

Abstract
Major recent advances and previous data have led to a plausible model of how key proteins mediate neurotransmitter release. In this model, the SNARE proteins syntaxin-1, SNAP-25 and synaptobrevin form tight complexes that bring the membranes together and are crucial for membrane fusion. NSF and SNAPs disassemble SNARE complexes and ensure that fusion occurs through an exquisitely regulated pathway that starts with Munc18–1 bound to a ‘closed’ conformation of syntaxin-1. Munc18–1 also binds to synaptobrevin, forming a template to assemble the SNARE complex when Munc13–1 opens syntaxin-1 while bridging the vesicle and plasma membranes. Synaptotagmin-1 and complexin bind to partially assembled SNARE complexes, likely stabilizing them and preventing fusion until Ca2+-binding to synaptotagmin-1 causes dissociation from the SNARE complex and induces interactions with phospholipids that help trigger release. Although fundamental questions remain about the mechanism of membrane fusion, these advances provide a framework to investigate the mechanisms underlying presynaptic plasticity.
Keywords: neurotransmitter release, membrane fusion, SNAREs, Munc18, Munc13, Synaptotagmin, Complexin, NSF, SNAPs
INTRODUCTION
The fascinating capacity of the brain to perform an immense variety of tasks depends on the ability of neurons to communicate with each other at synapses. Communication occurs primarily through neurotransmitters that are packaged in vesicles at presynaptic terminals and are released by Ca2+-evoked exocytosis. This process involves tethering of synaptic vesicles to specialized areas of the plasma membrane called active zones, one or more priming reactions that leave the vesicles ready for release, and fusion of the vesicle and plasma membranes when an action potential causes Ca2+ influx into the terminal (147). Exocytosis is typically triggered in a very fast, synchronous manner (< 1 ms after Ca2+ influx) but can also exhibit a slower, asynchronous mode (147). Neurotransmitter release does not merely constitute a fast means to transfer signals between neurons; each of the steps leading to release can be regulated and each form of regulation can provide a mechanism to encode information. Indeed, changes in release during widely varied short- and long-term plasticity processes shape the properties of neural networks and underlie diverse forms of information processing in the brain (121).
Research for over three decades has shown that neurotransmitter release is governed by a sophisticated protein machinery formed by core proteins that have homologues in most other types of intracellular membrane fusion, as well as by specialized components. The exquisite regulation of release depends on the specialized components and on unique properties of the core components that are not generally shared by their homologues. A major challenge in this field is not only to elucidate how membrane fusion occurs but, perhaps even more important, to understand how fusion is controlled by diverse mechanisms during presynaptic plasticity. Moreover, this system can be viewed as a four-dimensional jigsaw puzzle where the pieces change shape as a function of time, with a delicate balance between stimulating and inhibitory interactions that fine tune the probability that a vesicle fuses upon Ca2+ influx. The past few years have been very exciting, as multiple new findings have brought us close to a clear understanding of the basic steps that lead to release and have allowed initial discoveries on the molecular basis for key forms of presynaptic plasticity.
Figure 1 presents a model where I attempt to integrate our current knowledge of the molecular mechanisms underlying release (see the legend of Figure 1 for a summary of the key features of this model). The model is unavoidably complex, which is expected given the nature of this system, but I hope that the discussions in this review will help understand the molecular basis for each step and to what extent this basis is well established. Below I devote particular attention to recent developments but also outline fundamental concepts that emerged from older studies. I also emphasize the importance of weak interactions for the molecular transitions that lead to release and the difficulties of establishing the biological relevance of weak interactions observed in vitro even when defined in detail by structural studies. While I focus mostly on the neuronal exocytotic machinery, I also describe some seminal insights that have emerged from studies of yeast vacuolar fusion and illustrate which features of the neurotransmitter release mechanism are conserved and which are unique. Clearly, it is impossible to cover the large amount of literature available in these fields. Recent reviews have covered these areas, including multiple aspects not discussed here (15, 18, 41, 104, 126, 130, 148, 189).
Figure 1.

Working model of the basic steps that lead to neurotransmitter release. (a) Diagram showing the localization of synaptobrevin (red) and synaptotagmin-1 (blue) on a synaptic vesicle, and of αSNAP (brown) bound to a 1:2 complex between SNAP-25 (green) and syntaxin-1 (SNARE motif yellow; Habc domain orange) on the plasma membrane. Helices formed by the SNAREs are represented by cylinders. αSNAP binding to the syntaxin-1-SNAP-25 complex hinders binding to synaptobrevin and SNARE complex formation. (b) Diagram illustrating that, even if trans-SNARE complexes between synaptobrevin, syntaxin-1 and SNAP-25 are formed, binding of αSNAP to these complexes prevents fusion, ensuring that neurotransmitter release does not occur through non-regulated pathways. (c) Diagram showing syntaxin-1 adopting a closed conformation that binds tightly to Munc18–1 (violet). This binary complex constitutes the starting point of the pathway that leads to neurotransmitter release. Closed syntaxin-1 may be available on the plasma membrane or may form after NSF dissociates an αSNAP-bound syntaxin-1-SNAP-25 complex (from panel a) or an αSNAP-bound trans-SNARE complex (from panel b). (d-e) The conserved C-terminal region of Munc13–1 (cyan) bridges the vesicle and plasma membranes through respective interactions involving the C2C domain and the C1-C2B region, and opens syntaxin-1 by binding to the linker between the syntaxin-1 Habc domain and SNARE motif. This action likely facilitates binding of Munc18–1 to synaptobrevin, forming a template complex (d) that initiates SNARE complex assembly upon binding of SNAP-25 to syntaxin-1 and synaptobrevin (e). Synaptotagmin-1 likely facilitates assembly by binding to SNAP-25 (not shown in d for simplicity). The synaptotagmin-1 C2B domain binds to the partially assembled SNARE complex through the primary interface and to the plasma membrane through the polybasic region (e). Munc13–1 facilitates SNARE complex assembly but also limits the number of SNARE complexes that form and hinders C-terminal zippering by bridging the two membranes in an approximately perpendicular orientation that characterizes a loose primed state (LS). This inhibitory action may be aided by formation of Munc13–1 clusters. (f) Further but not complete C-terminal zippering of the SNARE complex, which is favored by binding of complexin (pink), forces the two membranes closer together and Munc13–1 must bridge the membrane in a slanted orientation, forming a tight primed state (TS) that has a much higher probability of release upon Ca2+ influx than LS. Complexin and synaptotagmin-1 stabilize this state and prevent disassembly of the trans-SNARE complexes by NSF/αSNAP, but hinder final zippering to prevent premature fusion. (g-h) Ca2+ binding to synaptotagmin-1 causes dissociation from the SNARE complex, relieving the inhibition and thus allowing final C-terminal zippering and synaptic vesicle fusion. The dissociated synaptotagmin-1 molecules, or other synaptotagmin-1 molecules that were not bound to the SNAREs (shown in the middle) accelerate fusion through interactions with the lipids, perhaps because they perturb the bilayers, bridge the two membranes and/or induce membrane curvature. The increase in vesicular release probability caused by accumulation of Ca2+ and DAG during repetitive stimulation is proposed to arise because Ca2+ and DAG bind to the C2B and C1 domains of Munc13–1, respectively, and favor the slanted orientation, shifting the equilibrium from LS to TS. The following features are not shown for simplicity: (b, e-h) the linker region between the two SNAP-25 SNARE motifs, which remains anchored on the plasma membrane through plamitoylation; (f-h) the N- and C-terminal regions of complexin, which bind to the membranes; (e-h) Munc18–1, which may remain bound to the N-terminal region of syntaxin-1; (e-f) additional synaptotagmin-1 molecules that may bind to the SNARE complex through the tripartite interface or form a synaptotagmin-1 oligomeric ring that has been proposed to prevent fusion before Ca2+ influx and is dissociated upon Ca2+ binding.
MEMBRANE FUSION
Experimental and theoretical studies suggest that physiological membrane fusion requires approach of the two membranes, bending of the bilayers to destabilize their packing, formation of an intermediate called stalk where the two proximal bilayer leaflets have fused, and merger of the distal leaflets to form a fusion pore (Figure 2a) (31). This mechanism is strongly supported by X-ray diffraction data showing that stalk intermediates can be formed with diverse lipids compositions (2). While some variations are possible, depending on how the membranes are perturbed, this mechanism offers a useful framework to consider how proteins may induce membrane fusion. Theoretical calculations indicate that the energy required for membrane fusion ranges from 40 to 100 kBT [e.g (33), but note that this energy should depend on the lipid composition and that a key consideration is not only how much energy can be provided by proteins to induce fusion but also how they can apply this energy on the membranes.
Figure 2.

The SNARE complex and membrane fusion. (a) The stalk model of membrane fusion, which postulates that the two membranes have to be brought into proximity, the bilayers bend and the proximal leaflets fuse to yield the so-called stalk intermediate, then the distal leaflets fuse to form the fusion pore (31). (b) Ribbon diagrams of the NMR structure of the syntaxin-1 Habc domain (orange) (50) and the crystal structure of the SNARE complex formed by the SNARE motifs of syntaxin-1 (yellow), SNAP-25 (green) and synaptobrevin (red) (150) (PDB IDs 1BR0 and 1SFC, respectively). N and C indicate N- and C-termini, respectively. (c) Model postulating that formation of the SNARE complex brings the vesicle and plasma membranes together, inducing membrane fusion. C indicates the C-termini of synaptobrevin and syntaxin-1. (d) Diagram illustrating the formation of extended membrane-membrane interfaces induced by SNARE complex formation, which have been observed by cryo-EM (61). In (c,d), the Habc domain is not shown.
CORE MEMBRANE FUSION MACHINERY
SNARE architecture and the SNARE complex
Most types of intracellular membrane traffic require soluble N-ethylmaleimide sensitive factor attachment protein receptors (SNAREs), which are characterized by ∼65 residue sequences called SNARE motifs (67). The neuronal SNAREs that mediate neurotransmitter release are the synaptic vesicle protein synaptobrevin/VAMP (for vesicle-associated membrane protein), and the plasma membrane proteins syntaxin-1 and SNAP-25. Synaptobrevin and syntaxin-1 each contain one SNARE motif that precedes a C-terminal transmembrane (TM) region, whereas SNAP-25 contains two SNARE motifs and is anchored on the plasma membrane through palmitoylation. The three proteins form a highly stable ‘SNARE complex’ (141) consisting of a bundle of four parallel α-helices (113, 150) (Fig. 2b). SNARE complexes formed between two membranes before fusion are called trans-SNARE complexes and those on one membrane resulting after fusion are called cis-SNARE complexes. Synaptobrevin is often classified as a v-SNARE (for vesicle SNARE), and syntaxin-1 and SNAP-25 as t-SNAREs (for target membrane SNAREs) (142). The SNARE four-helix bundle is stabilized by layers of four hydrophobic residues, but there is a central polar layer formed by one arginine from synaptobrevin and three glutamines from syntaxin-1 and SNAP-25, leading to their classification as R- and Q-SNAREs, respectively (47). Although SNARE complex formation occurs with some level of specificity, SNARE motifs are promiscuous and can form alternative four-helix bundles that represent kinetic traps, as exemplified by the tendency of syntaxin-1 and SNAP-25 to form a 2:1 complex [reviewed in (128)] (Figure 1a). Moreover, SNARE motifs can also bind in antiparallel fashion, which also hinders proper SNARE complex assembly (79).
In addition to a SNARE motif and a TM region, syntaxin-1 contains an N-terminal region that includes a short sequence at the very N-terminus called the N-peptide (73) and an autonomously folded three-helix bundle called Habc domain (50) (Fig. 2b). In isolated syntaxin-1, the Habc domain binds intramolecularly to the SNARE motif, forming a ‘closed conformation’ that hinders SNARE complex assembly (44). This feature constitutes a fundamental mechanism to control neurotransmitter release, as the closed conformation of syntaxin-1 binds tightly to Munc18–1 (44, 102) to initiate the pathway that leads to SNARE complex formation (see below). The importance of this control mechanism is highlighted by the fact that a so-called LE mutation that opens syntaxin-1 (44) increases the vesicle release probability and the speed of neurotransmitter release (1, 52), and can partially rescue the phenotypes caused by deletion of various proteins involved in different aspects of synaptic transmission (75, 125, 153). These results suggest that the number of SNARE complexes dictates at least in part the release probability. Note also that, while all SNAREs form SNARE complexes (67), the closed conformation is not generally conserved among syntaxin-1 homologues [e.g. (45, 46)].
Reconstitution of SNARE-dependent membrane fusion
The realization that the membrane proximal SNARE motifs of synaptobrevin and syntaxin-1 bind in a parallel fashion led to the proposal that formation of the SNARE complex brings the synaptic vesicle and plasma membranes together and induces membrane fusion (58) (Figure 2c). In this model, the energy of formation of this highly stable complex provides the energy to drive membrane fusion. The model was supported by pioneering reconstitution assays showing lipid mixing between synaptobrevin-containing liposomes and syntaxin-1-SNAP-25-containing liposomes (165) and subsequent research has provided ample evidence that the neuronal SNAREs alone can cause membrane fusion [reviewed (18, 67, 148, 189)]. This research has included the development of diverse additional methods that monitor fusion between small unilamellar vesicles (SUVs) and giant unilamellar vesicles (GUVs) (93), between single pairs of vesicles (78, 187), between vesicles and supported or suspended lipid bilayers (42, 60, 76), and between nanodiscs and vesicles (10, 136), planar bilayers (9) or cells (174).
Each one of these methods has its own advantages but it is also important to consider their limitations when interpreting the results. Bulk assays with liposomes that use fluorescence resonance energy transfer (FRET) are relatively easy to perform and have provided critical insights into the mechanisms underlying neurotransmitter release (88, 155, 165) and other types of membrane traffic, particularly yeast vacuolar fusion (101, 143). However, these assays suffer from a slow time resolution, which hinders dissection of steps such as docking and fusion, or detection of intermediates, and do not mimic the flat nature of the plasma membrane. Single vesicle-single vesicle fusion assays provide a faster time resolution and have also yielded important insights (39, 79, 81, 82). Some data have suggested that fusion of single vesicles with flat bilayers occurs faster than between two vesicles under comparable conditions (74), perhaps because the former mimic the geometry of synaptic vesicle fusion better or because of unknown technical reasons. It is also important to note that, in both bulk and single vesicle fusion assays monitored by fluorescence spectroscopy, it is critical to monitor not only lipid mixing but also content mixing to ensure that actual fusion without leakiness takes place, as large amounts of lipid mixing can occur with very little content mixing (25, 78, 196).
