

Abstract
Background:
Intestinal ischemia-reperfusion (I/R) injury is a severe disease associated with high mortality. Stimulator of interferon genes (STING) is an intracellular protein that is activated by cytosolic DNA and is implicated in I/R injury, resulting in transcription of type I interferons (IFN-α and IFN-β) and other pro-inflammatory molecules. H151 is a small molecule inhibitor of STING that has not yet been studied as a potential therapeutic. We hypothesize that H151 reduces inflammation, tissue injury and mortality after intestinal I/R.
Methods:
In vitro, RAW264.7 cells were pre-treated with H151 then stimulated with recombinant murine (rm) CIRP, and IFN-β levels in the culture supernatant were measured at 24 hours after stimulation. In vivo, male C57BL/6 mice were subjected to 60-min intestinal ischemia via superior mesenteric artery occlusion. At the time of reperfusion, mice were intraperitoneally instilled with H151 (10 mg/kg BW) or 10% Tween-80 in PBS (vehicle). Four hours after reperfusion, the small intestines, lungs and serum were collected for analysis. Mice were monitored for 24 hours after intestinal I/R to assess survival.
Results:
In vitro, H151 reduced rmCIRP-induced IFN-β levels in a dose-dependent manner. In vivo, intestinal levels of pIRF3 were increased after intestinal I/R and decreased after H151 treatment. There was an increase in serum levels of tissue injury markers (LDH, AST) and cytokine levels (IL-1β, IL-6) after intestinal I/R, and these levels were decreased after H151 treatment. I/R-induced intestinal and lung injury and inflammation were significantly reduced after H151 treatment, as evaluated by histopathologic assessment, measurement of cell death, chemokine expression, neutrophil infiltration, and myeloperoxidase activity. Finally, H151 improved the survival rate from 41% to 81% after intestinal I/R.
Conclusion:
H151, a novel STING inhibitor, attenuates the inflammatory response and reduces tissue injury and mortality in a murine model of intestinal I/R. H151 shows promise as a potential therapeutic in the treatment of this disease.
Keywords: Intestinal ischemia/reperfusion, H151, STING, Inflammation, pIRF3, IFN
INTRODUCTION
Intestinal ischemia-reperfusion (I/R) injury is a potentially fatal disease which results from acute mesenteric ischemia, trauma, volvulus, incarcerated hernia, myocardial infarction, congestive heart failure, cardiopulmonary bypass, intestinal transplantation, renal or hepatic disease, major abdominal or cardiovascular surgery, as well as neonatal intestinal injury.1–3 Primary treatment of intestinal I/R is focused at restoration of mesenteric blood flow, which results in a complex immune response consisting of pathological activation of both innate and adaptive immune systems.
Multiple mechanisms contribute to this overwhelming inflammatory response. For one, intestinal I/R is associated with intestinal barrier loss, which results in bacterial translocation, causing the release of pathogen-associated molecular patterns (PAMPs) into circulation.3 Intestinal I/R also results in widespread necrosis of intestinal epithelial cells and enterocytes thus releasing intracellular components, or damage-associated molecular patterns (DAMPs), from dying cells.4 Both PAMPs and DAMPs interact with membrane-bound or intracellular receptors to amplify pro-inflammatory signaling, thus exacerbating tissue injury.5 Additionally, Paneth cells are activated and damaged, resulting in impaired immune response to invading pathogens, along with the release of cytokines and chemokines.6 Resultant neutrophilic infiltration produces reactive oxygen species, which cause additional bystander tissue injury and inflammation.7 Overall, the ensuing inflammatory response leads to extraintestinal organ dysfunction, most commonly acute lung injury (ALI), which contributes to the significant morbidity and mortality of intestinal I/R injury.8,9 Despite numerous attempts to reduce inflammation and subsequent organ injury in intestinal I/R, there remains an unmet medical need for efficacious therapeutics for the treatment of intestinal I/R.10–12
Stimulator of interferon genes (STING) is a 379-amino acid protein expressed in both innate and adaptive immune cells (e.g., macrophages, dendritic cells, natural killer cells, T and B lymphocytes), as well as in nonhematopoietic-derived cells, including endothelial cells and epithelial cells.13 STING has been shown to be activated by PAMPs (e.g., lipopolysaccharides [LPS]) and DAMPs (e.g., extracellular cold-inducible RNA-binding protein [eCIRP]) via the degradation and release of mitochondrial DNA (mtDNA) in the intracellular compartment.14,15 Cyclic GMP-AMP synthase (cGAS) identifies aberrant cytosolic DNA species such as mtDNA, and catalyzes the production of cyclic GMP-AMP (cGAMP), which then activates STING.16,17 After activation, STING undergoes palmitoylation and clustering, which allows it to bind tank-binding kinase 1 (TBK1) and relocate to perinuclear regions of the cell. Here, STING delivers TBK1 to endolysosomal compartments where TBK1 phosphorylates interferon regulatory factor 3 (IRF3) and NF-κB, thus initiating transcription of type I interferons (IFNα, IFNβ) and other pro-inflammatory molecules.18 This pathway has been shown to mediate inflammation and organ damage in intestinal I/R injury.19,20 However, therapeutic approaches targeting the STING pathway have not been attempted in intestinal I/R injury.
H151 is a small molecule inhibitor which covalently and irreversibly binds to human and murine STING in order to block the activation-induced palmitoylation and clustering of STING, ultimately inhibiting the downstream signaling pathway.21 H151 has been shown to ameliorate inflammation and organ injury in several disease states, but has not yet been studied in ischemia-reperfusion injury.15,22–24 In this study, we hypothesize that H151 attenuates the inflammation, tissue injury and mortality seen in intestinal I/R. To investigate this, we first assessed the efficacy of H151 in inhibiting STING signaling in macrophages in vitro, then we established a model of intestinal I/R and studied the in vivo effects of H151 treatment on organ damage, inflammation, and survival.
METHODS
Treatment of macrophages with H151
Murine macrophage cell line RAW264.7 cells were obtained from American Type Culture Collection (ATCC), and cultured in Dulbecco’s modified Eagle medium (DMEM; Life Technologies Corporation, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA), 1% penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA), and 1% glutamine (Thermo Fisher Scientific, Waltham, MA). Cells were maintained in a humidified incubator with 5% CO at 37°C. Prior to experiments, DMEM culture media was removed and replaced with OPTI-MEM (Life Technologies Corporation, Grand Island, NY) devoid of FBS. Cells were pre-treated with various doses of H151 (0.25, 0.5, 1.0, 2.0 μM), and incubated for 1 hour. One hour after H151 treatment, cells were stimulated with 1 μg/mL recombinant mouse (rm) CIRP. Cells were then incubated, and culture supernatant was collected 24 hours after rmCIRP stimulation.
Experimental animals
Adult male C57BL/6 mice (8–12 weeks old, 20–25 g body weight [BW]) were purchased from Charles River Laboratory (Wilmington, MA), housed in temperature- and light-controlled environments and fed standard laboratory mouse diets. Mice acclimated to this environment for at least 7 days prior to experiments. Every attempt was made to limit the number of animals used. All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Feinstein Institutes for Medical Research and were performed in accordance with the National Institutes of Health and the Guide for the Care and Use of Laboratory Animals.
Animal model of intestinal I/R
Intestinal I/R was performed as previously described.10–12 In brief, mice underwent induction of general anesthesia using 2–4% inhalational isoflurane, and general anesthesia was maintained with 2% inhalational isoflurane. The ventral abdomen was shaved, then disinfected with betadine and alcohol antiseptic solutions. The animals were placed on a water-based heating pad to maintain core body temperatures. A midline laparotomy incision was performed to expose the abdomen, and the superior mesenteric artery (SMA) was identified and isolated. A vascular microclip was placed across the SMA to occlude mesenteric blood flow for 60 minutes. Upon completion of 60 minutes of ischemia, the vascular microclip was removed to initiate reperfusion. At the time of reperfusion, mice randomly designated to the H151 treatment group received 10 mg/kg BW H151 in 10% Tween-80 in PBS (total volume 225 μL) via intraperitoneal (i.p.) administration prior to abdominal closure. This dose is consistent with previous studies of H151 in vivo,21,25 and was selected after preliminary experiments which assessed various doses of H151 in vivo (data not shown). Mice randomly designated to the vehicle control group received an equivalent volume of 10% Tween-80 in PBS via intraperitoneal administration at time of reperfusion. After closure of the abdomen, all mice undergoing intestinal I/R received a subcutaneous bolus of 1 mL normal saline, as well as 0.05 mg/kg BW subcutaneous buprenorphine. At 4 hours after reperfusion, blood and organs were collected under general anesthesia, and mice were euthanized. Sham mice did not undergo operation and underwent blood and organ harvest under general anesthesia maintained with inhalational isoflurane. To assess survival, after the intestinal I/R procedure mice randomly received intraperitoneal H151 or vehicle, as described above. Mice were evaluated every four hours for 24 hours. Only male mice were used in this study, as sex-dependent differences in intestinal I/R are well documented, and females exhibit significantly less intestinal I/R induced tissue damage and inflammation.26–28
H151, a STING-selective inhibitor
H151 was synthesized by Invivogen (purity ≥ 95%) and provided as a lyophilized powder.21 The powder was solubilized in DMSO to a concentration of 10 mg/mL. The desired concentration of H151 was prepared in sterile 10% Tween-80 in PBS prior to use in mice.
