

Abstract
Condensed states of proteins, including liquid-like membraneless organelles and solid-like aggregates, contribute in fundamental ways to the organisation and function of the cell. Perturbations of these states can lead to a variety of diseases through mechanisms that we are now beginning to understand. We define protein condensation diseases as conditions caused by the disruption of the normal behaviour of the condensed states of proteins. We analyze the problem of the identification of targets for pharmacological interventions for these diseases and explore opportunities for the regulation of the formation and organisation of aberrant condensed states of proteins.
Subject terms: Drug discovery, Chemical biology, Protein folding
In this review, the authors define protein condensation diseases as conditions caused by aberrant liquid-like or solid-like states of proteins, and describe opportunities for therapeutic interventions to restore the normal phase behaviour of proteins. The review accompanies the related collection of articles published in Nature Communications focusing on possible therapeutic approaches involving liquid-liquid phase separation.
Introduction
By folding into their native states, proteins perform myriad molecular functions that are essential for the maintenance of cellular homoeostasis1. The phenomenon of protein folding is a prominent example of the ability of biological systems to self-assemble by bringing together reactive groups in complex arrangements that enable sophisticated biochemical functions.
In recent years, it has also emerged that the ability of proteins to organise themselves into functional forms extends beyond native states. Numerous proteins have been shown to undergo a liquid-liquid phase separation process leading to the formation of membraneless organelles, which are complex biomolecular assemblies resembling a dense liquid-like state2,3, also referred to as the droplet state4. Furthermore, many proteins can also form a highly ordered solid-like state, known as the amyloid state5, which in certain cases can be functional6,7. Since in the cell most proteins are typically expressed at concentrations at which they can form condensed states8,9, the droplet and amyloid states could be considered as fundamental states of proteins along with the native state4 (Fig. 1).
Fig. 1. Protein condensation diseases are conditions caused by the aberrant conversion of proteins between the native, amyloid and droplet states.
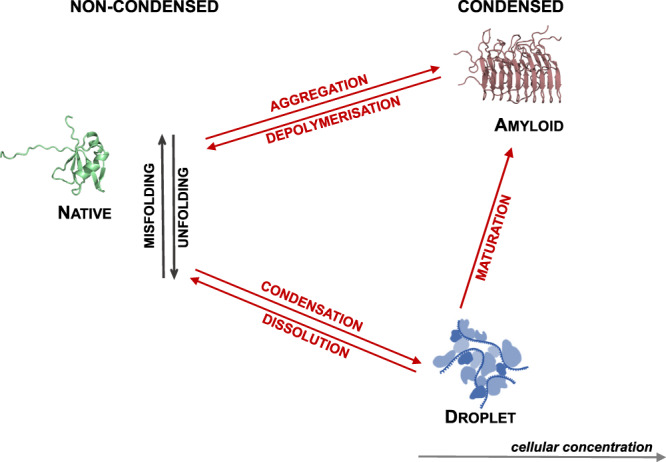
Under cellular conditions, many proteins, in addition to the native state, can populate two condensed states, the liquid-like droplet state and the solid-like amyloid state4,10. Protein condensation diseases are the consequence of the failure of the protein homoeostasis system to regulate the balance between the different protein states (Fig. 2). A list of currently known protein condensation diseases is provided in Table 1.
Proteins in condensed states can perform a wide range of biological functions by increasing the efficiency of cellular processes and by reducing biological noise10,11. The increase in the local concentrations of different cellular components in condensed states accelerates enzymatic reactions, such as in the cases of the premelanosome protein (Pmel17) in melanin synthesis12 and of cyclic GMP-AMP synthase (cGAS) in innate immune signalling13. Liquid–liquid phase separation can amplify signals by low-affinity effectors and ligands by facilitating the formation of signalling clusters, such as in T cell receptors14 or Wnt signalling15. Droplets can serve as non-membrane bound cellular compartments, such as the nucleolus16 or facilitate their formation through nucleation of polymerisation reactions, such as microtubulin for centrosome formation17. The assembly and disassembly of condensates promote morphological changes in developmental processes, such as the pattern specification process3. Condensates may orchestrate components of cellular pathways, such as in the cases of p53-binding protein 1 (53BP1) droplets, which concentrate components for DNA repair18 or of heterochromatin protein 1 (HP1) droplets, which induce gene silencing19. Furthermore, an increasing number of cellular processes have been associated with solid-like scaffolds6,7. In particular, signalling complexes in the innate immune system, such as inflammasomes, faddosomes, myddosomes often form solid-like condensates20,21 to recruit downstream signalling components.
In this work, we first characterise protein condensation diseases as disorders caused by aberrant liquid- or solid-like states of proteins. We then address the problem of identifying possible targets for drug discovery in order to restore the normal phase behaviour or proteins.
Regulation of protein condensation by the protein homoeostasis system
The balance between the condensed states and the native state of proteins must be highly regulated for optimal functions. The protein homoeostasis system controls in multiple ways the process of protein condensation, including the reversible formation of the droplet state from the native state, its irreversible maturation to the amyloid state, as well as the irreversible aggregation of the native state to the amyloid state5,22 (Fig. 2).
Fig. 2. Protein condensation and protein homeostasis.
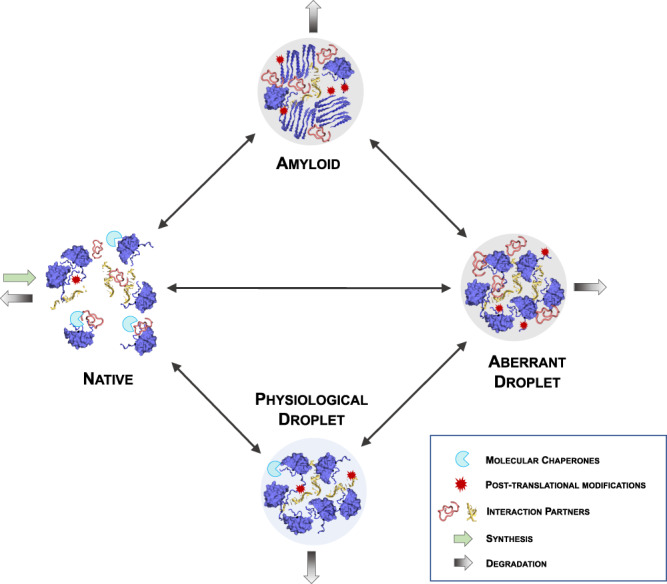
The protein homoeostasis system regulates the formation, clearance, composition, interactions, localisation and biophysical properties of protein condensates146,147. Although the complete mapping of the protein homoeostasis system that controls protein condensates is still far from complete, several examples have already been identified. The formation and dissolution of the droplet state are regulated by post-translational modifications23,24 and the availability of interaction partners25. The re-localisation within a cell of solid-like condensates may revert them to the liquid-like state by making available suitable interaction partners34,117. Molecular chaperones may interfere with misfolded protein intermediates and inhibit the formation of the amyloid state either from the native state through the deposition pathway or from the droplet state through the condensation pathway27. Autophagy contributes to stress granule clearance29, and the liquid-liquid phase separation of p62 with its ubiquitinated substrates may lead to autophagosome formation30.
The assembly and dissolution of the droplet state in response to specific cellular conditions is often regulated through post-translational modifications23,24 (Fig. 2). The protein kinase Sky1, for example, controls stress granule disassembly through the phosphorylation of the nucleocytoplasmic mRNA shuttling protein Npl325. Alternative mechanisms for stress granule clearance are provided by molecular chaperones26, in particular in the case of aberrant condensates containing misfolded proteins27 (Fig. 2). The two mechanisms are linked, as Sky1 overexpression can compensate chaperone defects in stress granule disassembly pathways25. The level of ubiquitination also controls stress granule formation, for example, depletion of the deubiquitylases USP5 and USP13 resulted in accelerated stress granule assembly and delayed the return to normal conditions28.
