

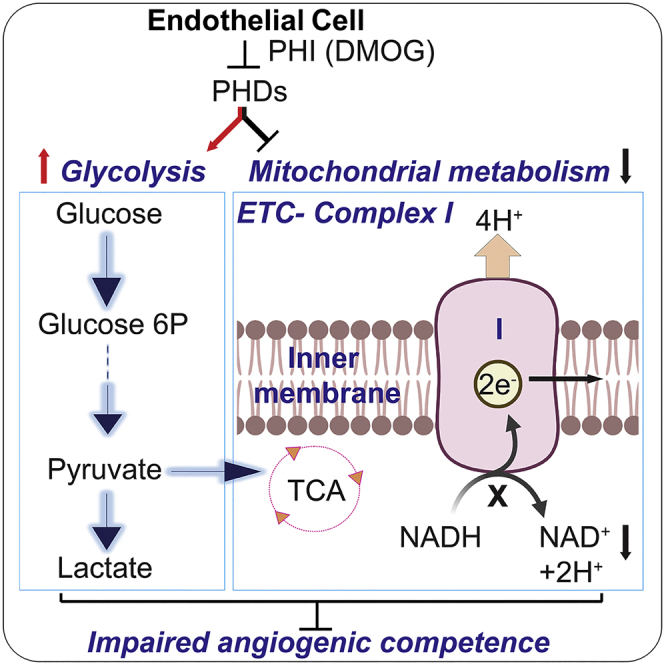
Summary
Endothelial cell (EC) metabolism has emerged as a driver of angiogenesis. While hypoxia inactivates the oxygen sensors prolyl-4 hydroxylase domain-containing proteins 1–3 (PHD1-3) and stimulates angiogenesis, the effects of PHDs on EC functions remain poorly defined. Here, we investigated the impact of chemical PHD inhibition by dimethyloxalylglycine (DMOG) on angiogenic competence and metabolism of human vascular ECs. DMOG reduced EC proliferation, migration, and tube formation capacities, responses that were associated with an unfavorable metabolic reprogramming. While glycolytic genes were induced, multiple genes encoding sub-units of mitochondrial complex I were suppressed with concurrent decline in nicotinamide adenine dinucleotide (NAD+) levels. Importantly, the DMOG-induced defects in EC migration could be partially rescued by augmenting NAD+ levels through nicotinamide riboside or citrate supplementation. In summary, by integrating functional assays, transcriptomics, and metabolomics, we provide insights into the effects of PHD inhibition on angiogenic competence and metabolism of human vascular ECs.
Subject areas: Biological sciences, Metabolomics, Transcriptomics
Graphical abstract
Highlights
-
•
DMOG impairs angiogenic competence of human vascular endothelial cells
-
•
DMOG induces an unfavorable metabolic reprogramming for angiogenesis
-
•
Suppressed mitochondrial metabolism and NAD+ contribute to angiogenic defects by DMOG
Biological sciences; Metabolomics; Transcriptomics
Introduction
Endothelial cells (ECs) line the blood vessels and play a critical role in providing oxygen and nutrients to all tissues in multicellular organisms. In adult tissues, normal ECs remain in quiescence, but injury can induce EC activation and angiogenesis, allowing delivery of oxygen and nutrients to hypoxic tissues. Recently, metabolism has emerged as a central regulator of angiogenesis in parallel to well-established angiogenic growth factors (Falkenberg et al., 2019). ECs are highly glycolytic and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3)-driven glycolysis controls vessel sprouting (De Bock et al., 2013). Furthermore, fatty acid oxidation promotes EC proliferation through DNA synthesis (Schoors et al., 2015), while glutamine metabolism replenishes the mitochondrial tricarboxylic acid (TCA) cycle and maintains EC sprouting (Huang et al., 2017; Kim et al., 2017). Finally, mitochondrial respiratory chain complex III was recently shown to be required for EC proliferation by sustaining amino acid availability (Diebold et al., 2019).
Since ECs reside in direct contact with oxygen, it is not surprising that they are equipped with various O2 sensing mechanisms able to regulate EC function with critical impact in surrounding tissues. A key pathway mediating hypoxia signaling is oxygen-dependent degradation of hypoxia-inducible factors (HIFs), heterodimeric transcription factors made up of an oxygen-labile α-subunit (HIF-α), and a stable β-subunit (HIF-β). Under normoxia, HIF-α undergoes proline hydroxylation, a post-translational modification mediated by 2-oxoglutarate, iron dioxygenases called prolyl-4-hydroxylase domain (PHD) proteins 1–3 (Epstein et al., 2001; Schofield and Ratcliffe, 2004). Proline hydroxylation allows recognition and ubiquitination of HIF-α by the tumor suppressor protein von Hippel-Lindau followed by proteasomal degradation (Maxwell et al., 1999). Conversely, in hypoxia, PHDs are inactivated and HIF-α subunits are stabilized and induce the expression of genes promoting adaptation to low O2 such as metabolic and angiogenic genes (Majmundar et al., 2010; Semenza, 2012). It is now well established that HIFs control angiogenesis in embryonic vascular development and disease settings such as cancer and vascular diseases (Krock et al., 2011a).
Besides sensing oxygen and metabolic stimuli such as amino acids, α-ketoglutarate, succinate, or fumarate (Bargiela et al., 2018; Duran et al., 2013; Pan et al., 2007), PHDs have emerged as regulators of metabolism. For instance, loss of PHD1 reduces mitochondrial respiration in muscle (Aragonés et al., 2008) and enhances glucose flux through the oxidative pentose phosphate pathway in neurons (Quaegebeur et al., 2016). PHD2 deficiency induces anaerobic glycolysis in macrophages (Guentsch et al., 2016), while PHD3 loss enhances mitochondrial fat metabolism in muscle (Yoon et al., 2020). Nevertheless, the impact of PHD inhibition in EC metabolism remains poorly defined. Given the importance of endothelial oxygen sensing and metabolism for vascular growth and function, we hypothesized that PHD inhibition may regulate angiogenic competence via reprogramming of EC metabolism. To induce chemical PHD inhibition, we used dimethyloxalylglycine (DMOG), a cell-permeable inhibitor of 2-oxoglutarate-dependent dioxygenase (2OGDD) enzyme superfamily, which includes PHDs. By integrating functional assays, transcriptomics, and metabolomics, we investigated the angiogenic competence and metabolic profile of EC subjected to DMOG.
Results
DMOG impairs EC proliferation, migration, and tube formation
In response to angiogenic stimuli, EC proliferation and migration are prerequisite processes for the formation of blood vessels (Adams and Alitalo, 2007; Carmeliet and Jain, 2011; Herbert and Stainier, 2011). To define the impact of PHD inhibition on these essential angiogenic processes, we exposed human primary pulmonary artery endothelial cells (HPAEC) to the 2OGDD inhibitor, DMOG. As expected, treatment with DMOG (1 mM) for 24 h led to nuclear stabilization of HIF-1α and HIF-2α as shown by immunofluorescence staining and immunoblot analysis (Figures S1A and S1B) without affecting cell viability based on Annexin V/propidium iodide (PI) staining (Figure S1C). To assess the effect of DMOG on HPAEC proliferation, we first used the MTT colorimetric assay, which is based on cell metabolic activity. We observed a ∼40% reduction in proliferation of ECs treated with DMOG compared to controls (Figure 1A). This effect was validated by BrdU immunofluorescence labeling, which showed reduction in number of BrdU positive nuclei per high-power field (hpf) by 84% in DMOG-exposed cells compared to vehicle-treated cells (p < 0.001) (Figure 1B). Furthermore, flow cytometric cell cycle analysis by DNA staining with propidium iodide indicated suppression of cell cycle progression in DMOG-treated cells (Figure 1C). Specifically, treatment with DMOG increased by ∼6% the proportion of cells in G0/G1 phase (p < 0.01) and reduced by ∼48% the proportion of cells in S phase (p < 0.0001). We also observed a reduction of cells in G2/M phase, which though did not reach statistical significance (p = 0.057).
Figure 1.

DMOG suppresses endothelial cell proliferation, migration, and tube formation
(A) Representative bright field images of formazan crystal formed after 3 h incubation of MTT with vehicle or DMOG-treated (1 mM) HPAEC. Right graph shows relative HPAEC proliferation assessed by MTT assay. Scale bar, 100μm.
(B) Representative images of BrdU immunostaining. Right graph shows semi-quantitative analysis of BrdU positive cells/hpf. Scale bar, 50μm.
(C) Representative histograms of cell cycle analysis for control and DMOG-treated cells. Right side graph demonstrates relative percentages of cell populations in G0/G1, S, and G2/M cell cycle phases.
(D) Representative images of 2D scratch wound assay of control and DMOG-treated cells and semi-quantitative analysis of healed area after 24 h. Scale bar, 200μm.
