however, was limited by the use of cultured cells that might be transcriptionally different from freshly isolated primary cells. Moreover, the use of a single cardiac fibroblast culture condition could not reveal the changes in the gene expression, chromatin accessibility, and motif enrichment that happen during cardiac fibroblast activation and differentiation. The use of fresh primary cardiac fibroblasts immediately after isolation at different time points after MI enabled us to predict some unique roles of individual TFs in specific post-MI activation and differentiation events.
We identified a possibly inhibitory role of Tcf21 in myofibroblasts-mediated post-MI ECM remodelling. The requirement of Tcf21 in the epithelial-to-mesenchymal transition (EMT) during the cardiac fibroblast specification of epicardial cells has been reported [42]. Mouse embryos lacking Tcf21 had a reduced number of cardiac fibroblasts. The deletion of Tcf21 in cardiac fibroblast progenitor cells promoted their differentiation into vascular smooth muscle cells [42–44]. Thus, it is possible that Tcf21 plays a role in preventing the myofibroblast differentiation of cardiac fibroblasts. In our study, it was reported that the expression of Tcf21 is regulated by C/EBPs [45], the target motif of which was also found enriched in cardiac fibroblasts. Interestingly, retinoic acid signalling, which likely involves Rora, was also reported to be critical during the prenatal differentiation of cardiac fibroblast [43], perhaps through promoting the expression of Tcf21 [46]. Indeed, a plausible positive regulation of Tcf21 expression by Rora was identified here. We also found that Tcf21 and Rora possibly have an inhibitory effect in the regulation of cardiac fibroblast proliferation. The possible anti-proliferative and/or anti-myofibroblast differentiation roles of Tcf21 and Rora, together with their stronger expression in cardiac fibroblasts from the uninjured myocardium suggest that Tcf21 and Rora may be important in maintaining the undifferentiated state of adult cardiac fibroblasts through inhibiting the expression of pro-proliferative and pro-myofibroblast genes. Their downregulation in cardiac fibroblasts after MI may facilitate the activation and differentiation of cardiac fibroblasts.
Similar to Tcf21, Nf1 is also expressed in the epicardial precursors of cardiac fibroblasts [47]. However, unlike Tcf21, the loss of Nf1 resulted in increased EMT of epicardial cells and their proliferation. Indeed, we found that many of the genes possibly inhibited by Nf1 were genes regulating cell motility, a major feature of EMT. It is possible that the increased expression of Nf1 in cardiac fibroblasts after MI is to prevent an over-activation of cell motility.
Fosl1 is a member of the Fos family which is a part of a large TF family named AP-1 that also includes the Jun family and ATF family [48]. Here, we found that Fosl1 possibly promotes cardiac fibroblast proliferation and migration after MI. Previous studies have also identified the roles of AP-1 family members in regulating cell proliferation [49] and myofibroblast differentiation [50,51]. It is very interesting that despite the increased enrichment of AP-1 motifs in cardiac fibroblasts after MI, a reduction in the expression of most AP-1 family genes was observed except for Fosl1 which, however, was expressed at a rather low level compared to some genes encoding other AP-1 components. To become functional, AP-1 subunits form heterodimers through the interaction of bZIP domains, which likely lead to their context-dependent functionality. It is possible that certain mechanisms increasing the efficiency of dimerization between specific AP-1 subunits are activated during the post-MI differentiation of cardiac fibroblasts, which allows these AP-1 subunits to work more efficiently even at a lower expression level.
In this study, we also identified the possible roles of Stat1 in promoting the expression of anti-apoptotic genes, and inhibiting the expression of genes preventing wound healing, a process involving collagen synthesis. A study comparing foetal and adult human cardiac fibroblasts identified Stat1 as an important TF responsible for the adult phenotype of cardiac fibroblasts [52]. Loss of Stat1 in cardiac fibroblasts led to reduced cell size and collagen gene expression, and increased apoptosis. Thus, Stat1 may contribute to the increased ECM protein production by cardiac fibroblasts and their relative resistance to apoptosis after MI [1].
Our GRN analysis also identified the possibly positive regulation of cardiac fibroblast proliferation, migration, and ECM remodelling by Tead2. Tead is a component of the Hippo pathway. Tead and its cofactor Yap1 were recently shown to promote both the fate determination of embryonic cardiac fibroblasts and the proliferation and myofibroblast differentiation of adult cardiac fibroblasts [53,54], which is consistent with our analysis.
