

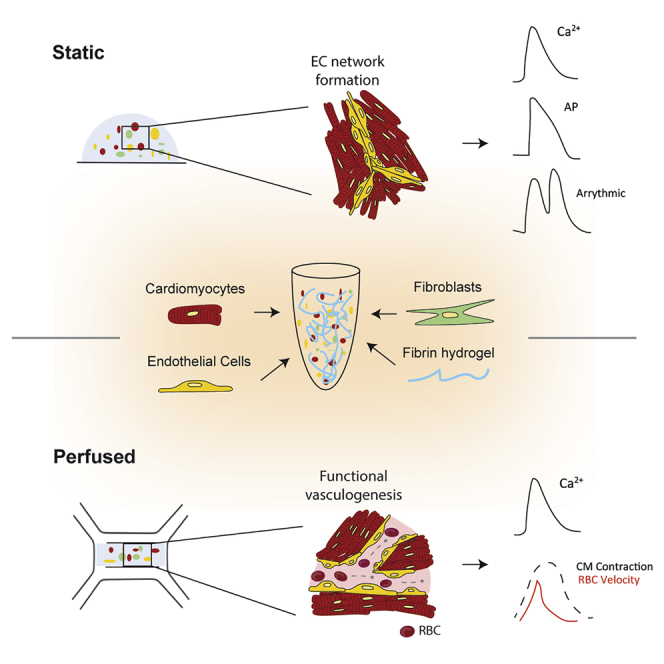
Summary
In this study, we report static and perfused models of human myocardial-microvascular interaction. In static culture, we observe distinct regulation of electrophysiology of human induced pluripotent stem cell derived-cardiomyocytes (hiPSC-CMs) in co-culture with human cardiac microvascular endothelial cells (hCMVECs) and human left ventricular fibroblasts (hLVFBs), including modification of beating rate, action potential, calcium handling, and pro-arrhythmic substrate. Within a heart-on-a-chip model, we subject this three-dimensional (3D) co-culture to microfluidic perfusion and vasculogenic growth factors to induce spontaneous assembly of perfusable myocardial microvasculature. Live imaging of red blood cells within myocardial microvasculature reveals pulsatile flow generated by beating hiPSC-CMs. This study therefore demonstrates a functionally vascularized in vitro model of human myocardium with widespread potential applications in basic and translational research.
Keywords: cardiac physiology, stem cell-derived models, microvasculature, electrophysiology, cardiomyocyte, endothelial cell, tissue engineering, E-C coupling, organ-on-chip, microphysiological systems
Graphical abstract
Highlights
-
•
Generation of perfusable microvasculature embedded in beating human myocardium
-
•
Distinct roles for endothelial cells and fibroblasts regulation of e-c coupling
-
•
Simultaneous quantification of contractility and intra-microvascular RBC velocity
Motivation
In vivo, capillaries communicate with beating myocardium to regulate heart homeostasis, with disruption of communication between microvascular endothelial cells (MVECs) and cardiomyocytes (CMs) causing transition from healthy to disease phenotype. However, much remains unknown about the molecular mechanisms related to CM-EC communication. This had led to a lack of treatment options for diseases in which the myocardial microvascular is compromised, such as cardiac microvascular dysfunction (CMD). Our understanding has been limited by a lack of in vitro models that recapitulate microvascular architectures and the contractile and hemodynamic biomechanics of the beating heart.
The microvasculature of the heart supplies nutrients required to maintain heartbeat. When these vessels can become dysfunctional, cardiac disease occurs. In this study, King et al. use human stem cells and organ-on-a-chip technology to reproduce these vessels. They then demonstrate regulation of essential aspects of heart function by microvasculature.
Introduction
The beating myocardium is one of the most densely vascularized tissues in the body, with cardiac muscle fibers being flanked by capillaries on all sides (Perbellini et al., 2018). The myocardial microvascular network maintains heart homeostasis via bidirectional exchange of nutrients and metabolites while acting as a source of paracrine cardioregulatory molecules to modulate cardiomyocyte (CM) contractility according to the body’s energetic demands (Brutsaert, 2003). Despite communication between myocardium and endothelium being fundamental to development, healthy physiology, and pathophysiology, much remains unknown about their interaction. This is evidenced by the lack of etiological understanding or treatment options for cardiac microvascular dysfunction (CMD), a prevalent phenomenon in which disruption of myocardial microvascular structure and function causes myocardial ischemia and heart failure (Berry and Duncker, 2020). These gaps in our knowledge persist due to a lack of experimental models that replicate the physiological complexity of the myocardial-microvascular microenvironment. After isolation from the myocardium, primary CMs undergo rapid remodeling and/or dedifferentiation (Nippert et al., 2017), reducing their physiological relevance and preventing their use in development of advanced in vitro models (King et al., 2021). However, the recent emergence of pluripotent stem cells as a source of human CMs with potentially unlimited in vitro viability (Karbassi et al., 2020), alongside microfluidic-based protocols for producing functional in vitro vasculature (Osaki et al., 2018; Pollet and den Toonder, 2020), has opened up new avenues for modeling the myocardial microvasculature (King et al., 2021).
Pluripotent stem cell-derived CMs (PSC-CMs) encapsulated in three-dimensional (3D) biomaterials can generate tissue-like preparations, which provide cells a more physiologically relevant biomechanical microenvironment compared with traditional cell culture techniques (Tulloch et al., 2011; Rodrigues et al., 2018). This may benefit the study of cardiac non-myocytes, such as endothelial cells (ECs) and fibroblasts (FBs), which are adept mechanosensors (Fang et al., 2019; Saucerman et al., 2019). In vivo, cardiac microvascular ECs (CMVECs) exist within a specific biomechanical context, including mechanical stimulation by beating CMs and pulsatile intra-microvascular shear stresses produced throughout the cardiac cycle (King et al., 2021). We therefore designed a 3D hydrogel-based myocardial-endothelial co-culture platform that allows direct contact between beating CMs and ECs, thereby permitting communication via mechanical interaction. To approximate biomechanics related to microvascular circulation, we further develop this co-culture by incorporating microfluidic perfusion and vasculogenic growth factor supplementation to promote organization of perfusable vessels. Both platforms demonstrate significant microvascular regulation of iPSC-CM electrophysiology, illustrating the importance of accounting for ECs when modeling human myocardium in vitro. We therefore demonstrate a model of functionally vascularized human myocardium with potential applications in basic and translational cardiac experimentation.
Results
3D hydrogel co-culture enables biomimetic mechanical communication between myocardium and endothelium
3D hydrogel scaffolds promote physiologically relevant cell behaviors due to their biomimetic mechanical and biochemical properties (Blache and Ehrbar, 2018). We utilized a fibrin hydrogel scaffold for two complementary purposes: to facilitate transmission of mechanical stimuli generated by beating iPSC-CMs to non-myocytes and to permit 3D mobility and remodeling necessary for spontaneous vasculogenesis. In vivo, fibrin is the primary constituent of blood clots and serves as the platform for ECs to conduct wound healing and vascular remodeling (Morin and Tranquillo, 2013). It is therefore well suited to our application of promoting myocardial vasculogenesis. 3D culture of induced PSC (iPSC)-CMs in fibrin (and other hydrogels) has also become a gold standard of measuring contractility in cardiac tissue engineering, as it enables production of 3D tissue-like cultures that shorten anisotropically via synchronized contractility (Eder et al., 2016; Ronaldson-Bouchard et al., 2018). For this study, we have encapsulated hiPSC-CMs, hCMVECs, and human left ventricular FBs (hLVFBs) at physiologically relevant ratios (1:1:0.1) (Pinto et al., 2016), in 5 mg/mL fibrin (Figure 1A), with cell density optimized toward 3D confluence after 1 week co-culture (Figure S1). Immunofluorescent microscopy reveals biomimetic physical association between CMs, ECs, and FBs, with ECs displaying spontaneous organization into connected networks embedded within human iPSC (hiPSC)-CMs (Figure 1B). We then used live imaging of human umbilical vein ECs expressing red fluorescent protein (RFP-HUVEC) to visualize mechanical interaction between beating CMs and ECs (Figure S1B; Video S1). Heterocellular mechanical interaction was further demonstrated using the myosin inhibitor blebbistatin, which supresses CM contractility and caused a corresponding and statistically significant reduction in EC displacement after 5 min (Figure S1C). We also observed mechanical stimulation of EC via modulation of CM beating rate through electrical field stimulation, with endothelial displacement mirroring CM beating frequency at rates ranging from 0.75 to 2 Hz (Figure S1D). As such, 3D hydrogel co-culture mimics the native physical association between myocardium and microvascular endothelium in that every time CMs beat, ECs experience mechanical stimuli.
Figure 1.

Endothelial and fibroblast 3D co-culture modulates iPSC-CM excitation-contraction coupling and reduces pro-arrhythmic substrate
(A) Schematic of cellular encapsulation of hiPSC-CMs, hCMVECs, and hLVFBs in fibrin hydrogel.
(B) Physical cell-cell association between CMs (actin (α-sarcomeric)), ECs (CD31), and FBs (vimentin) after 1 week of co-culture. Scale bar: 50 μm.
