

Abstract
The phenotypic transformation of astrocytes in Alzheimer’s disease (AD) is still not well understood. Recent analyses based on single-nucleus RNA sequencing of postmortem Alzheimer’s disease (AD) samples are limited by the low number of sequenced astrocytes, small cohort sizes, and low number of differentially expressed genes detected. To optimize the detection of astrocytic genes, we employed a novel strategy consisting of the localization of pre-determined astrocyte and neuronal gene clusters in publicly available whole-brain transcriptomes. Specifically, we used cortical transcriptomes from 766 individuals, including cognitively normal subjects (Controls), and people diagnosed with mild cognitive impairment (MCI) or dementia due to AD. Samples came from three independent cohorts organized by the Mount Sinai Hospital, the Mayo Clinic, and the Religious Order Study/Memory and Aging Project (ROSMAP). Astrocyte- and neuron-specific gene clusters were generated from human brain cell-type specific RNAseq data using hierarchical clustering and cell-type enrichment scoring. Genes from each cluster were manually annotated according to cell-type specific functional categories. Gene Set Variation Analysis (GSVA) and Principal Component Analysis (PCA) were used to establish changes in these functional categories among clinical cohorts. We highlight three novel findings of the study. First, individuals with the same clinical diagnosis were molecularly heterogeneous. Particularly in the Mayo Clinic and ROSMAP cohorts, over 50% of Controls presented down-regulation of genes encoding synaptic proteins typical of AD, whereas 30% of patients diagnosed with dementia due to AD presented Control-like transcriptomic profiles. Second, down-regulation of neuronal genes related to synaptic proteins coincided, in astrocytes, with up-regulation of genes related to perisynaptic astrocytic processes (PAP) and down-regulation of genes encoding endolysosomal and mitochondrial proteins. Third, down-regulation of astrocytic mitochondrial genes inversely correlated with the disease stages defined by Braak and CERAD scoring. Finally, we interpreted these changes as maladaptive or adaptive from the point of view of astrocyte biology in a model of the phenotypical transformation of astrocytes in AD. The main prediction is that early malfunction of the astrocytic endolysosomal system, associated with progressive mitochondrial dysfunction, contribute to Alzheimer’s disease. If this prediction is correct, therapies preventing organelle dysfunction in astrocytes may be beneficial in preclinical and clinical AD.
Keywords: Alzheimer’s disease, astrocytes, hierarchical clustering, MCI, mitochondria, perisynaptic astrocyte processes, RNA seq
Graphical Abstract
Introduction
‘Reactive’ GFAP-overexpressing astrocytes are found in the vicinity of amyloid-β plaques in postmortem brains from patients with Alzheimer’s disease (AD), in both the autosomal-dominant Alzheimer’s disease (ADAD) and the sporadic late-onset (AD) variants, as well as in mouse models of ADAD. The robust morphological transformation of astrocytes points to major phenotypical and, hence, functional alterations in astrocytes in AD (reviewed in (Perez-Nievas & Serrano-Pozo, 2018)).
However, despite non-negligible research in the last decade, knowledge about phenotypical alterations in human astrocytes from AD patients is still limited due, in part, to scarce and conflicting human astrocyte-specific omics data. Prior studies include transcriptomics of laser-microdissected astrocytes (Sekar et al., 2015; Simpson et al., 2011), co-expression-based gene clustering of whole-brain AD transcriptomes (B. Zhang et al., 2013), and single-cell RNA sequencing (scRNAseq) analyses (Grubman et al., 2019; Mathys et al., 2019). Recently, the study of phenotypic changes of astrocytes in AD has been approached with network analysis of postmortem immunohistochemical data obtained from perusal of the literature (Viejo et al., 2021).
Laser-microdissected astrocytes either presented an insufficient number of differentially-expressed genes (DEG) for pathway analysis (Sekar et al., 2015), or normalization anomalies, as suggested by the finding that 98% of the DEGs were down-regulated (Simpson et al., 2011). The problem may lie in the low RNA yields of laser microdissection, exacerbated by the poor RNA quality after long post-mortem intervals. As an alternative to laser microdissection, clustering statistics based on gene co-expression identified function-specific gene modules using 1647 whole-brain AD transcriptomes (B. Zhang et al., 2013). Cellular localization of modules was established using cell-specific markers, and causality between nodes was inferred with Bayesian statistics. Using this approach, alterations of glutamate and amino acid metabolism in an astrocytic module of 260 genes were discovered to be well-ranked for causal relevance in AD, but no further changes were unraveled.
Finally, although scRNAseq improves the cellular resolution of transcriptomics to the point of unraveling populations within a given cell-type, two challenges arise in studying astrocytes from postmortem brain samples with this technique. One is the low numbers of astrocytes per patient being sequenced, which renders astrocytic datasets underpowered (discussed in (Liddelow, Marsh, & Stevens, 2020). The second challenge is to analyze a sufficiently large number of subjects to ensure that patient heterogeneity, a critical factor for drug development in AD (Devi & Scheltens, 2018), manifests itself. For example, (Mathys et al., 2019) and (Grubman et al., 2019) examined only 48 and 12 subjects, respectively, including patients and Controls.
The main goal of this study was to extract consensus data of astrocytic changes from bulk transcriptomic data from three large independent clinical AD databases, encompassing 766 subjects. ‘Consensus data’ was defined as data detected in at least two out of the three databases. Identification of astrocyte-specific genes was optimized by taking a reverse approach as compared to previous studies: instead of determining a posteriori which self-organized gene modules might be astrocytic, we localized pre-determined astrocytic gene clusters in whole-brain AD transcriptomes with two-dimensional hierarchical clustering. The workflow is depicted in Fig. 1. First, we classified brain genes as cell-specific or non-specific using RNAseq data from astrocytes, neurons, microglia, endothelial cells and oligodendrocytes isolated from aged healthy human brains (Y. Zhang et al., 2016), and a combination of two algorithms: hierarchical clustering, and a recently proposed univariate cell-type enrichment τ scoring (Kryuchkova-Mostacci & Robinson-Rechavi, 2017). Second, astrocytic and, for comparison, neuronal genes, were manually annotated according to functions. Third, neuronal and astrocytic gene clusters were localized in three AD whole-brain transcriptomes generated by the Mount Sinai Hospital (MtSINAI) (B. Zhang et al., 2013) the Mayo Clinic (MAYO) (Allen et al., 2016), and the Religious Order Study and the Rush Memory and Aging project (ROSMAP), which also contains subjects diagnosed with MCI (Bennett et al., 2018). These databases present the advantages of including large cohorts (78–219 subjects), which greatly facilitates the use of multivariate systems approaches, and of having been generated by two different approaches, microarrays and RNAseq, thus diminishing technique-associated bias. Finally, alteration of astrocytic and neuronal functional categories in AD and MCI groups vs Controls was statistically established using gene set variation analysis (GSVA), and principal component analysis (PCA).
Figure 1. Workflow for the identification of altered astrocytic functions in AD and MCI (See Methods for details).

Step 1–3: Genes from human-brain cortex were classified as cell-specific or enriched using RNAseq data from cells isolated from aged healthy human brain cortices (Step 1), the univariate cell-type enrichment τ scoring (Step 2), and hierarchical clustering (Step 3).
Step 4: The astrocytic and neuronal gene clusters were manually categorized into functions and subfunctions.
Step 5: Control, AD, and MCI whole-brain transcriptomes from the MtSINAI, MAYO and ROSMAP databases were hierarchically sorted according to the cell-type specific clusters.
Step 6–7: Alterations of astrocytic and neuronal functional categories in AD and MCI groups vs Controls were statistically established using gene set variation analysis (GSVA, Step 6), and principal component analysis (PCA, Step 7).
Step 8: To compare patients with mice, the published transcriptome of APP/PS1 astrocytes was reanalyzed using our functional categorization and GSVA. The figure in Step 1 was created using cell art adapted from Servier Medical Art (https://smart.servier.com/).
We present the most comprehensive transcriptomic analysis of human AD astrocytes to date. As advocated in a recent consensus article about reactive astrocytes (Escartin et al., 2021), the impact of pathway alterations suggested by omics was interpreted in the context of astrocyte biology, instead of resorting to simplistic categorizations of astrocytes as ‘neuroprotective’ or ‘neurotoxic’ from the point of view of neurons. In this vein, our analysis points to dysfunction of the endolysosomal system/mitochondrion axis as a key driver of the phenotypical transformation of reactive astrocytes in AD.
Material & Methods
Transcriptome datasets
Cell-type specific RNAseq datasets generated from healthy human-brain cortical tissue (Y. Zhang et al., 2016) were downloaded from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) under the accession number GSE73721. Data were pre-aligned to gene symbols by the authors using human genome version 19. Data consisted of transcriptomic data from brain cells isolated from temporal cortical lobes of juvenile (8–18 years old) and adult (21–63 years old) non-demented individuals. Transcriptomes from 12 astrocytic, one neuronal, five oligodendrocytic, three microglial, and two endothelial-cell samples from individual patients were included (Supplementary file 1, tab 2). Principal component analysis (PCA, computed in R, R Foundation for Statistical Computing, Vienna, Austria) revealed that the transcriptomic data of astrocytes isolated from young (<47 years old) and old (>47 years old) were separated along principal component 1 (PC1, Supplementary file 1, tab 3). Since the goal of the study was to identify cell-specific signatures in aged subjects, data from young individuals were discarded, retaining a total of six astrocyte transcriptomes from 47–63-year-old subjects, together with all 11 transcriptomes from the remaining cell types. A threshold was set for gene expression such that a given transcript should present a count > 1 in reads per kilo base per million mapped reads (RPKM) in at least one sample. Genes with no counts > 1 were discarded from the analysis. 11077 genes were above this threshold (Supplementary file 1, tab 4).
The MtSINAI database containing microarray data of samples from the prefrontal cortex of 101 Controls and 129 AD patients obtained from the Harvard Brain Tissue Resource Center was downloaded from GEO under accession number GSE44770 (B. Zhang et al., 2013). The Rosetta gene identifiers were converted to the corresponding gene symbols using the Rosetta/Merck Human 44k 1.1 microarray platform table under accession number GPL4372. When multiple Rosetta identifiers mapped to the same gene, the identifier with the greatest variance across samples was selected. The MAYO database, which contains data from the temporal cortex of 78 Control samples and 82 AD samples, was downloaded from Synapse.org under Synapse ID syn3163039 (Allen et al., 2016). The data were downloaded as MayoRNAseq_RNAseq_TCXCounts_normalized.tsv, pre-aligned to Ensemble Gene Identifier. Ensemble Gene IDs were then converted to HGCN gene symbols using biomaRt v. 2.32.1 in R. When multiple Ensemble Gene IDs mapped to the same gene symbol, the one with greatest variance was selected. The ROSMAP database (Bennett et al., 2018) contains data from the dorsolateral prefrontal cortex obtained from autopsied non-demented individuals or patients diagnosed with AD. The data were downloaded from Synapse.org under synapse ID syn8456629 as ROSMAP_DLPFC_netResidualExpression.tsv. The dataset contained 200 Control cases, 157 MCI cases, and 219 AD cases. In the MCI group, there was no stratification into MCI and MCI-AD. The dataset as posted by the authors had been adjusted via voom (mean-variance modelling at the observational level) normalization to remove bias associated with batch number, RNA integrity, and other data acquisition and processing co-variates. Missing transcript values in the dataset were imputed using a nearest neighbor averaging method using the impute package in R.
Univariate cell-type enrichment score
To determine the cell-specificity of human genes listed in the brain-cell transcriptomics database (Y. Zhang et al., 2016), we used the τ method, reported to be particularly robust compared with other scoring approaches to identify tissue specificity of a gene among different magnitudes of expression and sizes of datasets (Kryuchkova-Mostacci & Robinson-Rechavi, 2017). The τ enrichment score was computed as:
where xi is the mean expression of the gene within the ith cell type and n is the number of cell types. We then defined the cell type enriched in the gene as that with the greatest average expression among all cell types. Top τ scores per gene are in Supplementary file 1, tab 5. A score of τ≥ 0.8 indicates that the gene is specific for a given cell type, and a score of 0.8 > τ ≥ 0.6 that the gene is enriched in that cell type. For simplicity, henceforth we refer to genes with τ ≥ 0.6 as ‘enriched’, although note that the ‘enriched’ pool also includes specific genes. Finally, τ < 0.6 indicates that the gene is non-specific, although it is assigned to the cell type with the greatest average expression.
Hierarchical clustering
The cell-type specific data from (Y. Zhang et al., 2016) (Supplementary file 1, tab 4) were z-scored for each gene across samples. Gene and sample hierarchical clustering were conducted using a Euclidian distance measure, and the average agglomeration method in R. Hierarchical clustering offers a clear visualization of data, facilitating a transparent analysis. A recent ranking of gene-module detection methods shows that agglomerative hierarchical clustering and WGCNA perform similarly (Saelens, Cannoodt, & Saeys, 2018). It is worth nevertheless emphasizing that the primary statistical tool used in the present study was not hierarchical clustering, but gene set variation analysis (GSVA, see below). In hierarchical clustering, gene clusters associated with neurons and astrocytes were determined by cutting the dendrogram at a height of 3.76 to achieve that at least 70% of the genes in the neuron and astrocyte clusters have the highest τ for that cell type (results from hierarchical clustering are in Supplementary file 1, tabs 6-8). Corresponding cell-specific gene clusters in whole tissue data sets (MtSINAI, MAYO, and ROSMAP) were identified by sorting the genes of whole-tissue data according to cell-type specific gene clusters. Whole-tissue subject samples (columns) were then clustered within each Control, MCI, or AD cohort (Euclidian distance and Ward.D2 agglomeration) to identify molecularly defined sub-groups of samples.
