

Abstract
We have previously described generation of mesenchymal stem cells (MSCs) from human embryonic and induced pluripotent stem cells. One of the central questions in stem cell biology is to understand how stem cells regulate the decision to self-renew vs. differentiate, at the molecular level. In the current studies we used loss-of-function and gain-of-function analyses in primary human MSCs to demonstrate that BMI1 is a critical regulator for self-renewal and multipotency in this interesting cell type. Knockdown of BMI1 in MSCs reduced self-renewal by upregulation of p16INK4A and increased apoptosis. Knockdown of p16INK4A partially rescued the self-renewal defect in MSCs with loss of BMI1. Overexpressed BMI1 reduced apoptosis and increased cell proliferation by repressing p16INK4A. Loss of BMI1 resulted in deregulation of PPARγ, an adipogenic factor, and imprinted gene network (IGN), which blocks osteogenesis. Knockdown of PPARγ or IGN in BMI1 defect models restored osteogenesis. Overexpression of BMI1 repressed transcripts of RUNX2 and PPARγ, in osteogenesis and adipogenesis, respectively, which lead to decreased lineage specification potential in MSCs. These data show that BMI1 regulates cell proliferation, apoptosis, and differentiation of human MSCs.
Keywords: Adipogenesis, cell cycle, epigenetics, iPSCs, mesenchymal stem cells, osteogenesis, polycomb-repressive complex 1, proliferation, stem cell differentiation
INTRODUCTION
Mesenchymal stem cells (MSCs) have gained extraordinary attention due to their unique characteristics and high potential in regenerative medicine. We have previously described generation of MSCs from human embryonic and induced pluripotent stem cells [1, 2], as well as from human bone marrow and adipose tissue [3-5]. MSCs are distinguished by self-renewal and the potential to differentiate to multiple cell types of mesodermal origin. Remarkable safety records for transplanted MSCs in pre-clinical and clinical trials throughout the world make MSCs major players in regenerative medicine [6-15], and they have been approved to be administered as drugs in several countries. Recent failures or only partial successes in a handful of phase III trials, however, raise controversy concerning the efficacy of MSCs [16]. For this cell type, the understanding of basic cell biology lags behind clinical application. To advance basic science studies and to render the therapeutic potential of MSCs reliable and reproducible, underlying mechanisms such as self-renewal and differentiation must be better understood. Important questions to answer are: how do MSCs decide between self-renewal vs. differentiation into daughter cells, and what are the core regulatory mechanisms to govern these decisions at the molecular level? Precise regulation of gene expression is critical for the development and maintenance of tissues [17, 18]. Several layers of regulatory units participate in the control of gene expression via post-transcriptional, translational, post-translational, and epigenetic control [19]. Understanding important master switches that control cascades of regulatory events in pluripotent and multipotent stem cells is therefore of utmost importance.
One of the central questions in stem cell biology is to understand how stem cells regulate self-renewal [19]. To self-renew, stem cells must proliferate, repress the expression of differentiation-related genes and reduce expression of genes related to apoptosis [19]. Recently, polycomb group (PcG) proteins have been associated with the regulation of several types of adult stem cells. PcG proteins were first identified as body-patterning regulators to repress Hox genes in Drosophila [20]. Since then, evidence indicates that PcG proteins act as global epigenetic transcriptional repressors and key components of cell differentiation. In mammals, two main complexes of PcG exist, and are called polycomb repressive complexes (PRC) [19, 21, 22]. PRC1 (BMI1, RING1A, RING1B, CBX, PHC) has monoubiquitylation activity, specifically acting on H2AK119, which is the histone marker associated with repressive chromatin status. PRC2 (EZH2, EED, SUZ12) catalyze trimethylation on H3K27, a well-known repressive marker. It is still ambiguous as to how PcG proteins repress specific loci in the genome because PcG complexes have no known DNA binding activity. How these complexes are silencing gene expression remains to be investigated.
BMI1, which is a polycomb ring finger oncogene [23] and a component of polycomb repressive complexes (PRC), is known to repress Hox genes and cell cycle regulators. It contains RING finger domain in its N-terminal end, a central helix-turn-helix motif and no known enzjmiatic activity. BMI1 regulates chromatin structures, cell cycle, genomic imprinting, cell fate transition, homeostasis, and tumorigenesis [19] and is necessary for self-renewal in hematopoietic (HSC) [24, 25], neural (NSC) [26, 27], lung (LSC) [28], prostate stem cells (PSC) [29] and mouse MSCs [30], but its role in human bone marrow (BM) MSCs remained to be illustrated. In the current studies, loss-of-function and gain-of-function analyses in primary human MSCs demonstrated that BMI1 is critical for MSC self-renewal and multipotency.
RESULTS AND DISCUSSION
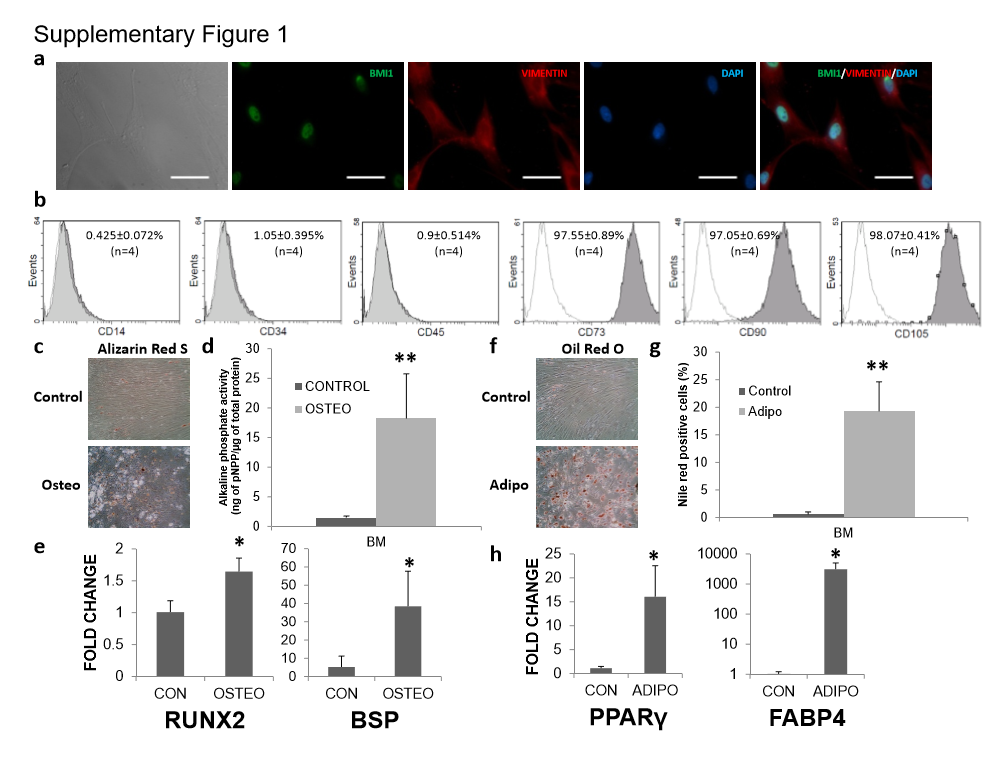
Human adult BM-MSCs expressed BMI1 in the nucleus (supplementary Fig. 1a) and were positive for CD73, CD90 and CD105 antigens and negative for CD14, CD34 and CD45 surface markers (see supplementary Fig. 1b). With induction of osteogenic media for 14 days, MSCs became alizarin Red S positive (supplementary Fig. 1c) and showed 17-fold higher expression of alkaline phosphate, an early osteogenesis marker, than the undifferentiated MSCs (supplementary Fig. 1d). Quantitative real time-PCR (qRT-PCR) analysis of osteoblasts revealed a 1.6-fold higher RUNX2, a master regulator of osteogenesis [31], mRNA level and 7.3-fold higher bone sialoprotein (BSP) mRNA level in the differentiated cells than in the control (supplementary Fig. 1e). Upon induction of adipogenesis, Oil Red O staining, adipocyte marker, was positive (supplementary Fig. 1f) and almost 20% of the differentiated MSCs became positive for Nile Red Staining (supplementary Fig. 1g). A 14-fold increase in PPARγ, a master regulator of adipogenesis [31], and a 3000-fold increase in Fatty acid binding protein 4 (FABP4) mRNA were observed in the differentiated adipocytes (supplementary Fig. 1h).