Stress granule clearance in mammalian cells can be also reduced by inhibition of autophagy, or by impairment of valosin-containing protein (VCP, the human ortholog of CDC48), which plays a critical role in protein quality control29. Droplet clearance by autophagy involves liquid-liquid phase separation of the ubiquitinated substrate and the ubiquitin-binding protein p6230. p62 condensates are further regulated by the death-domain-associated protein DAXX31 and contribute to the oxidative stress response mediated by the transcription factor Nrf232. p62 condensates and their interactions with the nuclear receptor Nur77 are also critical for the removal of damaged mitochondria33. Interactions with nuclear transport receptors regulate cellular localisation and condensate assembly, as it was shown for the TAR DNA-binding protein 43 (TDP-43) and the RNA-binding protein senataxin (SETX) in spinal cord motor neurons34.
Protein condensation diseases
As a counterpart to the wide range of the cellular processes described above, it is becoming increasingly clear that failures in the regulation of condensed states may lead to dysfunctional protein assemblies that could be involved in a range of pathological processes22,35,36.
Numerous pathological conditions have been mechanistically linked to the formation of aberrant liquid-like22,35,37,38 and solid-like5,39 condensates (Table 1). It is thus becoming increasingly clear that aberrant protein condensation likely has a causative nature in a wide range of human diseases. These pathologies, which can be collectively defined as protein condensation diseases, originate in alterations of the physiological states of proteins (Fig. 1), due to the failure of regulating the formation, clearance, composition, interactions and localisation of protein condensates (Fig. 2). In the following, we discuss examples of protein condensation diseases as conditions caused by the disruption of the normal behaviour of the condensed states of proteins.
Table 1.
Examples of currently known diseases associated with aberrant protein condensation
| Disease | Protein | Missense mutations | Type | Classification |
|---|---|---|---|---|
| Primary immune-deficiency syndromes122 | IRAK4 | R12C, R20W | LoF | Amyloid to native |
| Primary immune-deficiency syndromes122 | MYD88 | S34Y, S98C | LoF | |
| Autoimmune lymphoproliferative syndrome123 | FAS | R250Q, A257D, D260Y, T270K, E272K | LoF | |
| Amyotrophic lateral sclerosis52,102,124 | FUS | G156E, G187S, G225V, G230C, G399V, P525L, R244C, R216C, R521G | LoF | Droplet to amyloid |
| Amyotrophic lateral sclerosis50,125,126 | TDP-43 | A321V, G298S, M337V, A315T | LoF | |
| Amyotrophic lateral sclerosis101,127–130 | HNRNPA1 | D262V, D214V, P298L, D290V | LoF | |
| Amyotrophic lateral sclerosis53 | TIA1 | P362L, A381T, E384K | LoF | |
| Amyotrophic lateral sclerosis114 | UBQLN2 | P506A, P506S, P506T, P497H, P497S, | GoF | |
| Amyotrophic lateral sclerosis131 | CHCHD10 | S59L | LoF | |
| Frontotemporal dementia131 | CHCHD10 | S59L | LoF | |
| Alzheimer’s disease132 | TAU | P301L, P301S, A152T | LoF | |
| Progranulin deficiency54 | TDP-43 | LoF | ||
| Limb-girdle muscular dystrophy 1G133 | HNRNPDL | D378H, D378N | LoF | |
| Huntington disease134 | HTT | 43QP | LoF | |
| Distal hereditary motor neuronopathy72 | HSPB3 | R116P | LoF | |
| Cervix cancer77 | UTX | S781Y | LoF | |
| Autosomal-dominant distal myopathy135 | MATR3 | S85C | LoF | Droplet to native |
| Prostate cancer46 | SPOP | F133V, W131G | LoF | |
| Rett syndrome40 | MECP2 | P225R, R306C, P322L | LoF | |
| Atopic dermatitis42 | FLG | Tail-deficient mutants | LoF | |
| Mental retardation autosomal dominant 5 | SYNGAP1 | T1305A | LoF | |
| Inclusion of body myopathy with early-onset Paget’s disease29 | VCP | A232E | LoF | |
| Frontotemporal dementia29 | VCP | A232E | LoF | |
| Amyotrophic lateral sclerosis 14 without FTD29 | VCP | R155H | LoF | |
| Paget’s disease47 | SQSTM1 | M404V | LoF | |
| Dementia47 | SQSTM1 | M404V | LoF | |
| Usher syndrome136 | MYO7A | K2021R, L2186P, G2187D | LoF | |
| Skin cancer136 | MYO7B | E1288K | LoF | |
| Amyotrophic lateral sclerosis114 | UBQLN2 | T487I, P497L | GoF | Native to droplet |
| Amyotrophic lateral sclerosis34 | SETX | L389S, R2136H | GoF | |
| Central nervous system cancer119 | DDX3X | T275M, G302V, G325E, M370R | GoF | |
| Dilated cardiomyopathy137 | RBM20 | R636S | GoF | |
| Inherited taupathy43 | TAU | P301L | GoF | |
| Respiratory syncytial virus infection45 | M2-1 | GoF | ||
| Multisystem proteinopathy47 | TIA1 | N357S | GoF | |
| Noonan syndrome, Leopard syndrome108 | PTPN11 | G464A | GoF | |
| Juvenile myelomonocytic leukaemia108 | PTPN11 | D61G, E76A, E76K, Q506P | GoF | |
| Liver cancer108 | PTPN11 | Y279C | GoF | |
| Lung cancer138 | KEAP1 | R320Q | LoF | |
| Skin cancer138 | KEAP1 | R470C | LoF |
Disease-associated missense mutations shown to alter protein condensation are listed. Loss-of-function (LoF) and gain-of-function (GoF) mutations are classified based on the original studies. Misfolding diseases classified as native to amyloid are reviewed elsewhere39, and diseases that could be classified as amyloid to droplet are not currently known.
Perturbations that induce the disassembly of liquid-like condensates may compromise their physiological functions. For example, with the condensation of methyl CpG binding protein 2 (MeCP2) being critical for heterochromatin assembly, it has been reported that mutations that disrupt this process lead to transcriptional dysregulation in Rett syndrome40 (Table 1). Mutations of MeCP2 associated with Rett syndrome can also impair the formation of the RNA-binding fox-1 (Rbfox) condensates, compromising their splicing functions41. It has also been shown that the failure in the formation of keratophyalin granules compromises skin defence mechanisms in atopic dermatitis42.
Conversely, the droplet state can potentially concentrate harmful conformations or pathogenic material. For example, liquid-like droplets can stabilise cytotoxic assemblies of tau, which promote tau aggregation in Alzheimer’s disease43 (Table 1). It has also been reported that viral replication can take place in virus-induced inclusion bodies44, as observed in respiratory syncytial viral infections45.
More generally, shifting the phase boundary either towards the formation of condensates or towards their disassembly can have pathological consequences. Cancer-causing mutations in the speckle-type POZ protein (SPOP), by reducing its tendency to phase separate, lead to a failure in its co-localisation with DAXX, thus dysregulating ubiquitin-dependent protein homoeostasis46 (Table 1). In contrast, mutations in p62, by disturbing stress granule clearance, lead to multisystem proteinopathy and Paget’s disease47. A loss of liquid-like properties of the condensates of A-kinase anchoring protein (AKAP95) may cause tumorigenesis by compromising splicing functions48.
A wide range of disorders is caused by the shifting of the droplet state towards the amyloid state49. The irreversible maturation of granules of RNA-binding proteins, including TDP-4350, heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1)51, fused in sarcoma (FUS)52 and T-cell intracellular antigen 1 (TIA)53, can results in loss of function, as for example in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). The conversion of the droplet state into the amyloid state may lead to loss of function by amyloid fibril formation, as well as the formation of promiscuous intermediates causing cytotoxicity53. Protein aggregation may be induced by a deficiency of an interaction partner, such as in the case of progranulin, whose down-regulation contributes to microglial toxicity of TDP-4354. Droplet maturation, however, may also be required for physiological functions. For example, the innate immune mechanism involving the virus-induced inflammasome formed by NOD-like receptor family pyrin domain containing 6 (NLRP6) undergoes solidification after the recruitment of apoptosis-associated speck-like protein (ASC) for downstream activation55. Likewise, an acquisition of solid-like behaviour of the condensates of the active-zone scaffold proteins SYD-2 and ELKS-1 is required for synaptic functions56.