Data normalization within each cell type
A recognized caveat of bulk transcriptomic analyses is that DEG may reflect changes in cell-type composition rather than real changes in cell-specific transcriptomes. This is relevant to AD because there is neuronal death (Gomez-Isla et al., 1997), and microglial proliferation (Marlatt et al., 2014). Since neurons are the most predominant cell type in the brain (40% in the human neocortex (Pelvig, Pakkenberg, Regeur, Oster, & Pakkenberg, 2003)), a decrease of the neuronal pool, with no change in the astrocyte pool (Marlatt et al., 2014), arguably results in the artificial up-regulation of astrocyte genes. Several approaches have been employed to correct for cellular composition, such as the use of the so-called ‘high fidelity’ genes primarily expressed by one cell type (Kelley, Nakao-Inoue, Molofsky, & Oldham, 2018). Here, we reasoned that reliance on a subset of highly cell-specific genes could lead to artifacts should their expression change in the course of the disease, because there is no assurance that they are stably expressed as house-keeping genes. Likewise, recent de-convolution approaches such as CIBERSORTx and MuSiC proposed to identify cell type composition and contribution to bulk RNAseq or microarray datasets (Steen, Liu, Alizadeh, & Newman, 2020; Wang, Park, Susztak, Zhang, & Li, 2019) did not work well with our data, for we did not find strong agreement among methods with regards to neuron and astrocyte densities in Control brains, and changes thereof in AD, when applied to our datasets. Therefore, we elected to remove gross variance due to changes in cell density by variance stabilizing normalization using limma in R, which assumes a so-called affine transformation of gene expression within each sample, consisting of an offset and scaling factor selected to minimize inter-sample variance. What variance stabilizing normalization does is to bring genes from different samples onto the same scale in a manner that is blind to the sample group/identity. Removal of gross variance is a well-established approach to normalize transcriptomic data, based on the premise that only a negligible proportion of genes change their expression following perturbations (Lin, Du, Huber, & Kibbe, 2008). We reasoned that a bulk shift in the distribution of gene expressions with respect to zero in a given cell population reflected changes in the proportion of that cell type among brain cell populations. Thus, correcting gene expression according to such a shift would unmask the real changes in gene expression. Gross variance was separately corrected for astrocytic and neuronal gene clusters because their respective raw data presented an opposite shift (positive and negative) in the distribution of differential gene expressions in Controls vs AD samples (Results).
Cell function annotation and gene set variation analysis
Neuronal and astrocytic gene clusters generated by combining τ (Kryuchkova-Mostacci & Robinson-Rechavi, 2017) and hierarchical clustering of transcriptomes from cells from healthy brains (Y. Zhang et al., 2016), were organized into functional categories and subcategories (Supplementary file 2). Categories were manually curated by one expert and cross-validated by another expert using information from GeneCards, perusal of Medline and PubMed, and open-source platforms such as GO, KEGG, and Reactome. Manual curation was indispensable for two reasons. First, over 60% of the genes in the astrocytic cluster were not annotated in open-source platforms. This means a substantial loss of usable gene data if we only relied on genes annotated in open-source platforms. Second, annotation in such platforms represents ‘canonical’ pathways with little consideration for the fact that different CNS cell types are molecularly and functionally distinct (Y. Zhang et al., 2016). This limitation is critical for astrocytes. For example, unlike neuronal compartments such as ‘synapses’, ‘dendrites’ and ‘spines’, a highly specialized astrocytic compartment termed ‘perisynaptic astrocyte processes’ (PAP), concentrating RNA transcripts related to glutamate and GABA metabolism, energy metabolism, as well as the ribosomal machinery to perform local RNA translation (Sakers et al., 2017), is not a category in current KEGG and GO databases.
The genes were organized into general categories and subcategories using a mixed set of criteria including general pathways (e.g., carbohydrate metabolism or amino acid metabolism) and subcellular compartments with a clear functional specialization (e.g., mitochondria, peroxisome, or lysosome). Such flexibility allowed us to optimize gene inclusion, while generating gene sets with enough genes for statistical purposes. Although most genes were assigned to only one category, around 100 genes were assigned to two categories (in blue, Supplementary file 2, tab ‘astrocyte cluster 4’). Most of those genes (96) were PAP genes, because we reasoned that their presence in PAP does not exclude relevance in other subcellular compartments. For example, ALDOC and ACO2 are, plausibly, ubiquitous enzymes relevant for general glycolysis and mitochondrial Tricarboxylic Acid (TCA) cycle throughout the cell. Likewise, genes related to ‘TNFalpha signaling’ are in ‘stress responses’/’cytokines’ as well as in ‘gliotransmission’, for TNFalpha has been described to modulate glutamate exocytosis (Santello, Bezzi, & Volterra, 2011). Overlap between groups was less than 5% except for ‘PAP and gliotransmission’: 17.8% genes in ‘gliotransmission’ were in ‘PAP’, and 10.3% of PAP genes were in ‘gliotransmission’. For neurons, each gene was assigned to only one category.
Using our novel annotation database, the GSVA package in R was used to identify the enrichment of each gene set across all samples. GSVA is a generalized gene set enrichment method that detects variations of pathway activity over a sample population in an unsupervised manner (Hanzelmann et al., 2013). Statistical differences in enrichment scores for each gene set between subject groups were computed by comparing the true differences in means against a null distribution obtained by permuting the gene labels and re-computing the GSVA 1000 times. False discovery rate adjusted p-values (qFDR) were computed using the method of Benjamini & Hochberg (Hanzelmann et al., 2013). Gene sets with FDR adjusted p value qFDR < 0.05 were considered significant.
Relationships between clinical stages and GSVA scores
The ROSMAP dataset includes Braak and CERAD (Consortium to Establish a Registry for Alzheimer’s disease) pathological scores, APOE genotype, and sex for each sample. To determine the relationship between these phenotypes and the GSVA enrichment scores for each sample, we used the stats R package to fit linear models between enrichment scores and either Braak or CERAD for each gene set. The MAYO and MtSINAI databases do not include supplementary information per subject other than the clinical classification.
Figure preparation
The graphical abstract was made using BioRender.
Results
Hierarchical clustering identifies groups of cell-type specific genes
To identify human astrocyte-specific genes, we calculated the metric τ (Kryuchkova-Mostacci & Robinson-Rechavi, 2017) for every gene in the transcriptome data from isolated brain cells generated by Zhang et al. 2016 (Supplementary file 1, tab 5 ‘τ scores’ ). Cell type-specific (τ ≥ 0.8) and cell type-enriched (τ ≥ 0.6) genes represented 30% and 60% of the 11,077 genes, respectively.
In parallel, because cell-specific functions are arguably performed by co-expressed genes (Weirauch, 2011), we reasoned that cell-type specific genes would be identified by clustering analysis. Thus, we applied hierarchical clustering (Methods) to the cell-type specific datasets, generating 194 clusters that contained 1–1651 genes (Fig. 2A; Supplementary file 1, tab 6 ‘Hierarchical clustering’ and tab 7 ‘Cluster description’). We found that genes from the same cell type segregated together, defining clusters of highly co-expressed genes, plausibly representing cell-specific functions. The dendrogram height of hierarchical clustering was set to identify gene clusters that were highly enriched for individual brain cell types. The criteria for ‘enrichment’ were that the cluster contained more than 100 genes, of which over 70% corresponded to a unique cell type with τ ≥ 0.6 (Supplementary file 1, tab 8 ‘cluster selection’). Five clusters fulfilled these criteria, corresponding to astrocytes (cluster # 4), endothelial cells (cluster # 5), neurons (cluster # 13), microglia (cluster # 17) and oligodendrocytes (cluster # 164). There were other clusters containing over 77% of total of neuronal (e.g., clusters # 10, 116) or microglial (e.g., clusters # 30, 15) genes (Supplementary file 1, tab 8 ‘cluster selection’), but they were excluded from the analysis because less than 54% of their genes were enriched (τ ≥ 0.6) for that cell type, suggesting lower cell type specificity.
Figure 2. Identification of cell-specific gene clusters in whole-brain transcriptomes by two-step hierarchical clustering.
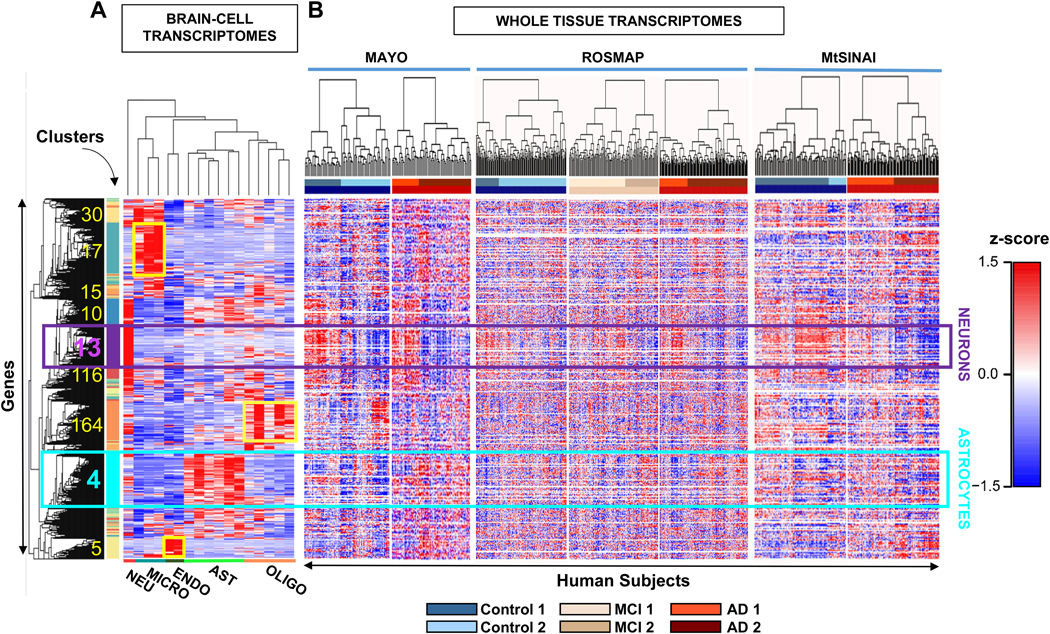
A. Hierarchical clustering was performed on transcriptomes of brain cell populations isolated from individual healthy subjects. Each column is a sample from a single subject and each row is a z-scored gene. In squares, the number and location of clusters with the highest z scores, that coincide with the highest τ scores for a given cell type (cluster information in Supplementary file 1, tabs 6-8). The turquoise and purple squares are the main astrocytic and neuronal clusters (cluster # 4 and cluster # 13), respectively. Microglia, oligodendrocyte, and endothelial-cell clusters are 17, 164 and 5, respectively (shown in yellow). Clusters 30, 15, 10 and 116 are highly enriched in microglial or neuronal genes, but fall below the criteria established for cell-specificity (Methods).
B. Whole-brain transcriptomes from individual subjects of Control (blue), MCI (beige) and AD (red) groups from MAYO, ROSMAP, MtSINAI were hierarchically clustered. The rows from the cell-type clustering in A. were kept fixed to identify cell-specific clusters. The figure was generated with raw data before correction for cell composition. The clustering segregated all groups with the same clinical diagnosis into at least 2 subgroups (defined as 1 and 2) showing opposite changes in geneexpression.
We continued with astrocyte cluster # 4 (1651 genes) and neuronal cluster # 13 (1258 genes) for two reasons: (i) a cluster size of over 1000 genes is sufficient for functional categorization and gene set analysis, and (ii) since molecular changes in neurons are better characterized in AD than changes in astrocytes, neurons served as a positive control to assure correct data mining.
In the neuronal cluster # 13, 99.9 % of the genes were neuronal according to τ score, of which 54.1% of genes were neuron-specific (τ ≥0.8), meaning that they encode proteins that perform functions highly specific to neurons. Indeed, the top 10 ranked genes based on τ (τ > 0.98) were related to neurotransmission (SYNPR, GABRA1, CNR1, SYT1, GABRG2 and GAD1), regulation of membrane potential (KCN2), microtubules (INA), and cell adhesion (RELN). The remaining 45.9% (τ < 0.8) were neuron-enriched or bulk genes plausibly supporting neuron-specific functions, since they were co-expressed with neuron specific genes (Supplementary file 1, tab 8 ‘cluster selection’; Supplementary file 2, tab 2 ‘Neurons cluster 13’).
