

Summary
Glycyrrhiza uralensis Fisch is a medicinal plant widely used to treat multiple diseases in Europe and Asia, and its efficacy largely depends on liquiritin and glycyrrhizic acid. The regulatory pattern responsible for the difference in efficacy between wild and cultivated G. uralensis remains largely undetermined. Here, we collected roots and rhizosphere soils from wild (WT) G. uralensis as well as those farmed for 1 year (C1) and 3 years (C3), generated metabolite and transcript data for roots, microbiota data for rhizospheres and conducted comprehensive multi‐omics analyses. We updated gene structures for all 40 091 genes in G. uralensis, and based on 52 differentially expressed genes, we charted the route‐map of both liquiritin and glycyrrhizic acid biosynthesis, with genes BAS, CYP72A154 and CYP88D6 critical for glycyrrhizic acid biosynthesis being significantly expressed higher in wild G. uralensis than in cultivated G. uralensis. Additionally, multi‐omics network analysis identified that Lysobacter was strongly associated with CYP72A154, which was required for glycyrrhizic acid biosynthesis. Finally, we developed a holistic multi‐omics regulation model that confirmed the importance of rhizosphere microbial community structure in liquiritin accumulation. This study thoroughly decoded the key regulatory mechanisms of liquiritin and glycyrrhizic acid, and provided new insights into the interactions of the plant's key metabolites with its transcriptome, rhizosphere microbes and environment, which would guide future cultivation of G. uralensis.
Keywords: G. uralensis, transcriptomics, rhizosphere microbes, liquiritin, glycyrrhizic acid, regulation patterns
The transcripts, metabolite profiles and rhizosphere microbial communities of wild and cultivated Glycyrrhiza uralensis Fisch were characterized and compared. The accumulations of both liquiritin and glycyrrhizic acid in wild G. uralensis were higher than those of cultivated G. uralensis. Significant gene expression differences were observed for G. uralensis with different growth status (wild and cultivated), especially for those genes involved in biosynthesis of liquiritin and glycyrrhizic acid. Rhizosphere microbial community structures have profound influences on the accumulation of liquiritin for G. uralensis.

Introduction
Licorice is one of the most important crude drugs in Asia and Europe, having been used in traditional medicine for 1000 of years (Kitagawa, 2002). Licorice comes from the roots and rhizomes (medicinal parts) of several Glycyrrhiza species including Glycyrrhiza uralensis Fisch, Glycyrrhiza inflata Bat and Glycyrrhiza glabra L. as recorded in Chinese Pharmacopoeia (Committee, 2010). G. uralensis Fisch is a perennial medicinal herb native to Asia's semi‐arid regions and widely cultivated for its therapeutic usefulness in inflammation, cold, asthma and liver and lung problems (Kao et al., 2014; Manach et al., 2010). The presence of flavonoids and triterpenoids (Rui et al., 2015), among which liquiritin and glycyrrhizic acid are the principal bioactive constituents, gives licorice its distinct pharmacological properties (Kojoma et al., 2011). However, knowledge about how molecular mechanisms stimulate the accumulation of liquiritin and glycyrrhizic acid in G. uralensis Fisch is still limited.
Liquiritin, the main flavonoid compound in licorice, is synthesized in the cytoplasm via a flavone biosynthetic pathway (Stefan et al., 2011; Zhang et al., 2021) that is controlled and regulated by phenylalanine ammonia‐lyase (PAL), cinnamate 4‐hydroxylase (C4H) and 4‐coumarate CoA ligase (4CL) to form 4‐coumaroyl‐CoA (Zhang et al., 2017). Under the catalysis of chalcone synthase (CHS), 4‐coumaroyl‐CoA is condensed with three molecules of malonyl‐CoA synthesized from acetyl‐CoA by acetyl‐CoA carboxylase (ACC). This process also necessitates the activity of chalcone reductase (CHR) to produce isoliquiritigenin, which is subsequently transformed to liquiritigenin under the catalysis of chalcone isomerase (CHI) (Zhao et al., 2017), and finally to form liquiritin catalysed by UDP‐glucosyltransferase (UGT). Although several functional genes for liquiritin biosynthesis have been successfully characterized in G. uralensis, many other important biosynthetic genes remain unknown.
Glycyrrhizic acid is a bioactive triterpenoid saponin synthesized via the mevalonate (MVA) pathway and regulated by a number of key enzymes (Haralampidis et al., 2002). The universal terpenoid precursors, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), can be obtained via the MVA and methylerythritol phosphate (MEP) pathways (Skorupinska‐Tudek et al., 2008). IPP and DMAPP are then condensed to produce prenyl diphosphate precursors: geranyl diphosphate (GPP), farnesyl diphosphate (FPP) (Ramilowski et al., 2013; Srivastava et al., 2018). The early stages of triterpenoid saponin biosynthesis involve the dimerization of two FPP molecules to squalene catalysed by squalene synthase (SQS), which is then catalysed by squalene epoxidase (SQE) to produce 2,3‐oxidosqualene (Mochida et al., 2017). The β‐amyrin synthase (BAS) catalyses the first reaction of the pathway committed exclusively to glycyrrhizic acid biosynthesis and plays a crucial role in directing intermediates to synthesize triterpene β‐amyrin (Seki et al., 2008). Following that, cytochrome P450 monooxygenases (CYP450s) including CYP72A154 and CYP88D6 catalyse oxidation steps to modify the β‐amyrin skeleton (Li et al., 2010; Seki et al., 2008; Seki et al., 2011). A series of UGTs participate in the multiple glycosylation of triterpenoid skeleton, resulting in the formation of glycyrrhizic acid (Nomura et al., 2019). Currently, the majority of the key genes involved in glycyrrhizic acid biosynthesis have been successfully characterized in the genome of G. uralensis (Mochida et al., 2017). However, it has yet to be determined how the transcription levels of these key genes coordinate with the accumulation of glycyrrhizic acid.
The content of liquiritin and glycyrrhizic acid in G. uralensis varies greatly between wild and cultivated forms (Chengcheng et al., 2019). Because wild licorice resources are becoming increasingly scarce, molecular breeding is being used to improve the potency of cultivated licorice. Molecular breeding to improve productivity (mainly liquiritin and glycyrrhizic acid) requires an understanding of its molecular basis and regulation mechanism. However, the underlying causes of the differences in accumulation of liquiritin and glycyrrhizic acid in the field between wild and cultivated G. uralensis are still unknown. The root microbiota was also found to be capable of influencing G. uralensis growth and production (Wei et al., 2018). However, little is known about how microbial communities influence the accumulation of G. uralensis metabolites. Furthermore, the interaction between gene expression, microbial community and metabolite accumulation in G. uralensis remains undetermined.
Multi‐omics studies could deepen our understanding of how various abiotic and biotic factors affect the accumulation of principal bioactive constituents. RNA‐Seq technology provides an unbiased approach to identifying species‐specific differential expression (Wang et al., 2009). The advancement of metagenomic sequencing technology has facilitated the investigation of microbial communities in specific environments. The combination of metabolite production, expression patterns and microbial communities will bring new insights into the comprehensive microbe–plant–metabolite regulation patterns for G. uralensis, paving the way for improved cultivation. However, no comprehensive microbe–plant–metabolite study on the medicinal plant G. uralensis has been conducted to date.
Here, we collected 25 wild G. uralensis, 25 cultivated G. uralensis for 1 year and 25 cultivated G. uralensis for 3 years, measured the root metabolites using high‐performance liquid chromatography (HPLC), sequenced the transcript of root samples by RNA sequencing, and performed amplicon metagenomic sequencing on rhizosphere and bulk soil samples. We generated metabolite profiles, transcriptomes and rhizosphere microbial community structures for wild‐type G. uralensis (WT) and cultivated G. uralensis that are grown for 1 year (C1) and 3 years (C3), profiled the important metabolic pathways in G. uralensis, characterized new genes involved in liquiritin and glycyrrhizic acid biosynthesis, and deciphered the compositional characteristics of microbes colonizing G. uralensis roots. We focused on the following key questions: (1) to what extent the contents of principal bioactive constituents of wild G. uralensis differ from those of cultivated the cultivated, and whether the expression of corresponding key genes coordinates with these metabolic patterns. (2) How abiotic and biotic factors affect the accumulation of principal bioactive constituents. (3) How the combination of functional gene regulation, secondary metabolic accumulation and rhizosphere microbial communities in concert regulate the synthesis of pharmacological active components of G. uralensis. Results have shown that: firstly, comparative metabolite analysis led to the identification of significantly different contents of liquiritin and glycyrrhizic acid, both of which were higher in wild G. uralensis. The updated gene structures and co‐expression analyses have charted the route‐map of both liquiritin and glycyrrhizic acid biosynthesis, while gene expression patterns in the corresponding biosynthetic pathways differ between wild and cultivated G. uralensis. Secondly, the multi‐omics regulation networks have shed light on comprehensive microbe–plant–metabolite regulation patterns, especially the intricate relationships between rhizosphere microbes and metabolite synthesis. Thirdly, a holistic regulation model of how biotic and abiotic stresses affect the microbe–plant–metabolite regulation patterns in G. uralensis was established, which revealed that cultivation time, pH, gene expression and microbial communities were important predictors of liquiritin accumulation. Taken together, our study has provided a global profile of microbe–plant–metabolite regulation for G. uralensis, which may lead to a better understanding of G. uralensis metabolism, as well as facilitate its cultivation for therapeutic needs.
