

Summary
Mechanisms of neutrophil involvement in severe coronavirus disease 2019 (COVID-19) remain incompletely understood. Here, we collect longitudinal blood samples from 306 hospitalized COVID-19+ patients and 86 controls and perform bulk RNA sequencing of enriched neutrophils, plasma proteomics, and high-throughput antibody profiling to investigate relationships between neutrophil states and disease severity. We identify dynamic switches between six distinct neutrophil subtypes. At days 3 and 7 post-hospitalization, patients with severe disease display a granulocytic myeloid-derived suppressor cell-like gene expression signature, while patients with resolving disease show a neutrophil progenitor-like signature. Humoral responses are identified as potential drivers of neutrophil effector functions, with elevated severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-specific immunoglobulin G1 (IgG1)-to-IgA1 ratios in plasma of severe patients who survived. In vitro experiments confirm that while patient-derived IgG antibodies induce phagocytosis in healthy donor neutrophils, IgA antibodies predominantly induce neutrophil cell death. Overall, our study demonstrates a dysregulated myelopoietic response in severe COVID-19 and a potential role for IgA-dominant responses contributing to mortality.
Keywords: COVID-19, neutrophil, IgA, G-MDSC, transcriptomics, NETosis, degranulation, SARS-CoV-2
Graphical abstract
Highlights
-
•
Clustering analyses identify dynamic switches between 6 distinct neutrophil states
-
•
Neutrophil states improve severity prediction models above available clinical data
-
•
IgA1:IgG1 ratios are elevated in patients who died relative to intubated survivors
-
•
IgG drives antibody-dependent neutrophil phagocytosis while IgA induces cell death
LaSalle et al. utilize bulk transcriptomics of neutrophils, plasma proteomics, and high-throughput antibody profiling of a large cohort of hospitalized COVID-19 patients to explore the relationship between neutrophil states and disease severity. They identify granulocytic myeloid-derived suppressor cell-like signatures and plasma IgA1-to-IgG1 ratios as predictive of disease severity and mortality.
Introduction
While our understanding of coronavirus disease 2019 (COVID-19) continues to grow, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) still causes a wide range of disease severity. Thus far, many studies of SARS-CoV-2 have shown that severe COVID-19 patients present with broad immune dysfunction (lymphopenia, hyper-inflammation, delayed antibody production, impaired interferon responses).1, 2, 3, 4, 5, 6, 7 Neutrophil hyperactivation has been described in severe COVID-19 and suggests that a dysregulated myeloid compartment underlies severe disease.5,8, 9, 10, 11, 12, 13 Finally, others have proposed that emergency myelopoiesis, which leads to an abundance of immature neutrophils, is a prominent feature of severe COVID-19 associated with poor prognosis.9,14,15 Yet, neutrophils are understudied as analyses focusing on peripheral blood mononuclear cells (PBMCs) cannot examine this crucial component of the immune system. The effects of dysregulated humoral responses on neutrophil responses are not well understood, as many of their effector functions such as antibody-dependent neutrophil phagocytosis (ADNP)16,17 (direct pathogen removal) or NETosis (cell death program in which neutrophils release neutrophil extracellular traps [NETs] consisting of chromatin modified with anti-microbial proteins18,19) are driven by antibody-Fc receptor interactions.20 Small case studies indicate the importance of NETs in COVID-19-associated myocardial infarctions and immunothrombosis.21,22 Here, we present a longitudinal study of a large cohort of hospitalized COVID-19 patients that combines unbiased, bulk transcriptomic analysis of enriched blood neutrophils with plasma proteomics, cell-free DNA (cfDNA) measurements, and high-throughput antibody profiling in order to understand neutrophil dynamics during the immune response to SARS-CoV-2 infection.
Results
Longitudinal profiling of neutrophils from COVID-19 patients
From March–May 2020, we enrolled 384 patients who presented to the Massachusetts General Hospital’s emergency department (ED) with suspected COVID-19 based on presentation of acute respiratory distress. Subsequently, 306 tested positive for COVID-19. We stratified disease acuity into five categories based on the World Health Organization COVID-19 outcome scale as previously described:23 A1, death within 28 days; A2, intubation, mechanical ventilation, and survival to 28 days; A3, hospitalized requiring supplemental oxygen; A4, hospitalized without requiring supplemental oxygen; and A5, discharged without requiring admission within 28 days. We classified A1–A2 as severe, and A3–A5 as non-severe. Outcomes (acuityMax [AMax], severityMax) were defined as the most severe disease level within 28 days of enrollment (Table S1). We took blood draws on days 0 (n = 374) upon ED admission (likely day 7–8 post-infection), 3 (n = 212), and 7 (n = 143) for all who remained hospitalized. Few patients received therapies other than supplemental oxygen or mechanical ventilation (STAR Methods). Dexamethasone was not administered to patients for COVID-19 at this time. We took 8 blood draws from healthy controls. Using negative selection to enrich for neutrophils, we obtained 781 samples from 388 individuals. We performed bulk RNA sequencing, and after quality control,24 we retained 698 samples from 370 patients (Table S1). Additionally, we analyzed 1,472 plasma proteins using the Olink platform (published),23 quantified cfDNA, and performed high-throughput antibody profiling (partially published)25, 26, 27 (Figure 1A; STAR Methods).
Figure 1.

SARS-CoV-2 infection induces distinct neutrophil profiles
(A) Schematic of cohort and study methodology.
(B) Correlation heatmap of clinical variable correlations with absolute neutrophil counts (ANCs) on day 0, 3, or 7 with q < 0.05 in COVID-19+ patients.
(C) Ordinal correlation between ANC quintile and acuityMax (AMax) for COVID-19+ patients.
(D) Comparison of CIBERSORTx total, mature, and immature neutrophil fractions on days 0, 3, and 7 for COVID-19+ patients by Kruskal-Wallis test (STAR methods).
(E) Uniform manifold approximation and projection (UMAP) plots of bulk RNA-seq samples that passed quality control.
(F) Volcano plot of genes DE between COVID-19+ and COVID-19– patients hospitalized with respiratory disease on day 0. Colored circles indicate log2(fold change)>0.5 (log2(FC)) and p < 10−4
(G and H) Gene set enrichment analysis (GSEA) for (G) signaling pathways and (H) cellular processes from MSigDB for samples on day 0 from (F).
(I) Boxplots of CIBERSORTx total, mature, and immature neutrophil percentages in severe and non-severe patients with Wilcoxon rank-sum p values.
COVID-19 induces an interferon response signature in neutrophils followed by an expansion of immature neutrophils
Similar to previous studies,28, 29, 30, 31, 32, 33 we analyzed ordinal correlations between clinical absolute neutrophil count (ANC) quintile and clinical parameters associated with severity. We observed positive correlations between ANC and creatinine, lactate dehydrogenase (LDH), C-reactive protein (CRP), and D-dimer, consistent with the role of neutrophils in inflammation and thrombosis9,34 (Figure 1B; Table S1). Additionally, we found robust ordinal correlations between ANC and acuity (and, accordingly, intubation) that increased from day 0 to 7 (Figure 1C).
Since enrichment did not result in 100% neutrophil purity (Figures S1A and S1B), we next determined the cell-type composition of our bulk RNA sequencing (RNA-seq) samples using CIBERSORTx.35 We used a published whole-blood single-cell dataset, which captures relevant cell types including immature neutrophils,36,37 to deconvolve our bulk data.9 (Figures S1C and S1D; Table S1; STAR Methods). We found that lower estimated total neutrophil (NeuTotal) content (sum of mature and immature neutrophils) was associated with lower clinical ANC (Fisher’s test p = 1.2 × 10−17). Overall, we estimated a mean of 75% NeuTotal (Figure S1E), concordant with flow cytometry estimates in separate healthy controls (Figure S1B). Among COVID-19 patients, the estimated NeuTotal increased from day 0 to 7 (Figure 1D), driven largely by expansion of immature neutrophils.14 Also, intubation status correlated with NeuTotal on days 3 and 7 (Table S1). Dimensionality reduction revealed groupings based on disease status and immature neutrophil fraction (Figure 1E).
Next, to identify genes and programs induced during COVID-19 infection, we performed differential expression (DE) and gene set enrichment analysis (GSEA) between COVID-19+ and similarly symptomatic COVID-19– respiratory disease patients on day 0 (Figures 1F–1H; Table S1). To correct for non-neutrophil contamination, we added CIBERSORTx cell-type fractions and an immunoglobulin score as model covariates (Figures S1F–S1I; STAR Methods). GSEA revealed strong anti-viral signatures enriched in COVID-19+ samples, such as response to interferon gamma (IFNγ) and IFNα, Toll-like receptor (TLR) signaling, and cytokine production (Figures 1G and 1H).
Finally, to identify neutrophil expression correlates of COVID-19 severity, we compared CIBERSORTx cell-type fractions across severe and non-severe patients (Figure 1I). NeuTotal was significantly elevated in severe patients across all time points, consistent with our observation of elevated ANC in severe disease. On day 0, this difference was driven by mature neutrophils, whereas on days 3 and 7, it was due to immature neutrophils (Figure 1I).
Unbiased NMF clustering defines neutrophil states during SARS-CoV-2 infection
We next sought to identify neutrophil gene expression subtypes and their associations with disease outcomes using Bayesian non-negative matrix factorization (NMF) clustering38 (STAR Methods). We clustered samples with NeuTotal>50% (n = 635, 91% of samples) to reduce artifacts of cell-type contamination and identified six robust neutrophil subtypes (Figures 2A and S2A; Table S2). We denote samples with <50% NeuTotal as Neu-Lo, which were significantly enriched for non-intubated patients (Table S2).
Figure 2.

Severe outcomes are associated with transitions between neutrophil states
(A) Heatmap of marker genes for all patients grouped by subtype using NMF clustering (STAR Methods).
(B) UMAPs of scRNA data from Bonn Cohort 2.9
(C) Network diagram displaying relationships between NMF subtype marker genes and published neutrophil signature genes.
(D) Volcano plot of genes DE between COVID-19+ severe and non-severe patients. Colored points indicate log2(FC) > 0.5 and p < 10−4.
(E) Bar plots of proportions of COVID-19+ samples in each NMF cluster. Bar heights indicate percentages of COVID-19+ samples for each time point separately. ∗q < 0.05, ∗∗q < 0.01 by Fisher’s exact test for each time point separately, with false discovery rate (FDR) correction across all days.
(F and G) GSEA for genes DE between COVID-19+ severe and non-severe patients.
See also Figures S2 and S3 and Table S2.
Two subtypes (NMF3, NMF6) had high expression of IFN-stimulated genes (ISGs). NMF3 markers included Fc and complement receptors and inflammatory caspases. NMF3 samples were enriched for patients requiring intubation compared with all other clusters (Table S2). On the other hand, NMF6 had high granzyme levels and distinct ISGs. Consistent with these markers, the estimated T/natural killer (NK) proportion was highest in NMF6 (Table S2).
NMF1 and NMF4 were composed of predominantly immature neutrophils. NMF1 was enriched for neutrophil granule components (ELANE, AZU1, DEFA1B, DEFA4), suggestive of a neutrophil-progenitor-like state, while NMF4 had a more activated (CEACAM8, CD24) profile and different neutrophil granule components (MPO, CHIT1, MMP8, LYZ). On day 7, NMF4 had higher D-dimer than NMF1, potentially implicating NMF4 in thrombosis (Table S2). We performed DE analysis and GSEA on NMF1 versus NMF4 samples, which revealed enrichment of neutrophil degranulation signatures and reactive oxygen species (ROS) generation pathways in NMF4 (Figure S2B; Table S2). NMF1 samples were enriched for the electron transport chain pathway and oxidative phosphorylation, suggesting that pro-neutrophils may be storing energy for differentiation.39
Finally, NMF2 and NMF5 shared transcriptional similarities with myeloid-derived suppressor cells (MDSCs). NMF2 displayed nuclear factor κB (NF-κB) activation (NFKB2, BCL3) and MMP25 expression, while NMF5 had a granulocytic MDSC-like gene expression signature (ARG1, CD177, MCEMP1, S100A12) and interleukin (IL)-1B signaling (IL1R1, IL1R2, IL1RAP). On day 3, NMF2 and NMF5 had significantly higher fractions of samples from intubated patients, and NMF2 patients had higher CRP and LDH (Table S2).
Our NMF signatures were similar to those identified by single-cell RNA-seq (scRNA-seq) in COVID-19 and sepsis patients9,40 (Figures 2B, S2C, and S2D). Additionally, we built a network displaying the relationships between our NMF signatures and published neutrophil gene signatures in COVID-19,9 cancer,41 sepsis,40 and non-COVID-19 acute respiratory distress syndrome (ARDS)42 (Figures 2C and S2E; STAR Methods). Network branches revealed signature groups of immature, mature, G-MDSC-like, and ISG neutrophils (STAR Methods). This analysis suggests that NMF marker genes may represent neutrophil subtypes in multiple disease contexts.