In astrocyte cluster # 4, 80% of the genes were astrocytic according to τ (Supplementary file 1, tabs 7 and 8 ‘Cluster description’ and ‘Cluster selection’; Supplementary file 2, tab ‘Astrocytes cluster 4’). Further, 30% had τ ≥ 0.8, including genes that encode typical astrocyte proteins such as GFAP (τ = 0.95), glutamine synthase (GLUL, τ = 0.95), glutamate decarboxylase (GLUD2, τ = 0.91), glutamate transporters (EAAT1/SLC1A3, τ = 0.83), lactate dehydrogenase (LDHB, τ = 0.91), and enzymes involved in mitochondrial fatty-acid oxidation (ACADVL, τ = 0.92). Enriched genes with τ between 0.6–0.8, or bulk genes below 0.6, arguably necessary for astrocyte functions, are the aquaporins AQP1 (τ = 0.739) and AQP4 (τ = 0.744), and the chaperone clusterin CLU (τ =0.373), which was identified by GWAS as a risk factor in AD (Han, Qu, Zhao, & Zou, 2018). These findings indicated that the group of genes gathered in cluster #4 is relevant to astrocyte biology.
Identification of cell-type specific gene clusters in AD databases
We next searched for the astrocytic and neuronal gene clusters in three AD whole-tissue transcriptomic databases (heatmaps of z-scores in Fig. 2B). Unexpectedly, hierarchical clustering of subjects (columns) revealed heterogeneity in groups with the same clinical diagnosis. Subjects were divided into at least two broad molecular groups within each of the Control, MCI, and AD cohorts across the three databases, as shown by the division of the dendrograms into two large groups in each cohort. We refer to type 2 subjects as those showing blue z-scores within the neuron gene cluster, suggesting massive down-regulation of neuronal genes, as compared to type 1 subjects, in which genes with red z-scores predominated. Type 1 sub-cohorts included Control1, MCI1 and AD1, while type 2 included Control2, MCI2 and AD2. Numbers of Control1/Control2 patients were 33/45 in MAYO, 51/149 in ROSMAP, and 81/20 in MtSINAI. Numbers of AD1/AD2 patients were 28/54 in MAYO, 70/149 in ROSMAP, and 66/63 in MtSINAI. MCI1/MC2 numbers were 99/58 in ROSMAP. Heterogeneity in MtSINAI data has been recently reported and replicated in ROSMAP (Neff, 2021).
The most profound down-regulation of genes within the neuronal gene cluster was observed in AD2. Because down-regulation of neuronal genes due to loss of synapses and neuronal demise is a hall-mark of AD (Gomez-Isla et al., 1997), we reasoned that AD2 was advanced AD, and Control1 a bona fide control composed of subjects that were neither demented, nor preclinical or pre-symptomatic at the time of death. Because Control1 and AD2 appeared to be the extreme groups across the continuum of AD phenotypes, they were selected for further analysis.
Since our goal was to identify changes in astrocytic functions, it was critical to eliminate gross variability among samples within the astrocytic and neuronal gene clusters due to global changes in cell composition. To do so, we applied variance stabilizing normalization (Methods) to all samples. Prior to normalization, the distributions of gene-expression differences between AD2 and Control1 shifted to the left in the neuronal cluster, suggesting depletion of neurons in AD2, while the distribution of astrocytic genes shifted to the right, suggesting a relatively higher content of astrocytes in AD2. The normalization shifted the peaks of the distributions of differences in gene expression between AD2 and Control1 to zero, thus validating this step of data processing (Fig. 3A). After the normalization, the global down-regulation of all neuronal genes in cluster # 13 in type 2 groups was attenuated, but intra-cohort heterogeneity persisted such that type 2 clusters still presented a prominent down-regulation of neuronal genes as compared to type 1 clusters (Fig. 3B and Supplementary file 3). In addition, new gene clusters previously masked were unraveled by hierarchical clustering. Thus, there were three neuronal (Fig. 3B, N-a, b, c), and four astrocytic gene subgroups (Fig. 3B, A-a, b, c, d) with opposite expressions patterns in type 1 vs type 2 subjects. For example, N-b genes were globally downregulated in AD2 and Control2, as compared to Control1 and AD1. Importantly, N-b contained genes related to synaptic function, including glutamatergic and GABAergic neurotransmission (e.g., GABRB3; GABRB2; GAD1; PDYN; SYN2; GABRG2; SYN1 NAPB; GRIA2; SLC17A7; GRIK2), supporting that this cohort is a bona fide AD. The normalized expression of cluster # 4 genes in the three AD databases is in Supplementary file 4. It is worth stressing that although changes in functions follow the same trend (up or down) in Control1/MC1/AD1 and Control2/MCI2/AD2 subjects, there are quantitative differences within type1 and type 2 groups; however, for simplicity, we limited herein the statistical comparison of all the sub-cohorts vs Control1.
Figure 3. Neuronal and astrocytic gene sub-clusters in the three clinical databases.
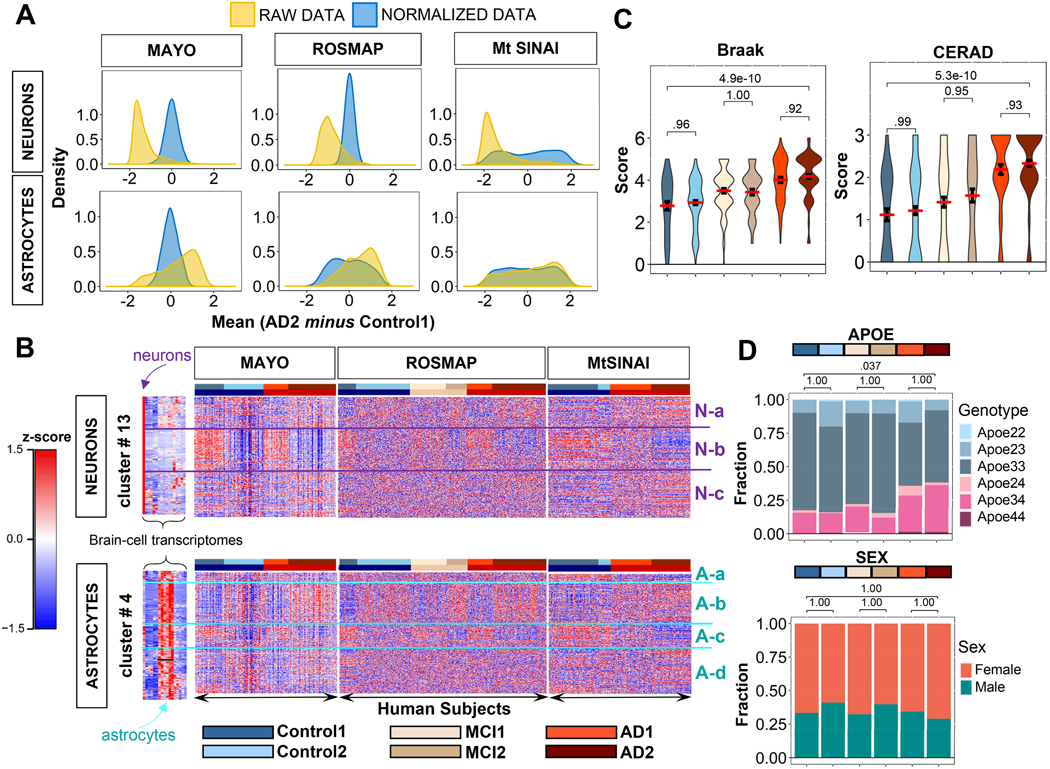
A. Distributions of gene expression differences between AD2 and Control1 cohorts before (yellow) and after (blue) variance stabilizing normalization, yielding zero-centered distributions (Methods).
B. Hierarchical clustering of Control (blue), MCI (beige), and AD (red) subjects from Fig. 2B using normalized data in rows. For both astrocytic and neuronal genes, subjects self-organized into at least two distinct subgroups (defined as 1 and 2) within each group with the same clinical diagnosis, while genes self-organized into up to four groups (Na-c and Aa-d). In MAYO and ROSMAP, clusters showed opposite directions of change in type 1 and 2 subjects. In MtSINAI, Control2 was like Control1, and AD1 like AD2, although with less pronounced changes.
C. Violin plots of pathological stages according to CERAD and Braak scoring shows no significant difference in pathological stages between sub-groups within each cohort in ROSMAP (means ± SEM, one-way ANOVA with Tukey post-hoc).
D. Distribution of APOE genotypes and sex in the six groups. While 3/3 was the predominant genotype across all groups, and 3/4 was more abundant in AD, there were no differences in frequency of APOE4 alleles between type 1 and 2 subgroups of subjects with the same clinical diagnosis. Likewise, females were more abundant than males in all groups, but not in type 2 subjects as compared to type 1 (p values determined by pairwise Fisher’s Exact test with Bonferroni adjustment).
Because the ROSMAP database included clinical covariate data, such as CERAD and Braak scores, APOE genotype and sex, we examined whether type 1 or type 2 sub-cohorts were associated with specific covariates to gain insight into why they were molecularly heterogeneous. We found that Braak and CERAD scores were significantly higher in AD groups than in Controls (Fig. 3C), confirming the clinical diagnosis. However, type 1 and type 2 subjects with the same clinical diagnoses did not significantly differ in their pathological stages according to CERAD and Braak scoring; nor were type 2 groups more enriched in APOE4 subjects or females (Fig. 3D).
GSVA reveals altered functions in AD and MCI vs Control1
We next identified changes in astrocytic and neuronal functions using the normalized expression data. Because most of the astrocytic genes were not annotated in existing pathways in open-source databases, we manually curated the astrocytic cluster in 17 functional categories (gene sets) and 135 subcategories, and the neuronal cluster into 15 functional categories and 79 subcategories (Methods, Supplementary file 2).
GSVA was then used to compare the gene sets corresponding to different functional categories among cohorts, yielding an enrichment score for each subject and gene set. We started by comparing the extreme cases, AD2 and Control1, using two criteria. First, probability values were computed using a permutation analysis across genes (cut-off for statistical significance was set at FDR-adjusted q < 0.05, Methods). Second, we considered that a function was altered in AD2 as compared to Control1 if such alteration was detected in at least two out of the three databases (consensus criterion).
For neurons, seven functions were altered in AD2 vs Control1 (Fig. 4A, statistics in Supplementary file 5, extended graphs in Supplementary file 6): ‘neurotransmission’ and ‘gene expression’ were altered in the three databases, and ‘synaptic plasticity’, ‘regulation of membrane potential’, ‘neural development’, ‘mitochondria’ and ‘stress response’ in MAYO and ROSMAP, but not MtSINAI. ‘Synaptic plasticity’, ‘neurotransmission’, ‘neural development’ and ‘membrane potential’ were down-regulated, and ‘gene expression’, ‘mitochondria’ and ‘stress response’ up-regulated. In MAYO and ROSMAP, the altered functions in Control2 and MCI2 followed trends like in AD2, and AD1 and MCI1 like in Control1 (Fig. 4B). In MtSINAI, we found no statistically significant difference between Control1 and Control2, and AD groups were similar, although changes in AD2 were more dramatic, as compared to Control1 (Fig. 4B).
Figure 4. GSVA unravels neuron-enriched functions altered in AD.

Differences in the general functional categories among cohorts in MAYO, ROSMAP and MtSINAI databases were examined by GSVA.
A. Comparison between AD2 and Control1. Functions are ranked according to the difference in enrichment scores (ES). Significantly changed functions (qFDR < 0.05) are labeled in red.
B. Enrichment scores of functional categories significantly changed in at least 2 out of 3 databases. (#) qFDR< 0.05 (&) qFDR <0.01, and (*) qFDR < 0.001, vs Control1.
For astrocytes, five functions were altered in AD2 vs Control1 (Fig. 5A, statistics in Supplementary File 5): ‘PAP’, ‘plasticity’ ‘stress response’ were up-regulated in three/three databases, ‘mitochondria’ was down-regulated in three/three databases, while ‘endolysosomal system’ was down-regulated in two/three databases. Because ‘PAP’ are specialized astrocyte-neuron contacts and, hence, this category includes many genes related to gliotransmission, and since ‘gliotransmission’ was highly significantly changed in ROSMAP (qFDR<0.001), and trending in MAYO (qFDR=0.07), henceforth ‘PAP’ and ‘gliotransmission’ were considered together. As with neurons, Control2 and MCI2 showed the same direction of change as AD2 compared to Control1 (Fig. 5B). Likewise, the enrichment of these gene sets in AD1 resembled that of Control1, particularly in MAYO and ROSMAP, while AD1 appeared to be a milder version of AD2 in MtSINAI.
Figure 5. GSVA unravels astrocyte-enriched functions altered in AD.

Differences in the functional categories among cohorts in MAYO, ROSMAP and MtSINAI databases were examined by GSVA.
A. Comparison between AD2 and Control1. Functions are ranked according to the difference in enrichment scores (ES). Significantly changed functions (qFDR < 0.05) are labeled in red.
B. Enrichment scores of functional categories significantly changed in at least 2 out of 3 databases. (#) qFDR < 0.05, (&) qFDR <0.01, and (*) qFDR < 0.001, vs Control1.
Co-variance of gene sets with one another and with pathological covariates
Correlations between altered functions and pathological stages.
Since CERAD and Braak scores were available for the ROSMAP data, we examined relationships between general functions and pathological stages by regressing enrichment scores for each gene set and sample against the corresponding CERAD and Braak scores, regardless of clinical diagnoses (Methods). Data from all the individuals in the three groups (Control, MCI and AD) were used in the analysis. For neurons, we found no statistically significant correlations between Braak and CERAD stages for any of the seven top dysregulated categories described in (Fig. 4A and Supplementary file 7). The lack of correlation can be interpreted to indicate similar gene dysregulation in all stages, suggesting that neuronal dysfunction is an early event in AD pathology, such that it appeared in subjects with no (i.e., Control2) or incipient (i.e., MCI2) cognitive deficits, and low CERAD and Braak scores.