(C) Representative traces of action potential morphology in spontaneous co-cultures measured by sharp microelectrode.
(D and E) Spontaneous beating rate.
(E) Minimum diastolic potential.
(F) Membrane potential at threshold of action potential upstroke.
(G) Diastolic depolarization (membrane potential depolarization between minimum and threshold potentials).
(H) 50% of action potential duration.
(I) 90% of action potential duration.
(J) Maximum rate of action potential upstroke.
(K) Maximum rate of action potential repolarization.
(L) Representative traces of iPSC-CM Ca2+ handling.
(M) Calcium transient amplitude (F1/F0).
(N) Time to calcium transient peak.
(O) Time to 80% decay of calcium transient.
(P) Example traces of rhythmic versus arrhythmic iPSC-CMs during 1 Hz electrical stimulation.
(Q) iPSC-CM arrhythmogenesis while electrically simulating at 1 Hz.
(R) iPSC-CM arrhythmogenesis while electrically simulating at 1 Hz and 1 μm isoprenaline treatment.
(S) iPSC-CM latency after high-frequency pacing protocol.
Data are shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Each datapoint represents 1 cell, 3 beats/cell, N = 5.
Live imaging of hiPSC-CM-GCaMP (Green) and RFP-HUVEC (Orange) demonstrates direct cell-cell contact and endothelial displacement during the cardiac cycle after 7 days co-culture.
ECs and FBs exert distinct modulation of iPSC-CM electrophysiology in mechanically biomimetic 3D co-culture
To assess the effect of mechanically biomimetic heterocellular co-culture on CM function, we characterized hiPSC-CM excitation-contraction (e-c) coupling in co-culture with ECs alone, FBs alone, and ECs and FBs together (Figures 1C–1S). FBs significantly alter iPSC-CM electrophysiology (Kane and Terracciano, 2018; Goldfracht et al., 2019; Beauchamp et al., 2020; Giacomelli et al., 2020), meaning that their inclusion would help to delineate endothelial-specific cardioregulation. By impaling hiPSC-CMs with sharp microelectrodes to quantify membrane potential (Figure 1C), we observed that EC co-culture significantly raised minimum diastolic potential (Figure 1E), threshold potential (Figure 1F), and diastolic depolarization of iPSC-CMs (Figure 1G). In contrast, FB co-culture demonstrated reduced diastolic depolarization compared with CM-only control (Figure 1G).
A notable difference between iPSC-derived and primary adult atrial or ventricular CMs is automaticity: in vivo, only the CMs of the sino-atrial node beat spontaneously (Irisawa et al., 1993). We observed the spontaneous beating rate of iPSC-CMs to be significantly reduced from an average of 73 to 48 beats per minute (BPM) in CM-EC and further reduced to 35 BPM in CM-EC-FB (Figure 1G). Despite reduction in beating rate, heterocellularity induced no changes in action potential (AP) kinetics, including rate of upstroke (Figure 1H), rate of repolarization (Figure 1I), and spontaneous AP duration (APD) (Figures 1H–1J) compared with CM-only control. However, differences in APD were observed between CM-EC and CM-FB co-cultures, with FB co-culture inducing a shorter duration.
To examine the contribution of heterocellularity to CM calcium handling, CMs differentiated from an hiPSC line, which has been genetically modified to express the calcium indicator GCaMP6f (Huebsch et al., 2015), were used in co-culture (Figure 1L). Amplitude of iPSC-CM calcium transients was increased in CM-FB and CM-EC-FB co-cultures (Figure 1M). Compared with CM-only control, calcium transient time to peak was prolonged in CM-EC, abbreviated in CM-FB, and unchanged in CM-EC-FB (Figure 1N). With respect to calcium transient relaxation kinetics, both CM-EC and CM-EC-FB conditions induced prolonged transients, while CM-FB was unchanged compared with control (Figure 1O). We then investigated whether changes in iPSC-CM electrophysiology would confer any differences in pro-arrhythmic substrate, as quantified by inability of a recorded cell to follow a 1 Hz field stimulus (Figure 1P). We observed reduced incidence of pro-arrhythmic substrate under 1 Hz field stimulation in both CM-EC and CM-EC-FB conditions (Figure 1Q). However, under arrhythmia provocation with 1 μM isoproterenol, reduced arrhythmia was maintained only in CM-EC co-culture Figure 1R). After further arrhythmia provocation using a high-frequency pacing protocol, CM-EC-FB demonstrated prolonged latency after cessation of stimulation compared with CM-only control (Figure 1S).
To determine whether direct physical contact was important in cardiac heterocellular communication, we performed a 3D co-culture in which each cell type was cultured in an isolated hydrogel but within the same cell culture dish (Figure 2A). We observed that non-myocyte regulation of spontaneous beating rate (Figure 2B), Ca2+ transient amplitude (Figure 2C), and Ca2+ transient kinetics (Figures 2D and 2E) were lost when cells were physically separated. These data suggest the requirement of direct physical cell-cell association to enable biomimetic heterocellular communication in vitro. Taken together, these results indicate that ECs and FBs exert significant and distinct regulatory influence on hiPSC-CM electrophysiology. Given the physiological relevance of CM-EC-FB co-culture, and the beneficial functional effects described above, this condition was brought forward to subsequent heart-on-a-chip experiments.
Figure 2.

Direct contact co-culture is required for non-myocyte regulation of iPSC-CM electrophysiology
(A) Experimental outline showing combined encapsulation of all cells (direct contact) or separation of each cell type (indirect contact).
(B) Quantification of iPSC-CM spontaneous beating rate.
(C) iPSC-CM calcium transient amplitude.
(D) iPSC-CM calcium transient time to peak.
(E) iPSC-CM calcium transient time to 80% decay.
Data are shown as mean ± SEM. Each datapoint represents 1 dish, containing average of 3 cells, 3 beats/cell, N = 3.
Generation of perfusable myocardial microvasculature in microfluidic heart on a chip
All ECs in the body are polarized, with a luminal surface exposed to the circulation and an abluminal side in contact with extracellular matrix and parenchymal cells (Fang et al., 2019). Biomechanical conditions within the beating heart produce pulsatile blood flow of varying magnitudes within the myocardial capillaries (Ashikawa et al., 1986). However, the consequence of fluctuating hemodynamics on CM and EC biology is largely unknown, as experimental models that reproduce this dynamic have not yet been described. To approximate myocardial microvascular flow dynamics in vitro, we have developed a microfluidic heart on a chip, which allows exposure of 3D hydrogel-based co-cultures to media perfusion (Figure 2A). Within our microfluidic heart-on-a-chip platform, generation of functional, perfusable cardiac microvasculature was first validated in the absence of CMs via co-culture of HUVECs and hLVFBs (Figure S2). To promote spontaneous vasculogenesis, we supplemented culture media with pro-vasculogenic growth factors VEGF and ANG-1 (Figure 3C), which have previously been demonstrated to induce formation of functional microvasculature in vitro (Jeon et al., 2014). Confocal microscopy confirmed the presence of a connected endothelial network with open lumens after 5 days of co-culture (Figure S2). We then visualized the flow within the microvasculature via perfusion of 40 kDa fluorescein isothiocyanate (FITC)-dextran (Figure S2) and via live imaging of red blood cells (RBCs) through microvasculature (Video S2), confirming the presence of a tight, non-leaky endothelium. Induction of myocardial microvascularization within the previously described beating, 3D myocardial co-culture was demonstrated via presence of hCMVEC vessels with open lumens, confirming the functional vasculogenic potential of ECs sourced from human heart tissue (Figures 3E and 3F; Video S3). Myocardial microvasculature was then shown to be functionally perfusable via addition of RBCs to the perfusing solution (Figures 3F; Video S4). To assess the effect of vascularization on iPSC-CM function, we performed optical mapping for Ca2+ cycling while field stimulating cultures at 1 Hz (Figure 3G). Vascularization of hiPSC-derived myocardium was associated with abbreviation of both time to peak (Figure 3I) and time to 50% relaxation (Figure 3H) of the Ca2+ transient. The effect of perfusion versus static culture was also investigated in vascularized cultures, with perfusion increasing Ca2+ transient amplitude (Figure 3H) and abbreviating time to peak (Figure 3H). As such, we observe that by providing appropriate environmental conditions, i.e., biomimetic mechanical substrate and fluid flow, it is possible to induce assembly of functionally perfusable microvasculature in beating myocardium, with consequent modulation of iPSC-CM electrophysiology.
Figure 3.

Generation of perfusable myocardial microvasculature via microfluidic and vasculogenic culture
(A) Schematic of heart-on-a-chip (i) layout, (ii) design, (iii) cell seeding, and (iv) connection to perfusion.
(B) Representative image of microfluidic chip connected to syringe for perfusion (tubing shortened for display).
(C) Vasculogenic culture protocol. Ri, Rho/ROCK inhibitor; VEGF, vascular endothelial growth factor; Ang-1, angiopoietin 1.