Transcriptionally distinct neutrophil states are associated with COVID-19 severity
To identify neutrophil states, genes, and pathways associated with COVID-19 severity, we performed DE analysis between severe and non-severe patients for each time point (Figure 2D; Table S2). To understand the dynamics of neutrophil subtypes, we first explored how NMF cluster membership varied across severity and over time (Figures 2E and S2F), and second, we performed GSEA using neutrophil gene signatures (Figure 2F). On day 0, COVID-19+ samples were most frequently assigned to NMF3, but proportionally more samples were from severe patients. We assessed ISG neutrophil expression over time in the Schulte-Schrepping data and also found enrichment early in the course of infection (Figures S2G and H; STAR Methods). In our cohort on day 3, severe samples were more evenly distributed across NMF clusters 1 to 5, with NMF2 (NF-κB+) and NMF5 (G-MDSC) significantly enriched for severe samples. In agreement, the signature most enriched in severe patients by GSEA was NMF5 (G-MDSC) at all time points (Figure 2F). Additionally, in scRNA data from hospitalized COVID-19 patients,36 a subset of whom were treated with immunosuppressive dexamethasone, we found that NMF5 was significantly enriched in patients that died and was significantly lower in dexamethasone-treated patients 7 days post-intensive care unit (ICU) admission (Figures S2I–S2K; Table S2). Finally, on day 7, NMF4 (immature activated) was enriched for severe samples. Non-severe samples were enriched for NMF1 (pro-neutrophil [pro-Neu]) (all days) and NMF6 (ISG+) (day 0). GSEA also indicated NMF1 (pro-Neu) as the most enriched neutrophil signature in non-severe samples. In addition, non-severe samples had higher frequencies of Neu-Lo (days 3 and 7), indicating a resolution of neutrophil activation.
We next performed gene and pathway-level analyses using GSEA (Figure 2G; Table S2). Across all 3 days, the pathways most enriched in severe patients included neutrophil degranulation, hypoxia, tumor necrosis factor α (TNF-α) signaling via NF-κB, ROS metabolic processes, and neutrophil migration (Figure 2G). Many of the top genes enriched in severe patients across days 0, 3, and 7 are involved in IL-1β signaling (IL1R1, IL1R2) and neutrophil degranulation (ARG1, CD177, MCEMP1). Major histocompatibility complex (MHC) class II genes were strongly associated with non-severe disease, as previously observed for monocytes,40,43 but we cannot rule out MHC expression in other cell types (Table S2). Of note, the gene sets “ARDS Up - Juss” and “ARDS Down - Juss” were consistently enriched in severe and non-severe patients, respectively, and the “ARDS Up - Juss” gene set significantly overlapped with NMF5 signature genes, suggesting that G-MSDC signatures are associated with both COVID-19 ARDS and non-COVID-19 ARDS (Figures S3A and S3B). We searched for genes and pathways with diverging expression patterns over time between severe and non-severe patients (Figure S3C; Table S2; STAR Methods). SERPINB2, a gene involved in Th1/Th2 modulation during lentiviral infections,44 increased over time in severe patients but slightly decreased over time in non-severe patients. ZBTB16, a glucocorticoid response negative feedback gene, was more highly expressed on day 0 in severe patients, but its expression decreased over time, compared with non-severe patients. On the pathway level, granulocyte chemotaxis remained high in severe patients but decreased over time in non-severe patients (GSEA q = 3.3 × 10−3). Furthermore, the TNF-α signaling via NF-κB metagene score increased with time in severe patients but was constant in non-severe patients (GSEA q = 3.7 × 10−15). These pathway results agree with the neutrophil subtype analysis, highlighting the role of neutrophil activation in severe COVID-19.
Neutrophil states are among the most powerful predictors of COVID-19 severity as early as day 0 of hospitalization
We hypothesized that neutrophil subtype metagene scores could improve predictive models of COVID-19 severity upon patient presentation to the ED, as the NMF5 (G-MDSC) signature correlated with acuity on day 0 (Figure 3A). We built three nested logistic regression models for predicting severityMax using data from day 0 (Figure 3B). Model 1 included only patient characteristics, model 2 added clinical laboratory values, and model 3 added these and NMF and ARDS neutrophil gene set scores (STAR Methods; Table S3). Adding neutrophil subtype scores resulted in a marked improvement (area under the curve [AUC]: 0.960, likelihood ratio test [LRT] p = 7.93 × 10−6), demonstrating that neutrophil subtypes may significantly improve clinical predictive models of COVID-19 severity.
Figure 3.

Neutrophil metabolism and dysregulated IFN signaling are associated with severity and acuity
(A) Boxplots of NMF5 metagene score for healthy and COVID-19+ samples grouped by AMax.
(B) Receiver operating characteristic (ROC) curve for performance of logistic regressions predicting COVID-19 severity on day 0. Significance of model improvement determined by likelihood ratio tests.
(C) ROC curve of performance of a LASSO model of COVID-19 severity on day 0 (STAR Methods) with median AUC curve across cross-validation repeats in red.
(D) Bar plot of inclusion frequency for each variable in the LASSO model.
(E) GSEA for genes DE between COVID-19+ patients with AMax1 (death) or AMax2 (intubation, survival).
(F) GSEA enrichment plots for gene sets with genes ranked by DE in (E).
(G) GSEA enrichment plots for NMF3 and NMF6 gene signatures with genes ranked by DE in (E).
We next identified which subset of features was most important for predicting severity by performing feature selection with a least absolute shrinkage and selection operator (LASSO) logistic regression model of COVID-19 severity on day 0 (Figure 3C; STAR Methods). Across all 100 5-fold repeats of cross-validation, the two features that were always included in the model were the highest NMF5:G-MDSC score quintile and the highest LDH quintile (Figure 3D; Table S3).
Longitudinal analyses reveal diverging pathway dynamics between survivors and non-survivors
To test whether any neutrophil genes or pathways could predict survival of the most severe patients upon intubation, we performed DE analysis and GSEA (Figure 3E; Table S3) between AMax1 (death within 28 days) and AMax2 (intubated, survived) samples. On day 0, the most enriched pathways in patients who died were the Juss et al.42 non-COVID-19 ARDS neutrophil signature (adjusted p value [padj] = 7.7 × 10−26) and neutrophil degranulation (padj = 1.1 × 10−15). Interestingly, we observed that several metabolic pathways switched from being enriched in AMax1 patients at day 0 to being enriched in AMax2 patients at day 7. On day 0, the IFNα and IFNγ response pathways were enriched in AMax2 patients, but on days 3 and 7, the signatures were more enriched in AMax1 patients (Figures 3F and S3D). Prior work has shown that IFN signaling is delayed or dysregulated in COVID-19.3 Another study in macaques demonstrated age stratification in IFN signaling, with stronger type I IFN responses in juveniles;45 in contrast, we find higher IFN signaling in older patients at later time points (Figure S3E), which could indicate that peak IFN in younger patients occurred pre-hospitalization, whereas IFN in older patients is delayed.3
In accordance with the IFN response signatures, we also observed that enrichment of the NMF3 (PD-L1+ISG+) and NMF6 (ISG+) signatures switched from patients who survived on day 0 to patients who died on days 3 and 7 (Figures 3G and S3F). Interestingly, the metabolic pathways distinguishing NMF1 (pro-Neu) and NMF4 (immature activated) followed the opposite trend. Though NMF cluster membership was not associated with AMax, GSEA and pathway enrichment suggest that the metabolic differences underlying the NMF clusters are associated with survival (Table S3).
NETosis is implicated in severe COVID-19 pathology through transcriptomics, proteomics, and circulating cfDNA
Several studies have reported associations between SARS-CoV-2 infection and NETs.46, 47, 48, 49 To look for NETosis associations with outcomes, we defined a NETosis metagene score (STAR Methods; Figures S3G and S3H).50 We found a significant enrichment of our score on days 3 and 7 in severe patients (Figure 4A). Additionally, across NMF clusters, we found higher scores in the immature clusters NMF1 and NMF4 (Figure 4A).
Figure 4.

Transcriptomics, proteomics, and cell-free DNA (cfDNA) analyses identify neutrophil effector function signatures associated with severe COVID-19 outcomes
(A) Boxplots of NETosis metagene score over time split by severityMax (top) and across NMF clusters (bottom).
(B) Olink plasma proteomics values over time split by severityMax.
(C) Citrullinated histone H3 in patient plasma. H = healthy (n = 6), n = 32 non-severe, and n = 46 severe patients.
(D–F) cfDNA concentration, arranged by (D) day and severityMax, (E) COVID-19 status, and (F) ANC.
(G) Pathway metagene score for REACTOME_NEUTROPHIL_DEGRANULATION.
(H) SomaScan protein expression Z scores.
(I) Expression of ARG1 and CD274.
p values for Wilcoxon rank-sum tests (A [top], B–E, and G–I).
Many factors promoting NETosis, such as histone modification, are post-transcriptional51 and would not be captured by RNA-seq, while other markers are expressed during neutrophil development and are not reflective of effector functions in real time. Therefore, we next searched for protein markers of NETosis in matched plasma proteomic data.23 Known protein markers of NETosis were significantly associated with severe disease across all time points and varied across neutrophil subtypes, including MPO, CXCL8, TNF, PADI4, HGF, and CD177 (Figure 4A; Table S3). These results agree with previous proteomic studies, which have associated MPO,52,53 CXCL8,14 and HGF54,55 with severe COVID-19. We next performed an ELISA for citrullinated histone H3 (CitH3), a specific marker of NETs, in matched plasma samples (n = 78 patients, plus n = 6 healthy controls). In agreement with the NETosis expression data, we found significant elevation of CitH3 on days 3 and 7 in severe patients (Figure 4C). The distribution of CitH3 across neutrophil subtypes mirrored the transcriptomic results, except NMF5, which showed higher CitH3 (Figure 4C). The NETosis metagene score and the CitH3 ELISA measurement were weakly positively correlated (ρ = 0.32) as expected, as the transcription of NETosis genes and the initiation of NETosis are temporally separated (Figure S3I).
We next measured levels of cfDNA in the plasma. Prior cfDNA methylation studies in COVID-19 have identified neutrophils as a major source of cfDNA.34 Concentration of cfDNA was significantly associated with COVID-19 status and severity across time points and correlated with ANC, but we did not observe significant differences between AMax1 and 2 patients (Figures 4D–4F; Table S3). Furthermore, cfDNA was elevated in NMF4 (immature activated) versus NMF1 (pro-Neu) samples, suggesting that NMF4 neutrophils may release greater amounts of NETs (Figure S3J).
Neutrophil degranulation signatures and immunosuppressive gene expression are associated with severity and distinguish neutrophil subtypes
Uncontrolled neutrophil degranulation can cause tissue damage and pathologic inflammation.56 Therefore, we defined a neutrophil degranulation metagene with the REACTOME_Neutrophil_Degranulation gene set. As expected, the metagene was highly enriched in severe patients across all time points, though it was only enriched in AMax1 versus AMax2 patients on day 0 (Figure 4G; Table S3). Of note, the neutrophil degranulation metagene score was highly enriched in the NMF4 (immature activated) subtype over the NMF1 (pro-Neu) subtype, which may support the possibility that fewer NMF1 neutrophils have granules and that NMF4 neutrophils may be more capable of effector functions (Table S3). Additionally, metagene scores for neutrophil granules (azurophilic, specific, tertiary) were enriched in severe patients at all time points (Table S3).
Since transcriptomics provide no insight into whether neutrophils have released their granules, we searched for protein markers of neutrophil granules using matched SomaScan23 plasma proteomic data. Indeed, we found elevated levels of ELANE, CTSG, LCN2, and PRTN3 in severe patient plasma at all time points (Figure 4H). Due to sample collection and technical constraints, it was not possible to perform neutrophil degranulation assays. However, transcriptomics and proteomics data both demonstrate that degranulation markers are associated with COVID-19 severity.
Neutrophils have been shown to suppress T cell activation, with some studies demonstrating T cell suppression only in fully differentiated neutrophils.57 Therefore, we investigated the associations between T cell suppression genes and severity or neutrophil NMF subtype. ARG1, which suppresses T cells by depleting L-arginine, was enriched in severe patients (Figure 4I) and had the highest expression in NMF5 (G-MDSC) and NMF4 (immature activated) (Table S3). CD274, encoding PD-L1, which suppresses T cells through PD-1, was enriched in severe patients on days 3 and 7 (Figure 4I) and was highest expressed in NMF3 (PD-L1+ ISG+) (Table S3). NMF1 (pro-Neu) showed low expression of both genes, consistent with the finding that progenitor neutrophils do not display MDSC functionality.57 We note that these are transcriptomic analyses and not functional assays due to technical constraints of preserving neutrophils, though the roles of these genes have been validated mechanistically.58, 59, 60
Antibody isotype profiles are major drivers of neutrophil effector functions in COVID-19
Neutrophils enact many effector functions in an Fc receptor-dependent manner,61 and thus antibodies influence neutrophil behavior. In total, 53 out of 300 assayed samples from day 0 had detectable viremia62 (Table S1), suggesting that circulating immune complexes (ICs) and Fc repertoire may influence whether neutrophils effect NETosis or phagocytosis. We measured the levels of antibody isotypes and subclasses for SARS-CoV-2 antigens and non-SARS-CoV-2 viral antigens in matched longitudinal plasma samples as published27,63 (Table S4). Importantly, our time course corresponds to days of hospitalization, not days post-infection; thus, timing is not standardized across patients. We found significantly higher levels of SARS-CoV-2 spike (S)-specific IgA1 antibodies in severe patients on day 7 (Figure 5A). Though there was no difference in S-specific IgG1 antibodies (Table S4), we found several differences in neutrophil Fc receptor expression (including FCAR) across severity and subtypes that could impact effector functions (Figures S4A and S4B). In particular, mature neutrophil subtypes NMF3 and NMF5 displayed higher expression of most Fc receptors, though NMF4 neutrophils expressed FCAR just as highly. NMF 4 had higher expression of Fc receptors (FCGR2A, FCGR3B, and FCAR) and thus potentially more ability to enact antibody-mediated effector functions, than NMF1 (Figure S4B).
Figure 5.

Antibody profiles are major drivers of neutrophil function
(A) Plasma SARS-CoV-2 spike (S) protein-specific IgA1 log10(MFI) values.