For astrocytes, of the five functions that were significantly altered in AD2 vs Control1 (Fig. 5A), the following ones significantly correlated with Break/CERAD according to p-values: ‘mitochondria’ (0.0000203/0.000742), ‘stress response’ and ‘plasticity’ (0.0768/0.00967) (Supplementary file 8). In contrast, ‘PAP’, ‘gliotransmission’, and ‘endolysosomal system’ did not correlate with pathological stages, suggesting that, as we reasoned with neurons, they represent early manifestations of alterations in astrocytic functions. For example, there was a progressive decline in the expression of nuclear-encoded mitochondrial genes from CERAD 1 to 3 and from Braak 1 to 6, while the expression of genes involved in ‘endolysosomal system’ was altered at early CERAD and Braak stages, before the detection of cognitive deficits in subjects.
Correlations among altered functions.
We examined correlations among all neuronal and astrocytic gene sets by regressing enrichment scores for each gene set against each other gene set. Hierarchical clustering of correlation coefficients and adjusted p-values are in Supplementary file 9 for ROSMAP, MAYO, and MtSINAI. In neurons, the strongest direct correlations were among highly related functions such as ‘membrane potential’, ‘neurotransmission’ and ‘synaptic plasticity’. Likewise, in astrocytes, the strongest correlations were between ‘PAP’ and ‘gliotransmission’. The strongest anti-correlations point to intertwined changes between: (i) morphological remodeling of PAP and loss of synaptic functions, and (ii) organelle dysfunction and ‘plasticity’ (which is enriched in survival pathways, see below).
Alteration of functional subcategories in astrocytes
We next aimed to gain further insight into which factors drive the functional alterations in AD astrocytes by searching for functional subcategories with statistically significant differences in enrichment scores between AD2 and Control1 (qFDR< 0.05) in at least two out of three of the databases (statistics in Supplementary file 10).
The category ‘PAP’ includes different categories related to morphology, metabolism, and gene expression. The consensus subcategory significantly upregulated was ‘PAP morphology’ (qFDR <0.001, AD2 vs Control1, in MAYO and ROSMAP). Because PAP are specialized in the functional refinement of adjacent synapses at a micro scale (Sakers et al., 2017), increased expression of PAP-related genes in AD astrocytes may be a compensatory reaction to locally optimize compromised synaptic functions.
Subcategories in ‘plasticity’ include annotations associated with morphological remodeling (e.g., ‘adherens junction’, ‘ECM’, ‘cytoskeleton’, ‘ciliogenesis’, ‘TGFbeta signaling’, ‘metalloproteases’), and pathways involved in brain development whose role in adult astrocytes is not well understood, although they are characteristically up-regulated in reactive astrocytes, perhaps as a mechanism of astro- or neuroprotection (e.g., ‘axon outgrowth’, ‘synaptogenesis’, ‘WNT signaling’, ‘hippo signaling’, ‘smoothened signaling’, ‘hedgehog signaling’, ‘homeobox signaling’, ‘notch signaling’, ‘FGF signaling’, ‘pluripotency’, and ‘glial cell fate commitment’). Significantly dysregulated pathways in AD2 vs Control1 were ‘ECM’, upregulated in the three databases, and ‘hippo signaling’ and ‘ciliogenesis’ (i.e., the process of generation of a microtubule-based and centriole-derived cilium), respectively upregulated and down-regulated in two databases—note that the fact that the general trend of a given general category is towards up-regulation does not preclude that some subcategories are down-regulated.
‘Stress responses’ includes ‘AMPK signaling’, ‘antioxidant response’, ‘cell death’, ‘chaperone’, ‘DNA repair’, ‘hypoxia’, ‘complement system’ and ‘cytokines’. No subcategory was dysregulated according to qFDR in at least two databases. However, it is worth noting genes related to TNFalpha in ‘cytokines’ were significantly dysregulated as the subcategory ‘TNFalpha’ of ‘gliotransmission’ in MAYO and ROSMAP, and trending in MtSINAI (qFDR=0.093).
In ‘endolysosomal system’, encompassing genes related to ‘endoplasmic reticulum’, ‘Golgi’, ‘lysosome’ and ‘vesicles’, no single subcategories were significantly dysregulated, while in ‘mitochondria’, ‘ETC’ and ‘TCA cycle’ were significantly downregulated in AD2 vs Control1 in three and two databases, respectively. For ‘ETC’, qFDR was < 0.001 in MAYO, < 0.001 in ROSMAP, and < 0.012 in MtSINAI. For ‘TCA cycle’, qFDR was <0.001 in MAYO, <0.39 in ROSMAP, and < 0.01 in MtSINAI. All in all, the dysregulated subcategories in astrocytes point to alterations in mitochondrial respiration concomitant to morphological remodeling.
PCA confirms main functional changes in AD astrocytes
GSVA was dependent on gene sets organized into pre-determined functions. For this reason, we also used PCA as an alternative, unsupervised approach to gain independent insight into the predominant molecular changes of AD astrocytes. First, we examined whether expression of genes contained in the astrocytic cluster # 4 segregated cohorts in ROSMAP. PCA segregated the Control1, MC1 and AD1 groups to the left, and the Control2, MCI2 and AD2 groups to the right, according to the principal component 1 (PC1, horizontal axis, Fig. 6A). Violin plots of PC1 scores per group confirmed this tendency.
Figure 6. PCA identifies top altered functions in AD astrocytes.

A. PCA analysis of astrocyte-enriched genes in ROSMAP. Dot plots and violin plots along PC1 (mean ± SEM) show differential expression of astrocyte genes in type 2 vs type 1 subjects.
B. Top varying genes in PC1 (up-regulated and down-regulated), color-coded for function according to the circos plot, inform about functions altered in AD astrocytes. The vertical axis represents the relative contribution (importance/weight) of each gene for the scoring of individual samples in panel A along PC1.
C. Scoring of MtSINAI and MAYO databases using genes overlapping with ROSMAP PCA in A. discriminates Control and AD cohorts along PC1, as shown by the dot and violin plots (means ± SEM).
Second, we examined the functions performed by the proteins encoded by the top 50 dysregulated genes in PC1, representing the maximally co-varying astrocytic genes (Supplementary file 11). Mirroring our results with GSVA, genes related to ‘plasticity’ and ‘PAP/gliotransmission’ were over-represented in the up-regulated portion, whereas genes related to ‘endolysosomal system’, ‘gene expression’, and ‘signaling’ were predominant in the down-regulated portion (Fig. 6B). Genes related to ‘mitochondria’ and ‘stress responses’ appeared in both sections, indicating mixed direction of dysregulation in these functions. Unlike GSVA, PCA also detected changes in ‘signaling’, as suggested by the presence of genes related to this function in the up and down segments of the top PC1 genes.
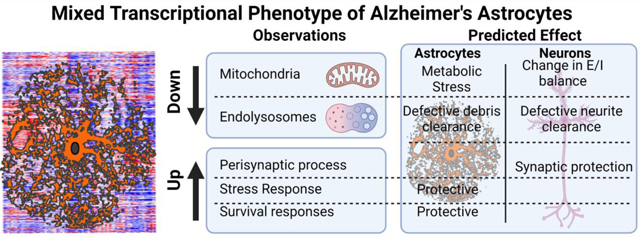
Third, we asked if the changes detected in AD astrocytes in ROSMAP with PCA were reproducible. Thus, we determined the capacity of the PC1 genes to segregate cohorts in MAYO and MtSINAI. As in ROSMAP, Control1 subjects were mostly negative, and AD2 cases were mostly positive in MAYO and MtSINAI, thus validating the discriminating capacity of the maximally co-varying genes in PC1 (Fig. 6C). AD1 segregated with Control1 in MAYO, but not in MtSINAI, where AD1 aligned with AD2, confirming less intra-cohort heterogeneity in MtSINAI than in the other two databases, as shown with GSVA. Altogether, these results point to multi-factorial changes in AD astrocytes, encompassing changes in astrocyte-neuron interactions and organelle dysfunction (model in Fig. 7A and description in Discussion).
Figure 7. Model of cortical human AD astrocytes and comparison with mice.

A. The model revolves around two predictions.
First prediction: Amyloid-β drives the phenotypical change of cortical astrocytes in AD by direct and indirect mechanisms. Direct: Amyloid-β causes endolysosomal and mitochondrial dysfunction in astrocytes, resulting in an adaptive astroprotective shift of gliotransmission to a GABA-predominant mode as a strategy to preserve mitochondrial respiration. Indirect: Synaptic dysfunction caused by amyloid-β in neurons augments the production of the PAP machinery and synaptogenic factors in astrocytes―a phenomenon of enhanced astrocytic plasticity to protect neurons.
Second prediction: The phenotypical transformation of astrocytes in AD happens in early and late stages. Control2 subjects, cognitively normal, would be at stage a, and AD2 patients, diagnosed with dementia, at stage b. In stage a, the astroprotective change in glutamate/GABA fluxes may preserve neural-circuit homeostasis, because the GABA produced by MAO-B may locally compensate for the deficits in inhibitory tone that disrupts circuitry synchronization (Lee et al., 2020). By contrast, deficits in the endolysosomal system would result in partial phagocytosis of amyloid-β (Sollvander et al., 2016) and dystrophic neurites (Gomez-Arboledas et al., 2018), thus exacerbating amyloid-β accumulation, and hindering neuronal repair due to lack of debris elimination. In stage b, the mechanisms triggered to protect mitochondria may fail, and cortical astrocytes become deeply malfunctional, as suggested by the reduction in TCA cycle and ETC transcripts, despite the increase in protective stress responses and pro-survival pathways aimed at preventing astrocyte demise. For simplicity, only predominantly altered functions are depicted. ECM, extracellular matrix; ETC: electron transport chain, GLU: glutamate; oAβ: oligomeric amyloid-β; PAP: perisynaptic astrocyte processes; TCA: tricarboxylic acid. Figure adapted from Servier Medical Art (https://smart.servier.com/).
B. GSVA of the transcriptome of astrocytes isolated from APP/PS1 mouse cortices using our manually curated functional categorization shows down-regulation of astrocyte-enriched functions.
C. Left, GSVA of the transcriptome of APP/PS1 astrocytes using gene sets consisting of the top/bottom 50 dysregulated human astrocyte genes in PC1 from the ROSMAP database, and the top/bottom 50 DEGs from APP/PS1 astrocytes (Orre et al, 2014). Human astrocyte-enriched genes dysregulated in AD (up and down) are down-regulated in APP/PS1 astrocytes, confirming B. Right, GSVA of PC1 AD and mouse APP/PS1 DEG gene sets in ROSMAP, MAYO and MtSINAI whole-brain transcriptomes shows that both the up and down-regulated mouse gene pools tend to be upregulated in AD databases. Discussion of the result of this comparison can be found in the main text.
Comparison with other human studies
Comparisons among whole-tissue transcriptomic analysis (B. Zhang et al., 2013), scRNAseq studies (Grubman et al., 2019) (Mathys et al., 2019) and the present study are in (Supplementary file 11). The greatest concordance was found between our study and (B. Zhang et al., 2013). Thus, there was an 11% overlap between cluster # 4 and the astrocyte cluster in the Zhang study. Importantly, 94.9 % of the common genes were concentrated in the UP portion of our PC1 list, and were particularly enriched in the 50 top UP PC1, which thus presented a 50% overlap with Zhang’s list. Common genes were related to ‘gliotransmission’ (e.g., glutamate transporter SLC1A3), ‘plasticity’ (e.g., hippo Signaling TP53BP2), ‘stress response ‘(e.g., antioxidant PON2), and ‘mitochondria’ (e.g., amino acid metabolism HADHB). By contrast, there was scarce representation of astrocytic genes from (B. Zhang et al., 2013) in the down-regulated portion of our cluster # 4, where most of the genes related to mitochondrial metabolism and the endolysosomal system lie. Since the astrocytic cluster was generated by co-expression within AD transcriptomes in (B. Zhang et al., 2013), we reason that down-regulated astrocytic genes may not co-express any longer with up-regulated ones, thus explaining why down-regulation of organelle-related genes was not detected in (B. Zhang et al., 2013). In summary, our study confirms the relevance of glutamate and amino acid metabolism in AD pathogenesis reported in large-scale network analysis (B. Zhang et al., 2013), and links it to mitochondrial dysfunction, which, we stress, presented a marked negative correlation with pathological stages according to regressions with Braak/CERAD.
As to scRNASeq studies, only 1.1% of Cluster # 4, and 0.3% of the top 50 PC1 genes, overlap with the 69 astrocytic DEG listed in (Mathys et al., 2019) (Supplementary file 11). Although the low number of DEG hinders statistically sound pathway analysis, it is worth noting that the top GO Biological Process of astrocyte DEGs in (Mathys et al., 2019) is ECM (GO:0030198), which coincides with our detection of consensus up-regulation of the ECM functional category. Indeed, common genes between top 50 PC1 and (Mathys et al., 2019) include PAP genes PREX2 and PLXNB1.