Runx1 has been shown to promote the proliferation of mesenchymal cells, but delay their differentiation into myofibroblasts [55]. A recent study showed that zebrafish with global Runx1 KO led to reduced cardiac fibrosis and a smaller number of myofibroblasts after cardiac injury [56]. It is possible that Runx1 plays an important function in the development of cardiac fibrosis through balancing cardiac fibroblast proliferation and myofibroblast differentiation, which is supported by our results showing that Runx1 very likely promotes cardiac fibroblast proliferation and ECM remodelling. Moreover, we found that the GO term ‘cartilage development’ was enriched in genes that are possibly positively regulated by Runx1, which is consistent with a previous study showing the important role of Runx1 in the development of cartilage, a tissue that shares some gene signatures with matrifibrocytes [57]. Runx1 is the only TF whose expression remains several folds higher in matrifibrocytes than in quiescent cardiac fibroblasts, accompanied by significantly higher motif enrichment. Unlike other TFs that seem to coregulate the expression of many genes, quite a few genes are possibly solely regulated by Runx1. Thus, Runx1 may also uniquely promote the matrifibrocyte differentiation in more stabilized infarct scars by upregulating the expression of selected chondrogenic genes.
One limitation of the motif enrichment and GRN analyses in this study is that the validation of TF binding to the target motifs and experiments validating the function of these TFs are lacking. However, our study revealed the possible roles of some key TFs in post-MI activation and differentiation of cardiac fibroblasts, each of which can be specifically targeted in the future research to explore their functional roles in greater detail.
Conclusion
Overall, our transcriptome and open chromatin analysis show that chromatin accessibility may play an important role in the post-MI activation and differentiation of cardiac fibroblasts. This integrated analysis also suggests the presence of mechanisms other than chromatin accessibility in the regulation of gene expression in cardiac fibroblasts. Finally, our motif analysis and GRN construction work identified several potential TFs that are likely coordinated to mediate the sequential activation and differentiation of cardiac fibroblasts through programming the transcriptome of cardiac fibroblasts.
Supplementary Material
Funding Statement
This work was supported by Louisiana Board of Regents BOR.Fu.LEQSF(2019-22)-RD-A-01 (X.F.), NIH/NIGMS P20GM130555 (X.F.), NIH/NIDDK 1R15DK122383-01 (X.F.), USDA/NIFA 2020-67015-30823 (X.F.), NIH/NICHD R01HD102533 (Z.J.), and USDA/NIFA 2019-67016-29863 (Z.J.);Louisiana Board of Regents [BOR.Fu.LEQSF(2019-22)-RD-A-01];National Institute of Child Health and Human Development [R01HD102533];National Institute of Food and Agriculture [2019-67016-29863];National Institute of Food and Agriculture [2020-67015-30823];National Institute of General Medical Sciences [P20GM130555];National institute of diabetes and digestive and kidney diseases [1R15DK122383-01].
Disclosure statement
The authors declare no competing interests.
