

Abstract
Glioblastoma (GBM) is the most aggressive and lethal form of brain tumor in human adults. The myeloid lineage cells, including macrophages, microglia, myeloid-derived suppressor cells, and neutrophils are the most frequent types of cells in the GBM tumor microenvironment (TME) that contribute to tumor progression. Emerging experimental evidence indicates that symbiotic interactions between cancer cells and myeloid cells are critical for tumor growth and immunotherapy resistance in GBM. This review covers the molecular mechanisms for how cancer cells shape a myeloid cell-mediated immunosuppressive TME, and reciprocally, how such myeloid cells affect tumor progression and immunotherapy efficiency in GBM. Moreover, this review discusses tumor-T-cell symbiosis and summarizes immunotherapeutic strategies intercepting this co-dependency in GBM.
Tumor-immune symbiotic interaction in glioblastoma
Glioblastoma (GBM) is the most common and lethal form of brain tumor in human adults, with a median survival of approximately 14–16 months following initial diagnosis [1,2]. The current standard-of-care for GBM patients includes maximal surgical resection followed by radiotherapy and/or chemotherapy, which offers minimal clinical benefits [1,2]. Moreover, clinical trials for targeted therapies (e.g., therapies targeting the receptor tyrosine kinase signaling) have also failed to improve patient outcomes, which is largely due to the inter/intra-tumoral genetic heterogeneity and instability, and insufficient target engagement within the brain [3].
GBM can be highly infiltrated with immune cells in the tumor microenvironment (TME) [4–6] (see Glossary). The most frequent immune population within the GBM TME are myeloid lineage cells, which include tumor-associated macrophages and microglia (TAMs), myeloid-derived-suppressor cells (MDSCs), and neutrophils [7–10]. Increasing evidence underscores that these myeloid cells not only promote GBM tumor growth, but provoke an immunosuppressive TME to induce resistance of immunotherapies, including immune checkpoint inhibitor (ICI) therapies [7,9,11–15]. In-depth studies focusing on the immune landscape have revealed that immune cells in GBM are multifaceted [4,16,17]. For example, the proportion and functional status of immune cells varies based on the genetic background, molecular state (e.g., mesenchymal, classical, and proneural), and disease stage of GBM [18–20]. Upon infiltration into the TME, immune cells are educated by cancer cells to promote tumor progression, inhibit anti-tumor immunity, and induce immunotherapy resistance [15,21,22]. Together, these findings vastly expand our knowledge of the context-dependent symbiotic interaction between cancer cells and immune cells in GBM. In this review, we discuss the molecular mechanisms by which cancer cells shape an immunosuppressive TME via regulating the biology of myeloid cells and lymphocytes, and, reciprocally, the mechanisms by which immune cells affect GBM progression and immunotherapy efficiency. Moreover, we discuss the therapeutic potential of targeting tumor-myeloid cell symbiosis in combination with immunotherapies (e.g., ICI therapies) for treatment of GBM.
GBM-TAM crosstalk
TAMs are the most abundant type of cells in the GBM TME (accounting for up to 50% of total live cells of the whole tumor mass) and composed of two major subpopulations, including bone marrow-derived macrophages (hereafter referred to as macrophages) and microglia (Box 1) [23]. Emerging evidence demonstrates that the context-dependent symbiotic interaction between cancer cells and TAMs is critical for GBM tumor growth via regulating distinct cytokines, chemokines, metabolites, and other factors [21]. Here, we discuss different molecular mechanisms underlying the tumor-TAM crosstalk in GBM (Figure 1 and Table 1).
Box 1. Bone marrow-derived macrophages and brain resident microglia in GBM.
Lineage-tracing experiments in mice have demonstrated that monocytes can migrate to the brain and differentiate into macrophages during the GBM progression. In contrast, microglia originate from yolk sac progenitors during embryonic development [21,122]. The morphological differences between macrophages and microglia have been observed under the high-resolution open-skull 2-photon microscopy [123]. Single-cell RNA sequencing (scRNA-seq) and cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) have shown that microglia usually occur in newly diagnosed GBM tumors. In contrast, macrophages are more prevalent in recurrent GBM tumors and in the hypoxic regions of tumors. In addition, tumor-associated macrophages and microglia (TAMs) are highly plastic cells and can be polarized toward both immunosuppressive and immunostimulatory phenotypes, thus exhibiting distinct effects [21]. Moreover, TAMs show a phenotype-specific spatial distribution in which immunosuppressive and immunostimulatory TAMs are enriched in the tumor periphery and core, respectively [19,124].
The characterization of heterogeneous populations of microglia and macrophages in GBM tumors is still an arduous task due to the lack of specific markers. Integrin alpha 4 (ITGA4, also known as CD49D) has been identified as a specific macrophage marker to distinguish them from microglia in GBM [125]. Moreover, scRNA-seq and multicolor fluorescence-activated cell sorting (FACS) analyses have revealed that P2RY12, TMEM119, CX3CR1, and HexB are highly expressed in tumor-associated microglia; on the contrary, FCGR2B, CLEC10A, CD1C, CD1B, CD207, and CD209 are highly expressed in macrophages [20,118,126,127]. By taking advantage of advanced technologies (e.g., CyTOF, spatial tissue characterization, and scRNA-seq), more compelling evidence supports that microglia and macrophages have distinct transcriptomic profiles and expression signatures in GBM [4,16,122,128].
Figure 1. Molecular mechanisms underlying the cancer cell-TAM crosstalk in GBM.
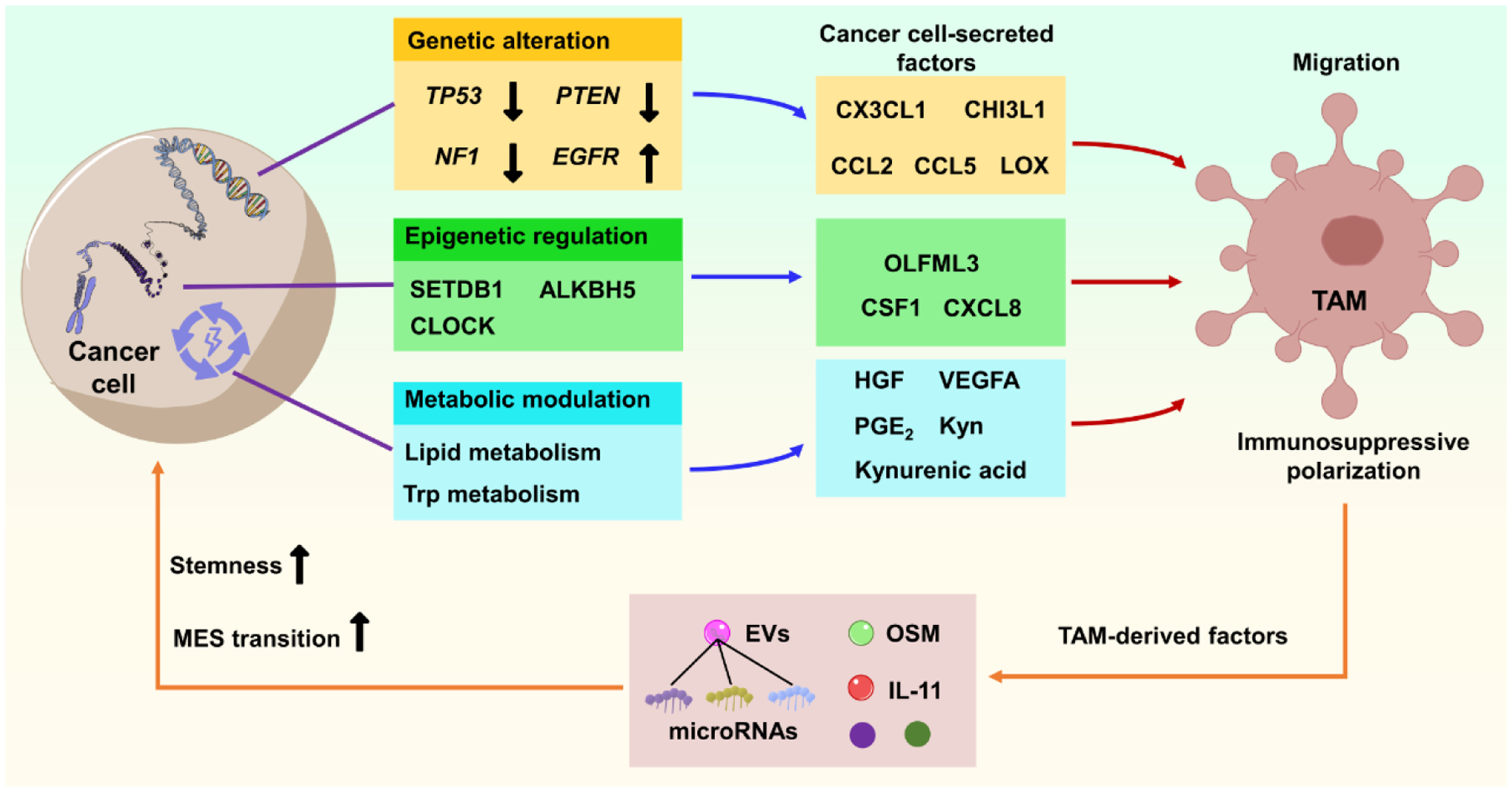
Genetic alteration (e.g., mutation/deletion of PTEN, TP53, NF1, and amplification/mutation of EGFR), epigenetic regulation (e.g., regulation of epigenetic factors SETDB1, ALKBH5, and CLOCK) and metabolic regulation (e.g., regulation of lipid and Trp metabolism) in cancer cells would trigger the expression and secretion of various cytokines and other factors (as indicated) that promote TAM infiltration and immunosuppressive polarization. Reciprocally, GBM-educated TAMs convert cancer cells toward a more aggressive state (e.g., inducing MES transition and increasing stemness) via secretion of distinct factors (e.g., OSM and IL-11) and EVs, thus promoting tumor progression. Abbreviations: ALKBH5, alkB homologue 5; CCL2/5, C-C motif chemokine ligand 2/5; CHI3L1, chitinase-3-like 1; CLOCK, circadian locomoter output cycles protein kaput; CSF1, macrophage-colony stimulating factor; CX3CL1, C-X3-C motif chemokine ligand 1; CXCL8, C-X-C motif chemokine ligand 8; EGFR, epidermal growth factor receptor; EVs, extracellular vesicles; GBM, glioblastoma; HGF, hepatocyte growth factor; IL-11, interleukin 11; Kyn, kynurenine; LOX, lysyl oxidase; MES, mesenchymal; NF1, neurofibromin 1; OLFML3, olfactomedin-like 3; OSM, oncostatin M; PGE2, prostaglandin E2; PTEN, phosphatase and tensin homolog; SETDB1, SET domain bifurcated histone lysine methyltransferase 1; TAM, tumor-associated macrophage and microglia; Trp, tryptophan; VEGFA, vascular endothelial growth factor A.