(B) Heatmaps displaying the signed (by FC) −log10(p) comparing levels of antigen-specific antibody isotypes between AMax1 and AMax2. Rows indicate antigens: SARS-CoV-2 (S, S1, S2, N, and receptor-binding domain [RBD]), human coronavirus OC43, influenza hemagglutinin (HA), and cytomegalovirus (CMV).
(C) Schematics for functional assays.
(D) Background-corrected antibody-dependent neutrophil phagocytosis (ADNP) assay.
(E) Log10 ratio of S-specific IgG1 to IgA1 MFI.
(F) Boxplots of background-corrected ADNP log10(MFI) values for severe patients on day 7, separated by IgG/IgA ratios.
(G) Paired-line plots of ADNP log10(MFI) values showing effects of SARS-CoV-2 S-specific IgG or IgA from day 7 plasma samples (n = 12 per condition).
(H) Reactive oxygen species luminescence of neutrophils exposed to IgG:S or IgA:S ICs or PBS. Color bars display the −log10(p) between IgG and IgA at each time point, with gray indicating no significant difference (n = 12 per condition).
(I) Representative microscopy images of neutrophil morphologies. PC, phase contrast; DAPI, DNA stain; NE, neutrophil elastase. Scale bars are indicated for each row of images.
(J) Mean percentage of cells undergoing any form of cell death quantified by fluorescence microscopy (controls n = 2 each, IgG/IgA n = 6 each).
(K) SYTOX Green Nucleic Acid Stain log10(RFU) from neutrophils exposed to free IgG or IgA (n = 12 per condition).
(L) MFI FC values of surface markers of neutrophil degranulation (controls n = 2 each, IgG/IgA n = 15 each).
p values for Wilcoxon rank-sum tests (A and L).
In contrast to severe versus non-severe comparisons, we found significantly higher IgG1, IgG2, and IgG3 antibodies for a variety of SARS-CoV-2 antigens in AMax2 versus AMax1 patients on day 3 (Figure 5B). On day 7, IgG1 antibodies for all five SARS-CoV-2 antigens were significantly higher in AMax2 patients, consistent with previous work linking delayed or diminished humoral responses to fatal COVID-19.7 We also observed associations between S-specific IgA1 antibodies and neutrophil NMF states, as well as ANC (Table S4).
To test whether antibody profiles impact neutrophil phagocytosis, we performed an ADNP assay using patient antibodies and donor neutrophils64 (Figure 5C; Table S4; STAR Methods). Though ADNP was not associated with severity (Table S4), we did find significantly higher ADNP in AMax2 versus AMax1 patients on days 3 and 7 (Figure 5D). Decreased phagocytosis could indicate an inability of neutrophils to clear debris from blood.65 To understand why ADNP levels were divergent, we evaluated differences in the antibody repertoires. On day 0, the majority of severe patients had higher S-specific IgA1 titers compared with S-specific IgG1; however, over time, intubated survivors shifted toward higher S-specific IgG1, whereas non-survivors maintained higher S-specific IgA1 titers (Wilcoxon rank-sum test, day 0, not significant [NS], day 3 p = 0.0058, day 7 p = 0.0090; Figure 5E). We did not observe this trend when comparing severe and non-severe disease (Table S4). We then stratified samples into two categories: higher S IgG1 titer or higher S IgA1 titer. Among severe patients on day 7, ADNP was significantly elevated in the higher S IgG1 group (Figure 5F), and the same trend was found across all samples (Table S4).
Recent studies have demonstrated that while IgG antibodies can induce neutrophil phagocytosis, IgA:virus ICs are potent inducers of NETosis.66 Antibody isotypes interact through different receptors, with IgA binding FcαR and IgG binding FcγR. In addition, changes in Fc-glycosylation can alter antibody interactions with FcRs.67, 68, 69 Therefore, we sought to determine whether neutrophil effector functions were differentially impacted by the plasma IgG/IgA ratio or whether antibodies from severe patients differentially modulate neutrophil functions. Thus, we separately purified IgG and IgA fractions from day 7 plasma samples from severe COVID-19 survivors, non-survivors, and non-severe patients (n = 12 each) and performed ADNP, ROS generation, neutrophil cell death, and degranulation assays using healthy donor (HD) neutrophils (Figure 5C; Table S4).
For the isotype-specific ADNP experiment, we generated IgG:S (SARS-CoV-2 S) and IgA:S ICs and incubated them each with HD neutrophils to assess phagocytosis. In all categories (non-severe, severe survivors, death), only IgG:S ICs robustly triggered ADNP (Figure 5G; Table S4). Next, we incubated HD neutrophils with ICs of both isotypes and measured the ROS production by neutrophils as a function of time (STAR Methods). Across all three categories, IgG:S induced higher ROS generation than IgA:S (Figure 5H). Notably, IgG:S ICs from severe survivors induced significantly higher ROS production than the non-severe group (Figure S4C). This may be related to distinct IgG glycosylation patterns in severe COVID-19 patients.70 To validate our finding, we performed GSEA on neutrophil RNA-seq samples from severe patients on day 7 comparing patients with IgA1>IgG1 with patients with IgA1<IgG1. The ROS pathway was enriched in samples with IgA1<IgG1, consistent with the ROS release assay (Figure S4D). In addition, we found that IFNα response and regulation of membrane potential pathways were enriched in samples with IgA1>IgG1. Changes in membrane potential are associated with components of neutrophil activation such as chemotaxis and NETosis.71
Next, we tested whether free IgA or IgG antibodies from patient serum could trigger neutrophil cell death and the release of DNA, thereby potentially causing microvascular thrombosis. We incubated HD neutrophils with free IgA or IgG antibodies, and after 3 h, cells were fixed and stained to quantify neutrophil cell death (apoptosis, necrosis, NETosis) following stimulation. As a positive control, we used phorbol 12-myristate 13-acetate (PMA) to induce NETosis (Figure 5I; STAR Methods; Table S4). This method is not able to distinguish between true NETosis, “incomplete” NETosis,72 and secondary necrosis (apoptosis or necrosis without being cleared). However, subsequent time-lapse microscopy experiments of PMA-treated cells revealed both true NETosis and necrosis (Videos S1 and S2). We found significantly elevated neutrophil cell death in donor cells treated with IgA compared with IgG antibodies (Figures 5J and S4E). We then used a high-throughput method by incubating HD neutrophils with free IgA or IgG antibodies in the presence of a live-cell-impermeable nucleic acid dye (Sytox Green), to quantify DNA released by neutrophils through all forms of cell death. We found strikingly higher levels of Sytox Green signal (which may correspond with NETosis or necrosis as the morphology resembled that of PMA-treated cells) from healthy neutrophils incubated with IgA than IgG, regardless of the patient severity (Figure 5K). This observation on neutrophil cell death and release of DNA into circulation could contribute to the understanding of why severe patients with high IgA1/IgG1 ratios in plasma were less likely to survive intubation.
Finally, we performed a flow cytometry-based neutrophil degranulation assay, treating neutrophils with free IgA or IgG antibodies from patient serum and staining for several components of neutrophil granules73 (STAR Methods; Table S4). We found large fold changes in mean fluorescence intensity (MFI) in IgA-treated neutrophils over IgG-treated neutrophils for CD11b (gelatinase granules), CD45 (secretory vesicles), CD63 (azurophil granules), and CD66b (specific granules) (Figure 5L), as well as a small increase in CD14 (secretory vesicles) (Table S4). We found a slight increase in CD35 (secretory vesicles) in IgG-treated neutrophils over IgA-treated neutrophils, and we did not observe any difference in the levels of CD15 (specific granules) (Table S4). Taken together, these functional assays further illustrate how an elevated plasma IgA1/IgG1 ratio could contribute to the systemic inflammatory consequences of severe COVID-19.
Plasma proteomics identifies neutrophil-driven secreted proteins and potential ligand-receptor interactions driving phenotypes
To further understand the role of neutrophils in COVID-19 in relation to other blood and immune cells, we analyzed the plasma proteome using our Olink dataset for this cohort. We began by searching for protein markers of neutrophil NMF clusters (Figure 6A; Table S5). NMF5 (G-MDSC) in particular had strong upregulation of markers of severity and neutrophil activation such as S100A12, HGF, IL-1RL1, IL-1R2, DEFA1/1B, PADI4, and TGFB1 (Figure 6B). Of note, transforming growth factor β (TGF-β) has been shown to influence B cells to class switch to IgA when stimulated with LPS,74 and TGF-β signaling has been implicated in impaired anti-viral responses in severe COVID-19,75 illustrating two ways in which NMF5 neutrophils could potentially contribute to disease severity. NMF4 (immature) had the highest levels of ACE2, potentially indicating tissue damage, while NMF3 (PD-L1+ ISG+) showed enrichment for IFNL1, CXCL10, and IFNG.
Figure 6.

Alterations in the plasma proteome are associated with neutrophil subtypes and antibody profiles
(A) Heatmap displaying scaled expression values for subtype-enriched proteins.
(B) DE proteins. Colored points indicate q < 0.05.
(C and D) Scatterplot comparing the log2(FC) values for neutrophil RNA-seq with the log2(FC) of the plasma proteomic data between (C) COVID-19+/− patients or (D) COVID-19+ severe and non-severe patients. Colored points indicate log2(FC) > 1.25 in mRNA and protein.
(E) DE proteins in matched plasma samples between samples with IgA > IgG or IgA < IgG. Colored points indicate q < 0.05.
(F) NPX (normalized protein expression) values for selected plasma proteins.
p values for Wilcoxon rank-sum tests.
See also Table S5.
Next, we sought to determine which severity-associated proteins in the plasma were expressed by neutrophils by comparing the DE results between severe and non-severe patients on RNA and protein levels (Figures 6C and 6D; Table S5; STAR Methods). We identified several components of neutrophil granules (CD177, MMP8, MMP9, ARG1, S100A12, TGFA), factors involved in clotting (F3, SERPINE1), chemoattraction (CXCL8, IL-4R), and inflammation (FKBP5, FCAR, IL-18R1, CLEC4D) upregulated in severe disease in both data types, suggesting that neutrophils are key contributors to the severity-associated plasma proteome.
Next, we searched for plasma proteins that were DE between patients with higher IgG1 or IgA1 titers (Figures 6E and 6F; Table S5). The top protein associated with higher IgA1 was IFNL1. While no study, to our knowledge, has linked IFNλ signaling with IgA isotype switching, IFNλ signaling is mainly targeted to epithelial cells, and IgA antibodies are typically found at mucosal surfaces rather than in plasma.76 Higher IgA1 was also associated with high plasma AGER (also known as RAGE), consistent with prior work.77 Many other plasma proteins associated with COVID-19 severity were enriched in IgA1-high samples, such as IFNG, CXCL10, and CXCL8, which have been associated with severity.14,78,79 On the other hand, within severe samples, IgG-high samples were enriched for FETUB, a protein involved in fatty acid metabolism that can suppress inflammation and which has been shown to be depleted in severe COVID-19,80 and CCL17, a Th2 chemokine that may be involved in the activation of class-switch recombination.81
Finally, we sought to determine whether any other soluble proteins could potentially contribute to neutrophil phenotypes or severity. We performed a hypothesis-generating ligand-receptor (L-R) interaction analysis between plasma ligands and receptors DE between NMF clusters (Figure 7A), and we tested the relationship between L-R pairs and outcomes, for each time point separately (Figures 7B, S5, and S6A; Table S5; STAR Methods). Among the more severe subtypes, NMF5 (G-MDSC) had the highest expression of IL1R1 and the highest levels of the ligands IL-1RN and IL-1B in plasma. These IL-1 family ligands show high mRNA expression in neutrophils, suggesting that the G-MDSC-like phenotype may be driven by a feedback loop of neutrophil-derived IL-1B. NMF4 (immature activated) had the highest expression of ITGB1 and ITGAV, which both interact with many ligands, the majority of which were expressed highest in native lung cells. In particular, IL1R1 on neutrophils was associated with both NMF5 and severe disease, and the F3-TFPI interaction (implicated in coagulation) was associated with NMF4 and severe disease, consistent with the many other indicators that NMF4 is involved in NETosis (Figure 7C).
Figure 7.

Ligand-receptor interactions in plasma are potential drivers of neutrophil phenotype and severity
(A) Ligand-receptor (L-R) analysis for DE ligands in plasma and receptors on neutrophils between NMF clusters for all COVID-19+ samples (STAR Methods).
(B) L-R analysis for DE ligands in plasma and receptors on neutrophils between COVID-19+ severe and non-severe samples on day 0.
(C) Table highlighting overlap between neutrophil NMF subtype L-R interactions and severity interactions.
See also Figures S5–S7 and Table S5.
In the less severe-specific subtypes, potential interactions between NMF1 (pro-Neu) neutrophils and plasma ligands featured many growth factor signaling pathways, and the majority of ligands had highest expression in native lung cells. NMF3 (PD-L1+ ISG+) showed strong upregulation of receptors involved in migration and activation (CCR1, CXCR2, SELL, CCR3) and their ligands (CCL8, CCL7, CD34). As expected, the IFNGR2/IFNG interaction was identified in this cluster. A higher fraction of ligands mapped back to monocytes for NMF3 than any other cluster. CCR3 interactions were identified in both NMF3 and non-severe disease, and CD74 interactions were associated with both NMF1 and non-severe disease (Figure 7C).
Similarly, the L-R interaction analysis for DE ligands and receptors between severe and non-severe patients revealed several interactions driving severity, including the neutrophil ligands IL-1RN, MMP9, VEGFA, PLAU, and IL-1B. Of note, we found at least one potential interaction within the uPA/uPAR system in severe patients across all three days. PLAU/uPA, which was expressed by neutrophils in COVID-19 bronchoalveolar lavage (BAL) fluid, has been shown to amplify neutrophil NF-κB responses, which can result in lung injury.82 In addition, we searched for our circulating neutrophil NMF signatures in the BAL fluid of patients with severe COVID-19 in an external cohort.83 We found similar patterns of enrichment between patients who died and patients who survived, suggesting that our neutrophil subtypes may be relevant to COVID-19 lung pathology (Figures S6B–S6E).