Western blotting
Lungs and small intestines were harvested from mice 4 hours after intestinal I/R, flash-frozen in liquid nitrogen, stored at −80°C, and crushed over dry ice to a fine powder, then homogenized in RIPA buffer (1X TBS [pH 7.5], 50mM EDTA, 50mM EGTA, Triton-X1000[1%], 2mM Na Orthovanadates [pH 7.6], 0.2mM PMSF) containing a protease inhibitor and phosphatase inhibitor using high-frequency sonication. Samples were then centrifuged at 12,000 ×g for 14 minutes at 4°C and supernatant was collected for further analysis. Protein concentration was determined by detergent compatible (DC) protein assay (Bio-Rad, Hercules, CA). Equal amounts of tissue homogenates were fractionated on SDS-PAGE (Invitrogen, Waltham, MA) and transferred to nitrocellulose membrane. The membranes were blocked by incubation with 0.1% casein in 0.2× PBS and incubated at 4°C overnight with the following primary antibodies: IRF3 (Cell Signaling Technology, Danvers, MA; catalog 4302S), pIRF3 (Cell Signaling Technology, Danvers, MA; catalog 4947S), and β-actin antibody (Sigma-Aldrich, St. Louis, MO; catalog A5441) in 0.2× PBS with 0.1% casein and 0.1% Tween 20. After washing, the blots were subsequently incubated with the corresponding fluorescent secondary antibody (LI-COR, Lincoln, NE). Bands were detected using the Odyssey FC Dual-Mode Imaging system 2800 (LI-COR, Lincoln, NE).
Determination of organ injury markers
Whole blood samples were drawn prior to euthanasia via cardiac puncture using a heparinized syringe, and centrifuged at 3,000 ×g for 10 minutes at 4°C. The supernatant containing the serum was collected, stored at 4°C and analyzed within 24 hours. Serum levels of aspartate aminotransferase (AST), and lactate dehydrogenase (LDH) were determined using specific colorimetric enzymatic assays according to manufacturer’s instructions (Pointe Scientific, Canton, MI).
Enzyme-linked immunosorbent assay (ELISA)
Serum was collected as described above and stored at −80°C prior to analysis. Serum was analyzed by ELISA kits specific to IL-6 (Catalogue No: 555220, BD Biosciences, San Jose, CA), and IL-1β (Catalogue No: 88–7013; Thermo Fisher Scientific, Waltham, MA).
Intestinal and lung immunohistochemical staining of Gr-1-positive neutrophils
Neutrophil infiltration into tissues was assessed by immunohistochemical staining of Gr-1-positive neutrophils. Paraffin-embedded sections of lung and small intestine were dewaxed in xylene and rehydrated in a graded series of ethanol. The slides were heated at 95°C for 30 min in 0.92% citric acid buffer (Vector Laboratories, Burlingame, CA). After cooling, the slides were incubated with 2% H2O2/60% methanol and blocked in Tris-buffered saline containing 2.5% horse serum. The anti-Gr-1 antibody (Catalogue No: 108402; BioLegend, San Diego, CA) was applied and incubated overnight. The detection was carried out using the NovaRED substrate of an immunohistochemistry kit (Vector Laboratories). Gr-1 positive neutrophils were counted per high power field (HPF; lung ×200, intestine ×100) using ImageJ software, and the number of neutrophils per HPF was determined by averaging the counts of three HPFs.
Measurement of myeloperoxidase (MPO)
Lung and intestinal tissues flash frozen in liquid nitrogen after collection were crushed over dry ice into a fine powder, and homogenized in 1 mL of potassium phosphate buffer containing 0.5% hexadecyltrimethylammonium bromide using high-frequency sonication. Two freeze-thaw cycles were performed, then samples were centrifuged, and the supernatant was collected. Protein concentration was determined by DC protein assay (Bio-Rad, Hercules, CA). Samples were added to a 96-well plate into phosphate buffer containing o-dianisidine hydrochloride and H2O2. Light absorbance was read at 460 nm over a period of 5 minutes. MPO activity, defined as 1 unit = change in absorbance per minute, is expressed as units per gram of protein.
Real-time quantitative polymerase chain reaction (qPCR)
Total RNA was extracted from frozen, crushed tissue using Trizol reagent (Invitrogen, Carlsbad, CA), and underwent reverse transcription to cDNA with reverse transcriptase (Applied Biosystems, Foster City, CA). The qPCR reaction was carried out in a 20 μL final volume containing 0.25 μL each of forward and reverse primers, 2 μL cDNA, 7.5 μL DEPC-treated water, and 10 μL Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). Amplification and analysis were conducted using a StepOnePlus real-time PCR machine (Applied Biosystems, Foster City, CA). Mouse β-actin mRNA levels were used as an internal control for normalization. Relative expression of mRNA was calculated using the 2–ΔΔCt method, and results are expressed as fold change in comparison with sham tissues. The primers used for qPCR are: MIP-2, 5’-CCCTGGTTCAGAAAATCATCCA-3’ (forward), and 5’-GCTCCTCCTTTCCAGGTCAGT-3’ (reverse); β-actin, 5’-CGTGAAAAGATGACCCAGATCA-3’ (forward), and 3’ TGGTACGACCAGAGGCATACAG-3’ (reverse).
Histological evaluation of tissue injury
Samples of lungs and small intestines were collected 4h after reperfusion and stored in 10% formaldehyde prior to being embedded in paraffin, cut into 5 μm sections and stained with hematoxylin and eosin (H&E). Sections of lung and intestinal tissues were evaluated under light microscopy in a blinded fashion to assess for injury using validated systems. Small intestines were evaluated per high power field (HPF; ×100) and scored according to a system previously created specifically for murine intestinal I/R injury. Scores range from 0–4 and consider multiple features of injury including loss of villus height, villus to crypt ratio, infiltration of lymphocytes, and degree of necrosis.29 Lungs were evaluated per HPF (×200) and scored according to the system established by the American Thoracic Society. Scores ranged from 0–1 and consist of weighted consideration of each of the following features: neutrophils in the alveolar space, neutrophils in the interstitial space, hyaline membranes, proteinaceous debris filling the airspaces, alveolar septal thickening.30 For all tissues, three HPF per sample were scored; scores for each sample are expressed as averages of the individual scores per HPF.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
Cellular apoptosis in lung and intestinal tissues was determined using a commercially available TUNEL assay kit (Roche Diagnostics, Indianapolis, IN). Paraffin-embedded sections of lung and small intestine were dewaxed in xylene and rehydrated in serial dilutions of ethanol and equilibrated in Tris buffered saline. The sections were digested with proteinase K for 20 minutes at room temperature. After washing, the tissue sections were incubated with an enzyme solution containing terminal deoxynucleotidyl transferase enzyme and fluorescence labeled nucleotides according to manufacturer’s instructions. Slides were examined under a fluorescent microscope (Nikon Elipse Ti-S, Melville, NY). TUNEL positive cells were counted per HPF (lung ×200; intestine ×100) using ImageJ software, and the number of apoptotic cells per HPF was determined by averaging the counts of 3 HPF.
Statistical analysis
Data are expressed in the figures as mean ± SEM. All data was assessed for normality using the Kolmogorov-Smirnov test. If the assumption of normality was violated, the Kruskal-Wallis test was used for comparison of multiple groups. If the assumption of normality was preserved, the Brown-Forsythe test was used to assess differences in variances between groups, and when such difference was detected, the Brown-Forsythe and Welch ANOVA was used. For groups with similar variances, one-way analysis of variance (ANOVA) using Student-Newman-Keuls (SNK) post hoc analysis was used for comparison of multiple groups. Survival rates were analyzed by the Kaplan-Meier estimator and compared using a log-rank hazard ratio. Significance was considered for p ≤ 0.05. Data analysis was performed using GraphPad Prism graphing and statistical software (GraphPad Software, LLC, San Diego, CA).
RESULTS
H151 inhibits STING activation in vitro and in vivo
eCIRP has been shown to mediate intestinal I/R injury, and to activate STING in macrophages.10,11,14,31 Thus, as an initial experiment to assess H151’s effects in the setting of eCIRP stimulation, RAW264.7 cells were pre-treated with H151 at various doses (0.25, 0.5, 1.0, 2.0 μM), 1 hour prior to rmCIRP exposure. At 24 hours after rmCIRP stimulation, there was a dose-dependent decrease in IFN-β in the culture supernatant of cells pre-treated with H151 compared to rmCIRP-treated cells alone(Figure 1A). In cells pretreated with 1 μM and 2 μM H151, there was a 57% and 74% reduction of IFN-β respectively(Figure 1A). Therefore, H151 inhibits eCIRP-induced activation of STING in vitro.
Figure 1. H151 inhibits STING activation in vitro and in vivo.