The overlap between the astrocyte cluster listed in (Grubman et al., 2019) and cluster # 4 is 12.7%, and 1.3% considering only the top 50 PC1 genes, although the glutamate transporter SLC1A3 is common to both lists. Considering functions, among the most dysregulated GO Biological Process in (Grubman et al., 2019), including ‘central nervous system myelination’ (GO:0022010), ‘axonogenesis’ (GO:0007409) and ‘chemical synaptic transmission’ (GO:0007268), only the latter two functions are equivalent to our detected up-regulation of developmental pathways and gliotransmission-related genes.
It is worth noting that the astrocytic DEG listed in (Mathys et al., 2019) and (Grubman et al., 2019) only share 32 genes, representing 47% of the DEG in (Mathys et al., 2019) and 4.5 % of those in (Grubman et al., 2019) (Supplementary file 11). Moreover, only 2.3% and 14.6% of the astrocytic cluster from (B. Zhang et al., 2013) overlap with (Mathys et al., 2019) and (Grubman et al., 2019). The only partial equivalence among studies may be due to several factors, including: (i) differences in methodologies, (ii) classifications of disease (e.g., clinical in (B. Zhang et al., 2013), (Grubman et al., 2019) and our study, but associated to amyloid-β pathology regardless of clinical diagnosis in (Mathys et al., 2019), (iii) brain regions sampled (prefrontal or temporal cortex in (Mathys et al., 2019) (B. Zhang et al., 2013) and in our case, but entorhinal cortex in (Grubman et al., 2019)), and (iv) the total number of subjects analyzed (1647 in (B. Zhang et al., 2013), 48 in (Mathys et al., 2019), 12 in (Grubman et al., 2019), and 766 herein.)
Finally, the main consistent finding between our study and the network analysis of proteins detected immunohistochemically in AD brains (Viejo et al., 2021) is the detection of up-regulation of ECM and oxidative stress (included in ‘stress responses’ in our case’). As with the rest of studies, the main difference is that the study by (Viejo et al., 2021) does not include down-regulation of proteins related to endolysosomal systems and mitochondrial functions such as TCA and ETC associated to AD astrocytes, because their analysis is centered on up-regulated proteins. Nor is subject heterogeneity reported in any of the studies discussed above.
Comparison with cortical APP/PS1 astrocytes
We concluded our analysis by asking whether changes identified in cortical AD astrocytes are recapitulated in cortical astrocytes isolated from 15–18-month-old APP/PS1 (Orre et al., 2014). Comparison with recent snRNAseq mouse data (Habib et al., 2020) was not appropriate because this latter study was carried out in the hippocampus.
We performed the following head-to-head comparisons between human and mouse data. First, we examined the changes of the functional categories identified in the human astrocytic cluster # 4 in the APP/PS1 vs WT (n=4 per group). GSVA revealed down-regulation of all functional categories (Fig. 7B). That is, APP/PS1 astrocytes mimic the down-regulation in ‘endolysosomal system’ and ‘mitochondria’ detected in human AD astrocytes, but not the up-regulation of ‘PAP’, ‘stress responses’ and ‘plasticity’.
Second, we examined the expression of the top ranked down- and up-regulated PC1 genes of human AD astrocytes in the mouse transcriptomes (gene sets 1 and 3, Fig. 7C-left panel), and the expression of the 50 most downregulated and upregulated DEG in APP/PS1 astrocytes (Orre et al., 2014) in the clinical databases (gene sets 2 and 4, Fig. 7C-right panel). Fig. 7C shows the hierarchical clustering of GSVA-generated enrichment scores. To help interpret the comparisons, it is worth stressing that, in addition to a difference in species, human gene sets 1 and 3 consisted of astrocyte-enriched genes, while the mouse data consisted of the entire mouse astrocyte transcriptome, encompassing enriched and ubiquitous genes. Thus, gene set 2 is enriched in typical homeostatic astrocytic genes related to neuronal support (e.g., FZD10, HES5, SOX9), and gene set 4 in inflammation-related genes (e.g., TREM2, IL1β, CCL3, CCL4, CCL5, C14). These genes are predominantly microglial, according to τ scores, and except for C1S, did not coincide with functionally related human astrocyte-enriched genes in cluster # 4 subcategories such as ‘cytokines’ and ‘complement’ (Supplementary file 12).
Both human clusters segregated with the down-regulated mouse gene set 2 (Fig. 7C left panel), confirming the global down-regulation of astrocyte-enriched genes in APP/PS1 astrocytes, as opposed to the up-regulation of generic inflammation-related genes. As also concluded in (Orre et al., 2014), this divergence points to a loss of homeostatic astrocytic functions and the adoption of a microglia-like phenotype, suggesting that cortical APP/PS1 astrocytes may undergo a phenotypical involution with time.
As expected, AD astrocyte gene sets 1 and 3 were respectively downregulated and upregulated in AD2 cohorts (Fig. 7C right panel), while the mouse gene set 2, containing neuron support genes, downregulated in APP/PS1 mice tended to be upregulated in AD2 cohorts. This might mean that the loss of homeostatic genes may be more advanced in APP/PS1 mice at 15–18 months of age than in AD2 patients. On the other hand, gene set 4 containing inflammation-related genes was largely upregulated in AD2, suggesting a predominant expression of typical microglial genes in AD2, plausibly upregulated in both microglia and astrocytes. Collectively, the comparison of human and mouse data suggests that there are both shared and distinctive changes in astrocytes in human AD and 15–18-month APP/PS1 mice. This factor should be taken into consideration when using animal models of AD.
Discussion
Because of the challenges associated with the isolation of astrocytes or their nuclei, and to profit from the wealth of large publicly available human-brain transcriptomes, the goal of this study was to gain insight into what happens to astrocytes in AD using systems-biology tools. The approach consisted of the re-compartmentalization of whole-brain transcriptomes from Control, MCI, and AD subjects into contributions from individual cell types by using pre-clustered cell-specific gene sets. The study has four main findings.
First, unbiased hierarchical clustering of patient cohorts reveals major intra-cohort heterogeneity, i.e., relationships are not robust between gene profiles, clinical diagnosis, and pathological stage according to CERAD and Braak scoring, as recently reported in (Neff, 2021). In other words, some non-demented subjects present AD-like astrocytes and neurons, while some demented subjects present healthy-like ones, particularly in MAYO and ROSMAP.
Second, GSVA and PCA show that AD astrocytes—so defined because they appear in patients with the typical down-regulation of neuronal synaptic functions documented in AD—undergo a profound phenotypical transformation encompassing up-regulation of genes related to ‘PAP’, ‘gliotransmission’, ‘plasticity’ and ‘stress responses’, and down-regulation of genes encoding organelle proteins (e.g., nuclear-encoded mitochondrial genes related to TCA and ETC). The detection of down-regulated astrocytic genes that may have gone unnoticed in large-scale clustering (B. Zhang et al., 2013), for their expression change in opposite direction to most genes in the original cluster, is an asset of the study.
Third, according to regression analyses between functions and CERAD or Braak scores, some astrocytic functions may be altered early in the AD continuum (e.g., ‘PAP’, ‘gliotransmission’ and ‘endolysosomal system’), while other functions may be increasingly dysregulated in parallel with the deposition of neuritic plaques and neurofibrillary tangles (e.g., mitochondrial functions, plasticity-related pathways, and stress responses).
Fourth, multivariate statistics and hierarchical clustering show that cortical astrocytes from human AD and from a 15–18-month-old mouse model of ADAD (Orre et al., 2014) are distinct transcriptional entities.
Limitations
A limitation of the study is the potential differences in gene expression due to different cortical regions being used in the three databases. However, the fact that consensus differences were found in astrocytic functional categories associated with AD supports the possibility that reactive astrocytes share common changes across brain areas, and that our method is robust enough to detect them. Likewise, our analysis with a single astrocytic gene cluster does not reveal astrocyte populations, as recently described in a mouse model of AD using snRNAseq (Habib et al., 2020), although it is tempting to speculate that the four astrocytic gene sub-clusters identified in the clinical cohorts (Aa-d, Fig. 3) might represent distinct transcriptomic states. Arguably, future studies and approaches such as spatial transcriptomics will reveal local and regional differences of human astrocytes in AD.
Another limitation of our study is that pathway dysregulation during AD may result in the remodeling of cell-type specific gene clusters, such that gene clusters identified in healthy brain transcriptomes may no longer exist in AD. However, quantification of network reorganization in AD subjects using a metric called modular differential connectivity showed equal or enhanced connectivity in 95.5% of the modules (B. Zhang et al., 2013), suggesting that clusters identified by co-expression analyses in healthy cells are, for the most part, preserved in AD. That said, we recognize that our analysis may overlook changes in transcripts ubiquitous in all cell types (e.g., glycolysis-related transcripts), as well as the up-regulation in astrocytes of transcripts typical of non-astrocytic cells, namely microglia, including the inflammatory response common to both glial cells described in 15–18-month-old APP/PS1 transgenic mice (Orre et al., 2014). Hence, our results may be just the tip of the iceberg in terms of defining the totality of changes in human AD astrocytes.
Because transcriptomic data are cross-sectional and descriptive, they do not demonstrate cause-effect relationships between pathway dysregulation and pathology, or unequivocally prove that changes are adaptive (meant to compensate for dysregulated functions and maintain or restore homeostasis), maladaptive (irreversibly contributing to dyshomeostasis and neurodegeneration), or epiphenomena with scarce bearing on disease onset and progression. Finally, transcriptomic data does not directly inform about changes in proteins and metabolites. Hence, predictions, based upon these data need validation in appropriate models.
Key implications and predictions
Use of molecular data for subject stratification in clinical trials.
A disconnect between presence of AD pathology and cognition has been long recognized (Neuropathology Group. Medical Research Council Cognitive & Aging, 2001). Neurocognitive measurements, atrophy patterns assessed with neuroimaging, neurofibrillary tangles assessed postmortem, CSF levels of amyloid-β1–42 and Tau, as well as Tau neuroimaging, have revealed subtypes of AD (Devi & Scheltens, 2018; Dujardin et al., 2020). The clinical and biological heterogeneity in AD, which is increasingly being recognized as a major obstacle to establishing drug efficacy in clinical trials, starts to be addressed with molecular tools. An example is the recent detection of three molecular classes in an extended MtSINAI cohort (MMBB-AD), using RNAseq transcriptomics of four brain regions (Neff, 2021). They found the largest global change between AD patients and non-demented controls in the parahippocampal gyrus (PHG), with AD patients presenting three molecular subtypes termed A, B and C. Although our analysis was performed in cortical samples rather than the PHG, our AD2 group might correspond to their ‘C typical’ AD class, and our AD1 to their ‘A atypical’ AD class. The reason is that both AD2 and C present down-regulation of synaptic networks as compared to AD1 and A. No further comparison between our study and (Neff, 2021) can be performed with regards to astrocytes because, among the only three astrocytic genes reported to be upregulated in the ‘C typical’ AD class’, only GNA12 appears in our astrocytic cluster # 4—although it is indeed upregulated in ROSMAP (Supplementary data 13).
What are Control2 and AD1? The terms ‘resistance’ and ‘resilience’ have been coined to describe two distinct clinical scenarios in AD consisting of avoiding pathology (‘resistance’) vs coping with pathology (‘resilience’) (reviewed in (Arenaza-Urquijo & Vemuri, 2018)). Thus, resistant subjects would be cognitively normal without significant amyloid-β or Tau pathology, despite their being at high-risk considering factors such as age or APOE genotype. By contrast, resilient subjects would remain cognitively normal despite significant AD pathology. We speculate that our AD1 subjects were perhaps ‘resilient’ to AD pathology, which they had, according to high CERAD and Braak scores, but that they developed dementia due to comorbidities (e.g., Lewy bodies, vascular disease, TDP43 deposits). However, the possibility exists that the tissues used for transcriptomic analysis and neuropathological assessment of neuritic plaques and neurofibrillary tangles were not the same. This means that the transcriptomic analysis in demented AD1 subjects with a healthy-like molecular profile might have been performed in cortical tissue spared from ongoing pathology, such that AD1 subjects were not truly resilient to AD pathology. By contrast, regardless of where omics and neuropathological assessments were performed, Control2 subjects with AD-like molecular profiles in ROSMAP and MAYO were cognitively normal. Thus, Control2 subjects might be high-risk, presymptomatic, resistant subjects in whom the appearance of neuritic plaques, Tau tangles, and cognitive impairment was delayed despite the decreased expression of synaptic genes, plausibly caused by the incipient accumulation of soluble oligomeric amyloid-β (Marsh & Alifragis, 2018). Whatever the case, our study supports the use of omics-based molecular phenotyping and clustering statistics as unbiased tools to stratify patients in clinical trials in the spirit of personalized medicine.
Early dysregulation of the endolysosomal system in astrocytes.