Results
Generating metabolomic, transcriptomic and microbiomic resources for G. uralensis
To survey the gene expression, metabolic and microbial profile of wild and cultivated types of G. uralensis, we collected root tissues and soils of 25 wild, 25 C1 and 25 C3 G. uralensis from representative locations (Figure 1a, Table S1). We used HPLC to determine the contents of liquiritin and glycyrrhizic acid in each root sample (Figure 1b), and performed RNA sequencing on the same root samples (Figure 1c). In addition, we performed 16s rRNA sequencing on microbes in the rhizospheres and soils (Figure 1d).
Figure 1.
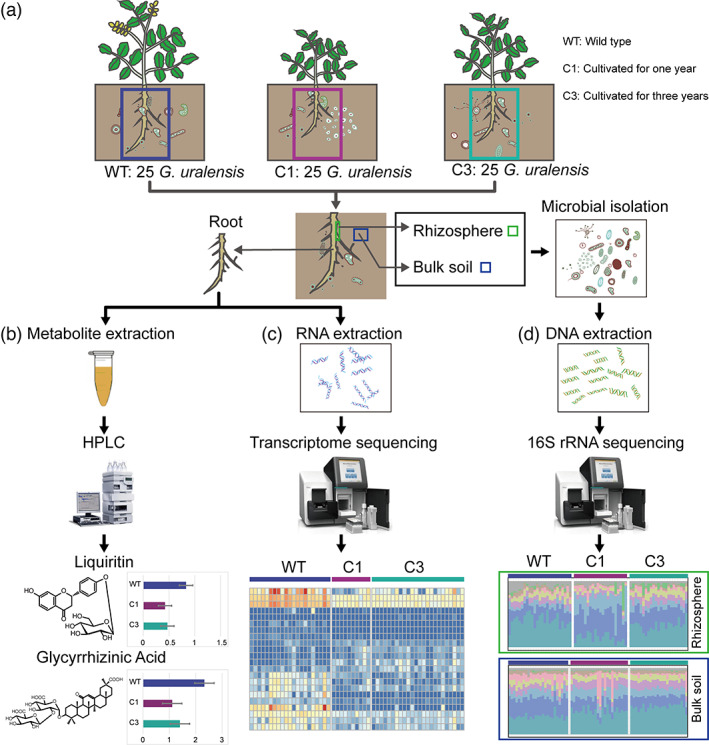
Overview of the analysis pipeline. (a) 25 samples were retrieved for each of the three different G. uralensis: wild (WT), cultivated G. uralensis that were grown for 1 year (C1) and 3 years (C3). The underground part of G. uralensis plant was collected and divided into three parts, (b) one for the sequencing of metabolites in roots, (c) one for transcriptome sequencing of roots and (d) the other for microbes collecting and sequencing in the rhizosphere and soil. [Colour figure can be viewed at wileyonlinelibrary.com]
We performed a comparative phytochemical analysis of wild and cultivated G. uralensis to uncover the accumulation of liquiritin and glycyrrhizic acid, which are important in defining the pharmaceutical qualities of the medicinal plant. The HPLC experiment revealed that wild G. uralensis accumulated more liquiritin and glycyrrhizic acid than cultivated G. uralensis (t‐test, P < 0.01; Figure 2a, b). For cultivated G. uralensis, there was no statistical difference in the content of liquiritin and glycyrrhizic acid between C1 and C3 G. uralensis.
Figure 2.
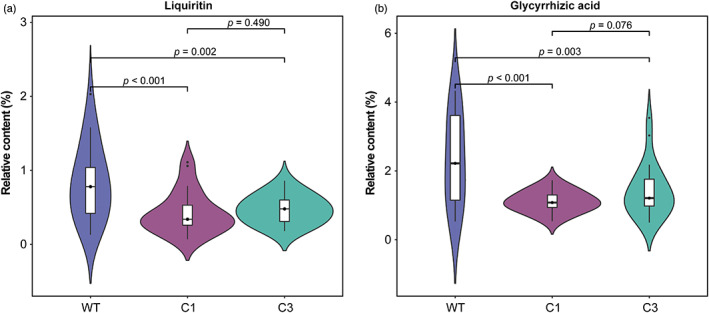
Differences in the accumulation of metabolites in G. uralensis with different growth statuses. (a) The accumulation difference of liquiritin in three types of G. uralensis: wild (WT), cultivated G. uralensis that were grown for 1 year (C1) and 3 years (C3). (b) The accumulation difference of glycyrrhizic acid in wild, C1 and C3 G. uralensis. [Colour figure can be viewed at wileyonlinelibrary.com]
To determine whether the requisite biosynthetic genes follow a similar type‐specific (wild and cultivated) expression pattern, we sequenced, assembled and annotated the transcriptome of root tissues (Figure 1c). We obtained 2 758 648 920 high‐quality reads from 55 samples in total (Table S2), yielding 40 091 unique assembled transcripts (details in Methods). When comparing with the previous whole‐genome sequencing assembly and annotation of the G. uralensis genome, we found that the additional 1052 newly annotated genes were the result of improvements in defining gene structure based on transcriptomic evidence, which was not obtained in the previous work (Mochida et al., 2017).
Altering root microbiota can influence G. uralensis growth and yield (Li et al., 2018). To better understand the pattern of microbial recruitment on G. uralensis roots, we performed 16s rRNA sequencing on microbial communities extracted from the rhizosphere and associated bulk soil samples (Figure 1d). We yielded 14 740 616 high‐quality reads for 69 rhizosphere samples and 14 921 664 high‐quality reads for 73 bulk soil samples (Table S2) and obtained 13 820 operational taxonomic units (OTUs) in these 142 samples.
Comparative transcriptomics between wild and cultivated G. uralensis
We compared the expression profiles of wild, C1 and C3 G. uralensis, and characterized 8369 differentially expressed genes (DEGs) that were significantly up‐ or down‐regulated, with 104 DEGs showing distinct expression profiles in all of these three types of G. uralensis (Figure 3a). The number of DEGs was higher between wild and cultivated (C1 and C3) G. uralensis than between C1 and C3 G. uralensis. Compared with wild G. uralensis, there were 3298 and 3450 up‐regulated genes, 3094 and 3621 down‐regulated genes in C1 and C3 G. uralensis, respectively (Figure 3b). However, only 269 DEGs were found between the C1 and C3 G. uralensis (Figure 3a, b). Concisely, the expression difference between wild and cultivated G. uralensis was greater than the difference between cultivated G. uralensis of different growth years. The functional enrichment analysis revealed that DEGs were significantly enriched in functions associated with the synthesis of secondary metabolites, such as flavonoid, phenylpropanoid and diterpenoid (Figure S1a, b and c).
Figure 3.
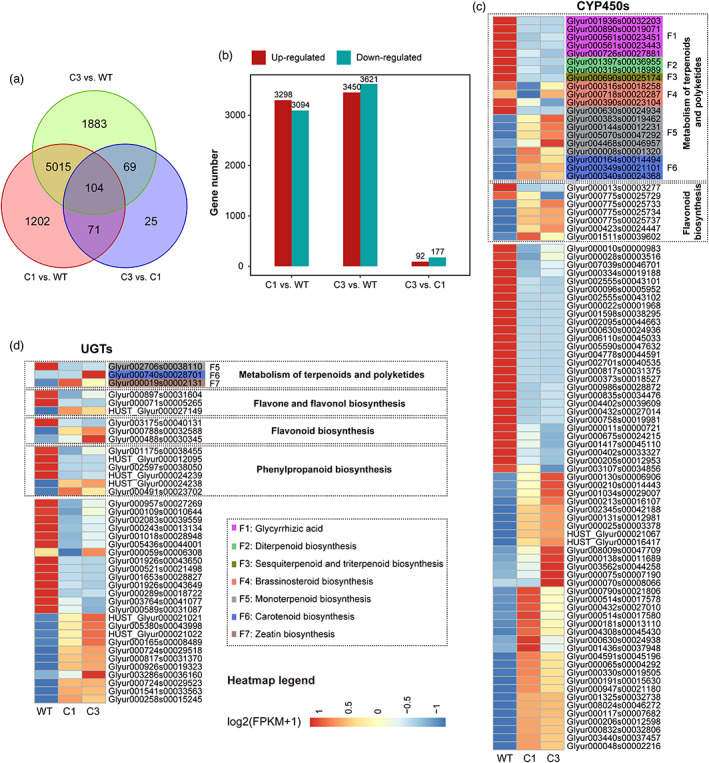
The comparison of differentially expressed genes between wild, C1 and C3 G. uralensis. (a) Venn diagram analysis of differential expression genes in the C1 vs. WT, C3 vs. WT, C3 vs. C1 groups. (b) The number of significantly up‐ and down‐regulated genes (P‐value < 0.05 and at least twofold change) in C1 vs. WT, C3 vs. WT, C3 vs. C1 groups. (c), (d) Expression patterns of genes encoding P450s (c) and UDP‐dependent glycosyltransferases (d) in the wild, C1 and C3 G. uralensis. The heatmap shows log2 (FPKM+1) values (FPKM, fragments per kilobase pair of transcript per million fragments mapped). The heatmaps of (c) and (d) were scaled in the row direction based on FPKM of genes. WT: wild type; C1: cultivated for 1 year; C3: cultivated for 3 years. [Colour figure can be viewed at wileyonlinelibrary.com]
To gain insights into the molecular mechanisms underlying the characteristics of secondary metabolites, we investigated growth type‐specific expression patterns of P450 mono‐oxygenases (CYP450s) and UGTs potentially involved in the flavonoids and terpenoids. Our findings revealed that these 88 CYP450s had significantly different patterns of gene expression in wild and cultivated G. uralensis, with 20 of them involved in the metabolism of terpenoids and polyketides (Figure 3c). These CYP450‐encoding DEGs for terpenoids metabolism, which included five glycyrrhizic acid biosynthesis genes, three brassinosteroid biosynthesis genes, two diterpenoid biosynthesis genes and one gene involving sesquiterpenoid and triterpenoid biosynthesis, were found to be more expressed in wild G. uralensis than in cultivated G. uralensis. CYP450‐encoding DEGs for carotenoid biosynthesis, on the other hand, were expressed more in cultivated G. uralensis. Carotenoids serve as precursors of important hormones such as abscisic acid (ABA), which is synthesized in response to stress (Nambara and Marion‐Poll, 2005). There were seven CYP450‐encoding DEGs with flavonoid biosynthesis functions, two of which were more highly expressed in wild G. uralensis and five of which were up‐regulated in C1 and C3 G. uralensis (Figure 3c). For the UGTs, encoding DEGs involved in carotenoid biosynthesis and zeatin biosynthesis were down‐regulated in wild G. uralensis (Figure 3d). In terms of newly annotated DEGs, two newly annotated genes were involved in CYP450s, and six newly annotated genes were involved in UGTs. These newly annotated DEGs for CYP450s and UGTs may aid in the investigation of glycyrrhizic acid and liquiritin biosynthetic pathways. Collectively, the expression of CYP450 and UGT genes, may actively promote the synthesis and accumulation of terpenoids and flavonoids.