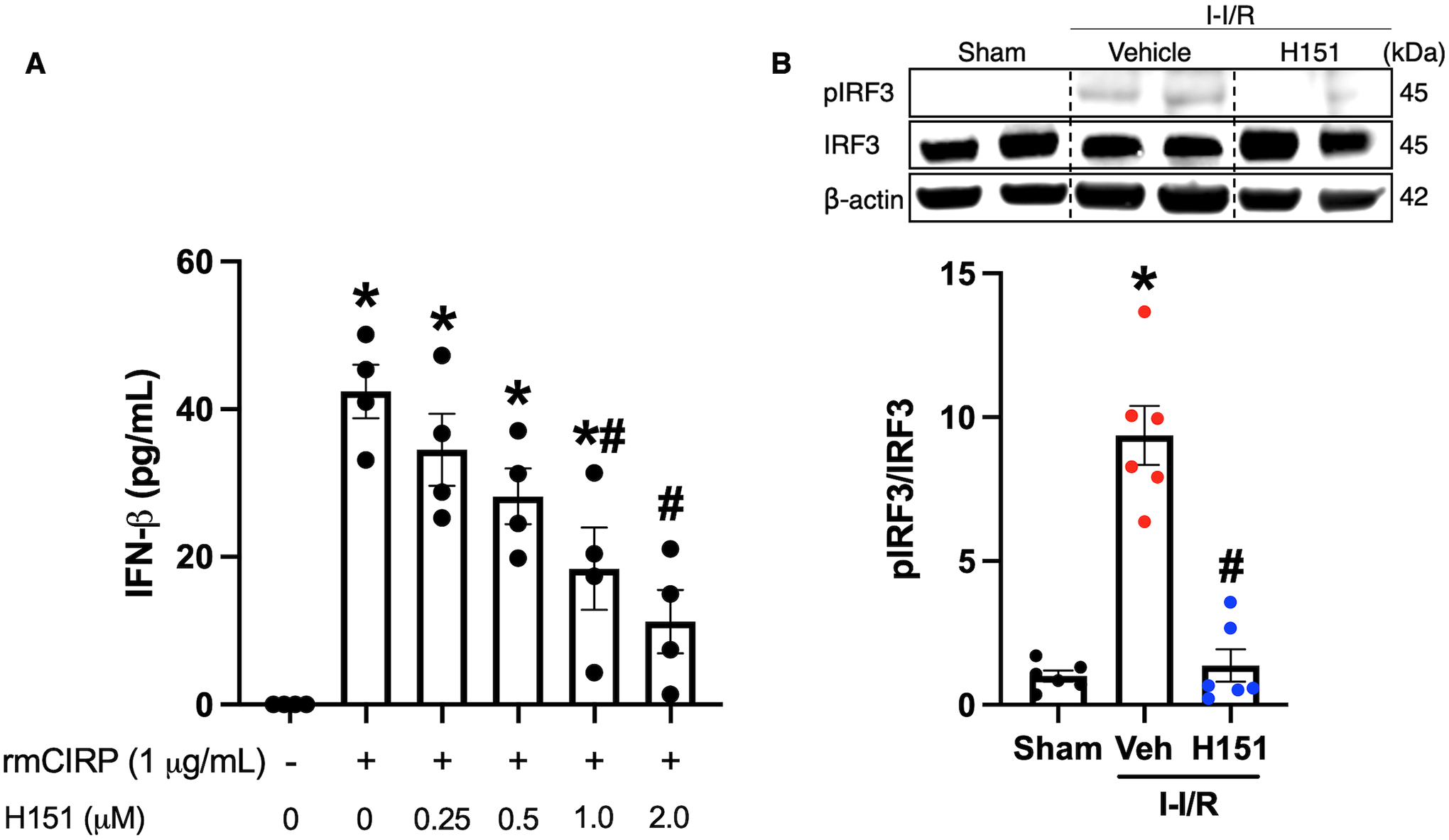
(A) RAW264.7 cells were pre-treated with H151 at various doses (0.25, 0.5, 1.0, 2.0 μM) one hour prior to stimulation with rmCIRP (1 μg/mL). Cells were then incubated for 24 hours, and culture supernatant was collected. IFN-β levels in the cell culture supernatant were measured using ELISA. Data are expressed as mean ± SEM and compared by ANOVA and SNK tests (*p < 0.05 vs control, #p < 0.05 vs rmCIRP alone; n = 4/group). (B) Intestines were collected from mice 4 hours after intestinal reperfusion and administration of intraperitoneal H151 or vehicle. Intestine tissue homogenates were assessed for pIRF3 and IRF3 proteins by Western blot. The blots were stripped and incubated with anti-β-actin antibodies to serve as the loading control. Representative Western blots for pIRF3, IRF3, and β-actin are shown. Each blot was quantified by densitometric analysis. pIRF3 expression in each sample was normalized to total IRF3 expression and the mean values of the sham group were standardized as 1 for comparison. Data are expressed as mean ± SEM and compared by the Kruskal-Wallis test (*p < 0.05 vs sham, #p < 0.05 vs vehicle, n = 6/group). I-I/R, intestinal-ischemia/reperfusion.
Based on these in vitro results, we next wanted to evaluate whether STING was inhibited by H151 in intestinal tissue after intestinal I/R. In order to do this, mice were subjected to 60 minutes intestinal ischemia, and received intraperitoneal H151 or vehicle at the time of reperfusion. Four hours after reperfusion, the small intestines were collected, and the levels of pIRF3 and total IRF3 were assessed by Western blotting. In intestinal tissue, pIRF3 levels were increased 9-fold after intestinal I/R compared to sham (p < 0.001), and were decreased by 85% (p < 0.001) in H151-treated mice compared to vehicle (Figure 1B). Therefore, H151 successfully inhibited STING in intestinal tissue after intestinal I/R.
Together, these data demonstrate that STING is activated in vitro after eCIRP stimulation and in vivo after intestinal I/R, and that H151 successfully inhibits STING’s downstream signaling in both conditions.
H151 treatment attenuates organ injury and pro-inflammatory cytokines after intestinal I/R
Serum LDH and AST were significantly increased in mice after intestinal I/R injury, by 9-fold (p < 0.001) and 3-fold (p < 0.001), respectively, in vehicle-mice as compared to sham mice (Figure 2A, B). H151 treatment was shown to be protective in intestinal I/R mice, and reductions were seen in LDH and AST by 57% (p < 0.05) and 30% (p < 0.05), respectively (Figure 2A, B). Serum IL-1β and IL-6 also significantly increased in mice after intestinal I/R injury as compared to sham, and treatment with H151 markedly decreased their levels by 56% (p < 0.05) and 42% (p = 0.089) respectively (Figure 2C, D). Thus, H151 treatment after intestinal I/R reduced systemic biomarkers of organ injury and inflammation.
Figure 2. Treatment with H151 attenuates organ injury markers and pro-inflammatory cytokines after intestinal I/R.
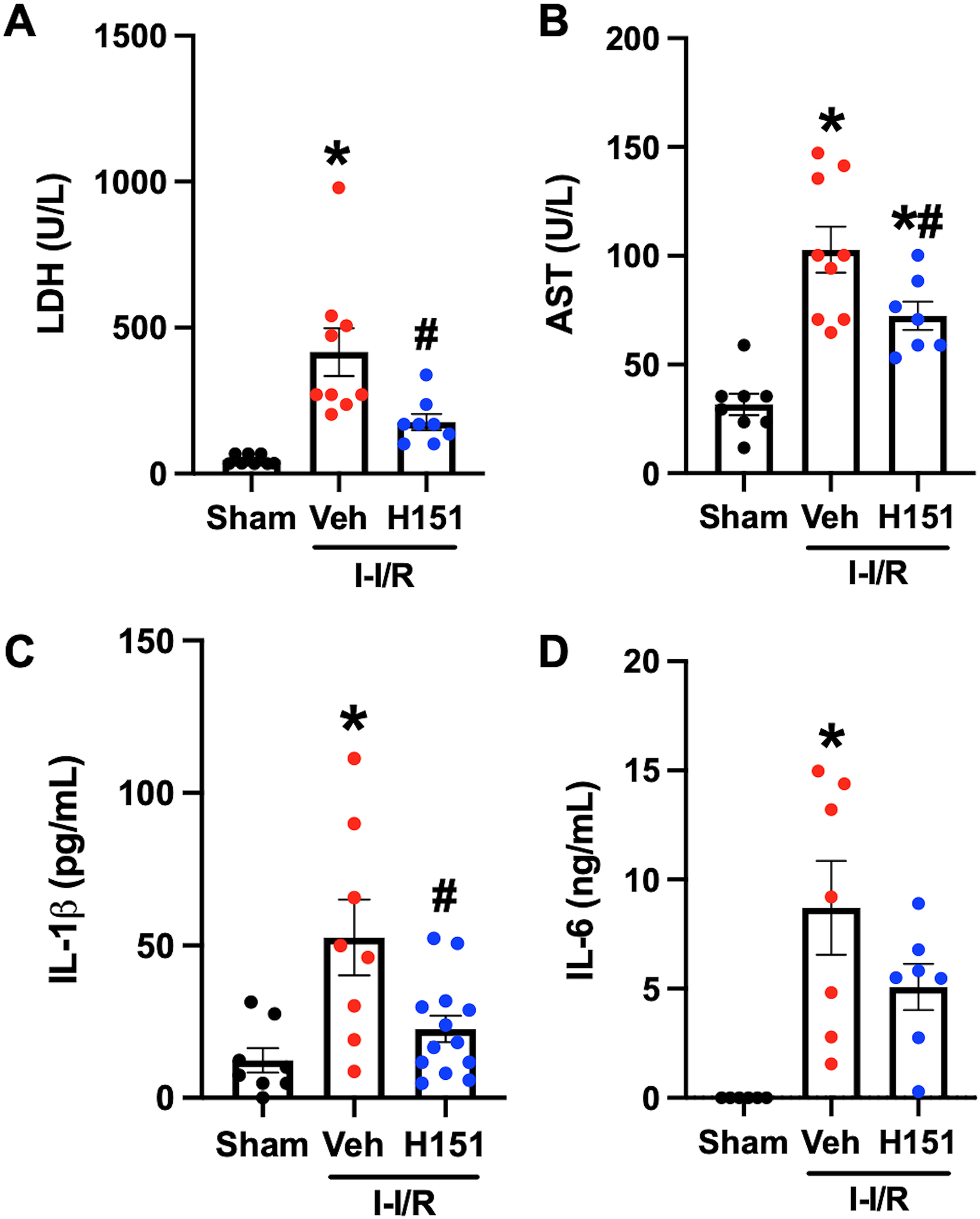
Intestinal ischemia was induced in mice by SMA occlusion, followed by reperfusion and resuscitation. Blood was collected from mice 4 hours after intestinal reperfusion and administration of intraperitoneal H151 or vehicle. Serum (A) LDH (n = 8–9/group) and (B) AST (n = 8–9/group) were measured using specific colorimetric enzyme assays. Serum (C) IL-1β (n = 8–13/group) and (D) IL-6 (n = 6–7/group) were measured using ELISA. Data are expressed as mean ± SEM and compared by (A) Kruskal-Wallis test, (B-C) ANOVA and SNK tests, and (D) Brown-Forsythe and Welch ANOVA (*p < 0.05 vs sham, #p < 0.05 vs vehicle). I-I/R, intestinal-ischemia/reperfusion.