(E) Representative images of tubes formed at indicated time points in control and DMOG-treated cells and semi-quantitative analysis of different parameters at 20 h time point. Scale bar, 200μm. Data are pooled from 3 independent experiments and represented as mean ± SEM. Statistics were determined by two-tailed t-test. ∗∗, p < 0.01; ∗∗∗∗, p < 0.0001; ns, not statistically significant. See also Figure S1.
Next, we conducted a 2D scratch-wound assay to determine the effect of DMOG on migratory capacity of HPAEC. As shown in Figure 1D, we found a ∼49% reduction in closure of the wounded gap in DMOG-treated ECs compared to vehicle (p < 0.0001). In addition, we assessed the effect of DMOG on EC tube formation. Quantitative analysis of several angiogenic parameters including total meshes area and segments length revealed that DMOG significantly disrupted EC tube formation. (Figure 1E). Taken together, our findings show that DMOG impairs angiogenic competence of HPAEC.
DMOG induces a broad transcriptional response in HPAEC
We next performed RNA-seq analysis to define the transcriptional response of HPAEC to DMOG. HPAEC were exposed to 1mM DMOG or vehicle (0.5% DMSO) for 24 h prior to analysis (4 DMOG and 4 vehicle-treated samples). RNA-seq identified 4,092 differentially expressed protein coding genes (DEGs), of which 1,000 were upregulated and 3,092 were downregulated (Figure S2A). Heatmap of the top 50 among the significantly upregulated and downregulated genes showed a distinct transcriptional profile following treatment with DMOG (Figure S2B). Hallmark analysis of DEGs showed that the transcriptional response to DMOG treatment mimicked hypoxia (Figure 2A), while HIF-1 signaling was among the top enriched pathways detected by KEGG pathway analysis (Figure 2B).
Figure 2.

DMOG mimics transcriptional response to hypoxia and regulates the expression of genes involved in metabolism, cell cycle, and angiogenesis
Bubble charts for top 20 enriched Hallmark (A) and KEGG (B) pathways of upregulated (top) or downregulated DEGs (bottom) by DMOG. Heat maps of RNA-seq data showing significantly altered genes involved in glycolysis and TCA cycle (C), mitochondrial electron transport complex I and II (D), cell cycle (E), and angiogenesis (F). Each column corresponds to a sample and each row corresponds to a specific gene. KEGG, Kyoto Encyclopedia of Genes and Genomes. See also Figures S2 and S3.
Importantly, Hallmark and KEGG pathways showed significant alteration of genes involved in cellular metabolism (Figures 2A and 2B). Specifically, exposure to DMOG significantly induced genes involved in glucose metabolism (Figures 2C and S2C), as indicated by the significant upregulation of the glucose transporters solute carrier family 2 member 1 (SLC2A1) and solute carrier family 2 member 3 (SLC2A3) and glycolytic genes hexokinase 1 (HK1), hexokinase 2 (HK2), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), and lactate dehydrogenase A (LDHA). In contrast, DMOG suppressed several genes involved in mitochondrial metabolism (Figures 2C, 2D, and S2C). Among TCA cycle genes, we observed downregulation of isocitrate dehydrogenase (IDH3A), succinyl-CoA ligase (SUCLA2), and succinate dehydrogenase (SDHC) by DMOG. Furthermore, genes encoding sub-units of mitochondrial complex I (MC1), which regenerates NAD+, were significantly suppressed by DMOG (Figures 2D and S2C) leading to reduction in the protein abundance and activity of MC1 (Figures S2D and S2E). Finally, succinate dehydrogenase complex assembly factor 3 and 4 (SDHAF3 and SDHAF4) of mitochondrial complex II were significantly downregulated, whereas no significant alterations were noted in genes encoding sub-units of mitochondrial complexes III, IV, and V (Figures 2D and S2C).
A comparative analysis of the RNA-seq data from DMOG-treated cells with publicly available RNA-seq data from HPAECs subjected to hypoxia (0.2% O2) confirmed a significant overlap between the two datasets (Figure S3A). Hallmark pathway analysis of overlapping DEGs revealed significant enrichment for hypoxia and glycolysis by both hypoxic exposure and DMOG (Figure S3B). Besides the expected upregulation of glycolytic genes, hypoxia also suppressed several genes encoding sub-units of mitochondrial complex I (Figure S3C). On the other hand, the majority of detected transcriptional responses were distinct for each stimulus, which could be due to differences in HIF activation (for instance, balance between HIF-1 and HIF-2 isoforms) or activation of HIF-independent pathways.
Consistent with the reduced proliferation capacity of HPAEC treated with DMOG, cell cycle was identified among the enriched pathways in the transcriptome of DMOG-treated cells. Indeed, hypoxia suppresses genes involved in DNA replication (Manalo et al., 2005) and DMOG reduced the expression of genes encoding DNA-polymerases, primases, and microsomal maintenance (MCM) proteins 2–7, components of a DNA helicase required for DNA replication and cell proliferation (Maiorano et al., 2006) (Figure 2E). Furthermore, mRNA levels of classic angiogenic genes were significantly altered by treatment with DMOG (Figure 2F). Among genes involved in VEGF signaling, vascular endothelial growth factor A (VEGFA), vascular endothelial growth factor B (VEGFB), vascular endothelial growth factor C (VEGFC), and fms-related receptor tyrosine kinase 1 (FLT1) were significantly upregulated, while kinase insert domain receptor (KDR) was downregulated in DMOG-treated cells compared to control. Significant alterations were also observed in the expression of genes in Notch signaling pathway as indicated by downregulation of delta-like protein 1 (DLL1) and hes family bHLH transcription factor 2 (HES2) and upregulation of delta-like canonical Notch ligand 4 (DLL4), notch receptor 1 (NOTCH1), transducin-like enhancer of split 1 (TLE1), hes family bHLH transcription factor 1 (HES1), and hes family bHLH transcription factor 4 (HES4) in the setting of DMOG exposure. Finally, among angiopoietins and angiopoietin-like proteins, DMOG induced the mRNAs of angiopoietin 2 (ANGPT2) and angiopoietin-like 4 (ANGPTL4) (Figure 2F).
DMOG induces metabolic reprogramming in HPAEC
To get insights into how DMOG exposure affects EC metabolism, we assessed the metabolome of cell lysates and media of HPAEC treated with DMOG compared to vehicle (5 DMOG-treated and 5 vehicle-treated samples). Unbiased metabolomic profiling by liquid chromatography/mass spectrometry (LC/MS) and gas chromatography/mass spectrometry (GC/MS) platforms showed significant alterations in 126 metabolites for cell lysates (70 upregulated and 56 downregulated) and 45 metabolites for cell media (19 upregulated and 26 downregulated) in the setting of DMOG treatment (Figure S4A, Supplemental Table sheet S1 and 2). Biochemical importance plot generated by random forest classification showed key differences in amino acid, nucleotide, lipid, and glucose metabolism (Figures S4B and S4C). Accordingly, metabolites set enrichment analysis using Small Molecule Pathway Database detected Warburg effect, citric acid, lipid, amino acid, and nucleotide metabolism among the most frequently modified pathways by DMOG (Figure 3A).
Figure 3.

DMOG alters the endothelial cell metabolome
(A) Shown are the top 25 downregulated (upper graph) and upregulated (lower graph) metabolic pathways detected by metabolites set enrichment analysis in DMOG-treated cells compared to control. Scaled intensity values indicating relative levels of metabolites related to glycolysis (B) and TCA cycle (C). (D) NAD+/NADH ratio in cells treated with vehicle or DMOG. Scaled intensity values indicating relative levels of lipid metabolites (E), nucleotides (F), and amino acids (G). n = 5 independent samples per condition. All statistical data are represented as mean ± SEM and statistics were determined by a Welch’s two sample t-test. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; ns, not significant. G6P, glucose-6-phosphate; FBP, fructose 1,6 bisphosphate; DHAP, dihydroxyacetone phosphate; PEP, phosphoenolpyruvate; AKG, alpha-ketoglutarate; DPA, docosapentaenoate; DHLA, dihomolinolenate; ALC, acetylcarnitine; CHOP, choline phosphate; GPC, glycerophosphorylcholine; PEA, phosphoethanolamine; GPEA, glycerylphosphorylethanolamine; G3P, glycerol 3-phosphate; 5′-AMP, adenosine-5′-monophosphate; 5′-ADP, adenosine-5′-diphopshate; 5′-CMP, cytidine 5′-monophosphate; CDP, cytidine diphosphate; 2′,3′-cCMP, cytidine 2′,3′-cyclic monophosphate; 5′-UDP, uridine-5-diphosphate; UTP, uridine 5′-triphosphate. See also Figure S4 and Table S1.