Table 1.
Effect and mechanism of cancer cell-derived factors in regulating immune cell biology
| Cancer cell-Derived factors | The mechanism for regulation in cancer cells | Targeting immune cells | Effect and mechanism in regulating immune cell biology | Ref |
|---|---|---|---|---|
| LOX | PTEN mutation/deletion activates YAP1 to upregulate LOX | Macrophage | Increasing macrophage migration via activation of the PYK2 signaling | [3] |
| CCL2 | TP53 mutation activates NF-κB to upregulate CCL2 and TNFα | Macrophage and microglia | Increasing macrophage and microglia migration | [134] |
| TNFα | ||||
| CCL5 | Nf1 deficiency promotes CCL5 and CX3CL1 production | Macrophage and microglia | Increasing macrophage and microglia migration | [135] |
| CX3CL1 | ||||
| CHI3L1 | CHI3L1 is regulated by the PI3K/AKT/mTOR pathway | Macrophage | Increasing macrophage migration and immunosuppressive polarization | [27] |
| CSF2 | Unknown | Macrophage and microglia | Increasing macrophage and microglia migration; Decreasing macrophages and microglia apoptosis | [31] |
| SLIT2 | Unknown | Macrophage and microglia | Increasing macrophage and microglia migration and immunosuppressive polarization via ROBO1/2-mediated PI3Kγ activation | [33] |
| P-selectin | Unknown | Microglia | Increasing microglia immunosuppressive polarization via the P-selectin-PSGL-1 axis | [34] |
| SPP1 | Unknown | Macrophage | Increasing macrophage migration and immunosuppressive polarization | [32] |
| OLFML3 | CLOCK-BMAL1 complex transcriptionally upregulate OLFML3 | Microglia | Increasing microglia infiltration | [40] |
| LGMN | CLOCK-BMAL1 complex transcriptionally upregulate LGMN; OLFML3 upregulates LGMN via the HIF1α signaling | Microglia | Increasing microglia infiltration and immunosuppressive polarization via the CD162 signaling | [41] |
| CSF1 | SETDB1 upregulates CSF1 via the AKT/mTOR signaling | Macrophage | Increasing macrophage migration | [42] |
| CXCL8 | Neutrophil-derived NETs interact with the receptor for advanced glycation end-products on cancer cells to upregulate CXCL8 by activating the ERK/NF-κB signaling pathway | Neutrophil | Increasing neutrophil migration | [77] |
| ALKBH5 demethylates stabilizes lncRNA NEAT1 to promote CXCL8 generation via the NEAT1/paraspeckle axis | Macrophage and microglia | Increasing macrophage migration | [45] | |
| Extracellular lipid | Lipid loading augments hypoxia-mediated secretion of pro-tumorigenic factors (VEGF and HGF) | Macrophage | Increasing macrophage migration | [49] |
| PGE2 | ARS2 directly activates MAGL to upregulate PGE2 | Macrophage | Increasing macrophage immunosuppressive polarization | [51] |
| Kyn | Trp-catabolic enzymes activate Kyn pathway | Macrophage | Increasing macrophage migration by activating the AHR-CCR2 axis; Increasing macrophage immunosuppressive function by upregulating CD39 | [54] |
| MIF | Unknown | MDSC | Increasing MDSC immunosuppressive function | [59] |
| CXCL1/2 | Unknown | MDSC | Increasing MDSC migration by upregulating S100A9-ERK1/2 and p70S60k | [63] |
| miR-1246 | Hypoxia increases miR-1246 expression and packaging by upregulating POU5F1 and hnRNPA1 | MDSC | Promoting MDSC differentiation and activation by activating the DUSP3/ERK pathway | [64] |
| PD-L1 | Wnt ligand and activated EGFR promote PD-L1 expression by inducing the binding of β-catenin/TCF/LEF to the CD274 promoter | T-cell | Inhibiting T-cell activation and infiltration | [94] |
| ICOSLG | ICOSLG expression is increased in mesenchymal GSCs in a TNFα/NF-κB dependent manner | Treg | Increasing Treg infiltration and IL-10 production | [97] |
Abbreviations: AHR, aryl hydrocarbon receptor; AKT, protein kinase B; ALKBH5, alkB homologue 5; ARS2, arsenite-resistance protein 2; BMAL1, aryl hydrocarbon receptor nuclear translocator like; CCL2/5, C-C motif chemokine ligand 2/5; CCR2, C-C motif chemokine receptor 2; CHI3L1, chitinase-3-like 1; CLOCK, circadian locomoter output cycles protein kaput; CX3CL1, C-X3-C motif chemokine ligand 1; CSF1/2, macrophage-colony stimulating factor1/2; CXCL8, C-X-C motif chemokine ligand 8; DUSP3, dual specificity phosphatase 3; EGFR, epidermal growth factor receptor; Erk, extracellular signal-regulated kinase; GBM, glioblastoma; HGF, hepatocyte growth factor; HIF1α, hypoxia-inducible factor 1-alpha; HMGB1, high mobility group box protein 1; hnRNPA1, heterogeneous nuclear ribonucleoprotein A1; IL, interleukin; Kyn, kynurenine; LEF, lymphoid enhancerbinding factor; LGMN, legumain; LOX, lysyl oxidase; MAGL, monoacylglycerol lipase; MDSC, myeloid-derived suppressor cells; mTOR, mammalian target of rapamycin; NEAT1, nuclear enriched abundant transcript 1; NF1, neurofibromin 1; NF-κB, nuclear factor kappa B; OLFML3, olfactomedin-like 3; PD-L1, programmed death-ligand 1; PGE2, prostaglandin E2; PI3K, phosphoinositide 3-kinase; POU5F1, POU class 5 homeobox 1; PTEN, phosphatase and tensin homolog; PYK2, protein tyrosine kinase 2 beta; RAGE, receptor for advanced glycation endproducts; SETDB1, SET domain bifurcated histone lysine methyltransferase 1; SRC, proto-oncogene tyrosine-protein kinase Src; TCF, T cell-specific factor; TNFα, tumor necrosis factor alpha; Treg, regulatory T cell; Trp, tryptophan; VEGFA, vascular endothelial growth factor A; YAP1, yes-associated protein 1.
Genetic alterations
Genetic alterations in cancer cells can regulate macrophage and microglia biology [21]. As such, isocitrate dehydrogenases (IDH) mutations are associated with higher infiltration of microglia, while IDH wild-type GBMs harbor relatively higher levels of intratumoral macrophages [20]. In contrast, PTEN mutations in cancer cells induce an increased infiltration of macrophages into the GBM TME, but do not affect microglia [3]. However, it should be noted that GBM tumorigenesis is not triggered by a single genetic alteration, but by combined alterations in a series of core signaling pathways [24,25]. For example, mesenchymal GBM is enriched for mutational alterations in PTEN, TP53, NF1, and RB1 [26]. Loss of PTEN, TP53, and NF1 in cancer cells upregulates lysyl oxidase (LOX); CCL2 and TNFα; and CCL5 and CX3CL1, which, in turn, triggers the infiltration of macrophages and/or microglia into the GBM TME [21]. Further evidence demonstrates that activation of the PI3K/AKT/mTOR signaling [27] or loss of NF1 [28] in cancer cells increase the expression and secretion of chitinase-3-like protein 1 (CHI3L1), which, in turn, increases macrophage infiltration and immunosuppressive polarization. These findings highlight the role of genetic alterations in affecting TAM biology in mesenchymal GBM, which is supported by the observation that mesenchymal GBM patient tumors harbor significantly higher TAMs compared to other tumor subtypes (e.g., classical and proneural) [29]. Within the GBM tumor, more macrophages surround mesenchymal-like cancer cells than other subtypes of cancer cells (e.g., oligodendrocyte progenitor-like cancer cells) [17,30]. Together, we posit that inactivation of PTEN, TP53, and NF1 in mesenchymal cancer cells might regulate TAM biology by secreting distinct chemokines and factors (Table 1). However, further studies are needed to investigate whether the loss of RB1, which is highly mutated in mesenchymal GBM, affects TAM biology. In contrast, some chemokines and factors (e.g., CSF2, SLIT2, P-selectin, and SPP1) are highly expressed in mesenchymal GBM and can trigger TAM infiltration and immunosuppressive polarization. However, it is unclear whether their expression is regulated by the core genetic alterations observed in mesenchymal GBM [31–34]. Together, these findings suggest that genetic alterations in cancer cells enhance the expression and secretion of different chemokines and factors, which, in turn, regulate TAM biology in the GBM TME. Mirroring cancer cell actions, TAMs promote GBM progression by reprograming cancer cells into a more aggressive state (Figure 1) [9,18,35]. For example, TAM-secreted cytokines, such as oncostatin M (OSM), interleukin-11 (IL-11), shift cancer cells toward a mesenchymal and stem cell-like state by activating the STAT3 pathway [17,18,35]. Additionally, TAM-derived extracellular vesicles (EVs) exhibit a similar pro-tumor effect. Experimental evidence demonstrates that EV-containing microRNAs (e.g., miR-27a-3p, miR-22-3p, and miR-221-3p) trigger a proneural-to-mesenchymal transition by regulating the CHD7-RelB/STAT3 pathway [36]. Therefore, blocking the genetic alteration-mediated tumor-TAM crosstalk will be a promising therapeutic strategy for GBM.