To test our hypothesis that BMI1 regulates self-renewal of MSCs, we constructed lentiviral vectors that contained shRNAs targeting BMI1 (shBMI1) or scrambled (SCRAMBLE) under the control of human U6 polIII promoter along with the selection of puromycin and BMI1 overexpressing vectors with coexpression of eGFP driven by the MND-U3 promoter (Fig. 1a) and its functions were examined by qRT-PCR (Fig. 1b) and western blot (data not shown). Previous works have indicated derepression of p14ARF/p16INK4A locus upon loss of BMI1, which leads to self-renewal defects in various adult stem cells such as HSC [24, 25], NSC [26], and LSC [28], p21CIP1 for NSC [27] or p57KIP2 for LSC [28]. As expected, multiple cyclin-dependent kinase inhibitors (CDKIs) were derepressed by knockdown of BMI1 in MSCs (Fig. 1c). Among the CDKIs, p16, p27, and p57 were significantly upregulated.
Fig. (1). BMI1 expression regulates cell proliferation and apoptosis in human adult bone-marrow mesenchymal stem cells.

(a) Schematic diagram of shRNA targeting BMI1 expressing and BMI1 expressing lentiviral vectors (b) mRNA expression of BMI1 isolated from MSCs 72 hours after the transduction of specific shRNAs targeting BMI1 lentivirus (shBMI1, P=2.3x10-5) or BMI1 overexpressing vectors (BMI1, P=0.0031). Expression was normalized to GAPDH and shown as fold change relative to SCRAMBLE samples set to 1. pLKO.1-scramble was used as vector control. n=4. (c) Derepression and hyper-repression of cyclin-dependent kinase inhibitors by shBMI1 and overexpression of BMI1, respectively. Samples were collected 72 hours after transduction and applied to quantitative real-time polymerase chain reaction (qRT-PCR), normalized as in (b). p16 (P=0.007), p27 (P=0.006), and p57 (P=0.05) was upregulated upon shBMI1. p15 (P=0.025) was downregulated by BMI1 overexpression. n=4. (d) MTT cell growth assay shows cell proliferation differences between shBMI1 and overexpression of BMI1. Cell growth is shown as fold change relative to day 0 of SCRAMBLE samples set to 1. For the BMI1 overexpression, at day 4 (P=0.039) outgrowth was observed, day 6 (P=0.033), day 8 (P=0.009). For shBMI1, at day 8 (P=0.002) cell proliferation was reduced. n=3. (e) Colony-forming unit-fibroblast assay (minimum 50 cells per colony) in knockdown of BMI1 showed reduced colony forming capacity (P=0.004) but overexpression of BMI1 increase the number of colonies. n=3. (f) MSCs require BMI1 to avoid apoptosis. Annexin V+ cells increase over in vitro cultures at shBMI1 MSCs but overexpression of BMI1 significantly reduces apoptosis. Date is matched to (e). n=3. BMI1 at day 6 (P=0.029) and day 9 (P=0.036). *: P<0.05, **: P<0.01, ***: P<0.001, ****: P<0.0005. Data shown as mean ± SEM.
Next, we tested whether upregulation of p16, p27 or p57 by loss of BMI1 affects cell proliferation. By MTT assay (Fig. 1d), BMI1 overexpressing MSCs outgrew SCRAMBLE MSCs from day 4 but shBMI1 samples showed defects in cell proliferation throughout the experiments. Colony-forming unit-fibroblast (CFU-F) assay is one of well-known methods for self-renewal in MSCs. shBMI1 samples had shown decreased colony formation as compared to SCRAMBLE MSCs, but BMI1 overexpressing samples had increased numbers of colonies (Fig. 1e). Interestingly, the size of colonies from BMI1-overexpressing samples was enlarged, which may represent outgrowth of MSCs by repression of cell cycle check point or reduction of programmed cell death.
To test the hypothesis that BMI1 regulates apoptosis, Annexin V staining was performed on the in vitro cultures. shBMI1 populations slightly increased apoptosis over the culture period, although differences were not statistically significant (Fig. 1f). Interestingly, overexpression of BMI1 dramatically reduced apoptosis, which may explain the enlarged colony formation in the CFU-F assay. These results suggest that knockdown of BMI1 may reduce the self-renewal potential of MSCs.