(D) Representative image of cellular distribution in microfluidic chips (EC = RFP-HUVEC). Scale bar: 1 mm.
(E) Confocal z stack demonstrating myocardial microvasculature with open lumen. White asterisk represents open luminal space (EC = hCMVEC). Scale bar: 50 μm (Video S3).
(F) Live-cell image of perfused erythrocyte in myocardial microvasculature lumen (EC = RFP-HUVEC). Scale bar: 50 μm (Video S4).
(G) Representative Ca2+ transient traces in heart on a chip under vascularized and/or perfused conditions.
(H–J) Quantification of Ca2+ transient (H) amplitude and (I and J) kinetics under vascularized and perfused conditions.
Data are shown as mean ± SEM. Each datapoint represents 1 chip, containing average of 3 regions of interest (ROIs; 5–15 cells), 3 beats/ROI.
Spontaneous vasculogenesis was induced using RFP-HUVEC (Orange) and hLVFB. Red blood cells were perfused through microvasculature to demonstrate vessel functionality.
Myocardial microvasculature was established in a Heart-on-a-chip model after 5 days vasculogenic co-culture with HCMEC (CD31, Yellow) and iPSC-CM (Alpha-Actinin, Red).
Functionality of microvasculature in Heart-on-a-chip was demonstrated via perfusion of red blood cells. Co-culture was performed with RFP-HUVEC (orange) and iPSC-CM-GCaMP (Green).
Beating iPSC-CMs generate pulsatile intra-microvascular flow profile
Epimyocardial microvascular flow profiles have been observed in animal models as early as 1972 using open chest transillumination microscopy (Hellberg et al., 1972), revealing highly pulsatile flow during the cardiac cycle. However, due to technical complexity and limitation to animal epimyocardium, this technique has not been widely adopted, and the relationship between contractility and microvascular flow remains poorly understood. We therefore hypothesized that our vascularized heart-on-a-chip cultures would serve as a platform for studying human myocardial microvascular flow dynamics. After inducing microvascularization within a beating culture over 5 days, we introduced RBCs to the media and acquired bright-field time series images at 100 fps. By selecting a field that includes both an open lumen and contracting hiPSC-CMs, we could quantify RBC velocity and myocyte contractility simultaneously (Figure 4; Video S5). This approach reveals pulsatile intra-microvascular RBC velocity over the course of the iPSC-CM contractile cycle, mirroring the pulsatility observed by early transillumination microscopy studies (Hellberg et al., 1972; Ashikawa et al., 1986). By supporting the propagation of mechanical forces through 3D matrix, along with microfluidic perfusion and high spatial and temporal resolution microscopy, this approach illustrates an avenue for the study of myocardial contribution to microvascular flow, thereby opening the door to experimental interrogation of this relationship in a human platform.
Figure 4.

Beating iPSC-CMs induce pulsatile intra-microvascular flow profile
Simultaneous recording and quantification of iPSC-CM contractility (via MUSCLEMOTION [Sala et al., 2018]) and erythrocyte velocity (via TRACKMATE [Tinevez et al., 2017]) reveals surge in erythrocyte velocity as iPSC-CMs contract (Video S5).
Red blood cell velocity through myocardial microvasculature was quantified using TrackMate Fiji/ImageJ plugin.
Discussion
In this study, we have described 3D models of the human myocardium that demonstrate the capacity of microvascular ECs to fundamentally alter myocardial function in vitro, including regulation of spontaneous beating rate, membrane potential, calcium handling, and pro-arrhythmic substrate (Figure 1). We also report a method of promoting vasculogenesis in 3D co-culture via incorporation of microfluidic perfusion and growth factor supplementation, resulting in functionally perfusable myocardial microvasculature (Figure 3). The data presented within this study build on an increasing body of research that demonstrates that iPSC-CMs are highly amenable to formation of 3D, tissue-like in vitro models, which in turn allows inclusion of ECs and other non-myocytes to approximate the native heterocellularity, cellular organization, and biomechanics of myocardial tissue.
Non-myocytes as fundamental regulators of iPSC-CM electrophysiology
Inclusion of cardiac non-myocytes in in vitro studies such as disease modeling may be essential to understand disease states that pivot around specific myocardial cells, such as CMD. A key takeaway from the electrophysiology data presented in this study is the distinct cardioregulatory roles for ECs and FBs. The EC-specific modulation of iPSC-CM function includes higher minimum diastolic (Figure 1E) and threshold (Figure 1F) potentials, increased diastolic depolarization (Figure 1G), prolonged calcium transient kinetics (Figures 1N and 1O), and reduced pro-arrhythmic substrate (Figures 1Q and 1R). Meanwhile FB-specific changes include reduced diastolic depolarization (Figure 1G), increased calcium transient amplitude (Figure 1M) and calcium influx kinetics Figure 1N). In vivo, ventricular CMs exhibit minimum diastolic potential closer to −70 mV, with little to no diastolic depolarization (Kane and Terracciano, 2017). EC depolarization of iPSC-CM membrane potential in our model brings the CMs closer to a sino-atrial node-like phenotype, which exhibits minimum diastolic potential closer to −50 mV and characteristic spontaneous diastolic depolarization (Kane and Terracciano, 2017). Interestingly, this shift is prevented when FBs are also added to co-culture, with no differences in membrane potential parameters being observed with respect to CM-only control in CM-EC-FB co-culture (Figures 1D–1F). As such, the inclusion of FBs may help maintain a more broadly relevant polarized membrane potential in hiPSC-CMs.
Despite this shift, EC co-culture reduces, rather than elevates, rates of automaticity. An explanation for this observation would require focused investigation into the electrophysiological machinery involved in coordinating both mechanisms of automaticity, i.e., the calcium and voltage clocks (Kim et al., 2015; Yaniv et al., 2015).
An interesting observation from the present study was that some, but not all, non-myocyte-specific phenotypes are present in CM-EC-FB tri-culture. In tri-culture, FB-associated effects dominate when observing membrane potentials, calcium transient amplitude, and calcium influx kinetics, whereas EC-associated effects dominate when observing calcium efflux kinetics and pro-arrhythmic substrate under paced conditions. Additionally, some behaviors are only evident when both non-myocytes are present, such as pronounced reduction of spontaneous beating rate (Figure 1D) and prolonged latency after high-frequency pacing (Figure 1S). These data build on previous studies that emphasize the importance distinct myocyte-non-myocyte interactions in iPSC-derived models, such as the demonstration by Giacomelli et al. of combined iPSC-EC and iPSC-FB co-culture promoting a mature ultrastructure, e-c coupling, and metabolism of iPSC-CMs (Giacomelli et al., 2020) and a study by Ravenscroft et al., which reports the requirement of both primary cardiac ECs and FBs for a mature pharmacological response in embryonic stem cell-derived CMs (ESC-CMs) (Ravenscroft et al., 2016).
An important question within the field of cardiac cell biology and electrophysiology is the extent of electrotonic coupling between CMs and non-myocytes via gap junctional connexins (Vasquez et al., 2011). As non-myocytes account for the majority of cells in the human myocardium (Pinto et al., 2016), and as their behavior is dramatically altered during disease-associated myocardial remodeling (Lim et al., 2015; Mayourian et al., 2018), any potential contribution to homeostasis via electrical continuity with CMs is important to identify. Historical focus has been placed on assessing the extent of CM-FB electrical interaction (Ongstad and Kohl, 2016), with in vitro studies demonstrating CM membrane potential modification in contact with FBs, providing evidence for an active role of FBs in myocardial electrophysiology (Rook et al., 1989; Kohl et al., 1994; Nagaraju et al., 2019). On the other hand, electrotonic coupling between ECs and CMs is not well studied, despite evidence of functional smooth muscle-endothelial gap junctions having been already demonstrated (Fleming, 2000). While this study does not specifically investigate myocyte-non-myocyte electrotonic coupling, our data demonstrate that both FBs and ECs significantly modulate iPSC-CM membrane potential, providing further incentive to investigate the potential CM-EC coupling.
Reproducing the myocardial microvascular microenvironment
Along with the importance of heterocellularity, this study also demonstrates the role of microenvironmental culture conditions in the regulation of myocardial function, as different iPSC-CM calcium-handling phenotypes were observed under static co-culture (Figures 1L and 1M), static co-culture with vasculogenic media (Figures 2G–2J), and perfused co-culture with vasculogenic media (Figures 2G–2J). While stem cell-derived models of the heart have grown dramatically in recent years, these data emphasize the need to pursue physiological complexity, i.e., biomechanics and heterocellularity, to maximize the relevance of human in vitro systems. 3D static “cardiac tissue mimetics” composed of neonatal rat ventricular (NRMV) CMs and HUVECs were recently used to elucidate vasculogenic signaling mechanisms via characterization of endothelial network organization (Wagner et al., 2020). Meanwhile, other studies have created functional vascular structures, with both microfluidic and bioprinted models demonstrating perfusable myocardium with either acellular vascular channels (Xiao et al., 2014; Skylar-Scott et al., 2019) or a living endothelium (Sakaguchi et al., 2013; Zhang et al., 2016; Ellis et al., 2017; Noor et al., 2019). However, the approach demonstrated here couples beating CMs in direct contact with perfusable, living microvasculature while allowing cellular resolution microscopy for quantification of electrophysiology, such as calcium transients and contractility, and physiological phenomena, such as CM contractile contribution to blood flow. In vivo, myocardial capillaries have an average lumen diameter of ∼5 μm (Kassab and Fung, 1994) and are embedded in the mechanically dynamic beating myocardium, making their study and the visualization of microvasculature flow prohibitively difficult. Within our model of de novo human myocardial microvascular formation, CM contribution to pulsatile and dynamic intra-microvascular flow is apparent and easily quantified with high temporal resolution (Figure 4). This approximation of a biologically and mechanically complex microenvironment represents an avenue to study human cardiac muscle, the myocardial microvascular endothelium, microvascular flow dynamics, and/or the interaction between these distinct components.