Comparison of gene expression patterns of liquiritin and glycyrrhizic acid biosynthesis pathways between wild and cultivated G. uralensis
To account for differences in liquiritin and glycyrrhizic acid accumulation in wild and cultivated G. uralensis, the expression profiles of genes involved in the liquiritin and glycyrrhizic acid synthetic pathways were compared. Several of them were found to be more highly expressed in wild G. uralensis, whereas others were not (Figure 4), implying that they may play different roles in the regulation of liquiritin and glycyrrhizic acid biosynthesis.
Figure 4.
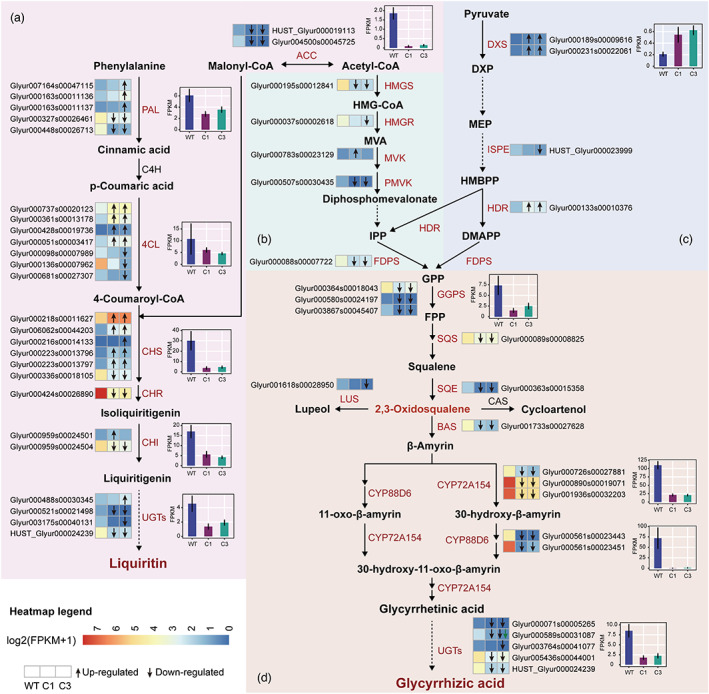
Schematic models for the biosynthesis of liquiritin and glycyrrhizic acid. (a) The liquiritin biosynthesis pathway, (b) the MVA pathway, (c) the MEP pathway. (d) The glycyrrhizic acid biosynthesis pathway. Enzymes encoded by the differentially expressed genes were marked in red and the heatmaps showed log2 (FPKM+1) values of each DEGs. Each row of the heatmap represented one gene and each column represented one a group (from left to right, WT, C1, C3). The green arrow in the heatmap indicated that the up (arrow up) or down (arrow down) regulation of C3 compared to C1, while the black arrow in the heatmap indicated that up (arrow up) or down (arrow down) regulation of C1 and C3 compared to the wild, respectively. The bar box showed the average FPKM of genes encoding the same enzymes. WT: wild type; C1: cultivated for 1 year; C3: cultivated for 3 years. [Colour figure can be viewed at wileyonlinelibrary.com]
Among all genes involved in liquiritin biosynthesis, 27 DEGs displayed distinct gene expression patterns in wild, C1 and C3 G. uralensis (Figure 4a). The conversion of acetyl‐CoA's carboxylation to malonyl‐CoA requires the catalysis of ACC enzymes (Galdieri and Vancura, 2012), for which a new gene (HUST_Glyur000019113) was identified. In addition, we identified a new UGT‐encoding gene (HUST_Glyur000024239) involved in liquiritin biosynthesis. Many genes in the liquiritin pathway encoded the same enzyme but in different expression directions. For example, despite the fact that three 4CL genes are more specifically expressed in wild G. uralensis, other four 4CL genes up‐regulated in both C1 and C3 G. uralensis (Figure 4a), indicating that the direction and magnitude of these genes' transcriptional responses were not conservative. Despite this, the overall expression level of genes encoding the same key enzymes (including ACC, PAL, 4CL, CHS, CHR, CHI and UGTs) of wild G. uralensis was significantly higher than that of cultivated G. uralensis (t‐test, P < 0.05; Figure 4a), which was consistent with the accumulation pattern of liquiritin in G. uralensis. These findings suggested that the liquiritin biosynthesis of G. uralensis was accomplished through the collaborative function of many classes of enzymes, which would explain why the total liquiritin content in wild G. uralensis was higher than that in cultivated G. uralensis.
The high glycyrrhizic acid content was most likely due to the constant and high expression of genes encoding key enzymes in the glycyrrhizic acid pathway, which were differentially expressed in wild, C1 and C3 G. uralensis (Figure 4b‐d). IPP and DMAPP are important intermediates in the formation of terpenoid backbones that can be synthesized via the MVA or MEP pathways (Zheng et al., 2014). In our transcriptome data, we discovered genes encoding essential enzymes for both pathways, indicating that both pathways are active in G. uralensis. DXS and HDR, which are involved in the biosynthesis of IPP and DMAPP in the MEP pathway, were expressed more in C1 and C3 G. uralensis (Figure 4c). The expression of the ISPE‐encoding gene (HUST Glyur000023999) was not significantly different between wild G. uralensis and C1 G. uralensis, but it was significantly down‐regulated in C3 G. uralensis. The expressions of DEGs for HMGS, HMGR and PMVK in the MVA pathway were higher in wild G. uralensis. This suggested that the regulation of the parallel but compartmentally separated biosynthesis pathways of IPP and DMAPP may differ between wild and cultivated G. uralensis, with the MVA pathway being more active in wild G. uralensis. In addition, DEGs encoding FDPS, GGPS, SQS and SQE involved in the synthesis of 2,3‐oxidosqualene had a higher expression level in wild G. uralensis than in cultivated G. uralensis. Furthermore, DEGs encoding BAS, CYP72A154, CYP88D6 and UGTs were involved in the final stage of glycyrrhizic acid biosynthesis, and they were found to be expressed more in wild G. uralensis compared to cultivated G. uralensis (Figure 4d). The high expression of these key enzyme genes in wild G. uralensis might increase the accumulation of principal bioactive constituents that contribute to the efficacy of roots in therapeutic practice, which was consistent with previous studies showing that wild G. uralensis was more effective than cultivated G. uralensis (Chengcheng et al., 2019).
Comparison of gene expression patterns related to hormone signalling pathways
Hormone signalling transduction pathways, such as jasmonic acid (JA) signalling pathway, ethylene signalling pathway, cytokinine signalling pathway and abscisic acid (ABA) signalling pathway, regulate plant growth in response to abiotic stress and respond to microbial attack and symbiosis (Devireddy et al., 2020). The up‐regulated expression of these pathways in G. uralensis was expected to enhance stress resistance. Most of the DEGs associated with these signalling pathways were up‐regulated in cultivated G. uralensis when compared to wild G. uralensis, especially the JA and ABA signalling pathways (Figure S2). Up‐regulation of genes encoding key proteins JAZ, AHP, PYR/PYL and PP2C, might promote the metabolism of jasmonic acid, cytokinin and carotenoid in cultivated G. uralensis. The biosynthesis of jasmonic acid, carotenoid and cytokinin could play an important role in plant acclimation, coordinating the overall response of plants to stress (Devireddy et al., 2020). As a result, high expression of synthetic genes for these hormones in cultivated G. uralensis could lead to two consequences: regulating growth, as well as restricting the biosynthesis of liquiritin and glycyrrhizic acid.