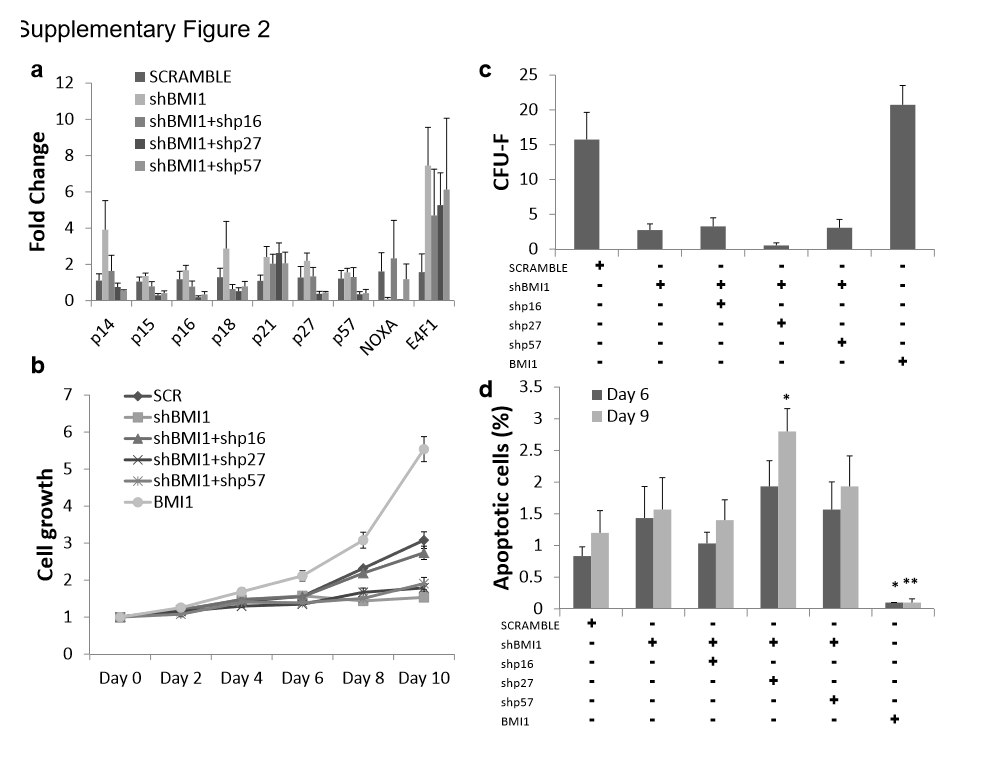
We next determined the ftmctional roles of elevated CDKI expression on the self-renewal defect in BMI1 knockdown MSCs. Compared to shBMI1 samples, knockdown of p16, p27, or p57 in BMI1 defect MSCs significantly reduced their transcriptional expression (supplementary Fig. 2a). Interestingly, shRNA for p27 (shp27) or for p57 (shp57) showed significant reduction of p15 and p16 mRNA levels in BMI1 knockdown MSCs. However the expression of p21 was not changed upon introduction of shRNAs against p16, p27 and p57, which may contribute to reduced cell proliferation. Unexpectedly, shp16 shRNA alone could rescue cell proliferation in the BMI1 knockdown MSCs. These data are in accordance with observances made in HSC [24, 25] and fibroblast [32]. CFU-F assay, however, showed different results in that shp16 in BMI1 knockdown MSCs could not rescue colony formation and the result was not dependent on apoptosis. shp27 in BMI1 knockdown MSCs increased apoptosis significantly over the duration of culture, which may explain even lower CFU-F potentials in rescue experiments (supplementary Fig. 2c).
To test the hypothesis that simultaneous deregulation of CDKIs caused reduction of cell growth in BMI1 knockdown MSCs, we examined combinations of CDKI shRNAs but none rescued cell proliferation and CFU-F formation (data not shown).
Knockdown of p16 can rescue the cell growth defects partially in BMI1 knockdown MSCs, suggesting that other factors exist in regulating cell proliferation. We hypothesized that multiple genes deregulated by loss of BMI1 may contribute to the proliferation defect in BMI1 knockdown MSCs. Among the candidates, expression of NOXA, which was shown to rescue CD4 T cell survival in a Bmi1−/− model [33], and E4F1, which was a Bmi1-interacting partner and was reported to restore repopulating potential of Bmi1−/− HSCs [34], was investigated (supplementary Fig. 2a). Interestingly, in the current studies gene expression of NOXA was reduced in shBMI1 MSC samples but upregulated in shp16 populations. E4F1 was highly elevated upon shBMI1 introduction but its expression was independent on shp16, shp27, or shp57, in accordance with data seen in HSCs by by Chagraoui et al. [34].
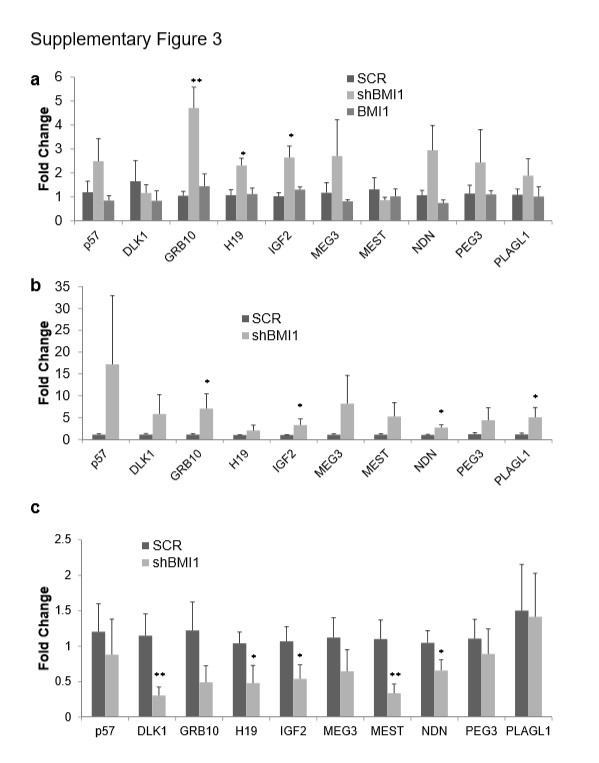
Recently, imprinted gene network (IGN) was shown to rescue self-renewal defects in a Bmi1-deficient mouse lung stem cell model [28]. In the current studies mRNA expression of IGN network genes such as DLK1, GRB10, H19, IGF2, MEG3 (as also known as Gtl2), MEST, NDN, PEGS, PLAGL1 and p57 in shBMI1 MSCs were transcriptionally upregulated in BMI1 knockdown MSCs (supplementary Fig. 3a). Among IGN, GRB10, H19 and IGF2 were 4.5-, 2.1-, 2.6-fold elevated, respectively, and other members of IGN were deregulated in BMI1 knockdown MSCs.
Multiple sets of shRNA expression vectors were used to examine other factors that could regulate cell proliferation (Fig. 2a). After introduction of shBMI1, expressions of IGN, p16 and E4F1 were upregulated, and their corresponding targeting shRNAs reduced transcription of each. However, loss of IGN or E4F1 did not confer the restoration of cell growth defects in shBMI1 samples (Fig. 2b). Interestingly, shp16 samples outgrew SCRAMBLE samples in the cell proliferation assay from Day 6 onward. This phenomenon was confirmed by CFU-F assay. The number of colonies formed for shBMI1 was higher than those obtained from SCRAMBLE samples in BMI1 knockdown MSCs. MSCs transduced by vectors carrying shRNAs for whole IGN or E4F1 grew slightly better and formed a slightly higher number of colonies but the differences were not statistically significant. An apoptosis study showed increased cell death in shIGNs samples, though again, these differences were not statistically significant.