Author contribution
X.F. and J.S. conceived the study; C.L., Q.L., H.M., L.W., and Y.L. collected samples. C.L., J.S., S.D., and R.L. analyzed data. C.L., J.S., J.F., and X.F. interpreted the data, assembled the results, and wrote the manuscript with inputs from all authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Fu X, Khalil H, Kanisicak O, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018;128(5):2127–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kanisicak O, Khalil H, Ivey MJ, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016;7(1):12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kaur H, Takefuji M, Ngai CY, et al. Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ. Res. 2016;118(12):1906–1917. [DOI] [PubMed] [Google Scholar]
- [4].Pfeffer JM, Pfeffer Ma, Fletcher PJ, et al. Progressive ventricular remodeling in rat with myocardial infarction. Am J Physiol. 1991;260:H1406–1414. [DOI] [PubMed] [Google Scholar]
- [5].Fu X, Liu Q, Li C, et al. Cardiac fibrosis and cardiac fibroblast lineage-tracing: recent advances. Front Physiol. 2020;11:416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Khalil H, Kanisicak O, Prasad V, et al. Fibroblast-specific tgf-β-smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017;127(10):3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Meng Q, Bhandary B, Bhuiyan MS, et al. Myofibroblast-specific tgfbeta receptor ii signaling in the fibrotic response to cardiac myosin binding protein c-induced cardiomyopathy. Circ. Res. 2018;123(12):1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dobaczewski M, Bujak M, Li N, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010;107(3):418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Molkentin JD, Bugg D, Ghearing N, et al. Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation. 2017;136(6):549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tsompana M, Buck MJ.. Chromatin accessibility: a window into the genome. Epigenetics Chromatin. 2014;7(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Buenrostro JD, Wu B, Litzenburger UM, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561):486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Acharya A, Baek ST, Banfi S, et al. Efficient inducible cre‐mediated recombination in tcf21cell lineages in the heart and kidney. Genesis. 2011;49(11):870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yamamoto M, Shook NA, Kanisicak O, et al. A multifunctional reporter mouse line for cre‐and flp‐dependent lineage analysis. Genesis. 2009;47(2):107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Van Berlo JH, Kanisicak O, Maillet M, et al. C-kit+ cells minimally contribute cardiomyocytes to the heart. Nature. 2014;509(7500):337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fu X, Zhao J-X, Zhu M-J, et al. Amp-activated protein kinase α1 but not α2 catalytic subunit potentiates myogenin expression and myogenesis. Mol. Cell. Biol. 2013;33(22):4517–4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ming H, Sun J, Pasquariello R, et al. The landscape of accessible chromatin in bovine oocytes and early embryos. Epigenetics. 2021;16(3):300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Li R, Hu K, Liu H, et al. Onestoprnaseq: a web application for comprehensive and efficient analyses of rna-seq data. Genes (Basel). 2020;11(10):1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of chip-seq (macs). Genome Biol. 2008;9(9):R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Assenov Y, Ramírez F, Schelhorn SE, et al. Computing topological parameters of biological networks. Bioinformatics. 2008;24(2):282–284. [DOI] [PubMed] [Google Scholar]
- [22].Wiese R, Eiglsperger M, Kaufmann M. Yfiles — visualization and automatic layout of graphs. In: Jünger M, Mutzel P, editors. Graph drawing software. Berlin Heidelberg: Springer Berlin Heidelberg; 2004. p. 173–191. [Google Scholar]
- [23].Hood R, Allen C. Cellularity of bovine adipose tissue. J. Lipid Res. 1973;14(6):605–610. [PubMed] [Google Scholar]
- [24].Elbarbary RA, Lucas BA, Maquat LE. Retrotransposons as regulators of gene expression. Science. 2016;351(6274):aac7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ichiyanagi K. Epigenetic regulation of transcription and possible functions of mammalian short interspersed elements, sines. Genes Genet. Syst. 2013;88(1):19–29. [DOI] [PubMed] [Google Scholar]
- [26].Estécio MR, Gallegos J, Dekmezian M, et al. Sine retrotransposons cause epigenetic reprogramming of adjacent gene promoters. Mol. Cancer Res. 2012;10(10):1332–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chow JC, Ciaudo C, Fazzari MJ, et al. Line-1 activity in facultative heterochromatin formation during x chromosome inactivation. Cell. 2010;141(6):956–969. [DOI] [PubMed] [Google Scholar]
- [28].Sasaki T, Nishihara H, Hirakawa M, et al. Possible involvement of sines in mammalian-specific brain formation. Proc. Natl. Acad. Sci. U. S. A. 2008;105(11):4220–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Faulkner GJ, Kimura Y, Daub CO, et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009;41(5):563–571. [DOI] [PubMed] [Google Scholar]
- [30].Robertson KD. DNA methylation and chromatin - unraveling the tangled web. Oncogene. 2002;21(35):5361–5379. [DOI] [PubMed] [Google Scholar]