H151 reduces intestinal neutrophil infiltration and chemokine expression after intestinal I/R
Immunohistochemical staining of intestinal tissue sections was performed using anti-Gr-1 antibodies to identify neutrophil infiltration in order to assess tissue inflammation. Presence of anti-Gr-1 neutrophils was quantified using ImageJ software. There was an 11-fold increase in Gr-1-neutrophil infiltration in mice subjected to intestinal I/R when compared to sham mice (p < 0.001), and a 75% decrease in Gr-1-neutrophils in H151-treated mice as compared to vehicle (p < 0.001; Figure 3A, B). To confirm neutrophil infiltration in these tissues, MPO activity was assessed. MPO activity was increased 12-fold in the intestines of mice after intestinal I/R, and reduced by 33% (p < 0.05) after H151 treatment (Figure 3C). To determine the possible mechanism of tissue neutrophil infiltration, chemokine expression in lung and intestinal tissue was assessed by qPCR measurement of macrophage inflammatory protein-2 (MIP-2) mRNA expression. MIP-2 mRNA expression was increased 180-fold in the intestines of mice after intestinal I/R, and was decreased after H151 treatment by 72% (p = 0.056) (Figure 3D). Therefore, H151 decreases chemokine expression, and resultant neutrophil infiltration in intestines after intestinal I/R.
Figure 3. H151 reduces intestinal neutrophil infiltration and chemokine expression after intestinal I/R.
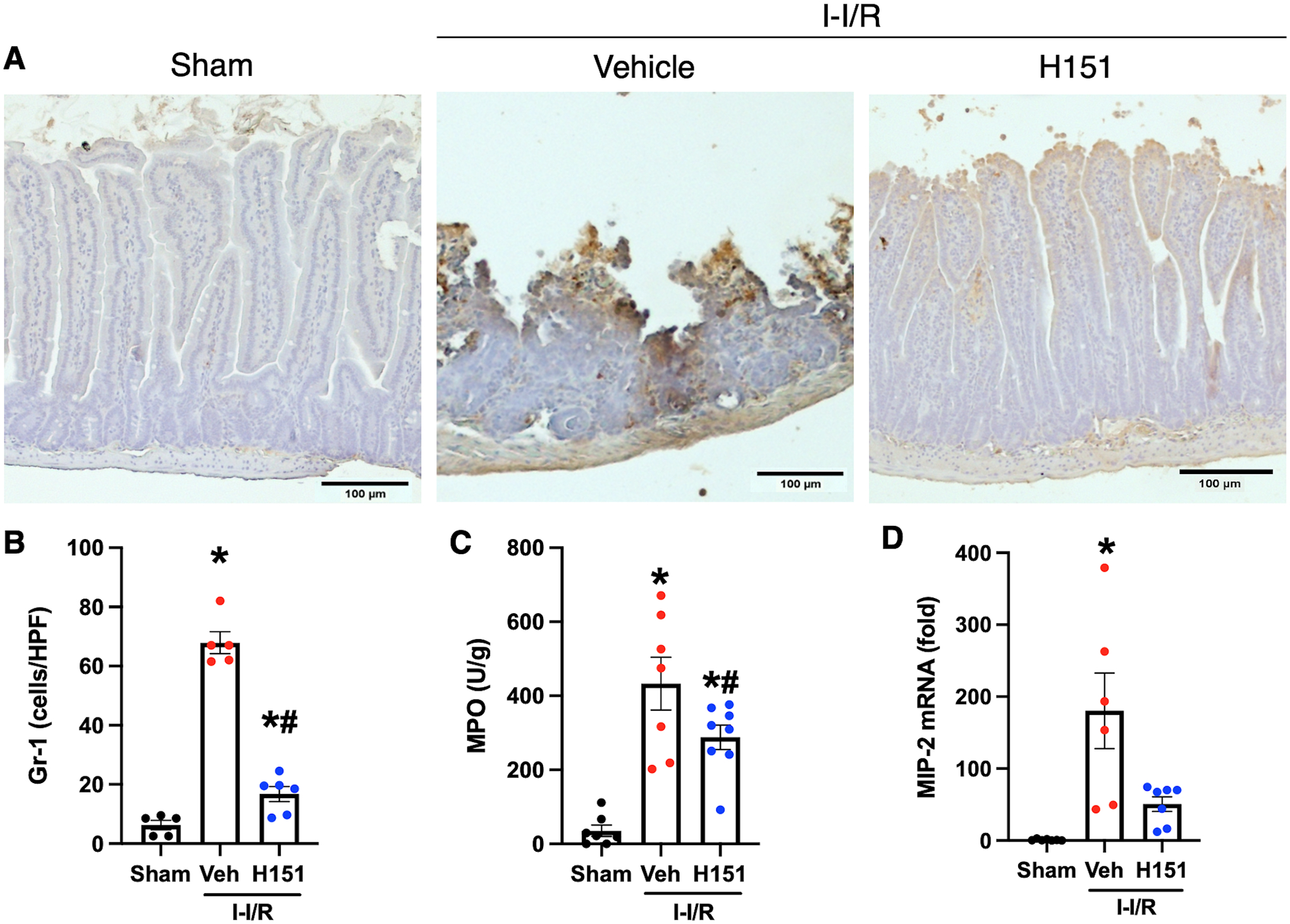
Four hours after reperfusion, intestines were collected. Immunohistochemical staining of Gr-1 neutrophils in intestinal tissue sections was performed. (A) Representative images of anti-Gr-1 immunohistochemical stains of intestinal tissue at are shown at 100x, (scale bar: 100μm). (B) Gr-1 neutrophil infiltration was quantified using ImageJ software (n = 5–6/group). (C) MPO activity in intestinal tissue was measured and is expressed in units per gram of protein (n = 7–8/group). (D) The mRNA levels of MIP-2 in intestines were measured by RT-qPCR, and normalized to β-actin (n = 6–7/group). Data are expressed as mean ± SEM and compared by (B, C) ANOVA and SNK tests, and (D) Brown-Forsythe and Welch ANOVA (*p < 0.05 vs sham, #p < 0.05 vs vehicle; n = 5–6/group). I-I/R, intestinal-ischemia/reperfusion.
H151 reduces intestinal tissue injury after I/R
Histological analysis of hematoxylin and eosin (H&E) stained sections of intestinal tissue was performed to assess structural damage after intestinal I/R and to evaluate the protective effect provided by H151 treatment. Tissue injury was scored according to a validated system, ranging from zero to four, taking into consideration loss of villus height, villus to crypt ratio, infiltration of lymphocytes, and degree of necrosis. Significantly greater tissue injury was observed in the intestines of vehicle-treated mice when compared to samples from sham mice, and tissue injury was significantly decreased in H151-treated mice as compared to the vehicle group. These histological changes were reflected by a 45% reduction in injury scores in H151-treated mice when compared to vehicle-treated mice (p < 0.05; Figure 4A, B).
Figure 4. H151 reduces intestinal injury and cell death after intestinal I/R.
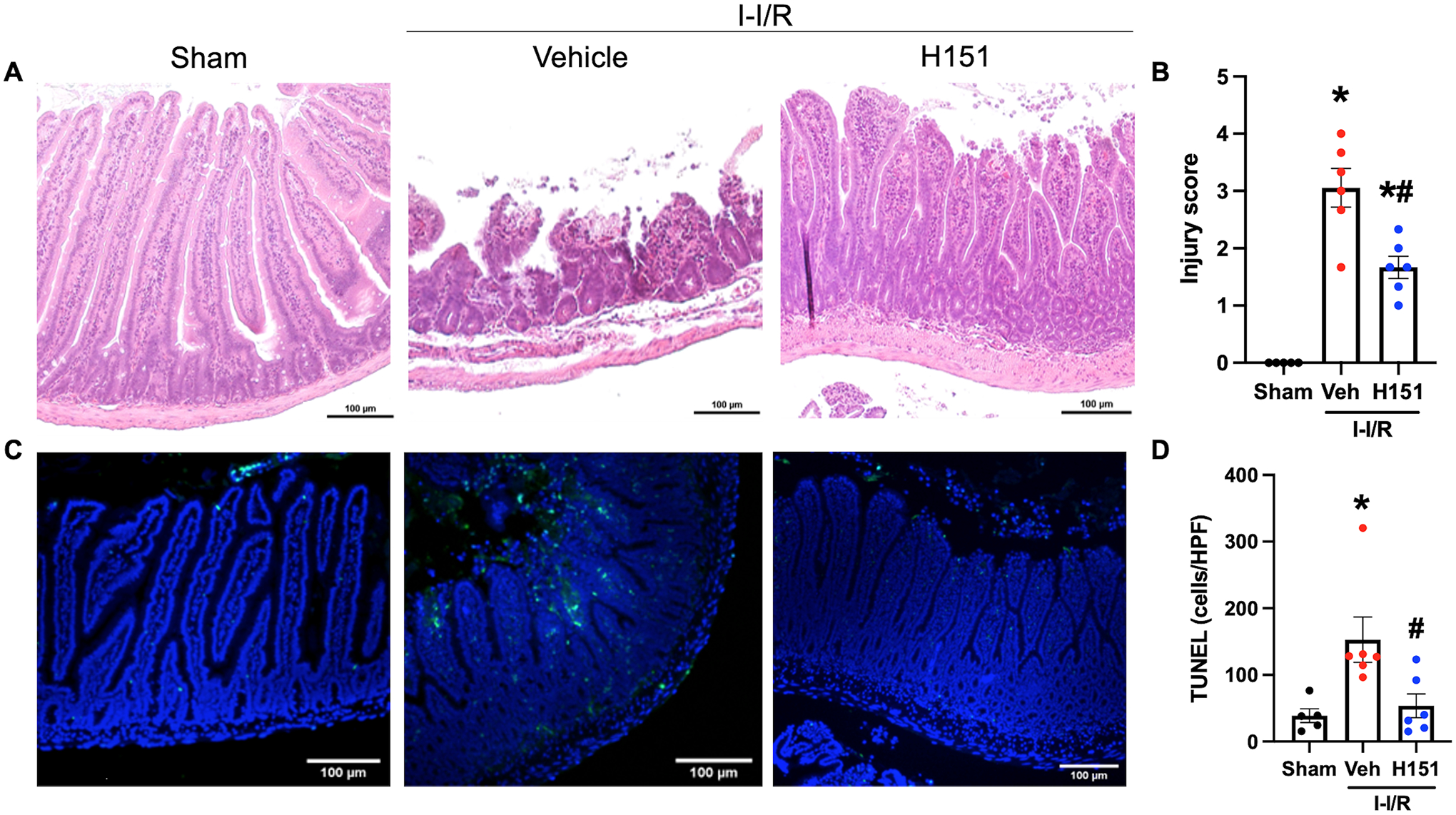
Organ injury was assessed by histological analysis using a validated scale of intestinal injury after I/R. (A) Representative images of H&E stained intestinal tissue at 100x (scale bar: 100μm). (B) Intestinal injury scores were calculated from zero to four with increasing score reflecting greater damage as assessed by blunting of the villi, decreased villi to crypt ratio, lymphocytic infiltration, and degree of necrosis (n = 5–6/group). TUNEL staining was performed to evaluate cell death. (C) Representative images of TUNEL stained sections at 100x (scale bar: 100μm). (D) TUNEL-positive cells were quantified using ImageJ software, and are expressed as cells/HPF (n = 5–6/group). Data are expressed as mean ± SEM and compared by (B) ANOVA and SNK tests, and (D) the Kruskal-Wallis test (*p < 0.05 vs sham, #p < 0.05 vs vehicle). I-I/R, intestinal-ischemia/reperfusion.