In addition to hormone coding genes, 73 DEGs participated in plant‐pathogen interaction and had type‐specific expression patterns in different G. uralensis (Figures S1, S1). These differential expression patterns could represent different types of G. uralensis' responses to their respective environmental microbes. These DEGs in G. uralensis might play important roles in pathogen infection limitation.
Taxonomic features of the rhizosphere microbes of wild and cultivated G. uralensis
Plant–microbe interaction has played important role in plant growth, which was supported by this study on the medicinal plant G. uralensis. Plants recruit specific root‐associated microbes, which allow plants to deliver photosynthates and root exudates to their root microbiome, thereby stimulating plant growth and productivity (Lareen and Schafer, 2016). Many plant rhizosphere microbes are beneficial, such as nitrogen‐fixing bacteria that promote plant growth (PGPR), whereas others, such as pathogenic microorganisms, are harmful to plant growth (Goswami et al., 2016). The plant rhizosphere recruits various microorganisms from soil, which assists them in dealing with specific abiotic or biotic stresses (Wille et al., 2019). There were significant differences in microbial diversity between bulk soil and the rhizosphere of G. uralensis, with bulk soil having higher microbial diversity than the rhizosphere (t‐test based on Shannon diversity, P < 0.05; Figure S4). The reduced diversity of the microbial community in the rhizosphere suggested that the G. uralensis rhizosphere might selectively recruit bulk soil microbes. C1 G. uralensis had significantly lower rhizosphere microbial diversity than wild and C3 G. uralensis (t‐test based on Shannon diversity, P < 0.01; Figure S5), but there was no statistically significant difference between wild and C3 G. uralensis.
Different types of G. uralensis recruited a particular set of rhizosphere microbes. We compared the taxonomic features of rhizosphere microbes on wild and cultivated G. uralensis to better understand the marker taxa driven by different types of G. uralensis and their habitat characteristics. We obtained 194 type‐specific marker rhizosphere microbial genera for different types of G. uralensis (Figure S6), reflecting type‐specific rhizosphere microbe recruitment patterns of G. uralensis. Nine plant pathogen genera had significant differences as regard to relative abundances in different types of G. uralensis (Kruskal–Wallis test, P < 0.05), with the majority being more enriched in C1 G. uralensis (Figure S7). The enrichment patterns of many marker pathogens, such as Agrobacterium, Arthrobacter and Streptomyces, were positively correlated with the expression patterns of genes involved in the plant‐pathogen interaction (Figure S8). The stress of more abundant pathogens colonized on C1 G. uralensis might increase the expression of key genes involved in the plant–pathogen interaction.
The growth status (wild or cultivated, different cultivation years) plays a significant role in determining which PGPRs were recruited. At the species level of rhizosphere microbial communities, we obtained 34 PGPR marker species that were enriched in either wild G. uralensis or cultivated G. uralensis. Representative PGPRs such as Bacillus badius and Bacillus firmus, were highly abundant in the wild G. uralensis (Kruskal–Wallis test, P < 0.05; Figure S9), and have been shown to assert a positive impact on crops by enhancing both above and below ground biomass (Azarias Guimar Es et al., 2012; Igiehon and Babalola, 2018). The PGPR Rhizobium leguminosarum was enriched in C3 G. uralensis rhizosphere (Figure S9), which can promote plant growth by producing plant hormones and fixing nitrogen. The PGPR Stenotrophomonas geniculate and Streptomyces purpeofuscus, which have been shown to mobilize zinc (Zn) through acidification of medium via gluconic acid production (Costerousse et al., 2018), were enriched in C1 G. uralensis (Figure S9).
Correlation of metabolite production, gene expression and microbial community of G. uralensis
We applied weighted gene co‐expression network analysis (WGCNA) to identify co‐expressed gene modules and co‐abundant microbial modules, revealing a link between gene expression, microbial community, and liquiritin and glycyrrhizic acid biosynthesis. We identified 32 co‐expressed gene modules (Figure S10a) and 10 co‐abundant microbial modules (Figure S10b), with seven gene modules (gene module 1, 2, 3, 4, 5, 23 and 29) and four microbial modules (microbial module 1, 2, 3 and 4) being positively or negatively correlated with liquiritin and glycyrrhizic acid accumulation, temperature and pH (Figure S10a, b). Among these modules, 80 DEGs in five gene modules (gene module 1, 2, 3, 4 and 5) and 39 marker microbes in four microbial modules (microbial module 1, 2, 3 and 4) were coordinated to metabolic traits and environmental factors (Figure 5a). Two co‐expressed DEGs with conserved domains PF12515 and PF03372 in gene module 1 were positively correlated with the accumulation of liquiritin and glycyrrhizic acid (Figure 5a). Six DEGs in gene module 2 were found to be negatively correlated with the glycyrrhizic acid accumulation, suggesting its critical role in glycyrrhizic acid consumption. Five DEGs in gene module 3 exhibited significant negative correlations with pH (Figure 5b). DEGs that encoded enzymes involved in MEP and MVA pathways, such as ACC, HDR and SQS in gene module 4, were co‐expressed with other ten DEGs related to the biosynthesis of other secondary metabolites such as carotenoid, brassinolide and zeatin (Figure 5a). DEGs involved in liquiritin and glycyrrhizic acid biosynthesis in gene module 5 were co‐expressed with a set of DEGs related to stress tolerance, such as the AUX/IAA signalling pathway and pathogen defence (Figure 5a). Many of these DEGs involved in glycyrrhizic acid biosynthesis were negatively correlated with temperature, while several DEGs related to stress tolerance were positively correlated with temperature (Figure 5b), indicating that G. uralensis had a complex expression regulation pattern in the coordination of metabolite biosynthesis and accumulation.
Figure 5.
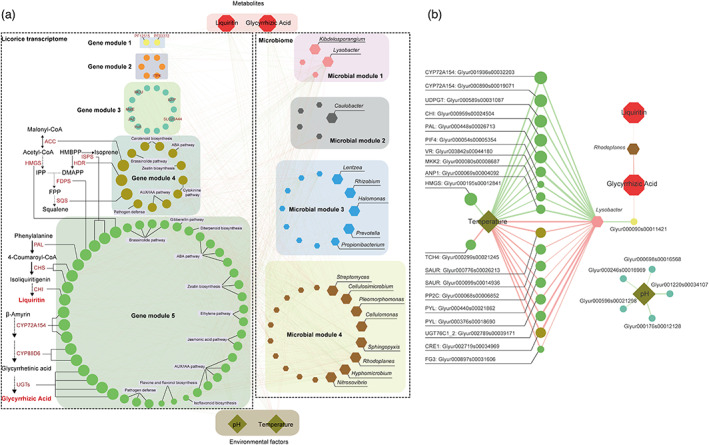
Multi‐omics network about the comprehensive microbe–plant–metabolite associations. (a) Gene co‐expressed modules associated metabolites and environmental factors. (b) Microbial co‐abundance modules associated metabolites and environmental factors. Highly correlated modules (P‐value < 0.05) were linked with a line. [Colour figure can be viewed at wileyonlinelibrary.com]
We then look into how rhizosphere microbes influenced gene expression and metabolite accumulation in G. uralensis. Environmental factors, the accumulation of liquiritin and glycyrrhizic acid, and genes involved in the liquiritin and glycyrrhizic acid biosynthesis, were all correlated with the marker microorganisms in microbial modules 1, 2, 3 and 4. The relative abundance of Lysobacter, a type of rhizosphere promoting bacteria, was not only positively correlated with temperature, but also with DEGs co‐expressed in module 5 (Figure 5b). This microbe was found to be positively correlated with genes involved in hormone signal pathways (such as PYL, PP2C and CRE1) and negatively correlated with genes involved in liquiritin and glycyrrhizic acid synthesis (such as CYP72A154, CHI and PAL), implying a potential role in enhancing hormone metabolism while decreasing liquiritin and glycyrrhizic acid synthesis. The comparative analysis revealed that the Lysobacter was remarkably abundant in rhizosphere of cultivated G. uralensis than in wild G. uralensis (Figure S11a). In addition, Rhodoplanes was found to be positively correlated with both liquiritin and glycyrrhizic acid accumulation, suggesting its potential role in stimulating the production of these metabolites. The wild G. uralensis presented an over‐representation of Rhodoplanes compared with cultivated G. uralensis (Figure S11b). Though genes in key biosynthetic pathways have been implicated in the biosynthesis of liquiritin and glycyrrhizic acid, the rhizosphere microbes identified here may also play an important role in regulating liquiritin and glycyrrhizic acid accumulation.
Effects of biotic and abiotic drivers on metabolite accumulation
We used structural equation modelling (SEM) to determine the effects of biotic components (microbial diversity, microbial community structure and plant gene expression) and abiotic factors (temperature, pH and growth time) on the accumulation of metabolites (liquiritin and glycyrrhizic acid). The model was built on the assumption that abiotic drivers (temperature, pH and growth time) could drive the accumulation of liquiritin and glycyrrhizic acid, but that biotic components (microbial diversity, community structure and plant gene expression) could also drive the accumulation of metabolites indirectly. Our structural equation model revealed that growth time was the most important driver of liquiritin and glycyrrhizic acid accumulation (Figure 6), implying that the longer growth time, the higher contents of pharmacological active components. Besides, pH was a strong positive driver of liquiritin accumulation but had no significant effect on glycyrrhizic acid accumulation. Temperature influenced liquiritin accumulation by altering the structure of the microbial community and gene expression related to liquiritin biosynthesis. We discovered that certain gene expressions were responsible for liquiritin accumulation: The differential gene expression of liquiritin biosynthetic pathway profoundly affected the accumulation of liquiritin, but differential gene expression of glycyrrhizic acid biosynthetic pathway had no significant impact on glycyrrhizic acid accumulation. This disparity could be attributed to the complicated regulation of metabolite biosynthesis and accumulation. These results emphasized how abiotic factors (such as temperature and pH), microbial community (structure and diversity) and gene expression of G. uralensis interact to influence the accumulation of liquiritin and glycyrrhizic acid.