- [31].Martinowich K, Hattori D, Wu H, et al. DNA methylation-related chromatin remodeling in activity-dependent bdnf gene regulation. Science. 2003;302(5646):890–893. [DOI] [PubMed] [Google Scholar]
- [32].Yin Y, Morgunova E, Jolma A, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356(6337):eaaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016;17(9):551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144(3):327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Golan-Lagziel T, Lewis YE, Shkedi O, et al. Analysis of rat cardiac myocytes and fibroblasts identifies combinatorial enhancer organization and transcription factor families. J Mol Cell Cardiol. 2018;116:91–105. [DOI] [PubMed] [Google Scholar]
- [36].Grossman SR, Engreitz J, Ray JP, et al. Positional specificity of different transcription factor classes within enhancers. Proc. Natl. Acad. Sci. U. S. A. 2018;115(30):E7222–e7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Albrecht-Schgoer K, Schgoer W, Holfeld J, et al. The angiogenic factor secretoneurin induces coronary angiogenesis in a model of myocardial infarction by stimulation of vascular endothelial growth factor signaling in endothelial cells. Circulation. 2012;126(21):2491–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fuda NJ, Ardehali MB, Lis JT. Defining mechanisms that regulate rna polymerase ii transcription in vivo. Nature. 2009;461(7261):186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Boettiger AN, Levine M. Synchronous and stochastic patterns of gene activation in the Drosophila Embryo. Science. 2009;325(5939):471–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Guhaniyogi J, Brewer G. Regulation of mrna stability in mammalian cells. Gene. 2001;265(1–2):11–23. [DOI] [PubMed] [Google Scholar]
- [41].Hao S, Baltimore D. The stability of mrna influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 2009;10(3):281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Acharya A, Baek ST, Huang G, et al. The bhlh transcription factor tcf21 is required for lineage-specific emt of cardiac fibroblast progenitors. Development. 2012;139(12):2139–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Billings SE, Pierzchalski K, Butler Tjaden NE, et al. The retinaldehyde reductase dhrs3 is essential for preventing the formation of excess retinoic acid during embryonic development. FASEB J. 2013;27(12):4877–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sazonova O, Zhao Y, Nürnberg S, et al. Characterization of tcf21 downstream target regions identifies a transcriptional network linking multiple independent coronary artery disease loci. PLoS Genet. 2015;11(5):e1005202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Huang GN, Thatcher JE, McAnally J, et al. C/ebp transcription factors mediate epicardial activation during heart development and injury. Science. 2012;338(6114):1599–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Braitsch CM, Combs MD, Quaggin SE, et al. Pod1/tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev. Biol. 2012;368(2):345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Baek ST, Tallquist MD. Nf1 limits epicardial derivative expansion by regulating epithelial to mesenchymal transition and proliferation. Development. 2012;139(11):2040–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Eferl R, Wagner EF. Ap-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer. 2003;3(11):859–868. [DOI] [PubMed] [Google Scholar]
- [49].Shaulian E, Karin M. Ap-1 in cell proliferation and survival. Oncogene. 2001;20(19):2390–2400. [DOI] [PubMed] [Google Scholar]
- [50].Avouac J, Palumbo K, Tomcik M, et al. Inhibition of activator protein 1 signaling abrogates transforming growth factor β-mediated activation of fibroblasts and prevents experimental fibrosis. Arthritis Rheum. 2012;64(5):1642–1652. [DOI] [PubMed] [Google Scholar]
- [51].Seidenberg J, Stellato M, Hukara A, et al. The ap-1 transcription factor fosl-2 regulates autophagy in cardiac fibroblasts during myocardial fibrogenesis. Int. J. Mol. Sci. 2021;22(4):1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Jonsson MKB, Hartman RJG, Ackers-Johnson M, et al. A transcriptomic and epigenomic comparison of fetal and adult human cardiac fibroblasts reveals novel key transcription factors in adult cardiac fibroblasts. JACC. 2016;1:590–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Xiao Y, Hill MC, Zhang M, et al. Hippo signaling plays an essential role in cell state transitions during cardiac fibroblast development. Dev. Cell. 2018;45(2):153–169.e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Xiao Y, Hill MC, Li L, et al. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev. 2019;33(21–22):1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kim W, Barron DA, San Martin R, et al. Runx1 is essential for mesenchymal stem cell proliferation and myofibroblast differentiation. Proc. Natl. Acad. Sci. U. S. A. 2014;111(46):16389–16394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Koth J, Wang X, Killen AC, et al. Runx1 promotes scar deposition and inhibits myocardial proliferation and survival during zebrafish heart regeneration. Development. 2020;147(8). DOI: 10.1242/dev.186569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kimura A, Inose H, Yano F, et al. Runx1 and runx2 cooperate during sternal morphogenesis. Development. 2010;137(7):1159–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.