Apoptosis was assessed using TUNEL staining of intestinal sections, and TUNEL-positive nuclei were counted using ImageJ software. Intestinal sections from vehicle-treated mice demonstrated a 4-fold increase of TUNEL-positive cells (p < 0.05) when compared to sham mice, whereas intestinal sections from H151-treated mice demonstrated a 65% reduction in number of TUNEL-positive cells per HPF when compared to vehicle-treated mice (p < 0.05; Figure 4C, D). Notably, while intestinal sections from sham mice demonstrated physiologic apoptosis at the tips of villi, greater apoptosis was observed in the lamina propria after I/R injury (Figure 4C, D). As assessed by these three methods, H151 treatment significantly reduces the local tissue injury that results from intestinal I/R.
H151 reduces neutrophil infiltration and chemokine expression in lungs after intestinal I/R
In order to assess acute lung inflammation Gr-1 neutrophil infiltration, MPO activity, and MIP-2 mRNA expression were also assessed in the lungs as described above. A 19-fold increase in Gr-1-neutrophil infiltration was observed in the lungs of mice subjected to intestinal I/R when compared to sham mice (p < 0.001), and a 72% decrease in Gr-1-neutrophils was observed in H151-treated mice as compared to vehicle (p < 0.001; Figure 5A, B). MPO activity was increased 6-fold in the lungs of mice after intestinal I/R, and decreased 38% after H151 treatment (Figure 5C). MIP-2 mRNA expression was increased 15-fold in the lungs of mice subjected to intestinal I/R, and was decreased in the lungs by 53% after H151 treatment (p = 0.09) (Figure 5D). Together these data demonstrate that H151 decreases chemokine expression, neutrophil presence and activity in both intestines and lungs after intestinal I/R.
Figure 5. H151 reduces neutrophil infiltration and chemokine expression in lungs after intestinal I/R.
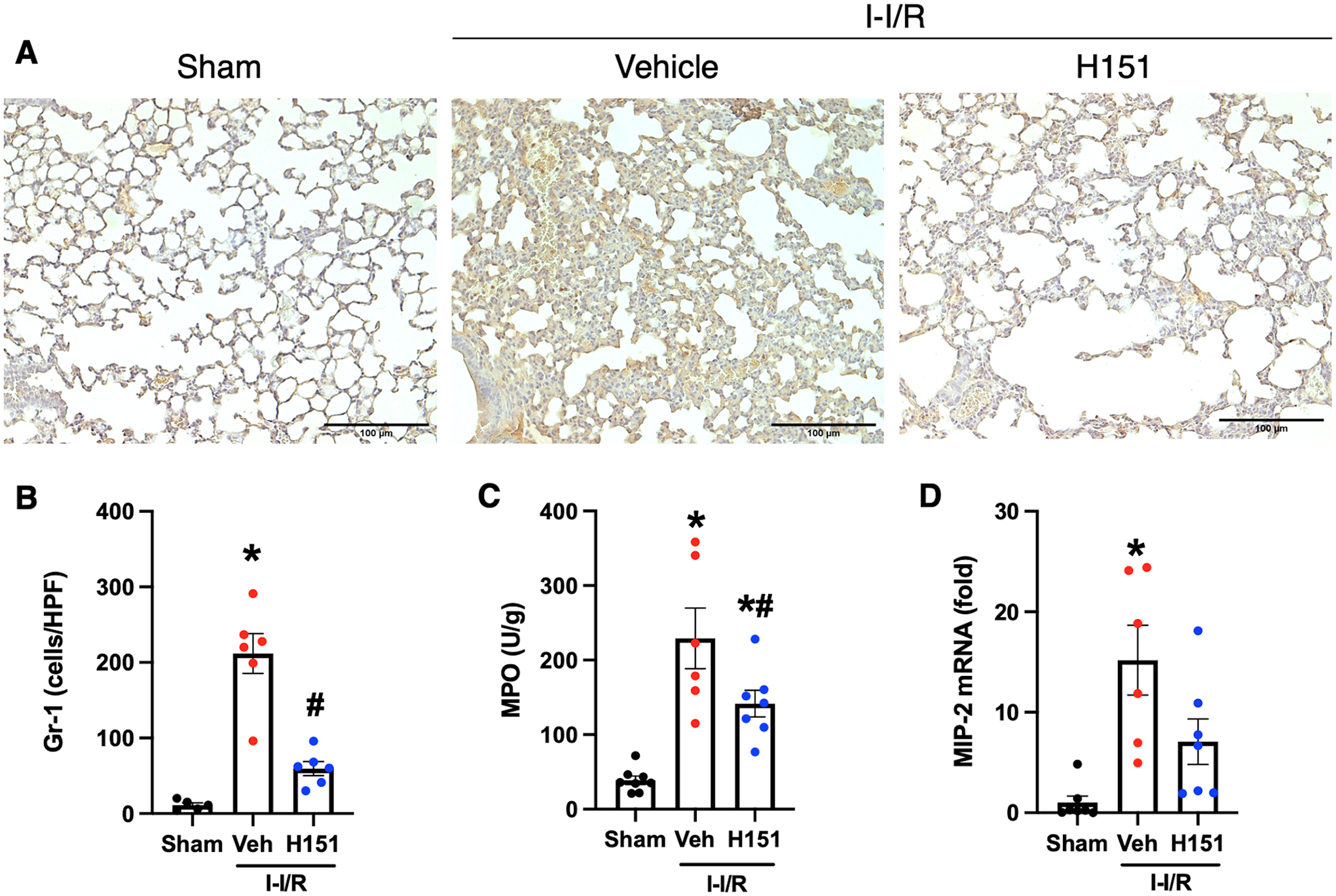
Four hours after reperfusion, lungs were collected. Immunohistochemical staining of Gr-1 neutrophils in lung tissue sections was performed. (A) Representative images of anti-Gr-1 immunohistochemical stains of lung tissue at are shown at 200x, (scale bar: 100μm). (B) Gr-1 neutrophil infiltration was quantified using ImageJ software (n = 5–6/group). (C) MPO activity in lung tissue was measured and is expressed in units per gram of protein (n = 7–8/group). (D) The mRNA levels of MIP-2 in lungs were measured by RT-qPCR, and normalized to β-actin (n = 6–7/group). Data are expressed as mean ± SEM and compared by ANOVA and SNK tests (*p < 0.05 vs sham, #p < 0.05 vs vehicle). I-I/R, intestinal-ischemia/reperfusion.
H151 reduces acute lung injury after intestinal I/R
To assess extraintestinal organ injury after intestinal I/R and H151 treatment, histological analysis of H&E sections of lung tissue was performed. ALI was evaluated according to a validated scoring system, ranging from zero to one, which considers neutrophilic infiltrate in the alveolar and interstitial spaces, presence of hyaline membranes, proteinaceous debris filling the airspaces, and alveolar septal thickening. Significantly greater ALI was observed in mice subjected to intestinal I/R as compared to sham mice, and ALI was significantly decreased in mice treated with H151 as compared to vehicle. This difference is reflected by a 65% reduction in injury scores in H151-treated mice when compared to vehicle-treated mice (p < 0.001; Figure 6A, B).
Figure 6. H151 treatment reduces acute lung injury and apoptosis after intestinal I/R.