Figure 6.
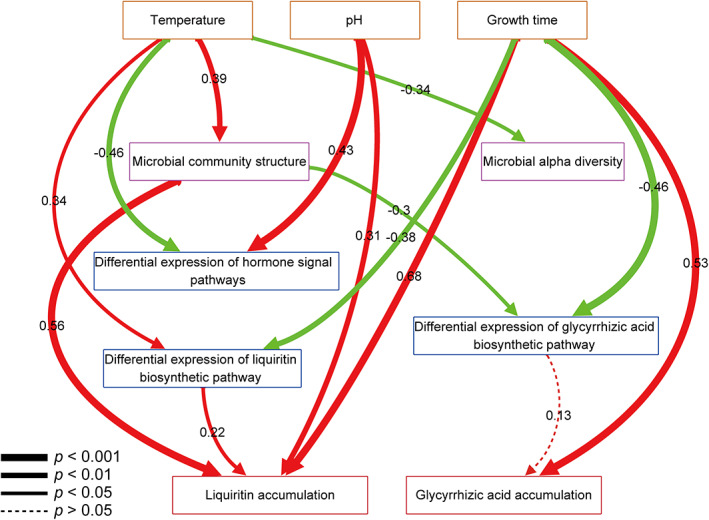
Structural equation model showing the influences of abiotic and biotic factors on metabolites accumulation. Paths were shown in red if positive or in green if negative. Path width corresponds to the degree of significance as shown in the lower left. [Colour figure can be viewed at wileyonlinelibrary.com]
Discussion
Research on the production of medicinal bioactive constituents using metabolomic, transcriptomic and microbiomic resources for medicinal plants is still lacking (De Luca et al., 2012). As liquiritin and glycyrrhizic acid are the marker components for evaluating the quality of G. uralensis, stimulating the accumulation of these two principal bioactive constituents has become the target of molecular breeding of G. uralensis. Complex metabolic pathways, gene regulatory networks and the rhizosphere microbes are essential for improved liquiritin and glycyrrhizic acid accumulation in cultivated G. uralensis. Here, we measured the root metabolites, sequenced the root transcript and sequenced the rhizosphere and bulk soil microbes for G. uralensis under three growth conditions: wild, one‐year cultivation and three‐year cultivation. We exploited liquiritin and glycyrrhizic acid regulation patterns in wild and cultivated G. uralensis using a combination of metabolite content, gene expression and microbial community to identify regulation of many genes and microbes on liquiritin and glycyrrhizic acid metabolism.
Extensive studies have been conducted to elucidate the genetic information and secondary metabolites of cultivated G. uralensis (Gafner et al., 2011; Mochida et al., 2017), and wild type provides a resource of genetic diversity for improved cultivar plant breeding programmes. We confirmed that compared with cultivated G. uralensis, wild G. uralensis produced significantly more liquiritin and glycyrrhizic acid (Figure 2). Two of the most important factors influencing gene expression are plant type and developmental stage. Through further analysis of transcriptome data, we found that G. uralensis had significant expression differences between different growth types (wild and cultivated), as well as between different cultivation times (cultivated 1 year and cultivated 3 years), especially those genes involved in biosynthesis of secondary metabolism. DEGs were greater between wild and cultivated G. uralensis than between cultivated G. uralensis of different growth years, indicating that plant types may have a greater effect on gene expression than cultivation time. In addition, we revealed the molecular mechanism underlying the difference in liquiritin and glycyrrhizic acid content between wild and cultivated G. uralensis. Compared with previously reported data (Mochida et al., 2017), we demonstrated that the majority of genes involved in liquiritin and glycyrrhizic acid biosynthesis were highly expressed in wild G. uralensis compared with cultivated G. uralensis (Figure 4). Previous studies have shown that plant domestication is often accompanied by a decrease of secondary metabolites, resulting in better quality in the wild than in cultivated plants (Kliebenstein, 2009; Van Bakel et al., 2011), which could explain why the content of liquiritin and glycyrrhizic acid, as well as the expressions of corresponding biosynthetic genes, were lower in cultivated G. uralensis than in wild G. uralensis. In wild G. uralensis, there was a considerable increase in expression of BAS, CYP72A154 and CYP88D6 when compared to C1 and C3 G. uralensis (Figure 4), which corresponded to the accumulation pattern of glycyrrhizic acid in roots (Figure 2). The low expression of these genes may limit glycyrrhizic acid synthesis in cultivated G. uralensis. In addition, due to the influence of alternative substrate competitors (the carotenoid biosynthesis pathways and diterpenoid biosynthesis pathways), the synthesis of glycyrrhizic acid can be limited in the branch products of geranylgeranyl diphosphate (GGPP). Three major OSC enzymes in G. uralensis are BAS, lupeol synthase (LUS) and cycloartenol synthase (CAS), which are responsible for branching of glycyrrhizic acid, betulinic acid and phytosterols, respectively (Hayashi et al., 2000). Glycyrrhizic acid production may be further inhibited by the 2,3‐oxidosqualene competitive effect between LUS, CAS and the BAS gene. Plant domestication has also unleashed the expression of genes in G. uralensis that are engaged in defence processes like pathogen interaction and stress tolerance, which had different expression patterns in different forms of G. uralensis. For example, the expression of PYL and PP2C in the ABA signalling pathway were significantly up‐regulated in cultivated G. uralensis compared to wild G. uralensis (Figure S2). Thus, increasing the expression of genes at different phases of the biosynthetic route while inhibiting the expression of genes in competing pathways could be an effective technique for increasing the production of liquiritin and glycyrrhizic acid.
This study also established an updated gene structure for G. uralensis, which was based on high‐quality transcript assembly and annotation, and from which we were able to identify essential genes responsible for the accumulation of active ingredients. We also found five CYP450s involved in the manufacture of glycyrrhizic acid in the wild G. uralensis with high expression levels. Notably, two newly annotated genes encoding ACC and UGT, which played key roles in liquiritin biosynthesis, were found to be substantially expressed in wild G. uralensis (Figure 4), giving fresh prospects for exploring regulation of liquiritin biosynthesis. The two genes revealed here, as well as other newly annotated genes including ISPE, CYP450 and UGT, were found to be involved in secondary metabolite synthesis, which had not previously been reported using only genomic data (Mochida et al., 2017). In summary, we discovered new genes that may encode enzymes involved in the biosynthesis of liquiritin and glycyrrhizic acid. Therefore, the newly annotated genes we identified are valuable resources for further research on the pharmaceutical quality of G. uralensis and the improvement of cultivated G. uralensis. These results would be used to characterize new genes associated with liquiritin and glycyrrhizic acid biosynthesis, as well as ways of increasing their production in cultivated G. uralensis.
Co‐expressed G. uralensis' genes and co‐abundant rhizosphere microbes were linked to the accumulation of liquiritin and glycyrrhizic acid, as well as environmental factors (Figure 5). Previous studies have identified genes involved in the liquiritin and glycyrrhizic acid synthesis pathways (Mochida et al., 2017; Seki et al., 2011), but none of the previous studies have uncovered the metabolic regulatory pathways and microbial interaction patterns. Our study revealed a regulatory microbe–plant–metabolite network for microorganisms and genes related to liquiritin and glycyrrhizic acid at the molecular level. The key genes involved in liquiritin and glycyrrhizic acid biosynthesis, such as CYP72A154, CHI and PAL encoding genes, tended to co‐express and were strongly negatively correlated with the abundance of bacteria Lysobacter (Figure 5b), indicating that Lysobacter enrichment may inhibit liquiritin and glycyrrhizic acid biosynthesis. Moreover, Rhodoplanes was found to be more abundant in the rhizosphere of wild G. uralensis compared to cultivated G. uralensis, and it was linked to the accumulation of liquiritin and glycyrrhizic acid. Therefore, the influence of these bacterial groups on roots might have a broader impact on the entire pathway of liquiritin and glycyrrhizic acid synthesis. In addition, we detected many different marker microorganisms in wild and cultivated G. uralensis, especially PGPR, including Rhizobium, Streptomyces, Nitrosovibrio, etc., which also had significant correlations with gene expression or accumulation of liquiritin and glycyrrhizic acid. Manipulation of the rhizosphere environment to target the interaction of these PGPRs with G. uralensis would be extremely beneficial in improving liquiritin and glycyrrhizic acid production.
The SEM modelling has enabled us to establish a multi‐omics model that describes the comprehensive microbe–plant–metabolite regulation pattern for G. uralensis (Figure 6). Liquiritin accumulation would be determined by gene expression, microbial community structure and environmental conditions. The fact that microbial community structure had a positive effect on liquiritin accumulation suggested that modulation of microbial community structure may alter the liquiritin accumulation. Temperature influenced liquiritin accumulation, acted through microbial community structure and gene expression of liquiritin biosynthetic pathway. However, neither gene expression nor microbial community had a dominant effect on glycyrrhizic acid accumulation. It must be emphasized that a substantial fraction of the cumulative variations in metabolites remain unexplained in the model, implying that additional research into other factors is required.