Epigenetic alternations
Although genetic alterations in mesenchymal GBM play an important role in regulating TAM biology, tumor subtype features are fluid and dynamic. Multiple subtypes may co-exist in the same tumor [5], and the subtypes can shift during tumor progression and upon therapeutic interventions [37]. The cellular and molecular heterogeneity of the GBM TME suggests that in addition to genetic alterations, alternative mechanisms may contribute to the symbiotic interaction between cancer cells and TAMs. Glioma stem cells (GSCs) are intrinsically immune suppressive of both adaptive and innate immunity [38]. GSCs can escape immune surveillance by recruiting immunosuppressive PD-L1+ macrophages. Independent of genetic selection, the infiltration of these immunosuppressive macrophages is triggered by epigenetic changes in GSCs following serial transplantation through immunocompetent hosts [39], suggesting a crucial role of cancer cell epigenetic regulation in influencing TAM biology. This hypothesis is reinforced further by a growing body of evidence highlighting the essential role of several epigenetic factors in regulating the GBM-TAM crosstalk (Figure 1 and Table 1). First, a gain-of-function screen of epigenetic regulators identified circadian regulator CLOCK as a key hit in GSCs that promotes microglia infiltration into the TME by transcriptionally upregulating olfactomedin-like 3 (OLFML3) and legumain (LGMN). In addition, CLOCK-regulated LGMN polarizes microglia toward an immunosuppressive phenotype, which, in turn, promotes tumor progression and suppresses anti-tumor immunity [41]. Second, SET domain bifurcated 1 (SETDB1) is a member of the methyltransferase family that can activate the AKT/mTOR pathway in cancer cells to upregulate the expression and secretion of CSF1, which, in turn, induces macrophage infiltration and immunosuppressive polarization [42]. As a result, these macrophages mediate the oncogenic effect of SETDB1 in GBM mouse models. Conversely, depletion of macrophages using liposomal clodronate impairs SETDB1 overexpression-induced tumor growth [42]. Finally, N6-methyl-adenosine (m6A) is one of the most abundant RNA modifications during GBM tumorigenesis [43]. This process can be erased by alkB homologue 5 (ALKBH5) demethylase [44]. Functionally, depletion or inactivation of ALKBH5 in cancer cells suppresses the expression and secretion of CXCL8, thus impairing hypoxia-induced macrophage recruitment and immunosuppression, as well as GBM tumor growth [45]. Together, these findings suggest that epigenetic regulation involves in regulating the tumor-TAM symbiosis in GBM, and highlight a therapeutic potential for targeting such epigenetic regulators.
Metabolism
From another angle, metabolic dysregulation is a hallmark of cancer [46]. Cancer cell metabolism not only ensures sufficient energy for maintaining tumor potential but also regulates TAM biology [47,48], thus inducing a metabolism-dependent GBM-TAM symbiotic interaction (Figure 1 and Table 1). Lipid metabolism is one of the mechanisms that regulates this symbiosis. For example, the loading of lipids in cancer cells upregulates the expression and secretion of pro-tumor factors (e.g., VEGFA and HGF) under hypoxic conditions, which, in turn, triggers the infiltration of macrophages in vitro and in GBM mouse models [49]. Moreover, peroxidation of polyunsaturated fatty acid arachidonic acid (AA) by cyclooxygenase-2 (COX-2) in cancer cells leads to the production of prostaglandin E2 (PGE2) [50,51], which can promote TAM immunosuppressive polarization in GBM [51]. Unfortunately, the clinical application of COX-2 inhibitors in GBM patients is limited [50]. Recent studies have demonstrated that PGE2 production can also be triggered by activation of arsenite-resistance protein 2 (ARS2)-induced monoacylglycerol lipase (MAGL) signaling in GSCs, and inhibition of the ARS2-MAGL axis impairs GBM progression and TAM immunosuppressive polarization [51]. Thus, these findings suggest that deciphering the mechanism of fatty acid metabolism during the GBM-TAM symbiosis may reveal new therapeutic targets for GBM. Tryptophan (Trp) metabolism is an important mechanism contributing to therapy resistance across many cancer types, including GBM [52]. The Trp-catabolic enzymes, such as indoleamine-2,3-dioxygenase 1 (IDO1), IDO2, and tryptophan-2,3-dioxygenase (TDO2), can mediate the first step of the kynurenine (Kyn) pathway and are upregulated in glioma cells [53]. The metabolites (e.g., Kyn and kynurenic acid) of this pathway can activate the aryl hydrocarbon receptor (AHR) on immune cells, including TAMs, thus affecting their function in the GBM TME [54]. Specifically, AHR is essential for TAM recruitment and Trp-induced TAM activation in GBM. Suppressing AHR genetically and pharmacologically inhibits GBM progression by impairing TAM infiltration and immunosuppressive polarization [20,54].
In line with the metabolic changes in cancer cells, aberrant amino acid metabolism in TAMs also contributes to tumor growth. GBM is featured by a highly acidic TME due to severe hypoxia. To survive in such a low pH environment, myeloid cells (e.g., TAMs and MDSCs) catabolize arginine to polyamines, thus maintaining an immunosuppressive TME to promote GBM tumor growth [55]. Depletion of arginine or administration of polyamine inhibitor (e.g., difluoromethylornithine) synergizes with radiation to improve the survival of GBM-bearing mice [55,56]. Together, these findings highlight that cancer cell and/or TAM metabolism (e.g., fatty acid, Trp, and arginine metabolism) can promote tumor growth via regulating the GBM-TAM symbiosis.
GBM-MDSC crosstalk
MDSCs are a highly heterogeneous population of myeloid cells contributing to tumor immunosuppression (Box 2) [57]. Emerging evidence demonstrates a clear GBM-MDSC symbiosis, where cancer cells contribute to MDSC infiltration and activation (Figure 2 and Table 1). During tumor progression, cancer cells secrete CCL20, IL-8, CXCL1, CXCL2, and macrophage migration inhibitory factor (MIF) to recruit MDSCs from the bone marrow [58,59]. In line with cancer cell-derived chemokines, TAM-derived CCL2 also attracts CCR2+Ly6C+ monocytic MDSC (M-MDSC) into the TME in response to cancer cell-secreted soluble factors (e.g., osteoprotegerin and CCL20), thus resulting in local immunosuppression [58,60,61]. However, the number of infiltrating Ly6G+ PMN-MDSCs in CCL2- and CCR2-deficient mice is unchanged [73,74], suggesting that the CCL2-CCR2 signaling does not contribute to infiltration of PMN-MDSCs in GBM. Further evidence demonstrates that M-MDSCs express high levels of the MIF receptor CD74. Targeting M-MDSCs with the MIF-CD74 interaction inhibitor Ibudilast significantly abrogates MDSC-induced immunosuppression [59]. Another MIF receptor CXCR4 also participates in the recruitment of M-MDSCs into the GBM TME via the SDF-1α-CXCR4 signaling [62]. Together, these findings highlight the molecular mechanism underlying GBM-induced M-MDSC recruitment. Although the infiltration of PMN-MDSCs in GBM has remained poorly understood, a recent study revealed that CXCL1 and CXCL2 could simultaneously enhance the infiltration of M-MDSCs and PMN-MDSCs into the TME in vivo [63].
Box 2. Myeloid-derived suppressor cells in GBM.
Myeloid-derived suppressor cells (MDSCs) in human are defined by human leukocyte antigen (HLA)-DR and further classified into three subsets with different phenotypic and morphological features, including early-stage MDSCs (e-MDSCs, Lin−HLA-DR−CD33+ cells, a mixed population of immature progenitor cells), polymorphonuclear MDSCs (PMN-MDSCs, CD11b+CD14+CD66b+ cells or CD11b+CD14−CD15+ cells), and monocytic MDSCs (M-MDSCs, CD11b+CD14+HLA-DR−/loCD15− cells). In murine models, M-MDSCs (CD11b+Ly6G−Ly6Chi cells) and PMN-MDSCs (CD11b+Ly6G+Ly6Clo cells) have been defined correspondingly. However, the e-MDSC population has not yet been clearly determined [57,58,70].
In human GBM tumor tissues, MDSCs (CD15loCD16−HLA-DR− cells, total MDSCs) are a smaller population of cells compared to TAMs, which account for 4%–8% of CD45+ cells [129]. Although the biology of e-MDSCs is less clear in GBM, both M-MDSCs and PMN-MDSCs are increased in the peripheral blood of GBM patients. PMN-MDSCs show a higher frequency in GBM tumors, where they account for more than 60% of total tumor-infiltrating MDSCs [130]. The composition of MDSC subsets es in GBM appears to be context-dependent. A recent study with phenotypic characterizations of monocytes has demonstrated that IDH-mutated GBM patient tumors harbor more PMN-MDSCs than that of IDH-WT tumors [131]. However, M-MDSCs are more relevant compared to PMN-MDSCs in GBM mouse models, implicating that the function of different MDSC subsets in GBM may vary from mouse to human [132]. Additionally, the composition of GBM MDSC subsets may vary between male and female. For example, more M-MDSCs are observed in male tumors, whereas PMN-MDSCs are enriched in female GBM tumors. Depletion of PMN-MDSCs via intraperitoneal injection of anti-Ly6G antibodies improve the survival of GBM-bearing female mice exclusively [133]. However, due to lack of specific markers, the functional differences among these MDSC subsets in GBM are ongoing.
Figure 2. Cancer cell-immune crosstalk and interactions in GBM.