The category ‘endolysosomal system’ was found down-regulated in AD2 vs Control1 by GSVA. Further, genes related to endoplasmic reticulum (e.g., FAF2, PIGX), endocytosis (e.g., ATXN2, MEGF8, RAB11FIP2), and autophagy (ATG9A) ranked among the top 50 PC1 downregulated in ROSMAP (Fig. 6B), and were significantly down-regulated in at least two of the three databases according to the Wilcoxon test (Supplementary file 4). These findings support cumulative evidence of inefficient phagocytosis and degradation of amyloid-β protofibrils (Funato et al., 1998; Sanchez-Mico et al., 2020; Sollvander et al., 2016) and dystrophic neurites (Gomez-Arboledas et al., 2018) in astrocytes in mouse and in vitro models of AD. The decreased production of exosomes from astrocytes exposed to amyloid-β (Abdullah et al., 2016) also points to malfunction of the astrocytic endolysosomal system in AD models. Thus, our study and others do not support the popular notion that astrocytes may efficiently clear amyloid-β plaques, as initially suggested by evidence in non-primary cultured astrocytes (Wyss-Coray et al., 2003), which, arguably, are endowed with greater motility and morphological plasticity than primary or in situ astrocytes. Moreover, targeted enhancement of lysosomal biogenesis by overexpression of the transcription factor EB (TFEB) in astrocytes enhances Tau clearance, and reduces pathological hallmarks in the hippocampus of PS19 tauopathy mice (Martini-Stoica et al., 2018), suggesting that the improvement of lysosomal function in astrocytes may reverse Tau pathology in AD. Along these lines, TFEB has been reported to regulate AD risk genes in an astrocyte subpopulation detected in AD brains by snRNAseq (Grubman et al., 2019).
Early and concomitant alteration of neuron-astrocyte contacts.
This prediction is based on two findings: the inverse correlation of changes in synaptic functions and PAP, and the lack of correlation between these functional categories with pathological stages, suggesting that they change early in AD. Accordingly, both PAP and synaptic genes are dysregulated in non-demented subjects with a type 2 molecular profile (Control2), which, as noted earlier, may represent preclinical AD. Specifically, GSVA showed up-regulation of ‘PAP morphology’, whose leading gene EZR encodes ezrin, a protein involved in PAP motility, and in the anchoring of the astrocyte membrane to neurons or the extracellular matrix (Derouiche & Geiger, 2019). Also, genes related to integrin and cell motility (e.g., FERMT2, PREX2) were part of the top genes detected by PCA as defining AD astrocytes (Fig. 6B). We posit that up-regulation of PAP genes is an adaptive change to improve the coverage of neurons to counteract the down-regulation of synaptic elements directly caused by oligomeric amyloid-β (Marsh & Alifragis, 2018). This scenario is further supported by the up-regulation of genes encoding for synaptogenic factors such as thrombospondin and glypicans (THSD1, GPC4) in the three databases (Supplementary file 4).
Mitochondrion dysfunction in AD astrocytes may impact gliotransmission.
It is generally assumed that the well-established mitochondrion dysfunction (Swerdlow, 2018) and reduced glucose metabolism in AD (often referred to as ‘hypometabolism’ (Hoffman et al., 2000)) are neuronal phenomena. However, our analysis reveals down-regulation of nuclear-encoded mitochondrial genes encoding TCA cycle and ETC components in astrocytes, suggesting deficiencies in energy-generating mitochondrial pathways. For example, one of the consensus downregulated genes, ATPAF1, is a component of the ATP synthase (complex V), and it is known that impairment of the ETC beyond complexes I-III increases reactive-oxygen species generation while decreasing ATP production (Shi & Gibson, 2007). In vitro and ex vivo studies link mitochondrial dysfunction in astrocytes to amyloid-β. Thus, in co-cultures of rat hippocampal neurons and astrocytes, amyloid-β causes loss of mitochondrial potential and transient mitochondrial depolarization, concomitant to activation of NADPH oxidase and decrease glutathione production, in astrocytes, but not neurons (Abramov, Canevari, & Duchen, 2004). The latter study also shows that loss of membrane potential and depolarization of astrocytic mitochondria is prevented by antioxidants, and by the administration of glutamate as a substrate for the mitochondrial complex I. This suggests that amyloid-β-elicited mitochondrial dysfunction in astrocytes is caused by oxidative stress and deficits in substrate supply, such that increasing the supply of fuels amenable for mitochondrial oxidation including aromatic amino acids such as glutamate may be astroprotective. Based on this evidence, we interpret the consensus up-regulation of the mitochondrial glutamate transporter SLC25A18, and the plasma membrane glutamate transporters SLC1A3, SLC7A11 and SLC6A11 (Supplementary file 4) to indicate that AD astrocytes may take up glutamate to counteract the damage caused by amyloid-β to mitochondrial respiration. Along these lines, HADH8, which encodes an enzyme involved in mitochondrial β-oxidation, was found upregulated in AD astrocytes by PCA.
Likewise, the consensus up-regulation in AD astrocytes of the monoamine oxidase MAO-B, a dopamine-degrading enzyme located in the outer mitochondrial membrane, may be interpreted as an attempt to reverse respiratory deficits. This is supported by the recent discovery that MAO-B increases the polarization of the inner mitochondrial membrane and ATP production by shuttling electrons through the inter-membrane space (Graves et al., 2020). MAO-B up-regulation in AD is relevant for two reasons. First, the MAO-B ligand (11)C-deuterium-L-deprenyl may serve as a PET-based biomarker of early astrocyte dysfunction in AD, as documented in ADAD (Rodriguez-Vieitez et al., 2016). Second, MAO-B may affect the excitatory/inhibitory balance of neural circuits by producing GABA from putrescine, as shown in APP/PS1 mouse hippocampus (Jo et al., 2014). In support of this scenario, the genes encoding the GABA transporter SLC6A11/GAT3, which may extrude GABA coupled to glutamate uptake, and SLC7A2, which imports the putrescine precursor L-arginine, are consensus dysregulated genes (Supplementary file 4).
Taken together, the data support that impaired mitochondrial respiration in astrocytes switches gliotransmission to a GABA-dominant mode. What may the clinical impact of this switch be? A detrimental effect of astrocytic GABA has been proposed based on the beneficial effects of the MAO-B inhibitor selegiline on electrophysiological readouts in APP/PS1 mouse hippocampi (Jo et al., 2014); however, there is no benefit of selegiline in patients (Birks & Flicker, 2003), thus raising doubts as to whether APP/PS1 mice appropriately model the impact of astrocytic GABA in AD.
Alternatively, astrocytic GABA might counteract the loss of GABAergic tone that has been causally related with neuronal hyperactivity, desynchronization of neural circuits, and amyloid-β production (Lee, Gerashchenko, Timofeev, Bacskai, & Kastanenka, 2020). Since our regression analyses reveal that mitochondrial dysregulation in astrocytes progressively increases in advanced pathological stages, and since the up-regulation of MAO-B detected by PET in prodromal ADAD is transient (Rodriguez-Vieitez et al., 2016), we reason that the two strategies used by the astrocytic mitochondria to preserve membrane potential and ETC (i.e., enhanced supply of substrates such as aromatic amino acids like glutamate, and of electrons via MAO-B) eventually fail. As a consequence, glutathione production might be decreased and NADPH oxidase activity increased (Shi & Gibson, 2007), perhaps exacerbating oxidative stress and overall astrocyte malfunction in advanced AD.
Pathological-stage dependent up-regulation of ‘plasticity’ and ‘stress response’ pathways might explain why, unlike neurons, astrocytes survive in AD.
The progressive up-regulation of the subcategory ‘ECM’, which includes integrins and proteoglycans, is not surprising considering the indisputable adoption of a ‘reactive’ morphology by astrocytes in AD, consisting of engrossment of primary and secondary processes due to over-expression of the intermediate filament protein GFAP, and increased production of ECM proteins (Escartin, Guillemaud, & Carrillo-de Sauvage, 2019). More intriguing is the consensus up-regulation of ‘hippo signaling’, an evolutionarily conserved network with a central role in the regulation of cell proliferation, cell fate, organ growth and regeneration (Misra & Irvine, 2018), and no reported role in healthy adult astrocytes, despite the fact that some of the genes in this pathway are highly specific to human adult astrocytes according to τ (e.g., WWC1 τ=0.91; WWOX τ =0.86, and YAP1 τ=0.88). YAP1 was, moreover, detected by PCA as a relevant gene in the astrocytic phenotype in AD. The up-regulation of ‘hippo signaling’ in AD astrocytes, as well as the global up-regulation of genes involved in brain development may be a manifestation of the well documented phenomenon of re-activation of developmental pathways in reactive astrocytes that some authors interpret as a failed attempt at reprogramming into neural stem cells (Torper & Gotz, 2017), but we interpret to be a protective response to facilitate the survival of defective astrocytes. In addition, the recent observation that YAP1 regulates scar-border formation in spinal cord injury in mice (Xie et al., 2020), suggests that hippo signaling is coordinated with the increased production of ECM in AD astrocytes. Finally, the consensus dysregulation of the subcategory ‘TNF-alpha’ (Supplementary file 10), and the consensus up-regulation of members of ‘TNFalpha’ TNFRSF11B, TNFSF13 and TRAF3IP2, as well as of members of the IL17 family (Il17D, IL17RB, and IL17RD), and of complement factors (CD59, C1S) (Supplementary file 4), might be interpreted as detrimental ‘neuroinflammation’, although evidence also supports modulatory or protective actions of these factors (Masgrau, Guaza, Ransohoff, & Galea, 2017). First, TNFα may facilitate the release of glutamate by improving vesicle docking at the astrocyte plasma membrane prior to exocytosis (Santello et al., 2011), such that TNFα signaling may help adjust gliotransmission in AD astrocytes. Second, the finding that Nrf2/IL17D axis acts as an antioxidant protective pathway in stress responses induced by tumorigenic stimuli and viral infections (Saddawi-Konefka et al., 2016), supports an astroprotective role of members of the IL17 family in AD. Also, members of the complement system have been shown to facilitate amyloid-β phagocytosis (Iram et al., 2016).
Model of the phenotypical transformation of cortical astrocytes in AD
The key predictions from our analysis are summarized in Fig. 7A. We posit that amyloid-β drives the phenotypical change of astrocytes in AD in several stages by causing dysfunction of the endolysosomal/mitochondrial axis, which, in turn, prompts a change in the balance of excitatory/inhibitory neurotransmission. Impairment of mitophagy due to endolysosomal malfunction may further exacerbate mitochondrial dysregulation. Control2 subjects (cognitively normal/AD-like molecular profile) would be at stage a, and AD2 patients (demented, AD molecular profile) at stage b. In both a and b, the phenotypical change is complex and mixed with regards to possible clinical impact, for changes that may disrupt the homeostasis of astrocytes themselves and/or astrocyte-neuron communications coexist with changes that may protect astrocytes and/or their interactions with neurons. The model thus represents a departure from simplistic neuroprotective/neurotoxic classifications of reactive astrocytes from the point of view of neurons. As recently discussed (Escartin et al., 2021), an ‘astrocyte-centric’ stance focused on the clarification of the maladaptive or adaptive nature of pathway changes in the complex astrocyte biology should be adopted. What is then the net impact of the mixed functional changes of reactive astrocytes in AD? In short, we posit that, despite organelle dysfunction, astrocytes manage in early disease stages to partially preserve their functions, including the modulation of neural circuits, through adaptive changes, while they become globally malfunctional in advanced stages. Thus, the early changes in astrocyte-neuron interplay may delay the onset of clinical symptoms, while detrimental phenomena would prevail in stage b. If these predictions are correct, therapies aimed at protecting and restoring the functions of the endolysosomal system and mitochondria to halt the transformation of astrocytes to stage b might help arrest the progression of AD. Even if GABA were detrimental, as reported in (Jo et al., 2014), preservation of mitochondrial respiration is therapeutically indicated, because increased GABA production, whatever its effect, would be the result of mitochondrial impairment. In summary, the present study points to prevention of organelle dysfunction in astrocytes as a therapeutic strategy in AD. These predictions need to be tested in transgenic mice or human cellular models that appropriately recapitulate the complex transformation of astrocytes in the AD continuum.
Conclusions
Our systems-biology based identification of astrocytic clusters in three independent datasets was aimed to extract consensus data of the main pathway changes in reactive astrocytes in AD. Arguably, the large number of subjects analyzed allowed us to detect molecular heterogeneity of subjects with the same clinical diagnosis by hierarchical clustering. We highlight that manually curated functional annotations were implemented to circumvent deficits in astrocyte-specific annotations in current platforms. The findings, supported by two independent statistical analysis, have led to a model of stage-dependent astrocyte dysfunction in AD caused by amyloid-β-elicited damage of the endolysosomal and mitochondrial systems. Defective mitophagy due to lysosomal malfunction may further exacerbate the mitochondrial dysfunction caused by Aβ. Future studies may clarify whether the highly significant negative correlation between down-regulation of nuclear-encoded mitochondrial genes and Braak and CERAD scores means that mitochondrion stress in astrocytes contribute to the progression of AD.
Supplementary Material
Tabs:
1. Tab description.
2. Transcriptomic databases of human-brain cells obtained from (Y. Zhang et al., 2016).
3. PCA of transcriptomic data of astrocytes isolated from young (<47 years old) and old (>47 years old) shows that they are significantly different.
4. Specific data used for τ scoring and hierarchical clustering.
5. Top τ scores per gene, indicating which cell type presents that score, and pie charts of the distribution of specific and enriched genes among brain cells. Astrocytes (8%), microglia (7.6%) and neurons (7.2%) present the largest percentage of cell-specific genes, whereas neurons (21%) contained the largest pool of cell-enriched genes, followed by microglia (13.4%) and astrocytes (9.4%). In contrast, endothelial cells and oligodendrocytes present the lowest percentages of cell-enriched (6%) and specific (3%) genes.