Fig. (2). shRNAs for p16 partially rescues the proliferative defect of BMI1 knockdown MSCs.

(a) mRNA expression of shRNAs for Imprinted Gene Network (IGN), p16 cell-cycle dependent kinases inhibitors and E4F1 in shBMI1 MSCs. Samples were isolated 72 hours including 48 hours of 5 μg/ml puromycin selection after transduction of lentiviral vectors and determined by qRT-PCR, normalized as in Fig 1b. pLKO.1-scramble was used as a vector control (SCRAMBLE). shBMI1; shRNAs for BMI1. shRNA for maternal IGN; p57, GRB10, MEG3 and H19. shRNA for paternal IGN, DLK1, IGF2, MEST, NDN, PEG3 and PLAGL1. shp16; shRNA for p16. shE4F1; shRNA for E4F1. n=3. (b) MTT cell growth assay shows cell proliferation defect in BMI1 knockdown MSCs were rescued by p16 knockdown. Normalization as in Fig 1e. P-values were calculated based on shBMI1 (see supplementary Table 2). n=3. (c) Colony-forming unit-fibroblast assay following knockdown of IGN, p16 and E4F1 shows p16 knockdown overcome growth defect by shBMI1. n=3. (d) Increased apoptosis on shBMI1 MSCs was observed. n=3. *: P0.05, **: P<0.01, ***: P<0.001, ****: P<0.0005. Data shown as mean ± SEM. See also supplementary Fig. (2).
To test whether DNA damage may contribute to the cell growth defect in BMI1 knockdown MSCs, hypoxia (5% O2), shown to reduce reactive oxygen species [35], and shRNA for CHEK2, known to rescue phenotypes of BMI1−/− mice [35], were examined in cell growth, CFU-F and apoptosis assays. Unfortunately, neither could rescue the growth defect in BMI1 knockdown MSCs (data not shown).
BMI1 represses Hox gene expression, well-known regulators of differentiation in Drosophila [19, 21] and BMI1−/− HSCs preferentially differentiate into B-cell lineage at the expense of T lymphopoiesis due to derepressed Ebf1 and Pax5, which are B-cell lineage regulators [36]. In MSCs, however, BMI1-interacting genes involved in multipotency and lineage specification remain elusive. The functional role of BMI1 in impacting the multipotency of MSCs had only been examined at mouse MSCs [30] but not human MSCs. We tested the functional roles of BMI1 during the differentiation into bone and fat. Alizarin Red S staining for osteogenesis revealed that shBMI1 blocked osteogenesis (Fig. 3a). Alkaline phosphate (ALP) activity, an early marker for osteogenesis, was reduced in shBMI1 populations (Fig. 3b). qRT-PCR of RUNX2, the master regulator of osteogenesis, osteocalcin (OCN) and BSP showed a 3-fold, 11.6-fold and 8.3-fold upregulation of mRNA expression in knockdown of BMI1 without osteogenic media induction, respectively (Fig. 3c), which may suggest that RUNX2, OCN and BSP are targets of BMI1. In osteogenic media, expression of OCN and BSP were restored close to the levels in SCRAMBLE samples, but RUNX2 transcript levels were reduced, which might be the reason why osteogenesis was decreased. In contrast to derepressed RUNX2, which may lead to increased osteogenesis, in shBMI1 without osteogenic induction, blocked osteogenesis as shown by reduced Alizarin Red S staining and ALP levels may imply that different factors deregulated in BMI1 knockdown MSCs undoubtedly exist. PPARγ, a master regulator of adipogenesis, has been shown to prevent osteogenesis [31]. Almost 6-fold upregulation of PPARγ expression in shBMI1 samples without differentiation induction was observed, which also suggests that PPARγ is one of the targets of BMI1. Its expression was maintained in osteogenic differentiation, which may lead to the blocked osteogenesis that we observed in the BMI1 knockdown MSCs. These results are consisted with mouse MSC data, which showed osteopenic phenotypes in Bmi1-null mice [30].
Fig. (3). Derepression of developmental regulator network via BMI1 knockdown decreases osteogenic and adipogenic differentiation.

(a-c) Osteogenesis in shBMI1 MSCs was tested. Control; no osteogenic induction, Osteo; osteogenic induction (a) Images from Alizarin Red S staining for osteoblasts. MSCs were cultured with osteogenic media for 14 days. (b) Alkaline phosphate activity (normalized by whole protein concentration) in shBMI1 was reduced (P=0.01), n=6. (c) mRNA levels of RUNX2, OCN and BSP in osteogenic differentiation, normalized as in Fig 1c, were reduced upon shBMI1 and derepression of RUNX2, PPARγ and OCN was observed without an osteogenic induction. n=3. PPARγ mRNA levels, well-known osteogenic inhibitor, were upregulated with and without osteogenic induction. (d-f) Adipogenesis in shBMI1 was evaluated 14 days after the treatment of adipogenic cocktails. Adipo; adipogenic induction (d) Images from Oil Red O staining for adipogenesis. (e) Nile Red+ cells by flow cytometry showed slight reductions in shBMI1 (P=0.049). n=4. (f) mRNA expressions of PPARγ and FABP4, normalized as in Fig 1b, shows that derepression of these transcripts were observed without adipogenic inductions. Gene expression of RUNX2, MSX2 and MITR , well-known osteogenic-related factors for blocking adipogenesis were evaluated. n=3. (g-j) PPARγ and IGN were necessary to rescue osteogenesis in shBMI1 MSCs (g) Images from Alizarin Red S staining for osteoblasts. Vector transduced MSCs were cultured with osteogenic media for 14 days. (h) Alizarin Red S was measured by acetic acid precipitation, normalized by whole protein concentration. P-value was calculated based on SCRAMBLE. shBMI1 (P=0.004), shBMI1+shIGN (P=0.04). AU; arbitrary unit. n=4. (i) Alkaline phosphate activity, normalized by whole protein concentration. p-value was calculated based on SCRAMBLE. shBMI1 (P=0.007), shBMI1+shIGN (P=0.0008). n=4. (j) mRNA levels of RUNX2, OCN and BSP in osteogenic differentiation, normalized to scramble osteogenic induction as 1, were shown. PPARγ mRNA levels, well-known osteogenic inhibitor, were upregulated upon shBMI1 and slight reduction in shIGN samples against shBMI1 alone. IGN transcripts were evaluated. n=4. *: P<0.05, **: P<0.01, ***: P<0.001, ****: P<0.0005 Data shown as mean ± SEM.