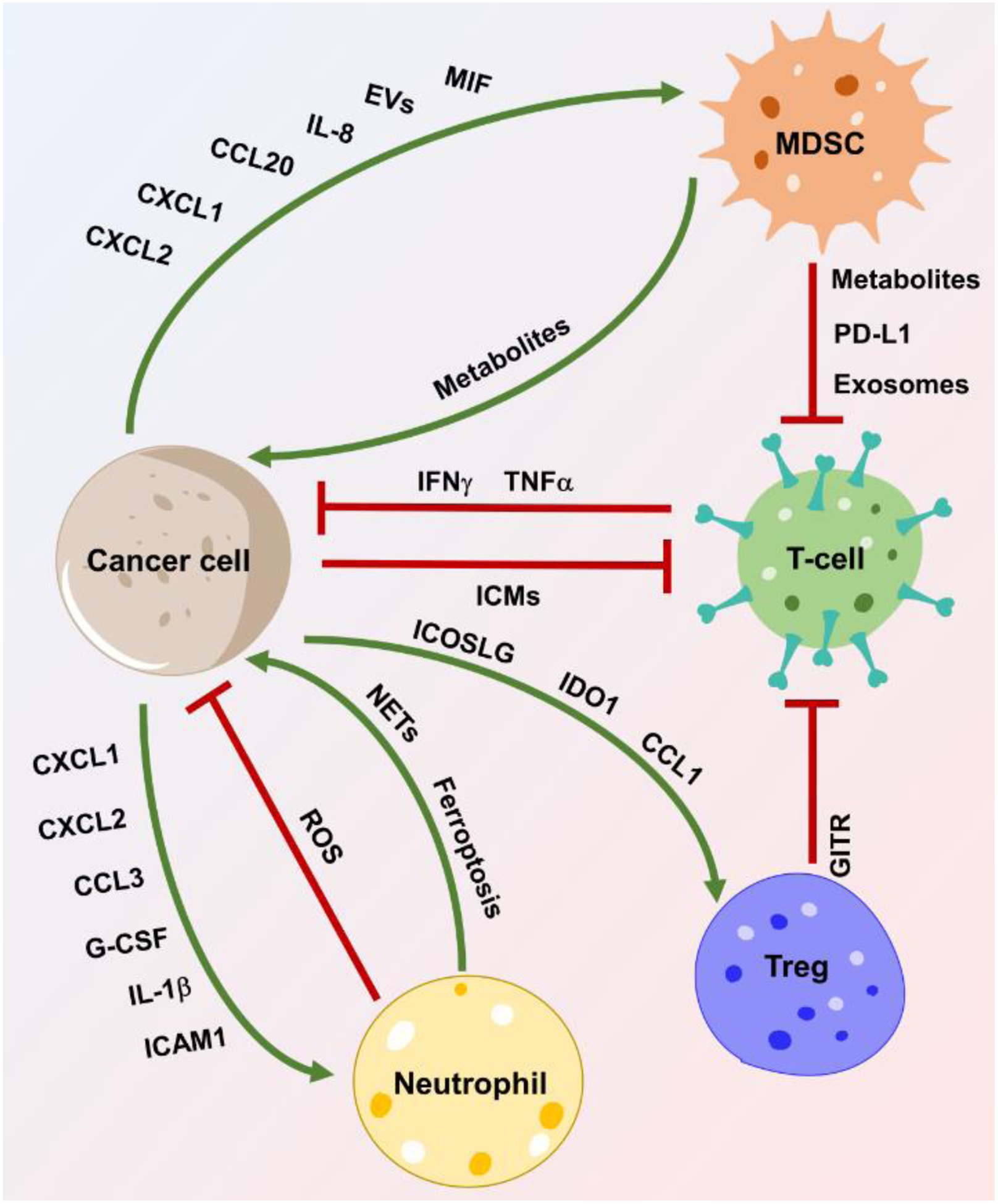
GBM-immune crosstalk (including GBM-MDSC, GBM-neutrophil, GBM-T cell, and CSC-Treg) and the immunosuppressive function of MDSCs and Tregs on T-cells in GBM. Crosstalk between two cell types is achieved by expression and/or secretion of a variety of chemokines, cytokines, exosomes, metabolites, or other factors as indicated. Neutrophils promote tumor growth via NET formation or ferroptosis-mediated necrosis in GBM. Abbreviations: CCL1, chemokine ligand 1; CCL1/3, chemokine ligand 3; CCL20, chemokine ligand 20; CXCL1/2, C-X-C motif chemokine ligand 1 and 2; EVs, extracellular vesicles; GBM, glioblastoma; G-CSF, granulocyte colony-stimulating factor; GITR, glucocorticoid-induced TNFR-related protein; ICAM1, intracellular adhesion molecule 1; ICMs, immune checkpoint molecules; ICOSLG, inducible T-cell costimulator ligand; IDO1, indoleamine-2,3-dioxygenase 1; IL-1β, interleukin-1 beta; IL-8, interleukin-8; MDSC, myeloid-derived suppressor cell; MIF, macrophage migration inhibitory factor; NETs, neutrophil extracellular traps; PD-L1, programmed death-ligand 1; ROS, reactive oxygen species; Treg, regulatory T-cell.
Once MDSCs infiltrate into the TME, they are further activated by different cytokines (e.g., M-CSF, GM-CSF, IL-6, IL-10, TGF-β, B7-H1, and INFγ) [58] and EVs to promote tumor growth directly and indirectly (Figure 2). First, GBM-derived exosomes promote MDSC expansion by transporting different miRNAs (e.g., miR-1246, miR-29a, and miR-92a) under hypoxic conditions [64,65]. Mechanistically, hypoxia-induced heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) facilitates miRNA packaging into exosomes. The secreted exosomes are then taken up by local MDSCs, which, in turn, promotes MDSC activation via the dual-specificity phosphatase 3 (DUSP3)/ERK pathway [64]. Upon activation, MDSCs express PD-L1 in a HIF-1α-dependent manner, thus resulting in T-cell exhaustion and inhibition of anti-tumor immunity [66]. Second, GBM-associated MDSCs produce exosomes containing PD-L1, thus inducing a rapid increase of PD-L1 in B-cells via caveolae-mediated endocytosis. As a result, these B-cells convert to regulatory B-cells (Bregs) to inhibit anti-tumor immunity and promote GBM tumor growth [67]. Third, MDSCs can function as an intermediate between cancer cells and T-cells, where GBM-derived EVs induce the expansion of M-MDSCs, which in turn, suppress T-cell-mediated anti-tumor immunity in GBM [68]. Finally, metabolism regulates the pro-tumor function of MDSCs in GBM. MDSCs can generate polyamines and fatty acids to maintain their pro-tumor and immunosuppressive function in GBM [55,69]. As a result, inhibition of the arginine-ornithine-polyamine axis reduces the survival of myeloid cells, including MDSCs, activates anti-tumor immunity, and impairs GBM tumor growth [55]. Together, these findings highlight a vital role of MDSCs in regulating anti-tumor immunity and tumor growth in GBM mouse models.
GBM-neutrophil crosstalk
Neutrophils are the most abundant circulating leukocytes and are essential for innate and adaptive immune responses [8]. Since classical neutrophils and PMN-MDSCs share the same set of phenotypic cell surface markers (e.g., CD11b+CD14−CD15+/CD66b+ and CD11b+Ly6G+Ly6Clo for human and mouse, respectively), functional analysis is required to distinguish them [58,70]. PMN-MDSCs display strong immunosuppressive activity by suppressing T-cells, whereas neutrophils do not [58]. Due to lack of effective identification and isolation methods, researchers tend to investigate the function of total neutrophils (CD66b+ and Ly6G+for human and mouse, respectively) in GBM, which may contain both classical neutrophils and PMN-MDSCs [71,72]. Here, we discuss the recent findings highlighting the role of cancer cell-neutrophil crosstalk in GBM progression (Figure 2).
Recent studies have revealed that neutrophil infiltration in GBM is associated with tumor genetic backgrounds and molecular states. For example, IDH mutation in GBM tumors suppresses neutrophil infiltration [4], whereas TERT mutation promotes such immune cell infiltration [73]. Neutrophils infiltrate into the early stage of mesenchymal GBM tumors, where chemokines (e.g., CXCL1, CCL3, CXCL2, G-CSF, IL-1β, and ICAM1) are secreted by cancer cells [74]. In addition, several other cancer cell-derived factors (e.g., IL-8, CXCL3, CXCL5, and osteopontin) have been shown to function as potent neutrophil chemokines [75–77]. However, it is unclear whether the expression of these chemokines is associated with genetic mutations found in GBM. Together, these findings highlight that cancer cells regulate neutrophil infiltration in a context-dependent manner.
Neutrophils also manifest substantial plasticity in the GBM TME although their roles in tumor progression remain controversial. On the one hand, neutrophils may exhibit anti-tumor properties via releasing reactive oxygen species (ROS) [78]. On the other hand, neutrophils promote GBM progression through distinct mechanisms. First, neutrophils promote ferroptosis-mediated tumor necrosis during GBM progression. Interestingly, neutrophils are spatially and temporally correlated with necrosis in the TME, where neutrophils transfer myeloperoxidase-containing granules into cancer cells to induce ferroptosis, thus inducing necrosis via accumulation of lipid peroxides [71]. Depleting neutrophils or inhibiting ferroptosis diminishes neutrophil-induced cancer cell necrosis and tumor aggressiveness [71]. Second, neutrophils induce therapy resistance by upregulating GSC self-renewal and mesenchymal transition. For example, in an irradiated GBM model, Ly6G+ neutrophils and MDSCs promote the conversion of cancer cells into GSCs via regulating the NOS2-NO-ID4 signaling axis [79]. Moreover, antiangiogenic therapy increases neutrophil infiltration in GBM, which, in turn, upregulates GSC proliferation, migration, and mesenchymal transition via activation of the S100A4 signaling [80]. Third, neutrophils promote GBM progression by releasing DNA into the extracellular space to form neutrophil extracellular traps (NETs) [81]. Consequently, NETs interact with the receptor for advanced glycation end-products (RAGE) on cancer cells to promote CXCL8 expression and secretion by upregulating the ERK/NF-κB signaling pathway (Table 1). The secreted CXCL8 further binds to CXCR2 on neutrophils to form additional NETs, thus inducing a positive feedback loop to promote GBM progression [77].
The opposite effects of neutrophils in GBM may relate to tumor stages. Neutrophils can infiltrate into the early stage of tumors in preclinical models, and depletion of neutrophils at this initial stage promotes tumor growth and reduces survival of GBM-bearing mice. In contrast, depletion of neutrophils at the late stage of tumor has no such effect [74]. This phenomenon is also supported by functional studies showing that neutrophils from healthy mice suppress the tumorgenicity of GSCs in a syngeneic mouse model, whereas neutrophils from tumor-bearing mice promote tumor progression and immunosuppression [74]. Mechanistically, the anti-tumor effects of neutrophils at the early tumor stage primarily rely on their cytotoxicity, trogoptosis, and immune-stimulating activity [72]. However, tumor-educated neutrophils change their phenotypes to exhibit a pro-tumor function [74]. Further studies revealing the molecular mechanisms underlying the context-dependent GBM-neutrophil symbiosis will help to develop therapeutic strategies.