Assays that monitor fusion by electrophysiological methods (9, 60, 174) offer even faster time resolution [in the 10 μs time scale] than fluorescence-based methods and hence are most promising as optimization of the reconstitution systems recapitulate synaptic vesicle fusion more accurately. These methods have allowed the observation of fusion pores and indicate that one SNARE complex is sufficient to form a fusion pore, but the size of the fusion pore increases with the number of SNARE complexes (9, 60). These observations agree with the notion that the number of assembled SNARE complexes dictates release probability. Moreover, comparable data were obtained in nanodisc-liposome, nanodisc-flat bilayer and liposome-flat bilayer assays (9, 60, 136), and there is evidence that a minimum of 2–3 SNARE complexes is normally required for neurotransmitter release but even a single SNARE complex can support release (103, 139). This correlation and the consistency of the data are important to support the biological relevance of the results, as the mere observation of membrane fusion in vitro does not mean that the underlying mechanism resembles that occurring in vivo.
A different picture emerged from cryo-electron microscopy (cryo-EM) images of bulk SNARE-dependent fusion reactions between liposomes or between liposomes and GUVs showing the formation of large, extended interfaces (13, 39, 61) (illustrated by the diagram of Figure 2d) that constitute intermediates in the fusion pathway (171). Because fusion and the disappearance of the extended interfaces in these experiments are much slower than that of neurotransmitter release, it is unlikely that these intermediates occur in vivo. Indeed, it appears that other components of the release machinery may limit the formation of these extended interfaces (39, 53). It is unclear whether the extended interfaces can lead to the fusion pores observed in liposome-flat bilayer fusion assays (60), but it is possible that fusion might occur by more than one mechanism. Thus, the observed fusion pores were estimated to involve a few SNARE complexes, which may lead initially to the formation of small, point-of-contact interfaces, whereas formation of a large number of SNARE complexes might lead to the extended interfaces. Hence, formation of the extended interfaces observed by cryo-EM might arise because the large amount of SNAREs present in the liposomes. Note that, although the SNARE-to-lipid ratios used in these experiments (13, 39, 61) were comparable to physiological levels, other key components of the release machinery such as Munc18–1 and Munc13–1 may limit the number of SNARE complexes that are formed in a primed vesicle [(177), see below], preventing the formation of extended interfaces.
In this context, the use of nanodiscs as one of the fusion partners is attractive because it allows good control of the number of SNARE complexes that can mediate fusion. However, synaptobrevin was normally incorporated into the nanodiscs, which does not mimic the geometry of synaptic vesicle fusion. Moreover, data obtained with small nanodiscs need to be interpreted with caution given the limited number of lipid molecules that can be incorporated in each nanodisc. For instance, experiments with 6 nm nanodiscs led to the conclusion that exocytotic fusion pores are composed of both lipids and proteins (10), but such nanodiscs are expected to contain only 30–40 lipids in each leaflet and it is therefore highly unclear whether the results can be extrapolated to physiological fusion pores. Geometrical considerations become even more important as other components of the release machinery are incorporated in reconstitutions assays because the actions of large proteins such as Munc13–1 (see below) may be impossible to recapitulate with nanodiscs even if they have diameters of 30 nm.
How do the SNAREs induce membrane fusion?
Although it is now well established that SNARE complexes can induce membrane fusion, the underlying mechanism remains highly enigmatic. Original models envisaged long, continuous helices formed by the SNARE motif, the short juxtamembrane region and the TM region of both synaptobrevin and syntaxin-1 coming progressively together and forcing membrane fusion as the SNARE complex ‘zippers’ from the N- to the C-terminus (58, 150, 165). Although such cartoons are widespread in the literature, the strong bend of the helices in the juxtamembrane sequences depicted in these models is highly unlikely based on conformational grounds. These sequences are expected to remain largely unstructured when the four helix bundle zippers fully between two membranes (as depicted in the diagram of Figure 2c, left). Note also that the free energy of SNARE complex assembly is estimated to be between 35 and 68 kBT (51, 83) and, therefore, formation of a few SNARE complexes or perhaps even just one can in principle overcome the energy required for membrane fusion, but it is unclear how this energy is applied to the membranes. Most of the free energy is spent when the four-helix bundle is fully zippered, which should bring the two membranes within a distance of few nanometers. However, bringing two membranes into contact does not necessarily lead to fusion. It thus seems likely that the SNAREs do more than bringing the membranes together to cause fusion, and the likely culprits are the juxtamembrane regions. These sequences contain abundant basic residues that are expected to interact with the negatively charged head groups and thus help bring the membranes even closer. Moreover, the synaptobrevin juxtamembrane region contains tandem tryptophan residues that can insert into lipid bilayers and perturb their packing. Indeed, liposome fusion in reconstitution assays is strongly affected by the position of these tryptophan residues with respect to the TM region (63), and Ca2+-evoked neurotransmitter release is considerably impaired by mutation of the two tryptophans to alanine (96). As discussed below, other proteins (Sec17 and synaptotagmin-1) likely accelerate membrane fusion also by perturbing lipid bilayers, suggesting that proteins must perform two key actions to fuse membranes: bring them into close proximity and perturb bilayer packing to facilitate the formation of non-bilayer intermediates that lead to fusion (169).
NSF and SNAPs
N-ethylmaleimide sensitive factor (NSF) is a soluble protein that was shown in 1988 to be required for transport of cargo vesicles to Golgi cisternae and was proposed to assemble into a multisubunit ‘fusion machine’ (91). Soluble NSF attachment proteins (SNAPs) were later shown to mediate binding of NSF to membrane sites (167), and to form a complex with synaptobrevin, syntaxin-1 and SNAP-25, leading to their designation as SNAREs [for SNAP receptors, (142)]. It was later shown that the SNAREs form a complex by themselves (the SNARE complex) and that NSF disassembles the SNARE complex through its ATPase activity, with the help of SNAPs (141). These and other results (8, 97) led to the current view that the main function of NSF/SNAPs is to disassemble the cis-SNARE complexes that result after membrane fusion to recycle the SNAREs. NSF/SNAPs also disassemble 2:1 syntaxin-1/SNAP-25 complexes and improperly assembled SNARE complexes with antiparallel helices (32, 88). Moreover, NSF/SNAPs were recently shown to disassemble trans-SNARE complexes in vitro (115, 185), which correlates with the finding that primed synaptic vesicles can be de-primed and N-ethylmaleimide impairs such de-priming (59). Not surprisingly, NSF and αSNAP completely inhibit SNARE-mediated liposome fusion in reconstitution assays (85, 88). In fact, αSNAP potently inhibits fusion by itself, in part because it binds to syntaxin-1-SNAP-25 heterodimers, preventing binding to synaptobrevin (Fig. 1a), and in part because it blocks fusion by pre-formed trans-SNARE complexes (111, 144) (Fig. 1b).
Cryo-EM structures of the 20S complex formed by NSF, αSNAP and the neuronal SNAREs provided seminal insights into the SNARE complex disassembly mechanism (26, 65, 166, 190) (Figure 3a). NSF contains an N-terminal domain that binds to αSNAP and two nucleotide binding domains of the AAA family, which are called D1 and D2 and mediate the formation of hexameric rings by NSF. NSF interacts directly with the N-terminus of SNAP25 through a pore formed by the D1 ring (166). Two to four αSNAP molecules bound to the SNAREs were observed in these structures, although the higher stoichiometry may be favored on membranes because of co-localization. When four αSNAP molecules are bound, they cover almost the entire surface of the SNARE four-helix bundle, interacting with the SNAREs primarily through electrostatic interactions and suboptimal packing, which explains the lack of specificity of the NSF/SNAP machinery. Suboptimal packing arises in part because the elongated αSNAP structure has a right-handed twist whereas the SNARE four-helix bundle has a left-handed twist, suggesting that αSNAP binding might already destabilize the interactions between the SNAREs. SNARE complex disassembly is believed to involve a power stroke when ATP hydrolysis by the D1 domain causes large conformational changes that propagate to the NSF N-terminal domain and αSNAP, ultimately tearing the SNARE four-helix bundle apart (65, 190). A hydrophobic N-terminal loop of αSNAP that inserts into membranes and accelerates disassembly of membrane-anchored cis-SNARE complexes (170) is located next to the C-terminus of the SNARE four-helix bundle in the structure of the 20S complex (Figure 3a), indicating that the acceleration arises because simultaneous interactions of four αSNAP molecules with the membrane and the SNAREs can cooperate to form the 20S complex.
Figure 3.
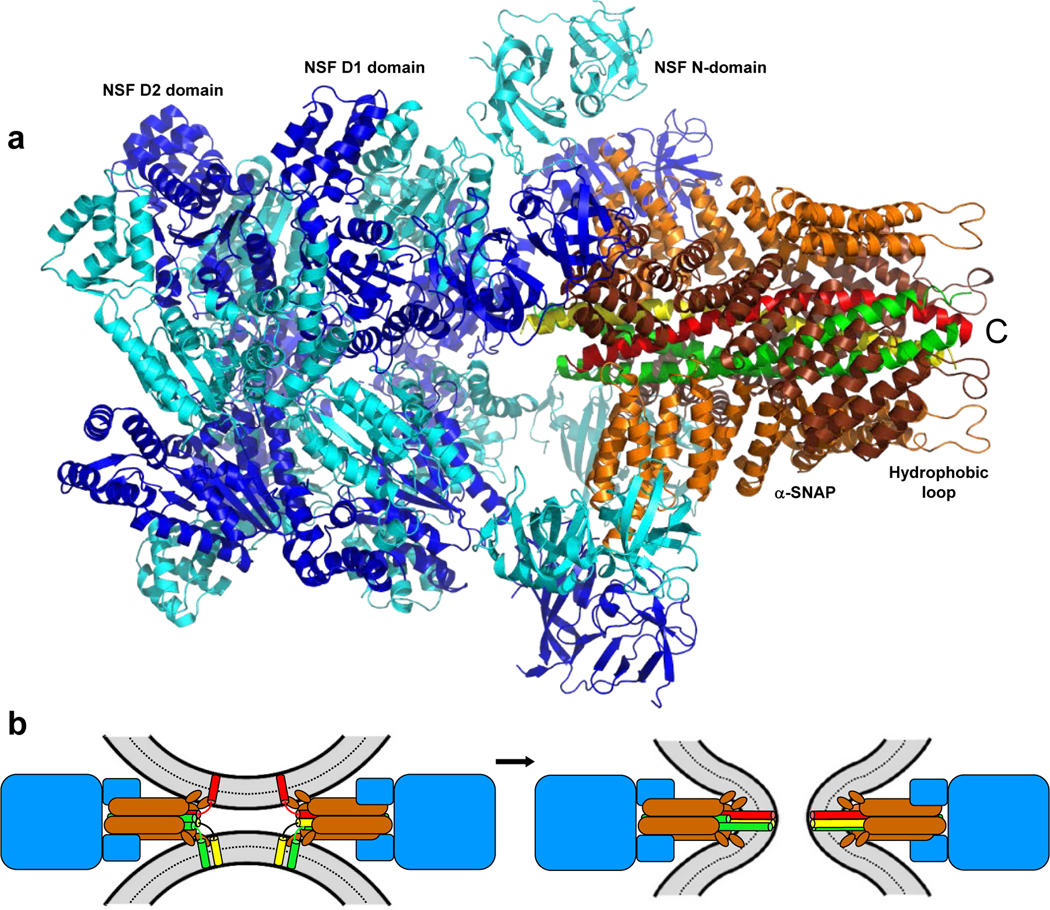
The 20S complex. (a) Cryo-EM structure of the 20S complex formed by NSF, αSNAP, synaptobrevin (red), syntaxin-1 (yellow) and SNAP-25 (green) (190) (PDB ID 3J96). The four molecules of αSNAP that bind around the SNARE four-helix bundle are colored in orange and brown in an alternative fashion. Similarly, the six molecules of NSF are colored in blue and cyan in an alternative fashion. The positions of the D1, D2 and N-domains of NSF, as well as the N-terminal hydrophobic loop of αSNAP, are indicated. (b) Model proposing that the core machinery that mediates yeast vacuolar fusion is a 20S complex formed by Sec18 (blue), Sec17 (orange) and the four vacuolar SNAREs (yellow, green and red) (143). Note that the small ellipse at the membrane-proximal end of each αSNAP molecule represents its N-terminal hydrophobic loop and insertion of the loop into the bilayers is postulated to make a key contribution to membrane fusion.
The core membrane fusion machinery re-defined?
The research summarized above has led to the general belief that the SNAREs form the core of the intracellular membrane fusion machinery, but recent studies of yeast vacuolar fusion suggest that this picture may need to be revised. Vacuolar fusion requires three membrane-anchored (Nyv1, Vam3 and Vti1) and one soluble SNARE (Vam7) (168). Systematic studies with reconstituted proteoliposomes have shown that the Rab protein Ypt7 and a complex of six proteins called HOPS (for homotypic fusion and vacuole protein sorting) mediate tethering of the two membranes (195), and that HOPS orchestrates assembly of the vacuolar SNARE complex in a manner that is resistant to Sec18 and Sec17, the yeast homologues of NSF and SNAPs, respectively (175). Intriguingly, HOPS and Sec17-Sec18 acted synergistically (101), suggesting that Sec17-Sec18 may have an important function(s) beyond disassembling SNARE complexes. Moreover, Sec17 was shown to rescue the arrest of vacuole fusion caused by deleting 22 residues of the C-terminus of the Vam7 SNARE motif (132). More recently, Sec17 together with Sec18 were shown to rescue liposome fusion even when three SNAREs are crippled so that it is impossible to zipper the C-terminus of the SNARE complex (143). Such rescue depended critically on the hydrophobic N-terminal loop of Sec17. These results show that C-terminal zippering of the vacuolar SNARE complex to draw the membranes tightly together is not essential for membrane fusion and suggest that, as long as SNARE complexes bring two membranes into proximity, perturbation of the lipid bilayers by the hydrophobic loop of Sec17 can induce fusion. Under some conditions, both ATP and the non-hydrolyzable ATP analogue ATPγS supported rescue of liposome fusion, but only ATPγS supported fusion under the most stringent conditions (143). This finding suggests that binding of ATP to Sec18 without hydrolysis is crucial to form a complex with Sec17 and the SNAREs that resembles the 20S complex and that most likely constitutes the true ‘fusion machine’ envisaged in 1988 (91) (Figure 3b), rather than the SNARE complex alone. It is tempting to speculate on the beautiful possibility that ATP hydrolysis by this fusion machine is slow in the trans configuration that bridges two membranes before fusion and is accelerated after fusion because of the change in geometry, leading to disassembly of the 20S complex and SNARE recycling.