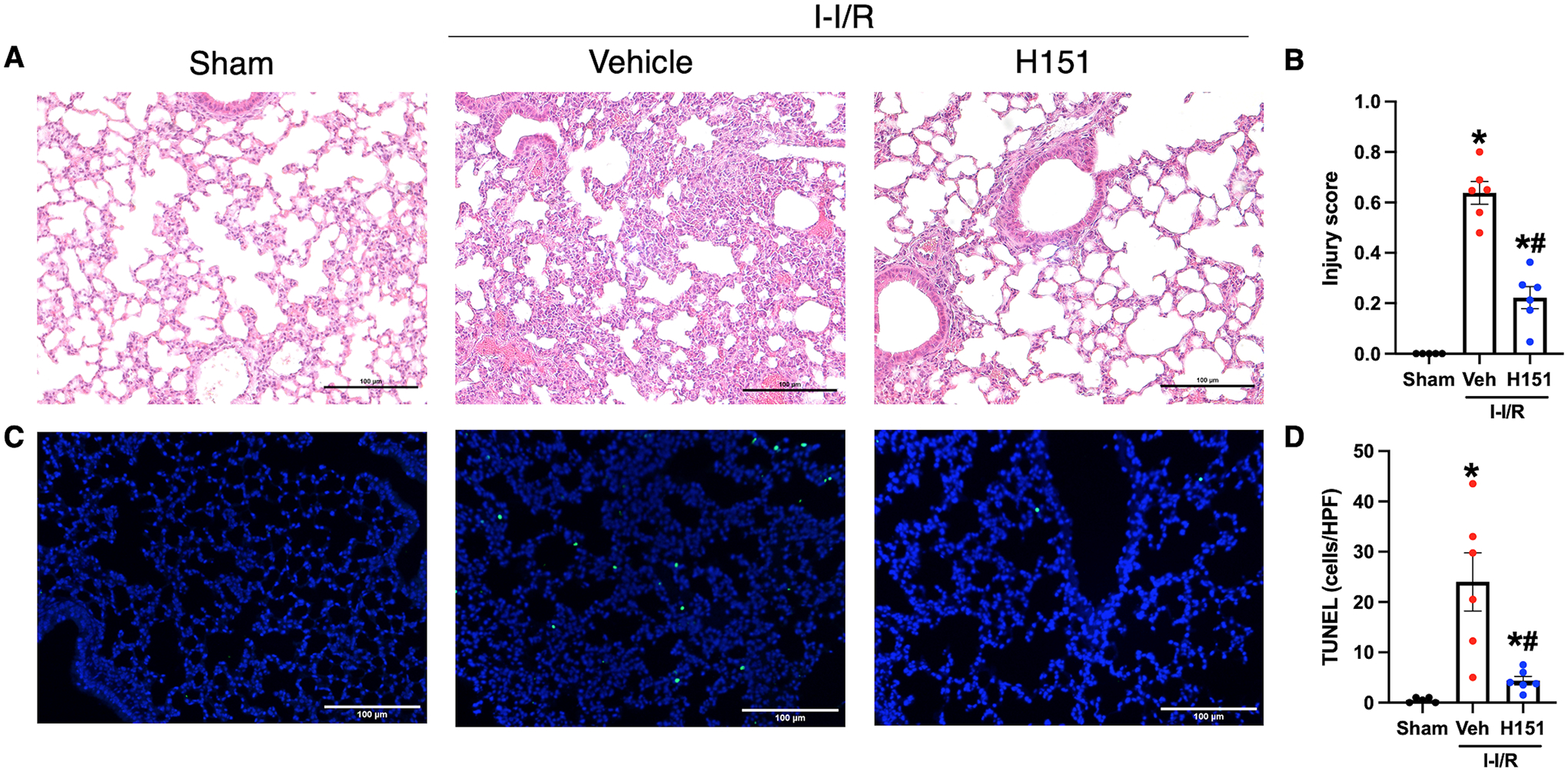
Histological analysis was performed on H&E stained sections of lung tissue, and acute lung injury was assessed using a validated grading scale. (A) Representative images of H&E stained lung tissue at 200x (scale bar: 100μm). (B) Acute lung injury score calculated from zero to one with greater scores reflecting greater injury, taking into consideration proteinaceous debris in the airspaces, thickening of alveolar septa, presence of hyaline membranes, and lymphocytic infiltration (n = 5–6/group). TUNEL staining of lung tissue sections was performed to evaluate cell death. (C) Representative images of TUNEL stained sections at 100x. Scale bar: 100μm. (D) TUNEL-positive cells were quantified using ImageJ software and are expressed as cells/HPF (n = 5–6/group). Data are expressed as mean ± SEM and compared by (B) ANOVA and SNK tests, and (D) Brown-Forsythe and Welch ANOVA (*p < 0.05 vs sham, #p < 0.05 vs vehicle). I-I/R, intestinal-ischemia/reperfusion.
Lung tissue apoptosis was also assessed using TUNEL staining. Lungs from vehicle-treated mice demonstrated a 48-fold increase in TUNEL-positive cells as compared to lungs from sham mice (p < 0.01), and lung sections from H151-treated mice demonstrated an 82% reduction in number of TUNEL-positive cells per HPF when compared to vehicle-treated mice (p < 0.05; Figure 6C, D). These results indicate that H151 significantly ameliorates intestinal I/R-induced ALI.
H151 improves survival after intestinal I/R injury
As the above data demonstrates a protective effect of H151 treatment, we performed a survival study to evaluate the potential survival benefit of H151. Analogous to the earlier experiments, mice were randomly assigned to receive intraperitoneal H151 or vehicle at the time of reperfusion after 60 minutes of SMA occlusion. All mice were then monitored for survival for 24 hours. H151 increased the 24-hour survival after intestinal I/R from 41% to 81% (p < 0.05; Figure 7). Thus, H151 is beneficial for the survival outcomes during intestinal I/R injury.
Figure 7. H151 improves survival after intestinal I/R.
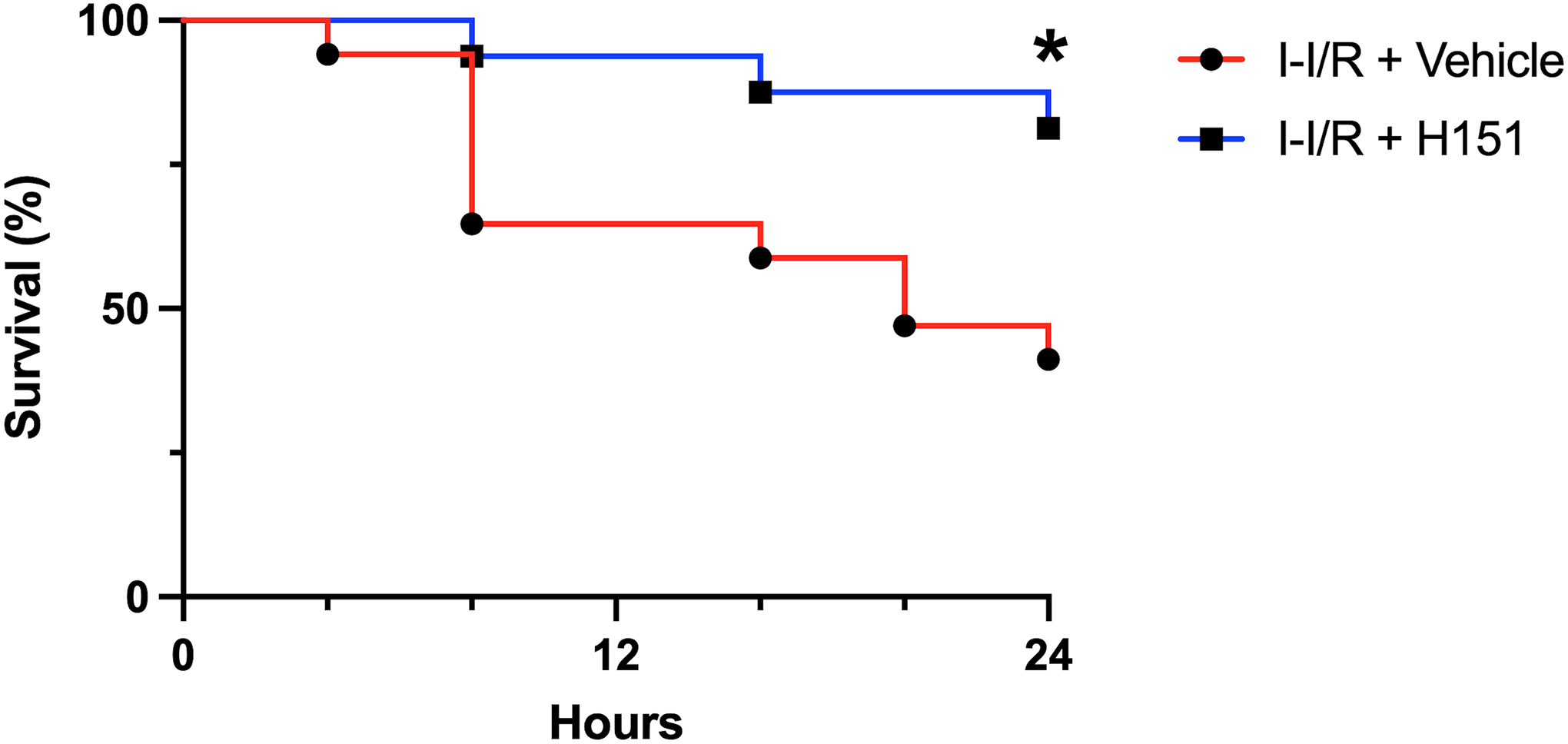
Intestinal I/R was induced by occluding the SMA for 60 minutes. At the time of reperfusion, mice received intraperitoneal injection of either H151 (10 mg/kg BW) or vehicle. Kaplan-Meier curves demonstrate survival rates over a 24-hour period of monitoring following intestinal I/R for vehicle- and H151-treated mice. Survival rates were analyzed by the log-rank test (n = 16–17 per group; *p < 0.05). I-I/R, intestinal-ischemia/reperfusion.