This work validates the capacity of primary human CMVECs and primary human ventricular FBs to form functional vasculature, both in the absence (Figure S2) and presence (Figures 3 and 4) of CMs. In the context of representative in vitro vascular models, the cell source is becoming increasingly recognized as an important determinant of physiologically relevant in vitro behavior, with ECs isolated from heart, lung, liver, and kidney having recently been shown to retain tissue-specific function for multiple passages in vitro (Marcu et al., 2018). As such, the list of organ/tissue-specific ECs validated for spontaneous microvascular formation in tissue-like in vitro microfluidic models is growing (Ewald et al., 2021).
A platform that includes a perfusable endothelium embedded in contractile myocardium could be valuable to a wide array of in vitro experimental cardiology. For example, during development, shear-stress-dependent EC-CM communication is a critical regulator of heathy myocardial morphogenesis(McCormick and Tzima, 2016; Garoffolo and Pesce, 2019), yet very little is known about how shear stress modulates EC-CM interaction in the healthy or diseased adult heart (King et al., 2021). Although this study falls short of shear stress quantification in myocardial microvasculature, the demonstration of high temporal and spatial resolution for live RBC imaging in perfusable myocardial microvasculature may be compatible with existing methods of approximating wall shear stress via particle tracking velocimetry (Ha et al., 2012). Focused investigation on the relationship between endothelial mechanosensing and cardiac contractility may shed light on poorly understood disease states such as CMD, where impeded microvascular flow and impaired contractility co-exist (Berry and Duncker, 2020) and for which human models are not available (Sorop et al., 2020).
In summary, this study emphasizes the importance of microenvironmental conditions in the myocardium, including multicellularity and biomechanics. We demonstrate significant and distinct regulation of iPSC-CM electrophysiology via 3D co-culture with CMVECs and FBs. We then report spontaneous assembly of a perfusable microvascular network within beating myocardium via microfluidic perfusion in a heart-on-a-chip model, allowing visualization of the interaction between myocardial contractility and microvascular flow dynamics in a fully human model.
Limitations of the study
Both the static and perfused models of human myocardium presented in this study have inherent limitations. In 3D multicellular preparations, the electrophysiological syncytium prohibits isolation of the contribution of specific electrophysiological mechanisms, e.g., ion channels and transporters, via the traditional patch-clamp approach. As such, our observations on non-myocyte regulation of iPSC-CM e-c coupling did not extend to mechanistic interrogation of individual components of the e-c coupling apparatus. In a 3D multicellular platform, this would best be approached via chemical interrogation; for example, a potential next step to study non-myocyte regulation of automaticity would be selective inhibition of key automaticity components, such as ivabradine to inhibit the cardiac pacemaker channel HCN4 (Chobanyan-Jürgens et al., 2018), ORM to inhibit the sodium-calcium exchanger (Jost et al., 2013), or barium to inhibit the inward rectifying potassium channel (Imoto et al., 1987).
Another difficulty associated with multicellular models is control of cell-type distribution. As CMs, ECs, and FB cells are mixed together before seeding and undergo spontaneous organization during culture, proximity of a given CM relative to non-myocardial cells is not known when performing electrophysiological experiments. In the present study, we assume that CMs and non-myocytes are equally distributed when acquiring APs. However, it is likely that variability in terms of connections and proximity of myocytes and non-myocytes is a significant determinant of electrophysiological phenotype.
In the perfusable model demonstrated in this study, the key limitation is lack of characterization of microvascular structure, i.e., network distribution, extent of vascularization, lumen sizes, timeline of vasculogenesis, etc. The aim of the present study was to establish models of the human myocardium that would be useful to probe the nature of non-myocyte regulation of myocardial function. However, characterization of the process of vascularization and the nature of the final vascular network would greatly enhance the value of the platform. As vessel architectures within this model are ultimately determined by spontaneous cellular behavior, the result is a morphologically diverse vascular network containing vessels with lumen sizes in the order of both capillaries (<10 μm) and arterioles (>50 μm) (Video S6). Many factors influence vessel architecture, including (but not limited to) hemodynamics (Baeyens et al., 2015; Kim et al., 2016), quantity and timing of growth factor supplementation (Shin et al., 2011; Jeon et al., 2014), extracellular matrix stiffness (Mason et al., 2013), density (Ghajar et al., 2008), and parenchymal cell population (Newman et al., 2011), and study of the influence of these parameters and a thorough characterization of microvascular morphological diversity is an obvious next step.
Demonstration of variability in vascular network via live imaging of red blood cell flow through 8 and 80 μm lumens.
Another parameter that has not been characterized in detail in this study but that would add valuable insight is the persistence of the microvascular network. Here, we adopt a previously reported VEGF- and Ang-1-mediated vasculogensis protocol, initially reported by Jeon et al. to induce formation of a stand-alone microvascular network (Jeon et al., 2014) and subsequently in a tumor environment (Jeon et al., 2015), which reports functional vasculature for up to 6 days. While we demonstrate the use of our model to study electrophysiological phenomena, which occur on millisecond timescales, evaluation of the stability of myocardial microvasculature over time would determine whether it would be suitable for the study of biological processes that occur over longer timescales, such as EC-mediated myocardial remodeling and plasticity (Tirziu and Simons, 2009).
Additionally, an important limitation of this model is the lack of control of flow direction through the vascular network. Media are perfused in the channels flanking (parallel to) the central 3D culture channel, meaning perfusion through the cell culture and embedded microvasculature occurs passively, i.e., flow is not oriented perpendicular to the central cell culture; flow instead enters the central region as the entire interior of the chip is perfused. For this reason, variability in flow dynamics and vessel orientation are present. As such, this model is likely best suited to the evaluation of local events on discreet sections of myocardial microvasculature, as demonstrated in Figure 4, where variability in vessel lumen size and orientation can be identified and accounted for.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CD31 | Abcam | ab32457 |
| Chicken polyclonal anti-Vimentin | Invitrogen | pa1-10003 |
| Mouse monoclonal anti-Alpha-Actinin | Sigma | a7811 |
| Biological samples | ||
| Human Left Ventricular Tissue | Tissue Donation | ICHTB HTA license: 12275, REC Approval 17/WA/0161 |
| Chemicals, peptides, and recombinant proteins | ||
| VEGF | Peprotech | 100-20 |
| Ang-1 | Peprotech | 130- 06 |
| Experimental models: Cell lines | ||
| GCaMP6f iPSC | Conklin Lab | Huebsch et al., 2015 |
| RFP-HUVEC | Angio-Proteomie | cAP-0001 |
| HCMEC | Promocell | (C-12285) |
| Software and algorithms | ||
| Musclemotion | Fiji Plugin | Sala et al., 2018 |
| Trackmate | Fiji Plugin | Tinevez et al., 2017 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Cesare Terracciano (c.terracciano@imperial.ac.uk).
Materials availability
This Study did not generate new unique reagents.
Experimental model and subject details
iPSC-CM differentiation and maintenance
All biological procedures were carried out under sterile conditions in a Biomat 2 class II microbial safety cabinet. Cells were incubated in an incubator (Sanyo, MCO-5M) at 37°C and 5% CO2. The iPSC line used for this project was human, male, GCaMP6f-expressing line genetically modified and provided to by the Conklin Lab, Gladstone Institute USA (GM25256, Coriell Institute). Differentiation of iPSC to CM was performed via previously described modulation of Wnt/beta-catenin pathway modulation(Lian et al., 2012), with previously reported high purity (>95%) (Jabbour et al., 2021). In brief, hiPSCs were cultured in Essential 8 Medium (Thermofisher, A1517001) on Matrigel (Corning, 356,231) coated 6 well plates until 90–100% confluence. Differentiation was initiated via Wnt activation using 6 μM CHIR99021 (R&D system, 4423) in RPMI 1640 (Thermofisher, R8758) with B27 supplement minus insulin (Thermofisher, A1895601) (RB-) for 48 h, followed by RB- for 24 h. Wnt inactivation was achieved via 48 h of 2.5 μM C59 (Tocris, 5148) in RB-. Differentiation media was replaced every 48 h, and transition to B27 supplement containing insulin (Thermofisher, 17504044) was done after onset of beating. On day 11 after Wnt activation, metabolic selection was performed by removing glucose from differentiation media for 96 h (Thermo Fisher Scientific, 11879020). Purified cardiomyocytes were dissociated for 3 hours at 37°C using 200 U/mL collagenase type II in Hanks Balanced Salt Solution without calcium or magnesium (HBSS−/−, Thermofisher, 14175095) plus 5 μM Y-27632 RHO/ROCK inhibitor (Ri) (Stemcell Technologies, 72302) and were replated at a density of 0.3 x 106 cells/cm2 in RPMI1604 + B27 + 10% fetal bovine serum (FBS) (Thermofisher, 10500064) + 5 μM Ri on Matrigel coated plates. Every 2–3 days afterwards cells were fed with RPMI/B27 until at least day 45, when they were used for experiments.