Finally, we built an additional logistic regression model for predicting severityMax on day 0 of hospitalization following SARS-CoV-2 infection (model 4), incorporating the top five plasma proteins associated with NMF5 neutrophils and the top five neutrophil-expressed severity-associated proteins on day 0 (STAR Methods). The model was able to perfectly predict severityMax and was overfit (Figure S7A). Thus, we again performed feature selection with a LASSO model (Figure S7B). Again, the two features that were included by the model every time were the highest NMF5:G-MDSC score quintile and the highest LDH quintile, confirming the NMF5 expression score as one of the strongest predictors of COVID-19 severity (Figure S7C). Strikingly, several neutrophil-related gene expression and protein signatures are included in the model more frequently than ALC, ANC, CRP, pre-existing lung conditions, D-dimer, and other powerful clinical predictors, suggesting that neutrophil features have a strong association with severe COVID-19.
Discussion
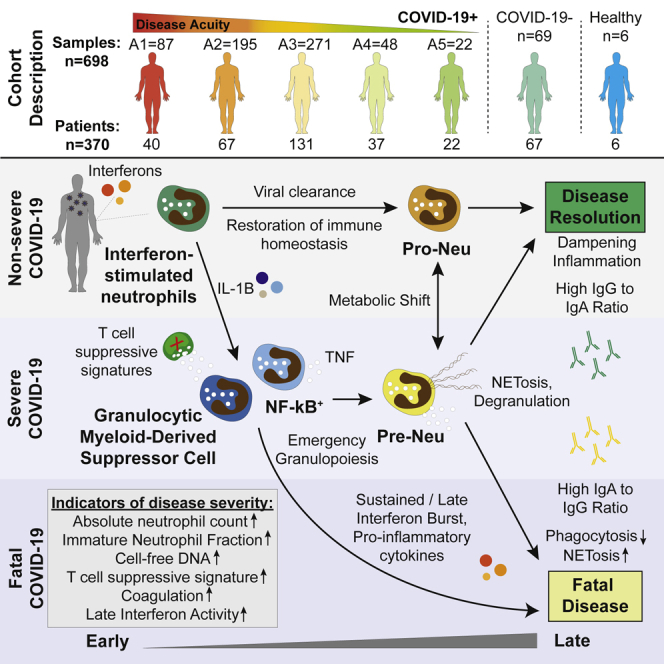
Here, we present a comprehensive characterization of circulating neutrophils from hospitalized COVID-19 patients. We first used unbiased NMF clustering to define six neutrophil subtypes associated with COVID-19 and SARS-CoV-2− respiratory disease. Our network analysis across diseases demonstrates that there is a common set of neutrophil states that exists across sepsis,40 cancer,41 and acute viral infection, each having distinct associations with severity. Therefore, potential therapeutic interventions targeting specific states may be applicable across diseases. Future studies will be required to isolate phenotypically distinct cell populations and assess their regulatory or inflammatory properties. Additionally, our analysis of longitudinal samples allowed us to distinguish signatures associated with outcome at initial hospitalization from those that developed over time. We observed that all patients have IFN-driven neutrophil signatures upon hospitalization, but this signature decreases over time and is replaced either by a G-MDSC-like signature in severe patients or a neutrophil progenitor signature in non-severe patients. Furthermore, we observed that patients who died maintained higher levels of IFN on days 3 and 7 of hospitalization, potentially indicating that the rate of change of the IFN response may be a biomarker of severe disease. Finally, multimodal analysis integrating transcriptomics and proteomics from matched plasma revealed a potential feedback loop of neutrophil IL-1B signaling in severe patients.
Our evolving understanding of the differential impact of IgA and IgG antibodies on neutrophil effector functions has potential therapeutic implications. The observation that patients who died maintained a higher IgA1/IgG1 ratio than patients who were intubated but survived directly implicates humoral responses in fatal COVID-19. SARS-CoV-2 infection can begin in nasal passages and trigger a strong mucosal IgA response.84 We hypothesize that IgA-enriched humoral responses may promote systemic circulating neutrophil dysregulation with higher rates of neutrophil cell death including NETosis. While IgA-induced NETosis would be beneficial in mucosal linings by preventing viral entry, it would be ineffective or harmful in other locations, as circulating neutrophils perform protective phagocytic functions in response to IgG antibodies. Many studies have shown that NETosis is a defining feature of severe disease,48,85,86 and we find signatures of NETosis in plasma and demonstrate that neutrophil cell death can be induced by IgA antibodies, which may occur in patients with high IgA1/IgG1 ratios in plasma. Potential therapeutics have been suggested for use in autoimmune disease to inhibit NETosis such as PAD4 inhibition or recombinant human thrombomodulin,87 and similar strategies could be applied in severe COVID-19. Additionally, clinical trials targeting IL-1B aimed at decreasing NETosis are underway (ClinicalTrials.gov: NCT04594356). Lastly, we hypothesize that infusion of convalescent plasma enriched for IgG and depleted for IgA may have a stronger impact on patient recovery than non-enriched plasma.
While manipulation of the antibody landscape could hold promise for effective interventions, the drivers of humoral responses in COVID-19 are still poorly understood. In this study, we identify a strong association between higher IgA1/IgG1 ratios in plasma and circulating IFNL1, though no study to date connected type III IFN with isotype switching to IgA. Future studies should aim to determine which plasma cells are responsible for IgA secretion in response to SARS-CoV-2. A recent study suggested that TNF-α-secreting cells could be responsible for the loss of germinal centers in the secondary lymphoid organs of severe COVID-19 patients.88 Though neutrophils produce lower levels of TNF-α than inflammatory macrophages, the robust enrichment of the TNF-α signaling via NF-κB pathway in neutrophils suggest that neutrophils may also play a role in the loss of germinal centers and weakening of humoral responses in severe COVID-19. Recent studies in patients treated with TNF-α blockers for autoimmunity demonstrated clinical benefit, but results from full-scale clinical trials are still needed.89, 90, 91
In summary, our study elucidates how circulating neutrophils and their interactions with soluble factors may drive COVID-19 severity, providing insight into this crucial and abundant cell type. We propose a model of SARS-CoV-2 infection in which antibody profiles drive neutrophils either to aid in disease resolution through phagocytosis or contribute to tissue damage via NETosis. Further, we hypothesize that therapies that simultaneously aim to modulate the levels of suppressive G-MDSC-like neutrophils and prevent excessive NETosis in circulation have the potential to aid with disease resolution in severe patients.
Limitations of the study
First, we performed bulk RNA-seq due to limitations at sample collection, so the neutrophil subtype gene signatures reflect a mixture of neutrophil states. Second, samples were enriched for neutrophils via negative selection, and high purity of samples could not be guaranteed. We used estimated cell-type proportions as covariates in all analyses, but the expression of contaminating cell-type-specific genes cannot be excluded. Third, our time course data were collected on days 0, 3, and 7 of hospitalization, but patients were infected for varying amounts of time prior to enrollment. Fourth, we only collected longitudinal samples from hospitalized patients, so we were unable to study pre-hospitalization or non-hospitalized patients. Fifth, samples at later time points were biased toward sicker patients with longer hospital stays. Sixth, our findings need to be validated in external cohorts with similar multimodal data structures. Seventh, our study provides insights into circulating factors, yet future studies should focus on longitudinal immunity occurring at mucosal barriers. Eighth, many of our conclusions are drawn from transcriptomics and cannot provide functional information. Though others have demonstrated the functional activity of specific genes, future studies will investigate their roles in COVID-19. Ninth, our neutrophil cell death assays relied on imaging at a fixed endpoint, which did not allow us to distinguish between NETosis, “incomplete” NETosis, and secondary necrosis. Future experiments will be needed to confirm the mode of cell death and the impact on disease resolution in vivo. Finally, samples were collected in March–May 2020, and treatments such as dexamethasone or tocilizumab may affect neutrophils during the course of disease; furthermore, antibody profiles elicited by COVID-19 vaccines may modulate neutrophil function.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Anti-Human IgG1-Fc PE | Southern Biotech | CAT# 9054-09; RRID: AB_2796628 |
| Mouse Anti-Human IgG2-Fc PE | Southern Biotech | CAT# 9060-09; RRID: AB_2796635 |
| Mouse Anti-Human IgG3-Fc PE | Southern Biotech | CAT# 9210-09; RRID: AB_2796701 |
| Mouse Anti-Human IgM-Fc PE | Southern Biotech | CAT# 9020-09; RRID: AB_2796577 |
| Mouse Anti-Human IgA1-Fc PE | Southern Biotech | CAT# 9130-09; RRID: AB_2796656 |
| Pacific Blue(TM) anti-human CD66b antibody | Biolegend | CAT# 305112; RRID: AB_2563294 |
| CD3 Pacific Blue | Biolegend | CAT# 317314; RRID: AB_571909 |
| CD11b APC | Biolegend | CAT# 301310; RRID: AB_314162 |
| CD14 APC/Cy7 | Biolegend | CAT# 367108; RRID: AB_2566710 |
| CD15 PE | Biolegend | CAT# 301905; RRID: AB_314197 |
| CD16 PE | Biolegend | CAT# 302007; RRID: AB_314207 |
| CD19 FITC | Biolegend | CAT# 302206; RRID: AB_314236 |
| CD20 FITC | Biolegend | CAT# 302304; RRID: AB_314252 |
| CD35 PE | Biolegend | CAT# 332404; RRID: AB_2890765 |
| CD45 Alexa Fluor 700 | Biolegend | CAT# 368514; RRID: AB_2566374 |
| CD56 PE-Cy5 | Biolegend | CAT# 318308; RRID: AB_604105 |
| CD63 FITC | Biolegend | CAT# 353006; RRID: AB_10898319 |
| CD66b APC | Biolegend | CAT# 305118; RRID: AB_2566607 |
| Human TruStain FcX | Biolegend | CAT# 422302; RRID: AB_2818986 |
| Rabbit anti-neutrophil elastase | Abcam | CAT# ab131260 |
| Donkey anti-rabbit IgG (H+L) Alexa Fluor 488 | Invitrogen | CAT# A21206; RRID: AB_2535792 |
| Biological samples | ||
| Patient samples used in this study are detailed in Table S1 | Massachusetts General Hospital | N/A |
| Healthy control blood samples | Stemexpress | CAT# PBEDT020F |
| Chemicals, peptides, and recombinant proteins | ||
| SARS-CoV-2 receptor binding domain (RBD) | Aaron Schmidt, Ragon Institute | N/A |
| SARS-CoV-2 nucleocapsid (N) protein | Aalto BioReagents | CAT# CK 6404-b |
| SARS-CoV-2 spike protein (S) | Eric Fischer, Dana Farber Cancer Institute | N/A |
| SARS-CoV-2 subunit 1 and 2 of the spike protein (S1 and S2) | Sino Biological | CAT#: 40591-V08B1; 40590-V08B |
| hCoV-OC43 RBD | Aaron Schmidt, Ragon Institute | N/A |
| hCoV-OC43 spike protein (S) | Sino Biological | CAT#: 40607-V08H1 |
| hCoV-HKU1 spike protein (S) | Immune Tech | CAT#: IT-002-025p |
| SARS-CoV-1, MERS spike proteins (S) | Jason McLellan, University of Texas | N/A |
| HA A/Michigan/45/2015 (H1N1) | Immune Tech | IT-003-00105DTMp |
| HA A/Singapore/INFIMH-16-0019/2016 (H3N2) | Immune Tech | IT-003-00434DTMp |
| HA B/Phuket/3073/2013 | Immune Tech | IT-003-B11DTMp |
| SYTOX™ Green Nucleic Acid Stain | Invitrogen | CAT# S7020 |
| Critical commercial assays | ||
| EasySep™ Direct Human Neutrophil Isolation Kit | STEMCELL technologies | CAT# 19666 |
| Qubit dsDNA High Sensitivity Assay Kit | Invitrogen | CAT# Q32854 |
| High-Sensitivity DNA Bioanalyzer Kit | Agilent | CAT# 5067-4626 |
| Nextera XT Library Prep kit | Illumina | CAT# FC-131-1024 |
| LEGEND MAX™ Human Myeloperoxidase ELISA Kit (BioLegend, Cat#440007) | Biolegend | CAT# 440007 |
| Citrullinated Histone H3 (Clone 11D3) ELISA Kit | Cayman Chemical | CAT# 501620 |
| Deposited data | ||
| Neutrophil bulk RNAseq analyzed data | This paper | GEO: GSE212041https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE212041 |
| Olink COVID-19 plasma proteomic data | Olink | https://info.olink.com/mgh-covid-study-overview-page; https://doi.org/10.5281/zenodo.7076472 |
| Somalogic COVID-19 plasma proteomic data | Filbin et al. 202123 | Mendeley Data: https://doi.org/10.17632/nf853r8xsj; https://dx.doi.org/10.5281/zenodo.7076472 |
| COVID-19 Neutrophil scRNA-Seq data | Schulte-Schrepping et al. 20209 | EGA: EGAS00001004571 |
| Sepsis Neutrophil scRNA-seq data | Reyes et al. 202140 | https://singlecell.broadinstitute.org/single_cell/study/SCP1492/ |
| Dexamethasone COVID-19 Neutrophil scRNA-seq data | Sinha et al. 202136 | GEO: GSE157789 |
| BAL scRNA-Seq data | Bost et al. 20205 | GEO: GSE145926 and GSE149443 |
| ARDS Neutrophil RNA-Seq | Juss et al. 201642 | GEO: GSE76293 |
| Human Protein Atlas Blood cell RNA-Seq | Uhlen et al. 201992 | https://www.proteinatlas.org/download/rna_blood_cell.tsv.zip |
| Lung cancer single-cell neutrophil RNA-seq data | Zilionis et al. 201941 | GEO: GSE127465 |
| BAL Neutrophil scRNA-seq data | Wendisch et al. 202183 | EGA: EGAS00001004928 and EGAS00001005634 |
| Oligonucleotides | ||
| RT primer (DNA oligo) | IDT | 5′–AAGCAGTGGTATCAACGCAGAGTACT30VN-3′ |
| TSO primer (RNA oligo with LNA) | Qiagen | 5′-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3′ |
| ISPCR (DNA oligo) | IDT | 5′-AAGCAGTGGTATCAACGCAGAGT-3′ |
| Software and algorithms | ||
| Code and Data from this manuscript | This manuscript | https://doi.org/10.5281/zenodo.7030528; https://doi.org/10.5281/zenodo.7076472 |
| IntelliCyt ForeCyt (v8.1) | Sartorius | https://intellicyt.com/products/software/ |
| FlowJo (v10.7.1) | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo |
| Prism 9.2.0 (283) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| GTEx-TOPMed RNA-Seq pipeline | Broad Institute | https://github.com/broadinstitute/gtex-pipeline/ |
| STAR v2.5.3a | Dobin et al. 201393 | https://github.com/alexdobin/STAR/releases/tag/2.5.3a |
| RSEM v1.3.0 | Li et al. 201194 | https://github.com/deweylab/RSEM/releases/tag/v1.3.0 |
| RNA-SeQC 2 | Graubert et al. 202124 | https://github.com/getzlab/rnaseqc |
| CIBERSORTx | Newman et al. 201935 | https://cibersortx.stanford.edu |
| DESeq2 v1.30.1 | Love et al. 201495 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Fgsea | Korotkevich et al. 201696 | http://bioconductor.org/packages/release/bioc/html/fgsea.html |
| Seurat v4.0.4 | Hao and Hao et al. 202197 | https://cran.r-project.org/web/packages/Seurat/index.html |
| Other | ||
| FluoSpheres™ NeutrAvidin™-Labeled Microspheres, 1.0 μm, yellow-green fluorescent (505/515), 1% solids | Invitrogen | CAT# F8776 |
| MagPlex microspheres | Luminex corporation | CAT# MC12001-01 |
| CaptureSelect IgA Affinity Matrix | ThermoScientific | CAT# 1942880005 |
| Protein A/G Agarose | ThermoFisher | CAT# 20424 |
| Luminol | Sigma-Aldrich | CAT# 123072 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Moshe Sade-Feldman (msade-feldman@mgh.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
MGH patients cohort description
Between March to May 2020 during the peak of the COVID-19 pandemic, we enrolled a total of 384 patients 18 years or older who presented in Massachusetts General Hospital Emergency Department (ED) with acute respiratory distress and clinical concern for COVID-19. The study was approved by the Mass General Brigham Institutional Review Board under protocol 2017P001681, with an approval for a waiver of informed consent in compliance with the 45CFR 46, 2018 Common rule. Out of the 384 patients enrolled in this study, 306 tested positive for SARS-CoV-2 (COVID-19+), while 78 patients that were admitted to the ED with similar symptoms tested negative (COVID-19–) and were used as controls in this study. Additionally, we collected blood samples from 8 healthy donors.