Alterations in glucose metabolism
DMOG-treated EC showed significant alterations in several glucose-derived metabolites (Figure 3B). Compared to vehicle, treatment with DMOG induced significant reduction in levels of 6-carbon glycolytic intermediates, glucose-6-phosphate (G6P) (3.2-fold, p < 0.01), and fructose 1,6 bisphosphate (FBP) (7-fold, p < 0.01), while significant increase was noted for 3-carbon glycolytic intermediates, dihydroxyacetone phosphate (DHAP) (1.3-fold, p < 0.05), and phosphoenolpyruvate (PEP) (1.5-fold, p < 0.01). Finally, pyruvate level was reduced by 10-fold (p < 0.001), while lactate level was increased by 1.46-fold (p < 0.05) in DMOG-treated cells.
Impaired mitochondrial metabolism
DMOG-treated cells displayed decreased levels for several TCA cycle intermediates compared to the control (Figure 3C). Specifically, citrate, alpha-ketoglutarate, fumarate, and malate levels were significantly reduced by DMOG. Furthermore, DMOG-treated cells showed significantly reduced NAD+/NADH ratio, known to limit carbon entry and flow through the TCA cycle (Martinez-Reyes and Chandel, 2020) (Figure 3D). Interestingly, acetyl-CoA levels remained unchanged despite the significant reduction in pyruvate levels, suggesting either reduced carbon-flux of TCA cycle and/or contribution of other pathways in the replenishment of acetyl-CoA pool.
Impaired lipid metabolism
Lipids function as structural components of membrane, signaling molecules for proliferation and apoptosis, and support TCA cycle (Falkenberg et al., 2019). After DMOG treatment, significant increase was observed in several lipids including, oleate (1.5-fold, p < 0.001) and linoleate (1.5-fold, p < 0.05), choline (1.7-fold, p < 0.001), glycerophosphorylcholine (GPC) (5.3-fold, p < 0.001), glycerophosphoethanolamine (GPEA) (2.4 -fold, p < 0.001), and glycerol 3-phosphate (G3P) (1.6-fold, p < 0.001) (Figure 3E). These changes are consistent with the increased intracellular levels of total fatty acids reported in hypoxic cancer cells (Valli et al., 2015) and may suggest activation of phospholipases contributing to alterations in complex lipid metabolism.
Alterations in nucleotide metabolism
Metabolomic profiling revealed significant alterations in the levels of inosine, and several purine and pyrimidine metabolites (Figure 3F). Specifically, treatment with DMOG caused significant reduction in inosine, adenine, adenosine-5′-diphopshate (ADP), adenosine-5′-monophosphate (AMP), and guanosine levels. Among pyrimidine metabolites, cytidine 5′-monophosphate (CMP), cytidine diphosphate (CDP), cytidine 2′,3′-cyclic monophosphate (2′,3′-cCMP), uridine, uridine-5-diphosphate (UDP), and uridine 5′-triphosphate (UTP) were significantly reduced by DMOG treatment. The resulting diminished production in nucleotide pool is likely to suppress EC vessel sprouting as reported by Schoors et al. (2015).
Alterations in amino acid metabolism
DMOG exposed HPAEC showed significant changes in the level of amino acids (Figure 3G). Out of 20 amino acids, 13 were significantly altered post DMOG treatment. Specifically, the levels of 4 amino acids (aspartate, glutamate cysteine, and proline) were significantly reduced, while 9 amino acids (glutamine, histidine, isoleucine, leucine, methionine, phenylalanine, tryptophan, tyrosine, and valine) showed significant increase following treatment with DMOG. Importantly, increased glutamine levels in conjunction with the diminished glutamate indicated diminished glutamine utilization, a response that can impair angiogenesis (Huang et al., 2017; Kim et al., 2017). Furthermore, significant reduction was noted in levels of aspartate, known to regulate cell proliferation by supporting the biosynthesis of purines and pyrimidines (Birsoy et al., 2015; Sullivan et al., 2015).
In summary, these findings suggest that treatment of HPAEC with DMOG induces a switch to anaerobic glycolysis, impairs TCA cycle, and alters broadly lipid and amino acid levels with concurrent reduction in purine and pyrimidine levels. These metabolic changes may contribute to the impaired angiogenic capabilities of HPAEC in the setting of DMOG treatment.
Pyruvate or aspartate supplementation and treatment with DCA fail to rescue DMOG-induced defects in angiogenic competence of HPAEC
Among the metabolic alterations induced by DMOG, we wished to define the role of the observed reductions in pyruvate and aspartate levels. Pyruvate is essential in maintaining carbon flux in TCA cycle and reduced pyruvate levels have been reported to impair mitochondrial metabolism (Diers et al., 2012; Kim et al., 2018; Wang et al., 2018). Aspartate functions as a precursor for nucleotide synthesis and inhibition of aspartate production from mitochondria or its transport to cytoplasm reduces cell proliferation (Alkan et al., 2018; Sullivan et al., 2015). We therefore examined whether supplementation of these metabolites was sufficient to rescue the angiogenic defects induced by DMOG. Nevertheless, the effect of DMOG on proliferation, migration, and tube formation abilities of HPAEC (Figures S5A, S5B, S5C, S5D, and S5E) remained unchanged by supplementation with methyl pyruvate (2, 1, and 0.5 mM) or methyl aspartate (5, 2, and 0.5 mM). In the absence of DMOG, methyl pyruvate did not influence endothelial angiogenic competence (Figures S5A, S5B, and S5E), while methyl aspartate at high concentrations (5 and 2 mM) reduced cell proliferation. To examine whether suppression of oxidative respiration by pyruvate dehydrogenase kinases contributes to the DMOG-induced angiogenic defects, we used dichloroacetate (DCA), a non-selective PDK inhibitor. In fact, treatment with DCA did not significantly modify the effects of DMOG on EC proliferation and migration (Figures S6A, S6B, and S6C), whereas exacerbated the defects in tube formation (Figure S6D).
Citrate supplementation or nicotinamide riboside partially rescues DMOG-induced defects in cell migration and tube formation ability of HPAEC
Because the collapse of TCA cycle intermediates in the setting of blocked glutamine metabolism may contribute in DMOG-induced angiogenic defects, we next focused on citrate, an important substrate in TCA cycle, which has been shown to promote cell proliferation (MacPherson et al., 2017). Therefore, we directly supplemented cells with membrane-permeable form of citrate (trimethyl citrate) (Hayek et al., 2019). Citrate supplementation (0.5 mM) improved the metabolic activity-based MTT assay (Figure 4A) but had no effect on DMOG-induced alterations in cell proliferation and cell cycle progression as indicated by BrdU incorporation assay and flow cytometric analysis, respectively (Figures 4B, 4C and S7A). Nevertheless, citrate supplementation partially rescued the DMOG-induced defects on migration and tube formation abilities (Figures 4D and 4E) as indicated by a 22% increase in healing area of 2D scratch wound assay (p < 0.0001) (Figure 4D) and improvement of multiple tube formation-related parameters (Figure 4E). To investigate the molecular mechanism by which citrate improved the DMOG-induced angiogenic defects, we examined the HIF pathway. Notably, treatment with citrate did not alter HIF-1α and HIF-2α protein levels in the setting of DMOG exposure (Figures S7B and S7C). Because citrate is a metabolic intermediate of reductive carboxylation of glutamine, a process known to recycle NADH to NAD+ under impaired mitochondrial metabolism (Gaude et al., 2018), we next examined the impact of citrate supplementation on DMOG-induced reduction in NAD+ levels. By performing LC/MS, we found that citrate supplementation in DMOG-treated cells raised NAD+ levels by 2.1-fold (Figure S5D). In the absence of DMOG, citrate supplementation did not affect NAD+ levels in HPAEC. Furthermore, boosting NAD+ levels by adding the NAD+ precursor nicotinamide riboside (NR) (200 μM) mirrored the effects of citrate and improved the DMOG-induced defects in migration and tube formation but without affecting EC proliferation (Figures 5A–5D). In summary, these findings identify citrate and NAD+ depletion as significant metabolic alterations contributing to the impaired EC migration and tube formation capacity in the context of PHD inhibition.
Figure 4.

Citrate supplementation partially rescues the DMOG-induced defects in endothelial migration and tube formation capacity
(A) Representative bright field images of formazan crystal formed after MTT incubation with control, DMOG (1mM), DMOG + citrate and citrate (0.5mM)-treated HPAEC. Right graph shows relative HPAEC proliferation calculated by MTT assay. Scale bar, 100μm.
(B) Representative images of BrdU immunostaining under the conditions indicated in A. Right graph shows semi-quantitative analysis of BrdU positive cells per hpf. Scale bar, 50μm.
(C) Quantitative analysis of cell cycle showing relative percentage of cell population in G0/G1, S, and G2/M cell cycle phase.
(D) Representative images of 2D scratch wound assay of control or DMOG-treated cells and semi-quantitative analysis of healed area after 24 h. Scale bar, 200μm.
(E) Representative images of tubes formed at the indicated time points in control, DMOG, DMOG + citrate, and citrate-treated cells and semi-quantitative analysis of different parameters at 20 h time point. Scale bar, 200μm. Data are pooled from 3 independent experiments and represented as mean ± SEM. Statistics were determined by one-way ANOVA with Sidak correction for multiple comparisons. ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; ns, not significant. Asterisks above bars indicate significant difference between control and treated group, whereas asterisks above lines indicate significant difference between DMOG and DMOG + citrate treated groups. See also Figures S5–S7.