Endothelial cell culture
In this study hCMVEC were used for all co-culture experiments which report quantification of iPSC-CM electrophysiology, and fixed immunofluorescence microscopy. hCMVEC were purchased from Promocell (C-12285) and maintained in microvascular growth medium (Promocell, C-22120). Visualisation of EC in live imaging co-culture experiments was performed using RFP-HUVEC, which were purchased from Angio-Proteomie and maintained in endothelial growth medium (Promocell, C-22111). For clarity, all experiments using RFP-HUVEC are labeled such in figures and figure legends. All EC were grown on 1% gelatin until 90% confluence before 1:3 passaging. For experiments, passages 5–7 were used.
Fibroblast isolation and culture
Human left ventricular fibroblasts were isolated in-house from donated human hearts via explant technique. (ICHTB HTA licence: 12275, REC Approval 17/WA/0161). Male and female donors between the ages of 25 and 65 were used for this study. In brief, tissue culture plastic Petri dishes were coated with 5 μg/mL fibronectin (Sigma, F1141) at 37°C for 30 min. Left ventricular free wall samples were washed in sterile PBS +5% P/S, before dissection into 1–5 mm3 explants. Explants were then washed in 0.05% Trypsin-EDTA for 2 min, before quenching in 20% FBS in DMEM. Explants were then transferred to fibronectin-coated dishes along with 0.5 mL fibroblast maintenance media (DMEM +10% FBS, 1% P/S and 1% L-glutamine) and were allowed to adhere to dishes for 1 h at 37°C. Maintenance media was then topped up, and replaced every 2-3 days to allow fibroblast outgrowth. When fibroblasts reached 70–100% confluence they were washed 2X with sterile PBS, then detached with 0.05% Trypsin-EDTA for 5 min at 37°C, before transferring as passage 1 to tissue culture flasks. Passage 2–3 were used for all experiments.
Method details
Design and microfabrication of the heart-on-a-chip platform
The microfluidic chip was designed and built to perfuse cell culture medium to feed 3D microtissues. It has an overall footprint of 2 mm by 3 mm and a height of 100 μm. Two arrays of triangular micropillars define the central region representing the cell culture compartment (1000 μm wide) which is flanked by 2 side perfusion compartments. The gap between micropillars is 100 μm to allow cell-laden gel confinement during injection (Huang et al., 2009; Ugolini et al., 2018). The inlet leads to a binary tree manifold that guarantees equal hydraulic resistance in all branches. Two dedicated wells at the end of the cell culture chamber facilitate field stimulation of cultures for functional experiments.
The device layout was drawn using computer-assisted design (CAD) software (AutoCAD, Autodesk Inc.). The corresponding photomask for photolithography was printed at high resolution (64,000 DPI) on a polyester film (JD Photodata, UK). The mold was produced on silicon wafers by photolithography techniques. In detail, a 4″ silicon wafer (100 mm diameter, 0-100 Ω-cm, 500 μm thickness, test grade, University wafer) was treated with a thin film of Ti-Prime adhesion promoter (Micro-Chemicals, GmbH). SU8-2050 negative photoresist (Micro-Chemicals, GmbH) was spin-coated according to supplier’s recommendation to a 100μm thick layer, followed by soft baking. The high-resolution transparency mask containing the Heart-on-a-chip design was aligned on the wafer and the negative photoresist was exposed to 200 mJ/cm2 of collimated UV light. The photoresist underwent postexposure bake and was developed in SU8 Developer (Micro-Chemicals GmbH). Finally, it was hard-baked for 2 min at 120°C.
PDMS-based Heart-on-a-chip device was produced by soft lithography. A two-part PDMS kit (Sylgard 184, Dow Corning) was used to create the liquid polymer. Uncured liquid PDMS (10:1 elastomer to crosslinker ratio) was poured onto the mold to a thickness of 5mm. The cell culture chamber was peeled off and access points were created for inlet and outlets. Subsequently, it was bonded to a glass no. 1.5 coverslip (VWR, 631-0138) by oxygen plasma bonding at 1 mBar 100 W for 1 min (Harrick Plasma Inc), followed by 2 h bonding at 60°C. The final assembled device was further cured at 80°C for 15 min to finalize the bonding process. The ports to access the inlet and control channels permit the connection of 22-gauge stainless steel couplers (Instech Laboratories, SC22/15), which fit tightly into both the ports on the chip and the inside of the tubing.
Hydrogel formation and cell culture protocol
For 3D co-culture, cells were encapsulated in fibrin hydrogels at a ratio of 1:1:0.1 of hiPSC-CM: hCMEC: FB. In static culture, unless otherwise stated CM density was maintained constant at 7.5×106 cells/ml across all conditions, with additional cells being added as per co-culture condition. In Heart-on-a-chip experiments, where cell seeding through tight microfluidic channels results in significant cell loss, cell suspensions of 100×106 cells/ml were found to result in 3D confluence in the final culture. Polymerisation of fibrin hydrogels was achieved via addition of 1 U/mL of thrombin (Sigma, T7572-250UN) to cell suspension containing 5 mg/mL fibrinogen (Sigma-Aldrich, F8630) immediately before seeding, then incubating at 37°C for 10 min before fully hydrating with media. Co-culture media is a 1:1 mixture of 50% microvascular endothelial growth media (Promocell, C-22022) and iPSC-CM maintenance media, which consisted of high glucose DMEM (Thermofisher, 41966029), 10% FBS, 1% penicillin/streptomycin (Sigma, P0781), 0.1% human insulin solution (Sigma-Aldrich, I9278) and 0.1% aprotinin from bovine lung (Sigma-Aldrich, A1153). 5 μM Ri was added for the first 24 h of culture. For static experiments, co-culture media was changed every 2 days, with experiments conducted between day 6 and 8. Heart-on-a-chip vasculogenic culture protocol was adapted from Jeon et al (Jeon et al., 2014). Co-culture was supplemented with 50 ng/mL vascular endothelial growth factor (Peprotech, 100-20), and 100ng/mL Angiopoietin 1 (Peprotech, 130-06) on day 3-5, with experiments being performed on day 5. Media perfusion rate of 0.5 μL/min was maintained using a syringe pump (Harvard Apparatus, PHD2000 Series) throughout culture and experimental recordings.
Fluorescence microscopy
Immunofluorescence microscopy was used to visualise cell morphologies in co-culture. Samples were washed with phosphate-buffered saline (PBS) for 5 min twice before fixation with 4% paraformaldehyde (Alfa Aesar, 43368.9M) for 30 min. Samples were then blocked and permeabilised using PBS containing 3% bovine serum albumin (BSA) (Sigma-Aldrich, A3059) and 0.5% triton (Sigma-Aldrich, X100) for 24 h at 4°C with gentle rocking. Primary and secondary antibodies were prepared in PBS with 1% BSA and incubated for 24 h each at 4°C, with three 15-min PBS washes in between incubations. Hoechst (Life Technologies, H3570) was used to stain the nuclei for 10 min at RT then washed with PBS for 15 min. Images were captured using a Zeiss LSM-780 inverted confocal microscope and processed using Fiji/ImageJ.
Live cell microscopy was performed using a Zeiss Axio Observer widefield microscope. For images with multiple channels, images were captured sequentially and overlayed post-hoc in Fiji/ImageJ. To measure EC displacement, fluorescence from RFP-HUVEC in co-culture with beating hiPSC-CM was captured at 100 FPS, before processing with the Fiji plugin MuscleMotion(Sala et al., 2018). For quantification (Figure S1C) each datapoint represents average of all dishes at specified timepoint.
Sharp microelectrode
All electrophysiology experiments were conducted at 37°C in freshly prepared Tyrode’s solution (10 mM NaCl, 4.5 mM KCl, 10 mM glucose, 10 mM HEPES, 1 mM MgCl2 and 1.8 mM CaCl2, pH 7.4). iPSC-CM in 3D co-culture were impaled with sharp microelectrodes to record spontaneous action potentials. ≥30MΩ pipettes (30–0058, Harvard Apparatus) were pulled using a pipette puller (P-97, Sutter Instruments). Internal recording solution contained 2M KCl, 0.1mM EGTA, 5mM HEPES, pH = 7.2. Membrane potential was recorded via Axon Instruments Digidata 1440A digitiser and Multi-Clamp 700A amplifier, and analyzed with pClamp (Clampfit, Molecular Devices). Each datapoint represents one cell, containing the average values from three beats per cell.