DISCUSSION
The primary treatment of intestinal I/R is directed at correcting the underlying etiology, restoring intestinal blood flow, resecting unsalvageable bowel, and supportive measures.1,32 However, there are currently no available clinical therapeutics that mitigate the ensuing intestinal damage and systemic inflammation that occurs after reperfusion, despite the evidence that subsequent extraintestinal organ dysfunction, most commonly ALI, contributes significantly to the morbidity and mortality of intestinal I/R. Furthermore, as the inflammatory response is triggered both by widespread intestinal cell damage as well as translocation of bacterial components from the intestinal lumen, multiple complex pathways are at play.7 Because STING is activated by both PAMPs and DAMPs, and is involved in initiating the immune response to bacterial infections in addition to cellular damage, it is a promising therapeutic target in intestinal I/R that has yet to be studied thoroughly.16,19 The purpose of our study was to determine whether inhibition of STING with H151 could provide therapeutic benefit in intestinal I/R.
First, we demonstrated that H151 inhibits eCIRP-induced activation of STING in macrophages in a dose-response fashion. Because eCIRP is a potent DAMP that mediates inflammation and injury in intestinal I/R and has been shown to activate STING signaling, this result suggested that H151 may be protective in this model in vivo.10,11,14,31 Second, we demonstrated STING pathway activation in intestinal tissues after intestinal I/R as demonstrated by increased levels of pIRF3, consistent with prior studies.19,20 Next, we demonstrated that inhibiting STING with H151 results in decreased intestinal and lung inflammation and injury, and decreased systemic inflammation after intestinal I/R. Notably, we also demonstrated improved survival after H151 administration. Taken together, inhibiting STING activation reflects a novel therapeutic avenue to safeguard patients from intestinal I/R injury.
STING has been known to modulate the immune response to viruses and bacteria, contribute to the pathogenesis of autoinflammatory conditions, and drive tumorigenesis in cancers associated with inflammation.18 The role of STING in acute inflammation and critical illness is less known, and is a highly active area of ongoing research. Recently, our lab demonstrated that STING exacerbates the effects of eCIRP, a DAMP known to amplify inflammation in critical illnesses including sepsis, ischemia-reperfusion injury, and hemorrhagic shock.14 STING activation has also been found to play a critical role in the development of sepsis-associated acute lung injury, and recently, in ischemia-reperfusion injury.33,34 Specifically, Zhang et al. showed that STING activation by mtDNA promotes enterocyte injury by inducing necroptosis, and Wu et al. showed that STING mediates intestinal I/R injury and associated ALI through induction of lipid peroxidation.19,20 However, our current study is the first to attempt to inhibit this pathway therapeutically in intestinal I/R and associated ALI.
In addition to demonstrating increased levels of pIRF3 in the intestines after intestinal I/R, consistent with previous literature, we were able to demonstrate inhibition of STING signaling in vitro and in vivo using H151, a covalent STING antagonist.14 H151 was initially described to effectively inhibit STING signaling and subsequent inflammation when administered intraperitoneally as a pre-treatment prior to administration of a known STING agonist. H151 binds to STING covalently at a cysteine residue within the transmembrane domain spanning the membrane of the endoplasmic reticulum, inhibiting the palmitoylation and clustering of STING which is necessary for downstream signaling to occur.21 Since its initial description, H151 has shown to be effective at reducing STING’s pro-inflammatory signaling in models of lupus, psoriasis, drug-induced acute kidney injury, and post-infarction cardiac fibrosis.22–25 This is the first study to describe H151’s potential therapeutic benefit in a model of intestinal I/R. In the intestines of mice treated with H151 after intestinal I/R, pIRF3 levels were decreased when compared to vehicle-treated mice, indicating H151’s successful inhibition of STING in the small intestine. Lung and intestinal MIP-2 expression and subsequent neutrophilic infiltration were also reduced after H151 treatment, resulting in less severe tissue injury as measured by histologic assessment and cell death. The protective effects of H151 were evident systemically, as H151-treated mice demonstrated reduced levels of organ injury markers LDH and AST, and reduced levels of cytokines IL-1β and IL-6 when compared to vehicle-treated mice. Finally, treatment with H151 conferred a survival benefit at 24-hours after intestinal I/R. These results highlight the potential of H151 in the treatment of ischemia-reperfusion injury, and associated ALI.
ALI is the most common complication after intestinal I/R, and though we demonstrated that H151 reduces intestinal I/R-induced ALI, the specific mechanisms underlying this therapeutic benefit are unclear. Many pathways have been implicated in the development of ALI after intestinal I/R, yet the specific role of STING activation, and therapeutic inhibition of STING, are unknown.35,36 Though primary reduction of intestinal injury by inhibition of STING with H151 may account for the amelioration of ALI observed in this study, we hypothesize that H151 also reduces the systemic inflammatory response to intestinal injury, thus reducing secondary organ injury. Previously, Wu et al. demonstrated that STING mediates vascular endothelial cell-mediated immune cell chemotaxis and adhesion in the lungs, and that inhibition of STING in a model of LPS-induced ALI is protective through this mechanism, in addition to reducing expression of pro-inflammatory chemokines and cytokines.15 This mechanism, and others, may contribute to H151’s reduction of intestinal I/R-associated ALI, and further study is necessary to test the hypothesis that H151 reduces secondary organ injury through systemic mechanisms in addition to reducing primary intestinal injury.
Furthermore, it remains unknown which cell types predominantly mediate the therapeutic benefit of H151, and which mechanisms downstream of STING account for the protection demonstrated here. Since STING is ubiquitously expressed in immune (macrophages) and non-immune (epithelial, endothelial) cells, it is reasonable that activation of STING across cell types contributes to inflammation and tissue damage.13,18 In this study, we established that H151 inhibits eCIRP-induced activation of STING in macrophages, as evidenced by a dose-dependent decrease in IFN-β after H151 treatment. The most widely studied mode of action of STING-mediated pathway for inflammation and tissue injury is through the release of type I IFNs (IFNα and IFNβ).13 In line with this fact, our previous study revealed that blocking the receptor of type I IFN, i.e., IFN-α receptor (IFNAR) by anti-IFNAR antibodies protected mice from hemorrhagic shock-induced inflammation and acute lung injury.37 In our intestinal I/R model, we assessed the levels of expression of the transcription factor, pIRF3, which governs type I IFN expression, and found that H151 treatment significantly decreased pIRF3 expression, indicating one of the mechanisms of action of H151 to ameliorate inflammation and ALI in intestinal I/R injury. However, because multiple mechanisms are mediated by STING in the pathophysiology of intestinal I/R, such as necroptosis, lipid peroxidation, and production of reactive oxygen species19,20, we hypothesize that multiple deleterious outcomes driven by the STING pathway underly the inflammatory response and tissue injury observed in this model, which illuminates the benefit of this therapeutic strategy.
This study is not without limitations. First, only male mice were used in animal experiments, based on evidence that mice exhibit sexually dimorphic responses to intestinal I/R.26,27 In humans, females are also protected against intestinal I/R injury in comparison to males.28 Study of intestinal I/R across the sexes is warranted to identify potential differences in STING activation between males and females, and to replicate the therapeutic benefit of H151 in both sexes. Additionally, though we demonstrated a survival benefit at 24 hours, we did not study this at later time points. The first 24 hours after diagnosis and treatment of intestinal I/R are critical, so we believe this acute survival benefit to be valuable. However, long term outcomes after H151 treatment in this setting must be assessed. Lastly, the absorption, distribution, and safety of H151 remain unknown. H151 is solubilized in 10% Tween-80, a weak detergent which poses the risk of tissue damage, which was used as a vehicle control in this study. The findings from this vehicle control group are consistent with our lab’s previous investigations in intestinal I/R when isotonic crystalloid is administered intraperitoneally as a vehicle control.10–12 Therefore, we are confident that the use of 10% Tween-80 as a vehicle control did not result in a more severe model when compared to historical data from our lab. At this time, there are no known off-target effects of H151 solubilized in 10% Tween-80, however this has yet to be studied thoroughly. H151 is a highly specific antagonist of STING, and binds to a single cysteine residue thereby preventing the palmitoylation required for STING’s downstream signaling. Haag et al. showed that a chemically similar precursor of H151, C-178, precisely acts through this mechanism, but does not affect the palmitoylation of other proteins such as calnexin and transferrin receptor. Furthermore, no off-target effects were demonstrated when healthy mice received C-178 twice-daily for two weeks.21 However, these studies must be replicated with H151, which is effective against human and murine STING, whereas C-178 is only effective against murine STING. Additionally, STING has been shown to have variable functions in different conditions related to acute and chronic inflammation, as well as tumorigenesis. Though H151 acts in a highly specific fashion, the potential adverse effects of inhibiting STING must also be explored.
In conclusion, we have demonstrated that STING is activated in vitro by eCIRP and in vivo after intestinal I/R. We additionally showed that H151, a small molecule inhibitor of STING, successfully attenuates intestinal and extra-intestinal organ damage and inflammation after intestinal I/R. Treatment with H151 also improved the survival in mice after intestinal I/R. Future work should focus on the mechanisms by which STING mediates intestinal I/R-induced ALI, as ALI is the most common complication after intestinal I/R and contributes significantly to morbidity and mortality. Besides providing value in the broad areas of inflammatory diseases, our findings may lead to a substantial therapeutic impact on a particular disease condition whose data is seemingly limited. These findings will implicate future study on higher primate models or in human intestinal cells to implement H151 as a potential therapeutic candidate in human intestinal I/R injuries. Additionally, studies of the properties of H151 are necessary prior to further translational studies in larger animals or humans, and eventual drug development. Nonetheless, STING inhibition with H151 shows promise as a therapeutic strategy in the treatment of intestinal I/R injury.