Perturbing the interplay between membrane-bound organelles and condensates may lead to additional disease mechanisms57–59. Ribonucleoprotein granule biogenesis is modulated by the contact sites with the endoplasmic reticulum (ER), which regulate the fusion and fission of processing bodies (P-bodies) and stress granules60. This process couples the ER translational capacity with the generation of membraneless organelles. In a similar vein, the ER forms a compartment with TIS granules, which through interactions between 3’ untranslated RNA regions modulates the expression of membrane proteins61. In addition, interactions with ER membranes affect the size of Whi3 membraneless organelles, thereby limiting the local concentration increase on the ER surface62. Via modulating protein concentrations, ER-linked STING protein condensates influence innate immunity responses63. Although growing evidence demonstrates the biological importance of condensate-organelle communications, only a few disease-associated mutations have been directly linked to this process. As annexin A11 enhances RNA transport in neurons by tethering RNP granule cargos to lysosomes64, ALS-associated mutations of annexin A11 disrupt its interactions with lysosomes and impair its adaptor function64.
Classification of protein condensation diseases
To identify links between condensate-forming proteins (Supplementary data set: Table S1) and human disease, we searched for pathologies associated with genes encoding these proteins. Our analysis indicates that up to a third of human diseases can be associated with genes that encode condensate-forming proteins (Supplementary data set: Table S2), and that missense mutations in these genes accumulate in the droplet-promoting regions of the corresponding proteins (Supplementary data set: Table S3). The aim of these rankings is to help future studies identify diseases in which protein condensation has a causative nature, and corresponding possible targets for pharmacological intervention (Tables S1, S2 and S3).
The top disease categories based on gene-disease associations (Supplementary data set: Table S2) include abnormal tissue morphology changes, such as breast, liver, colorectal, prostate, lung tumours, stomach carcinoma, glioblastoma. These aberrant condensates lead to dysregulation of gene-expression programs46, cell division or failure of DNA repair processes18. The liquid-like properties of droplets can also promote morphological changes by concentrating selected components for cancer development and metastasis65. Top-ranking protein condensation diseases also include nervous system disorders, such as schizophrenia, bipolar and autistic disorders, depression, epilepsy, as well as Alzheimer’s and Parkinson’s diseases. Most of these neurological disorders are associated with genes encoding proteins forming synaptic condensates66. As synaptic plasticity requires in many cases a liquid-liquid phase separation of synaptic proteins67, aberrant protein condensation was shown to compromise synaptic functions66,68. In addition, according to our analysis, aberrant condensates of cytoskeletal14 and signalling proteins69 are likely to contribute to these neurological disorders. We also identified cardiovascular protein condensation diseases, such as myocardial ischaemia, atrial fibrillation, myocardial failure, atherosclerosis and cardiomyopathy (Supplementary data set: Table S2). Troponin, a key marker of myocardial infarction, and proteins controlling the circadian clock were associated with nuclear condensates70,71. Aberrant phase separation can perturb nuclear functions, as demonstrated for small heat shock proteins associated with cardiac myopathy72, and contribute to different muscular dystrophies, as illustrated by the case of the membraneless compartmentalisation of Z-disk proteins, which is essential for myofibrillogenesis73. We also identified digestive system disorders (Supplementary data set: Table S2), such as liver cirrhosis, hepatitis, alcoholic intoxication, that involve genes encoding condensate-forming proteins. These include cytosolic glutathione-S-transferases, the urea cycle enzyme carbamoyl phosphate synthase I, several enzymes involved in amino acid metabolism, and cholesterol transport, as components of cellular bodies formed in response to stress74,75. Aberrant protein condensation of metabolic enzymes is associated in our analysis with a wide range of disorders, including diabetes mellitus and metabolic syndrome (Supplementary data set: Table S2). Energy stress was shown to modulate localisation and condensation of glycolytic enzymes76. We also identified immune system disorders (polyarthritis, asthma) and viral infections (influenza) associated with genes encoding PYD and CARD domain-containing proteins, the condensation of which is required for innate immune signalling13,55 (Supplementary data set: Table S2).
Next, based on the analysis of disease-associated missense variants, we identified over 600 disorders that can be classified as protein condensation diseases (Supplementary data set: Table S3), as most contributing mutations are in droplet-promoting regions of experimentally identified condensate-forming proteins (Supplementary data set: Table S1). This classification included rare multisystem disorders such as the Kabuki77, Werner and Rubinstein-Taybi syndromes, which have a high fraction of droplet-associated mutations and involve various biological pathways (Supplementary data set: Table S4). Thus, we systematically analysed the genes associated with 3178 orphan diseases from the Orphanet database (https://www.orpha.net) and found that over 2168 orphan diseases (i.e. over two-thirds) have a considerable contribution from genes encoding droplet-forming proteins (Supplementary data set: Table S2). Furthermore, we identified 140 rare disorders for which most missense mutations are associated with known droplet-forming proteins (Supplementary data set: Table S3). This analysis indicates that many orphan diseases are likely to be associated with protein condensation, which can offer mechanisms for targeting these pathologies. This observation can for example be exploited for screening compound libraries for these disorders, including by using fluorescent markers of components forming aberrant condensates.
Interactions within protein condensates in health and disease
We are only beginning to understand the molecular forces that drive liquid-liquid phase separation by finely tuning the balance between the native and condensed states in the cellular environment4,78–80. The formation of the liquid-like condensed state has been initially associated with the presence of disordered regions81 and of prion-like domains82 in RNA-binding proteins. However, increasing numbers of structured proteins, ranging from metabolic enzymes77 to signalling complexes83, have also been observed to undergo liquid-liquid phase separation. These observations suggest that the inter-molecular interactions driving condensate formation could have a more generic nature and be more widespread in the proteome8,9,84.
According to our current understanding of the protein condensation process, liquid-like condensates are stabilised and regulated by disordered interactions4,78–80,85, while the formation of solid-like aggregates requires the self-assembly of inter-backbone hydrogen-bonding networks into highly ordered amyloid structures86. The process of liquid–liquid phase separation can be driven by a wide range of sequence motifs including electrostatic (π–π87 and charge–π88) and hydrophobic89 interactions. Organisation of such non-canonical motifs into patterns, such as those of aromatic and charged residues, was observed to enable phase separation88,90 Perturbing interaction patterns modulates the conformational propensity of a protein sequence91, which can shift the droplet state to the native state88. Along these lines, linker regions contribute to phase separation by influencing the number of accessing binding, such as in the case of the adaptor protein Nck92.
The multivalent interactions driving liquid–liquid phase separation exhibit strong dependence on the cellular context79,93, including the pH94 and salt concentration78. Cellular localisation and concentration of interaction partners, including RNA, are critical for promoting the formation and controlling the properties of condensates95,96. Together with hydrophobic interactions, aromatic interactions are important under high salt conditions, while electrostatic interactions dominate the process under low salt conditions78. Post-translational modifications and allosteric effects of the flanking regions can provide a further layer of regulation to switch the motifs on and off19,68. For example, phosphorylation regulates the formation of FMRP and caprin-1 condensates to control mRNA deadenylation97, dual-specificity kinases are important regulators of condensate homoeostasis23,24, and histone H1 acetylation antagonises chromatin phase separation98.