Optical mapping – Calcium Imaging
Co-culture containing iPSC-CM which express the calcium biosensor GCaMP6f were used to record calcium transients. At time of encapsulation, co-cultures were plated directly onto 7 mm glass bottom dishes (MatTek) for static cultures. On day of recording, cell culture media was switched to Tyrodes solution, co-cultures were transferred to a warmed microscope stage (37°C) and field stimulated (1 Hz, 20 mV, 10 ms, bipolar pulse) for 5 min acclimatisation before recording. Fluorescence was excited via a 470nM LED and recorded using a long pass filter of 530 ± 35 nm on an inverted Nikon Eclipse TE2000 microscope with a 40X oil objective, and an ORCA-Flash4.0 camera (Hamamatsu) at acquisition rate of 250 frames per second. When stated, 1 μM isoprenaline hydrochloride (Sigma, I6504-100MG) containing D-Isoascorbic acid (Sigma, 856061) was perfused in 37°C Tyrode’s solution to stimulate β-adrenergic receptors. Calcium transient analysis was automated using a Matlab script developed in-house. For static co-culture (Figures 1M–1O and S2), each datapoint represents one dish, which contains the average of three beats from three ROI’s, each containing ∼5–20 cells. In β-adrenergic stimulation experiments (Figure S6), each datapoint represents average of all dishes at specified timepoint.
Spontaneous beating rate
iPSC-CM spontaneous beating rate was quantified using the same GCaMP6f calcium indicator, electrical stimulation parameters and microscope setup described in the Calcium Imaging methods. After a 5-min acclimatisation period, three 10 s recordings were obtained from three different ROI’s within the same dish. The average value ∗ 6 was used to determine beats per minute. Each datapoint represents one dish.
Pro-arrhythmic substrate
iPSC-CM pro-arrhythmic substrate was determined as the ability to follow 1 Hz electrical field stimulation (Figure 1P). Beating behavior was acquired using the same GCaMP6f calcium indicator, electrical stimulation parameters and microscope setup described in the Calcium Imaging methods. After a 5-min acclimatisation period, 10 s recordings were obtained and any deviation from 1:1 beat:stimulus ratio was designated as arrhythmic. Each datapoint represents one dish.
To determine iPSC-CM latency, co-cultures were subjected to a stepped electrical pacing protocol, which involved 1 min pacing at the following frequencies: 0.5, 1, 1.5, 2, 3, 4, 5 Hz, before cessation of stimulus. Latency was defined as the duration before first spontaneous beat. Each datapoint represents one dish.
Flow visualisation and quantification
RBC and FIT-C Dextran (10 μM, 40 kDa. Sigma, FD40S) were used to visualise flow in microfluidic chips. For RBC, Sprague Dawley rats with a body mass between 300 and 500 g, were sedated with isoflurane (4% isoflurane and 4 L/min O2) and sacrificed using cervical dislocation and dissection of the carotid arteries. Whole blood was collected 1:1 in 7.4 mM EDTA to prevent coagulation. RBCs were then washed by centrifugation at 10000 RPM in SAGM solution containing 0.87% w/v Sodium Chloride (Fisher Scientific UK Ltd, 27810.295), 0.017% w/v Adenine (Sigma, A8626), 0.9% w/v Glucose (Fisher Scientific UK Ltd,G/0500/53), 0.525% w/v Mannitol (Sigma, M4125) in MilliQ water three times, before resuspension to 50% haematocrit. A 1:10 dilution with perfusing solution was then used for flow visualisation. RBC velocity was quantified using 100 FPS brightfield image series captured with a Zeiss Axio Observer widefield microscope, before processing with Fiji plugin TrackMate (Germanguz et al., 2011). Fiji plugin MuscleMotion (Sala et al., 2018) was used for parallel quantification of iPSC-CM contractility.
Quantification and statistical analysis
Data were plotted as mean ± standard error of the means using GraphPad Prism 8. Shapiro-Wilk’s test was used to assess normality. To compare normally distributed data, one-way ANOVA was used followed by Tukey’s multiple comparison. When data were not normally distributed, Kruskal Wallis test and Dunn’s multiple comparison test were used. Pearson’s chi-squared test was used to compare conditional datasets. For all comparisons, p value with significant difference was indicated as follows (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Acknowledgments
We would like to acknowledge Prof. Bruce Conklin of the Gladstone Institute for kindly providing the GCaMP6f iPSC line, Dr. Bryan Hassell for his contribution to calcium analysis, Dr. Akemi Nogiwa Valdez for proofreading, and Mr. Stephen Rothery (Facility for Imaging by Light Microscopy, Imperial College London) for assistance with confocal and wide-field microscopy. For funding this work through an MRes + PhD studentship to O.K. (FS/16/56/32732), we thank the British Heart Foundation. Human tissue samples were provided by the Cardiovascular Research Centre Biobank at the Royal Brompton and Harefield NHS Foundation Trust, UK (NRES ethics number for biobank samples: 09/H0504/104 + 5; Biobank approval numbers: NP001-06-2015 and MED_CT_17_079). Current/additional author affiliations are as follows: O.K.: Human Safety, Bayer Crop Science, Sophia-Antipolis, France; D.C.-M.: 3B’s Research Group – Biomaterials, Biodegradables and Biomimetics, University of Minho, Braga, Portugal; A.S.: UPMC Mercy Hospital, Pittsburgh, PA, USA; F.K.: Institute of Physiology and Pathophysiology, Heidelberg University, Heidelberg, Germany; B.X.W.: Department of Metabolism, Digestion and Reproduction, Faculty of Medicine, Imperial College London, London, UK.
Author contributions
Concept of study by O.K. and C.M.T. Microfluidic chip design and fabrication by D.C.-M. and M.R. A.S. performed static arrhythmogenesis study. F.K. performed co-culture density study. I.S. provided calcium analysis support. B.X.W. provided cardiac FBs. B.D. performed latency experiment. J.F. and A.M.R. provided theoretical and practical support on endothelial biology. D.H. and M.M.S. provided support with microfluidic chip fabrication. All other experiments performed by O.K.
Declaration of interests
The authors declare no competing interests.
Published: August 29, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.crmeth.2022.100280.
Contributor Information
Oisín King, Email: ok314@ic.ac.uk.
Cesare M. Terracciano, Email: c.terracciano@imperial.ac.uk.