Collectively, our results suggested that soil properties, rhizosphere microbial communities, gene expression of G. uralensis were associated with the accumulation of liquiritin and glycyrrhizic acid, which could affect the accumulation of liquiritin and glycyrrhizic acid directly or indirectly. The enrichment of liquiritin and glycyrrhizic acid biosynthesis observed in wild G. uralensis, and this could result in higher medicinal quality. This finding should be a key consideration in future microbe–plant–metabolite interactions research that aims to integrate medicinal plants metabolomes, transcriptomes and microbiomes to understand how environmental factors, rhizosphere microbial communities and plant gene expression in concert impact metabolite accumulation and medicinal plant quality.
Conclusion
Glycyrrhiza uralensis is an important perennial medicinal plant with a long history of medicinal use in Europe and Asia, but the microbe–plant–metabolite regulation patterns for G. uralensis remain largely undetermined. In this study, we collected roots and rhizosphere soils from G. uralensis under three different growth conditions: wild, cultivated for 1 year and cultivated for 3 years. Based on these samples, we have explored the regulatory networks of the liquiritin and glycyrrhizic acid in the wild and cultivated G. uralensis at the metabolite, transcriptome and microbiome levels. We have first identified the differences of G. uralensis under different growth statuses at the metabolite and transcriptome levels: we have confirmed that wild G. uralensis accumulated significantly more liquiritin and glycyrrhizic acid than cultivated G. uralensis; we have updated gene structures of G. uralensis and supplemented the route‐map of both liquiritin and glycyrrhizic acid biosynthesis; and we have found that biosynthesis genes of glycyrrhizic acid, including BAS, CYP72A154 and CYP88D6, were up‐regulated in wild G. uralensis compared to cultivated G. uralensis. Secondly, based on multi‐omics network analysis, we have obtained a better understanding of the comprehensive microbe–plant–metabolite associations. This finding was exemplified by rhizosphere microbes Lysobacter and Rhodoplanes, which were found to be strongly associated with the gene expression and accumulation of liquiritin and glycyrrhizic acid, respectively. Thirdly, based on SEM modelling, it was discovered that growth time has a positive effect on the accumulation of liquiritin and glycyrrhizic acid, whereas rhizosphere microbial community structure exerted strong effects on liquiritin accumulation.
To our knowledge, this is the first attempt to combine metabolite production, expression profiles and microbial communities to study their regulation patterns on pharmacological active components of G. uralensis, as well as to explore how biotic and abiotic stresses affect the microbe–plant–metabolite regulation patterns in this medicinal plant. Taken together, the mining of plant metabolomic, transcriptomic and microbiomic resources, as well as establishing microbe–plant–metabolite regulation patterns, are all useful in guiding future cultivation of G. uralensis.
Materials and methods
Plant and soil materials
The roots of wild and cultivated (C1 and C3) G. uralensis were collected in August 2018 from Shiji Quan (106.86 N 37.99 W) and Tianji Zhang (107.27 N 37.81 W) villages in Yanchi County, Wuzhong City, Ningxia, China, respectively, and identified as G. uralensis Fisch. By Prof. Yinghua Wang (Ning Xia Institute for Drug Control). As the wild G. uralensis Fisch is mostly unaffected by human activities, it is hard to determine the actual growth time for every sample. On the other hand, the wild G. uralensis Fisch samples used in this study have grown for roughly 3–5 years, based on manual inspection by medicinal plant experts and local residents. The C1 and C3 G. uralensis Fisch were cultivated in two separate fields for 1 year and 3 years in the Tianji Zhang village, respectively. The study included 75 select G. uralensis, and each root sample was cut off and divide into three parts. One part was frozen immediately in liquid nitrogen and stored at −80 °C before RNA extraction, one part was collected in aseptic centrifuges to collect rhizosphere microorganisms, and the other parts were immediately collected in zipper bags and then dried naturally at room temperature for the determination of liquiritin and glycyrrhizic acid. In addition, the bulk soil of each root pit was collected in the germ‐free centrifuge tubes for soil microbial research. We measured the temperature and the pH of each root pit during sampling process. Detailed sampling information of G. uralensis was shown in Table S1.
Total RNA extraction, library preparation and transcriptome sequencing
Total RNA was extracted from root tissues of three different G. uralensis Fisch (wild, C1 and C3). The complete frozen roots were pulverized in liquid nitrogen, and RNAs were extracted using TRIzol® Reagent. NanoDrop 2000 was used to check and measure the purity and concentration of the isolated RNA. The quality of the isolated RNA was identified by agarose gel electrophoresis, and the integrity of the isolated RNA was detected by Agilent2100 Bioanalyzer. A total amount of 5 μg RNA per sample (concentration ≥ 200 ng/μL and OD260/280 between 1.8 and 2.2) was used as input material for the sample preparations. The total RNA was purified by Oligo (dT) and was enriched with PCR for preparing the sequencing library. The cDNA library was prepared by TruseqTM RNA sample prep Kit and was quantified by TBS380. The cDNA library was sequenced on the Illumina Hiseq platform to generate paired‐end reads of 150 bp.
Data processing, transcriptome assembly and functional annotation
The raw RNA reads were filtered to obtain the high‐quality clean reads by removing adaptor sequences, ambiguous nucleotides (N) and low‐quality bases. The raw reads with ambiguous nucleotides rate greater than 10%, a quality value less than 20 and length shorter than 50 bp were discarded using Trimmomatic (v0.38) (Bolger et al., 2014). The clean reads were aligned to the G. uralensis genome (available at http://ngs‐data‐archive.psc.riken.jp/Gur‐genome/download.pl) using HISAT2 (v2‐2.1.0; Kim et al., 2019) with default parameters. The mapped sam files were converted to bam files and sorted using samtools (0.1.19; Li et al., 2009). The StringTie (v1.3.6; Pertea et al., 2016) was used to assemble the transcript, and the following options were used: ‐f 0.3, ‐j 3, ‐c 5 and ‐g 100. These assemblies were then merged with StringTie (v1.3.6) and GffCompare (v0.11.2) with default parameters were used to compare them to the reference genome. The protein coding potential of new transcripts (class_code = u) were predicted by CPC2 (v0.1; Kang et al., 2017) and TransDecoder (v5.5.0). Finally, the expression levels of genes were calculated using StringTie (v1.3.6) with ‐e ‐A parameters and the read count information was extracted using the prepDE.py script. Metabolic pathway analysis was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.genome.jp/kegg/; Kanehisa et al., 2015). The functional enrichment analysis was analysed by KOBAS (v3.0; Xie et al., 2011) software with an adjusted P‐value <0.05.
Differential expression analysis and functional enrichment
The differential gene expression analysis was analysed by DESeq2 (Love et al., 2014) using the gene‐level read count information. Genes showing an absolute fold change (FC) > 2 with an adjusted P‐value <0.05 were considered as differentially expressed between wild, C1 and C3 G. uralensis. In addition, functional‐enrichment analysis was carried out using KOBAS (v3.0) to determine which DEGs were significantly enriched in metabolic pathways at Bonferroni‐corrected P‐value <0.05.
Microbial DNA extraction and 16S rRNA gene sequencing
To isolate rhizosphere microbial communities, the rhizoplane samples were first soaked with ddH2O and then centrifuged (10 min at 2350 g) to collect all of the sediment from the root surfaces, which were treated as rhizosphere microbial samples. The microbial DNA of rhizosphere and soil samples were extracted using HiPure Soil DNA Kit B (Magen, China) following the manufacturer's instructions. The DNA concentration was monitored by using Qubit® dsDNA HS Assay Kit. The 20–30 ng isolated DNA of each sample was used to perform 16S rRNA gene V3‐V4 region tagged amplicon. The primers used for amplification were ‘CCTACGGRRBGCASCAGKVRVGAAT’ (forward primer) and ‘GGACTACNVGGGTWTCTAATCC’ (reverse primer). The concentration of the library was validated by Qubit3.0 Fluorometer and was then sequenced on the Illumina MiSeq platform. In total, 14 740 616 and 14 921 664 high‐quality 16S rRNA amplicons for 69 rhizosphere samples and 73 soil samples were obtained and analysed (Table S2).
Microbial data analysis
The quality of raw data from the 16S rRNA gene sequencing was filtered using mothur (v.1.39.5; Schloss et al., 2009). Hence, the reads with ambiguous base calls (N) and the length shorter than 300 bp or longer 500 bp were removed. The clean reads were used to analyse the taxonomy using QIIME (v1.9.1) pipeline. The operational taxonomic units (OTUs) were clustered at 97% sequence similarity. Microbial composition at each taxonomic level was defined using the summarize_taxa function in QIIME (Caporaso et al., 2010). The alpha_rarefaction.py was selected to evaluate the alpha diversity of microbial communities among rhizosphere and soil samples. Diversity matrices were calculated using the vegan R library. Linear discriminate analysis effect size (LEfSe; Segata et al., 2011) was used to analyse the differentially taxonomical features among microbial communities from a different group. The significant taxonomical biomarkers were select with the P‐value of the Kruskal–Wallis test <0.05 and logarithmic LDA score >2.