6. Raw results of hierarchical clustering.
7. Cluster description.
8. Cluster selection.
Tabs:
1. Circo plots showing functional categories (inner ring) and subcategories (outer ring) of astrocytic gene cluster # 4 (1650 genes) and neuronal gene cluster # 13 (1258 genes). For astrocytes, there are 17 groups and 133 subgroups. For neurons, 15 groups and 76 subgroups.
2. Functional categorization of neuronal cluster # 13.
3. Functional categorization of astrocytic cluster # 4.
Gene expressions were normalized according to the probabilistic distribution of relative expressions to reach distribution peaks of zero (Methods). The normalization improved the resolution of astrocytic and neuronal gene clusters between and within cohorts.
The statistical comparison between AD2 and Control1 was performed by the Wilcoxon rank sum test. Statistical significance pFDR < 0.05. Consensus dysregulated genes are indicated in color: blue, down-regulated; yellow, up-regulated, and grey, unchanged.
Comparisons: ROSMAP: Control2 vs Control1 (pFDR1), MCI1 vs Control1 (pFDR2), MCI2 vs Control1 (pFDR3), AD1 vs Control1 (pFDR4), AD2 vs Control1 (pFDR4). MAYO and MtSINAI: Control2 vs Control1 (pFDR1), AD1 vs Control1 (pFDR2), AD2 vs Control1 (pFDR3). Tabs: 1. Functional categories in neurons. 2. Functional categories in astrocytes.
Enrichment scores of functional categories significantly changed in at least 2 out of 3 databases. (&) pFDR <0.01, (#) pFDR < 0.05 and (*) pFDR < 0.001, vs Control1.
Bolded numbers are the probability of a zero slope for a linear model. A. Braak scores. B. CERAD scores.
Bolded numbers are the probability of a zero slope for a linear model. A. Braak scores. B. CERAD scores.
A, C, E. Hierarchical clustering of Pearson correlation coefficients. The enrichment scores of all functions across all samples were correlated with one another (Methods). The clustering segregated correlations in two main branches that grouped down-regulated (branch 1) and up-regulated functions (branch 2), respectively. Functions found altered in at least two out of the three databases are labeled in blue (down-regulated) or red (up-regulated) squares. Strongest direct correlations were among highly related biological processes such as ‘membrane potential’, ‘neurotransmission’ and ‘synaptic plasticity’ in neurons, and ‘PAP’ and ‘gliotransmission’ in astrocytes (orange scale). The strongest anti-correlations (i.e., concomitant changes in opposite directions, grey scale) were: (i) down-regulation of synaptic functions vs up-regulation of neuronal ‘mitochondria’, ‘stress response’ and ‘gene expression’ (dark green squares); (ii) decrease in synaptic functions vs up-regulation of ‘PAP/gliotransmission’ and ‘plasticity’ in astrocytes (light green squares); (iii) in astrocytes, increases in ‘PAP/gliotransmission’ vs down-regulation of ‘endolysosomal system’ and ‘gene expression’, and ‘plasticity’ vs ‘mitochondria’ (pink squares). In summary, anti-correlations (negative p-values) point to highly intertwined changes between morphological remodeling in astrocytes and neuronal dysfunction, and between survival responses and organelle dysfunction in astrocytes.
B, D, F. Heatmap of FDR adjusted p-values. In red, adj FDR p-values < 0.05
Comparisons: Control2 vs Control1 (pFDR1), AD1 vs Control1 (pFDR2), AD2 vs Control1 (pFDR3).
Tabs:
1. All functional subcategories/MAYO.
2. All functional subcategories/ROSMAP.
3. All functional subcategories/MtSINAI.
4. PAP/all databases.
5. Gliotransmission/all databases.
6. Plasticity/all databases.
7. Stress response/all databases.
8. Mitochondria in all data/all databases.
9. Endolysosomal system/all databases.
Astrocyte cluster # 4 ranked according to PC1 in ROSMAP, and comparison with DEG identified by snRNAseq in human AD astrocytes (Mathys et al., 2019) (Grubman et al., 2019). Tabs:
1. Venn diagrams.
2. Comparison of PC1/ROSMAP with (Mathys et al., 2019).
3. Comparison of PC1/ROSMAP with (Grubman et al., 2019).
4. Comparison of PC1/ROSMAP between (Mathys et al., 2019) and (Grubman et al., 2019).
Supplementary file 12. Comparison of the functional category of cluster # 4 of human astrocytes containing immunity-related genes with the immunity-related GO groups found significantly dysregulated in APP/PS1 mouse astrocytes (Orre et al., 2014). Only C1S (Complement Component 1, S Subcomponent) appears in both gene sets.
Highlights.
Detection of two molecular profiles in both control subjects and AD patients.
Use of astrocyte-specific annotations and pre-clustered gene sets.
Downregulation of endolysosome-related genes in early Braak/CERAD stages.
Progressive downregulation of mitochondrion genes along Braak/CERAD stages.
Astrocytic endolysosomes and mitochondria as therapeutic targets in AD.
Acknowledgements:
The authors thank Dr. Alberto Lleó of the Unitat de Memòria at the Hospital Sant Pau, Barcelona, Spain, for critical reading of the manuscript. This work was supported by Fundació Marato-TV3 (grant number 201414-30), and Grup de Recerca en demèncias: Sant Pau SGR2017547, Generalitat de Catalunya (E.G.), by funds from the Coins for Alzheimer’s Research Trust (L.B.W.) and startup funds from the Woodruff School of Mechanical Engineering at Georgia Tech (L.B.W.). L.D.W. and A.F.P were supported in part by the National Institutes of Health Cell and Tissue Engineering Biotechnology Training Grant (T32-GM008433). CNRS, CEA and France Alzheimer (C.E.). The results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.synapse.org). The authors thank the researchers in charge of the portal for their kind and patient assistance. Study data were provided by the following sources: The Mayo Clinic Alzheimers Disease Genetic Studies, led by Dr. Nilufer Ertekin-Taner and Dr. Steven G. Younkin, Mayo Clinic, Jacksonville, FL using samples from the Mayo Clinic Study of Aging, the Mayo Clinic Alzheimers Disease Research Center, and the Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, R01 AG003949, NINDS grant R01 NS080820, CurePSP Foundation, and support from Mayo Foundation. Study data includes samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. Study data were also provided by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago. Data collection was supported through funding by NIA grants P30AG10161 (ROS), R01AG15819 (ROSMAP; genomics and RNAseq), R01AG17917 (MAP), R01AG30146, R01AG36042 (5hC methylation, ATACseq), RC2AG036547 (H3K9Ac), R01AG36836 (RNAseq), R01AG48015 (monocyte RNAseq) RF1AG57473 (single nucleus RNAseq), U01AG32984 (genomic and whole exome sequencing), U01AG46152 (ROSMAP AMP-AD, targeted proteomics), U01AG46161(TMT proteomics), U01AG61356 (whole genome sequencing, targeted proteomics, ROSMAP AMP-AD), the Illinois Department of Public Health (ROSMAP), and the Translational Genomics Research Institute (genomic). ROSMAP data can be requested at www.radc.rush.edu. All data generated during this study are included in this published article and its supplementary information files. Codes are available upon reasonable request.
Footnotes
Declaration of interests: none.
Elena Galea: Conceptualization, Methodology, Data curation, Funding acquisition, Supervision, Writing (Original Draft, Review and Editing); Laura D. Weinstock: Methodology, Software, Data curation, Formal analysis, Writing (Review and Editing); Raquel Larramona-Arcas: Data curation; Writing (Review and Editing); Alyssa F. Pybus: Methodology, Software, Data curation, Formal analysis, Writing (Review and Editing); Lydia Giménez-Llort: Funding acquisition, Writing (Review and Editing); Carole Escartin: Writing (Major Review and Editing); Levi B. Wood: Conceptualization, Methodology, Software, Data curation, Formal analysis, Funding acquisition, Supervision, Writing (Original Draft, Review and Editing).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdullah M, Takase H, Nunome M, Enomoto H, Ito J, Gong JS, & Michikawa M. (2016). Amyloid-beta Reduces Exosome Release from Astrocytes by Enhancing JNK Phosphorylation. J Alzheimers Dis, 53(4), 1433–1441. doi: 10.3233/JAD-160292 [DOI] [PubMed] [Google Scholar]
- Abramov AY, Canevari L, & Duchen MR (2004). β-Amyloid Peptides Induce Mitochondrial Dysfunction and Oxidative Stress in Astrocytes and Death of Neurons through Activation of NADPH Oxidase. The Journal of Neuroscience, 24(2), 565–575. doi: 10.1523/jneurosci.4042-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, Carrasquillo MM, Funk C, Heavner BD, Zou F, Younkin CS, . . . Ertekin-Taner N. (2016). Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci Data, 3, 160089. doi: 10.1038/sdata.2016.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenaza-Urquijo EM, & Vemuri P. (2018). Resistance vs resilience to Alzheimer disease: Clarifying terminology for preclinical studies. Neurology, 90(15), 695–703. doi: 10.1212/WNL.0000000000005303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, & Schneider JA (2018). Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis, 64(s1), S161–S189. doi: 10.3233/JAD-179939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birks J, & Flicker L. (2003). Selegiline for Alzheimer’s disease. Cochrane Database Syst Rev(1), CD000442. doi: 10.1002/14651858.CD000442 [DOI] [PubMed]
- Derouiche A, & Geiger KD (2019). Perspectives for Ezrin and Radixin in Astrocytes: Kinases, Functions and Pathology. Int J Mol Sci, 20(15). doi: 10.3390/ijms20153776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi G, & Scheltens P. (2018). Heterogeneity of Alzheimer’s disease: consequence for drug trials? Alzheimers Res Ther, 10(1), 122. doi: 10.1186/s13195-018-0455-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin S, Commins C, Lathuiliere A, Beerepoot P, Fernandes AR, Kamath TV, . . . Hyman BT (2020). Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nature Medicine, 26(8), 1256–1263. doi: 10.1038/s41591-0200938-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, . . . Verkhratsky A. (2021). Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. doi: 10.1038/s41593-020-00783-4 [DOI] [PMC free article] [PubMed]
- Escartin C, Guillemaud O, & Carrillo-de Sauvage MA (2019). Questions and (some) answers on reactive astrocytes. Glia, 67(12), 2221–2247. doi: 10.1002/glia.23687 [DOI] [PubMed] [Google Scholar]
- Funato H, Yoshimura M, Yamazaki T, Saido TC, Ito Y, Yokofujita J, . . . Ihara Y. (1998). Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. Am J Pathol, 152(4), 983–992. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/9546359 [PMC free article] [PubMed] [Google Scholar]
- Gomez-Arboledas A, Davila JC, Sanchez-Mejias E, Navarro V, Nunez-Diaz C, Sanchez-Varo R, . . . Gutierrez A (2018). Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia, 66(3), 637–653. doi: 10.1002/glia.23270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, . . . Hyman BT (1997). Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol, 41(1), 17–24. doi: 10.1002/ana.410410106 [DOI] [PubMed] [Google Scholar]
- Graves SM, Xie Z, Stout KA, Zampese E, Burbulla LF, Shih JC, . . . Surmeier DJ (2020). Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat Neurosci, 23(1), 15–20. doi: 10.1038/s41593-019-0556-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman A, Chew G, Ouyang JF, Sun GZ, Choo XY, McLean C, . . . Polo JM (2019). A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nature Neuroscience, 22(12), 2087–+. doi: 10.1038/s41593-019-0539-4 [DOI] [PubMed] [Google Scholar]
- Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, . . . Schwartz M. (2020). Disease-associated astrocytes in Alzheimer’s disease and aging. Nature Neuroscience, 23(6), 701–706. doi: 10.1038/s41593-020-0624-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Qu J, Zhao J, & Zou X. (2018). Analyzing 74,248 Samples Confirms the Association Between CLU rs11136000 Polymorphism and Alzheimer’s Disease in Caucasian But Not Chinese population. Sci Rep, 8(1), 11062. doi: 10.1038/s41598-018-29450-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, & Guinney J. (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics, 14, 7. doi: 10.1186/1471-2105-14-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JM, Welsh-Bohmer KA, Hanson M, Crain B, Hulette C, Earl N, & Coleman RE (2000). FDG PET imaging in patients with pathologically verified dementia. J Nucl Med, 41(11), 1920–1928. [PubMed] [Google Scholar]
- Iram T, Trudler D, Kain D, Kanner S, Galron R, Vassar R, . . . Frenkel D. (2016). Astrocytes from old Alzheimer’s disease mice are impaired in Abeta uptake and in neuroprotection. Neurobiol Dis, 96, 84–94. doi: 10.1016/j.nbd.2016.08.001 [DOI] [PubMed] [Google Scholar]
- Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, . . . Lee CJ (2014). GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med, 20(8), 886–896. doi: 10.1038/nm.3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley KW, Nakao-Inoue H, Molofsky AV, & Oldham MC (2018). Variation among intact tissue samples reveals the core transcriptional features of human CNS cell classes. Nat Neurosci, 21(9), 1171–1184. doi: 10.1038/s41593-018-0216-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryuchkova-Mostacci N, & Robinson-Rechavi M. (2017). A benchmark of gene expression tissue-specificity metrics. Brief Bioinform, 18(2), 205–214. doi: 10.1093/bib/bbw008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YF, Gerashchenko D, Timofeev I, Bacskai BJ, & Kastanenka KV (2020). Slow Wave Sleep Is a Promising Intervention Target for Alzheimer’s Disease. Frontiers in Neuroscience, 14(705). doi: 10.3389/fnins.2020.00705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Marsh SE, & Stevens B. (2020). Microglia and Astrocytes in Disease: Dynamic Duo or Partners in Crime? Trends Immunol, 41(9), 820–835. doi: 10.1016/j.it.2020.07.006 [DOI] [PubMed] [Google Scholar]