GBM-T-cell crosstalk
T-cells are lymphocytes that play a vital role in anti-tumor responses. Naïve T-cells can be recruited and trained in the thymus for differentiation into different subpopulations, including innate T-cells (e.g., natural killer T-cells) and adaptive T-cells (e.g., cytotoxic CD8+, helper CD4+, and memory T-cells) [82]. Emerging evidence demonstrates that different subsets of T-cells can affect cancer cell biology via distinct mechanisms. For example, CD8+ cytotoxic T-cells can induce cancer cell apoptosis via cell-cell interaction or secretion of effector molecules. CD8+ T-cells tend to directly kill cancer cells by recognizing MHC-I and activating cytotoxic signals. However, cancer cells may adaptively express low MHC-I to escape recognition and attack from CD8+ T-cells [10]. In contrast, CD4+ T-cells recognize MHC-II molecules on antigen-presenting cells (e.g., DCs and macrophages) [83]. Nevertheless, a recent single-cell RNA sequencing (scRNA-seq) study proposed that glioma-infiltrating CD4+ T-cells also express cytotoxicity genes [84], where they can directly affect cancer cells by secreting TNFα and IFNγ [10]. In addition, CD4+ T-cells may help CD8+ T-cells to mediate anti-tumor immunity in GBM [10]. For instance, CD4+ T-cell-derived IL-2 activates CD8+ T-cells by promoting the expression of IL-2 receptor α subunit (CD25), which, in turn, exhibits an anti-tumor activity [85]. Finally, regulatory T-cells (Tregs) have been identified as a pro-tumor subpopulation of CD4+ T-cells in GBM tumor tissues and circulating system [86]. Tregs are composed of two subpopulations, including induced Tregs (iTregs) and natural Tregs (nTregs). In GBM, thymus-derived nTregs are the predominant Treg population, which are closely associated with tumor progression and immunotherapy resistance [87]. Although these emerging evidence supports the role of T-cell-mediated anti-tumor immunity in the TME, GBM is recognized as a “cold” tumor due to the bone marrow sequestration of T-cells [88–90].
T-cell receptor (TCR) sequencing on a multi-region of tumors has revealed that TCR repertoires within the TME are highly heterogeneous and spatially restricted [91]. These findings suggest a potential context-dependent regulation of cancer cells on T-cell biology (Table 1). Compared to other GBM subtypes, mesenchymal cancer cells and GSCs express higher inhibitory immune checkpoint molecules (ICMs), such as PD-L1 [92,93], which is triggered by the Wnt ligand and activated EGFR through promoting the binding of β-catenin/T-cell-specific factor (TCF)/lymphoid enhancer-binding factor (LEF) to the CD274 promoter [94]. Consequently, cancer cell-expressed or EV-delivered ICMs (e.g., PD-L1) suppress T-cell function and proliferation by ligating their corresponding receptors on T-cells [95,96]. Similarly, the Wnt/β-catenin signaling pathway helps GSCs to escape T-cell-mediated killing by suppressing MHC-I [93]. Alternatively, cancer cells and GSCs express various factors (e.g., ICOSLG, IDO1, and CCL1) to enhance Tregs expansion in the TME [61,88,97,98]. As a result, T-cells tend to be skewed toward Tregs rather than cytotoxic CD8+ T-cells in GBM [99]. Disrupting the GBM-T-cell crosstalk by inhibition of cancer cell-derived IDO1 improves the survival of GBM-bearing mice by suppressing Tregs [88,100]. Similarly, elimination of Tregs in GBM using anti-GITR (a GITR agonistic antibody that promotes Tregs differentiation into CD4+ effector T-cells) inhibits Treg-mediated immunosuppression and activates CD4+ T-cell-mediated anti-tumor immunity, thus killing cancer cells [99]. These findings highlight a direct regulation mechanism of cancer cells on T-cells in GBM. Additionally, myeloid cells may serve as a central hub to pass immunosuppressive signals from cancer cells to T-cells. For example, cancer cell-derived IL-6 and Kyn can upregulate PD-L1 expression on myeloid cells by activating the STAT3 pathway and activate TAMs via AHR, respectively, which, in turn, suppresses T-cell function in the TME [54,101]. Together, these findings reveals the molecular basis for how cancer cells symbiotically interact with T-cells and how this symbiosis affects tumor progression in GBM.
Impact of the GBM-immune symbiosis on the effectiveness of immunotherapies
Tumor-immune symbiosis is critical for regulating anti-tumor immunity in GBM. Therefore, inhibition of this symbiosis may affect the effectiveness of immunotherapies, including ICI therapies [21,38,102]. Indeed, recent evidence shows that myeloid cell infiltration in GBM correlates with increased immunotherapy resistance [7,14,103]. This section summarizes recent findings highlighting the role of targeting the symbiosis between cancer cells and immune cells (e.g., T-cells, TAMs, MDSCs, and neutrophils) to improve the effectiveness of immunotherapies, especially ICI therapies, in GBM (Figure 3, Key Figure).
Figure 3. Therapeutic approaches targeting tumor-immune symbiosis to improve the effectiveness of immunotherapies in GBM.
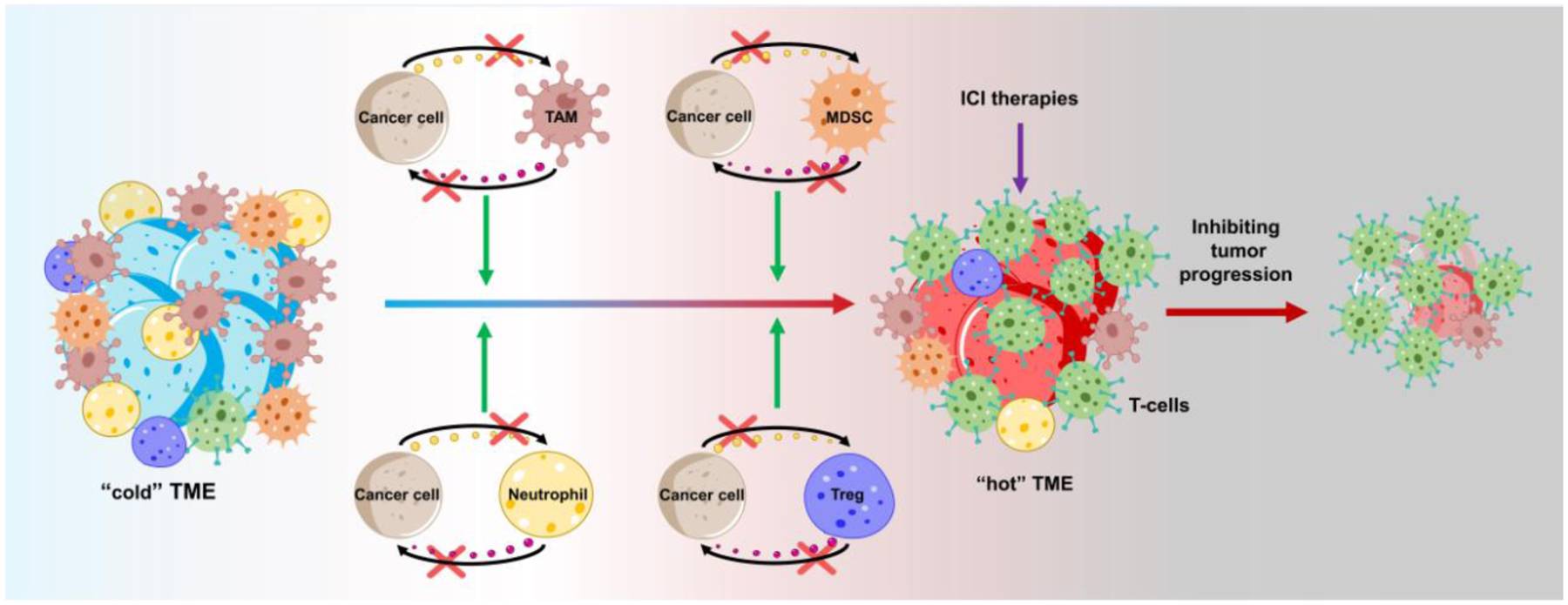
Due to extensive infiltration of immunosuppressive immune cells (e.g., TAMs, MDSCs, neutrophils, and Tregs), GBM is featured as a “cold” tumor with relatively low cytotoxic T-cells. Targeting the GBM-immune symbiosis (e.g., GBM-TAM, GBM-MDSC, GBM-neutrophil, GBM-T cell, and CSC-Treg) shifts the “cold” TME to “hot”, thus inhibiting tumor growth and synergizing with immunotherapies (e.g., ICI therapies) in GBM. Abbreviations: GBM, glioblastoma; ICI, immune checkpoint inhibitor; MDSC, myeloid-derived suppressor cell; TAM, tumor-associated macrophage and microglia; TME, tumor microenvironment.