Taken together, these observations suggest that the formation of the droplet state is mediated by disordered interactions, while that of the solid-like amyloid state by ordered interactions99 (Fig. 3). Neurogenerative diseases are thus in many cases associated with mutations that increase the multiplicity of binding modes by promoting interactions that promote both the droplet and amyloid states. Thus, regions that can sample both types of interactions can drive amyloid formation within condensates100. Charge–π interactions, for example, can lead to reversible amyloid formation, while the mutation of charged residues into hydrophobic ones can stabilise the amyloid state101. Familial mutations associated with neurodegenerative disorders may expand the repertoire of binding modes of a protein, such as in the case of FUS G156E102, enabling a gradual shift towards more ordered configurations of condensates. Indeed, ALS-associated and non-ALS-associated mutations of RNA-binding proteins can be distinguished on the basis of the sequence-based calculation of physico-chemical properties of proteins, including droplet and aggregate propensities, and diversity of interaction modes99.
Fig. 3. Inter-molecular interactions within protein condensates in health and disease.

A Interaction modes of residues in the prion-like domain of TDP-43 vary between disordered and ordered modes. The interaction motifs that promote the formation of the condensed states of this protein are influenced by their flanking regions. The TDP-43 amyloid-core region (residues 321–330, orange) and the flanking aggregation hot-spots (residues 312–320 and 331–342, yellow) sample both ordered and disordered interactions (multi-modal binding). In contrast, most residues outside these regions are droplet-promoting (residues 262–311 and 343–414, blue), which sample mostly disordered interactions (unimodal binding). B The droplet landscape of TDP-43 prion-like domain illustrates the conversion between droplet and amyloid states. The likelihood of aggregation within droplets depends on two features99, the residue-specific multiplicity of binding modes (MBM) and the probability of undergoing liquid–liquid phase separation (LLPS). The multiplicity of binding modes characterises the ability of sampling both disordered interactions, which bias towards the droplet state (blue, based on PDB: 2N3X148), and ordered interactions, which bias towards the amyloid state (PDB:7KWZ149, orange). Both properties can be predicted from the sequence using the FuzDrop method (https://fuzdrop.bio.unipd.it)8. Droplet-promoting regions (blue circles) have a low multiplicity of binding modes in contrast to the amyloid core (orange triangles) and aggregation hot-spots (yellow diamonds), which exhibit high multiplicity of binding modes (large y values)99. C The sequence of the amyloidogenic region of TDP-43 (residues 311–360) is shown corresponding to the solution structure (PDB: 2N3X). The amyloid core is shown by orange, the aggregation hot-spot by yellow and the flanking residues by blue. The liquid–liquid phase separation of the prion-like domain of TDP-43 depends on the presence of an α-helical structural element125.
The nature of the inter-molecular interactions stabilising the droplet and amyloid states can be illustrated using the example of the prion-like domain of TDP-43 (Fig. 3A). Depending on the sequences of flanking regions103, the residues of the amyloid core and the flanking regions exhibit a multiplicity of binding modes (Fig. 3A). This property turns these residues into aggregation hot-spots that induce the conversions of the liquid-like into the solid-like state. In contrast, residues that promote droplet formation exhibit unimodal interactions and mostly sample disordered interactions (Fig. 3A). Thus the multiplicity of binding modes is a key feature to characterise the likelihood of conversion between the droplet and amyloid states, together with the residue-specific probability of undergoing liquid–liquid phase separation, as represented by droplet landscapes99 (Fig. 3B).
Therapeutic opportunities for protein condensation diseases
The observation that condensate-forming proteins appear to be ubiquitous in human disease opens the way to the development of therapeutic strategies capable of modulating their condensation behaviour and restore their physiological states (Table 2 and Fig. 4).
Table 2.
Examples of therapeutic opportunities for protein condensation diseases
| Protein state perturbation | Therapeutic opportunities | Mechanism of action | Examples |
|---|---|---|---|
| Native to amyloid | Antibodies against protein aggregation |
Removal of protein aggregates Inhibition of the protein aggregation process |
Aβ112,113 |
| Small molecules against protein aggregation |
Stabilisation of the native state Inhibition of the protein aggregation process |
TTR111, Aβ109,110 | |
| Small molecules promoting protein degradation | Activation of autophagy and of the ubiquitin–proteasome system to remove aggregating proteins and protein aggregates | mTOR139, PKA140 | |
| Small molecules inhibiting protein synthesis | Inhibition of enzymes required for the production of aggregating proteins | Aβ141 | |
| Small molecules promoting the unfolded protein response | Activation of the unfolded protein response to remove aggregating proteins and protein aggregates, or to inhibit the formation of protein aggregates | PERK142 | |
| Molecular chaperones against protein aggregation | Activation of the protein homoeostasis system to remove protein aggregates or inhibit their formation | Hsp70143 | |
| Native to droplet | Small molecules against protein liquid-liquid phase separation |
Stabilisation of the native state Perturbation of protein interactions within droplets |
SHP2108, M2-145, tau106 |
| Small molecules promoting protein degradation | Activation of autophagy to remove droplet-forming proteins | tau106 | |
| Small molecules regulating post-translational modifications | Activation of the protein homeostasis system to inhibit droplet formation | FUS144 | |
| Molecular chaperones against protein liquid-liquid phase separation | Activation of the protein homeostasis system to inhibit droplet formation | FUS144 | |
| Droplet to native | Small molecules regulating post-translational modifications | Prevention of droplet dissolution | DYRK323 |
| Droplet to amyloid | Small molecules against protein aggregation | Inhibition of the protein aggregation process | α-synuclein145 |
| Molecular chaperones against protein aggregation | Stabilisation of folded protein domains within droplets | FUS116 | |
| Molecular chaperones regulating cellular localisation | Re-localisation of nuclear RNA-binding proteins | FUS, hnRNPA1, hnRNPA2117 | |
| Amyloid to native | Small molecules against the conversion to the native state | Stabilisation of the functional amyloid state | Currently not known |
| Amyloid to droplet | Small molecules promoting droplet hardening | Stabilisation of the functional amyloid state | Currently not known |
We anticipate that many approaches developed for the prevention of the conversion between the native and the amyloid states will be applicable to the conversion between the native and droplet states, as well as between the droplet and amyloid states. Since for diseases associated with the conversion of the amyloid state into the native or droplet state there are no currently known examples of therapeutic approaches, we suggested possible mechanisms of action (in italics).
Fig. 4. Examples of therapeutic opportunities for protein condensation diseases.

Small molecules and antibodies are shown by brown circles. Candidate drugs can: (i) directly bind short sequence motifs that drive the formation of condensed states or stabilise them (A, F, I), (ii) interfere with the regulation of the assembly and disassembly of condensed states (G), (iii) modulate the stability of the native state (D, E, H, J), (iv) modify the concentration of a protein or its partners via inhibiting synthesis or inducing degradation (B, C, L), or (v) re-localise the protein itself (K). Examples of currently investigated approaches are listed in Table 2.
Small molecules could be developed to modulate the interactions required for the stability of the droplet state. This is a mechanism of action that may for example be applicable to regulate cancer-driving super-enhancers104. Support for this type of approach is provided by the case of the steroidal alkaloid cyclopamine, which was shown to block the replication of the respiratory syncytial virus (RSV) by hardening the interactions within the condensates of the host proteins that drive viral RNA synthesis45. An appealing aspect of this strategy is that protein condensates can selectively partition small molecules. Mitoxantrone, for example, was observed to be selectively concentrated in nuclear condensates of the transcriptional coactivator MED1 and of nucleophosmin, driven by interactions of aromatic groups105. Similarly, small molecules can be used to shift the phase boundaries between the native and condensed states. The flavonoid compound myricetin was shown to inhibit droplet formation of the protein tau, resulting in decreased aggregation and toxicity106. The phase boundary of TDP-43 was modulated by multivalent interactions of an aromatic compound, bis-ANS107. Small molecules can be further used to specifically destabilise conformations that drive droplet formation, as in the case of allosteric inhibitors of the protein tyrosine phosphatase SHP2, which restored its normal MAPK activity108.