The finding that neuronal αSNAP strongly inhibits liposome fusion by pre-formed neuronal trans-SNARE complexes (111, 144) contrasts with the ability of Sec17 to rescue fusion and suggests that there is a fundamental difference between the fusion machinery of the two systems. It seems likely that the results obtained with the vacuolar proteins apply to most types of intracellular membrane fusion and that the 20S complex normally constitutes the core of the membrane fusion machinery, but the synaptic vesicle fusion machinery diverged to meet the stringent regulatory requirements of neurotransmitter release. This view is supported by the observation that synaptotagmin-1 and complexins, which are key regulators of the last steps that lead to evoked release, bind to the SNARE four-helix bundle (see below) and such binding would be impossible if αSNAP covers the surface of the bundle as it does in the 20S complex (144).
MUNC18–1 AND MUNC13–1
Orchestration of SNARE complex assembly
Assembly of the SNARE complex in a cellular environment where NSF/SNAPs strongly favor complex disassembly requires factors that can overcome this disassembly activity. Reconstitution experiments strongly suggest that Munc18–1 and Munc13s perform this function at neuronal synapses (88, 115). Munc18–1 is a member of the Sec1/Munc18–1 (SM) family of proteins that are required for all forms of SNARE-dependent membrane traffic (22), whereas Munc13s are large (ca. 200 kDa) multidomain proteins with a variable N-terminal region and a conserved C-terminal region (147). The functional importance of these proteins was demonstrated by the observation that Ca2+-triggered release, spontaneous release, and sucrose-induced release (which involves all the vesicles that are primed) are abolished in Munc18–1 KO mice and in Munc13–1/2 double KO (DKO) mice, as well as in nulls of the invertebrate homologue UNC-13 (4, 124, 158, 159). These essential functions were explained by reconstitution assays showing that fusion between liposomes containing synaptobrevin and synaptotagmin-1 (which stimulates fusion; see below) and syntaxin-1-SNAP-25-containing liposomes was abolished by NSF/αSNAP, but highly efficient fusion could be observed upon addition of Munc18–1 and a Munc13–1 C-terminal fragment (85, 88) (Figure 4a,b). The requirement of both proteins arises because both are necessary to orchestrate SNARE complex assembly in a NSF/αSNAP-resistant manner (115).
Figure 4.

Liposome fusion assays that recapitulate the dependence of synaptic vesicle fusion on Munc18–1, Munc13–1 and synaptotagmin-1. (a,b) Assays that monitored lipid mixing (a) and content mixing (b) of VSyt1-liposomes containing synaptobrevin (P:L ratio 1:500) and synaptotagmin-1 (P:L ratio 1:1,000) with T-liposomes containing syntaxin-1 (P:L ratio 1:800) bound to SNAP-25, in the presence of NSF and αSNAP. Under these conditions, there is no fusion without other additions (T + VSyt1, black curves). Inclusion of Munc18–1 and a Munc13–1 C-terminal fragment spanning the C1, C2B, MUN and C2C domains (C1C2BMUNC2C) leads to highly efficient Ca2+-dependent fusion (+M13 +M18, red curves), but there is no fusion when only one of these proteins is included (+M18, orange curves; +M13, blue curves) (85). Note that in the complete reaction with all the reagents there is some lipid mixing before Ca2+ addition, indicating that a few SNARE complexes are formed, but there is no content mixing, showing that there is not fusion. (c) Content mixing assays performed under analogous conditions but with V-liposomes containing only synaptobrevin (P:L ratio 1:500) in the presence of NSF, αSNAP, Munc13–1 C1C2BMUNC2C and WT Munc18–1 (T + V, black curve), Munc18–1 D326K (red curve) or Munc18–1 L348R (blue curve). The D326K mutation that unfurls the Munc18–1 loop leads to efficient fusion before Ca2+ addition, whereas the L348R mutation that impairs synaptobrevin binding to Munc18–1 strongly hinders fusion (140). Note that the Ca2+-dependent fusion observed with WT Munc18–1 is highly efficient even though synaptotagmin-1 was absent [compare black curve in panel (c) with red curve in panel (b)]. (d) Content mixing assays performed under analogous conditions but with VSyt1-liposomes containing synaptobrevin at 1:10,000 P:L ratio and synaptotagmin-1 at 1:1,000 ratio (T + VSyt1, red curve) or V-liposomes containing only synaptobrevin at 1:10,000 P:L ratio (T + V, black curve), in the presence of NSF, αSNAP, Munc13–1 C1C2BMUNC2C and WT Munc18–1. Note that inclusion of synaptotagmin-1 led to much more efficient fusion at this low synaptobrevin densities, and that experiments with VSyt1-lipsomes bearing synaptotagmin-1 with mutations in the Ca2+-binding sites of both C2 domains (T + VSyt1 C2A*B*, blue curve) led to similar fusion to that observed without synaptotagmin-1, showing that stimulation of fusion depends on Ca2+ binding to synaptagmin-1 (145).
This pathway of SNARE complex assembly is initiated by formation of a tight binary complex of Munc18–1 with the closed conformation of syntaxin-1 (44) (Fig. 1c). Munc18–1 is an arch-shaped protein with four domains (termed D1, D2, D3a and D3b), and closed syntaxin-1 binds to a cavity formed by domains 1 and 3a (102) (Figure 5a). The N-peptide of syntaxin-1 also contributes to binding (19) and likely plays a key role in release (73, 192) by helping keep Munc18–1 bound to syntaxin-1 during conformational changes that are required to form the SNARE complex. Munc18–1 also binds to synaptobrevin (178), which led to the proposal that Munc18–1 forms a template for SNARE complex assembly (108). This notion was made evident by two crystal structures of Vps33, the Munc18–1 homologue involved in yeast vacuolar fusion, one in a complex with the SNARE motif of the syntaxin-1 homologue Vam3 and the other in complex with the synaptobrevin homologue Nyv1 (7). The structures showed that simultaneous binding of both SNAREs to Vps33 would place the N-terminal sequences of the SNARE motifs of Vam3 and Nyv1 close to each other, in register to form the SNARE complex (Figure 5b).
Figure 5.

Structures of the SNARE complex assembly machinery. (a) Ribbon diagram of the crystal structure of Munc18–1 (violet) bound to closed syntaxin-1 (SNARE motif in yellow, N-terminal region in orange) (102) (PDB ID 3C98). The positions of the Munc18–1 domains (D1, D2, D3a and D3b), of the furled loop that covers the synaptobrevin binding site and of the syntaxin-1 N-peptide (N-pep) are indicated. (b) Superposition of crystal structures of the Munc18–1 homologue Vps33 (violet) bound to the SNARE motif of the syntaxin-1 homologue Vam3 (yellow) or the SNARE motif of the synaptobrevin homologue Nyv1 (red) (7) (PDB IDs 5BV0 and 5BUZ, respectively). (c) Domain diagram of Munc13–1. CaMb = calmodulin-binding region. (d) Model of the structure of a fragment spanning the C1 (salmon), C2B (blue) and MUN (cyan) domains of Munc13–1 built from the crystal structure of this fragment (177) and completing the Ca2+-binding region of the C2B domain with the crystal structure of this domain bound to Ca2+ (137) (PDB IDs 5UE8 and 3KWU, respectively). Zinc ions are shown as yellow spheres and Ca2+ ions as orange spheres. N indicates the N-terminus of the fragment, where the Munc13–1 N-terminal region should emerge. A homology model of the Munc13–1 C2C domain (blue) (116) is shown at its expected position at the C-terminus of the MUN domain. The DAG-binding site of the C1 domain, the Ca2+/PIP2-binding site of the C2B domain and the membrane-binding site of the C2C domain, predicted to bind to synaptic vesicles, are indicated. A peptide corresponding to the juxtamembrane region of synaptobrevin is shown in red in the position observed in the crystal structure of this peptide bound to the Munc13–1 MUN domain (163) (PDB ID 6A30). Note that the position of residue 92, which is close to the TM region, is far from the expected membrane-binding region of the C2C domain.
The binding mode between Vam3 and Vps33 is similar to that observed between the syntaxin-1 SNARE motif and Munc18–1 (Figure 5a), and it is natural to expect that synaptobrevin binds to the same site of Munc18–1 as that observed for binding of Nyv1 to Vps33, which involves a helical hairpin of domain 3a (Figure 5b). However, the loop of this hairpin of Munc18–1 is furled over the putative synaptobrevin binding site in the structure of the Munc18–1/syntaxin-1 complex (Figure 5a), suggesting that this loop hinders synaptobrevin binding and explaining the low affinity of synaptobrevin for Munc18–1 (178). Indeed, a mutation that unfurls the Munc18–1 loop (D326K) increases synaptobrevin binding and leads to a gain of function in liposome fusion assays in vitro (Figure 4c) and in C. elegans in vivo (140). Moreover, liposome fusion is impaired by a mutation in the helical hairpin of Munc18–1 that disrupts synaptobrevin binding (108) (Figure 4c), neurotransmitter release is strongly impaired by a phosphomimetic mutation in Munc18–1 (Y473D) that is expected to disrupt synaptobrevin binding (99), and optical tweezer experiments also provided strong support for the notion that Munc18–1 forms a template to assemble the SNARE complex much like Vps33 (68). Note however that the template function of Munc18–1 is hindered by at least two energy barriers, one imposed by the furled Munc18–1 loop and the other by the closed conformation of syntaxin-1, which is stabilized by Munc18–1 binding (29). As a consequence, Munc18–1 binding to syntaxin-1 strongly hinders assembly of the SNARE complex with synaptobrevin and SNAP-25 (19, 87), and assembly requires another factor, Munc13.
Munc13–1, the most abundant Munc13 isoform in the mammalian brain, contains a C2A domain and a calmodulin binding (CaMb) sequence (70) in the variable N-terminal region, whereas the conserved C-terminal region includes a diacylglycerol (DAG)/phorbol ester-binding C1 domain (122), a MUN domain homologous to tethering factors from various membrane compartments (11, 112) and two C2 domains, C2B and C2C (Figure 5c). Note that, while C2 domains act commonly as Ca2+-dependent phospholipids binding modules (127), only the C2B domain of Munc13–1 binds Ca2+, which induces binding to PIP2 (137). The C2A domain forms a homodimer by itself and a heterodimer with RIM (43, 86) (see below), and the C2C domain binds weakly to membranes in a Ca2+-independent manner (116). The finding that the LE mutation that opens syntaxin-1 partially rescues neurotransmitter release in unc-13 nulls in C. elegans suggested that UNC-13/Munc13s help open syntaxin-1 (125). Although subsequent studies revealed that the LE mutation rescues phenotypes from a variety of mutants in C. elegans (75, 153), this proposal was confirmed by multiple evidence showing that the Munc13–1 MUN domain accelerates the transition from the Munc18–1/closed syntaxin-1 complex to the SNARE complex (87, 162, 184). Interestingly, this catalysis involves a very weak interaction of the MUN domain with the linker region between the Habc domain and the SNARE motif of syntaxin-1 (162) that is barely detectable even by NMR spectroscopy, whereas a stronger albeit still weak interaction between the MUN domain and the syntaxin-1 SNARE motif appears to be irrelevant (90).
These results illustrate that weak interactions can play critical roles by lowering energy barriers that hinder crucial conformational transitions, and at the same time show the difficulties involved in pinpointing which weak interactions are biologically relevant. Moreover, the key weak interactions can cooperate with other weak interactions to enhance formation of macromolecular assemblies. Indeed, the MUN domain binds to the Munc18–1/syntaxin-1 complex and to the Munc18–1-syntaxin-1-synaptobrevin complex with higher affinity than to syntaxin-1, which likely involves MUN-Munc18–1 interactions (87, 138, 162, 164). The Munc13–1 MUN domain has also been reported to bind to SNAP-25 (72) but the significance of this finding is unclear, as binding required the cysteines residues of the SNAP-25 linker between the two SNARE motifs, which are expected to be palmitoylated in neurons and are not required for the highly efficient liposome fusion catalyzed by the Munc13–1 C-terminal fragment (Figure 4a,b). Note also that the MUN domain binds to the juxtamembrane region of synaptobrevin, but this interaction is most likely non-specific (140), as this region is highly promiscuous and is expected to strongly interact with the membrane (17), aided by its proximity. A crystal structure of the MUN domain bound to a peptide from the synaptobrevin juxtamembrane region suggested that this interaction helps to form the template complex (163), but molecular modeling shows that the C-terminus of the peptide would be oriented away from the vesicle membrane, indicating that the observed binding mode is incompatible with anchoring of synaptobrevin to the vesicle (Figure 5d). Hence, more research will be required to further elucidate how Munc18–1 and Munc13–1 organize SNARE complex assembly. Regardless of the actual mechanism, a benefit of this assembly pathway is that it prevents the formation of antiparallel SNARE complexes and 2:1 syntaxin-1-SNAP-25 heterodimers (79). It is still unclear to what extent formation of these antiparallel complexes hinders SNARE complex assembly, as reconstitution assays have shown that the neuronal SNAREs alone can drive membrane fusion (see above), but antiparallel complexes would likely be a nuisance for the exquisite regulation of synaptic vesicle fusion.