Figure 5.

Nicotinamide Riboside supplementation partially rescues the DMOG-induced defects in endothelial migration and tube formation capacity
(A) Representative bright field images of formazan crystal formed after incubation with MTT in control, DMOG (1mM), DMOG + NR and NR (200μM)-treated HPAEC. Right graph shows relative HPAEC proliferation assessed by MTT assay. Scale bar, 100μm.
(B) Representative images of BrdU immunostaining. Right graph shows semi-quantitative analysis of BrdU positive cells/hpf. Scale bar, 50μm.
(C) Representative images of 2D scratch wound assay and semi-quantitative analysis of healed area after 24 h. Scale bar, 200μm.
(D) Representative images of tubes formed at different time points in control, DMOG, DMOG + NR and NR-treated ECs and semi-quantitative analysis of different parameters at 20 h time point. Scale bar, 200μm. Data are pooled from 3 independent experiments and represented as mean ± SEM. Statistics were determined by one-way ANOVA with Sidak correction for multiple comparisons. ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001; ns, not significant. Asterisks above bars indicate significant difference between control and treated group, whereas asterisks above lines indicate significant difference between DMOG and DMOG + NR-treated groups. NR, nicotinamide riboside.
Discussion
Here, we discovered a previously unidentified role for PHD inhibition in suppressing angiogenic properties of human vascular endothelium, which may impair vascular homeostasis and regeneration in ischemic tissues. Our studies demonstrate that induction of EC pseudohypoxia by DMOG impairs EC proliferation, migration, and tube formation capacities. Although metabolic reprogramming induced by hypoxic signaling has been implicated in vascular biology (Krock et al., 2011a; Semenza, 2010), the role of PHD/HIF pathway in endothelial metabolism remains unclear. By combining unbiased RNA-seq and metabolomic analysis, we found that chemical PHD inhibition induces an unfavorable metabolic reprogramming for angiogenesis characterized by alterations in amino acid metabolism and suppression of mitochondrial metabolism. Furthermore, our mechanistic studies revealed that suppression of mitochondrial metabolism with reduced NAD+ regeneration may at least partially drive the defects in migration and tube forming capacity of EC in the context of PHD inhibition. These findings demonstrate a maladaptive metabolic reprogramming as important molecular signature that characterizes the response of vascular endothelium to chemical PHD inhibition.
In hypoxic tissues, parenchymal cells respond to oxygen deprivation via HIFs, which upregulate angiogenic genes leading to activation of EC and vessel growth (Hashimoto and Shibasaki, 2015; Krock et al., 2011a; Pugh and Ratcliffe, 2003). As oxygen sensors, PHDs have been also implicated in regulating angiogenesis mostly through their role in controlling the stability of HIFs (Fraisl et al., 2009). Using DMOG, a 2-oxoglutarate analog which competitively inhibits PHDs, we show that chemical PHD inhibition suppresses angiogenesis by reducing EC proliferation, migration, and tube formation. These findings were quite surprising, particularly since prior studies have shown that both HIF-1 and HIF-2 in EC promote angiogenesis in a cell-autonomous manner (Manalo et al., 2005; Skuli et al., 2009). Notably, germline deletion of Phd2 results in abnormal placental vascularization and embryonic lethality (Takeda et al., 2006), while heterozygosity for Phd2 did not alter capillary density despite the increase in HIF activity (Mazzone et al., 2009). Furthermore, our findings agree with prior study, which showed that exposure to hypoxia slowed EC growth, decreased entry of cell into S phase, and delayed wound repair (Shreeniwas et al., 1991). While our study did not find an increase in EC death by DMOG, other studies have shown that hypoxia may trigger EC apoptosis by induction of NFkB (Matsushita et al., 2000) or HIF-1-dependent stabilization of p53 (Stempien-Otero et al., 1999). On the other hand, several studies have reported that moderate hypoxia promotes EC proliferation, survival, and migration (Krock et al., 2011b). Therefore, inhibition of PHDs may differentially regulate angiogenesis depending on the context, means of inhibition, and cell-type affected.
Herein, we explored the transcriptional responses of EC to HIF-1 and HIF-2 stabilization with the prolyl-hydroxylase inhibitor DMOG. Compared to prior microarray-based studies, we employed RNA-seq as an unbiased approach to uncover a wide range of transcriptional responses that may drive endothelial functional alterations to chemical PHD inhibition. As expected, we found that DMOG exposure mimics hypoxia and regulates the expression of several angiogenic genes. Related to decreased EC proliferation following DMOG exposure, our RNA-seq analysis showed reduced transcription of genes associated with MCM complex formation, which is necessary for DNA synthesis and cell proliferation (Maiorano et al., 2006). This finding is consistent with the study by Hubbi, which reported that HIF-1α inhibits MCM protein complex activation leading to decreased DNA replication and cell cycle arrest (Hubbi et al., 2013). Consistently, we found suppression of transcripts encoding DNA polymerases DNA polymerase alpha 1, catalytic subunit (POLA1), DNA polymerase alpha 2, accessory subunit (POLA2), DNA polymerase epsilon 2, accessory subunit (POLE2), and DNA polymerase delta 3, accessory subunit (POLD3), an effect that may contribute to the antiproliferative action of DMOG on endothelial cells.
Endothelial metabolism has emerged as a critical regulator of EC homeostasis and growth (Fraisl et al., 2009). While ECs generate most of their ATP by glycolysis, PHD inhibition led to a broad metabolic reprogramming as demonstrated by our transcriptomic and metabolomic analysis. Besides the expected upregulation of glycolytic genes by DMOG, we found significant transcriptional suppression for several sub-units of MC1 complex and its product NAD+. Previously, hypoxia has been reported to inhibit MC1 in different cell types including HUVECs (Tello et al., 2011) by induction of NADH dehydrogenase [ubiquinone] 1 alpha subcomplex, 4-like 2 (NDUFA4L2). Nevertheless, we found no upregulation of NDUFA4L2 by DMOG, which suggests a distinct mechanism for suppression of MC1. Remarkably, the suppression of MC1 could be the primary driver for the low NAD+/NADH ratio, which is known to impair flow of TCA cycle and glycolytic flux. Furthermore, we found increase in NAD+ levels following repletion with citrate, that may be supported by cytosol-confined NADH recycling through malate dehydrogenase 1 (MDH1), a process reported to operate in cells with mitochondrial dysfunction (Gaude et al., 2018). Finally, the observed improvement of EC migration and tube formation by NR or citrate supplementation is probably due to NAD+ -induced enhancement of glycolytic flux, as glycolysis and not oxidative phosphorylation is the main regulator for EC migration.
Consistent with our findings is a recent study in which S-2-hydroxyglutarate (S-2HG), a metabolite with inhibitory effect on PHDs, lowered the proliferative capacity of EC, an effect also recapitulated by genetic PHD inactivation (Andrade et al., 2021). The precise metabolic mechanism by which chemical PHD inactivation impairs proliferation of human ECs remains to be defined but probably involves inability to generate the essential metabolites for macromolecule synthesis due to suppressed mitochondrial oxidation and glutamine metabolism. While reduced aspartate synthesis is critical for cell proliferation upon electron transport chain inhibition (Birsoy et al., 2015; Sullivan et al., 2015), its supplementation, like that of pyruvate, failed to improve the proliferative defect of DMOG-treated ECs. Furthermore, the inhibition of glutamine metabolism by DMOG is likely to play a major role in the reduced proliferative capacity of DMOG-treated cells, as glutamine supplies most carbons in TCA cycle and contributes to biomass synthesis in ECs (Teuwen et al., 2019). Interestingly, a recent study showed a HIF-independent mechanism by which DMOG impairs glutamine metabolism, which involves the inhibition of multiple enzymatic steps, including glutamate dehydrogenase (GDH) by inducing the generation of the metabolite N-oxalylglycine (NOG) (Fets et al., 2018). Finally, impaired lipid metabolism by DMOG may limit EC proliferation by reducing fatty acid carbons for DNA synthesis (Schoors et al., 2015).
Recent advances in our understanding of endothelial metabolism in health and disease have created an expectation that modulation of EC metabolism by hypoxia signaling-based therapeutics may have promising clinical potential. Our study shows that chemical PHD inhibition by DMOG induces an unfavorable metabolic reprogramming for angiogenic competence. Future studies should investigate the role of PHDs in the endothelium and define HIF-dependent vs independent effects on EC metabolism.