- Lin SM, Du P, Huber W, & Kibbe WA (2008). Model-based variance-stabilizing transformation for Illumina microarray data. Nucleic Acids Research, 36(2), e11–e11. doi: 10.1093/nar/gkm1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlatt MW, Bauer J, Aronica E, van Haastert ES, Hoozemans JJ, Joels M, & Lucassen PJ (2014). Proliferation in the Alzheimer hippocampus is due to microglia, not astroglia, and occurs at sites of amyloid deposition. Neural Plast, 2014, 693851. doi: 10.1155/2014/693851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh J, & Alifragis P. (2018). Synaptic dysfunction in Alzheimer’s disease: the effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen Res, 13(4), 616–623. doi: 10.4103/1673-5374.230276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini-Stoica H, Cole AL, Swartzlander DB, Chen F, Wan YW, Bajaj L, . . . Zheng H. (2018). TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J Exp Med, 215(9), 2355–2377. doi: 10.1084/jem.20172158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masgrau R, Guaza C, Ransohoff RM, & Galea E. (2017). Should We Stop Saying ‘Glia’ and ‘Neuroinflammation’? Trends Mol Med, 23(6), 486–500. doi: 10.1016/j.molmed.2017.04.005 [DOI] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng ZY, Gao F, Mohammadi S, Young JZ, . . . Tsai LH (2019). Single-cell transcriptomic analysis of Alzheimer’s disease (vol 570, pg 332, 2019). Nature, 571(7763), E1–E1. doi: 10.1038/s41586-019-1329-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra JR, & Irvine KD (2018). The Hippo Signaling Network and Its Biological Functions. Annu Rev Genet, 52, 65–87. doi: 10.1146/annurev-genet-120417-031621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff RA, et al. (2021). Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Science Advances, 7. doi: 10.1126/sciadv.abb5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuropathology Group. Medical Research Council Cognitive, F., & Aging S. (2001). Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet, 357(9251), 169–175. doi: 10.1016/s0140-6736(00)03589-3 [DOI] [PubMed] [Google Scholar]
- Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K, & Hol EM (2014). Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol Aging, 35(12), 2746–2760. doi: 10.1016/j.neurobiolaging.2014.06.004 [DOI] [PubMed] [Google Scholar]
- Pelvig DP, Pakkenberg H, Regeur L, Oster S, & Pakkenberg B. (2003). Neocortical glial cell numbers in Alzheimer’s disease. A stereological study. Dement Geriatr Cogn Disord, 16(4), 212–219. doi: 10.1159/000072805 [DOI] [PubMed] [Google Scholar]
- Perez-Nievas BG, & Serrano-Pozo A. (2018). Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front Aging Neurosci, 10, 114. doi: 10.3389/fnagi.2018.00114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Vieitez E, Saint-Aubert L, Carter SF, Almkvist O, Farid K, Schöll M, . . . Nordberg A. (2016). Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain, 139(Pt 3), 922–936. doi: 10.1093/brain/awv404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saddawi-Konefka R, Seelige R, Gross ET, Levy E, Searles SC, Washington A Jr., . . . Bui JD (2016). Nrf2 Induces IL-17D to Mediate Tumor and Virus Surveillance. Cell Rep, 16(9), 2348–2358. doi: 10.1016/j.celrep.2016.07.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saelens W, Cannoodt R, & Saeys Y. (2018). A comprehensive evaluation of module detection methods for gene expression data. Nat Commun, 9(1), 1090. doi: 10.1038/s41467-018-03424-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakers K, Lake AM, Khazanchi R, Ouwenga R, Vasek MJ, Dani A, & Dougherty JD (2017). Astrocytes locally translate transcripts in their peripheral processes. Proc Natl Acad Sci U S A, 114(19), E3830–E3838. doi: 10.1073/pnas.1617782114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Mico MV, Jimenez S, Gomez-Arboledas A, Munoz-Castro C, Romero-Molina C, Navarro V, . . . Vitorica J. (2020). Amyloid-beta impairs the phagocytosis of dystrophic synapses by astrocytes in Alzheimer’s disease. Glia. doi: 10.1002/glia.23943 [DOI] [PubMed]
- Santello M, Bezzi P, & Volterra A. (2011). TNFα controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron, 69(5), 988–1001. doi: 10.1016/j.neuron.2011.02.003 [DOI] [PubMed] [Google Scholar]
- Sekar S, McDonald J, Cuyugan L, Aldrich J, Kurdoglu A, Adkins J, . . . Liang WS (2015). Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes. Neurobiology of Aging, 36(2), 583–591. doi: 10.1016/j.neurobiolaging.2014.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, & Gibson GE (2007). Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Dis Assoc Disord, 21(4), 276–291. doi: 10.1097/WAD.0b013e31815721c3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JE, Ince PG, Shaw PJ, Heath PR, Raman R, Garwood CJ, . . . Ne MCFA (2011). Microarray analysis of the astrocyte transcriptome in the aging brain: relationship to Alzheimer’s pathology and APOE genotype. Neurobiology of Aging, 32(10), 1795–1807. doi: 10.1016/j.neurobiolaging.2011.04.013 [DOI] [PubMed] [Google Scholar]
- Sollvander S, Nikitidou E, Brolin R, Soderberg L, Sehlin D, Lannfelt L, & Erlandsson A. (2016). Accumulation of amyloid-beta by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol Neurodegener, 11(1), 38. doi: 10.1186/s13024-016-0098-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen CB, Liu CL, Alizadeh AA, & Newman AM (2020). Profiling Cell Type Abundance and Expression in Bulk Tissues with CIBERSORTx. Methods in Molecular Biology, 2117, 135–157. doi: 10.1007/978-1-0716-0301-7_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH (2018). Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. Journal of Alzheimers Disease, 62(3), 1403–1416. doi: 10.3233/Jad-170585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torper O, & Gotz M. (2017). Brain repair from intrinsic cell sources: Turning reactive glia into neurons. Functional Neural Transplantation Iv: Translation to Clinical Application, Pt A, 230, 69–97. doi: 10.1016/bs.pbr.2016.12.010 [DOI] [PubMed] [Google Scholar]
- Viejo L, Noori A, Merrill E, Das S, Hyman BT, & Serrano-Pozo A. (2021). Systematic review of human post-mortem immunohistochemical studies and bioinformatics analyses unveil the complexity of astrocyte reaction in Alzheimer’s disease. Neuropathol Appl Neurobiol. doi: 10.1111/nan.12753 [DOI] [PMC free article] [PubMed]
- Wang X, Park J, Susztak K, Zhang NR, & Li M. (2019). Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat Commun, 10(1), 380. doi: 10.1038/s41467-018-08023-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch M. (2011). Gene Coexpression Networks for the Analysis of DNA Microarray Data (Salvador PDMDPDFESAGA Ed. Vol. 1). Applied Statistics for Network Biology: Methods in Systems Biology, Volume 1. [Google Scholar]
- Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, . . . Husemann, J. (2003). Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nature Medicine, 9(4), 453–457. doi: 10.1038/nm838 [DOI] [PubMed] [Google Scholar]
- Xie C, Shen X, Xu X, Liu H, Li F, Lu S, . . . Huang Z. (2020). Astrocytic YAP Promotes the Formation of Glia Scars and Neural Regeneration after Spinal Cord Injury. J Neurosci, 40(13), 2644–2662. doi: 10.1523/JNEUROSCI.2229-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, . . . Emilsson V. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell, 153(3), 707–720. doi: 10.1016/j.cell.2013.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, . . . Barres BA (2016). Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron, 89(1), 37–53. doi: 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tabs:
1. Tab description.
2. Transcriptomic databases of human-brain cells obtained from (Y. Zhang et al., 2016).
3. PCA of transcriptomic data of astrocytes isolated from young (<47 years old) and old (>47 years old) shows that they are significantly different.
4. Specific data used for τ scoring and hierarchical clustering.
5. Top τ scores per gene, indicating which cell type presents that score, and pie charts of the distribution of specific and enriched genes among brain cells. Astrocytes (8%), microglia (7.6%) and neurons (7.2%) present the largest percentage of cell-specific genes, whereas neurons (21%) contained the largest pool of cell-enriched genes, followed by microglia (13.4%) and astrocytes (9.4%). In contrast, endothelial cells and oligodendrocytes present the lowest percentages of cell-enriched (6%) and specific (3%) genes.
6. Raw results of hierarchical clustering.
7. Cluster description.
8. Cluster selection.
Tabs:
1. Circo plots showing functional categories (inner ring) and subcategories (outer ring) of astrocytic gene cluster # 4 (1650 genes) and neuronal gene cluster # 13 (1258 genes). For astrocytes, there are 17 groups and 133 subgroups. For neurons, 15 groups and 76 subgroups.
2. Functional categorization of neuronal cluster # 13.
3. Functional categorization of astrocytic cluster # 4.
Gene expressions were normalized according to the probabilistic distribution of relative expressions to reach distribution peaks of zero (Methods). The normalization improved the resolution of astrocytic and neuronal gene clusters between and within cohorts.
The statistical comparison between AD2 and Control1 was performed by the Wilcoxon rank sum test. Statistical significance pFDR < 0.05. Consensus dysregulated genes are indicated in color: blue, down-regulated; yellow, up-regulated, and grey, unchanged.
Comparisons: ROSMAP: Control2 vs Control1 (pFDR1), MCI1 vs Control1 (pFDR2), MCI2 vs Control1 (pFDR3), AD1 vs Control1 (pFDR4), AD2 vs Control1 (pFDR4). MAYO and MtSINAI: Control2 vs Control1 (pFDR1), AD1 vs Control1 (pFDR2), AD2 vs Control1 (pFDR3). Tabs: 1. Functional categories in neurons. 2. Functional categories in astrocytes.
Enrichment scores of functional categories significantly changed in at least 2 out of 3 databases. (&) pFDR <0.01, (#) pFDR < 0.05 and (*) pFDR < 0.001, vs Control1.
Bolded numbers are the probability of a zero slope for a linear model. A. Braak scores. B. CERAD scores.
Bolded numbers are the probability of a zero slope for a linear model. A. Braak scores. B. CERAD scores.
A, C, E. Hierarchical clustering of Pearson correlation coefficients. The enrichment scores of all functions across all samples were correlated with one another (Methods). The clustering segregated correlations in two main branches that grouped down-regulated (branch 1) and up-regulated functions (branch 2), respectively. Functions found altered in at least two out of the three databases are labeled in blue (down-regulated) or red (up-regulated) squares. Strongest direct correlations were among highly related biological processes such as ‘membrane potential’, ‘neurotransmission’ and ‘synaptic plasticity’ in neurons, and ‘PAP’ and ‘gliotransmission’ in astrocytes (orange scale). The strongest anti-correlations (i.e., concomitant changes in opposite directions, grey scale) were: (i) down-regulation of synaptic functions vs up-regulation of neuronal ‘mitochondria’, ‘stress response’ and ‘gene expression’ (dark green squares); (ii) decrease in synaptic functions vs up-regulation of ‘PAP/gliotransmission’ and ‘plasticity’ in astrocytes (light green squares); (iii) in astrocytes, increases in ‘PAP/gliotransmission’ vs down-regulation of ‘endolysosomal system’ and ‘gene expression’, and ‘plasticity’ vs ‘mitochondria’ (pink squares). In summary, anti-correlations (negative p-values) point to highly intertwined changes between morphological remodeling in astrocytes and neuronal dysfunction, and between survival responses and organelle dysfunction in astrocytes.
B, D, F. Heatmap of FDR adjusted p-values. In red, adj FDR p-values < 0.05
Comparisons: Control2 vs Control1 (pFDR1), AD1 vs Control1 (pFDR2), AD2 vs Control1 (pFDR3).
Tabs:
1. All functional subcategories/MAYO.
2. All functional subcategories/ROSMAP.
3. All functional subcategories/MtSINAI.
4. PAP/all databases.
5. Gliotransmission/all databases.
6. Plasticity/all databases.
7. Stress response/all databases.
8. Mitochondria in all data/all databases.
9. Endolysosomal system/all databases.
Astrocyte cluster # 4 ranked according to PC1 in ROSMAP, and comparison with DEG identified by snRNAseq in human AD astrocytes (Mathys et al., 2019) (Grubman et al., 2019). Tabs:
1. Venn diagrams.
2. Comparison of PC1/ROSMAP with (Mathys et al., 2019).
3. Comparison of PC1/ROSMAP with (Grubman et al., 2019).
4. Comparison of PC1/ROSMAP between (Mathys et al., 2019) and (Grubman et al., 2019).
Supplementary file 12. Comparison of the functional category of cluster # 4 of human astrocytes containing immunity-related genes with the immunity-related GO groups found significantly dysregulated in APP/PS1 mouse astrocytes (Orre et al., 2014). Only C1S (Complement Component 1, S Subcomponent) appears in both gene sets.