Given the remarkable T-cell dysfunction in GBM, multiple therapeutic approaches (e.g., ICIs) have been developed to target the GBM-T-cell interaction. A randomized clinical trial suggested that newly diagnosed and relapsed GBM patients may benefit from anti-PD1 therapy [12,104]. However, a Phase III randomized study did not conclusively demonstrate that recurrent GBM patients benefit from the anti-PD1 therapy [102]. We propose that these controversial results may relate to context-dependent GBM-T-cell symbiosis. Indeed, recent studies have demonstrated that anti-PD1 therapy non-responders harbor more PTEN mutations, whereas responders contain more MAPK pathway alterations (e.g., PTPN11 and BRAF mutations) and higher phospho-ERK1/2 expression in cancer cells [103,105]. Given the critical role of PTEN mutation in triggering macrophage infiltration in GBM [3] and the evidence showing that tumors from ICI non-responders harbor higher infiltration of myeloid cells (including TAMs and monocytes) but less infiltration of T-cells compared to responder tumors from GBM mouse models and patients [7,105], it is plausible that anti-PD1 therapy resistance in PTEN-deficient tumors may relate to TAMs. This hypothesis is supported by emerging evidence demonstrating that blockade of TAM immunosuppressive function through distinct strategies can improve the effectiveness of ICIs in GBM. First, macrophages from non-responder tumors express higher immunosuppression-related genes (e.g., PD-L1). Functionally, inhibition of macrophage PD-L1 and its alternative binding partner CD80 restores the anti-tumor effect of ICI therapies (e.g., the combined anti-PD1 and anti-CTLA4 therapy) [7]. Second, the feature of TAM heterogeneity in GBM spurs researchers to develop new strategies to target specific TAM subpopulations. The studies using scRNA-seq and cytometry by time-of-flight (CyTOF) technologies have identified novel macrophage populations that can contribute to ICI therapy resistance in GBM. For example, a unique population of CD73+ macrophages has been identified, which exhibit an immunosuppressive function in GBM. Depleting CD73 in mice decreases the immunosuppressive CD206+Arg1+VISTA+PD1+CD115+ macrophage cluster, but increases the immunostimulatory iNOS+ myeloid cell clusters. As a result, the anti-tumor effect of combined anti-PD1 and anti-CTLA4 therapy is significantly improved by CD73 depletion [14]. Third, targeting the signaling axis (e.g., the ligand-receptor axis) responsible for the GBM-TAM crosstalk is an additional promising strategy for enhancing the effectiveness of ICI therapies in GBM. For example, inhibition of the IL-6 (cancer cell)-IL-6R (TAM) axis [106], the PROS1 (TAM)-AXL (GSC) axis [107], the MAGL-PGE2 (GSC)-KLF4 (TAM) axis [51], the SLIT2 (cancer cell)-ROBO1/2 (TAM) axis [33], the CSF1 (cancer cell)-CSF-1R (TAM) axis [108], and the CD47 (cancer cell)-SIRPα (TAM) axis [109,110] genetically and/or pharmacologically shows a robust synergy with ICI therapies in GBM mouse models. Finally, since immunostimulatory macrophages can be triggered by distinct cytokines (e.g., IL-12 and IL-23), the cytokine-targeted therapies may help to reconstruct the TME for improving ICI efficiency. For example, addition of oncolytic herpes simplex virus (oHSV) G47D expressing IL-12 significantly improves the efficacy of anti-CTLA4 and anti-PD1 therapy in GBM-bearing mice by increasing intratumoral immunostimulatory macrophages and enhancing T effector to Treg ratios [13].
Similar to blockade of the GBM-TAM crosstalk, targeting the GBM-MDSC/neutrophil symbiosis might also improve the effectiveness of ICI therapies in GBM [38]. For example, CCR2 is highly expressed in M-MDSCs, and inhibition of CCR2 using CCR2-deficient mice or its antagonist CCX872 synergizes with anti-PD1 therapy in the GBM-bearing mice [111]. Similarly, depletion of neutrophils using anti-Ly6G antibodies enhance the therapeutic efficiency of anti-PD1 therapy in a GBM mouse model [112]. These findings highlight that disrupting the GBM-myeloid cell symbiosis is a promising therapeutic strategy for improving the anti-tumor response to ICI therapies in GBM.
In addition to ICIs, adoptive T-cell therapy (e.g., CAR T-cell therapy) is an alternative strategy to eliminate cancer cells by engineered T-cells [113]. One such human trial on recurrent GBM patients utilized EGFRvIII-CAR T-cells. However, anti-tumor efficiency of this treatment is dampened by therapy-induced adaptive changes in the local TME and antigen loss in cancer cells [114]. To overcome these challenges, one strategy is to engineer EGFRvIII-CAR T-cells to co-express a bispecific T-cell engager (BiTE) against EGFR wild-type cancer cells [115]. The second strategy is to design a new CAR employing a toxin as the targeting entity. This approach is based on the cancer cell-binding potential of chlorotoxin (CLTX). As a result, CLTX-CAR T-cells efficiently limit tumor growth in the absence of off-target effects in GBM mouse models [116]. Together, these findings demonstrates that targeting the GBM-T-cell symbiosis is a promising strategy for improving the effectiveness of adoptive T-cell therapy.
Concluding remarks and future perspectives
The remarkable development in the field brings light to our understanding and ability to target the highly complicated TME and immune landscape of GBM [4,16,24,29,74,117,118]. Various inspiring findings, as discussed above, suggest that the crosstalk between cancer cells and immune cells is essential for regulating tumor progression and immunotherapy resistance in GBM. In the course of tumor progression, cancer cells recruit and educate immune cells (e.g., myeloid cells and T-cells). Reciprocally, such infiltrating immune cells increase cancer cell aggressiveness and GSC stemness, and induce immunotherapy resistance. These findings suggest that targeting the tumor-immune symbiosis is a promising therapeutic strategy for GBM. Moreover, cancer cells, GSCs, and immune cells in the GBM TME are highly dynamic and plastic with respect to different disease stages, molecular states, mutational statuses, and treatments [4,21,38], which results in context-dependent tumor-immune symbiosis and informs the development of personalized therapies for GBM.
Although ICI therapies exhibit a robust anti-tumor effect in various solid tumors, their applications in GBM have not yet been attained, likely due, at least in part, to the infiltration of immunosuppressive myeloid cells [7,14,102,103]. Based on its critical role in suppressing anti-tumor immunity, different approaches have been developed to enhance the response of GBM to ICI therapies in mouse models through regulating the GBM-myeloid cell crosstalk [21,38,119]. Hence, in-depth mechanism studies underlying the GBM-immune symbiosis are critical for identifying effective therapeutic targets. The current strategies of targeting the tumor-immune symbiosis aim to block the ligand-receptor interaction, which not only inhibit GBM progression but also reshape T-cell biology, thus improving the anti-tumor efficiency of ICI therapies. Nevertheless, we admit that our current knowledge of the tumor-immune symbiosis in GBM is still at an early stage (see Outstanding Questions). In addition to cancer cells, immune cells exhibit a remarkable heterogeneity in the TME. For example, recent unbiased scRNA-seq and CyTOF studies have revealed certain unique TAM subpopulations, including CD73high macrophages and a specific high-grade glioma-associated microglia (HGG-AM), that play an important role in GBM development [14,120,121]. Genetic studies confirmed that depletion of CD73 in GBM-bearing mice exhibits a robust anti-tumor effect and synergizes with ICI therapy [14]. Similarly, HGG-AM has pro-tumor effect via secreting IL-1β to promote GSC proliferation via apolipoprotein E (ApoE)-mediated NLRP1 inflammasome formation [121]. Supporting by this initial success, further comprehensive studies using scRNA-seq, CyTOF, and additional advanced technologies (e.g., advanced imaging, whole-exome sequencing, CRISPR KO screening, high throughput screening, brain tumor organoids, nanotechnology, tumor-on-a-chip system, and exosome delivery system) followed by detailed molecular studies are needed to identify context-dependent tumor-immune symbiosis and to reveal the molecular basis underlying its role in promoting tumor growth and immunotherapy resistance. As a result, these studies will lead to identification of novel and effective therapeutic strategies intercepting the context-dependent tumor-immune symbiosis in GBM.
Outstanding Questions.
What are regulatory and functional differences between the tumor-macrophage symbiosis and the tumor-microglia symbiosis in GBM? How can we effectively target these two co-dependencies specifically? Within each symbiosis, can we identify novel therapeutic vulnerabilities?
What are the detailed molecular mechanisms underlying the GBM-MDSC/neutrophil symbiosis? What are regulatory and functional differences between M-MDSCs and PMN-MDSCs in the GBM TME?
Can we develop therapeutic tools to target the context-dependent GBM-immune crosstalk (e.g., GBM-TAM, GBM-MDSC, GBM- neutrophil, GBM-T-cell, and myeloid cell-T-cell crosstalk) in GBM patients? Of the aforementioned, which should be prioritized for therapeutic implementation?
Is there context-dependent myeloid-myeloid symbiosis (e.g., macrophage-microglia, TAM-MDSC, TAM-neutrophil, and MDSC-neutrophil symbiosis) in GBM? If yes, what are the molecular mechanisms underlying them and how do we target them?
Whether and how the tumor-immune symbiosis affects T cell-mediated anti-tumor immunity and immunotherapies in GBM? Can we target the tumor-myeloid cell symbiosis to overcome the resistance of immunotherapies (e.g., ICI therapies) in GBM patients? Can we develop novel immunotherapies for GBM patients based on the context-dependent tumor-immune symbiosis?
Highlights.
Heterogeneity is a hallmark of glioblastoma (GBM), which includes cancer cell heterogeneity (e.g., distinct genetic and epigenetic alterations) and tumor microenvironment (TME) heterogeneity (e.g., distinct stromal cell types with different phenotypes). Heterogeneity generates context-dependent GBM-TME crosstalk that is critical for tumor growth and treatment resistance.
Among the TME, myeloid cells (e.g., macrophages, microglia, myeloid-derived suppressor cells, and neutrophils) are the most prominent and dominant cells that contribute to tumor progression and immunosuppression via symbiotically interacting with cancer cells and lymphocytes.
Blockade of the tumor-immune symbiosis inhibits tumor progression and improves the effectiveness of immunotherapies (e.g., immune checkpoint inhibitor therapies) in GBM.
Acknowledgments
This work was supported by NIH R00 CA240896 (P.C.), DoD Career Development Award W81XWH-21-1-0380 (P.C.), Cancer Research Foundation Young Investigator Award (P.C.), Lynn Sage Scholar Award (P.C.), American Cancer Society Institutional Research Grant IRG-21-144-27 (P.C.), philanthropic donation from Mindy Jacobson and the Bill Bass Foundation (P.C.), Northwestern University start-up funds (P.C.), and the Robert H. Lurie Comprehensive Cancer Center (P.C.).
Glossary
- Bone marrow-derived macrophages
a type of macrophages that originate from bone marrow-derived hematopoietic stem cells.
- CAR T-cell therapy
a highly personalized form of adoptive T-cell therapy that takes advantage of patient’s own T-cells and is engineered to express a CAR, which consists of the antigen-recognition site of an antibody fused with the cytoplasmic domains of the T-cell receptor chain and costimulatory receptors.