Membrane bridging by Munc13–1 and presynaptic plasticity
The crystal structure of a fragment spanning the C1, C2B and MUN domains of Munc13–1 revealed that the conserved C-terminal region of Munc13–1 has a banana-shaped architecture due to the highly elongated shape of the MUN domain (177) (Figure 5d). The C1 and C2B domains pack at one end of the MUN domain, with their respective DAG/phorbol ester- and Ca2+/PIP2-binding sites next to each other, suggesting that they can cooperate in binding to the plasma membrane. The C2C domain, which plays a crucial role in neurotransmitter release and binds weakly to membranes in a Ca2+-independent manner (116), was not present in the Munc13–1 fragment used for crystallography, but it is expected to emerge at the other end of the MUN domain (Figure 5d). These and other findings (85) led to the hypothesis that binding of the Munc13–1 C1-C2B to the plasma membrane and of the C2C domain to a synaptic vesicle can bridge the two membranes, which was strongly supported by the finding that point mutations in the predicted membrane-binding loops of the C2C domain strongly disrupted liposome clustering and fusion in vitro and neurotransmitter release in neurons (116). The finding that a single point mutation in this 200 kDa protein almost abolished release emphasizes the functional importance of this bridging activity.
Hence, there are at least two critical roles that underlie the essential nature of Munc13s for neurotransmitter release, membrane bridging and syntaxin-1 opening. Interestingly, the dramatic phenotypes observed in C. elegan unc-13 nulls can be partially rescued not only by syntaxin-1 bearing the LE mutation (125, 153) (see above) but also by UNC-18 (the C. elegans homologue of Munc18–1) bearing a gain-of-function P334A mutation that was designed to enhance binding of Munc18–1 to synaptobrevin (108) but likely stimulates SNARE complex assembly by a different mechanism (109, 153). Note also that the closed conformation of syntaxin-1, the furled loop of Munc18–1 and the inhibitory activity of αSNAP on fusion mediated by pre-formed SNARE complexes (see above) are features that are not conserved in the vacuolar fusion machinery. The picture that emerges is that the neurotransmitter release machinery has evolved to suppress any non-regulated fusion that could be mediated by the SNAREs alone, ensuring that the pathway that leads to exocytosis requires Munc18–1 and at the same time imposing energy barriers to ensure that release also depends strictly on Munc13s, thus enabling the main different modes of regulation of neurotransmitter release that depend on these proteins (Fig. 1). This view is also supported by recent evidence suggesting that neurotransmitter release is restricted to sites where Munc13–1 forms nanoclusters of about nine copies [(131); see also (120)]. Such clusters might be related to the ‘buttressed hypothesis’, which predicts that the Munc13 MUN domain forms oligomeric rings that provide a platform for SNARE complex assembly (129).
Munc13–1 acts as a master regulator of release through its multiple domains (Figure 5c). The C2A domain forms a homodimer that inhibits release (86) and disruption of this homodimer by RIMs, resulting in a Munc13–1/RIM heterodimer, releases this inhibition (12, 20, 38, 86). RIMs are Rab3 effectors that organize the active zone, and the Rab3/RIM interaction tethers synaptic vesicles to the active zone (147). The RIM/Munc13–1 interaction localizes Munc13–1 to the active zone (64, 191), and leads to the formation of a tripartite Munc13–1/RIM/Rab3A complex (43) that likely couples the vesicle priming machinery to Rab3- and RIM-dependent forms of long-term presynaptic plasticity (23, 24) [reviewed in (126)]. The CaMb sequence is involved in Ca2+-dependent short-term plasticity (70). The C1 domain mediates the augmentation of release caused by DAG and phorbol esters (122), and the C2B domain is also involved in Ca2+-dependent short-term plasticity (137). The importance of these regulatory processes was emphasized for instance by the observation that mice bearing a H567K mutation in the C1 domain of Munc13–1 exhibited normal evoked release but died 2–3 hours after birth (122).
The crystal structure of the Munc13–1 C1C2BMUN fragment provided key insights into the molecular mechanisms that underlie these regulatory processes (177). On one hand, the observation that the N-terminal helix of this fragment emerges next to the MUN domain (Figure 5d) suggests that regulation of release by the N-terminal region containing the C2A domain and the CaMb sequence may involve inhibitory interactions with the MUN domain that are released by RIM and calmodulin, respectively. On the other hand, the C1C2BMUN structure revealed that the region containing the C1 and C2B domains contains a large polybasic face that could mediate membrane binding, which would lead to an approximately perpendicular orientation of Munc13–1 with respect to the plasma membrane (177) (Figure 6a). In contrast, binding of the C1 domain to DAG and of the C2B domain to Ca2+/PIP2 would lead to a much more slanted orientation of Munc13–1 (Figure 6b). Note also that the Munc13–1 C1C2BMUNC2C fragment can bridge liposomes with similar efficiency in the absence and presence of Ca2+, yet liposome fusion is dramatically enhanced by Ca2+-binding to the C2B domain (Figure 4 a,b) (85). These observations led to a two state model whereby the Munc13–1 C1-C2B region mediates DAG and Ca2+-dependent presynaptic plasticity because it can bridge the vesicle and plasma membranes in two orientations: i) a perpendicular orientation that is critical to initiate SNARE complex formation but hinders full zippering of the complex and membrane fusion; and ii) a slanted orientation that facilitates full SNARE complex zippering and fusion, and is stabilized by DAG and Ca2+ (177) (Figure 6). The effects of mutations in the two proposed membrane binding faces of the C1C2B region on neurotransmitter release have recently supported this proposal and have shown that the polybasic face plays a key role in synaptic vesicle priming (21).
Figure 6.

Model of how the Munc13–1 C-terminal region controls vesicular release probability and presynaptic plasticity. The model postulates that the Munc13–1 C-terminal region (cyan) can bridge the vesicle and plasma membranes in two orientations: i) an approximately perpendicular orientation that is favored in the absence of Ca2+ and allows initiation of SNARE complex assembly, but hinders C-terminal zippering and membrane fusion (a); and ii) a slanted orientation that allows full zippering of the SNARE complex and membrane fusion, and that is favored by Ca2+-binding to the C2B domain and DAG binding to the C1 domain (b) (116, 177). These two orientations have been proposed to underlie the formation of two primed states (LS and TS), with TS having a much higher probability for release upon Ca2+ influx (104). The equilibrium can be shifted toward TS before Ca2+ influx by factors such as complexins (Figure 1e,f), and by accumulation of DAG and Ca2+ during repetitive stimulation, leading to enhanced release probability.
Interestingly, a large amount of data available on short-term presynaptic plasticity can be explained by a related model envisaging that the two states with distinct Munc13–1 orientations and different extents of SNARE complex assembly correspond to two primed states of synaptic vesicles referred to as loose state (LS) and tight state (TS) (104). In this model, treatment with hypertonic sucrose or other methods used to quantitate the readily-releasable pool of vesicles trigger release of vesicles on both states, but Ca2+-triggered release occurs with much higher probability from TS than from LS. The model is also supported by the finding that the H567K mutation, which unfolds the C1 domain, leads to decreased vesicle priming but increased release probability (122), and that deletion of the C1 domain or the C2B domain of UNC-13 enhances release in C. elegans, but deletion of both domains strongly impairs release (100).
The two states are incorporated in the overall model of Figure 1 (panels e,f) and likely reflect a tug-of-war between the tendency of the SNARE complex to fully zipper and the preference of multiple Munc13–1 molecules for the perpendicular orientation in the absence of Ca2+, which limits the number of SNARE complexes that can be assembled and the extent of their assembly. During repetitive stimulation, increases in DAG and intracellular Ca2+ levels are expected to shift the equilibrium toward TS, thus raising the release probability. This model explains the tight Ca2+ dependence of the liposome fusion data of Figure 4a,b, which is dominated by Ca2+-binding to the Munc13–1 C2B domain and is thus more related to Ca2+-dependent short term plasticity than to the normal Ca2+-dependence of evoked neurotransmitter release (85, 177). However, lowering the activity of the system by decreasing the synaptobrevin densities in the liposomes renders the system highly sensitive to Ca2+ binding to synaptotagmin-1 (145) (Figure 4d), the Ca2+ sensor that triggers release (see below). These findings illustrate that events that underlie evoked release and presynaptic plasticity are closely related, as postulated by the LS-TS model (104), and the reconstitutions may reflect different aspects depending on the conditions. Note also that under the conditions of Figure 4a,b there is some lipid mixing but no content mixing before addition of Ca2+, suggesting that some SNARE complexes are formed and catalyze lipid exchange, but they are not sufficient for fusion. However, some Ca2+-independent fusion can be triggered if SNARE complex assembly is facilitated by using the Munc18–1 D326K (Figure 4c) or P335A gain-of-function mutants (109, 140), or by anchoring SNAP-25 on the syntaxin-1-liposomes instead of adding it as a soluble protein (145). In vivo, the balance between LS and TS before Ca2+ influx is also expected to be influenced by a variety of factors, including synaptotagmin-1 and complexins as discussed below.
CONTROL OF Ca2+ TRIGGERING OF RELEASE BY SYNAPTOTAGMIN-1 AND COMPLEXINS
Complexins
Complexins are small soluble proteins that bind tightly to the SNARE complex (98). Triple KO of complexins-1–3, the major isoforms in mice, decreased spontaneous release and strongly impaired evoked neurotransmitter release without affecting sucrose-induced release (183), thus leading to a decreased vesicular release probability. Complexin nulls in invertebrates also exhibited impaired evoked release but spontaneous release was enhanced, particularly in Drosophila, and vesicle docking was decreased (62, 66, 95). These results suggested that complexins play dual inhibitory and active roles in release, a notion that was supported by further studies in mice [(96, 182)]. The differences in the results on spontaneous release observed in mammals and invertebrates might in principle arise from divergence among species, but it seems more likely that there is just a difference in the energetic balance between inhibitory and stimulatory activities in the different species (180).
Complexin-1 is largely unstructured in solution (105) but forms a central α-helix upon binding to the SNARE complex (30) (Figure 7a). This central helix interacts with a groove between syntaxin-1 and synaptobrevin, and is extended at the N-terminus into an accessory helix (AH) that does not contact the SNARE complex. Binding of the central helix to the SNARE complex is essential for all complexin functions (96, 182). Deletion of the N-terminal region preceding the AH abolishes complexin-1 function in mice, but function is mostly restored upon deletion of the N-terminal region and the AH (182). Thus, the AH inhibits neurotransmitter release and the N-terminal region is key to release this inhibition. The AH of invertebrate complexin-1 also inhibits release, but deletion of the N-terminal region did not impair release in C.elegans (62, 95, 118, 180). The AH was proposed to inhibit release by replacing the C-terminus of the synaptobrevin SNARE motif in the SNARE four helix bundle, thus preventing full zippering (54, 182), and a crystal structure of a complexin-1 triple mutant with the SNARE complex led to a related model (77). However, this structure is unlikely to be formed with wild type (WT) complexin-1 and the purported interactions underlying the crystal structure were not detected in solution (114, 154) [see discussions in (126, 154)]. Indeed, no interactions of the complexin-1 AH with the SNARE motifs of partially assembled SNARE complexes were observed by NMR spectroscopy, leading to a model whereby the AH inhibits release because electrostatic and/or steric hindrance with the vesicle membrane hinder C-terminal zippering of the SNARE complex (Figure 1f, 7f), which was supported by physiological data (154). Conversely, recent data suggested that the complexin-1 AH interacts with the C-terminus of SNAP-25, hindering SNARE complex zippering (92). However, the finding that the inhibitory activity of complexin is retained when the AH is replaced by a completely unrelated helical sequence in C. elegans (118) strongly suggests that the function of AH does not involve specific protein-protein interactions and is consistent with the notion that the AH helix inhibits release due to steric hindrance with the membrane. Regardless of the underlying mechanism, strong evidence supports the conclusion that complexin hinders C-terminal zippering of trans-SNARE complexes (84, 186). It is unclear how the complexin-1 N-terminal region releases the inhibition caused by the AH, but it may be due to interactions of this region with the SNARE complex and/or the membrane (80, 179).
Figure 7.

Coupling of synaptotagmin-1, SNARE and complexin function. (a) Domain diagram of complexin-1 with selected residue numbers indicated above and ribbon diagram of the crystal structure of the SNARE complex bound to a complexin-1 fragment (30) (PDB ID 1KIL). Synaptobrevin is in red, syntaxin-1 in yellow, SNAP-25 in green and the complexin-1 fragment in orange (accessory helix) and pink (central helix). (b) Ribbon diagrams of the NMR structures of the Ca2+-bound C2A and C2B domains of synaptotagmin-1 (49, 134) (PDB IDs 1BYN and 1K5W, respectively). Ca2+ ions are shown as orange spheres. In the diagram of the C2B domain, selected residues that were implicated in binding to the SNARE complex and/or PIP2 are represented by spheres and color coded (R322 and K325 blue; K324 and K326, cyan; E295 and Y338, magenta; R398 and R399, purple). (c-e) Ribbon diagrams of a representative conformer of the ensemble of NMR structures of the synaptotagmin-1 C2B domain bound to the SNARE complex via the polybasic region (16) (c), of the crystal structure of synaptotagmin-1 C2AB bound to the SNARE complex through the primary interface (193) (d) and of the crystal structure of synaptotagmin-1 C2AB bound to a complexin-1-SNARE complex through the tripartite interface (194) (e) (PDB IDs 2N1T, 5KJ7 and 5W5C, respectively). The C2A domain is not shown. (f) Model showing how the synaptotagmin-1 C2B domain can bind simultaneously to the plasma membrane through the polybasic region and to the SNARE complex through the primary interface while complexin-1 can bind to the opposite side of the SNARE complex. The model was constructed by superimposing the structures of panels (a) and (d). Models of the plasma membrane and a synaptic vesicle are represented by gray spheres, with a PIP2 molecule in the plasma membrane shown in red. The dashed lines indicate that the C-termini of the synaptobrevin and syntaxin-1 SNARE motifs should be close to the vesicle and plasma membranes, respectively; hence, the SNARE complex cannot be fully zippered as in the structure shown. Note also that the complexin-1 accessory helix points straight toward the vesicle in this orientation and hence should hinder C-terminal zippering of the SNARE complex. (g) Model of how the Ca2+-bound synaptotagmin-1 C2B domain is expected to bind to the plasma membrane in an approximately perpendicular orientation that allows insertion of both Ca2+-binding loops into the bilayer. This orientation is incompatible with binding to the SNARE complex in the three modes of panels (c-e). Note that R322 and K325 can readily bind to PIP2 whereas K324 and K326 point away from the membrane. The color codes of panels (a, b) are also used in panels (c-g). N and C indicated the N- and C-termini of the SNARE four-helix bundle, respectively.