Limitations of the study
This study has several limitations. First, like other studies, we used DMOG to inactivate PHDs and activate HIF signaling, but DMOG may have HIF-independent effects by inhibiting other members of 2-oxoglutarate-dependent dioxygenases such as JMJD2A and TET enzymes. Future studies should explore the effects of other chemical PHD inhibitors and compare them to genetic PHD inactivation. Second, we did not dissect the role of HIF-1α and HIF-2α on the DMOG-induced defects on angiogenic competence and EC metabolism. Finally, more studies should be performed to investigate the role of endothelial PHDs on angiogenic responses in vivo.
STAR★Methods
Key resources table
.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BrdU (Bu20a) Mouse mAb | Cell signaling Technology | Cat#5292S; RRID: AB_10548898 |
| HIF-1α | Novus Biologicals | Cat#NB100-449; RRID: AB_10001045 |
| HIF-2α | Novus Biologicals | Cat#NB100-122; RRID: AB_10002593 |
| Lamin B1 (D9V6H) Rabbit mAb | Cell signaling Technology | Cat# 13435; RRID: AB_2737428 |
| Goat Anti-Mouse IgG H&L (Alexa Fluor® 488) | Abcam | Cat#ab150113; RRID: AB_2576208 |
| Goat anti rabbit Alexa Fluor® 594 | Invitrogen | Cat#A32740; RRID: AB_2762824 |
| Goat anti-Rabbit IgG (H + L) Secondary Antibody [HRP] | Novus Biologicals | Cat#NB7160; RRID: AB_524669 |
| Anti-phospho-PDHE1-A type I (Ser293) | Sigma-Aldrich | Cat#ABS204; RRID: AB_11213668 |
| PDH E1 alpha Polyclonal Antibody | Thermo Scientific™ | Cat#18068-1-AP; RRID: AB_2162931 |
| Purified anti-GAPDH Antibody | BioLegend | Cat#631402; RRID: AB_2107422 |
| Chemicals, peptides, and recombinant proteins | ||
| EGM™-2 Endothelial Cell Growth Medium-2 BulletKit™ | Lonza | Cat#CC-3162 |
| Quality Biological Inc Trypsin (0.25%) – EDTA (0.02%) | Thermo Scientific™ | Cat#50983216 |
| Dimethyl sulfoxide | Sigma-Aldrich | Cat#D8418 |
| Dimethyloxalylglycine (DMOG) | Cayman Chemical | Cat# 71210 |
| Trimethyl citrate | Sigma-Aldrich | Cat#27502 |
| Methyl pyruvate | Thermo Scientific™ | Cat#A13966 |
| N-Methyl-L-aspartic acid | Santa Cruz | sc-212234 |
| Nicotinamide riboside | Cayman Chemical | Cat#23132 |
| Gibco™ DPBS, no calcium, no magnesium | Thermo Scientific™ | Cat#14190250 |
| Invitrogen™ Image-iT™ Fixative Solution (4% formaldehyde, methanol-free) | Thermo Scientific™ | Cat#R37814 |
| UltraPure™ DNase/RNase-Free Distilled Water | Thermo Scientific™ | Cat#10977023 |
| Sodium dichloroacetate (DCA) | Sigma-Aldrich | Cat#347795 |
| MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) | Thermo Scientific™ | Cat#M6494 |
| BD Bromodeoxyuridine (BrdU) | Thermo Scientific™ | Cat#BDB550891 |
| Vectashield Vibrance Antifade Mounting Medium with DAPI - 10 mL | Thermo Scientific™ | Cat#NC1601055 |
| PureLink™ RNase A | Thermo Scientific™ | Cat#12091021 |
| Propidium Iodide | Thermo Scientific™ | Cat# P3566 |
| Thermo Scientific™ RIPA Lysis and Extraction Buffer | Thermo Scientific™ | Cat#PI89901 |
| Pierce™ Protease Inhibitor Mini Tablets, EDTA-free | Thermo Scientific™ | Cat#A32955 |
| Pierce™ Phosphatase Inhibitor Mini Tablets | Thermo Scientific™ | Cat#A32957 |
| NuPAGE™ LDS Sample Buffer (4X) | Thermo Scientific™ | Cat#NP0007 |
| Novex™ Tris-Glycine SDS Running Buffer (10X) | Thermo Scientific™ | LC2675 |
| Novex™ Tris-Glycine Transfer Buffer (25X) | Thermo Scientific™ | Cat#LC3675 |
| PageRuler™ Plus Prestained Protein Ladder, 10 to 250 kDa | Thermo Scientific™ | Cat#26619 |
| Methanol (Certified ACS), Fisher Chemical™ | Thermo Scientific™ | A4124 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | Thermo Scientific™ | Cat#34095 |
| TWEEN® 20 | Sigma-Aldrich | Cat#P9416-100ML |
| Acetonitrile (LC-MS) | MilliporeSigma | Cat# 1000294000 |
| Water, BAKER ANALYZED™ LC/MS Reagent Grade | J.T. Baker | Cat#9831-02 |
| Critical commercial assays | ||
| Angiogenesis Assay Kit (In Vitro) | Abcam | Cat#ab204726 |
| Human NADH dehydrogenase ELISA Kit (Complex I) | Abcam | Cat#ab178011 |
| Complex I Enzyme Activity Microplate Assay Kit (Colorimetric) | Abcam | Cat#ab109721 |
| Pierce™ Rapid Gold BCA Protein Assay Kit | Thermo Scientific™ | Cat# A53226 |
| Deposited data | ||
| RNAseq data | This manuscript-GEO | GSE189504 |
| Experimental models: Cell lines | ||
| Primary Pulmonary Artery Endothelial Cells; Normal, Human (HPAEC) | ATCC | Cat# PCS-100-022 |
| Software and algorithms | ||
| Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| FIJI ImageJ | NIH | https://imagej.net/software/fiji/downloads |
| FastQC | Babraham Bioinformatics | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| edgeR | Bioconductor | http://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Xcalibur 4.1 | Thermo Fisher Scientific | https://assets.thermofisher.com/TFS-Assets/CMD/manuals/man-xcali-97928-xcalibur-41-quan-start-manxcali97928-en.pdf |
| TraceFinder 4.1 | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-software/lc-ms-data-acquisition-software/tracefinder-software.html |
| Other | ||
| Western Blotting Filter Paper, Extra Thick, 7 cm × 8.4 cm | Thermo Scientific™ | Cat#88605 |
| Novex™ WedgeWell™ 6%, Tris-Glycine, 1.0 mm, Mini Protein Gels | Thermo Scientific™ | Cat#XP00060BOX |
| Novex™ WedgeWell™ 6%, Tris-Glycine, 1.0 mm, Mini Protein Gels | Thermo Scientific™ | Cat#XP00062BOX |
| Novex WedgeWell 4 to 20%, Tris-Glycine, 1.0 mm, Mini Protein Gel, 10-well | Thermo Scientific™ | Cat#XP04200BOX |
| Novex WedgeWell 4 to 20%, Tris-Glycine, 1.0 mm, Mini Protein Gel, 12-well | Thermo Scientific™ | Cat#XP04202BOX |
| Nitrocellulose Membrane, 0.45 μm | Bio-rad | Cat#1620115 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Pinelopi P. Kapitsinou (pinelopi.kapitsinou@northwestern.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Cell culture
Human primary pulmonary artery endothelial cells (HPAEC) were purchased from ATCC (ATCC) and maintained in endothelial cell growth medium-2 (EGM-2, Lonza) supplemented with EGM-2 SingleQuot Kit (Lonza). HPAEC were cultured in multi-well cell culture plates and flasks coated with 0.1% gelatin. All experiments were performed in cells cultured for ≤7 passages. DMOG (Tocris) was dissolved in dimethyl sulfoxide (DMSO). Trimethyl citrate (Sigma), methyl pyruvate (Thermo Scientific™), methyl aspartate (Santa Cruz) and nicotinamide riboside (Cayman) were used for metabolic repletion experiments.
Method details
Proliferation assays
Cell proliferation was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described by Zirkel A et al. (Zirkel et al., 2018) and BrdU incorporation assay. Briefly, HPAEC were seeded in gelatin-coated 96-well culture plates. The next day, cells were treated with DMOG (1 mM), while control cells were treated with DMSO. After 20 h incubation, MTT solution (Invitrogen) was added for 4 h at 37°C. Subsequently, the intracellular formazan crystals were solubilized with DMSO and the absorbance of the colored solution was read at 540 nm. For BrdU labeling, cells were grown in 4-well chamber slides. Control and DMOG-treated cell were incubated with BrdU solution (BD Pharmingen™) for 3 h under optimal growth conditions. Post incubation, cells were washed 3 times with 0.1% tween 20 in PBS (PBST) and fixed with a methanol/acetone mixture (molar ratio 1:1) for 30 min. After a wash, cells were incubated with 1.5 M HCl for 25 min at room temperature. Next, cells were washed again with PBST and blocked with 3% BSA for 1 h. After blocking, cells were incubated with an anti-BrdU primary antibody (Cell Signaling Technology) overnight at 4°C. After 3 washes with PBST, cells were probed with a secondary antibody (Abcam) at 37°C for 1 h. Finally, cells were washed and mounted with Vectashield Vibrance Antifade Mounting Medium containing DAPI (Vector Laboratories, Inc.). Five random images per well were captured using a fluorescence microscope (Leica DM IL LED) and BrdU positive nuclei were counted. Minimum of 3 independent experiments were performed and analyzed.