In adipogenesis, knockdown of BMI1 in MSCs caused a slight reduction in Oil Red O staining (Fig. 3d) and it was confirmed by flow cytometric analysis of Nile Red staining (16% in scramble vs. 9% in shBMI1, p<0.05. Fig. 3e). Without adipogenic induction, mRNA expression of PPARγ and FABP4 were increased up to 5.7-fold and 3-fold in shBMI1 samples as compared to the SCRAMBLE control, respectively and expression remained elevated during adipogenesis (Fig. 3f). To test whether osteogenic factors are involved in reduction of adipogenesis, gene expression of RUNX2, MSX2, which was shown to diminish PPARγ expression and adipogenesis [37], and MITR, which was also reported to prevent PPARγ expression and adipogenesis [38], were evaluated. 3.4-, 1.7- and 2.3-fold upregulation of RUNX2, MSX2 and MITR transcripts were observed in shBMI1 samples, respectively, and RUNX2, MSX2 and MITR levels were stabilized with adipogenic differentiation.
To test the idea that PPARγ or other factors deregulated from BMI1 knockdown MSCs prevents osteogenesis, shRNAs for PPARγ (shPPARG) were applied to shBMI1 samples. Alizarin Red S staining showed that knockdown of PPARγ rescued osteogenesis (Fig. 3g). Quantification of Alizarin Red S revealed that shPPARG transduced samples partially rescued osteogenesis in shBMI1 samples (Fig. 3h) and ALP assay confirmed this phenomenon (Fig. 3i). 0.1 μM GW9662, a potent inhibitor of PPARγ protein, in osteogenesis of shBMI1 also showed upregulated Alizarin Red S staining and ALP (data not shown). This result points out that knockdown of PPARγ is sufficient to rescue osteogenesis partially but deregulated PPARγ itself by loss of BMI1 is not responsible for preventing osteogenesis. IGN has been known to be expressed in adult stem cells such as HSC, skeletal muscle satellite cells, and epidermal stem cells, and to be downregulated postnatally in response of differentiation [28, 39]. To test the hypothesis that adult stem cells share common stemness factors to regulate self-renewal and multipotency, gene expression of IGN in osteogenically [supplementary Fig. 3b) and adipogenically [supplementary Fig. 3c) differentiated shBMI1 samples were assessed in comparison to SCRAMBLE. Interestingly, mRNA expression of IGN in osteogenic shBMI1 samples still remained significantly upregulated but its transcripts in adipogenic shBMI1 samples decreased significantly, which implicates IGN as an osteogenic inhibitor. To answer this question, shIGNs were applied to BMI1 knockdown MSCs. Alizarin Red S staining showed elevated calcium deposition (Fig. 3g) and its quantification confirmed that it had slightly lower expression than shPPARG in BMI1 knockdown MSCs (P=0.04 against SCRAMBLE, n=4, Fig. 3h). On the contrary, ALP assay showed significantly reduced expression in shIGN samples along with shBMI1. Gene expression analysis of these samples showed that shIGN samples in BMI1 knockdown MSCs were not able to repress PPARγ transcripts, which may lead to a slight reduction of osteogenic rescue as compared to shPPARG.
To test whether BMI1 acts as a repressor of differentiation-related transcripts, the differentiation potential of BMI1 overexpressing MSCs was investigated. Osteogenesis in BMI1 overexpressing MSCs was reduced, as assessed by Alizarin Red S staining (Fig. 4a) and was confirmed by ALP assay (4.5-fold reduction between SCRAMBLE and overexpressed BMI1 samples, n=3, Fig. 4b). qRT-PCR showed a 24-fold reduction in RUNX2 transcripts but expression of OCN and BSP in BMI1 overexpressing MSCs were maintained as compared to SCRAMBLE without osteogenic induction (Fig. 4c) and remained closed to undifferentiated states with osteogenic media. PPARγ in osteogenesis was also reduced with and without induction media, which indicates that PPARγ is not responsible for reduction of osteogenesis. These results suggest that RUNX2 and PPARγ are targets of BMI1 by unknown mechanisms. Overexpression of BMI MSCs showed a decline in Oil Red O positive cells (Fig. 4d) and Nile Red staining confirmed that BMI1 prevented adipogenesis (Fig. 4e). PPARγ and FABP4 transcripts were decreased when BMI1 was overexpressed with no differentiation induction (Fig. 4f) but levels were upregulated with adipogenic induction. RUNX2 in adipogenesis decreased 7-fold lower than SCRAMBLE in the undifferentiated state but increased almost 5-fold compared to SCRAMBLE upon the adipogenic media; its levels were still lower than SCRAMBLE without differentiation induction.
Fig. (4). Hyper-repression of developmental regulator network via overexpression of BMI1 decreases osteogenic and adipogenic differentiation.

(a-c) Osteogenesis in BMI1 overexpressing MSCs was tested. Control; no osteogenic induction, Osteo; osteogenic induction. (a) Images from Alizarin Red S staining for osteogenesis. (b) Alkaline phosphate activity, normalized as in Fig. (3b), in BMI1 overexpressing MSCs was reduced (P=0.27), n=3. (c) Gene expression of RUNX2 (P=0.09) in osteogenesis, normalized as in Fig. (1c), was repressed upon overexpressed BMI1 samples without osteogenic induction. mRNA expression of PPARγ (P=0.017) was reduced by BMI1 overexpression with an osteogenic induction. (d-f) Adipogenesis in BMI1 overexpressing samples was evaluated 14 days after adipogenic induction (d) Images from Oil Red O staining for adipogenesis. (e) Nile Red+ cells by flow cytometry showed slight reductions in BMI1 overexpressing samples (P=0.011). n=4. (f) mRNA expression of PPARγ and FABP4, normalized as in Fig. (1b), shows that derepression of these transcripts were observed without adipogenic inductions. Gene expression of RUNX2 was shown. n=3. (g) A schematic depicting the model for self-renewal by BMI1. BMI1 represses cyclin-dependent kinase inhibitors to promote G1 to S phase cell cycle entry, which leads to cell proliferation. In addition, BMI1 regulates programmed cell death. For the regulation of differentiation into bone or fat, BMI1 represses RUNX2 and PPARγ transcripts, respectively. IGN, which was shown to prevent osteogenesis, was regulated by BMI1. *: P<0.05, **: P<0.01, ***: P<0.001, ****: P<0.0005 Data shown as mean ± SEM.