Supplemental information
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Ashikawa K., Kanatsuka H., Suzuki T., Takishima T. Phasic blood flow velocity pattern in epimyocardial microvessels in the beating canine left ventricle. Circ. Res. 1986;59:704–711. doi: 10.1161/01.RES.59.6.704. [DOI] [PubMed] [Google Scholar]
- Baeyens N., Nicoli S., Coon B.G., Ross T.D., Van den Dries K., Han J., Lauridsen H.M., Mejean C.O., Eichmann A., Thomas J.L., et al. Vascular remodeling is governed by a VEGFR3-dependent fluid shear stress set poin. Elife. 2015;4:1–35. doi: 10.7554/ELIFE.04645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauchamp P., Jackson C.B., Ozhathil L.C., Agarkova I., Galindo C.L., Sawyer D.B., Suter T.M., Zuppinger C. 3D co-culture of hiPSC-derived cardiomyocytes with cardiac fibroblasts improves tissue-like features of cardiac spheroids. Front. Mol. Biosci. 2020;7:14. doi: 10.3389/fmolb.2020.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry C., Duncker D.J. Coronary microvascular disease: the next frontier for cardiovascular research. Cardiovasc. Res. 2020;116:737–740. doi: 10.1093/cvr/cvaa035. https://pubmed.ncbi.nlm.nih.gov/32149331/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blache U., Ehrbar M. Inspired by nature: hydrogels as versatile tools for vascular engineering. Adv. Wound Care. 2018;7:232–246. doi: 10.1089/wound.2017.0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brutsaert D.L. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiol. Rev. 2003;83:59–115. doi: 10.1152/physrev.00017.2002. [DOI] [PubMed] [Google Scholar]
- Chobanyan-Jürgens K., Heusser K., Duncker D., Veltmann C., May M., Mehling H., Luft F.C., Schröder C., Jordan J., Tank J. Cardiac pacemaker channel (HCN4) inhibition and atrial arrhythmogenesis after releasing cardiac sympathetic activation. Sci. Rep. 2018;8:7748. doi: 10.1038/S41598-018-26099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder A., Vollert I., Hansen A., Eschenhagen T. Human engineered heart tissue as a model system for drug testing. Adv. Drug Deliv. Rev. 2016;96:214–224. doi: 10.1016/j.addr.2015.05.010. [DOI] [PubMed] [Google Scholar]
- Ellis B.W., Acun A., Can U.I., Zorlutuna P. Human IPSC-derived myocardium-on-chip with capillary-like flow for personalized medicine. Biomicrofluidics. 2017;11:024105. doi: 10.1063/1.4978468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald M.L., Chen Y.H., Lee A.P., Hughes C.C.W. The vascular niche in next generation microphysiological systems. Lab Chip. 2021;21:3244–3262. doi: 10.1039/D1LC00530H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Wu D., Birukov K.G. Comprehensive Physiology. Wiley; 2019. Mechanosensing and mechanoregulation of endothelial cell functions; pp. 873–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming I. Myoendothelial gap junctions. Circ. Res. 2000;86:249–250. doi: 10.1161/01.RES.86.3.249. [DOI] [PubMed] [Google Scholar]
- Garoffolo G., Pesce M. Mechanotransduction in the cardiovascular system: from developmental origins to homeostasis and pathology. Cells. 2019 doi: 10.3390/cells8121607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germanguz I., Sedan O., Zeevi-Levin N., Shtrichman R., Barak E., Ziskind A., Eliyahu S., Meiry G., Amit M., Itskovitz-Eldor J., et al. Molecular characterization and functional properties of cardiomyocytes derived from human inducible pluripotent stem cells. J. Cell Mol. Med. 2011;15:38–51. doi: 10.1111/j.1582-4934.2009.00996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghajar C.M., Chen X., Harris J.W., Suresh V., Hughes C.C.W., Jeon N.L., Putnam A.J., George S.C. The effect of matrix density on the regulation of 3-D capillary morphogenesis. Biophys. J. 2008;94:1930–1941. doi: 10.1529/biophysj.107.120774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomelli E., Meraviglia V., Campostrini G., Cochrane A., Cao X., Van Helden R.W., Garcia A.K., Mircea M., Kostidis S., Davis R.P., et al. Human-iPSC-derived cardiac stromal cells enhance maturation in 3D cardiac microtissues and reveal non-cardiomyocyte contributions to heart disease. Cell Stem Cell. 2020;26:862–879.e11. doi: 10.1016/j.stem.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfracht I., Efraim Y., Shinnawi R., Kovalev E., Huber I., Gepstein A., Arbel G., Shaheen N., Tiburcy M., Zimmermann W.H., et al. Engineered heart tissue models from hipsc-derived cardiomyocytes and cardiac ecm for disease modeling and drug testing applications. Acta Biomaterialia. 2019;92:145–159. doi: 10.1016/j.actbio.2019.05.016. [DOI] [PubMed] [Google Scholar]
- Ha H., Nam K.H., Lee S.J. Hybrid PIV–PTV technique for measuring blood flow in rat mesenteric vessels. Microvasc. Res. 2012;84:242–248. doi: 10.1016/J.MVR.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Hellberg K., Wayland H., Rickart A.L., Bing R.J. Studies on the coronary microcirculation by direct visualization. Am. J. Cardiol. 1972;29:593–597. doi: 10.1016/0002-9149(72)90158-0. [DOI] [PubMed] [Google Scholar]
- Huang C.P., Lu J., Seon H., Lee A.P., Flanagan L.A., Kim H.Y., Putnam A.J., Jeon N.L. Engineering microscale cellular niches for three-dimensional multicellular co-cultures. Lab Chip. 2009;9:1740–1748. doi: 10.1039/b818401a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebsch N., Loskill P., Mandegar M.A., Marks N.C., Sheehan A.S., Ma Z., Mathur A., Nguyen T.N., Yoo J.C., Judge L.M., et al. Automated video-based analysis of contractility and calcium flux in human-induced pluripotent stem cell-derived cardiomyocytes cultured over different spatial scales. Tissue Eng. Part C Methods. 2015;215:467–479. doi: 10.1089/ten.tec.2014.0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoto Y., Ehara T., Matsuura H. Voltage- and time-dependent block of Ik1 underlying Ba2+-induced ventricular automaticity. Am. J. Physiol. 1987;252:H325–H333. doi: 10.1152/AJPHEART.1987.252.2.H325. [DOI] [PubMed] [Google Scholar]
- Irisawa H., Brown H.F., Giles W. Cardiac pacemaking in the sinoatrial node. Physiol. Rev. 1993;73:197–227. doi: 10.1152/physrev.1993.73.1.197. [DOI] [PubMed] [Google Scholar]
- Jabbour R.J., Owen T.J., Pandey P., Reinsch M., Wang B., King O., Couch L.S., Pantou D., Pitcher D.S., Chowdhury R.A., et al. In vivo grafting of large engineered heart tissue patches for cardiac repair. JCI Insight. 2021;6:144068. doi: 10.1172/JCI.INSIGHT.144068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon J.S., Bersini S., Whisler J.A., Chen M.B., Dubini G., Charest J.L., Moretti M., Kamm R.D. Generation of 3D functional microvascular networks with human mesenchymal stem cells in microfluidic systems. Integr. Biol. 2014;6:555–563. doi: 10.1039/c3ib40267c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon J.S., Bersini S., Gilardi M., Dubini G., Charest J.L., Moretti M., Kamm R.D. Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc. Natl. Acad. Sci. USA. 2015;112:214–219. doi: 10.1073/pnas.1417115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost N., Nagy N., Corici C., Kohajda Z., Horváth A., Acsai K., Biliczki P., Levijoki J., Pollesello P., Koskelainen T., et al. ORM-10103, a novel specific inhibitor of the Na+/Ca2+ exchanger, decreases early and delayed afterdepolarizations in the canine heart. Br. J. Pharmacol. 2013;170:768. doi: 10.1111/BPH.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane C., Terracciano C.M. Human cardiac fibroblasts engage the Sarcoplasmic reticulum in induced pluripotent stem cell-derived cardiomyocyte excitation–contraction coupling. J. Am. Coll. Cardiol. 2018;72:1061–1063. doi: 10.1016/j.jacc.2018.06.028. [DOI] [PubMed] [Google Scholar]
- Kane C., Terracciano C.M.N. Concise review: criteria for chamber-specific categorization of human cardiac myocytes derived from pluripotent stem cells. Stem Cells. 2017;35:1881–1897. doi: 10.1002/stem.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbassi E., Fenix A., Marchiano S., Muraoka N., Nakamura K., Yang X., Murry C.E. Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020;17:341–359. doi: 10.1038/S41569-019-0331-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassab G.S., Fung Y.C. Topology and dimensions of pig coronary capillary network. Am. J. Physiol. 1994;267:H319–H325. doi: 10.1152/ajpheart.1994.267.1.h319. [DOI] [PubMed] [Google Scholar]
- Kim J.J., Yang L., Lin B., Zhu X., Sun B., Kaplan A.D., Bett G.C., Rasmusson R.L., London B., Salama G. Mechanism of automaticity in cardiomyocytes derived from human induced pluripotent stem cells. J. Mol. Cell. Cardiol. 2015;81:81–93. doi: 10.1016/j.yjmcc.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Chung M., Ahn J., Lee S., Jeon N.L. Interstitial flow regulates the angiogenic response and phenotype of endothelial cells in a 3D culture model. Lab Chip. 2016;16:4189–4199. doi: 10.1039/c6lc00910g. http://xlink.rsc.org/?DOI=C6LC00910G [DOI] [PubMed] [Google Scholar]
- King O., Sunyovszki I., Terracciano C.M. Vascularisation of pluripotent stem cell–derived myocardium: biomechanical insights for physiological relevance in cardiac tissue engineering. Pflugers Arch. 2021;473:1117–1136. doi: 10.1007/s00424-021-02557-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl P., Kamkin A.G., Kiseleva I.S., Noble D. Mechanosensitive fibroblasts in the sino-atrial node region of rat heart: interaction with cardiomyocytes and possible role. Exp. Physiol. 1994;79:943–956. doi: 10.1113/expphysiol.1994.sp003819. [DOI] [PubMed] [Google Scholar]
- Lian X., Hsiao C., Wilson G., Zhu K., Hazeltine L.B., Azarin S.M., Raval K.K., Zhang J., Kamp T.J., Palecek S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA. 2012;109:E1848–E1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S.L., Lam C.S., Segers V.F., Brutsaert D.L., De Keulenaer G.W. Cardiac endothelium–myocyte interaction: clinical opportunities for new heart failure therapies regardless of ejection fraction. Eur. Heart J. 2015;36:2050–2060. doi: 10.1093/eurheartj/ehv132. [DOI] [PubMed] [Google Scholar]