The C-terminal region that follows the central helix of complexin-1 plays both inhibitory and stimulatory roles in release (71). This region binds to membranes, likely helping to bind to synaptic vesicles and thus increasing the local concentration of complexin at the site of fusion (55, 133, 173), which may enhance both roles. In some isoforms, the C-terminal region is localized to membranes by farnesylation, which is key for the strong inhibitory activity of Drosophila complexin (180).
A potential explanation for the selective need of complexins for Ca2+-evoked release but not vesicle priming (183) was suggested by NMR data showing that complexin-1 binding stabilizes the C-terminal half of the SNARE four-helix bundle, which led to a model whereby priming occurs in two steps, the first one involving partial SNARE complex assembly and the second involving more fully assembled SNARE complexes that are stabilized by complexin (30). This early model is similar to that proposing two primed states of synaptic vesicles (LS and TS) (104) (Figure 1e,f, 6), with complexin-1 stabilizing TS. Interestingly, while priming as assessed by the standard treatment with 500 mM hypertonic was not impaired in complexin-1/2 DKO mice, release evoked by 250 mM sucrose was markedly impaired (179). These results suggest that, although 500 mM sucrose releases vesicles in both LS and TS, 250 mM sucrose is more discriminatory and preferentially releases vesicles in the state stabilized by complexin (TS). Note also that complexin-1 strongly inhibits disassembly of trans-SNARE complexes by NSF/αSNAP (115), suggesting that complexins may allow a larger number of SNARE complexes to remain assembled and thus favor a higher vesicular release probability.
Synaptotagmin-1 and Ca2+/phospholipid binding
Synaptotagmins are a family of proteins that contain tandem C2 domains and includes Ca2+ sensors involved in diverse forms of regulated secretion (172). Here I focus on the synaptic vesicle protein synaptotagmin-1, the Ca2+ sensor that triggers synchronous neurotransmitter release (48), but it is important to keep in mind its interplay with other Ca2+ sensors that may compete for binding to common targets. Thus, synchronous release is abrogated in synaptotagmin-1 KO mice but asynchronous release is enhanced, and this asynchronous component is almost abolished by knockdown of synaptotagmin-7 (5), showing that synaptotagmin-7 acts as a Ca2+ sensor in asynchronous release and illustrating a functional competition between synaptotagmins-1 and −7. Conversely, Synaptotagmin-1 KO mice exhibit an enhancement in spontaneous release that is not altered by synaptotagmin-7 knockdown (5). This increase likely arises because of an inhibitory activity of Ca2+-free synaptotagmin-1 (see below) and/or because of an interplay with other Ca2+ sensors such as Doc2b that remains controversial (35, 40, 107). Synaptic vesicle priming is impaired in synaptotagmin-1 KO mice (27) but is not abolished as in Munc18–1 KO or Munc13–1/2 DKO mice (158, 159). These results likely arise because Munc13 and Munc18–1 are essential for assembly of trans-SNARE complexes in the presence of NSF and αSNAP, whereas synaptotagmin-1 is not essential but markedly stimulates assembly (115).
The C2A and C2B domains of synaptotagmin-1 form most of its cytoplasmic region and bind three and two Ca2+-ions, respectively, through loops at the top of β-sandwich structures (49, 149, 156) (Figure 7b). These loops also mediate Ca2+-dependent phospholipid binding, which involves a Ca2+-induced switch in the electrostatic potential (135), coordination of Ca2+ by the phospholipid head groups and a combination of ionic and hydrophobic interactions between the loops and the lipids (28, 188). Mutations that increased or decreased the apparent Ca2+ affinity of synaptotagmin-1 binding to membranes led to parallel changes in the Ca2+ sensitivity of neurotransmitter release (48, 123), which demonstrated that synaptotagmin-1 is the major Ca2+ sensor that triggers release and that Ca2+/phospholipid binding is key for this function. Mutations in residues of the C2B domain that bind Ca2+ or phospholipids, including hydrophobic residues that insert into the lipid bilayer, cause stronger phenotypes than analogous mutations in the C2A domain because they cause dominant negative effects, but mutations in the C2A domain can also cause severe disruption of release (14, 89, 106, 146), showing that cooperation between the two C2 domains is crucial for synaptotagmin-1 function.
The synaptotagmin-1 tandem C2 domains can bind not only to one membrane but to two membranes simultaneously in the presence of Ca2+ due to the presence of multiple basic sequences on their surface, particularly that of the C2B domain (3) (Figure 7b). This observation led to the proposal that upon Ca2+ binding synaptotagmin-1 can cooperate with the SNAREs in bringing the membranes together, which was supported by diverse evidence (157, 181). Among the basic sequences, particularly important for synaptotagmin-1 function are R398,R399 at the bottom of the C2B domain, which were implicated in membrane bridging (3, 181), and a polybasic sequence on the side of the C2B domain β-sandwich, which binds to PIP2-containing membranes in the absence of Ca2+ (6). Both of these sequences also mediate binding to the SNARE complex in vitro (see below). Interestingly, the drastically different effects of distinct mutations in the polybasic region on neurotransmitter release correlate well with their distinct effects on Ca2+-dependent binding to PIP2-containing membranes (see below) but not with their indiscriminate effects on Ca2+-independent PIP2 binding (16, 160). Hence, although binding of synaptotagmin-1 to PIP2 likely stabilizes the primed states of synaptic vesicles (Figure 1e,f), what appears to be particularly critical for release is the Ca2+-dependent interaction with PIP2.
Synaptotagmin-1-SNARE-complexin coupling in neurotransmitter release
Unraveling how the actions of synaptotagmin-1, complexins and the SNAREs are coordinated to trigger fast neurotransmitter release upon Ca2+ influx has been hindered by the difficulty of distinguishing which among the many interactions that have been described between synaptotagmin-1 and the SNAREs are biologically relevant and which arise merely from the promiscuity of these proteins (126). This uncertainty also arose from the observation that a complexin-1 fragment spanning the accessory and central helices and a synaptotagmin-1 fragment spanning its two C2 domains (C2AB) bind simultaneously to the SNARE complex in solution, but compete for binding to membrane-anchored SNARE complex (152, 176). Elucidation of three synaptotagmin-1-SNARE complex structures (16, 193, 194) first added to the confusion because they revealed three different binding modes, but ultimately proved fundamental to understand the behavior of these proteins in vitro and to bring a clearer picture of how their functions are coupled in vivo.
A structure determined by NMR spectroscopy revealed a dynamic binding mode involving ionic interactions between the polybasic region of the synaptotagmin-1 C2B domain and a polyacidic area of the SNARE complex (16) (Figure 7c). In contrast, a crystal structure revealed binding of the C2B domain to the SNARE complex through a so-called primary interface involving two regions of the C2B domain, one formed largely by E295 and Y338, and another including R398,R399 (193) (Figure 7d). This interface was also observed in a subsequent crystal structure that included a complexin-1 fragment, but the crystals contained an additional interface involving binding of an α-helix at the bottom of the C2B domain to a groove of the SNARE complex, continuing the complexin-1 helix (referred to as tripartite interface) (194) (Figure 7e). A recent detailed analysis of syntaptotagmin-1-SNARE complex interactions in solution and on nanodiscs, together with previously available data, suggested that the key physiological interaction between synaptotagmin-1 and the SNARE complex is mediated by the primary interface (160). This study led to a model whereby this interaction occurs in the primed states of synaptic vesicles (Figures 1e,f) and is dissociated upon Ca2+ influx to initiate synaptic vesicle fusion (Figures 1f,g,h and 7f,g). This model challenges the long-held view that Ca2+ stimulates synaptotagmin-1-SNARE complex interactions and is supported by the following arguments.
Extensive evidence from initial studies showed that Ca2+ stimulates synaptotagmin-1-SNARE interactions in solution, but it is now clear that binding to soluble SNARE complex involves the polybasic and primary interfaces rather than the Ca2+ binding loops (160). Thus, such stimulation can be attributed to a Ca2+-induced increase in the positive electrostatic potential of the C2B domain. Ca2+ also stimulates binding of synaptotagmin-1 C2AB to SNARE complex anchored on membranes containing phosphatidyl serine (PS) (36), which also involves the polybasic and primary interfaces (160). This finding explains the competition of Ca2+-bound C2AB with the complexin-1 fragment for binding to membrane-anchored SNARE complex (152), as simultaneous binding would lead to steric clashes of the complexin-1 fragment with the membrane (16). Note however that this competition is most likely irrelevant because C2AB does not bind to SNARE complex anchored on nanodiscs containing PS and PIP2 in the presence of Ca2+ under physiological conditions including ATP (160), consistent with previous results (110). Moreover, Ca2+-dependent binding of C2AB to PS-PIP2-containing membranes occurs with very high affinity and is strongly disrupted by a R322E/K325E mutation but not by a K324E/K326E mutation in the polybasic region (160), in correlation with the effects of these mutations on neurotransmitter release (16). These results suggest that the very tight interaction of the C2B domain with PS-PIP2-containing membranes induced by Ca2+ involves an approximately perpendicular orientation incompatible with SNARE complex binding (Figure 7g). It is worth noting that the effects of the R322E/K325E and K324E/K326E mutations on release also correlated with disruption of SNARE complex binding through the polybasic interface in solution (16). However, it now appears that binding of the C2B domain polybasic region to the SNARE complex is not physiologically relevant, as it seems much more likely that the disruption of release caused by the R322E/K325E mutation arises because of impairment of the much tighter interaction of the C2B domain with PS-PIP2 -containing membranes.
In the absence of Ca2+, C2AB does bind to SNARE complex anchored on PS-PIP2-containing nanodiscs under physiological conditions including ATP (160). Such binding is dominated by the primary interface, likely because it allows simultaneous binding of the C2B domain polybasic region to PIP2 on the membrane (Figure 7f) as suggested by a low-resolution cryo-EM structure of C2AB bound to SNARE complex on lipid nanotubes (56). This arrangement is compatible with complexin-1 binding to the SNARE complex and is expected to orient the complexin-1 AH toward the vesicle membrane (Figure 7f), which would hinder final zippering of the SNARE complex and synaptic vesicle fusion. Hence, this model suggests a mechanism by which synaptotagmin-1 and complexin-1 promote the formation of a primed state where the SNARE complex is close to fully assembled and ready for release, but fusion is inhibited (Figure 1f) until the arrival of Ca2+ relieves the inhibition by inducing dissociation of synaptotagmin-1 from the SNARE complex (Figure 1g, 7g). The tight coupling between synaptotagmin-1 and complexin-1 functions predicted by this model is strongly supported by mutagenesis studies in Drosophila (69) [but see (34)]. The notion that Ca2+ dissociates synaptotagmin-1 from the SNARE complex was also suggested by the cryo-EM studies on lipid nanotubes mentioned above (56). Note also that the strong impairments of evoked neurotransmitter release caused by E295A/Y338W and R398Q/R399Q mutations designed to disrupt binding through the primary interface supported the physiological importance of this interface (193). However, while the R398Q/R399Q mutation abrogated the inhibition of spontaneous release caused by synaptotagmin-1 and increased asynchronous release, the E295A/Y338W mutant still clamped spontaneous release and did not increase asynchronous release. These findings were explained by the observation that the E295A/Y338W mutation actually enhances binding of synaptotagmin-1 to the SNARE complex, whereas the R398Q/R399Q mutation abrogates this interaction (160). Overall, these results support the notion that synaptotagmin-1 binding to the SNARE complex through the primary interface is critical to generate the primed state that is ready for fast release (Figure 1f), but this interaction needs to be released upon Ca2+ influx to allow release (Figure 1g,h).
The C2B domain-SNARE complex binding mode involving the tripartite interface (Figure 7e) was interesting because it provided an explanation for the finding that complexins are required for the dominant negative effect caused by mutations that abolish Ca2+ binding to the synaptotagmin-1 C2B domain (194). However, binding through the primary interface with concomitant binding of complexin-1 (Figure 7f) can also explain this observation. Indeed, a screen for mutations that abrogate this dominant negative effect in Drosophila yielded a large number of mutations in the primary interface but none in the tripartite interface (57). Conversely, a quintuple mutation that disrupts the primary interface did not abolish the dominant negative effect in mice (194). The basis for these different results is unclear. It is also worth noting that binding of the C2B domain to the complexin-1-SNARE complex through the tripartite interface could not be detected so far in solution by NMR spectroscopy at 85 μM concentration even after disrupting binding to the polybasic and primary interfaces (160). Moreover, in the arrangement of Figure 7f, it is difficult to envision how a second C2B domain can bind simultaneously to the SNARE complex through the tripartite interface and to PIP2 on the plasma membrane through the polybasic region. Thus, binding through the tripartite interface would require dissociation of the stronger interaction of the C2B domain with PIP2 on the plasma membrane, although it is plausible that binding to the tripartite interface is stabilized by interactions of C2B with the vesicle membrane (18). Overall, it seems premature to rule out the relevance of the tripartite interface, but also to accept it without further evidence.
An intriguing property of synaptotagmin-1 is the ability to form oligomeric rings of 11–26 units that are disrupted upon Ca2+ binding, which suggested a model whereby these rings inhibit release and Ca2+ disassembles the rings, activating neurotransmitter release (161). A concern about the significance of this property is that most of the ring images reported so far were obtained by negative stain EM, but such rings were not observed in cryo-electron tomography images of reconstituted liposomes containing synaptotagmin-1 [(53) and our unpublished results]. Nevertheless, the formation of ring-like structures has also been supported by the observation of symmetrical densities between synaptic vesicles and the plasma membrane in tomographic images of presynaptic terminals (117). Moreover, the relevance of synaptotagmin-1 rings is supported by the effects caused by a mutation (F349A) that disrupts ring formation on liposome fusion assays in vitro and on neurotransmitter release in neurons (119, 151). However, F349 is intimately involved in forming the tripartite interface between the synaptotagmin-1 C2B domain and the SNARE complex (194). Hence, the physiological effects of the F349A mutation might arise because of disruption of the tripartite interface.