In this study, our findings that loss of BMI1 reduces self-renewal capacity in MSCs emphasize the conserved role of BMI1 across adult tissues. Tissue-specific stem cells in a human use the same sets of genes but they are expressed differently in different tissues and the expression changes over time. Epigenetic regulations are thought to be critical in the control of the transcriptional circuitry in stem cells [19, 21, 22]. The balance between self-renewal and differentiation is a crucial matter in stem cells, and accumulating evidence indicates that polycomb group (PcG) proteins are one of the major players in the transcriptional network of self-renewal in adult stem cells [19, 21, 22]. BMI1, which is a component of polycomb repressive complexes (PRC) and known to repress Hox genes and cell cycle regulators such as p16 or p19 [18-22], is known to be required for self-renewal of HSCs [24, 25, 40], NSCs [26, 27], intestinal stem cells [27], prostate stem cells [29], and lung stem cells [28]. Human adult BM-MSCs are shown to support hematopoiesis, and can give rise to osteoblasts, adipocytes, chondrocytes, fibroblast, or muscle cells [4, 41, 42]. Although many transcription factors involved in the lineage specification of MSCs have been identified, molecular mechanisms of self-renewal remain to be answered. In human MSCs, tests of self-renewal are experimentally limited because there is no assay to confirm self-renewal potential in vitro [43, 44] and repopulating study after transplantation in a xenograft model is imperfect since human MSCs lack of homing capacity to the bone marrow niche [16, 45]. Therefore assays such as those described in the current studies are used, and a better understanding of regulatory processes at the stem cell level has been needed for human MSCs.
To self-renew, stem cells must antedate cell cycle and move forward through mitosis [19]. Simultaneously, one daughter cell must retain multipotency and cells should repair DNA damage and prevent apoptosis [19]. In this study, we divided characteristics of self-renewal as three aspects to; cell proliferation, apoptosis and repression of differentiation and differentiation-related genes during cell proliferation. Our results highlight that BMI1 is important for MSC proliferation, is crucial for apoptosis, and represses the expression in differentiation-related genes (Fig. 4g). Loss of BMI1 induces prevention of MSC proliferation by derepressing p16INK4A expression and increases cell death. Upregulated p16 mRNA levels clearly indicate that cell cycle was inhibited at the G1/S phase checkpoint (data not shown) and lead to reduction of cell proliferation [32] and knockdown of p16 was sufficient to restore cell proliferation in BMI1 knockdown MSCs. In contrast, BMI1 overexpression eases the outgrowth of MSCs by repressing CDKI transcripts and reducing apoptosis. In this study, we also demonstrated that BMI1 poises between osteo- and adipogenesis by regulating the expression of PPARγ and IGN. Knockdown of BMI1 causes premature activation of both lineage-specific genes and enhances adipogenic differentiation in comparison to th6e osteogenic lineage even though both osteogenic and adipogenic potential were reduced against scrambled MSCs. A similar role for BMI1 in differentiation was observed in mouse HSCs [36], where loss of Bmi1 caused deregulated lineage-specific genes and enhance B-cell lineage differentiation but not T-cell lineage. Among the prematurely activated genes, PPARγ, which suppresses osteogenic-related genes such as homeobox protein Dlx5, RUNX2 and Osterix [31, 46], and IGN are highly upregulated in osteogenesis and knockdown of these genes were sufficient to rescue osteogenesis. IGN was suggested as a shared regulatory mechanism in adult stem cells [39] and regulates self-renewal of lung stem cells [28]. Even though knockdown of BMI1 elevates expression of IGN, it was not sufficient to rescue cell proliferation upon the knockdown of IGN in BMI1 knockdown MSCs. However, as a developmental regulator, IGN plays a major role in the regulation of osteogenesis in BMI1 knockdown MSCs.
These observations taken together with cell proliferation and apoptosis support the regulation of self-renewal of MSCs by BMI even though the downstream pathway of cell proliferation in BMI1 knockdown MSCs is still unknown. Our results provide important insights into how BMI1 regulates self-renewal of MSC via control of proliferation, apoptosis, and how BMI1-mediated repression of differentiation-related genes is governed during differentiation.
MATERIALS AND METHODS
Cell Culture and In Vitro Differentiation
Human bone marrow aspirates (purchased from Lonza] were passed through 90 μm cell strainers for isolation of bone spicules and diluted with equal volume of phosphate-buffered saline (PBS) and centrifuged over Ficoll (GE Healthcare) for 30 minutes at 700g. Mononuclear cells and bone spicules were plated in plastic cell culture flasks with α-MEM (Hyclone) supplemented with 10 % Fetal Bovine Serum (Hyclone). 2 days after initial plating, nonadherent cells were removed.
Human adult bone marrow (BM) mesenchymal stem cells (MSCs) were maintained as previously described [47]. Briefly, BM-MSCs were cultured in α-MEM (Hyclone) supplemented with 10 % Fetal Bovine Serum (Hyclone), and 5000 units ml−1 penicillin/streptomycin (Invitrogen). Media was changed every two days. In vitro MSC differentiation was tested as previously described [1]. Oil Red O (Electron Microscopy Sciences) and Alizarin Red S (Ricca Chemical Company) were used for staining adipocytes and osteoblasts, respectively. For the quantification of Alizarin Red S, 10 % acetic acid was applied for 30 mins and supernatants were transferred into 1.5 ml tubes for 13,200 rpm centrifugation for 10 mins. Samples were read by EMax Endpoint ELISA Microplate Reader (Molecular Devices) with 405 nm wave-length. For the quantification of adipocyte positive cells, Nile Red (MP biomedical) staining was used according to the manufacturer’s protocol. Throughout the experiment, data was collected only from MSC batches that expanded to passage 10 without premature senescence in the control arm. MSCs from a minimum of 3 donors were used for the experiments and numbers of donor MSCs used in experiments were indicated as n in each figure. In each donor, at least duplicates of technical replicates are used in experiments.
Plasmid Construction
Primer sequences used in cloning are listed in the supplementary Table 2. For shRNA vectors, pLKO.1-shBMI1s were purchased from Open biosystems. Cyclin-dependent kinase inhibitors, NOXA, E4F1, IGN and PPARγ targeting shRNA primers were annealed and cloned into pLKO.1-scramble [48] (Addgene plasmid 1864) between AgeI and EcoRI and were confirmed by sequencing. pLKO.1-scramble (SCRAMBLE) was used as a vector control. For the overexpression of BMI1, mRNAs of BMI1 were isolated from BM-MSCs and cloned into TOPO TA plasmid (Invitrogen) with MfeI overhang at the 5’- and 3’-ends to create pCR2.1-BMI1 and were confirmed by sequencing. MfeI digested pCR2.1-BMI1 was cloned into EcoRI digested pCCLc-MND-PGK-eGFP (UC Davis stem cell program vector core) and was confirmed the direction by sequencing (BMI1).
Generation of shRNAs and BMI1 Expressing MSCs
Lentiviral transductions were conducted as previously described [49]. Briefly, 2.5 x 107 HEK-293T cells in T-225 flasks (Corning) were transfected with 25 μg of lentiviral vectors, 25 μg of pCMV-dR8.91, and 5 μg of pMDG-VSV-G using TrasIT-293 transfection reagent (Mirus) according to the manufacturer’s protocol. Three days after transfections, supernatants were centrifuged with Centricon plus-70 (Millipore) for 35 minutes at 3500 rpm and collected and filtered through 0.45 μm HV DURAPORE (Millipore), and viral titer was measured and stored at −80°C. For shRNA or BMI1 overexpression, BM-MSCs were transduced with lentiviral vectors (MOI 3). shRNAs expressing MSCs were subsequently selected by 5 ug ml−1 puromycin (Invivogen) for 2 days and the expression of BMI1 was checked by western blot or qRT-PCR. Transduction efficiency of overexpressed BMI1 in MSCs was checked by FC500 flow cytometry (Beckman-Coulter).