Small molecules can also be exploited to interfere with protein aggregation. The nucleation and elongation rates in the aggregation process of the Aβ peptide were inhibited by compounds that can be potentially developed as drugs for Alzheimer’s disease109. Small molecules may also stabilise the native conformations of aggregation-prone proteins, thus inhibiting the conversion between the native and amyloid states110,111. In addition, the inhibition of the formation of toxic oligomers and the removal of amyloid aggregates by degradation pathways and can be promoted by conformation-specific antibodies112,113.
Alternatively, activation of degradation pathways can be exploited for the removal of aberrant liquid-like condensates. The ubiquitination of Ras GTPase-activating protein-binding protein 1 (G3BP1) was shown to induce stress-granule disassembly via its interactions with the ubiquitin-dependent segregase valosin114,115. Valosin is known to activate autophagy, and its familial mutations lead to delayed droplet clearance29.
More generally, the modulation of the protein homoeostasis system can be explored for therapeutic purposes in protein condensation diseases. Molecular chaperone activation may stabilise aggregation-prone domains within droplets, as shown by the chaperoning the folded RNA-binding domain of FUS by the small heat shock protein HspB8, which inhibited the formation of aberrant condensates116. Cellular relocalisation may also prevent droplet aggregation, as shown by karyopherin-β2, which dissolves aberrant fibrillar hydrogels formed by FUS and hnRNPA1, and importin-α with karyopherin-β1 can revert TDP-43 aggregation117. Furthermore, as condensate assembly and biophysical properties are also regulated by the concentration of interaction partners118, modifying the expression level of these partners may offer a strategy to regulate the condensed states. For example, stress-granule hyper-assembly induced by medulloblastoma-associated DDX3 mutants can be reverted by depletion of other assembly factors119. In addition, one could activate or inhibit post-translational modifications that regulate the condensed states, such as those that stabilise the droplet state120, or promote formation of prion-like states121. Inhibitors of the dual-specificity kinase DYRK3 for example can prevent stress-granule dissolution23.
Outlook
An increasing body of experimental observations suggests that protein condensation diseases may be ubiquitous. The strategies for drug discovery (Table 2) and the range of corresponding possible targets (Tables S1–S3) that we discussed here may be investigated further in future studies, given the growing interest in this therapeutic area. Although drug discovery targeting aberrant condensed states is likely to require different approaches than those used for stoichiometric complexes, proof-of-principle interventions to restore the balance between the different states of proteins have been already reported (Table 2). We anticipate that a better understanding of the nature of these diseases, and of the factors that regulate protein condensation, will promote the development of increasingly effective pharmacological approaches.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
M.F. acknowledges the financial support of AIRC Foundation for cancer research I.G. 26229.
Source data
Author contributions
M.V. and M.F. performed the research and wrote the paper.
Peer review
Peer review information
Nature Communications thanks the other anonymous reviewer(s) for their contribution to the peer review of this work.
Data availability
Gene–disease associations were derived from the DisGeNet database (http://disgenet.org), missense mutation-disease associations from the Human Variants Database (https://www.iitm.ac.in/bioinfo/huvarbase). Experimentally observed condensate-forming proteins were derived from three public databases: PhaSepDB data set (http://db.phasep.pro), PhaSePro (https://phasepro.elte.hu), LLPSDB (http://bio-comp.org.cn/llpsdb). For protein sequences, we used the UniProt database (uniprot.org). For GO enrichment we used the STRING (string-db.org) database. A list of protein condensation diseases is available at https://fuxreiterlab.github.io/databases_protein.html. The structures mentioned in this work are publicly available under the PDB accession codes 2N3X (Solution Structure of TDP-43 Amyloidogenic Core Region) and 7KWZ (TDP-43 LCD amyloid fibrils). Source data are provided with this paper.
Competing interests
M.V. is a founder of Wren Therapeutics. M.F. declares no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Michele Vendruscolo, Email: mv245@cam.ac.uk.
Monika Fuxreiter, Email: monika.fuxreiter@unipd.it.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-022-32940-7.
References
- 1.Alberts, B. Molecular Biology of the Cell 6th edn (Garland Science, 2015).
- 2.Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brangwynne CP, et al. Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science. 2009;324:1729–1732. doi: 10.1126/science.1172046. [DOI] [PubMed] [Google Scholar]
- 4.Fuxreiter M, Vendruscolo M. Generic nature of the condensed states of proteins. Nat. Cell Biol. 2021;23:587–594. doi: 10.1038/s41556-021-00697-8. [DOI] [PubMed] [Google Scholar]
- 5.Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 6.Wu H. Higher-order assemblies in a new paradigm of signal transduction. Cell. 2013;153:287–292. doi: 10.1016/j.cell.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fowler DM, Koulov AV, Balch WE, Kelly JW. Functional amyloid–from bacteria to humans. Trends Biochem. Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Hardenberg M, Horvath A, Ambrus V, Fuxreiter M, Vendruscolo M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc. Natl Acad. Sci. USA. 2020;117:33254–33262. doi: 10.1073/pnas.2007670117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vecchi G, et al. Proteome-wide observation of the phenomenon of life on the edge of solubility. Proc. Natl Acad. Sci. USA. 2020;117:1015–1020. doi: 10.1073/pnas.1910444117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lyon AS, Peeples WB, Rosen MK. A framework for understanding the functions of biomolecular condensates across scales. Nat. Rev. Mol. Cell Biol. 2021;22:215–235. doi: 10.1038/s41580-020-00303-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stoeger T, Battich N, Pelkmans L. Passive noise filtering by cellular compartmentalization. Cell. 2016;164:1151–1161. doi: 10.1016/j.cell.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Fowler DM, et al. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006;4:e6. doi: 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du M, Chen ZJ. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science. 2018;361:704–709. doi: 10.1126/science.aat1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Case LB, Zhang X, Ditlev JA, Rosen MK. Stoichiometry controls activity of phase-separated clusters of actin signaling proteins. Science. 2019;363:1093–1097. doi: 10.1126/science.aau6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaefer KN, Peifer M. Wnt/Beta-catenin signaling regulation and a role for biomolecular condensates. Dev. Cell. 2019;48:429–444. doi: 10.1016/j.devcel.2019.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimobayashi SF, Ronceray P, Sanders DW, Haataja MP, Brangwynne CP. Nucleation landscape of biomolecular condensates. Nature. 2021;599:503–506. doi: 10.1038/s41586-021-03905-5. [DOI] [PubMed] [Google Scholar]
- 17.Woodruff JB, et al. The centrosome is a selective condensate that nucleates microtubules by concentrating tubulin. Cell. 2017;169:1066–1077. doi: 10.1016/j.cell.2017.05.028. [DOI] [PubMed] [Google Scholar]
- 18.Kilic S, et al. Phase separation of 53BP1 determines liquid‐like behavior of DNA repair compartments. EMBO J. 2019;38:e101379. doi: 10.15252/embj.2018101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larson AG, et al. Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature. 2017;547:236–240. doi: 10.1038/nature22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu A, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou F, et al. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alberti S, Hyman AA. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021;22:196–213. doi: 10.1038/s41580-020-00326-6. [DOI] [PubMed] [Google Scholar]
- 23.Wippich F, et al. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell. 2013;152:791–805. doi: 10.1016/j.cell.2013.01.033. [DOI] [PubMed] [Google Scholar]
- 24.Berchtold D, Battich N, Pelkmans L. A systems-level study reveals regulators of membrane-less organelles in human cells. Mol. Cell. 2018;72:1035–1049. doi: 10.1016/j.molcel.2018.10.036. [DOI] [PubMed] [Google Scholar]