- Cytometry by time-of-flight (CyTOF)
a technology that enables single-cell analysis of protein expression by using rare heavy metal isotopes-conjugated antibodies.
- Extracellular vesicles (EVs)
membrane-bound submicron vesicles released by cells into the tumor microenvironment, including exosomes (50–200 nm), microvesicles (100–1 μm), and large oncosomes (>1 μm).
- Glioma stem cells (GSCs)
a population of highly malignant and self-renewing glioma cells, which play a crucial role in tumor maintenance and therapeutic resistance in GBM.
- Immune checkpoint inhibitor (ICI)
inhibitors that block immune checkpoint molecules (e.g., PD1 and CTLA4) to activate anti-tumor immune responses.
- Indoleamine-2,3-dioxygenase 1 (IDO1)
an essential enzyme that catabolizes tryptophan to kynurenine, leading to immunosuppression via regulating Tregs and effector T-cells.
- Isocitrate dehydrogenases (IDH)
an energy metabolic enzyme involves in the Krebs cycle.
- Microglia
differentiated brain resident monocytes from yolk sac progenitors during embryonic development.
- Myeloid-derived-suppressor cells (MDSCs)
a population of immature myeloid cells from bone marrow that can induce immunosuppression and promote tumor progression.
- Neutrophils
a subset of granulocytes that act as immune system’s first line of defense in the body.
- Regulatory B-cells (Bregs)
a small population of B cells that can induce immunosuppression.
- Regulatory T-cells (Tregs)
a specific subpopulation of CD4+ T cells with strong immunosuppressive activity.
- Single-cell RNA sequencing (scRNA-seq)
an approach for the detection and quantitative analysis of RNA expression at the single-cell level, thus providing the information of states, phenotypes, and function of an individual cell.
- Symbiotic interaction
a type of interaction between two types of cells in which at least one cell type benefits.
- Tumor-associated macrophages and microglia (TAMs)
two types of macrophages (bone marrow-derived macrophages and brain-resident microglia) infiltrated in the GBM tumor tissues that display a protumor and immunosuppressive function.
- Tumor microenvironment (TME)
a group of stromal components (e.g., immune cells, molecules, fibroblasts, extracellular matrix, and blood vessels) that surround and feed tumor cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
No potential conflicts of interest were disclosed by the authors.
References
- 1.Wen PY et al. (2020) Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol 22, 1073–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R et al. (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet Oncology 10, 459–466 [DOI] [PubMed] [Google Scholar]
- 3.Chen P et al. (2019) Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 35, 868–884.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klemm F et al. (2020) Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 181, 1643–1660.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmitt MJ et al. (2021) Phenotypic Mapping of Pathologic Cross-Talk between Glioblastoma and Innate Immune Cells by Synthetic Genetic Tracing. Cancer Discov 11, 754–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Francis JM et al. (2014) EGFR Variant Heterogeneity in Glioblastoma Resolved through Single-Nucleus Sequencing. Cancer Discovery 4, 956–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aslan K et al. (2020) Heterogeneity of response to immune checkpoint blockade in hypermutated experimental gliomas. Nat Commun 11, 931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin Y-J et al. (2021) Roles of Neutrophils in Glioma and Brain Metastases. Frontiers in Immunology 12, 3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pombo Antunes AR et al. (2020) Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife 9, e52176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woroniecka KI et al. (2018) T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin Cancer Res 24, 3792–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Groot J et al. (2020) Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro-Oncology 22, 539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schalper KA et al. (2019) Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med 25, 470–476 [DOI] [PubMed] [Google Scholar]
- 13.Saha D et al. (2017) Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell 32, 253–267.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goswami S et al. (2020) Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat Med 26, 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ott M et al. (2021) The immune landscape of common CNS malignancies: implications for immunotherapy. Nat Rev Clin Oncol 18, 729–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ochocka N et al. (2021) Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat Commun 12, 1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neftel C et al. (2019) An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hara T et al. (2021) Interactions between cancer cells and immune cells drive transitions to mesenchymal-like states in glioblastoma. Cancer Cell 39, 779–792.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pombo Antunes AR et al. (2021) Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat Neurosci 24, 595–610 [DOI] [PubMed] [Google Scholar]
- 20.Friedrich M et al. (2021) Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat Cancer 2, 723–740 [DOI] [PubMed] [Google Scholar]
- 21.Xuan W et al. (2021) Context-Dependent Glioblastoma-Macrophage/Microglia Symbiosis and Associated Mechanisms. Trends Immunol 42, 280–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xuan W et al. (2021) Circadian regulation of cancer cell and tumor microenvironment crosstalk. Trends in Cell Biology 31, 940–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buonfiglioli A and Hambardzumyan D (2021) Macrophages and microglia: the cerberus of glioblastoma. Acta Neuropathologica Communications 9, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brennan CW et al. (2013) The Somatic Genomic Landscape of Glioblastoma. Cell 155, 462–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McLendon R et al. (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verhaak RGW et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17, 98–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen A et al. (2021) Chitinase-3-like 1 protein complexes modulate macrophage-mediated immune suppression in glioblastoma. J Clin Invest 131, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wood MD et al. (2018) Neurofibromin knockdown in glioma cell lines is associated with changes in cytokine and chemokine secretion in vitro. Sci Rep 8, 5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Lage M et al. (2019) Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathologica Communications 7, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaffes I et al. (2019) Human Mesenchymal glioblastomas are characterized by an increased immune cell presence compared to Proneural and Classical tumors. OncoImmunology 8, e1655360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sielska M et al. (2020) Tumour-derived CSF2/granulocyte macrophage colony stimulating factor controls myeloid cell accumulation and progression of gliomas. Br J Cancer 123, 438–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei J et al. (2019) Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J Clin Invest 129, 137–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geraldo LH et al. (2021) SLIT2/ROBO signaling in tumor-associated microglia and macrophages drives glioblastoma immunosuppression and vascular dysmorphia. J Clin Invest 131, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeini E et al. (2021) P-selectin axis plays a key role in microglia immunophenotype and glioblastoma progression. Nat Commun 12, 1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J et al. (2021) PI3Kγ inhibition suppresses microglia/TAM accumulation in glioblastoma microenvironment to promote exceptional temozolomide response. PNAS 118, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Z et al. (2020) Transfer of MicroRNA via Macrophage-Derived Extracellular Vesicles Promotes Proneural-to-Mesenchymal Transition in Glioma Stem Cells. Cancer Immunol Res 8, 966–981 [DOI] [PubMed] [Google Scholar]
- 37.Wang Z et al. (2021) The adaptive transition of glioblastoma stem cells and its implications on treatments. Sig Transduct Target Ther 6, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen P et al. (2021) Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Reports 34, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gangoso E et al. (2021) Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 184, 2454–2470.e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen P et al. (2020) Circadian Regulator CLOCK Recruits Immune-Suppressive Microglia into the GBM Tumor Microenvironment. Cancer Discov 10, 371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xuan W et al. (2022) Circadian Regulator CLOCK Drives Immunosuppression in Glioblastoma. Cancer Immunology Research DOI: 10.1158/2326-6066.CIR-21-0559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han S et al. (2020) SETDB1 promotes glioblastoma growth via CSF-1-dependent macrophage recruitment by activating the AKT/mTOR signaling pathway. Journal of Experimental & Clinical Cancer Research 39, 218. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Xie Q et al. (2018) N6-methyladenine DNA Modification in Glioblastoma. Cell 175, 1228–1243.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng G et al. (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49, 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dong F et al. (2021) ALKBH5 Facilitates Hypoxia-Induced Paraspeckle Assembly and IL8 Secretion to Generate an Immunosuppressive Tumor Microenvironment. Cancer Res 81, 5876–5888 [DOI] [PubMed] [Google Scholar]
- 46.Hanahan D (2022) Hallmarks of Cancer: New Dimensions. Cancer Discovery 12, 31–46 [DOI] [PubMed] [Google Scholar]
- 47.Cheng X et al. (2020) Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metabolism 32, 229–242.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qiu R et al. (2021) Metabolic Remodeling in Glioma Immune Microenvironment: Intercellular Interactions Distinct From Peripheral Tumors. Frontiers in Cell and Developmental Biology 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Offer S et al. (2019) Extracellular lipid loading augments hypoxic paracrine signaling and promotes glioma angiogenesis and macrophage infiltration. Journal of Experimental & Clinical Cancer Research 38, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jiang J et al. (2017) Prostaglandin E2 Signaling: Alternative Target for Glioblastoma? Trends in Cancer 3, 75–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yin J et al. (2020) ARS2/MAGL signaling in glioblastoma stem cells promotes self-renewal and M2-like polarization of tumor-associated macrophages. Nature Communications 11, 2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Platten M et al. (2019) Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov 18, 379–401 [DOI] [PubMed] [Google Scholar]
- 53.Panitz V et al. (2021) Tryptophan metabolism is inversely regulated in the tumor and blood of patients with glioblastoma. Theranostics 11, 9217–9233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takenaka MC et al. (2019) Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci 22, 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miska J et al. (2021) Polyamines drive myeloid cell survival by buffering intracellular pH to promote immunosuppression in glioblastoma. Sci Adv 7, eabc8929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hajji N et al. (2022) Arginine deprivation alters microglia polarity and synergises with radiation to eradicate non arginine auxotrophic glioblastoma tumors. J Clin Invest DOI: 10.1172/JCI142137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brandau S and Dorhoi A (2021) Myeloid-Derived Suppressor Cells, Springer. [Google Scholar]
- 58.Veglia F et al. (2021) Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol 21, 485–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alban TJ et al. (2020) Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front Immunol 11, 1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kumar V et al. (2016) The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol 37, 208–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang AL et al. (2016) CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res 76, 5671–5682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang X et al. (2021) BATF2 prevents glioblastoma multiforme progression by inhibiting recruitment of myeloid-derived suppressor cells. Oncogene 40, 1516–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu J et al. (2021) Regulation of tumor immune suppression and cancer cell survival by CXCL1/2 elevation in glioblastoma multiforme. Science Advances DOI: 10.1126/sciadv.abc2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qiu W et al. (2021) Exosomal miR-1246 from glioma patient body fluids drives the differentiation and activation of myeloid-derived suppressor cells. Molecular Therapy 29, 3449–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guo X et al. (2019) Glioma exosomes mediate the expansion and function of myeloid-derived suppressor cells through microRNA-29a/Hbp1 and microRNA-92a/Prkar1a pathways. Int J Cancer 144, 3111–3126 [DOI] [PubMed] [Google Scholar]