Clearly, further research will be required to establish whether the synaptotagmin-1 rings occur in vivo. If they do, ring formation (not shown in Figure 1 for simplicity) can cooperate with the inhibitory activity arising from the synaptotagmin-1-SNARE-complexin-1 assembly to prevent premature release. After Ca2+ disrupts this assembly (and the putative oligomeric ring), synaptotagmin-1 may cooperate with the SNAREs in inducing membrane fusion by helping bridge the membranes and/or inducing membrane curvature (3, 94). It is also plausible that synaptotagmin-1 accelerates fusion by perturbing the lipid bilayers upon insertion of its Ca2+-binding loops, much as the Sec17 hydrophobic loop likely stimulates vacuolar fusion (169). Reconstitution experiments suggest that, in addition to initiating membrane fusion, synaptotagmin-1 plays a key role in formation and expansion of the fusion pore (37, 81). However, the mechanism underlying the role of synaptotagmin-1 in fusion remains as one of the most crucial enigmas in this field.
Perspective
The astounding progress made in understanding neurotransmitter release allows formulation of detailed models of the molecular mechanisms underlying release that have a reasonable probability of standing the test of time, such as that of Figure 1. Although some details may change, it is now clear that the release apparatus is designed to tightly control fusion by limiting the number of SNARE complexes that are formed and by imposing additional energy barriers to prevent fusion before Ca2+ influx, in an exquisite interplay where most key components play inhibitory and stimulatory roles. Munc18–1 and Munc13s are critical to organize SNARE complex assembly, but also hinder formation of too many SNARE complexes. Synaptotagmin-1 and complexins likely help to form and/or stabilize a primed state with more fully assembled SNARE complexes but also prevent premature fusion before Ca2+ influx. Nevertheless, even after all these advances, fundamental questions remain about the basic steps leading to release. In particular, it is still unclear how the SNAREs cause membrane fusion and how synaptotagmin-1 accelerates fusion. The precise mechanisms by which complexins play dual roles, Munc13s open syntaxin-1 and the template complex transitions to the SNARE complex also remain enigmatic. And how does αSNAP prevent fusion by preformed SNAREs? In the more general context of intracellular membrane fusion, is the 20S complex the true fusion machine in most systems, or is the yeast vacuolar fusion an exception?
Further progress will continue to involve structural studies, biochemical and reconstitution assays, and electrophysiological experiments. A key challenge in structural studies is the dynamic nature expected for complexes formed as the system evolves toward membrane fusion. The power of cryo-EM to analyze dynamic systems will be helpful, but it is unclear whether it will be sufficient to elucidate the last steps leading to fusion. Reconstitution experiments with minimal systems have been informative, but the more key components are included, the higher the likelihood that the results are physiologically relevant. The amounts of each component can also be critical, and physiological P:L ratios may not necessarily be optimal to recapitulate the events that lead to release. For instance, reconstitutions including Munc18–1, Munc13–1, NSF and αSNAP initially yielded such highly efficient Ca2+-dependent fusion that, even with SNARE-to-lipid ratios lower than physiological levels, it was impossible to observe stimulation by synaptotagmin-1 (85) (Figure 4b,c). However, strong stimulation by synaptotagmin-1 was observed with much lower synaptobrevin-to-lipid ratio (1:10,000) (145) (Figure 4d). As progress continues and increasing attention is paid to the regulation of release by the active zone, which includes multiple large proteins such as Munc13s and RIMs (147), it will become increasingly important to reproduce the geometry of the synapse by analyzing fusion of vesicles to flat membranes, ideally by electrophysiological methods with fast time resolution [e.g. (60)]. Exciting discoveries in this field will undoubtedly keep emerging.
Acknowledgments
I would like to thank all the members of my laboratory, as well as Thomas Sudhof, Christian Rosenmund, Diana Tomchick, Shuzo Sugita, William Wickner, Reinhard Jahn, Axel Brunger and Yeon-Kyun Shin, for many fruitful discussions, and Guillaume David, Klaudia Jaczynska, Axel Brunger and William Wickner for insightful comments on the manuscript. Work on this field in my laboratory is supported by grant I-1304 from the Welch Foundation and NIH Research Project Award R35 NS097333.
References
- 1.Acuna C, Guo Q, Burre J, Sharma M, Sun J, Sudhof TC. 2014. Microsecond dissection of neurotransmitter release: SNARE-complex assembly dictates speed and Ca(2)(+) sensitivity. Neuron 82: 1088–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aeffner S, Reusch T, Weinhausen B, Salditt T. 2012. Energetics of stalk intermediates in membrane fusion are controlled by lipid composition. Proc. Natl. Acad. Sci. U. S. A 109: E1609–E18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arac D, Chen X, Khant HA, Ubach J, Ludtke SJ, et al. 2006. Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat. Struct. Mol. Biol 13: 209–17 [DOI] [PubMed] [Google Scholar]
- 4.Aravamudan B, Fergestad T, Davis WS, Rodesch CK, Broadie K. 1999. Drosophila UNC-13 is essential for synaptic transmission. Nat. Neurosci 2: 965–71 [DOI] [PubMed] [Google Scholar]
- 5.Bacaj T, Wu D, Yang X, Morishita W, Zhou P, et al. 2013. Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80: 947–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bai J, Tucker WC, Chapman ER. 2004. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat. Struct. Mol. Biol 11: 36–44 [DOI] [PubMed] [Google Scholar]
- 7.Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM. 2015. A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science 349: 1111–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banerjee A, Barry VA, DasGupta BR, Martin TF. 1996. N-Ethylmaleimide-sensitive factor acts at a prefusion ATP-dependent step in Ca2+-activated exocytosis. J. Biol. Chem 271: 20223–26 [DOI] [PubMed] [Google Scholar]
- 9.Bao H, Das D, Courtney NA, Jiang Y, Briguglio JS, et al. 2018. Dynamics and number of trans-SNARE complexes determine nascent fusion pore properties. Nature 554: 260–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bao H, Goldschen-Ohm M, Jeggle P, Chanda B, Edwardson JM, Chapman ER. 2016. Exocytotic fusion pores are composed of both lipids and proteins. Nat Struct Mol Biol 23: 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basu J, Shen N, Dulubova I, Lu J, Guan R, et al. 2005. A minimal domain responsible for Munc13 activity. Nat. Struct. Mol. Biol 12: 1017–18 [DOI] [PubMed] [Google Scholar]
- 12.Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, et al. 2001. Functional interaction of the active zone proteins Munc13–1 and RIM1 in synaptic vesicle priming. Neuron 30: 183–96 [DOI] [PubMed] [Google Scholar]
- 13.Bharat TA, Malsam J, Hagen WJ, Scheutzow A, Sollner TH, Briggs JA. 2014. SNARE and regulatory proteins induce local membrane protrusions to prime docked vesicles for fast calcium-triggered fusion. EMBO Rep 15: 308–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowers MR, Reist NE. 2020. The C2A domain of synaptotagmin is an essential component of the calcium sensor for synaptic transmission. PLoS One 15: e0228348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowers MR, Reist NE. 2020. Synaptotagmin: Mechanisms of an electrostatic switch. Neurosci Lett 722: 134834 [DOI] [PubMed] [Google Scholar]
- 16.Brewer KD, Bacaj T, Cavalli A, Camilloni C, Swarbrick JD, et al. 2015. Dynamic binding mode of a Synaptotagmin-1-SNARE complex in solution. Nat. Struct. Mol. Biol 22: 555–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brewer KD, Li W, Horne BE, Rizo J. 2011. Reluctance to membrane binding enables accessibility of the synaptobrevin SNARE motif for SNARE complex formation. Proc. Natl. Acad. Sci. U. S. A 108: 12723–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brunger AT, Leitz J, Zhou Q, Choi UB, Lai Y. 2018. Ca(2+)-Triggered Synaptic Vesicle Fusion Initiated by Release of Inhibition. Trends Cell Biol 28: 631–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D. 2008. Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. EMBO J 27: 923–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camacho M, Basu J, Trimbuch T, Chang S, Pulido-Lozano C, et al. 2017. Heterodimerization of Munc13 C2A domain with RIM regulates synaptic vesicle docking and priming. Nat Commun 8: 15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camacho M, Quade B, Trimbuch T, Xu J, Sari L, et al. Control of neurotransmitter release by two distinct membrane-binding faces of the Munc13–1 C1-C2B region. eLife, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carr CM, Rizo J. 2010. At the junction of SNARE and SM protein function. Curr. Opin. Cell Biol 22: 488–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castillo PE, Janz R, Sudhof TC, Tzounopoulos T, Malenka RC, Nicoll RA. 1997. Rab3A is essential for mossy fibre long-term potentiation in the hippocampus. Nature 388: 590–93 [DOI] [PubMed] [Google Scholar]
- 24.Castillo PE, Schoch S, Schmitz F, Sudhof TC, Malenka RC. 2002. RIM1alpha is required for presynaptic long-term potentiation. Nature 415: 327–30 [DOI] [PubMed] [Google Scholar]
- 25.Chan YH, van LB, Boxer SG. 2009. Effects of linker sequences on vesicle fusion mediated by lipid-anchored DNA oligonucleotides. Proc. Natl. Acad. Sci. U. S. A 106: 979–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang LF, Chen S, Liu CC, Pan X, Jiang J, et al. 2012. Structural characterization of full-length NSF and 20S particles. Nat Struct Mol Biol 19: 268–75 [DOI] [PubMed] [Google Scholar]
- 27.Chang S, Trimbuch T, Rosenmund C. 2018. Synaptotagmin-1 drives synchronous Ca(2+)-triggered fusion by C2B-domain-mediated synaptic-vesicle-membrane attachment. Nat Neurosci 21: 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chapman ER, Davis AF. 1998. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J. Biol. Chem 273: 13995–4001 [DOI] [PubMed] [Google Scholar]
- 29.Chen X, Lu J, Dulubova I, Rizo J. 2008. NMR analysis of the closed conformation of syntaxin-1. J. Biomol. NMR 41: 43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X, Tomchick DR, Kovrigin E, Arac D, Machius M, et al. 2002. Three-dimensional structure of the complexin/SNARE complex. Neuron 33: 397–409 [DOI] [PubMed] [Google Scholar]
- 31.Chernomordik LV, Kozlov MM. 2008. Mechanics of membrane fusion. Nat. Struct. Mol. Biol 15: 675–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi UB, Zhao M, White KI, Pfuetzner RA, Esquivies L, et al. 2018. NSF-mediated disassembly of on and off-pathway SNARE complexes and inhibition by complexin. eLife 7: e36497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cohen FS, Melikyan GB. 2004. The energetics of membrane fusion from binding, through hemifusion, pore formation, and pore enlargement. J. Membr. Biol 199: 1–14 [DOI] [PubMed] [Google Scholar]
- 34.Courtney NA, Bao H, Briguglio JS, Chapman ER. 2019. Synaptotagmin 1 clamps synaptic vesicle fusion in mammalian neurons independent of complexin. Nat Commun 10: 4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Courtney NA, Briguglio JS, Bradberry MM, Greer C, Chapman ER. 2018. Excitatory and Inhibitory Neurons Utilize Different Ca(2+) Sensors and Sources to Regulate Spontaneous Release. Neuron 98: 977–91 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dai H, Shen N, Arac D, Rizo J. 2007. A Quaternary SNARE-Synaptotagmin-Ca(2+)-Phospholipid Complex in Neurotransmitter Release. J. Mol. Biol 367: 848–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Das D, Bao H, Courtney KC, Wu L, Chapman ER. 2020. Resolving kinetic intermediates during the regulated assembly and disassembly of fusion pores. Nat Commun 11: 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng L, Kaeser PS, Xu W, Sudhof TC. 2011. RIM proteins activate vesicle priming by reversing autoinhibitory homodimerization of Munc13. Neuron 69: 317–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diao J, Grob P, Cipriano DJ, Kyoung M, Zhang Y, et al. 2012. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. eLife 1: e00109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diez-Arazola R, Meijer M, Bourgeois-Jaarsma Q, Cornelisse LN, Verhage M, Groffen AJ. 2020. Doc2 Proteins Are Not Required for the Increased Spontaneous Release Rate in Synaptotagmin-1-Deficient Neurons. J Neurosci 40: 2606–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dittman JS. 2019. Unc13: a multifunctional synaptic marvel. Curr Opin Neurobiol 57: 17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Domanska MK, Kiessling V, Stein A, Fasshauer D, Tamm LK. 2009. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J. Biol. Chem 284: 32158–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dulubova I, Lou X, Lu J, Huryeva I, Alam A, et al. 2005. A Munc13/RIM/Rab3 tripartite complex: from priming to plasticity? EMBO J 24: 2839–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, et al. 1999. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J 18: 4372–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dulubova I, Yamaguchi T, Gao Y, Min SW, Huryeva I, et al. 2002. How Tlg2p/syntaxin 16 ‘snares’ Vps45. EMBO J 21: 3620–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dulubova I, Yamaguchi T, Wang Y, Sudhof TC, Rizo J. 2001. Vam3p structure reveals conserved and divergent properties of syntaxins. Nat. Struct. Biol 8: 258–64 [DOI] [PubMed] [Google Scholar]
- 47.Fasshauer D, Sutton RB, Brunger AT, Jahn R. 1998. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc. Natl. Acad. Sci. U. S. A 95: 15781–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, et al. 2001. Synaptotagmin I functions as a calcium regulator of release probability. Nature 410: 41–49 [DOI] [PubMed] [Google Scholar]
- 49.Fernandez I, Arac D, Ubach J, Gerber SH, Shin O, et al. 2001. Three-dimensional structure of the synaptotagmin 1 c(2)b-domain. Synaptotagmin 1 as a phospholipid binding machine. Neuron 32: 1057–69 [DOI] [PubMed] [Google Scholar]
- 50.Fernandez I, Ubach J, Dulubova I, Zhang X, Sudhof TC, Rizo J. 1998. Three-dimensional structure of an evolutionarily conserved N-terminal domain of syntaxin 1A. Cell 94: 841–49 [DOI] [PubMed] [Google Scholar]