For each patient, medical history and clinical data were collected and are presented in Table S1 and as previously described.23 Samples were collected at three different time points: Day 0 upon admission to the ED (n = 374 samples); Day 3 (n = 212 samples) and Day 7 (n = 143 samples) for COVID-19+ hospitalized patients. In addition, in some cases up to day 28 post-admission to the ED, a fourth blood sample was collected upon a major change in clinical status, and was termed an event driven sample (n = 44 samples). Acuity categories were classified into five classes (A1-A5) using the WHO ordinal outcomes scale as recently described in Filbin et al. 2021,23 with the following classifications: A1 and A2 were classified as severe disease, with A1 defined as death within 28 days (n = 40 patients and 96 samples), and A2 for patients that survived within 28 days but required mechanical ventilation and/or intubation (n = 67 patients and 222 samples). Groups A3-A5 were defined as non-severe, with A3 classified as patients that required supplemental oxygen (n = 133 patients and 298 samples), A4 hospitalized but no need for supplemental oxygen (n = 41 patients and 45 samples), and A5 classified as patients that were discharged from ED in the first 24 h and did not return to the hospital within 28 days (n = 23 patients and 23 samples; Table S1). Primary disease severity outcomes following SARS-CoV-2 infection for each patient (AcuityMax or SeverityMax) were defined as the most severe disease level with 28 days of enrollment.
In total, the gender of 52.9% of patients was male. Of all 306 COVID-19+ patients enrolled, the following percentages of patients had past medical history: 15.7% heart disease, 36.3% diabetes, 47.7% hypertension, 30.7% hyperlipidemia, 21.6% chronic lung disease, 13.4% kidney disease, 8.2% immunocompromised. Of COVID-19+ patients, 1.9% were enrolled in trials of remdesivir versus placebo and 7.1% were enrolled in trials of anti-interleukin-6 (IL-6) receptor monoclonal antibody versus placebo. Overall, the primary outcomes described in this manuscript are not reporting any clinical trial results but rather the clinical status and outcomes resulting from the SARS-CoV-2 viral infection and hospitalization. Since this study included the enrollment of patients with an approval for a waiver of informed consent, demographic information, and other clinical parameters described in this study (e.g., blood counts, LDH, CRP etc.) are limited and reported in quintiles.
Method details
Neutrophil isolation and lysis
Blood samples were collected in EDTA vacutainer tubes and transported to the laboratory. Neutrophils were isolated from whole blood via negative selection using the EasySep Direct Human Neutrophil Isolation Kit (STEMCELL Technologies, Cat# 19666). All described procedures in this section were done at room temperature. Between 0.25 and 0.5 mL whole blood was lysed with ACK Lysis Buffer (ThermoFisher Scientific, Cat# A1049201) in a 15 mL conical tube and white blood cells were pelleted at 300 xg for 5 min. Following aspiration of the lysed red blood cells and resuspension of the pellet in 250 μL 1 mM EDTA in PBS, 50 μL each of the RapidSpheres and Isolation Cocktail were added to the cell suspension. Following a 5 min incubation, sample volumes were completed to 4 mL with 1 mM EDTA in PBS, mixed gently, and placed on an EasyEights™ EasySep™ Magnet (STEMCELL Technologies, Cat# 18103) for 5 min. Next, supernatants were transferred to new 15 mL conical tubes, 25 μL RapidSpheres were added, and the samples were gently mixed and incubated for 5 min. Samples were then placed on the magnet, and after 5 min incubation supernatants were transferred to new tubes, and were placed immediately on the magnet for a second incubation before the supernatants containing the enriched neutrophil populations were collected, pelleted, and resuspended in 1 mM EDTA in PBS. Cells were counted on a TC20™ Automated Cell Counter (Bio-Rad Laboratories, Inc., Cat# 1450102) with trypan blue staining for dead cell exclusion. Neutrophils were then lysed in TCL Buffer (QIAGEN, Cat# 1031576) with 1% 2-Mercaptoethanol at a concentration of 1000 cells/μL, flash-frozen on dry ice, and then stored at −80°C until use.
Neutrophil purity flow cytometry
Blood samples from healthy donors were collected in EDTA vacutainer tubes. 500mL of whole blood underwent lysis with ACK Lysis Buffer (ThermoFisher Scientific, Cat# A1049201) and was set aside, and 500mL of blood was taken through the neutrophil isolation protocol described above. Cells from both conditions were then stained for 30 min at RT with a panel of the following antibodies: CD3, CD14, CD16, CD19, CD20, CD56, CD66b and Human TruStain FcX. Sample acquisition was performed using a Sony MA900 instrument in flow cytometry mode. Data was analyzed in FlowJo and major cell lineages were quantified based on the percentage of live cells.
Patient matched plasma isolation
Following the aliquoting of 0.25–0.5 mL whole blood for neutrophil isolation, remaining blood volumes were diluted 1:2 with room temperature RPMI. Each diluted sample was then added carefully to a SepMate tube (STEMCELL Technologies, Cat# 85450 or 85,415) that had been prefilled with 15 mL Ficoll (VWR, Cat# 21008-918). Samples were spun at 1200 xg for 20 min at 20°C with maximum acceleration and the brake on. After centrifugation, the plasma layer was transferred into a clean conical tube and spun at 1000 xg for 5 min at 4°C to pellet any remaining cell debris. Without disturbing the pellet, each sample was aliquoted into 1.5 mL Cryovials (VWR, Cat# 66008-710) and frozen at −80°C until analysis.
Cell-free DNA (cfDNA) quantification
cfDNA was quantified using the Qubit dsDNA High Sensitivity Assay Kit (Invitrogen, Cat# Q32854). 98 μL of DNA dye was aliquoted into each well of a 96-well black clear bottom plate (Corning, Cat# 3904). Plasma samples which had been pre-aliquoted into 96-well Eppendorf PCR plates were thawed at RT, vortexed, and spun down briefly. 2 uL of plasma sample was added to each well of the assay plate. Fluorescence was quantified on a Cytation 5 Microplate reader at 523 nm.
Smart-Seq2 cDNA preparation
cDNA was prepared from bulk populations of 2x104 neutrophils per sample via the Smart-Seq2 protocol98 with some modifications to the reverse transcription step as previously described.99 20 μL (at a concentration of 1000 cells/μL) of neutrophil lysates were thawed on ice and plated into 96-well plates prior to centrifugation at 1500 rpm for 30 s. RNA was purified with Agencourt RNAClean XP SPRI beads (Beckman Coulter, Cat# A63987) and then the samples were resuspended in 4 μL of Mix-1 [Per 1 sample: 1 μl (10 μM) RT primer (DNA oligo) 5′–AAGCAGTGGTATCAACGCAGAGTACT30VN-3′; 1 μl (10 μM) dNTPs; 1μl (10%, 4 U/μl) recombinant RNase inhibitor; 1 μl nuclease-free water], denatured at 72 °C for 3 min and placed immediately on ice for 1 min before 7 μL of Mix-2 [Per 1 sample: 0.75 μl nuclease-free water; 2 μl 5X RT buffer (Thermo Fisher Scientific, Cat# EP0753); 2 μl (5 M) betaine; 0.9 μl (100 mM) MgCl2; 1 μl (10 μM) TSO primer (RNA oligo with LNA) 5′-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3′; 0.25 μl (40 U/μl) recombinant RNase inhibitor; 0.1 μl (200 U/μl) Maxima H Minus Reverse Transcriptase] was added. Reverse transcription reactions were performed at 50°C for 90 min, followed by 5 min incubation at 85°C. Then, 14 μL of Mix-3 [Per 1 sample: 1 μl nuclease-free water; 0.5 μl (10 μM) ISPCR primer (DNA oligo) 5′-AAGCAGTGGTATCAACGCAGAGT-3′; 12.5 μl 2X KAPA HiFi HotStart ReadyMix] was added to each well and the whole-transcriptome amplification step was performed at 98°C for 3 min, followed by 16 cycles of [98°C for 15 s, 67°C for 20 s, and 72°C for 6 min], and final extension at 72C for 5 min cDNA was purified using AgencourtAMPureXP SPRI beads (Beckman Coulter, Cat# A63881) as described,99 to remove all primer residue. Quality control was performed on samples prior to library construction and included: (1) concentration measurements via the Qubit dsDNA high sensitivity assay kit (Invitrogen, Cat# Q32854) on the Cytation 5 Microplate Reader (BioTek); (2) cDNA size distribution using the High-Sensitivity DNA Bioanalyzer Kit (Agilent, Cat# 5067-4626).
Library construction and sequencing
Libraries were generated using the Nextera XT Library Prep kit (Illumina, Cat# FC-131-1024) with custom indexing adapters99 in a 384-well PCR plate, followed by a cleanup step to remove residual primer dimers. Pooled libraries containing 384 samples were then sequenced on a NovaSeq S4 (Illumina) using paired-end 150-base reads. Additionally, 16 samples were sequenced on a NextSeq 500 sequencer (Illumina), using paired-end 38-base reads. This approach insured an appropriate coverage for all samples analyzed in this study.
Citrullinated histone H3 ELISA
Citrullinated Histone H3 ELISA was quantified in patient plasma using the Citrullinated Histone H3 (clone 11D3) ELISA kit (Cayman Chemical, 501620) according to manufacturer specifications. Plasma samples (which were previously diluted 1:2 with RPMI) were diluted 1:2 with Assay Buffer. Sample acquisition was performed using the Cytation 5 Microplate Reader (BioTek) at 450nm. The standard curve was fitted with a 4-parameter logistic curve-fitting algorithm using the dr4pl package in R.