2D scratch migration assay
Cell migration was assessed using a scratch wound healing assay (Keyes et al., 2016). HPAEC were seeded in 12-well plates and allowed to grow until monolayer formation. Scratches were created manually using pipette tip, washed with media followed by addition of DMOG or DMSO. To assess cell migration, bright field images were captured by at 0 and 24 h time points. Percentage of healed area was measured using Fiji software based on ImageJ (NIH).
Matrigel tube formation assay
Tube formation assay was performed using an angiogenesis assay kit (Abcam). Briefly, HPAEC treated with DMOG or DMSO for 24 h were trypsinized and resuspended in EGM-2 media containing either DMOG or DMSO and then added to Matrigel coated 96-well plates. Bright field images were captured at different time points (2, 4, 8, 20 h). Captured images were analyzed for total meshes area, mean mesh size, branching intervals, number of segments, number of master segments, total segment length, and total master segment length using the image J-based Angiogenesis Analyzer tool.
Cell cycle and Annexin V/PI cell death assays
Following 24 h treatment with DMOG, cells were trypsinized, centrifuged at 300g for 5 min. Cell pellets were resuspended in ice cold PBS, centrifuged and fixed overnight with 70% ethanol. Fixed cells were resuspended in propidium iodide (PI) staining buffer (Invitrogen) and incubated at 37°C for 30 min. Subsequently, cell cycle was assessed by a BD LSR Fortessa flow cytometer (BD Biosciences) using FACS Diva and FlowJo software.
Cell death was examined by a FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen™) following manufacturer’s protocol. ECs were trypsinized, centrifuged, and washed twice with cold PBS and resuspended in a Binding Buffer provided with the kit. For flow cytometric preparation, cell suspensions were incubated with 5 μL of Annexin V-FITC and 5 μL of PI for 15 min at room temperature in the dark. After incubation, 400 μL of Binding Buffer was added and applied to the BD LSR Fortessa flow cytometer (BD Biosciences). Data analysis was performed using FACS Diva and FlowJo software.
Nuclear protein isolation and immunoblotting
Nuclear protein was isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific) following manufacturer’s instructions. Nuclear protein extracts were separated on SDS-PAGE gel, transferred on nitrocellulose membrane and incubated with HIF-1α (Cayman) or HIF-2α (Novus Biologicals) antibodies at 4°C. After overnight incubation, nitrocellulose membrane was washed and incubated with secondary antibody (Novus Biologicals) for 90 min at 4°C. Blots were visualized by SuperSignal™ West Femto Chemiluminescent Substrate (Thermo Fisher Scientific) and chemiluminescence was detected on an iBright Western Blot Imaging Systems (Thermo Fisher Scientific).
HIF-1α and HIF-2α immunostaining
Cells were fixed with Image-iT™ Fixative Solution (Thermo Fisher Scientific) for 10 min at room temperature. Subsequently, cells were washed 3 times with PBS, permeabilized for 10 min with 0.1% tween 20 in PBS and then nonspecific antigens were blocked with 3% BSA for 2 h. After blocking, cells were incubated with primary antibody against HIF-1α (Cayman) or HIF-2α (Novus Biologicals) at 4°C overnight, followed by incubation with secondary antibody (Thermo Fisher Scientific) for 2 h at room temperature. Following 3 washes with PBS, cells were mounted with Vectashield Vibrance Antifade Mounting Medium containing DAPI (Vector Laboratories, Inc.). Images were captured by a fluorescence microscope (Leica DM IL LED) and analyzed using Fiji software.
RNA isolation and RNA-sequencing
Total RNA was extracted using RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. RNA integrity was checked with Agilent Technologies 2100 Bioanalyzer. Poly(A) tail-containing mRNAs were purified using oligo-(dT) magnetic beads with two rounds of purification. After purification, poly(A) RNA was fragmented using divalent cation buffer in elevated temperature. Poly(A) RNA sequencing library was prepared following Illumina’s TruSeq-stranded-mRNA sample preparation protocol. Quality control analysis and quantification of the sequencing library were performed using Agilent Technologies 2100 Bioanalyzer High Sensitivity DNA Chip. Paired-ended sequencing was performed on Illumina’s NovaSeq 6000 sequencing system. Adapter contaminated peaks, low quality bases and undermined bases were removed using Cutadapt (Kechin et al., 2017) and perl scripts. Sequence quality was verified using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). HISAT2 (Kim et al., 2015) was used to map reads to the genome of ftp://ftp.ensembl.org/pub/release-96/fasta/homo_sapiens/dna/ and mapped reads of each sample were assembled using StringTie (Pertea et al., 2015). Subsequently, all transcriptomes were merged to reconstruct a comprehensive transcriptome using perl scripts and gffcompare. Final transcriptome was analyzed using StringTie (Pertea et al., 2015) and edgeR (Robinson et al., 2010) to estimate the expression level of all transcripts. StringTie was used to perform expression level for mRNAs by calculating FPKM. The differentially expressed protein coding mRNAs were selected with fold change >1.5 and with adjusted p value <0.05 by R package edgeR. Volcano plot was generated by EnhancedVolcano (https://github.com/kevinblighe/EnhancedVolcano). Enrichment analyses were performed in EnrichR (https://maayanlab.cloud/Enrichr/) and pathfindR (Ulgen et al., 2019).
Raw mRNA read count data from human pulmonary artery endothelial cells exposed to normoxia or hypoxia (0.2% O2) for 24hrs (n = 3/condition) were retrieved from the GSE163827 dataset of GEO. Retrieved data were analyzed using DESeq2 Package in r studio and the differentially expressed mRNAs were selected with fold change >1.5 and with an adjusted p value <0.05. Finally, r studio was used to perform overlap analysis for DEGs between hypoxia and DMOG treatment.
Metabolomic profiling
To assess the effect of DMOG on HPAEC, global metabolic profiles were determined in cell pellets and supernatants derived from HPAEC grown in the presence of DMOG or vehicle. The metabolomic analysis was conducted by Metabolon, Inc (Durham, NC). Samples were prepared using the automated MicroLab STAR® system (Hamilton Company, USA). A recovery standard was added prior to the first step in the extraction process for QC purposes. For the maximum recovery of diverse metabolites, proteins were precipitated with methanol under vigorous shaking for 2 min (Glen Mills GenoGrinder 2000) followed by centrifugation. The resulting extract was divided into five fractions: one for analysis by UPLC-MS/MS with positive ion mode electrospray ionization, one for analysis by UPLC-MS/MS with negative ion mode electrospray ionization, one for analysis by UPLC-MS/MS polar platform (negative ionization), one for analysis by GC-MS, and one sample was reserved for backup. Samples were placed briefly on a TurboVap® (Zymark) to remove the organic solvent. For LC, the samples were stored overnight under nitrogen before preparation for analysis. For GC, each sample was dried under vacuum overnight before preparation for analysis. Several types of controls were analyzed in concert with the experimental samples: a pooled matrix sample generated by taking a small volume of each experimental sample served as a technical replicate throughout the data set; extracted water samples served as process blanks; and a cocktail of QC standards that were carefully chosen not to interfere with the measurement of endogenous compounds were spiked into every analyzed sample, allowing instrument performance monitoring and aiding chromatographic alignment. Instrument variability was determined by calculating the median relative standard deviation (RSD) for the standards that were added to each sample prior to injection into the mass spectrometers. Overall process variability was determined by calculating the median RSD for all endogenous metabolites (i.e., non-instrument standards) present in 100% of the pooled matrix samples.
Four major components were involved in the informatics system: Laboratory Information Management System (LIMS), the data extraction and peak-identification software, data processing tools for QC and compound identification, and a collection of information interpretation and visualization tools. LAN backbone, and a database server running Oracle 10.2.0.1 Enterprise Edition worked as the hardware and software foundation for the components of the informatics system.
Raw data were extracted, peak-identified and QC processed using Metabolon’s hardware and software. Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities based on authenticated standards that contains the retention time/index (RI), mass to charge ratio (m/z), and chromatographic data (including MS/MS spectral data) on all molecules present in the library. Furthermore, biochemical identifications are based on three criteria: retention index within a narrow RI window of the proposed identification, accurate mass match to the library +/− 0.005 amu, and the MS/MS forward and reverse scores between the experimental data and authentic standards. For quantification, peaks were quantified using area-under-the-curve and were normalized. Significantly altered metabolites (p < 0.05) were used to perform metabolite sets enrichment analysis by MetaboAnalyst (https://www.metaboanalyst.ca).