- 25.Shattuck JE, Paul KR, Cascarina SM, Ross ED. The prion-like protein kinase Sky1 is required for efficient stress granule disassembly. Nat. Commun. 2019;10:3614. doi: 10.1038/s41467-019-11550-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walters RW, Muhlrad D, Garcia J, Parker R. Differential effects of Ydj1 and Sis1 on Hsp70-mediated clearance of stress granules in Saccharomyces cerevisiae. RNA. 2015;21:1660–1671. doi: 10.1261/rna.053116.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mateju D, et al. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017;36:1669–1687. doi: 10.15252/embj.201695957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie X, et al. Deubiquitylases USP5 and USP13 are recruited to and regulate heat-induced stress granules through their deubiquitylating activities. J. Cell Sci. 2018;131:jcs210856. doi: 10.1242/jcs.210856. [DOI] [PubMed] [Google Scholar]
- 29.Buchan JR, Kolaitis R-M, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell. 2013;153:1461–1474. doi: 10.1016/j.cell.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaffagnini G, et al. p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J. 2018;37:e98308. doi: 10.15252/embj.201798308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y, et al. Cytoplasmic DAXX drives SQSTM1/p62 phase condensation to activate Nrf2-mediated stress response. Nat. Commun. 2019;10:3759. doi: 10.1038/s41467-019-11671-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kageyama S, et al. p62/SQSTM1-droplet serves as a platform for autophagosome formation and anti-oxidative stress response. Nat. Commun. 2021;12:16. doi: 10.1038/s41467-020-20185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng S-z, et al. Phase separation of Nur77 mediates celastrol-induced mitophagy by promoting the liquidity of p62/SQSTM1 condensates. Nat. Commun. 2021;12:5989. doi: 10.1038/s41467-021-26295-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bennett CL, et al. Senataxin mutations elicit motor neuron degeneration phenotypes and yield TDP-43 mislocalization in ALS4 mice and human patients. Acta Neuropathol. 2018;136:425–443. doi: 10.1007/s00401-018-1852-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alberti S, Dormann D. Liquid–liquid phase separation in disease. Annu. Rev. Genet. 2019;53:171–194. doi: 10.1146/annurev-genet-112618-043527. [DOI] [PubMed] [Google Scholar]
- 36.Banani SF, et al. Genetic variation associated with condensate dysregulation in disease. Dev. Cell. 2022;57:1776–1788. doi: 10.1016/j.devcel.2022.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mathieu C, Pappu RV, Taylor JP. Beyond aggregation: pathological phase transitions in neurodegenerative disease. Science. 2020;370:56–60. doi: 10.1126/science.abb8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsang B, Pritišanac I, Scherer SW, Moses AM, Forman-Kay JD. Phase separation as a missing mechanism for interpretation of disease mutations. Cell. 2020;183:1742–1756. doi: 10.1016/j.cell.2020.11.050. [DOI] [PubMed] [Google Scholar]
- 39.Chiti F, Dobson CM. Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu. Rev. Biochem. 2017;86:27–68. doi: 10.1146/annurev-biochem-061516-045115. [DOI] [PubMed] [Google Scholar]
- 40.Li CH, et al. MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature. 2020;586:440–444. doi: 10.1038/s41586-020-2574-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiang Y, et al. Rett syndrome linked to defects in forming the MeCP2/Rbfox/LASR complex in mouse models. Nat. Commun. 2021;12:5767. doi: 10.1038/s41467-021-26084-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quiroz FG, et al. Liquid-liquid phase separation drives skin barrier formation. Science. 2020;367:eaax9554. doi: 10.1126/science.aax9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kanaan NM, Hamel C, Grabinski T, Combs B. Liquid-liquid phase separation induces pathogenic tau conformations in vitro. Nat. Commun. 2020;11:2809. doi: 10.1038/s41467-020-16580-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heinrich BS, Maliga Z, Stein DA, Hyman AA, Whelan SP. Phase transitions drive the formation of vesicular stomatitis virus replication compartments. mBio. 2018;9:e02290–02217. doi: 10.1128/mBio.02290-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Risso-Ballester J, et al. A condensate-hardening drug blocks RSV replication in vivo. Nature. 2021;595:596–599. doi: 10.1038/s41586-021-03703-z. [DOI] [PubMed] [Google Scholar]
- 46.Bouchard JJ, et al. Cancer mutations of the tumor suppressor SPOP disrupt the formation of active, phase-separated compartments. Mol. Cell. 2018;72:19–36. doi: 10.1016/j.molcel.2018.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee Y, et al. TIA1 variant drives myodegeneration in multisystem proteinopathy with SQSTM1 mutations. J. Clin. Investig. 2018;128:1164–1177. doi: 10.1172/JCI97103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li W, et al. Biophysical properties of AKAP95 protein condensates regulate splicing and tumorigenesis. Nat. Cell Biol. 2020;22:960–972. doi: 10.1038/s41556-020-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ramaswami M, Taylor JP, Parker R. Altered ribostasis: RNA-protein granules in degenerative disorders. Cell. 2013;154:727–736. doi: 10.1016/j.cell.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gopal PP, Nirschl JJ, Klinman E, Holzbaur EL. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 rnp granules in neurons. Proc. Natl Acad. Sci. USA. 2017;114:E2466–E2475. doi: 10.1073/pnas.1614462114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim HJ, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murakami T, et al. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs rnp granule function. Neuron. 2015;88:678–690. doi: 10.1016/j.neuron.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mackenzie IR, et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron. 2017;95:808–816. doi: 10.1016/j.neuron.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J, et al. Neurotoxic microglia promote TDP-43 proteinopathy in progranulin deficiency. Nature. 2020;588:459–465. doi: 10.1038/s41586-020-2709-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen C, et al. Phase separation drives RNA virus-induced activation of the NLRP6 inflammasome. Cell. 2021;184:5759–5774. doi: 10.1016/j.cell.2021.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McDonald NA, Fetter RD, Shen K. Assembly of synaptic active zones requires phase separation of scaffold molecules. Nature. 2020;588:454–458. doi: 10.1038/s41586-020-2942-0. [DOI] [PubMed] [Google Scholar]
- 57.Zhang C, Rabouille C. Membrane-bound meet membraneless in health and disease. Cells. 2019;8:1000. doi: 10.3390/cells8091000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao YG, Zhang H. Phase separation in membrane biology: The interplay between membrane-bound organelles and membraneless condensates. Dev. Cell. 2020;55:30–44. doi: 10.1016/j.devcel.2020.06.033. [DOI] [PubMed] [Google Scholar]
- 59.Koppers M, Özkan N, Farías GG. Complex interactions between membrane-bound organelles, biomolecular condensates and the cytoskeleton. Front. Cell Dev. Biol. 2020;8:618733. doi: 10.3389/fcell.2020.618733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee JE, Cathey PI, Wu H, Parker R, Voeltz GK. Endoplasmic reticulum contact sites regulate the dynamics of membraneless organelles. Science. 2020;367:eaay7108. doi: 10.1126/science.aay7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma W, Mayr C. A membraneless organelle associated with the endoplasmic reticulum enables 3′ UTR-mediated protein-protein interactions. Cell. 2018;175:1492–1506. doi: 10.1016/j.cell.2018.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Snead WT, et al. Membrane surfaces regulate assembly of ribonucleoprotein condensates. Nat. Cell Biol. 2022;24:461–470. doi: 10.1038/s41556-022-00882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu X, et al. The STING phase-separator suppresses innate immune signalling. Nat. Cell Biol. 2021;23:330–340. doi: 10.1038/s41556-021-00659-0. [DOI] [PubMed] [Google Scholar]
- 64.Liao Y-C, et al. RNA granules hitchhike on lysosomes for long-distance transport, using annexin A11 as a molecular tether. Cell. 2019;179:147–164. doi: 10.1016/j.cell.2019.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Astro V, Chiaretti S, Magistrati E, Fivaz M, De Curtis I. Liprin-α1, ERC1 and LL5 define polarized and dynamic structures that are implicated in cell migration. J. Cell Sci. 2014;127:3862–3876. doi: 10.1242/jcs.155663. [DOI] [PubMed] [Google Scholar]