Antibody subclass and isotype measurements
SARS-CoV-2 and eCoV-specific antibody subclass/isotype levels were assessed using a 384-well based customized multiplexed Luminex assay, as previously described.25 SARS-CoV-2 receptor binding domain (RBD) (kindly provided by Aaron Schmidt, Ragon Institute), SARS-CoV-2 nucleocapsid (N) protein (Aalto BioReagents), and SARS-CoV-2 spike protein (S) (kindly provided by Eric Fischer, Dana Farber), SARS-CoV-2 subunit 1 and 2 of the spike protein (S1 and S2) (Sino Biological), as well as human eCoV antigens: hCoV-OC43 RBD (kindly provided by Aaron Schmidt, Ragon Institute), hCoV-OC43 spike protein (S) (Sino Biological), hCoV-HKU1 spike protein (S) (Immune Tech), SARS-CoV-1, MERS spike proteins (S) (kindly provided by Jason McLellan, University of Texas) were used to profile specific humoral immune response. A mix of HA A/Michigan/45/2015 (H1N1), HA A/Singapore/INFIMH-16-0019/2016 (H3N2), HA B/Phuket/3073/2013 (Immune Tech) was used as a control. Antigens were coupled to magnetic Luminex beads (Luminex Corp) by carbodiimide-NHS ester-coupling (Thermo Fisher). Antigen-coupled microspheres were washed and incubated with plasma samples at an appropriate sample dilution (1:500 for IgG1 and 1:100 for all other readouts) for 2 h at 37°C in 384-well plates (Greiner Bio-One). Unbound antibodies were washed away, and antigen-bound antibodies were detected by using a PE-coupled detection antibody for each subclass and isotype (IgG1, IgG2, IgG3, IgG4, IgA1, and IgM; Southern Biotech). After 1h incubation, plates were washed, and flow cytometry was performed with an IQue (Intellicyt), and analysis was performed on IntelliCyt ForeCyt (v8.1). PE median fluorescence intensity (MFI) is reported as a readout for antigen-specific antibody titers.
Antibody-dependent neutrophil phagocytosis (ADNP) assay
ADNP was conducted as previously described.64 SARS-CoV-2 Spike proteins were biotinylated using EDC (Thermo Fisher) and Sulfo-NHS-LC-LC biotin (Thermo Fisher) and coupled to NeutrAvidin beads (Thermo Fisher, Cat# F8775). To form immune complexes, antigen-coupled beads were incubated for 2 h at 37°C with serum and then washed to remove unbound antibodies. The immune complexes were incubated for 1 h with RBC-lysed whole blood. Following the incubation, neutrophils were stained for CD66b+ (Biolegend, Cat# 305112) and fixed in 4% PFA.
Flow cytometry was performed to identify the percentage of cells that had phagocytosed beads as well as the number of beads that had been phagocytosed (phagocytosis score = % positive cells × Median Fluorescent Intensity of positive cells/10000). Flow cytometry was performed with an IQue (Intellicyt) or LSRII(BD), and analysis was performed using IntelliCyt ForeCyt (v8.1) or FlowJo V10.7.1.
SARS-CoV-2 spike specific IgG and IgA isolation
IgA were purified from human plasma samples using CaptureSelect IgA Affinity Matrix (Thermo Fisher Scientific, Cat# 1942880005), and flowthrough was used to purify the IgG with Protein A/G Agarose (Thermo Fisher Scientific, Cat# 20424). For both, the capture matrices were washed three times with Binding Buffer (0.1 M phosphate, 0.15 M sodium chloride; pH 7.2) and incubated overnight with 1:5 diluted plasma samples. Antibodies bound to matrices were washed 3x with PBST by centrifugation and eluted with Elution Buffer (0.1 M glycine, pH 2-3). The antibodies were collected to tubes containing Neutralization Buffer (1 M Tris, pH 8-9) and used for further analysis. The presence of IgA and IgG was confirmed by ELISA.
Antibody-dependent neutrophil activation and ROS release
A high-binding 96-well plate was coated with SARS-CoV-2 Spike protein (5ug/ml) and blocked with 5% BSA. Isolated antibodies were added and incubated for 2h at RT; afterward, the plate was washed three times with PBS-Tween. Neutrophils were isolated from fresh blood using the EasySep™ Direct Human Neutrophil Isolation Kit (STEMCELL Technologies, Cat# 19666) and adjusted to the concentration of 106 cells/mL. Luminol (Sigma-Aldrich, Cat# 123072) was diluted in DMSO and added to neutrophils at the final concentration of 0.2 mg/mL. Cells with luminol were added to each well, and chemiluminescence was read immediately on a plate reader (for around two hours). ROS release was quantified as chemiluminescence count/second.
Neutrophil cell death imaging assay
Methods were adapted from a previous publication.66
Fibronectin glass slide coating
15-mm round glass coverslips (Electron Microscopy Sciences, Cat# 72228-01) were coated in 5 μg/cm2 of fibronectin (Sigma, Cat# F1141) diluted in PBS at room temperature for 5 min. The solution was removed and slides were allowed to dry for at least 45 min before use.
Enhanced neutrophil isolation
Fresh blood was collected from healthy donors, moved to a 50mL conical, and diluted 1:2 with room temperature RPMI. Diluted samples were added to a SepMate tubes (Stemcell Technologies, Cat# 85450) that had been prefilled with 16 mL Ficoll (VWR, Cat# 21008-918). Samples were spun at 1200 xg for 20 min at 20°C with maximum acceleration and the brake on. Plasma and PBMCs were removed, and the high density layer containing erythrocytes and granulocytes was moved to a 50mL tube. Samples then underwent two rounds of red blood cell lysis using ACK Lysis Buffer (ThermoFisher Scientific, Cat# A1049201) and centrifugation for 5 min at 1500g, RT. Pellets were resuspended in 500μL of 1 mM EDTA in PBS per 10mL of blood, and 250uL aliquots were moved to 15mL conicals. Negative selection for neutrophils was then performed with the EasySep™ Direct Human Neutrophil Isolation Kit (STEMCELL Technologies, Cat# 19666) with custom modifications. 75 μL each of the RapidSpheres and Isolation Cocktail were added to the cell suspension. Following a 5 min incubation, sample volumes were completed to 4 mL with 1 mM EDTA in PBS, mixed gently, and placed on an EasyEights™ EasySep™ Magnet (STEMCELL Technologies, Cat# 18103) for 5 min. Next, supernatants were transferred to new 15 mL conical tubes, 37.5 μL RapidSpheres were added, and the samples were gently mixed and incubated for 5 min. Samples were then placed on the magnet, and after 5 min incubation supernatants were transferred to new tubes, and were placed immediately on the magnet for a second incubation before the supernatants containing the enriched neutrophil populations were collected, pelleted, and resuspended in PBS. Cells were counted on a TC20™ Automated Cell Counter (Bio-Rad Laboratories, Inc., Cat# 1450102) with trypan blue staining for dead cell exclusion.
Cell death induction
15-mm fibronectin-coated glass coverslips were plated in individual wells of a 12-well plate. 400,000 neutrophils were added to each slide in RPMI+L-glu and allowed to settle for 1 h at 37°C. Media was gently removed and replaced with desired treatment (PBS, 100nM Phorbol 12-myristate 13-acetate (PMA, Sigma, Cat# P1585), or free IgG or IgA antibodies isolated from patient plasma). Cells were then incubated for 3 h at 37°C and 5% CO2.
Slide preparation
Media was gently removed from the slides and replaced with 4% paraformaldehyde (PFA) for 1 h. PFA was removed and slides were gently washed with PBS three times. Cells were then permeabilized with 0.5% Triton X-100 in PBS with 0.1% Tween (PBS-T) for 30 min. Fixed and permeabilized cells were then blocked for 30 min at RT in 10% FBS in PBS-T. Blocking buffer was removed and cells were incubated with primary rabbit anti-neutrophil elastase antibody (Abcam, Cat# ab131260) at a 1:100 dilution for 1 h at RT. Coverslips were then washed three times with PBS. Cells were then incubated with Alexa Fluor 488-conjugated donkey anti-rabbit antibody at a dilution of 1:1000 in PBS for 1 h at RT, protected from light. Coverslips were washed three times with PBS. Cells were incubated with Hoescht 33342 trihydrochloride trihydrate at 1ug/mL for 5 min at RT, protected from light. Coverslips were washed three times with PBS. Coverslips were then mounted onto glass slides in EverBrite Mounting Medium (BIOTIUM, Cat# 23001) and edges were sealed with clear nail polish.
Imaging and quantification
Cells were imaged with phase contrast and fluorescence microscopy using the Leica THUNDER Imager. Five random fields per condition were imaged at 20x magnification. We counted potential NETs based on mixing of decondensed chromatin and neutrophil elastase, whereas potentially apoptotic cells could be identified by membrane blebbing with phase contrast, and potential necrotic cells had distinct condensed nuclei with loss of segmentation and membrane swelling (Figure 5I).
Neutrophil PMA-treatment time-lapse microscopy
Fresh healthy donor neutrophils were isolated as described above using the enhanced neutrophil isolation protocol. Wells of an 8-well chambered coverslip (ibidi, Cat# 80807) were coated with 0.01% poly-L-lysine for 10 min, aspirated, washed twice with PBS, dried for 2 h, and rinsed once more. 75,000 neutrophils were plated in 200μL per well in RPMI+L-glu with 100nM SYTOX green and 20μg/mL Hoechst 33342. Cells were placed in the Leica THUNDER Imager chamber and allowed to settle for 15 min. Cells were imaged once per minute for 45 min at 20x magnification with phase contrast, 3 fields per well. PMA (or an equivalent volume of PBS) was added to each well for a final concentration of 100nM, and cells were imaged every minute for 300 min.
Sytox green cell death assay
Methods were adapted from a previous publication.46 All reagents used in this section were allowed to equilibrate to RT before use.
Poly-L-lysine plate coating: 96-well black clear bottom plates (Corning, Cat# 3904) were coated in 40μL of a 1:10 dilution of 0.01% poly-L-lysine (Sigma-Aldrich, Cat# P4707-50ML) in sterile water. Plates were incubated at 37°C for one hour and subsequently washed twice with sterile water, and were allowed to dry for at least two hours before use.
Enhanced neutrophil isolation: Enhanced neutrophil isolation was performed using the same protocol as the neutrophil cell death imaging assay.
Neutrophil cell death induction and quantification: Using the highly-enriched neutrophil samples, 50,000 cells were plated in each well of the poly-L-lysine-coated 96-well black clear bottom plates. Plates were then incubated for 20 min at 37°C and 5% CO2 to allow neutrophils to adhere. Supernatant was then gently removed and immediately replaced with 32μL RPMI + L-glu with 625nM SYTOX™ Green Nucleic Acid Stain (Invitrogen, Cat# S7020). 8μL of patient-isolated antibody was then added to each well for a total of 40μL per well with a final 1:5 dilution of free antibody and 500nM SYTOX Green. Plates were then incubated for 4 h at 37°C and 5% CO2. Cells were gently removed from the incubator and fluorescence was quantified on a Cytation 5 Microplate reader at 485nm and 523nm using the area scan setting from the bottom of the plate. Absorbance at 485nm was subtracted from the absorbance at 523nm to obtain corrected RFU values.
Neutrophil degranulation assay
400,000 neutrophils isolated from healthy donors using the enhanced neutrophil isolation protocol described above were plated per well of a 96-well U-bottom plate (Nunc, Cat# 168136). Cells were treated with the desired condition (10μg/mL LPS (Sigma, Cat# L4391), PBS, or free patient-derived IgA or IgG antibodies at an average concentration of 0.2 mg/mL) in duplicate for 12 h at 37°C and 5% CO2. Cells were then stained with two separate panels of cell surface markers of neutrophil degranulation which are translocated to the surface following exocytosis of granule contents. Panel 1: CD11b-APC (Clone ICRF44), CD14-APC/Cy7 (Clone 63D3), CD15-PE (Clone HI98). Panel 2: CD35-PE (Clone 9H3), CD45-Alexa Fluor 700 (Clone 2D1), CD63-FITC (Clone H5C6), CD66b-APC (Clone G10F5). Flow cytometry was performed using a Sony MA900 instrument. Neutrophils were gated in FlowJo based on forward (FSC) and side (SSC) scatter profiles, and MFI was averaged between duplicates.
Quantification and statistical analysis
RNA-seq alignment
A custom FASTA was generated from the Homo sapiens (human) genome assembly GRCh38 (hg38) following exclusion of ALT, HLA, and Decoy contigs according to documentation in the Broad Institute GTEx-TOPMed RNA-seq pipeline (https://github.com/broadinstitute/gtex-pipeline/), with an appended SARS-CoV2 genome. GENCODE v35 with the appended SARS-CoV2 GTF was used for annotation. Raw FASTQ files were aligned to the custom genome FASTA in the Terra platform with the Broad Institute GTEx pipeline using STAR v2.5.3a, and expression quantification based on a collapsed annotation was performed using RSEM v1.3.0.
Quality control
RNA-SeQC 224 (https://github.com/getzlab/rnaseqc) was used to calculate quality control metrics for each sample. Samples were excluded if they did not meet the following criteria: 1) percentage of mitochondrial reads less than 20%, 2) greater than 10,000 genes detected with at least 5 unambiguous reads, 3) median exon CV less than 1, 4) exon CV MAD less than 0.75, 5) exonic rate greater than 25%, 6) median 3′ bias less than 90%. This filtration kept 698 out of 781 samples (89.4%) (Table S1). Genes were included in the analysis if they were expressed at a level of 0.1 TPM in at least 20% of samples and if there were at least 6 counts in 20% of samples. In total, 20283 genes passed the filtration criteria.
Neutrophil fraction estimation and contamination control
CIBERSORTx35 was used to estimate the proportions of mature neutrophils, immature neutrophils, T/NK cells, B cells, plasmablasts, and monocytes in each sample. To generate the signature matrix for deconvolution, we utilized the single-cell RNA-seq data of PBMCs and neutrophils from whole blood from Cohort 2 of the Schulte-Schrepping et al dataset.9 Using the designations provided in the public data, we created pseudobulks for each cell type per patient by summing the counts of a given cell type, and we excluded pseudobulked cell type samples from individual patients if the cell type had less than 5000 counts. To generate the CIBERSORTx signature matrix, we set limits of 50–100 marker genes per cell type, and filtered for only hematopoietic genes. Following the generation of a signature matrix, we ran CIBERSORTx in “Impute Cell Fractions” in “relative” run mode to estimate the proportions of each cell type. In agreement with studies demonstrating lymphopenia in severe COVID-19,100 we found higher fractions of T/NK cells in non-severe patients across all time points (Table S1).