Extraction and measurement of NAD+
Cells were grown in 10 cm2 dishes. Following 24 h of treatment with DMOG or vehicle, media was aspirated, and cells were quickly rinsed with ice cold normal saline. Culture dishes were placed on dry ice and 600μL ice cold extraction buffer (40:40:20 acetonitrile/methanol/200 mM NaCl, 10 mM Tris-HCl, pH 9.2) were added directly to the cells. Cells were scraped and transferred to 1.5 mL tubes and were frozen in liquid nitrogen. Cells were subjected to two freeze thaw cycles with vortex of 15 s after each thaw. After lysis, cell lysates were centrifuged at 20,000xg for 15 min at 4°C. Metabolite containing supernatant was transferred to fresh 1.5 mL tube and protein pellets were used for protein quantification. The metabolite containing supernatants were dried down by a SpeedVac concentrator (Thermo Savant). After reconstitution in 60% acetonitrile, samples were analyzed by Thermo Fisher Scientific U3000 UHPLC equipped with Q ExactiveTM Q-Orbitrap MS.
NADH dehydrogenase (complex I) abundance
Semiquantitative measurement of NADH dehydrogenase was performed by Human NADH Dehydrogenase SimpleStep ELISA® Kit (Complex I) (abcam) following the kit protocol. Briefly, protein was isolated from cells cultured in 10 cm dishes in the presence of DMOG or vehicle for 24hrs. Samples with an equal amount of protein and standards were incubated with 50 μL of antibody cocktail in a 96 well plate for 1 h. Post incubation, the wells were washed three times with washing buffer. TBM development solution was added, and the plate was incubated for 10 min in the dark with shaking. 100 μL of Stop Solution was added to each well and the plate was read at 450 nm. A standard curve was plotted and the abundance of NADH dehydrogenase in samples was calculated.
Complex I enzyme activity assay
Mitochondrial Complex I enzyme activity was measured using Complex I Enzyme Activity Assay Kit (abcam) following manufacturer’s instructions . Briefly, after protein isolation from vehicle and DMOG-treated cells, an equal amount of protein was incubated in antibody-coated 96 well plate for 3 h at room temperature. Post incubation, the wells were washed three times with washing buffer and 200 μL assay buffer was added. The plate was read at 450 nm at room temperature for 30 min.
Quantification and statistical analysis
Statistical analyses were performed using the program “R” (http://cran.r-project.org/) or GraphPad Prism 9 (GraphPad Software, La Jolla, CA). Two-tailed t-test with Welch’s correction was used to calculate statistical significance between two groups, while for multiple comparisons statistical significance was calculated by one-way ANOVA with Sidak correction. A two-sided significance level of 5% was considered statistically significant. RNA-seq data was analyzed using the software R with package edgeR. Adjusted p values <0.05 were considered statistically significant. All the experiments were performed at least 3 times independently. Results are reported as mean ± SEM.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants R01DK115850 and R01DK132672 (P.P.K.). Metabolomics services were performed by the Metabolomics Core Facility at Robert H. Lurie Comprehensive Cancer Center of Northwestern University. We acknowledge the Northwestern University George M. O'Brien Kidney Research Core Center (NU GoKidney), an NIH/NIDDK funded program (P30 DK114857) for their core services and support. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author contributions
P.P.K. and R.T. conceived the study; R.T., N.C., and P.P.K. designed the experiments; R.T. and P.V.B. performed experiments; R.T., P.V.B., P.G., M.J.S., Y.Z., S.E.Q., N.C., and P.P.K. analyzed and interpreted data; P.G. developed and optimized the quantitative LC/MS assay for measurement of NAD+; R.T. and P.P.K. wrote the manuscript and all authors approved and commented on the manuscript.
Declaration of interests
The authors declare that they have no conflict of interest.
Published: October 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.105086.
Supplemental information
Shown are fold changes with corresponding p- and q-values. Statistically significant increased metabolites are marked in red while green is used for significantly decreased metabolites in DMOG treated group (n = 5). Data were collected using the Metabolon LC/MS and GC/MS platforms.
Data and code availability
-
•
The RNA-seq generated during this study are available on GEO: GSE189504. The RNA-seq data from HPAECs subjected to hypoxia are publicly available on GEO: GSE163827
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request
References
- Adams R.H., Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007;8:464–478. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- Alkan H.F., Walter K.E., Luengo A., Madreiter-Sokolowski C.T., Stryeck S., Lau A.N., Al-Zoughbi W., Lewis C.A., Thomas C.J., Hoefler G., et al. Cytosolic aspartate availability determines cell survival when glutamine is limiting. Cell Metabol. 2018;28:706–720.e706. doi: 10.1016/j.cmet.2018.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade J., Shi C., Costa A.S.H., Choi J., Kim J., Doddaballapur A., Sugino T., Ong Y.T., Castro M., Zimmermann B., et al. Control of endothelial quiescence by FOXO-regulated metabolites. Nat. Cell Biol. 2021;23:413–423. doi: 10.1038/s41556-021-00637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragonés J., Schneider M., Van Geyte K., Fraisl P., Dresselaers T., Mazzone M., Dirkx R., Zacchigna S., Lemieux H., Jeoung N.H., et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat. Genet. 2008;40:170–180. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- Bargiela D., Burr S.P., Chinnery P.F. Mitochondria and hypoxia: metabolic crosstalk in cell-fate decisions. Trends Endocrinol. Metab. 2018;29:249–259. doi: 10.1016/j.tem.2018.02.002. [DOI] [PubMed] [Google Scholar]
- Birsoy K., Wang T., Chen W.W., Freinkman E., Abu-Remaileh M., Sabatini D.M. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 2015;162:540–551. doi: 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P., Jain R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bock K., Georgiadou M., Schoors S., Kuchnio A., Wong B.W., Cantelmo A.R., Quaegebeur A., Ghesquiere B., Cauwenberghs S., Eelen G., et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–663. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- Diebold L.P., Gil H.J., Gao P., Martinez C.A., Weinberg S.E., Chandel N.S. Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat Metab. 2019;1:158–171. doi: 10.1038/s42255-018-0011-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diers A.R., Broniowska K.A., Chang C.F., Hogg N. Pyruvate fuels mitochondrial respiration and proliferation of breast cancer cells: effect of monocarboxylate transporter inhibition. Biochem. J. 2012;444:561–571. doi: 10.1042/BJ20120294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran R.V., MacKenzie E.D., Boulahbel H., Frezza C., Heiserich L., Tardito S., Bussolati O., Rocha S., Hall M.N., Gottlieb E. HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene. 2013;32:4549–4556. doi: 10.1038/onc.2012.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein A.C.R., Gleadle J.M., McNeill L.A., Hewitson K.S., O'Rourke J., Mole D.R., Mukherji M., Metzen E., Wilson M.I., Dhanda A., et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/S0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Falkenberg K.D., Rohlenova K., Luo Y., Carmeliet P. The metabolic engine of endothelial cells. Nat Metab. 2019;1:937–946. doi: 10.1038/s42255-019-0117-9. [DOI] [PubMed] [Google Scholar]
- Fets L., Driscoll P.C., Grimm F., Jain A., Nunes P.M., Gounis M., Doglioni G., Papageorgiou G., Ragan T.J., Campos S., et al. MCT2 mediates concentration-dependent inhibition of glutamine metabolism by MOG. Nat. Chem. Biol. 2018;14:1032–1042. doi: 10.1038/s41589-018-0136-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraisl P., Mazzone M., Schmidt T., Carmeliet P. Regulation of angiogenesis by oxygen and metabolism. Dev. Cell. 2009;16:167–179. doi: 10.1016/j.devcel.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Gaude E., Schmidt C., Gammage P.A., Dugourd A., Blacker T., Chew S.P., Saez-Rodriguez J., O'Neill J.S., Szabadkai G., Minczuk M., Frezza C. NADH shuttling couples cytosolic reductive carboxylation of glutamine with glycolysis in cells with mitochondrial dysfunction. Mol. Cell. 2018;69:581–593.e587. doi: 10.1016/j.molcel.2018.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guentsch A., Beneke A., Swain L., Farhat K., Nagarajan S., Wielockx B., Raithatha K., Dudek J., Rehling P., Zieseniss A., et al. PHD2 is a regulator for glycolytic reprogramming in macrophages. Mol. Cell Biol. 2016;37 doi: 10.1128/MCB.00236-16. e00236–00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T., Shibasaki F. Hypoxia-inducible factor as an angiogenic master switch. Front. Pediatr. 2015;3 doi: 10.3389/fped.2015.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayek I., Fischer F., Schulze-Luehrmann J., Dettmer K., Sobotta K., Schatz V., Kohl L., Boden K., Lang R., Oefner P.J., et al. Limitation of TCA cycle intermediates represents an oxygen-independent nutritional antibacterial effector mechanism of macrophages. Cell Rep. 2019;26:3502–3510.e3506. doi: 10.1016/j.celrep.2019.02.103. [DOI] [PubMed] [Google Scholar]