Immunocytofluorescence Analysis
MSCs were fixed in 4% paraformaldehyde for 15 mins, permeabilized with BD Perm/Wash buffer (BD) for 30 mins, and wash twice with PBS (Hyclone). Primary antibodies (listed in supplementary Table 3) were applied for 1 hr at room temperature, and washed twice with PBS, and secondary antibodies (see supplementary Table 3) were incubated for 1 hr at room temperature. Hoechst 33342 (Invitrogen) was added for 10 min. Images were taken by Nikon TiU microscope (Nikon).
Flow Cytometry Analysis
Antibodies used in flow cytometry experiments are listed in supplementary Table 3. Adherent MSCs were lifted by Cell Stripper (Mediatech) and washed twice with PBS and blocked with Staining buffer (BD). PE-conjugated antibodies were incubated in 1 hr at room temperature and wash twice with PBS. Flow cytometric experiments were performed with FC500. For the Nile red staining, 10 ug/ml of Nile red was incubated with adipocytes for 10 mins at the dark room, washed twice with PBS, and analyzed using an FC500.
Apoptosis Assay and Cell Proliferation Assay
Apoptosis assay was performed as previously described [50]. MTT assay (Roche) was performed according to the manufacturer’s protocol. Briefly, 500 MSCs were plated onto 48-well plate (duplicates) with 300 μl MSC culture media. Media was changed every two days. For the measurement of MTT, 30 μl MTT was incubated for 4 hours at 37°C and then 300 μl stop solution was followed for one hour at 37°C. Supernatants were replated into 96-well plate (triplicates) and 570 nm wave lengths were used to read the samples by EMax Endpoint ELISA Microplate Reader. 650 nm wavelengths were used as a background reference.
Colony-Forming Unit-Fibroblast (CFU-F)
CFU-F assay was performed according to the manufacturer’s protocol. Briefly, 150 cells were plated onto 100 mm Petri Dishes (5 replicates) with 13 ml MSC culture media. 14 days after incubation, colonies were stained with 0.5% crystal violet solution (Acros Organics) for 30 mins and washed four times with PBS. Then, Petri dishes were rinsed with tap water and dried. Stained colonies were counted based on the minimum of 50 cells.
Gene Expression Analysis
Total RNAs were isolated using RNA Stat-60 (Tel-test) according to the manufacturer’s protocol. QuantiTect Reverse transcription kit (Qiagen) was used to synthesize cDNA and QuantiTect SYBR Green PCR kit (Qiagen) was utilized to perform qRT-PCR on Applied Biosystems 7300 Real Time PCR system with primer sets corresponding to Supplementary Table 2. Samples were normalized by GAPDH.
Statistical Analysis
Data are shown as mean ± standard error unless otherwise indicated. Student's t-test (unpaired, two-tailed) was used to compare data within two groups, and the P-value was calculated by Excel (Microsoft). P<0.05 was considered as statistically significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
We thank Fernando Fierro for providing BM-MSCs. Y.J. was supported by a Howard Hughes Medical Institute Med into Grad scholarship and JN is partially supported by the California Institute for Regenerative Medicine (CIRM). This work was supported by the NIH Heart Lung and Blood Institute (RO1 HL073256 to JN), the NIH Office of the Director (Transformative Grant R01GM099688 to JN) and UC Davis Stem Cell Program start-up funds from the Dean's office.
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers web site along with the published article.
REFERENCES
- [1].Gruenloh W, Kambal A, Sondergaard C, et al. Characterization and in vivo testing of mesenchymal stem cells derived from human embryonic stem cells. Tissue Eng Part A 2011; 17(11-12): 1517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jung Y, Bauer G, Nolta JA. Concise review: induced pluripotent stem cell-derived mesenchymal stem cells: progress toward safe clinical products. Stem Cells 2012; 30(1): 42–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Meyerrose T, I R, M D, P H, G B, Nolta J. Establishment and transduction of primary human stromal/ mesenchymal stem cell monolayers. Nolta J, editor. Dordrecht, the Netherlands: Kluwer Academic Publishers; 2006. [Google Scholar]
- [4].Meyerrose T, Olson S, Pontow S, et al. Mesenchymal stem cells for the sustained in vivo delivery of bioactive factors. Adv Drug Deliv Rev 2010; 62(12): 1167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Meyerrose TE, De Ugarte DA, Hofling AA, et al. In vivo distribution of human adipose-derived mesenchymal stem cells in novel xenotransplantation models. Stem Cells 2007; 25(1): 220–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Xiong YY, Fan Q, Huang F, et al. Mesenchymal stem cells versus mesenchymal stem cells combined with cord blood for engraftment failure after autologous hematopoietic stem cell transplantation: a pilot prospective, open-label, randomized trial. Biol Blood Marrow Transplant 2014; 20(2): 236–42. [DOI] [PubMed] [Google Scholar]
- [7].Wang P, Li Y, Huang L, et al. Effects and Safety of Allogenic Mesenchymal Stem Cell Intravenous Infusion in Active Ankylosing Spondylitis Patients Who Failed NSAIDs: A 20-Week Clinical Trial. Cell Transplant 2014; 23(10): 1293–303. [DOI] [PubMed] [Google Scholar]
- [8].Suncion VY, Ghersin E, Fishman JE, et al. Does transendocardial injection of mesenchymal stem cells improve myocardial function locally or globally?: An analysis from the Percutaneous Stem Cell Injection Delivery Effects on Neomyogenesis (POSEIDON) randomized trial. Circ Res 2014; 114(8): 1292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Skrahin A, Ahmed RK, Ferrara G, et al. Autologous mesenchymal stromal cell infusion as adjunct treatment in patients with multidrug and extensively drug-resistant tuberculosis: an open-label phase 1 safety trial. Lancet Respir Med 2014; 2(2): 108–22. [DOI] [PubMed] [Google Scholar]
- [10].Karantalis V, DiFede DL, Gerstenblith G, et al. Autologous mesenchymal stem cells produce concordant improvements in regional function, tissue perfusion, and fibrotic burden when administered to patients undergoing coronary artery bypass grafting: The Prospective Randomized Study of Mesenchymal Stem Cell Therapy in Patients Undergoing Cardiac Surgery (PROMETHEUS) trial. Circ Res 2014; 114(8): 1302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sleeboom-Faulkner M. Experimental treatments: Regulating stem-cell therapies worldwide. Nature 2013; 495(7439): 47. [DOI] [PubMed] [Google Scholar]
- [12].Prockop DJ. Concise review: two negative feedback loops place mesenchymal stem/stromal cells at the center of early regulators of inflammation. Stem Cells 2013; 31(10): 2042–6. [DOI] [PubMed] [Google Scholar]