Given the levels of immunoglobulin genes from contaminating plasma cells, we created an immunoglobulin score for each sample to use as a covariate for regression (Figures S1F and S1I). To select genes, we chose the top 115 immunoglobulin genes which were differentially expressed in Day 0 COVID+ vs. COVID- patients (DESeq2, no covariates) and assigned each sample a score according to a previously described method.101 Briefly, the score was defined as the average log2(TPM+1) expression of the immunoglobulin gene set, minus the average log2(TPM+1) expression of a control gene set. The control gene set was selected by sorting the entire list of genes by aggregate counts across all samples, breaking the list into 25 bins, and for each gene in the immunoglobulin gene set, selecting 100 genes at random from the same expression bin. Using this method, the control gene set has a comparable distribution of expression levels relative to the immunoglobulin gene set and accounts for the varying complexity between samples.
Dimensionality reduction and visualization
PCA and UMAP were performed in R using prcomp() and umap() with default parameters. All box plots display median with first and third quartiles (hinges); whiskers extend from the hinges to the smallest or largest value within 1.5 ∗ IQR (interquartile range) of the hinge.
Differential expression analysis
Differential expression analyses were performed using the DESeq2 package in R.95 For each analysis i, we excluded genes with less than 5 counts in xi samples. To determine xi, we generated a curve plotting the required number of samples having ≥5 counts as the independent variable and the number of genes satisfying this condition as the dependent variable. We then selected the inflection point of this curve to be xi.
Gene set enrichment analysis
We performed gene set enrichment analysis using the fgsea package in R using the following pathway sets from MSigDB Release v7.2: H, C5 GO BP. We also performed a search of MSigDB using the keyword “neutrophil” and added the following pathways: BIOCARTA_NEUTROPHIL_PATHWAY, GO_AZUROPHIL_GRANULE, GO_AZUROPHIL_GRANULE_LUMEN, GO_AZUROPHIL_GRANULE_MEMBRANE, GO_FICOLIN_1_RICH_GRANULE, GO_NEGATIVE_REGULATION_OF_NEUTROPHIL_ACTIVATION, GO_NEGATIVE_REGULATION_OF_NEUTROPHIL_MIGRATION, GO_NEUTROPHIL_CHEMOTAXIS, GO_NEUTROPHIL_EXTRAVASATION, GO_NEUTROPHIL_MIGRATION, GO_POSITIVE_REGULATION_OF_NEUTROPHIL_MIGRATION, GO_REGULATION_OF_NEUTROPHIL_ACTIVATION, GO_REGULATION_OF_NEUTROPHIL_CHEMOTAXIS, GO_REGULATION_OF_NEUTROPHIL_DEGRANULATION, GO_REGULATION_OF_NEUTROPHIL_EXTRAVASATION, GO_REGULATION_OF_NEUTROPHIL_MEDIATED_CYTOTOXICITY, GO_REGULATION_OF_NEUTROPHIL_MIGRATION, GO_SPECIFIC_GRANULE, GO_SPECIFIC_GRANULE_LUMEN, GO_SPECIFIC_GRNAULE_MEMBRANE, GO_TERTIARY_GRANULE, HP_ABNORMAL_NEUTROPHIL_COUNT, HP_ABNORMALITY_OF_NEUTROPHIL_MORPHOLOGY, HP_ABNORMALITY_OF_NEUTROPHIL_PHYSIOLOGY, HP_ABNORMALITY_OF_NEUTROPHILS, HP_IMPAIRED_NEUTROPHIL_BACTERICIDAL_ACTIVITY, MARTINELLI_IMMATURE_NEUTROPHIL_DN, MARTINELLI_IMMATURE_NEUTROPHIL_UP, NICK_RESPONSE_TO_PROC_TREATMENT_DN, NICK_REPSONSE_TO_PROC_TREATMENT_UP, REACTOME_NEUTROPHIL_DEGRANULATION.
In addition to these pathways, we added gene sets corresponding to various neutrophil states and signatures: genes up- or down-regulated more than threefold in blood neutrophils from ARDS patients,42 single-cell neutrophil clusters in blood or lung tissue of patients with lung cancer,41 single-cell neutrophil clusters from blood of patients with sepsis,40 and single-cell neutrophil clusters from COVID-19 patients and healthy controls.9 For single-cell cluster markers, if there were more than 100 marker genes per cluster, gene sets were selected as the top 100 genes ranked by p value for enrichment in a given cluster. In addition, we included the NMF cluster gene markers from this study as neutrophil state gene sets. The GMT file containing all genes per pathway used in this analysis is available on Zenodo (https://doi.org/10.5281/zenodo.7076472), and the lists are included in Table S1.
NMF clustering analysis
In order to identify neutrophil subtypes, we performed NMF clustering of bulk RNA-Seq samples with CIBERSORTx estimated neutrophil fraction >50% (mature neutrophils and immature neutrophils combined) to reduce artifacts of cell type contamination. We used a previously described Bayesian NMF approach which identified 6 clusters.38,102,103 This method attempts to find a small number of gene sets, termed metagenes, that capture the greatest amount of variability. None of the NMF-derived signatures mapped to healthy control-derived scRNA clusters in external datasets.9,40
Sample pathway scoring
Bulk RNA-seq samples were scored for expression of genes in a gene set according to a previously described method used to control for sample complexity, as we anticipated that cells with higher complexity resulting from contamination from other cell types would have more genes detected and thus score higher for any gene set.101 Briefly, the score for each sample was defined as the average expression of the genes in the gene set minus the average expression of genes in a control gene set. To define the control gene set, all genes were ranked according to average expression across all samples and divided into 25 bins. Next for each gene in the gene set, 100 genes were selected from the same expression bin to create a gene set with comparable expression levels which is 100-fold larger. For the NETosis sample pathway score, we used the genes PADI4, MPO, ELANE, TNF, CXCL8, GSDMD, and TLR3. Our score correlated strongly with a previously-defined NETosis gene signature (Figure S3H).50
Clustering analysis for single-cell blood neutrophils from sepsis patients
The gene expression matrix was imported into R using Seurat 4.0.4. Cells were excluded with fewer than 100 genes. Data were normalized using the NormalizeData function and expression values were scaled using the ScaleData function in Seurat. 40 PCs were selected for building the neighborhood graph. Clustering was performed with the Louvain algorithm with a resolution of 0.6 which resulted in 6 clusters. Cluster markers were determined using the FindMarkers function in Seurat, and p value corrections were performed with the Benjamini-Hochberg method. Additionally, we scored cells for each NMF signature by taking the mean z-scored expression across signature genes.
Neutrophil state network analysis
Neutrophil state gene signatures were taken from the same GMT file used for GSEA analysis in Figure 2F. The network was built using the igraph package in R. Edges were drawn between nodes if the Jaccard index between the two gene signature lists was greater than 0.05. Edge width was scaled according to the overlap coefficient between the gene sets, and nodes were scaled according to gene set size and colored according to the number of neighbors in the graph. Roughly six signatures grouped together in a branch of the network we labeled “Immature Neutrophils”, which included NMF1 and NMF4; key genes in this intersection were ribosomal genes and development of neutrophil granules (NMF1 genes: DEFA4, AZU1, ELANE, CTSG, PRTN3. NMF4 genes: LTF, CAMP, MMP8, LYZ, CEACAM8). Roughly ten signatures grouped together as G-MDSC-like neutrophils, with genes such as S100A12, ARG1, CD177, MCEMP1, and GYG1 often shared. Finally, the interferon-stimulated neutrophils had the most distinctive gene expression patterns, sharing many ISGs including IFIT1, IFIT2, IFIT3, XAF1, OASL, PLSCR1, TNFSF13B, RSAD2, ISG15, DDX58, and several more. By examining the overlap of the NMF marker genes with these previously-defined neutrophil transcriptional signatures, we confirmed that multiple signatures across studies shared several genes, suggesting that these NMF signatures may represent neutrophil subtypes in multiple disease contexts.
Schulte-Schrepping single-cell RNA-seq reanalysis for early-late threshold
The single-cell fresh whole blood neutrophil data from Bonn cohort 2, originally analyzed by Schulte-Schrepping et al.,9 was reanalyzed for cluster membership according to day using day 11 as the threshold for late disease. For each cluster, we created a running metric for how many cells were classified as “early” by calculating the percentage of cells collected from Day 0 to Day x (Figure S2G).
Sinha dexamethasone analysis
We downloaded the merged COVID-19 scRNA data from the Sinha study36 and subsetted the data to the cells labeled as neutrophils. We then renormalized the data using the Seurat function NormalizeData, identified variable features using the Seurat function FindVariableFeatures, clustered cells using data from the first 15 principal components using the Louvain algorithm with a resolution of 0.6, resulting in 12 clusters. We then scored each cell for each of the NMF signatures by taking the mean z-scored expression for each of the signature genes. Finally, we grouped cells by time point (either 72 h or 7 days) and then compared each NMF signature score between cells from patients treated with dexamethasone vs. non-dexamethasone-treated patients as well as patients that survived vs. those that died using the Wilcoxon rank-sum test.
ARDS log fold-change comparisons
Log2(fold-change) (LFC) values in blood neutrophil microarray gene expression between non-COVID-19 ARDS patients and healthy volunteers was obtained from the study from Juss et al.42 Linear regression on the LFC values in ARDS vs. healthy volunteers and severe COVID-19 vs. mild COVID-19 was performed using the lm package in R. To generate a ranked list of genes based on the differences in LFC values, ARDS LFC values were z-scored, and mild vs. severe COVID-19 LFC values were z-scored on each individual day. GSEA was then performed on the lists using the difference in LFC z-score as the ranking metric. Finally, we assessed the over-representation of each NMF signature gene set in the ARDS Up and Down gene sets by counting the number of overlaps for each signature and evaluating the significance of each overlap using the hypergeometric test.
Day:Severity interaction analysis
To identify diverging patterns of gene expression between severity groups with time, we built models using DESeq2 for COVID-19+ samples on Days 0, 3, and 7. The full model included CIBERSORTx estimated cell type fractions, the immunoglobulin score, and the terms for Day, SeverityMax, and the Day:SeverityMax interaction term, while the reduced model did not include the interaction term, and we used the likelihood ratio test in DESeq2 to compare these models. Log(fold-change) values and p values were extracted to generate a ranked list of genes according to signed p values for GSEA.
Logistic regression models to predict severe COVID-19 on day 0
Logistic regression models were built using the glm package in R. In order to ensure the stability and interpretability of the coefficients in the model, we included only COVID-19+ patients on Day 0 who were not immediately discharged from the ED (AcuityMax1-4) and who had complete data for ANC, ALC, D-dimer, CRP, LDH, and BMI measured at Day 0. For patients with AcuityMax 1-4, 7 patients had missing clinical data, and these 7 missing patients were not biased towards a particular severity according to Fisher’s exact test. All parameters used were broken into discrete quintiles unless insufficient samples belonged to one category, in which case factor levels were combined in order to minimize the standard error of the coefficient estimation. We combined factor levels for age, LDH, and BMI, leaving 4 factor levels for age and LDH, and 5 factor levels for BMI (BMI was the only category scored from 0 to 5). Models were built according to three tiers of parameters. Model 1: clinical characteristics (age, gender, ethnicity, heart disease, diabetes, hypertension, hyperlipidemia, lung disease, kidney disease, immunocompromised status, BMI), Model 2: clinical characteristics plus clinical laboratory values (ANC, ALC, Creatinine, CRP, D-dimer, LDH), and Model 3: clinical characteristics plus clinical laboratory values plus neutrophil gene signature scores (NMF1, NMF2, NMF3, NMF4, NMF5, NMF6, ARDS Up - Juss, ARDS Down - Juss). ROC curves and AUC values were calculated using the pROC package in R. Significance of model improvement was determined using the likelihood ratio test using the lrtest package in R.
Feature selection for the best predictors on Day 0 of severity within 28 days among the variables used in Model 3 was performed using LASSO with the glmnet package in R with 100 repeats of 5-fold cross validation.23 The LASSO algorithm shrinks all coefficients and sets coefficients of less important (i.e. highly correlated) variables to zero, simplifying potential application of the model by decreasing the number of values measured. Model tuning was performed using the caret package in R. We ranked features according to the number of cross-validation folds in which they were selected for the LASSO model (Figure 3D). Features included in at least 98% of repeats were related to NMF5 score, LDH, IL1RL1 protein, S100A12 protein, age, AREG protein, creatinine, NMF1 score, ARDS UP score, and FKBP5 protein. Finally, we performed the same analyses using a fourth model (Model 4) which included clinical characteristics plus clinical laboratory values plus neutrophil gene signature scores plus neutrophil-expressed protein measurements in patient plasma (NMF5: TNC, TNFRSF10C, S100A12, HGF, F9; Severe: AREG, MMP8, IL1RL1, FKBP5, VSIG4).