- Herbert S.P., Stainier D.Y. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 2011;12:551–564. doi: 10.1038/nrm3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Vandekeere S., Kalucka J., Bierhansl L., Zecchin A., Brüning U., Visnagri A., Yuldasheva N., Goveia J., Cruys B., et al. Role of glutamine and interlinked asparagine metabolism in vessel formation. EMBO J. 2017;36:2334–2352. doi: 10.15252/embj.201695518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbi M.E., Kshitiz, Gilkes D.M., Rey S., Wong C.C., Luo W., Kim D.H., Dang C.V., Levchenko A., Semenza G.L. A nontranscriptional role for HIF-1alpha as a direct inhibitor of DNA replication. Sci. Signal. 2013;6:ra10. doi: 10.1126/scisignal.2003417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kechin A., Boyarskikh U., Kel A., Filipenko M. cutPrimers: a new tool for accurate cutting of primers from reads of targeted next generation sequencing. J. Comput. Biol. 2017;24:1138–1143. doi: 10.1089/cmb.2017.0096. [DOI] [PubMed] [Google Scholar]
- Keyes B.E., Liu S., Asare A., Naik S., Levorse J., Polak L., Lu C.P., Nikolova M., Pasolli H.A., Fuchs E. Impaired epidermal to dendritic T cell signaling slows wound repair in aged skin. Cell. 2016;167:1323–1338.e1314. doi: 10.1016/j.cell.2016.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B., Li J., Jang C., Arany Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017;36:2321–2333. doi: 10.15252/embj.201796436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B., Jang C., Dharaneeswaran H., Li J., Bhide M., Yang S., Li K., Arany Z. Endothelial pyruvate kinase M2 maintains vascular integrity. J. Clin. Invest. 2018;128:4543–4556. doi: 10.1172/JCI120912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krock B.L., Skuli N., Simon M.C. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. 2011;2:1117–1133. doi: 10.1177/1947601911423654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krock B.L., Skuli N., Simon M.C. Hypoxia-induced angiogenesis: good and evil. Genes Cancer. 2011;2:1117–1133. doi: 10.1177/1947601911423654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson S., Horkoff M., Gravel C., Hoffmann T., Zuber J., Lum J.J. STAT3 regulation of citrate synthase is essential during the initiation of lymphocyte cell growth. Cell Rep. 2017;19:910–918. doi: 10.1016/j.celrep.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Maiorano D., Lutzmann M., Mechali M. MCM proteins and DNA replication. Curr. Opin. Cell Biol. 2006;18:130–136. doi: 10.1016/j.ceb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Majmundar A.J., Wong W.J., Simon M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manalo D.J., Rowan A., Lavoie T., Natarajan L., Kelly B.D., Ye S.Q., Garcia J.G., Semenza G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- Martinez-Reyes I., Chandel N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020;11:102. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita H., Morishita R., Nata T., Aoki M., Nakagami H., Taniyama Y., Yamamoto K., Higaki J., Yasufumi K., Ogihara T. Hypoxia-induced endothelial apoptosis through nuclear factor-κB (NF-κB)–Mediated bcl-2 suppression. Circ. Res. 2000;86:974–981. doi: 10.1161/01.RES.86.9.974. [DOI] [PubMed] [Google Scholar]
- Maxwell P.H., Wiesener M.S., Chang G.-W., Clifford S.C., Vaux E.C., Cockman M.E., Wykoff C.C., Pugh C.W., Maher E.R., Ratcliffe P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Mazzone M., Dettori D., de Oliveira R.L., Loges S., Schmidt T., Jonckx B., Tian Y.M., Lanahan A.A., Pollard P., de Almodovar C.R., et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–851. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y., Mansfield K.D., Bertozzi C.C., Rudenko V., Chan D.A., Giaccia A.J., Simon M.C. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell Biol. 2007;27:912–925. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M., Pertea G.M., Antonescu C.M., Chang T.C., Mendell J.T., Salzberg S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh C.W., Ratcliffe P.J. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- Quaegebeur A., Segura I., Schmieder R., Verdegem D., Decimo I., Bifari F., Dresselaers T., Eelen G., Ghosh D., Davidson S.M., et al. Deletion or inhibition of the oxygen sensor PHD1 protects against ischemic stroke via reprogramming of neuronal metabolism. Cell Metabol. 2016;23:280–291. doi: 10.1016/j.cmet.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield C.J., Ratcliffe P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- Schoors S., Bruning U., Missiaen R., Queiroz K.C., Borgers G., Elia I., Zecchin A., Cantelmo A.R., Christen S., Goveia J., et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature. 2015;520:192–197. doi: 10.1038/nature14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G.L. Vascular responses to hypoxia and ischemia. Arterioscler. Thromb. Vasc. Biol. 2010;30:648–652. doi: 10.1161/ATVBAHA.108.181644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G.L. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shreeniwas R., Ogawa S., Cozzolino F., Torcia G., Braunstein N., Butura C., Brett J., Lieberman H.B., Furie M.B., Joseph-Silverstein J., Stern D. Macrovascular and microvascular endothelium during long-term hypoxia: alterations in cell growth, monolayer permeability, and cell surface coagulant properties. J. Cell. Physiol. 1991;146:8–17. doi: 10.1002/jcp.1041460103. [DOI] [PubMed] [Google Scholar]
- Skuli N., Liu L., Runge A., Wang T., Yuan L., Patel S., Iruela-Arispe L., Simon M.C., Keith B. Endothelial deletion of hypoxia-inducible factor–2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood. 2009;114:469–477. doi: 10.1182/blood-2008-12-193581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stempien-Otero A., Karsan A., Cornejo C.J., Xiang H., Eunson T., Morrison R.S., Kay M., Winn R., Harlan J. Mechanisms of hypoxia-induced endothelial cell death: role of p53 in apoptosis. J. Biol. Chem. 1999;274:8039–8045. doi: 10.1074/jbc.274.12.8039. [DOI] [PubMed] [Google Scholar]
- Sullivan L.B., Gui D.Y., Hosios A.M., Bush L.N., Freinkman E., Vander Heiden M.G. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. 2015;162:552–563. doi: 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Ho V.C., Takeda H., Duan L.-J., Nagy A., Fong G.-H. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol. Cell. Biol. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tello D., Balsa E., Acosta-Iborra B., Fuertes-Yebra E., Elorza A., Ordonez A., Corral-Escariz M., Soro I., Lopez-Bernardo E., Perales-Clemente E., et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metabol. 2011;14:768–779. doi: 10.1016/j.cmet.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Teuwen L.-A., Geldhof V., Carmeliet P. How glucose, glutamine and fatty acid metabolism shape blood and lymph vessel development. Dev. Biol. 2019;447:90–102. doi: 10.1016/j.ydbio.2017.12.001. [DOI] [PubMed] [Google Scholar]
- Ulgen E., Ozisik O., Sezerman O.U. pathfindR: an R package for comprehensive identification of enriched pathways in omics data through active subnetworks. Front. Genet. 2019;10:858. doi: 10.3389/fgene.2019.00858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valli A., Rodriguez M., Moutsianas L., Fischer R., Fedele V., Huang H.L., Van Stiphout R., Jones D., McCarthy M., Vinaxia M., et al. Hypoxia induces a lipogenic cancer cell phenotype via HIF1alpha-dependent and -independent pathways. Oncotarget. 2015;6:1920–1941. doi: 10.18632/oncotarget.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Wang Q., Yan G., Qiao Y., Zhu B., Liu B., Tang C. Hypoxia induces lactate secretion and glycolytic efflux by downregulating mitochondrial pyruvate carrier levels in human umbilical vein endothelial cells. Mol. Med. Rep. 2018;18:1710–1717. doi: 10.3892/mmr.2018.9079. [DOI] [PubMed] [Google Scholar]
- Yoon H., Spinelli J.B., Zaganjor E., Wong S.J., German N.J., Randall E.C., Dean A., Clermont A., Paulo J.A., Garcia D., et al. PHD3 loss promotes exercise capacity and fat oxidation in skeletal muscle. Cell Metabol. 2020;32:215–228.e217. doi: 10.1016/j.cmet.2020.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirkel A., Nikolic M., Sofiadis K., Mallm J.P., Brackley C.A., Gothe H., Drechsel O., Becker C., Altmuller J., Josipovic N., et al. HMGB2 loss upon senescence entry disrupts genomic organization and induces CTCF clustering across cell types. Mol. Cell. 2018;70:730–744.e736. doi: 10.1016/j.molcel.2018.03.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Shown are fold changes with corresponding p- and q-values. Statistically significant increased metabolites are marked in red while green is used for significantly decreased metabolites in DMOG treated group (n = 5). Data were collected using the Metabolon LC/MS and GC/MS platforms.
Data Availability Statement
-
•
The RNA-seq generated during this study are available on GEO: GSE189504. The RNA-seq data from HPAECs subjected to hypoxia are publicly available on GEO: GSE163827
-
•
This paper does not report original code
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request