- 66.Noman MZ et al. (2014) PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. Journal of Experimental Medicine 211, 781–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee-Chang C et al. (2019) Myeloid-Derived Suppressive Cells Promote B cell–Mediated Immunosuppression via Transfer of PD-L1 in Glioblastoma. Cancer Immunol Res 7, 1928–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Himes BT et al. (2020) The role of extracellular vesicles and PD-L1 in glioblastoma-mediated immunosuppressive monocyte induction. Neuro Oncol 22, 967–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Di Ianni N et al. (2021) Altered Metabolism in Glioblastoma: Myeloid-Derived Suppressor Cell (MDSC) Fitness and Tumor-Infiltrating Lymphocyte (TIL) Dysfunction. Int J Mol Sci 22, 4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bronte V et al. (2016) Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 7, 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yee PP et al. (2020) Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat Commun 11, 5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hedrick CC and Malanchi I (2021) Neutrophils in cancer: heterogeneous and multifaceted. Nat Rev Immunol DOI: 10.1038/s41577-021-00571-6 [DOI] [PubMed] [Google Scholar]
- 73.Gao M et al. (2021) TERT Mutation Is Accompanied by Neutrophil Infiltration and Contributes to Poor Survival in Isocitrate Dehydrogenase Wild-Type Glioma. Frontiers in Cell and Developmental Biology 9, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Magod P et al. (2021) Exploring the longitudinal glioma microenvironment landscape uncovers reprogrammed pro-tumorigenic neutrophils in the bone marrow. Cell Reports 36, [DOI] [PubMed] [Google Scholar]
- 75.Atai NA et al. (2011) Osteopontin is up-regulated and associated with neutrophil and macrophage infiltration in glioblastoma. Immunology 132, 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee SY et al. (2017) CD133 Regulates IL-1β Signaling and Neutrophil Recruitment in Glioblastoma. Mol Cells 40, 515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zha C et al. (2020) Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol Med 17, 154–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.SenGupta S et al. (2021) The Recruitment of Neutrophils to the Tumor Microenvironment Is Regulated by Multiple Mediators. Frontiers in Immunology 12, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jeon H-Y et al. (2019) Ly6G+ inflammatory cells enable the conversion of cancer cells to cancer stem cells in an irradiated glioblastoma model. Cell Death Differ 26, 2139–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liang J et al. (2014) Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res 20, 187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Papayannopoulos V (2018) Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18, 134–147 [DOI] [PubMed] [Google Scholar]
- 82.Wang H et al. (2021) Different T-cell subsets in glioblastoma multiforme and targeted immunotherapy. Cancer Letters 496, 134–143 [DOI] [PubMed] [Google Scholar]
- 83.Qian J et al. (2018) TLR2 Promotes Glioma Immune Evasion by Downregulating MHC Class II Molecules in Microglia. Cancer Immunol Res 6, 1220–1233 [DOI] [PubMed] [Google Scholar]
- 84.Mathewson ND et al. (2021) Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 184, 1281–1298.e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tay RE et al. (2021) Revisiting the role of CD4+ T cells in cancer immunotherapy—new insights into old paradigms. Cancer Gene Ther 28, 5–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.DiDomenico J et al. (2018) The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology 7, e1448329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li C et al. (2020) Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Molecular Cancer 19, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhai L et al. (2021) Tumor Cell IDO Enhances Immune Suppression and Decreases Survival Independent of Tryptophan Metabolism in Glioblastoma. Clin Cancer Res 27, 6514–6528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhao B et al. (2021) Investigation of Genetic Determinants of Glioma Immune Phenotype by Integrative Immunogenomic Scale Analysis. Front Immunol 12, 557994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chongsathidkiet P et al. (2018) Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med 24, 1459–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schaettler MO et al. (2021) Characterization of the Genomic and Immunologic Diversity of Malignant Brain Tumors through Multisector Analysis. Cancer Discov DOI: 10.1158/2159-8290.CD-21-0291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Berghoff AS et al. (2015) Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro-Oncology 17, 1064–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yang W et al. (2020) MHC class I dysfunction of glioma stem cells escapes from CTL-mediated immune response via activation of Wnt/β-catenin signaling pathway. Oncogene 39, 1098–1111 [DOI] [PubMed] [Google Scholar]
- 94.Du L et al. (2020) β-Catenin induces transcriptional expression of PD-L1 to promote glioblastoma immune evasion. Journal of Experimental Medicine 217, e20191115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ricklefs FL et al. (2018) Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci Adv 4, eaar2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mirzaei R et al. (2018) Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. OncoImmunology 7, e1478647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Iwata R et al. (2020) ICOSLG-mediated regulatory T-cell expansion and IL-10 production promote progression of glioblastoma. Neuro Oncol 22, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhai L et al. (2017) Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin Cancer Res 23, 6650–6660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Amoozgar Z et al. (2021) Targeting Treg cells with GITR activation alleviates resistance to immunotherapy in murine glioblastomas. Nat Commun 12, 2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ladomersky E et al. (2018) IDO1 Inhibition Synergizes with Radiation and PD-1 Blockade to Durably Increase Survival Against Advanced Glioblastoma. Clin Cancer Res 24, 2559–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lamano JB et al. (2019) Glioblastoma-Derived IL-6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin Cancer Res 25, 3643–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reardon DA et al. (2020) Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncology 6, 1003–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Arrieta VA et al. (2021) ERK1/2 phosphorylation predicts survival following anti-PD-1 immunotherapy in recurrent glioblastoma. Nat Cancer 2, 1372–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cloughesy TF et al. (2019) Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med 25, 477–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao J et al. (2019) Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med 25, 462–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lamano JB et al. (2019) Glioblastoma-Derived IL6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin Cancer Res 25, 3643–3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sadahiro H et al. (2018) Activation of the Receptor Tyrosine Kinase AXL Regulates the Immune Microenvironment in Glioblastoma. Cancer Res 78, 3002–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Przystal JM et al. (2021) Targeting CSF1R Alone or in Combination with PD1 in Experimental Glioma. Cancers 13, 2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.von Roemeling CA et al. (2020) Therapeutic modulation of phagocytosis in glioblastoma can activate both innate and adaptive antitumour immunity. Nat Commun 11, 1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hsu SPC et al. (2020) Rapamycin and hydroxychloroquine combination alters macrophage polarization and sensitizes glioblastoma to immune checkpoint inhibitors. J Neurooncol 146, 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Flores-Toro JA et al. (2020) CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. PNAS 117, 1129–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang P-F et al. (2020) Neutrophil depletion enhances the therapeutic effect of PD-1 antibody on glioma. Aging 12, 15290–15301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bagley SJ et al. (2018) CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro-Oncology 20, 1429–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.O’Rourke DM et al. (2017) A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Science Translational Medicine 9, eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Choi BD et al. (2019) CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol 37, 1049–1058 [DOI] [PubMed] [Google Scholar]
- 116.Wang D et al. (2020) Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Science Translational Medicine 12, eaaw2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Friebel E et al. (2020) Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 181, 1626–1642.e20 [DOI] [PubMed] [Google Scholar]
- 118.Woolf Z et al. (2021) Single-cell image analysis reveals a protective role for microglia in glioblastoma. Neuro-Oncology Advances 3, vdab031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lim M et al. (2018) Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol 15, 422–442 [DOI] [PubMed] [Google Scholar]
- 120.Van Hove H et al. (2019) A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22, 1021–1035 [DOI] [PubMed] [Google Scholar]
- 121.Liu H et al. (2021) Pro-inflammatory and proliferative microglia drive progression of glioblastoma. Cell Rep 36, 109718. [DOI] [PubMed] [Google Scholar]
- 122.Bian Z et al. (2020) Deciphering human macrophage development at single-cell resolution. Nature 582, 571–576 [DOI] [PubMed] [Google Scholar]
- 123.Chen Z et al. (2019) Intravital 2-photon imaging reveals distinct morphology and infiltrative properties of glioblastoma-associated macrophages. PNAS 116, 14254–14259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Landry AP et al. (2020) Distinct regional ontogeny and activation of tumor associated macrophages in human glioblastoma. Sci Rep 10, 19542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bowman RL et al. (2016) Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Reports 17, 2445–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Haage V et al. (2019) Comprehensive gene expression meta-analysis identifies signature genes that distinguish microglia from peripheral monocytes/macrophages in health and glioma. Acta Neuropathologica Communications 7, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li Q et al. (2019) Expression of Tmem119/Sall1 and Ccr2/CD69 in FACS-Sorted Microglia- and Monocyte/Macrophage-Enriched Cell Populations After Intracerebral Hemorrhage. Frontiers in Cellular Neuroscience 12, 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lu Y et al. (2021) Resolution of tissue signatures of therapy response in patients with recurrent GBM treated with neoadjuvant anti-PD1. Nat Commun 12, 4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.González-Tablas Pimenta M et al. (2021) Tumor cell and immune cell profiles in primary human glioblastoma: Impact on patient outcome. Brain Pathol 31, 365–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Raychaudhuri B et al. (2011) Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro-Oncology 13, 591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Raghavan JV et al. (2021) Immuno-phenotyping of IDH-mutant grade 3 astrocytoma and IDH-wildtype glioblastoma reveals specific differences in cells of myeloid origin. Oncoimmunology 10, 1957215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Raychaudhuri B et al. (2015) Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J Neurooncol 122, 293–301 [DOI] [PubMed] [Google Scholar]
- 133.Bayik D et al. (2020) Myeloid-Derived Suppressor Cell Subsets Drive Glioblastoma Growth in a Sex-Specific Manner. Cancer Discov 10, 1210–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ham SW et al. (2019) TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ 26, 409–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Guo X et al. (2019) Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell–specific chemokine recruitment of T cells and microglia. Neuro-Oncology 21, 1250–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]