Acknowledgements
We thank Bridgette Reilly and Fangming Zhang of the Center for Immunology and Inflammation at the Feinstein Institutes for Medical Research for providing technical support.
Conflicts of Interest and Source of Funding:
This study was supported by the National Institutes of Health (NIH) grants R01HL076179 (P.W.), R35GM118337 (P.W.), and R01GM129633 (M.A.). The authors declared that they have no competing interests.
References
- 1.Clair DG, Beach JM. Mesenteric Ischemia. N Engl J Med. 374(10):959–968, 2016. [DOI] [PubMed] [Google Scholar]
- 2.Young CM, Kingma SDK, Neu J. Ischemia-reperfusion and neonatal intestinal injury. J Pediatr. 158(2 Suppl):e25–28, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Lenaerts K, Ceulemans LJ, Hundscheid IHR, Grootjans J, Dejong CHC, Olde Damink SWM. New insights in intestinal ischemia-reperfusion injury: implications for intestinal transplantation. Curr Opin Organ Transplant. 18(3):298–303, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Denning NL, Aziz M, Gurien SD, Wang P. DAMPs and NETs in Sepsis. Front Immunol. 10:2536, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aziz M, Jacob A, Yang WL, Matsuda A, Wang P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J Leukoc Biol. 93(3):329–342, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grootjans J, Hodin CM, de Haan J, Derikx JPM, Rouschop KMA, Verheyen FK, van Dam RM, Dejong CHC, Burrman WA, Lenaerts K. Level of Activation of the Unfolded Protein Response Correlates With Paneth Cell Apoptosis in Human Small Intestine Exposed to Ischemia/Reperfusion. Gastroenterology. 140(2):529–539.e3, 2011. [DOI] [PubMed] [Google Scholar]
- 7.Grootjans J, Lenaerts K, Derikx JPM, Matthijsen RA, de Bruine AP, van Bijnen AA, van Dam RM, Dejong CHC, Burrman WA. Human intestinal ischemia-reperfusion-induced inflammation characterized: experiences from a new translational model. Am J Pathol. 176(5):2283–2291, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhan Y, Ling Y, Deng Q, Qiu Y, Shen J, Lai H, Chen Z, Huang C, Liang L, Li X, et al. HMGB1-Mediated Neutrophil Extracellular Trap Formation Exacerbates Intestinal Ischemia/Reperfusion-Induced Acute Lung Injury. The Journal of Immunology. 208(4):968–978, 2022. [DOI] [PubMed] [Google Scholar]
- 9.Moraes LB, Murakami AHF, Fontes B, Poggetti RS, van Rooijen N, Younes RN, Heimbecker AM, Birolini D. Gut ischemia/reperfusion induced acute lung injury is an alveolar macrophage dependent event. Journal of Trauma - Injury, Infection and Critical Care. 64(5):1196–1200, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Denning NL, Aziz M, Ochani M, Prince JM, Wang P. Inhibition of TREM-1 with an eCIRP-derived Peptide Protects Mice from Intestinal Ischemia-reperfusion Injury. Surgery. 168(3):478–485, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGinn JT, Aziz M, Zhang F, Yang WL, Nicastro JM, Coppa GF, Wang P. Cold-inducible RNA-binding protein-derived peptide C23 attenuates inflammation and tissue injury in a murine model of intestinal ischemia-reperfusion. Surgery. 164(6):1191–1197, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Royster W, Ochani M, Aziz M, Wang P. Therapeutic Potential of B-1a Cells in Intestinal Ischemia-Reperfusion Injury. Journal of Surgical Research. 268:326–336, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Couillin I, Riteau N. STING Signaling and Sterile Inflammation. Front Immunol. 12:753789, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen K, Cagliani J, Aziz M, Tan C, Brenner M, Wang P. Extracellular CIRP activates STING to exacerbate hemorrhagic shock. JCI Insight. 6(14):e143715, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu B, Xu MM, Fan C, Feng CL, Lu QK, Lu HM, Xiang CG, Bai F, Wang HY, Wu YW, et al. STING inhibitor ameliorates LPS-induced ALI by preventing vascular endothelial cells-mediated immune cells chemotaxis and adhesion. Acta Pharmacol Sin. Published online December 14, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao S, Luo J, Kadier T, Ding K, Chen R, Meng Q. Mitochondrial DNA Release Contributes to Intestinal Ischemia/Reperfusion Injury. Front Pharmacol. 13:854994, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu Q, Zhou Q, Wu J, Wu X, Ren J. The Role of Mitochondrial DNA in the Development of Ischemia Reperfusion Injury. Shock. 51(1):52–59, 2019. [DOI] [PubMed] [Google Scholar]
- 18.Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 15(12):760–770, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Wu J, Liu Q, Li X, Li S, Chen J, Hong Z, Wu X, Zhao Y, Ren J. mtDNA-STING pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis. 11(12):1050, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu J, Liu Q, Zhang X, Wu X, Zhao Y, Ren J. STING-dependent induction of lipid peroxidation mediates intestinal ischemia-reperfusion injury. Free Radic Biol Med. 163:135–140, 2021. [DOI] [PubMed] [Google Scholar]
- 21.Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G, Behrendt R, et al. Targeting STING with covalent small-molecule inhibitors. Nature. 559(7713):269–273, 2018. [DOI] [PubMed] [Google Scholar]
- 22.Hu S, Gao Y, Gao R, Wang Y, Qu Y, Yang J, Wei X, Zhang F, Ge J. The selective STING inhibitor H-151 preserves myocardial function and ameliorates cardiac fibrosis in murine myocardial infarction. Int Immunopharmacol. 107:108658, 2022. [DOI] [PubMed] [Google Scholar]
- 23.Pan Y, You Y, Sun L, Sui Q, Liu L, Yuan H, Chen C, Liu J, Wen X, Dai L, et al. The STING antagonist H-151 ameliorates psoriasis via suppression of STING/NF-κB-mediated inflammation. Br J Pharmacol. 178(24):4907–4922, 2021. [DOI] [PubMed] [Google Scholar]
- 24.Tumurkhuu G, Chen S, Montano EN, Laguna DE, De Los Santos G, Yu JM, Lane M, Yamashita M, Markman JL, Blanco LP, et al. Oxidative DNA Damage Accelerates Skin Inflammation in Pristane-Induced Lupus Model. Front Immunol. 11:554725, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong W, Lu L, Zhou Y, Liu J, Ma H, Fu L, Huang S, Zhang Y, Zhang A, Jia Z. The novel STING antagonist H151 ameliorates cisplatin-induced acute kidney injury and mitochondrial dysfunction. Am J Physiol Renal Physiol. 320(4):F608–F616, 2021. [DOI] [PubMed] [Google Scholar]
- 26.Wu M, Rowe JM, Fleming SD. Complement Initiation Varies by Sex in Intestinal Ischemia Reperfusion Injury. Front Immunol. 12:649882, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu M, Rowe JM, Fleming SD. Eicosanoid production varies by sex in mesenteric ischemia reperfusion injury. Clin Immunol. 220:108596, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hundscheid IHR, Schellekens DHSM, Grootjans J, et al. Females Are More Resistant to Ischemia-Reperfusion-induced Intestinal Injury Than Males: A Human Study. Ann Surg. 272(6):1070–1079, 2020. [DOI] [PubMed] [Google Scholar]
- 29.Stallion A, Kou TD, Latifi SQ, Miller KA, Dahms BB, Dudgeon DL, Levine AD. Ischemia/reperfusion: a clinically relevant model of intestinal injury yielding systemic inflammation. J Pediatr Surg. 40(3):470–477, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM; Acute Lung Injury in Animals Study Group. An Official American Thoracic Society Workshop Report: Features and Measurements of Experimental Acute Lung Injury in Animals. Am J Respir Cell Mol Biol. 44(5):725–738, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cen C, McGinn J, Aziz M, Yang WL, Cagliani J, Nicastro JM, Coppa GF, Wang P. Deficiency in cold-inducible RNA-binding protein attenuates acute respiratory distress syndrome induced by intestinal ischemia-reperfusion. Surgery. 162(4):917–927, 2017. [DOI] [PubMed] [Google Scholar]
- 32.Oldenburg WA, Lau LL, Rodenberg TJ, Edmonds HJ, Burger CD. Acute Mesenteric Ischemia: A Clinical Review. Archives of Internal Medicine. 164(10):1054–1062, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Ning L, Wei W, Wenyang J, Rui X, Qing G. Cytosolic DNA‐STING‐NLRP3 axis is involved in murine acute lung injury induced by lipopolysaccharide. Clin Transl Med. 10(7):e228, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Q, Wu J, Zhang X, Li X, Wu X, Zhao Y, Ren J. Circulating mitochondrial DNA-triggered autophagy dysfunction via STING underlies sepsis-related acute lung injury. Cell Death Dis. 12(7):673, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mura M, Andrade CF, Han B, Seth R, Zhang Y, Bai XH, Waddell TK, Hwang D, Keshavjee S, Liu M. Intestinal Ischemia-Reperfusion-Induced Acute Lung Injury and Oncotic Cell Death in Multiple Organs. Shock. 28(2):227–238, 2007. [DOI] [PubMed] [Google Scholar]
- 36.Li G, Zhang Y, Fan Z. Cellular Signal Transduction Pathways Involved in Acute Lung Injury Induced by Intestinal Ischemia-Reperfusion. Oxid Med Cell Longev. 9985701, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cagliani J, Yang WL, McGinn JT, Wang Z, Wang P. Anti-IFNAR1 Antibodies Attenuate Inflammation and Organ Injury Following Hemorrhagic Shock. J Trauma Acute Care Surg. 86(5):881–890, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]