- 66.Zeng M, et al. Phase transition in postsynaptic densities underlies formation of synaptic complexes and synaptic plasticity. Cell. 2016;166:1163–1175. doi: 10.1016/j.cell.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zeng M, et al. Reconstituted postsynaptic density as a molecular platform for understanding synapse formation and plasticity. Cell. 2018;174:1172–1187. doi: 10.1016/j.cell.2018.06.047. [DOI] [PubMed] [Google Scholar]
- 68.Milovanovic D, Wu Y, Bian X, De Camilli P. A liquid phase of synapsin and lipid vesicles. Science. 2018;361:604–607. doi: 10.1126/science.aat5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Su X, et al. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science. 2016;352:595–599. doi: 10.1126/science.aad9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stenström L, et al. Mapping the nucleolar proteome reveals a spatiotemporal organization related to intrinsic protein disorder. Mol. Syst. Biol. 2020;16:e9469. doi: 10.15252/msb.20209469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saitoh N, et al. Proteomic analysis of interchromatin granule clusters. Mol. Biol. Cell. 2004;15:3876–3890. doi: 10.1091/mbc.e04-03-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morelli FF, et al. Aberrant compartment formation by HSPB2 mislocalizes lamin A and compromises nuclear integrity and function. Cell Rep. 2017;20:2100–2115. doi: 10.1016/j.celrep.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sponga A, et al. Order from disorder in the sarcomere: FATZ forms a fuzzy but tight complex and phase-separated condensates with α-actinin. Sci. Adv. 2021;7:eabg7653. doi: 10.1126/sciadv.abg7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vu L, et al. Defining the caprin-1 interactome in unstressed and stressed conditions. J. Proteome Res. 2021;20:3165–3178. doi: 10.1021/acs.jproteome.1c00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Youn J-Y, et al. Properties of stress granule and P-body proteomes. Mol. Cell. 2019;76:286–294. doi: 10.1016/j.molcel.2019.09.014. [DOI] [PubMed] [Google Scholar]
- 76.Jang S, et al. Glycolytic enzymes localize to synapses under energy stress to support synaptic function. Neuron. 2016;90:278–291. doi: 10.1016/j.neuron.2016.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shi B, et al. UTX condensation underlies its tumour-suppressive activity. Nature. 2021;597:726–731. doi: 10.1038/s41586-021-03903-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krainer G, et al. Reentrant liquid condensate phase of proteins is stabilized by hydrophobic and non-ionic interactions. Nat. Commun. 2021;12:1085. doi: 10.1038/s41467-021-21181-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nott TJ, et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell. 2015;57:936–947. doi: 10.1016/j.molcel.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Murthy AC, et al. Molecular interactions underlying liquid-liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol. 2019;26:637–648. doi: 10.1038/s41594-019-0250-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lin Y, Protter DS, Rosen MK, Parker R. Formation and maturation of phase-separated liquid droplets by RNA-binding proteins. Mol. Cell. 2015;60:208–219. doi: 10.1016/j.molcel.2015.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bienz M. Head-to-tail polymerization in the assembly of biomolecular condensates. Cell. 2020;182:799–811. doi: 10.1016/j.cell.2020.07.037. [DOI] [PubMed] [Google Scholar]
- 84.Rana U, Brangwynne CP, Panagiotopoulos AZ. Phase separation vs aggregation behavior for model disordered proteins. J. Chem. Phys. 2021;155:125101. doi: 10.1063/5.0060046. [DOI] [PubMed] [Google Scholar]
- 85.Wu H, Fuxreiter M. The structure and dynamics of higher-order assemblies: amyloids, signalosomes, and granules. Cell. 2016;165:1055–1066. doi: 10.1016/j.cell.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Knowles TP, et al. Role of intermolecular forces in defining material properties of protein nanofibrils. Science. 2007;318:1900–1903. doi: 10.1126/science.1150057. [DOI] [PubMed] [Google Scholar]
- 87.Vernon RM, et al. π-π contacts are an overlooked protein feature relevant to phase separation. eLife. 2018;7:e31486. doi: 10.7554/eLife.31486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schmidt HB, Barreau A, Rohatgi R. Phase separation-deficient TDP43 remains functional in splicing. Nat. Commun. 2019;10:4890. doi: 10.1038/s41467-019-12740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Burke KA, Janke AM, Rhine CL, Fawzi NL. Residue-by-residue view of in vitro FUS granules that bind the C-terminal domain of RNA polymerase ii. Mol. Cell. 2015;60:231–241. doi: 10.1016/j.molcel.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Martin EW, et al. Valence and patterning of aromatic residues determine the phase behavior of prion-like domains. Science. 2020;367:694–699. doi: 10.1126/science.aaw8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tóth-Petróczy Á, et al. Assessing conservation of disordered regions in proteins. Open Proteom. J. 2008;1:46–53. doi: 10.2174/1875039700801010046. [DOI] [Google Scholar]
- 92.Banjade S, et al. Conserved interdomain linker promotes phase separation of the multivalent adaptor protein nck. Proc. Natl Acad. Sci. USA. 2015;112:E6426–E6435. doi: 10.1073/pnas.1508778112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Riback JA, et al. Composition-dependent thermodynamics of intracellular phase separation. Nature. 2020;581:209–214. doi: 10.1038/s41586-020-2256-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Franzmann TM, et al. Phase separation of a yeast prion protein promotes cellular fitness. Science. 2018;359:eaao5654. doi: 10.1126/science.aao5654. [DOI] [PubMed] [Google Scholar]
- 95.Maharana S, et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science. 2018;360:918–921. doi: 10.1126/science.aar7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ukmar-Godec T, et al. Lysine/RNA-interactions drive and regulate biomolecular condensation. Nat. Commun. 2019;10:2909. doi: 10.1038/s41467-019-10792-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim TH, et al. Phospho-dependent phase separation of FMRP and caprin1 recapitulates regulation of translation and deadenylation. Science. 2019;365:825–829. doi: 10.1126/science.aax4240. [DOI] [PubMed] [Google Scholar]
- 98.Gibson BA, et al. Organization of chromatin by intrinsic and regulated phase separation. Cell. 2019;179:470–484. doi: 10.1016/j.cell.2019.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vendruscolo M, Fuxreiter M. Sequence determinants of the aggregation of proteins within condensates generated by liquid-liquid phase separation. J. Mol. Biol. 2022;434:167201. doi: 10.1016/j.jmb.2021.167201. [DOI] [PubMed] [Google Scholar]
- 100.Sun Y, et al. The nuclear localization sequence mediates hnRNPA1 amyloid fibril formation revealed by cryoEM structure. Nat. Commun. 2020;11:6349. doi: 10.1038/s41467-020-20227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gui X, et al. Structural basis for reversible amyloids of hnRNPA1 elucidates their role in stress granule assembly. Nat. Commun. 2019;10:2006. doi: 10.1038/s41467-019-09902-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Patel A, et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- 103.Gianni S, et al. Fuzziness and frustration in the energy landscape of protein folding, function, and assembly. Acc. Chem. Res. 2021;54:1251–1259. doi: 10.1021/acs.accounts.0c00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sabari BR, et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018;361:eaar3958. doi: 10.1126/science.aar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Klein IA, et al. Partitioning of cancer therapeutics in nuclear condensates. Science. 2020;368:1386–1392. doi: 10.1126/science.aaz4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dai, B. et al. Myricetin slows liquid–liquid phase separation of tau and activates ATG5-dependent autophagy to suppress tau toxicity. J. Biol. Chem. 297, 101222 (2021). [DOI] [PMC free article] [PubMed]
- 107.Babinchak WM, et al. Small molecules as potent biphasic modulators of protein liquid-liquid phase separation. Nat. Commun. 2020;11:5574. doi: 10.1038/s41467-020-19211-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhu G, et al. Phase separation of disease-associated SHP2 mutants underlies MAPK hyperactivation. Cell. 2020;183:490–502. doi: 10.1016/j.cell.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Habchi J, et al. Systematic development of small molecules to inhibit specific microscopic steps of Aβ42 aggregation in Alzheimer’s disease. Proc. Natl Acad. Sci. USA. 2017;114:E200–E208. doi: 10.1073/pnas.1615613114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Heller GT, et al. Small-molecule sequestration of amyloid-β as a drug discovery strategy for Alzheimer’s disease. Sci. Adv. 2020;6:eabb5924. doi: 10.1126/sciadv.abb5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Coelho T, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79:785–792. doi: 10.1212/WNL.0b013e3182661eb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Linse S, et al. Kinetic fingerprints differentiate the mechanisms of action of anti-Aβ antibodies. Nat. Struct. Mol. Biol. 2020;27:1125–1133. doi: 10.1038/s41594-020-0505-6. [DOI] [PubMed] [Google Scholar]