- [13].Mohamadnejad M, Alimoghaddam K, Bagheri M, et al. Randomized placebo-controlled trial of mesenchymal stem cell transplantation in decompensated cirrhosis. Liver Int 2013; 33(10): 1490–6. [DOI] [PubMed] [Google Scholar]
- [14].Lv YT, Zhang Y, Liu M, et al. Transplantation of human cord blood mononuclear cells and umbilical cord-derived mesenchymal stem cells in autism. J Transl Med 2013; 11(1): 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Jiang PC, Xiong WP, Wang G, et al. A clinical trial report of autologous bone marrow-derived mesenchymal stem cell transplantation in patients with spinal cord injury. Exp Ther Med 2013; 6(1): 140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009; 4(3): 206–16. [DOI] [PubMed] [Google Scholar]
- [17].Beisel C, Paro R. Silencing chromatin: comparing modes and mechanisms. Nat Rev Genet. 2011; 12(2): 123–35. [DOI] [PubMed] [Google Scholar]
- [18].Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 2009; 10(10): 697–708. [DOI] [PubMed] [Google Scholar]
- [19].Sauvageau M, Sauvageau G. Polycomb group proteins: multifaceted regulators of somatic stem cells and cancer. Cell Stem Cell 2010; 7(3): 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet 2007; 8(1): 9–22. [DOI] [PubMed] [Google Scholar]
- [21].Surface LE, Thornton SR, Boyer LA. Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell 2010; 7(3): 288–98. [DOI] [PubMed] [Google Scholar]
- [22].Sawarkar R, Paro R. Interpretation of developmental signaling at chromatin: the Polycomb perspective. Dev Cell 2010; 19(5): 651–61. [DOI] [PubMed] [Google Scholar]
- [23].Alkema MJ, Wiegant J, Raap AK, Berns A, van Lohuizen M. Characterization and chromosomal localization of the human proto-oncogene BMI-1. Hum Mol Genet 1993; 2(10): 1597–603. [DOI] [PubMed] [Google Scholar]
- [24].Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003; 423(6937): 255–60. [DOI] [PubMed] [Google Scholar]
- [25].Park IK, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003; 423(6937): 302–5. [DOI] [PubMed] [Google Scholar]
- [26].Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003; 425(6961): 962–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fasano CA, Dimos JT, Ivanova NB, Lowry N, Lemischka IR, Temple S. shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell 2007; 1(1): 87–99. [DOI] [PubMed] [Google Scholar]
- [28].Zacharek SJ, Fillmore CM, Lau AN, et al. Lung stem cell self-renewal relies on BMI1-dependent control of expression at imprinted loci. Cell Stem Cell 2011; 9(3): 272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lukacs RU, Memarzadeh S, Wu H, Witte ON. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 2010; 7(6): 682–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang HW, Ding J, Jin JL, et al. Defects in mesenchymal stem cell self-renewal and cell fate determination lead to an osteopenic phenotype in Bmi-1 null mice. J Bone Miner Res 2010; 25(3): 640–52. [DOI] [PubMed] [Google Scholar]
- [31].Kawai M, Rosen CJ. PPARgamma: a circadian transcription factor in adipogenesis and osteogenesis. Nat Rev Endocrinol 2010; 6(11): 629–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999; 397(6715): 164–8. [DOI] [PubMed] [Google Scholar]
- [33].Yamashita M, Kuwahara M, Suzuki A, et al. Bmi1 regulates memory CD4 T cell survival via repression of the Noxa gene. J Exp Med 2008; 205(5): 1109–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chagraoui J, Niessen SL, Lessard J, et al. E4F1: a novel candidate factor for mediating BMI1 function in primitive hematopoietic cells. Genes Dev 2006; 20(15): 2110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu J, Cao L, Chen J, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 2009; 459(7245): 387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Oguro H, Yuan J, Ichikawa H, et al. Poised lineage specification in multipotential hematopoietic stem and progenitor cells by the polycomb protein Bmi1. Cell Stem Cell 2010; 6(3): 279–86. [DOI] [PubMed] [Google Scholar]
- [37].Ichida F, Nishimura R, Hata K, et al. Reciprocal roles of MSX2 in regulation of osteoblast and adipocyte differentiation. J Biol Chem 2004; 279(32): 34015–22. [DOI] [PubMed] [Google Scholar]
- [38].Chen YH, Yeh FL, Yeh SP, et al. Myocyte enhancer factor-2 interacting transcriptional repressor (MITR) is a switch that promotes osteogenesis and inhibits adipogenesis of mesenchymal stem cells by inactivating peroxisome proliferator-activated receptor gamma-2. J Biol Chem 2011; 286(12): 10671–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Berg JS, Lin KK, Sonnet C, et al. Imprinted genes that regulate early mammalian growth are coexpressed in somatic stem cells. PLoS One 2011; 6(10): e26410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Iwama A, Oguro H, Negishi M, et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity 2004; 21(6): 843–51. [DOI] [PubMed] [Google Scholar]
- [41].Jung Y, Bauer G, Nolta JA. Induced Pluripotent Stem Cell - Derived Mesenchymal Stem Cells: Progress Toward Safe Clinical Products. Stem Cells 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Parekkadan B, Milwid JM. Mesenchymal stem cells as therapeutics. Annu Rev Biomed Eng 2010; 12: 87–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell 2012; 10(6): 709–16. [DOI] [PubMed] [Google Scholar]
- [44].Scadden DT. Rethinking stroma: lessons from the blood. Cell Stem Cell 2012; 10(6): 648–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sackstein R, Merzaban JS, Cain DW, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med 2008; 14(2): 181–7. [DOI] [PubMed] [Google Scholar]
- [46].Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B. PPARgamma2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells. J Cell Biochem 2009; 106(2): 232–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Olson SD, Kambal A, Pollock K, et al. Examination of mesenchymal stem cell-mediated RNAi transfer to Huntington's disease affected neuronal cells for reduction of huntingtin. Molecular and cellular neurosciences. 2012; 49(3): 271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Stewart SA, Dykxhoorn DM, Palliser D, et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003; 9(4): 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kambal A, Mitchell G, Cary W, et al. Generation of HIV-1 resistant and functional macrophages from hematopoietic stem cell-derived induced pluripotent stem cells. Mol Ther 2011; 19(3): 584–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dao MA, Nolta JA. Cytokine and integrin stimulation synergize to promote higher levels of GATA-2, c-myb, and CD34 protein in primary human hematopoietic progenitors from bone marrow. Blood 2007; 109(6): 2373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.