- Marcu R., Choi Y.J., Xue J., Fortin C.L., Wang Y., Nagao R.J., Xu J., MacDonald J.W., Bammler T.K., Murry C.E., et al. Human organ-specific endothelial cell heterogeneity. iScience. 2018;4:20–35. doi: 10.1016/j.isci.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason B.N., Starchenko A., Williams R.M., Bonassar L.J., Reinhart-King C.A. Tuning three-dimensional collagen matrix stiffness independently of collagen concentration modulates endothelial cell behavior. Acta Biomater. 2013;9:4635–4644. doi: 10.1016/j.actbio.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayourian J., Ceholski D.K., Gonzalez D.M., Cashman T.J., Sahoo S., Hajjar R.J., Costa K.D. Physiologic, pathologic, and therapeutic paracrine modulation of cardiac excitation-contraction coupling. Circ. Res. 2018;122:167–183. doi: 10.1161/CIRCRESAHA.117.311589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick M.E., Tzima E. Pulling on my heartstrings: mechanotransduction in cardiac development and function. Curr. Opin. Hematol. 2016;23:235–242. doi: 10.1097/MOH.0000000000000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin K.T., Tranquillo R.T. In vitro models of angiogenesis and vasculogenesis in fibrin gel. Exp. Cell Res. 2013:2409–2417. doi: 10.1016/j.yexcr.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraju C.K., Dries E., Gilbert G., Abdesselem M., Wang N., Amoni M., Driesen R.B., Sipido K.R. Myofibroblast modulation of cardiac myocyte structure and function. Sci. Rep. 2019;9:8879. doi: 10.1038/S41598-019-45078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A.C., Nakatsu M.N., Chou W., Gershon P.D., Hughes C.C.W. The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol. Biol. Cell. 2011;22:3791–3800. doi: 10.1091/mbc.E11-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nippert F., Schreckenberg R., Schlüter K.D. Isolation and cultivation of adult rat cardiomyocytes. J. Vis. Exp. 2017;2017 doi: 10.3791/56634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor N., Shapira A., Edri R., Gal I., Wertheim L., Dvir T. 3D printing of personalized thick and perfusable cardiac patches and hearts. Adv. Sci. 2019;6:1900344. doi: 10.1002/advs.201900344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongstad E., Kohl P. Fibroblast-myocyte coupling in the heart: potential relevance for therapeutic interventions. J. Mol. Cel. Cardiol. 2016:238–246. doi: 10.1016/j.yjmcc.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaki T., Sivathanu V., Kamm R.D. Vascularized microfluidic organ-chips for drug screening, disease models and tissue engineering. Curr. Opin. Biotechnol. 2018:116–123. doi: 10.1016/j.copbio.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perbellini F., Watson S.A., Bardi I., Terracciano C.M. Heterocellularity and cellular cross-talk in the cardiovascular system. Front. Cardiovascular Med. 2018:143. doi: 10.3389/fcvm.2018.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto A.R., Ilinykh A., Ivey M.J., Kuwabara J.T., D’antoni M.L., Debuque R., Chandran A., Wang L., Arora K., Rosenthal N.A., et al. Revisiting cardiac cellular composition. Circ. Res. 2016;118:400–409. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollet A.M.A.O., den Toonder J.M.J. Recapitulating the vasculature using Organ-on-Chip technology. Bioengineering. 2020;7:E17. doi: 10.3390/bioengineering7010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravenscroft S.M., Pointon A., Williams A.W., Cross M.J., Sidaway J.E. Cardiac non-myocyte cells show enhanced pharmacological function suggestive of contractile Maturity in stem cell derived cardiomyocyte microtissues. Toxicol. Sci. 2016;152:99. doi: 10.1093/TOXSCI/KFW069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues I.C., Kaasi A., Maciel Filho R., Jardini A.L., Gabriel L.P. Cardiac tissue engineering: current state-of-the-art materials, cells and tissue formation. Einstein. 2018:eRB4538. doi: 10.1590/S1679-45082018RB4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaldson-Bouchard K., Ma S.P., Yeager K., Chen T., Song L., Sirabella D., Morikawa K., Teles D., Yazawa M., Vunjak-Novakovic G. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature. 2018;556:239–243. doi: 10.1038/s41586-018-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook M.B., Jongsma H.J., de Jonge B. Single channel currents of homo- and heterologous gap junctions between cardiac fibroblasts and myocytes. Pflugers Arch. 1989;414:95–98. doi: 10.1007/BF00585633. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K., Shimizu T., Horaguchi S., Sekine H., Yamato M., Umezu M., Okano T. In vitro engineering of vascularized tissue surrogates. Sci. Rep. 2013;3:1316–1317. doi: 10.1038/srep01316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala L., van Meer B.J., Tertoolen L.G.J., Bakkers J., Bellin M., Davis R.P., Denning C., Dieben M.A.E., Eschenhagen T., Giacomelli E., et al. MUSCLEMOTIONNovelty and significance. Circ. Res. 2018;122:e5–e16. doi: 10.1161/CIRCRESAHA.117.312067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saucerman J.J., Tan P.M., Buchholz K.S., McCulloch A.D., Omens J.H. Mechanical Regulation of Gene Expression in Cardiac Myocytes and Fibroblasts. Nat. Rev. Cardiol. 2019:361–378. doi: 10.1038/s41569-019-0155-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y., Jeon J.S., Han S., Jung G.S., Shin S., Lee S.H., Sudo R., Kamm R.D., Chung S. In vitro 3D collective sprouting angiogenesis under orchestrated ANG-1 and VEGF gradients. Lab Chip. 2011;11:2175–2181. doi: 10.1039/c1lc20039a. [DOI] [PubMed] [Google Scholar]
- Skylar-Scott M.A., Tan P.M., Buchholz K.S., McCulloch A.D., Omens J.H. Biomanufacturing of organ-specific tissues with high cellular density and embedded vascular channels. Sci. Adv. 2019;5:eaaw2459. doi: 10.1126/sciadv.aaw2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorop O., Van De Wouw J., Chandler S., Ohanyan V., Tune J.D., Chilian W.M., Merkus D., Bender S.B., Duncker D.J. Experimental animal models of coronary microvascular dysfunction. Cardiovasc. Res. 2020:756–770. doi: 10.1093/cvr/cvaa002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez J.Y., Perry N., Schindelin J., Hoopes G.M., Reynolds G.D., Laplantine E., Bednarek S.Y., Shorte S.L., Eliceiri K.W. TrackMate: An Open and Extensible Platform for Single-Particle Tracking. Methods. 2017;115:80–90. doi: 10.1016/j.ymeth.2016.09.016. [DOI] [PubMed] [Google Scholar]
- Tirziu D., Simons M. Endothelium as master regulator of organ development and growth. Vascul. Pharmacol. 2009;50:1–7. doi: 10.1016/j.vph.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulloch N.L., Muskheli V., Razumova M.V., Korte F.S., Regnier M., Hauch K.D., Pabon L., Reinecke H., Murry C.E. Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ. Res. 2011;109:47–59. doi: 10.1161/CIRCRESAHA.110.237206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugolini G.S., Visone R., Cruz-Moreira D., Mainardi A., Rasponi M. Methods in Cell Biology. Academic Press Inc.; 2018. Generation of functional cardiac microtissues in a beating heart-on-a-chip; pp. 69–84. [DOI] [PubMed] [Google Scholar]
- Vasquez C., Benamer N., Morley G.E. The cardiac fibroblast: functional and electrophysiological considerations in healthy and diseased hearts. J. Cardiovasc. Pharmacol. 2011;57:380–388. doi: 10.1097/FJC.0B013E31820CDA19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J.U.G., Pham Minh Duc, Nicin Luka, Hammer Marie, Bottermann Katharina, Yuan Ting, Sharma Rahul, John David, Muhly-Reinholz Marion, Tombor Lukas, et al. Dissection of heterocellular cross-talk in vascularized cardiac tissue mimetics. J. Mol. Cell. Cardiol. 2020;138:269–282. doi: 10.1016/j.yjmcc.2019.12.005. [DOI] [PubMed] [Google Scholar]
- Xiao Y., Zhang B., Liu H., Miklas J.W., Gagliardi M., Pahnke A., Thavandiran N., Sun Y., Simmons C., Keller G., Radisic M. Microfabricated perfusable cardiac biowire: a platform that mimics native cardiac bundle. Lab Chip. 2014;14:869–882. doi: 10.1039/c3lc51123e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaniv Y., Lakatta E.G., Maltsev V.A. From two competing oscillators to one coupled-clock pacemaker cell system. Front. Physiol. 2015:28. doi: 10.3389/fphys.2015.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Montgomery M., Chamberlain M.D., Ogawa S., Korolj A., Pahnke A., Wells L.A., Massé S., Kim J., Reis L., et al. Biodegradable scaffold with built-in vasculature for organ-on-a-chip engineering and direct surgical anastomosis. Nat. Mater. 2016;15:669–678. doi: 10.1038/nmat4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Live imaging of hiPSC-CM-GCaMP (Green) and RFP-HUVEC (Orange) demonstrates direct cell-cell contact and endothelial displacement during the cardiac cycle after 7 days co-culture.
Spontaneous vasculogenesis was induced using RFP-HUVEC (Orange) and hLVFB. Red blood cells were perfused through microvasculature to demonstrate vessel functionality.
Myocardial microvasculature was established in a Heart-on-a-chip model after 5 days vasculogenic co-culture with HCMEC (CD31, Yellow) and iPSC-CM (Alpha-Actinin, Red).
Functionality of microvasculature in Heart-on-a-chip was demonstrated via perfusion of red blood cells. Co-culture was performed with RFP-HUVEC (orange) and iPSC-CM-GCaMP (Green).
Red blood cell velocity through myocardial microvasculature was quantified using TrackMate Fiji/ImageJ plugin.
Demonstration of variability in vascular network via live imaging of red blood cell flow through 8 and 80 μm lumens.
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.