- 51.Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, et al. 2012. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337: 1340–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gerber SH, Rah JC, Min SW, Liu X, de WH, et al. 2008. Conformational switch of syntaxin-1 controls synaptic vesicle fusion. Science 321: 1507–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gipson P, Fukuda Y, Danev R, Lai Y, Chen DH, et al. 2017. Morphologies of synaptic protein membrane fusion interfaces. Proc Natl Acad Sci U S A 114: 9110–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giraudo CG, Garcia-Diaz A, Eng WS, Chen Y, Hendrickson WA, et al. 2009. Alternative zippering as an on-off switch for SNARE-mediated fusion. Science 323: 512–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gong J, Lai Y, Li X, Wang M, Leitz J, et al. 2016. C-terminal domain of mammalian complexin-1 localizes to highly curved membranes. Proc Natl Acad Sci U S A 113: E7590–E99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grushin K, Wang J, Coleman J, Rothman JE, Sindelar CV, Krishnakumar SS. 2019. Structural basis for the clamping and Ca(2+) activation of SNARE-mediated fusion by synaptotagmin. Nat Commun 10: 2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guan Z, Bykhovskaia M, Jorquera RA, Sutton RB, Akbergenova Y, Littleton JT. 2017. A synaptotagmin suppressor screen indicates SNARE binding controls the timing and Ca(2+) cooperativity of vesicle fusion. eLife 6: e28409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hanson PI, Heuser JE, Jahn R. 1997. Neurotransmitter release - four years of SNARE complexes. Curr. Opin. Neurobiol 7: 310–15 [DOI] [PubMed] [Google Scholar]
- 59.He E, Wierda K, van Westen R, Broeke JH, Toonen RF, et al. 2017. Munc13–1 and Munc18–1 together prevent NSF-dependent de-priming of synaptic vesicles. Nat Commun 8: 15915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heo P, Coleman J, Fleury JB, Rothman JE, Pincet F. 2021. Nascent fusion pore opening monitored at single-SNAREpin resolution. Proc Natl Acad Sci U S A 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hernandez JM, Stein A, Behrmann E, Riedel D, Cypionka A, et al. 2012. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science 336: 1581–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hobson RJ, Liu Q, Watanabe S, Jorgensen EM. 2011. Complexin Maintains Vesicles in the Primed State in C. elegans. Curr. Biol 21: 106–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu Y, Zhu L, Ma C. 2020. Structural Roles for the Juxtamembrane Linker Region and Transmembrane Region of Synaptobrevin 2 in Membrane Fusion. Front Cell Dev Biol 8: 609708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hu Z, Tong XJ, Kaplan JM. 2013. UNC-13L, UNC-13S, and Tomosyn form a protein code for fast and slow neurotransmitter release in Caenorhabditis elegans. eLife 2: e00967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang X, Sun S, Wang X, Fan F, Zhou Q, et al. 2019. Mechanistic insights into the SNARE complex disassembly. Sci Adv 5: eaau8164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huntwork S, Littleton JT. 2007. A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat. Neurosci 10: 1235–37 [DOI] [PubMed] [Google Scholar]
- 67.Jahn R, Scheller RH. 2006. SNAREs--engines for membrane fusion. Nat. Rev. Mol. Cell Biol 7: 631–43 [DOI] [PubMed] [Google Scholar]
- 68.Jiao J, He M, Port SA, Baker RW, Xu Y, et al. 2018. Munc18–1 catalyzes neuronal SNARE assembly by templating SNARE association. eLife 7: e41771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jorquera RA, Huntwork-Rodriguez S, Akbergenova Y, Cho RW, Littleton JT. 2012. Complexin controls spontaneous and evoked neurotransmitter release by regulating the timing and properties of synaptotagmin activity. J. Neurosci 32: 18234–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Junge HJ, Rhee JS, Jahn O, Varoqueaux F, Spiess J, et al. 2004. Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell 118: 389–401 [DOI] [PubMed] [Google Scholar]
- 71.Kaeser-Woo YJ, Yang X, Sudhof TC. 2012. C-terminal complexin sequence is selectively required for clamping and priming but not for Ca2+ triggering of synaptic exocytosis. J. Neurosci 32: 2877–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kalyana Sundaram RV, Jin H, Li F, Shu T, Coleman J, et al. 2021. Munc13 binds and recruits SNAP25 to chaperone SNARE complex assembly. FEBS Lett 595: 297–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Khvotchev M, Dulubova I, Sun J, Dai H, Rizo J, Sudhof TC. 2007. Dual modes of Munc18–1/SNARE interactions are coupled by functionally critical binding to syntaxin-1 N terminus. J. Neurosci 27: 12147–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim J, Shin YK. 2017. Productive and Non-productive Pathways for Synaptotagmin 1 to Support Ca(2+)-Triggered Fast Exocytosis. Front Mol Neurosci 10: 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koushika SP, Richmond JE, Hadwiger G, Weimer RM, Jorgensen EM, Nonet ML. 2001. A post-docking role for active zone protein Rim. Nat. Neurosci 4: 997–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuhlmann JW, Junius M, Diederichsen U, Steinem C. 2017. SNARE-Mediated Single-Vesicle Fusion Events with Supported and Freestanding Lipid Membranes. Biophys J 112: 2348–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kummel D, Krishnakumar SS, Radoff DT, Li F, Giraudo CG, et al. 2011. Complexin cross-links prefusion SNAREs into a zigzag array. Nat. Struct. Mol. Biol 18: 927–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kyoung M, Srivastava A, Zhang Y, Diao J, Vrljic M, et al. 2011. In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc. Natl. Acad. Sci. U. S. A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lai Y, Choi UB, Leitz J, Rhee HJ, Lee C, et al. 2017. Molecular Mechanisms of Synaptic Vesicle Priming by Munc13 and Munc18. Neuron 95: 591–607 e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lai Y, Diao J, Cipriano DJ, Zhang Y, Pfuetzner RA, et al. 2014. Complexin inhibits spontaneous release and synchronizes Ca2+-triggered synaptic vesicle fusion by distinct mechanisms. eLife 3: e03756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lai Y, Diao J, Liu Y, Ishitsuka Y, Su Z, et al. 2013. Fusion pore formation and expansion induced by Ca2+ and synaptotagmin 1. Proc. Natl. Acad. Sci. U. S. A 110: 1333–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee HK, Yang Y, Su Z, Hyeon C, Lee TS, et al. 2010. Dynamic Ca2+-dependent stimulation of vesicle fusion by membrane-anchored synaptotagmin 1. Science 328: 760–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li F, Pincet F, Perez E, Eng WS, Melia TJ, et al. 2007. Energetics and dynamics of SNAREpin folding across lipid bilayers. Nat. Struct. Mol. Biol 14: 890–96 [DOI] [PubMed] [Google Scholar]
- 84.Li F, Pincet F, Perez E, Giraudo CG, Tareste D, Rothman JE. 2011. Complexin activates and clamps SNAREpins by a common mechanism involving an intermediate energetic state. Nat. Struct. Mol. Biol 18: 941–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu X, Seven AB, Camacho M, Esser V, Xu J, et al. 2016. Functional synergy between the Munc13 C-terminal C1 and C2 domains. eLife 5: e13696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu J, Machius M, Dulubova I, Dai H, Sudhof TC, et al. 2006. Structural Basis for a Munc13–1 Homodimer to Munc13–1/RIM Heterodimer Switch. PLoS. Biol 4: e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ma C, Li W, Xu Y, Rizo J. 2011. Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat. Struct. Mol. Biol 18: 542–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ma C, Su L, Seven AB, Xu Y, Rizo J. 2013. Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339: 421–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE. 2002. The C(2)B Ca(2+)-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature 418: 340–44 [DOI] [PubMed] [Google Scholar]
- 90.Magdziarek M, Bolembach AA, Stepien KP, Quade B, Liu X, Rizo J. 2020. Re-examining how Munc13–1 facilitates opening of syntaxin-1. Protein Sci 29: 1440–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Malhotra V, Orci L, Glick BS, Block MR, Rothman JE. 1988. Role of an N-ethylmaleimide-sensitive transport component in promoting fusion of transport vesicles with cisternae of the Golgi stack. Cell 54: 221–7 [DOI] [PubMed] [Google Scholar]
- 92.Malsam J, Barfuss S, Trimbuch T, Zarebidaki F, Sonnen AF, et al. 2020. Complexin Suppresses Spontaneous Exocytosis by Capturing the Membrane-Proximal Regions of VAMP2 and SNAP25. Cell Rep 32: 107926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Malsam J, Parisotto D, Bharat TA, Scheutzow A, Krause JM, et al. 2012. Complexin arrests a pool of docked vesicles for fast Ca2+-dependent release. EMBO J 31: 3270–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Martens S, Kozlov MM, McMahon HT. 2007. How synaptotagmin promotes membrane fusion. Science 316: 1205–08 [DOI] [PubMed] [Google Scholar]
- 95.Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS. 2011. Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr. Biol 21: 97–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC. 2009. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science 323: 516–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mayer A, Wickner W, Haas A. 1996. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell 85: 83–94 [DOI] [PubMed] [Google Scholar]
- 98.McMahon HT, Missler M, Li C, Sudhof TC. 1995. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell 83: 111–19 [DOI] [PubMed] [Google Scholar]
- 99.Meijer M, Dorr B, Lammertse HC, Blithikioti C, van Weering JR, et al. 2017. Tyrosine phosphorylation of Munc18–1 inhibits synaptic transmission by preventing SNARE assembly. EMBO J [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Michelassi F, Liu H, Hu Z, Dittman JS. 2017. A C1-C2 Module in Munc13 Inhibits Calcium-Dependent Neurotransmitter Release. Neuron 95: 577–90 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mima J, Hickey CM, Xu H, Jun Y, Wickner W. 2008. Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones. EMBO J 27: 2031–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Misura KM, Scheller RH, Weis WI. 2000. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature 404: 355–62 [DOI] [PubMed] [Google Scholar]
- 103.Mohrmann R, de WH, Verhage M, Neher E, Sorensen JB. 2010. Fast vesicle fusion in living cells requires at least three SNARE complexes. Science 330: 502–05 [DOI] [PubMed] [Google Scholar]
- 104.Neher E, Brose N. 2018. Dynamically Primed Synaptic Vesicle States: Key to Understand Synaptic Short-Term Plasticity. Neuron 100: 1283–91 [DOI] [PubMed] [Google Scholar]
- 105.Pabst S, Hazzard JW, Antonin W, Sudhof TC, Jahn R, et al. 2000. Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J. Biol. Chem 275: 19808–18 [DOI] [PubMed] [Google Scholar]
- 106.Paddock BE, Wang Z, Biela LM, Chen K, Getzy MD, et al. 2011. Membrane penetration by synaptotagmin is required for coupling calcium binding to vesicle fusion in vivo. J. Neurosci 31: 2248–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Sudhof TC. 2011. Doc2 supports spontaneous synaptic transmission by a Ca(2+)-independent mechanism. Neuron 70: 244–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Parisotto D, Pfau M, Scheutzow A, Wild K, Mayer MP, et al. 2014. An extended helical conformation in domain 3a of Munc18–1 provides a template for SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) complex assembly. J. Biol. Chem 289: 9639–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Park S, Bin NR, Yu B, Wong R, Sitarska E, et al. 2017. UNC-18 and Tomosyn Antagonistically Control Synaptic Vesicle Priming Downstream of UNC-13 in Caenorhabditis elegans. J Neurosci 37: 8797–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Park Y, Seo JB, Fraind A, Perez-Lara A, Yavuz H, et al. 2015. Synaptotagmin-1 binds to PIP(2)-containing membrane but not to SNAREs at physiological ionic strength. Nat Struct Mol Biol 22: 815–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Park Y, Vennekate W, Yavuz H, Preobraschenski J, Hernandez JM, et al. 2014. alpha-SNAP interferes with the zippering of the SNARE protein membrane fusion machinery. J Biol Chem 289: 16326–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pei J, Ma C, Rizo J, Grishin NV. 2009. Remote homology between Munc13 MUN domain and vesicle tethering complexes. J. Mol. Biol 391: 509–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Poirier MA, Xiao W, Macosko JC, Chan C, Shin YK, Bennett MK. 1998. The synaptic SNARE complex is a parallel four-stranded helical bundle. Nat. Struct. Biol 5: 765–69 [DOI] [PubMed] [Google Scholar]
- 114.Prinslow EA, Brautigam CA, Rizo J. 2017. Reconciling isothermal titration calorimetry analyses of interactions between complexin and truncated SNARE complexes. eLife 6: e30286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Prinslow EA, Stepien KP, Pan YZ, Xu J, Rizo J. 2019. Multiple factors maintain assembled trans-SNARE complexes in the presence of NSF and alphaSNAP. eLife 8: e38880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Quade B, Camacho M, Zhao X, Orlando M, Trimbuch T, et al. 2019. Membrane bridging by Munc13–1 is crucial for neurotransmitter release. eLife 8: e42806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Radhakrishnan A, Li X, Grushin K, Krishnakumar SS, Liu J, Rothman JE. 2021. Symmetrical arrangement of proteins under release-ready vesicles in presynaptic terminals. Proc Natl Acad Sci U S A 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Radoff DT, Dong Y, Snead D, Bai J, Eliezer D, Dittman JS. 2014. The accessory helix of complexin functions by stabilizing central helix secondary structure. eLife 3: e04553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ramakrishnan S, Bera M, Coleman J, Krishnakumar SS, Pincet F, Rothman JE. 2019. Synaptotagmin oligomers are necessary and can be sufficient to form a Ca(2+) -sensitive fusion clamp. FEBS Lett 593: 154–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Reddy-Alla S, Bohme MA, Reynolds E, Beis C, Grasskamp AT, et al. 2017. Stable Positioning of Unc13 Restricts Synaptic Vesicle Fusion to Defined Release Sites to Promote Synchronous Neurotransmission. Neuron 95: 1350–64 e12 [DOI] [PubMed] [Google Scholar]
- 121.Regehr WG. 2012. Short-term presynaptic plasticity. Cold Spring Harb. Perspect. Biol 4: a005702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rhee JS, Betz A, Pyott S, Reim K, Varoqueaux F, et al. 2002. Beta phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 108: 121–33 [DOI] [PubMed] [Google Scholar]