Metabolite measurements
The Dionex U3000 HPLC (high‐performance liquid chromatography) system was used to carry out the metabolite determination with Ultimate C18 column (150 × 4.6 mm I.D., 5 μm). The acetonitrile (TEDIA, USA) in chromatographic (A) and 0.1% aqueous trifluoroacetic acid solution (B) were used as the mobile phase, and the detection wavelength was 237 nm. The gradient elution were as follows: 0–8 min, 19% A and 81% B; 8–35 min, 19–50% A and 81–50% B; 35–36 min, 100% A and 0% B. The metabolites (liquiritin and glycyrrhizic acid) were measured by comparing the area of the individual peaks with standard curves obtained from liquiritin and glycyrrhizic acid bought from National Institutes for Food and Drug Control (China).
Co‐occurrence network analysis
Weighted gene co‐expression network analysis (WGCNA; Langfelder and Horvath, 2008) was used to generate co‐expression and co‐occurrence networks based on the gene expression and microbial OTUs data, respectively. The module gene expression and OTU defined as the first principal component of a module was used to calculate the Pearson correlation between a module and a metabolic trait, respectively. First, module eigengenes (MEs) were defined as the first principal component of each gene module (or microbial module), and the expression (or relative abundance) of MEs serving as a representation of all genes (microbes) in a given module. The Pearson correlation between MEs and metabolic trait was calculated to identify the metabolic significant module. In addition, the gene (microbial) significance was defined as mediated P‐value of each gene (microbe) in the linear regression between gene expression and metabolic traits. Then, the module significance (MS) was defined as the average gene (microbial) significance of all the genes involved in the module. The metabolic information of top 2 gene (microbe) modules with significant positive or negative correlations with metabolic traits were selected to incorporate into the co‐expression network. Cytoscape (v3.3.0; Shannon et al., 2003) was utilized to visualize and clarify the co‐occurrence networks of transcriptome, metabolome and microbiome data.
Statistical analysis
The statistical analyses were carried out in R using the vegan libraries. Each variable's normality was checked, and the log transformed if necessary. The NMDS of the bray distance matrices was used to describe the structure of the microbial community. The Euclidean distance matrices were used in the NMDS of gene expression. We used SEM to investigate the direct and indirect effects of abiotic and biotic parameters on the content of liquiritin and glycyrrhizic acid. The hypothesized path structure was based on the proposition that abiotic drivers (pH, temperature and cultivation time) drive metabolite (liquiritin and glycyrrhizic acid) accumulation directly, but also indirectly by affecting the biotic drivers. Our hypotheses were as follows: (1) microbial alpha diversity and community structure were driven by abiotic factors (pH, temperature and cultivation time); (2) abiotic factors, microbial alpha diversity and community structure are driving the gene expression; (3) pH, temperature, cultivation time, microbial alpha diversity and community structure, gene expression drives liquiritin and glycyrrhizic acid accumulation. The lavaan SEM package (Rosseel, 2012) was used to fit the SEM model path.
Conflicts of interest
The authors declared that they have no conflict of interest.
Authors' contributions
KN and HB conceived of and proposed the idea, and designed the study. CFZ, CYC, XG and CYT performed the experiments and analysed the data. KN, HB and CFZ contributed to editing and proof‐reading the manuscript. All authors read and approved the final manuscript.
Supporting information
Figure S1 KEGG pathways with the most significant enrichment of DEGs in the three groups of G. uralensis.
Figure S2 Expression patterns of genes involved in plant hormone signal transduction pathways.
Figure S3 Expression patterns of genes involved in plant‐pathogen interaction.
Figure S4 The comparison of microbial community diversities between the rhizosphere and soil.
Figure S5 The comparison of rhizosphere microbial community diversity between wild, C1 and C3 G. uralensis.
Figure S6 Marker genera in rhizosphere samples of wild, C1 and C3 G. uralensis.
Figure S7 Marker pathogen genera in rhizosphere samples of wild, C1 and C3 G. uralensis.
Figure S8 The correlation between pathogen abundance and the expression of genes related to plant‐pathogen interaction in G. uralensis.
Figure S9 Marker PGPRs and pathogens at the species level in rhizosphere samples of wild, C1 and C3 G. uralensis.
Figure S10 Matrix of Module‐Trait Relationships (MTRs) and P‐values for selected traits.
Figure S11 Relative abundance comparison of Lysobacter and Rhodoplanes in three types of G. uralensis.
Table S1 The information about collected samples in this study.
Table S2 The sequencing information about collected samples in this study.
Acknowledgements
This work was partially supported by National Science Foundation of China (81774008, 32071465, 31871334 and 31671374) and the Ministry of Science and Technology’s National Key Research and Development Program of China (No. 2018YFC0910502). We were grateful for Yinghua Wang from Ningxia Institute for Drug Control for assistance in sample collection and quality check works.
Contributor Information
Hong Bai, Email: baihong@hust.edu.cn.
Kang Ning, Email: ningkang@hust.edu.cn.
Data availability statement
Transcriptome sequence data and microbial sequence data were deposited in the Sequence Read Archive (SRA) under the accession number BioProject PRJNA705545 and PRJNA705567, respectively.
References
- Azarias Guimar Es, A. , Duque Jaramillo, P.M. , Sim, O. , Abrah, O.N. , Brega, R. , Florentino, L.A. , Barroso Silva, K. et al. (2012) Genetic and symbiotic diversity of nitrogen‐fixing bacteria isolated from agricultural soils in the western Amazon by using cowpea as the trap plant. Appl. Environ. Microbiol. 78, 6726–6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J. G. , Kuczynski, J. , Stombaugh, J. , Bittinger, K. , Bushman, F. D. , Costello, E. K. , Fierer, N. et al. (2010). Qiime allows analysis of high‐throughput community sequencing data. Nat. Methods, 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chengcheng, W. , Zhichen, C. , Jingjing, S. , Shuyu, C. and Tan, M. (2019) Comparative metabolite profiling of wild and cultivated licorice based on ultra‐fast liquid chromatography coupled with triple quadrupole‐time of flight tandem mass spectrometry. Chem. Pharm. Bull. 67, 1104–1115. [DOI] [PubMed] [Google Scholar]
- Committee, N.P. (2010) Part 1. Pharmacopoeia of People's Republic of China. Beijing: Chemical Industry Press. [Google Scholar]
- Costerousse, B. , Schnholzer‐Mauclaire, L. , Frossard, E. and Thonar, C. (2018) Identification of heterotrophic zinc mobilization processes among bacterial strains isolated from wheat rhizosphere (Triticum aestivum L.). Appl. Environ. Microbiol. 84(1), e01715–e01717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca, V. , Salim, V. , Atsumi, S.M. and Yu, F. (2012) Mining the biodiversity of plants: a revolution in the making. Science, 336, 1658–1661. [DOI] [PubMed] [Google Scholar]
- Devireddy, A.R. , Zandalinas, S.I. , Fichman, Y. and Mittler, R. (2020) Integration of reactive oxygen species and hormone signaling during abiotic stress. Plant J. 105(2), 459–476. [DOI] [PubMed] [Google Scholar]
- Gafner, S. , Bergeron, C. , Villinski, J.R. , Godejohann, M. , Kessler, P. , Cardellina, J.H. , Ferreira, D. et al. (2011) Isoflavonoids and coumarins from Glycyrrhiza uralensis: antibacterial activity against oral pathogens and conversion of isoflavans into isoflavan‐quinones during purification. J. Nat. Prod. 74, 2514–2519. [DOI] [PubMed] [Google Scholar]
- Galdieri, L. and Vancura, A. (2012) Acetyl‐CoA carboxylase regulates global histone acetylation. J. Biol. Chem. 287, 23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami, D. , Thakker, J.N. , Dhandhukia, P.C. and Moral, M.T. (2016) Portraying mechanics of plant growth promoting rhizobacteria (PGPR): A review. Cogent Food Agric. 2, 1127500. [Google Scholar]
- Haralampidis, K. , Trojanowska, M. and Osbourn, A.E. (2002) Biosynthesis of triterpenoid saponins in plants. Adv. Biochem. Eng. Biotechnol. 75, 31. [DOI] [PubMed] [Google Scholar]
- Hayashi, H. , Hiraoka, N. , Ikeshiro, Y. , Kushiro, T. , Morita, M. , Shibuya, M. and Ebizuka, Y. (2000) Molecular cloning and characterization of a cDNA for Glycyrrhiza glabra cycloartenol synthase. Biol. Pharm. Bull. 23, 231–234. [DOI] [PubMed] [Google Scholar]