- 113.Sevigny J, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 114.Dao TP, et al. ALS-linked mutations affect UBQLN2 oligomerization and phase separation in a position-and amino acid-dependent manner. Structure. 2019;27:937–951. doi: 10.1016/j.str.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gwon Y, et al. Ubiquitination of G3BP1 mediates stress granule disassembly in a context-specific manner. Science. 2021;372:eabf6548. doi: 10.1126/science.abf6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Boczek EE, et al. HspB8 prevents aberrant phase transitions of FUS by chaperoning its folded RNA-binding domain. eLife. 2021;10:e69377. doi: 10.7554/eLife.69377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Guo L, et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell. 2018;173:677–692. doi: 10.1016/j.cell.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang H, et al. RNA controls polyQ protein phase transitions. Mol. Cell. 2015;60:220–230. doi: 10.1016/j.molcel.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Valentin-Vega YA, et al. Cancer-associated DDX3X mutations drive stress granule assembly and impair global translation. Sci. Rep. 2016;6:25996. doi: 10.1038/srep25996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Makwana KM, Sarnowski MP, Miao J, Lin Y-S, Del Valle JR. N-amination converts amyloidogenic tau peptides into soluble antagonists of cellular seeding. ACS Chem. Neurosci. 2021;12:3928–3938. doi: 10.1021/acschemneuro.1c00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li J, et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell. 2012;150:339–350. doi: 10.1016/j.cell.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yamamoto T, et al. Functional assessment of the mutational effects of human IRAK4 and MyD88 genes. Mol. Immunol. 2014;58:66–76. doi: 10.1016/j.molimm.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 123.Wang L, et al. The FAS-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat. Struct. Mol. Biol. 2010;17:1324–1329. doi: 10.1038/nsmb.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rhine K, et al. ALS/FTD-linked mutations in FUS glycine residues cause accelerated gelation and reduced interactions with wild-type FUS. Mol. Cell. 2020;80:666–681. doi: 10.1016/j.molcel.2020.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Conicella AE, Zerze GH, Mittal J, Fawzi NL. ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure. 2016;24:1537–1549. doi: 10.1016/j.str.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.French RL, et al. Detection of tar DNA-binding protein 43 (TDP-43) oligomers as initial intermediate species during aggregate formation. J. Biol. Chem. 2019;294:6696–6709. doi: 10.1074/jbc.RA118.005889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Molliex A, et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–133. doi: 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ryan VH, et al. Mechanistic view of hnRNPA2 low-complexity domain structure, interactions, and phase separation altered by mutation and arginine methylation. Mol. Cell. 2018;69:465–479. doi: 10.1016/j.molcel.2017.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Murray DT, et al. Structural characterization of the D290V mutation site in hnRNPA2 low-complexity–domain polymers. Proc. Natl Acad. Sci. USA. 2018;115:E9782–E9791. doi: 10.1073/pnas.1806174115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lu J, et al. CryoEM structure of the low-complexity domain of hnRNPA2 and its conversion to pathogenic amyloid. Nat. Commun. 2020;11:4090. doi: 10.1038/s41467-020-17905-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Baek M, et al. TDP-43 and PINK1 mediate CHCHD10S59L mutation–induced defects in drosophila and in vitro. Nat. Commun. 2021;12:1924. doi: 10.1038/s41467-021-22145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wegmann S, et al. Tau protein liquid–liquid phase separation can initiate tau aggregation. EMBO J. 2018;37:e98049. doi: 10.15252/embj.201798049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Batlle C, et al. hnRNPDL phase separation is regulated by alternative splicing and disease-causing mutations accelerate its aggregation. Cell Rep. 2020;30:1117–1128. doi: 10.1016/j.celrep.2019.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Peskett TR, et al. A liquid to solid phase transition underlying pathological huntingtin exon1 aggregation. Mol. Cell. 2018;70:588–601. doi: 10.1016/j.molcel.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gallego-Iradi M, et al. N-terminal sequences in matrin 3 mediate phase separation into droplet-like structures that recruit TDP43 variants lacking RNA binding elements. Lab. Invest. 2019;99:1030–1040. doi: 10.1038/s41374-019-0260-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.He Y, Li J, Zhang M. Myosin vii, USH1C, and ANKS4B or USH1G together form condensed molecular assembly via liquid-liquid phase separation. Cell Rep. 2019;29:974–986. doi: 10.1016/j.celrep.2019.09.027. [DOI] [PubMed] [Google Scholar]
- 137.Schneider JW, et al. Dysregulated ribonucleoprotein granules promote cardiomyopathy in RBM20 gene-edited pigs. Nat. Med. 2020;26:1788–1800. doi: 10.1038/s41591-020-1087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cloer E, et al. p62-dependent phase separation of patient-derived KEAP1 mutations and NRF2. Mol. Cell. Biol. 2018;38:e00644. doi: 10.1128/MCB.00644-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Myeku N, et al. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016;22:46–53. doi: 10.1038/nm.4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Boland B, et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2018;17:660–688. doi: 10.1038/nrd.2018.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kennedy ME, et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016;8:363ra150. doi: 10.1126/scitranslmed.aad9704. [DOI] [PubMed] [Google Scholar]
- 142.Moreno JA, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013;5:206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- 143.Nachman E, et al. Disassembly of tau fibrils by the human Hsp70 disaggregation machinery generates small seeding-competent species. J. Biol. Chem. 2020;295:9676–9690. doi: 10.1074/jbc.RA120.013478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Qamar S, et al. FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation-π interactions. Cell. 2018;173:720–734. doi: 10.1016/j.cell.2018.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sawner AS, et al. Modulating α-synuclein liquid–liquid phase separation. Biochemistry. 2021;60:3676–3696. doi: 10.1021/acs.biochem.1c00434. [DOI] [PubMed] [Google Scholar]
- 146.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 147.Hipp MS, Kasturi P, Hartl FU. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019;20:421–435. doi: 10.1038/s41580-019-0101-y. [DOI] [PubMed] [Google Scholar]
- 148.Jiang L-L, et al. Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Sci. Rep. 2016;6:1–11. doi: 10.1038/s41598-016-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li Q, Babinchak WM, Surewicz WK. Cryo-EM structure of amyloid fibrils formed by the entire low complexity domain of TDP-43. Nat. Commun. 2021;12:1620. doi: 10.1038/s41467-021-21912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Gene–disease associations were derived from the DisGeNet database (http://disgenet.org), missense mutation-disease associations from the Human Variants Database (https://www.iitm.ac.in/bioinfo/huvarbase). Experimentally observed condensate-forming proteins were derived from three public databases: PhaSepDB data set (http://db.phasep.pro), PhaSePro (https://phasepro.elte.hu), LLPSDB (http://bio-comp.org.cn/llpsdb). For protein sequences, we used the UniProt database (uniprot.org). For GO enrichment we used the STRING (string-db.org) database. A list of protein condensation diseases is available at https://fuxreiterlab.github.io/databases_protein.html. The structures mentioned in this work are publicly available under the PDB accession codes 2N3X (Solution Structure of TDP-43 Amyloidogenic Core Region) and 7KWZ (TDP-43 LCD amyloid fibrils). Source data are provided with this paper.