- 123.Rhee JS, Li LY, Shin OH, Rah JC, Rizo J, et al. 2005. Augmenting neurotransmitter release by enhancing the apparent Ca2+ affinity of synaptotagmin 1. Proc. Natl. Acad. Sci. U. S. A 102: 18664–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Richmond JE, Davis WS, Jorgensen EM. 1999. UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat. Neurosci 2: 959–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Richmond JE, Weimer RM, Jorgensen EM. 2001. An open form of syntaxin bypasses the requirement for UNC-13 in vesicle priming. Nature 412: 338–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rizo J 2018. Mechanism of neurotransmitter release coming into focus. Protein Sci 27: 1364–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rizo J, Sudhof TC. 1998. C2-domains, structure and function of a universal Ca2+-binding domain. J. Biol. Chem 273: 15879–82 [DOI] [PubMed] [Google Scholar]
- 128.Rizo J, Sudhof TC. 2012. The Membrane Fusion Enigma: SNAREs, Sec1/Munc18 Proteins, and Their Accomplices-Guilty as Charged? Annu. Rev. Cell Dev. Biol 28: 279–308 [DOI] [PubMed] [Google Scholar]
- 129.Rothman JE, Krishnakumar SS, Grushin K, Pincet F. 2017. Hypothesis - buttressed rings assemble, clamp, and release SNAREpins for synaptic transmission. FEBS Lett 591: 3459–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ryu JK, Jahn R, Yoon TY. 2016. Review: Progresses in understanding N-ethylmaleimide sensitive factor (NSF) mediated disassembly of SNARE complexes. Biopolymers 105: 518–31 [DOI] [PubMed] [Google Scholar]
- 131.Sakamoto H, Ariyoshi T, Kimpara N, Sugao K, Taiko I, et al. 2018. Synaptic weight set by Munc13–1 supramolecular assemblies. Nat Neurosci 21: 41–49 [DOI] [PubMed] [Google Scholar]
- 132.Schwartz ML, Merz AJ. 2009. Capture and release of partially zipped trans-SNARE complexes on intact organelles. J Cell Biol 185: 535–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Seiler F, Malsam J, Krause JM, Sollner TH. 2009. A role of complexin-lipid interactions in membrane fusion. FEBS Lett 583: 2343–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shao X, Fernandez I, Sudhof TC, Rizo J. 1998. Solution structures of the Ca2+-free and Ca2+-bound C2A domain of synaptotagmin I: does Ca2+ induce a conformational change? Biochemistry 37: 16106–15 [DOI] [PubMed] [Google Scholar]
- 135.Shao X, Li C, Fernandez I, Zhang X, Sudhof TC, Rizo J. 1997. Synaptotagmin-syntaxin interaction: the C2 domain as a Ca2+-dependent electrostatic switch. Neuron 18: 133–42 [DOI] [PubMed] [Google Scholar]
- 136.Shi L, Shen QT, Kiel A, Wang J, Wang HW, et al. 2012. SNARE proteins: one to fuse and three to keep the nascent fusion pore open. Science 335: 1355–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shin OH, Lu J, Rhee JS, Tomchick DR, Pang ZP, et al. 2010. Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat. Struct. Mol. Biol 17: 280–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Shu T, Jin H, Rothman JE, Zhang Y. 2020. Munc13–1 MUN domain and Munc18–1 cooperatively chaperone SNARE assembly through a tetrameric complex. Proc Natl Acad Sci U S A 117: 1036–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sinha R, Ahmed S, Jahn R, Klingauf J. 2011. Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc. Natl. Acad. Sci. U. S. A 108: 14318–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sitarska E, Xu J, Park S, Liu X, Quade B, et al. 2017. Autoinhibition of Munc18–1 modulates synaptobrevin binding and helps to enable Munc13-dependent regulation of membrane fusion. eLife 6: e24278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sollner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. 1993. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75: 409–18 [DOI] [PubMed] [Google Scholar]
- 142.Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, et al. 1993. SNAP receptors implicated in vesicle targeting and fusion. Nature 362: 318–24 [DOI] [PubMed] [Google Scholar]
- 143.Song H, Torng TL, Orr AS, Brunger AT, Wickner WT. 2021. Sec17/Sec18 can support membrane fusion without help from completion of SNARE zippering. eLife 10: e67578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Stepien KP, Prinslow EA, Rizo J. 2019. Munc18–1 is crucial to overcome the inhibition of synaptic vesicle fusion by alphaSNAP. Nat Commun 10: 4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stepien KP, Rizo J. 2021. Synaptotagmin-1-, Munc18–1-, and Munc13–1-dependent liposome fusion with a few neuronal SNAREs. Proc Natl Acad Sci U S A 118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Striegel AR, Biela LM, Evans CS, Wang Z, Delehoy JB, et al. 2012. Calcium binding by synaptotagmin’s C2A domain is an essential element of the electrostatic switch that triggers synchronous synaptic transmission. J. Neurosci 32: 1253–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sudhof TC. 2013. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80: 675–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sudhof TC, Rothman JE. 2009. Membrane fusion: grappling with SNARE and SM proteins. Science 323: 474–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sutton RB, Davletov BA, Berghuis AM, Sudhof TC, Sprang SR. 1995. Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell 80: 929–38 [DOI] [PubMed] [Google Scholar]
- 150.Sutton RB, Fasshauer D, Jahn R, Brunger AT. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395: 347–53 [DOI] [PubMed] [Google Scholar]
- 151.Tagliatti E, Bello OD, Mendonca PRF, Kotzadimitriou D, Nicholson E, et al. 2020. Synaptotagmin 1 oligomers clamp and regulate different modes of neurotransmitter release. Proc Natl Acad Sci U S A 117: 3819–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tang J, Maximov A, Shin OH, Dai H, Rizo J, Sudhof TC. 2006. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126: 1175–87 [DOI] [PubMed] [Google Scholar]
- 153.Tien CW, Yu B, Huang M, Stepien KP, Sugita K, et al. 2020. Open syntaxin overcomes exocytosis defects of diverse mutants in C. elegans. Nat Commun 11: 5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Trimbuch T, Xu J, Flaherty D, Tomchick DR, Rizo J, Rosenmund C. 2014. Re-examining how complexin inhibits neurotransmitter release. eLife 3: e02391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tucker WC, Weber T, Chapman ER. 2004. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304: 435–38 [DOI] [PubMed] [Google Scholar]
- 156.Ubach J, Zhang X, Shao X, Sudhof TC, Rizo J. 1998. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J 17: 3921–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.van den BG, Thutupalli S, Risselada JH, Meyenberg K, Holt M, et al. 2011. Synaptotagmin-1 may be a distance regulator acting upstream of SNARE nucleation. Nat. Struct. Mol. Biol 18: 805–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, et al. 2002. Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13-mediated vesicle priming. Proc. Natl. Acad. Sci. U. S. A 99: 9037–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, et al. 2000. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287: 864–69 [DOI] [PubMed] [Google Scholar]
- 160.Voleti R, Jaczynska K, Rizo J. 2020. Ca(2+)-dependent release of Synaptotagmin-1 from the SNARE complex on phosphatidylinositol 4,5-bisphosphate-containing membranes. eLife 9: e57154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang J, Bello O, Auclair SM, Wang J, Coleman J, et al. 2014. Calcium sensitive ring-like oligomers formed by synaptotagmin. Proc Natl Acad Sci U S A 111: 13966–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wang S, Choi UB, Gong J, Yang X, Li Y, et al. 2017. Conformational change of syntaxin linker region induced by Munc13s initiates SNARE complex formation in synaptic exocytosis. EMBO J 36: 816–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang S, Li Y, Gong J, Ye S, Yang X, et al. 2019. Munc18 and Munc13 serve as a functional template to orchestrate neuronal SNARE complex assembly. Nat Commun 10: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang X, Gong J, Zhu L, Wang S, Yang X, et al. 2020. Munc13 activates the Munc18–1/syntaxin-1 complex and enables Munc18–1 to prime SNARE assembly. EMBO J 39: e103631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, et al. 1998. SNAREpins: minimal machinery for membrane fusion. Cell 92: 759–72 [DOI] [PubMed] [Google Scholar]
- 166.White KI, Zhao M, Choi UB, Pfuetzner RA, Brunger AT. 2018. Structural principles of SNARE complex recognition by the AAA+ protein NSF. eLife 7: e38888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Whiteheart SW, Brunner M, Wilson DW, Wiedmann M, Rothman JE. 1992. Soluble N-ethylmaleimide-sensitive fusion attachment proteins (SNAPs) bind to a multi-SNAP receptor complex in Golgi membranes. J Biol Chem 267: 12239–43 [PubMed] [Google Scholar]
- 168.Wickner W 2010. Membrane fusion: five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Annu. Rev. Cell Dev. Biol 26: 115–36 [DOI] [PubMed] [Google Scholar]
- 169.Wickner W, Rizo J. 2017. A cascade of multiple proteins and lipids catalyzes membrane fusion. Mol Biol Cell 28: 707–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Winter U, Chen X, Fasshauer D. 2009. A conserved membrane attachment site in alpha-SNAP facilitates N-ethylmaleimide-sensitive factor (NSF)-driven SNARE complex disassembly. J Biol Chem 284: 31817–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Witkowska A, Heinz LP, Grubmuller H, Jahn R. 2021. Tight docking of membranes before fusion represents a metastable state with unique properties. Nat Commun 12: 3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wolfes AC, Dean C. 2020. The diversity of synaptotagmin isoforms. Curr Opin Neurobiol 63: 198–209 [DOI] [PubMed] [Google Scholar]
- 173.Wragg RT, Snead D, Dong Y, Ramlall TF, Menon I, et al. 2013. Synaptic vesicles position complexin to block spontaneous fusion. Neuron 77: 323–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wu Z, Auclair SM, Bello O, Vennekate W, Dudzinski NR, et al. 2016. Nanodisc-cell fusion: control of fusion pore nucleation and lifetimes by SNARE protein transmembrane domains. Sci Rep 6: 27287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Xu H, Jun Y, Thompson J, Yates J, Wickner W. 2010. HOPS prevents the disassembly of trans-SNARE complexes by Sec17p/Sec18p during membrane fusion. EMBO J 29: 1948–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Xu J, Brewer KD, Perez-Castillejos R, Rizo J. 2013. Subtle Interplay between Synaptotagmin and Complexin Binding to the SNARE Complex. J. Mol. Biol 425: 3461–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Xu J, Camacho M, Xu Y, Esser V, Liu X, et al. 2017. Mechanistic insights into neurotransmitter release and presynaptic plasticity from the crystal structure of Munc13–1 C1C2 BMUN. eLife 6: e22567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Xu Y, Su L, Rizo J. 2010. Binding of Munc18–1 to Synaptobrevin and to the SNARE Four-Helix Bundle. Biochemistry 49: 1568–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Xue M, Craig TK, Xu J, Chao HT, Rizo J, Rosenmund C. 2010. Binding of the complexin N terminus to the SNARE complex potentiates synaptic-vesicle fusogenicity. Nat. Struct. Mol. Biol 17: 568–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Xue M, Lin YQ, Pan H, Reim K, Deng H, et al. 2009. Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila Complexins orchestrates synaptic vesicle exocytosis. Neuron 64: 367–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Xue M, Ma C, Craig TK, Rosenmund C, Rizo J. 2008. The Janus-faced nature of the C(2)B domain is fundamental for synaptotagmin-1 function. Nat. Struct. Mol. Biol 15: 1160–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Xue M, Reim K, Chen X, Chao HT, Deng H, et al. 2007. Distinct domains of complexin I differentially regulate neurotransmitter release. Nat. Struct. Mol. Biol 14: 949–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Xue M, Stradomska A, Chen H, Brose N, Zhang W, et al. 2008. Complexins facilitate neurotransmitter release at excitatory and inhibitory synapses in mammalian central nervous system. Proc. Natl. Acad. Sci. U. S. A 105: 7875–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yang X, Wang S, Sheng Y, Zhang M, Zou W, et al. 2015. Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic-vesicle priming. Nat. Struct. Mol. Biol 22: 547–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yavuz H, Kattan I, Hernandez JM, Hofnagel O, Witkowska A, et al. 2018. Arrest of trans-SNARE zippering uncovers loosely and tightly docked intermediates in membrane fusion. J Biol Chem 293: 8645–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yin L, Kim J, Shin YK. 2016. Complexin splits the membrane-proximal region of a single SNAREpin. Biochem J 473: 2219–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Yoon T-Y, Okumus B, Zhang F, Shin YK, Ha T. 2006. Multiple intermediates in SNARE-induced membrane fusion. Proc. Natl. Acad. Sci. U. S. A 103:19731–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zhang X, Rizo J, Sudhof TC. 1998. Mechanism of phospholipid binding by the C2A-domain of synaptotagmin I. Biochemistry 37: 12395–403 [DOI] [PubMed] [Google Scholar]
- 189.Zhang Y, Hughson FM. 2021. Chaperoning SNARE Folding and Assembly. Annu Rev Biochem 90: 581–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Zhao M, Wu S, Zhou Q, Vivona S, Cipriano DJ, et al. 2015. Mechanistic insights into the recycling machine of the SNARE complex. Nature 518: 61–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhou K, Stawicki TM, Goncharov A, Jin Y. 2013. Position of UNC-13 in the active zone regulates synaptic vesicle release probability and release kinetics. eLife 2: e01180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhou P, Pang ZP, Yang X, Zhang Y, Rosenmund C, et al. 2013. Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. EMBO J 32: 159–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, et al. 2015. Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature 525: 62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhou Q, Zhou P, Wang AL, Wu D, Zhao M, et al. 2017. The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 548: 420–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zick M, Wickner W. 2016. Improved reconstitution of yeast vacuole fusion with physiological SNARE concentrations reveals an asymmetric Rab(GTP) requirement. Mol Biol Cell 27: 2590–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zick M, Wickner WT. 2014. A distinct tethering step is vital for vacuole membrane fusion. eLife 3: e03251 [DOI] [PMC free article] [PubMed] [Google Scholar]