- Igiehon, N.O. and Babalola, O.O. (2018) Rhizosphere microbiome modulators: contributions of nitrogen fixing bacteria towards sustainable agriculture. Int. J. Environ. Res. Public Health, 15, 574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M. , Sato, Y. , Kawashima, M. , Furumichi, M. and Tanabe, M. (2015) Kegg as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, Y.‐J. , Yang, D.‐C. , Kong, L. , Hou, M. , Meng, Y.‐Q. , Wei, L. and Gao, G. (2017) CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 45, W12–W16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, T.C. , Wu, C.H. and Yen, G.C. (2014) Bioactivity and potential health benefits of licorice. J. Agric. Food Chem. 62, 542–553. [DOI] [PubMed] [Google Scholar]
- Kim, D. , Paggi, J.M. , Park, C. , Bennett, C. and Salzberg, S.L. (2019) Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat. Biotechnol. 37, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa, I. (2002) Licorice root. A natural sweetener and an important ingredient in Chinese medicine. Pure Appl. Chem. 74, 1189–1198. [Google Scholar]
- Kliebenstein, D.J. (2009) Use of Secondary Metabolite Variation in Crop Improvement. In Plant‐derived Natural Products: Synthesis, Function, and Application( Osbourn, A.E. and Lanzotti, V. , eds). Springer US: New York, NY. [Google Scholar]
- Kojoma, M. , Hayashi, S. , Shibata, T. , Yamamoto, Y. and Sekizaki, H. (2011) Variation of glycyrrhizin and liquiritin contents within a population of 5‐year‐old licorice (Glycyrrhiza uralensis) plants cultivated under the same conditions. Biol. Pharm. Bull. 34, 1334–1337. [DOI] [PubMed] [Google Scholar]
- Langfelder, P. and Horvath, S. (2008) Wgcna: an R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareen, A. , Burton, F. and Schäfer, P. (2016) Plant root‐microbe communication in shaping root microbiomes. Plant Mol. Biol. 90(6), 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Luo, H.‐M. , Sun, C. , Song, J.‐Y. , Sun, Y.‐Z. , Wu, Q. , Wang, N. et al. (2010) Est analysis reveals putative genes involved in glycyrrhizin biosynthesis. BMC Genomics, 11, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Mohamad, O. , Ma, J. , Friel, A.D. , Su, Y. , Wang, Y. , Musa, Z. et al. (2018) Synergistic plant–microbe interactions between endophytic bacterial communities and the medicinal plant Glycyrrhiza uralensis F. Antonie Van Leeuwenhoek, 111(10), 1735–1748. [DOI] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manach, C. , Hubert, J. , Llorach, R. and Scalbert, A. (2010) The complex links between dietary phytochemicals and human health deciphered by metabolomics. Mol. Nutr. Food Res. 53, 1303–1315. [DOI] [PubMed] [Google Scholar]
- Mochida, K. , Sakurai, T. , Seki, H. , Yoshida, T. , Takahagi, K. , Sawai, S. , Uchiyama, H. et al. (2017) Draft genome assembly and annotation of Glycyrrhiza uralensis, a medicinal legume. Plant J. 89(2), 181–194. [DOI] [PubMed] [Google Scholar]
- Nambara, E. and Marion‐Poll, A. (2005) Abscisic acid biosynthesis and catabolism. Annu. Rev. Plant Biol. 56, 165–185. [DOI] [PubMed] [Google Scholar]
- Nomura, Y. , Seki, H. , Suzuki, T. , Ohyama, K. and Muranaka, T. (2019) Functional specialization of UDP‐glycosyltransferase 73P12 in licorice to produce a sweet triterpenoid saponin, glycyrrhizin. Plant J. 99(6), 1127–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea, M. , Kim, D. , Pertea, G.M. , Leek, J.T. and Salzberg, S.L. (2016) Transcript‐level expression analysis of RNA‐seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramilowski, J.A. , Sawai, S. , Seki, H. , Mochida, K. , Yoshida, T. , Sakurai, T. , Muranaka, T. et al. (2013) Glycyrrhiza uralensis transcriptome landscape and study of phytochemicals. Plant Cell Physiol. 54, 697–710. [DOI] [PubMed] [Google Scholar]
- Rosseel, Y. (2012) Lavaan: An R package for structural equation modeling. J. Stat. Software, 48, 1–36. [Google Scholar]
- Rui, Y. , Li‐Qiang, W. , Bo‐Chuan, Y. and Liu, Y. (2015) The pharmacological activities of licorice. Planta Med. 81, 1654–1669. [DOI] [PubMed] [Google Scholar]
- Schloss, P.D. , Westcott, S.L. , Ryabin, T. , Hall, J.R. , Hartmann, M. , Hollister, E.B. , Lesniewski, R.A. et al. (2009) Introducing mothur: Open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata, N. , Izard, J. , Waldron, L. , Gevers, D. , Miropolsky, L. , Garrett, W.S. and Huttenhower, C. (2011) Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki, H. , Ohyama, K. , Sawai, S. , Mizutani, M. , Ohnishi, T. , Sudo, H. , Akashi, T. et al. (2008) Licorice β‐amyrin 11‐oxidase, a cytochrome P450 with a key role in the biosynthesis of the triterpene sweetener glycyrrhizin. Proc. Natl. Acad. Sci. 105, 14204–14209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki, H. , Sawai, S. , Ohyama, K. , Mizutani, M. , Ohnishi, T. , Sudo, H. , Fukushima, E.O. et al. (2011) Triterpene functional genomics in licorice for identification of Cyp72A154 involved in the biosynthesis of glycyrrhizin. Plant Cell, 23, 4112–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N.S. , Wang, J.T. , Ramage, D. , Amin, N. et al. (2003) Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorupinska‐Tudek, K. , Poznanski, J. , Wojcik, J. , Bienkowski, T. , Szostkiewicz, I. , Zelman‐Femiak, M. , Bajda, A. et al. (2008) Contribution of the mevalonate and methylerythritol phosphate pathways to the biosynthesis of dolichols in plants. J. Biol. Chem. 283, 21024–21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava, P.L. , Shukla, A. and Kalunke, R.M. (2018) Comprehensive metabolic and transcriptomic profiling of various tissues provide insights for saponin biosynthesis in the medicinally important Asparagus racemosus. Sci. Rep. 8, 9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan, G. , Chantal, B. , Jacquelyn, R. , Villinski, M. and Pavel, G. (2011) Isoflavonoids and coumarins fromglycyrrhiza uralensis: Antibacterial activity against oral pathogens and conversion of isoflavans into isoflavan‐quinones during purification. J. Nat. Prod. 74, 2514–2519. [DOI] [PubMed] [Google Scholar]
- Van Bakel, H. , Stout, J.M. , Cote, A.G. , Tallon, C.M. , Sharpe, A.G. , Hughes, T.R. and Page, J.E. (2011) The draft genome and transcriptome of Cannabis sativa . Genome Biol. 12, R102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Gerstein, M. and Snyder, M. (2009) RNA‐Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, X. , Hao, Z. , Meng, Y. , Wu, Z. and Chen, B. (2018) Improved phosphorus nutrition by arbuscular mycorrhizal symbiosis as a key factor facilitating glycyrrhizin and liquiritin accumulation in Glycyrrhiza uralensis . Plant Soil, 439, 1–15. [Google Scholar]
- Wille, L. , Messmer, M.M. , Studer, B. and Hohmann, P. (2019) Insights to plant–microbe interactions provide opportunities to improve resistance breeding against root diseases in grain legumes. Plant Cell Environ. 42, 20–40. [DOI] [PubMed] [Google Scholar]
- Xie, C. , Mao, X. , Huang, J. , Ding, Y. , Wu, J. , Dong, S. , Kong, L. et al. (2011) KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Tian, S. , Qi, L. , Li, W. , Hou, J. , Yang, L. , Zhang, Z. et al. (2021) Gene polymorphism of chalcone isomerase influence the accumulation of flavonoids in licorice (Glycyrrhiza spp.). Genet. Resour. Crop. Evol. 68, 899–913. [Google Scholar]
- Zhang, F.S. , Wang, Q.Y. , Pu, Y.J. , Chen, T.Y. , Qin, X.M. and Gao, J. (2017) Identification of genes involved in flavonoid biosynthesis in sophora japonica through transcriptome sequencing. Chem. Biodivers. 14, e1700369. [DOI] [PubMed] [Google Scholar]
- Zhao, Y. , Lv, B. , Feng, X. and Li, C. (2017) Perspective on biotransformation and de novo biosynthesis of licorice constituents. J. Agric. Food Chem. 65, 11147–11156. [DOI] [PubMed] [Google Scholar]
- Zheng, X. , Xu, H. , Ma, X. , Zhan, R. and Chen, W. (2014) Triterpenoid saponin biosynthetic pathway profiling and candidate gene mining of the Ilex asprella root using RNA‐Seq. Int. J. Mol. Sci. 15, 5970–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 KEGG pathways with the most significant enrichment of DEGs in the three groups of G. uralensis.
Figure S2 Expression patterns of genes involved in plant hormone signal transduction pathways.
Figure S3 Expression patterns of genes involved in plant‐pathogen interaction.
Figure S4 The comparison of microbial community diversities between the rhizosphere and soil.
Figure S5 The comparison of rhizosphere microbial community diversity between wild, C1 and C3 G. uralensis.
Figure S6 Marker genera in rhizosphere samples of wild, C1 and C3 G. uralensis.
Figure S7 Marker pathogen genera in rhizosphere samples of wild, C1 and C3 G. uralensis.
Figure S8 The correlation between pathogen abundance and the expression of genes related to plant‐pathogen interaction in G. uralensis.
Figure S9 Marker PGPRs and pathogens at the species level in rhizosphere samples of wild, C1 and C3 G. uralensis.
Figure S10 Matrix of Module‐Trait Relationships (MTRs) and P‐values for selected traits.
Figure S11 Relative abundance comparison of Lysobacter and Rhodoplanes in three types of G. uralensis.
Table S1 The information about collected samples in this study.
Table S2 The sequencing information about collected samples in this study.
Data Availability Statement
Transcriptome sequence data and microbial sequence data were deposited in the Sequence Read Archive (SRA) under the accession number BioProject PRJNA705545 and PRJNA705567, respectively.