Plasma proteomic markers of neutrophil subtypes
To identify plasma proteins associated with neutrophil NMF subtypes, we performed a Wilcoxon rank-sum test for all of the 1472 proteins measured in the Olink plasma proteomic assay between samples from NMF clusteri versus all other clusters (including Neu-Lo). We used the updated Olink proteomics data (https://info.olink.com/broad-covid-study-overview-download) which had the following modifications: 1) scale correction factors were no longer used, and 2) limits of detection were calculated on a per plate basis rather than the whole project. This resulted in the recovery of 43 assays which were not included in the original version; using the new method, no assays had 100% of samples below the limit of detection. Results for each cluster were filtered for padj > 0.05, first selecting only positive markers (higher protein levels in clusteri), and next selecting only negative markers (lower protein levels in clusteri). The strongest positive markers were selected by filtering out all markers which did not satisfy the criteria that 1) the highest expression of the protein was in the given NMF cluster and 2) the step ratio, defined as the NPX difference between the given NMF cluster and the second-highest expressing cluster, was at least 0.1. A similar method filtering out markers that did not have the lowest expression in the given NMF cluster and markers with a step ratio for the second lowest cluster of at least 0.1 was used for negative markers. Heatmaps of the protein markers per cluster were generated with the pheatmap package in R, with genes ordered according to p value.
Comparison of differential expression and plasma proteomic data
To compare log2(fold-change) values on the plasma protein level and neutrophil RNA transcriptional level, we performed differential expression analyses for each. For plasma proteins, we fit linear models using the lm packages in R for each protein using the following clinical covariates: age, gender, ethnicity, heart disease, diabetes, hypertension, hyperlipidemia, pulmonary condition, kidney disease, immunocompromised status. For RNA-seq data, we used DESeq2 differential expression analysis in R with the same clinical covariates as well as the CIBERSORTx estimated cell type fractions. The LFC values were compared for COVID-19+ vs. COVID-19– samples, as well as severe vs. non-severe samples on Days 0, 3, and 7 separately.
Ligand-receptor interaction analysis
A curated ligand-receptor pair database from FANTOM5 was used to search for interactions between neutrophil receptors and plasma ligands on either the basis of SeverityMax or neutrophil NMF cluster.104 The database was filtered on ligand-receptor interactions identified as “literature-supported” or “putative”, and was further filtered for receptors with non-zero expression in granulocytes according to the Human Protein Atlas.92 To identify neutrophil receptors associated with specific NMF clusters, differential expression was performed using DESeq2 for NMF clusteri versus all other clusters irrespective of Day. Only positive gene markers were kept with padj < 0.05. Differentially expressed receptors which were not unique to a single NMF cluster were excluded. Similarly, differential expression of plasma proteins was performed using lm in R comparing NMF clusteri vs all other clusters (including Neu-Lo), and proteins were kept with padj < 0.05. Thus a list of potential interactions was generated using the database. To determine whether the neutrophil receptors and plasma proteins were differentially expressed within the same sample rather than the aggregated group, the percentage of samples within a given NMF cluster on a specific day which had higher than mean expression across all COVID-19+ samples of both neutrophil receptor and plasma protein were calculated. In Figures 7A, 7B, and S5, ligands matching with multiple receptors were then colored according to the interaction which had the highest percentage of above-mean expression, and secondary interactions were indicated with reduced line width. Plasma ligands were then mapped to the inferred cell-of-origin using single-cell data from bronchoalveolar lavage fluid from COVID-19 patients as previously described.5,23
Wendisch BAL scRNA-seq data analysis
We downloaded the BAL COVID-19 scRNA-seq data from the Wendisch study83 and subsetted the data to the cells labeled as neutrophils. We then renormalized the data using the Seurat function NormalizeData, identified variable features using the Seurat function FindVariableFeatures, and clustered cells using data from the first 15 principal components using the Louvain algorithm with a resolution of 0.6, resulting in 9 clusters. We then scored each cell for each of the NMF signatures by taking the mean z-scored expression for each of the signature genes. Finally, we compared each NMF signature score between cells from patients that survived vs. those that died using the Wilcoxon rank-sum test.
Acknowledgments
We acknowledge support from the Ragon Institute of MGH, MIT and Harvard, the Massachusetts Consortium on Pathogen Readiness (MassCPR), the NIH (3R37AI080289-11S1, R01AI146785, U19AI42790-01, U19AI135995-02, U19AI42790-01, 1U01CA260476-01, CIVIC75N93019C00052, and T32 GM007592), the American Lung Association, and the MGH Executive Committee on Research. T.J.L. was supported by T32GM007753 from the National Institute of General Medical Sciences (NIGMS). A.C.V. acknowledges funding from the COVID-19 Clinical Trials Pilot grant from the Executive Committee on Research at MGH, a COVID-19 Chan Zuckerberg Initiative grant (2020-216954), and funds from the Manton Foundation and the Klarman Family Foundation. We thank Mikael Pittet for his expertise in neutrophil biology. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or the NIH.
Author contributions
Conceptualization, T.J.L., A.L.K.G., S.S.F., P.K., G.A., N.H., and M.S.-F.; methodology, T.J.L., A.L.K.G., S.S.F., P.K., G.A., M.B.G., M.R.F., A.C.V., N.H., and M.S.-F.; validation, T.J.L., P.K., and M.S.-F.; formal analysis, T.J.L. and S.S.F.; investigation, T.J.L., A.L.K.G., P.K., I.G., K.R.K., K.M., J.T., M.R.-L., B.C.R., N.S., M.F.T., K.M.L.-P., B.M.L., B.N.M., N.C.C., H.K.K., C.L.L., J.D.M., E.M.B., P.B.L., O.-Y.R., A.M., A.S., R.P.B., B.A.P., M.B.G., M.R.F., A.C.V., and M.S.-F.; resources, M.B.G., M.R.F., A.C.V., G.A., N.H., and M.S.-F.; data curation, T.J.L., S.S.F., N.H., and M.S.-F.; writing – original draft, T.J.L., A.L.K.G., S.S.F., P.K., and M.S.-F.; writing – review & editing, T.J.L., A.L.K.G., S.S.F., P.K., I.G., G.A., M.B.G., M.R.F., A.C.V., N.H., and M.S.-F.; visualization, T.J.L., A.L.K.G., S.S.F., P.K., and M.S.-F.; supervision, G.A., N.H., and M.S.-F.
Declaration of interests
M.S.-F. received funding from Bristol-Myers Squibb. G.A. is a founder of Seromyx Systems Inc. N.H. holds equity in Biontech and holds equity in and advises Danger Bio.
Inclusion and diversity
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community.
Published: October 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2022.100779.
Contributor Information
Thomas J. LaSalle, Email: tlasalle@broadinstitute.org.
Nir Hacohen, Email: nhacohen@mgh.harvard.edu.
Moshe Sade-Feldman, Email: msade-feldman@mgh.harvard.edu.
Supplemental information
Data and code availability
The raw RNA sequencing data reported in this study cannot be deposited in a public repository because these data were collected at the beginning of the COVID-19 pandemic, and as such, a waiver of informed consent was approved by the Massachusetts General Hospital governing institutional review board, in compliance with the Code of Federal Regulation (45CFR 46, 2018 Common Rule). To protect the identity of individual subjects, public posting of raw sequencing data from the patients has not been approved; therefore, raw data is not provided. However, the read count matrix and TPM matrix used in this study are available in GEO under accession number GSE212041 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE212041). All code required to run the analyses in this manuscript is deposited in Zenodo (https://doi.org/10.5281/zenodo.7030528) based on the associated Github repository (https://github.com/lasalletj/COVID_Neutrophils). Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
References
- 1.Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tiwari-Heckler S., Rauber C., Longhi M.S., Zörnig I., Schnitzler P., Jäger D., Giese T., Merle U. Dysregulated host response in severe acute respiratory syndrome coronavirus 2-induced critical illness. Open Forum Infect. Dis. 2021;8:ofab019. doi: 10.1093/ofid/ofab019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lucas C., Wong P., Klein J., Castro T.B.R., Silva J., Sundaram M., Ellingson M.K., Mao T., Oh J.E., Israelow B., et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature. 2020;584:463–469. doi: 10.1038/s41586-020-2588-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Q., Meng Y., Wang K., Zhang X., Chen W., Sheng J., Qiu Y., Diao H., Li L. Inflammation and antiviral immune response associated with severe progression of COVID-19. Front. Immunol. 2021;12:631226. doi: 10.3389/fimmu.2021.631226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bost P., De Sanctis F., Canè S., Ugel S., Donadello K., Castellucci M., Eyal D., Fiore A., Anselmi C., Barouni R.M., et al. Deciphering the state of immune silence in fatal COVID-19 patients. Nat. Commun. 2021;12:1428. doi: 10.1038/s41467-021-21702-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Combes A.J., Courau T., Kuhn N.F., Hu K.H., Ray A., Chen W.S., Chew N.W., Cleary S.J., Kushnoor D., Reeder G.C., et al. Global absence and targeting of protective immune states in severe COVID-19. Nature. 2021;591:124–130. doi: 10.1038/s41586-021-03234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zohar T., Loos C., Fischinger S., Atyeo C., Wang C., Slein M.D., Burke J., Yu J., Feldman J., Hauser B.M., et al. Compromised humoral functional evolution tracks with SARS-CoV-2 mortality. Cell. 2020;183:1508–1519.e12. doi: 10.1016/j.cell.2020.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wauters E., Van Mol P., Garg A.D., Jansen S., Van Herck Y., Vanderbeke L., Bassez A., Boeckx B., Malengier-Devlies B., Timmerman A., et al. Discriminating mild from critical COVID-19 by innate and adaptive immune single-cell profiling of bronchoalveolar lavages. Cell Res. 2021;31:272–290. doi: 10.1038/s41422-020-00455-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schulte-Schrepping J., Reusch N., Paclik D., Baßler K., Schlickeiser S., Zhang B., Krämer B., Krammer T., Brumhard S., Bonaguro L., et al. Severe COVID-19 is marked by a dysregulated myeloid cell compartment. Cell. 2020;182:1419–1440.e23. doi: 10.1016/j.cell.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aschenbrenner A.C., Mouktaroudi M., Krämer B., Oestreich M., Antonakos N., Nuesch-Germano M., Gkizeli K., Bonaguro L., Reusch N., Baßler K., et al. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome Med. 2021;13:7. doi: 10.1186/s13073-020-00823-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zenarruzabeitia O., Astarloa-Pando G., Terrén I., Orrantia A., Pérez-Garay R., Seijas-Betolaza I., Nieto-Arana J., Imaz-Ayo N., Pérez-Fernández S., Arana-Arri E., Borrego F. T cell activation, highly armed cytotoxic cells and a shift in monocytes CD300 receptors expression is characteristic of patients with severe COVID-19. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.655934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reusch N., De Domenico E., Bonaguro L., Schulte-Schrepping J., Baßler K., Schultze J.L., Aschenbrenner A.C. Neutrophils in COVID-19. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.652470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ackermann M., Anders H.J., Bilyy R., Bowlin G.L., Daniel C., De Lorenzo R., Egeblad M., Henneck T., Hidalgo A., Hoffmann M., et al. Patients with COVID-19: in the dark-NETs of neutrophils. Cell Death Differ. 2021;28:3125–3139. doi: 10.1038/s41418-021-00805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meizlish M.L., Pine A.B., Bishai J.D., Goshua G., Nadelmann E.R., Simonov M., Chang C.H., Zhang H., Shallow M., Bahel P., et al. A neutrophil activation signature predicts critical illness and mortality in COVID-19. Blood Adv. 2021;5:1164–1177. doi: 10.1182/bloodadvances.2020003568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilk A.J., Lee M.J., Wei B., Parks B., Pi R., Martínez-Colón G.J., Ranganath T., Zhao N.Q., Taylor S., Becker W., et al. Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19. J. Exp. Med. 2021;218:e20210582. doi: 10.1084/jem.20210582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Powell R.L.R., Fox A., Itri V., Zolla-Pazner S. Primary human neutrophils exhibit a unique HIV-directed antibody-dependent phagocytosis profile. J. Innate Immun. 2019;11:181–190. doi: 10.1159/000494371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Worley M.J., Fei K., Lopez-Denman A.J., Kelleher A.D., Kent S.J., Chung A.W. Neutrophils mediate HIV-specific antibody-dependent phagocytosis and ADCC. J. Immunol. Methods. 2018;457:41–52. doi: 10.1016/j.jim.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 18.Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D.S., Weinrauch Y., Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 19.Takei H., Araki A., Watanabe H., Ichinose A., Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996;59:229–240. doi: 10.1002/jlb.59.2.229. [DOI] [PubMed] [Google Scholar]
- 20.Uribe-Querol E., Rosales C. Our current understanding of a universal biological process. Front. Immunol. 2020;11:1066. doi: 10.3389/fimmu.2020.01066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Middleton E.A., He X.Y., Denorme F., Campbell R.A., Ng D., Salvatore S.P., Mostyka M., Baxter-Stoltzfus A., Borczuk A.C., Loda M., et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. 2020;136:1169–1179. doi: 10.1182/blood.2020007008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blasco A., Coronado M.J., Hernández-Terciado F., Martín P., Royuela A., Ramil E., García D., Goicolea J., Del Trigo M., Ortega J., et al. Assessment of neutrophil extracellular traps in coronary thrombus of a case series of patients with COVID-19 and myocardial infarction. JAMA Cardiol. 2021;6:469. doi: 10.1001/jamacardio.2020.7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filbin M.R., Mehta A., Schneider A.M., Kays K.R., Guess J.R., Gentili M., Fenyves B.G., Charland N.C., Gonye A.L., Gushterova I., et al. Longitudinal proteomic analysis of plasma from patients with severe COVID-19 reveal patient survival-associated signatures, tissue-specific cell death, and cell-cell interactions. Cell Rep. Med. 2021;2 doi: 10.1016/j.xcrm.2021.100287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Graubert A., Aguet F., Ravi A., Ardlie K.G., Getz G. RNA-SeQC 2: efficient RNA-seq quality control and quantification for large cohorts. Bioinformatics. 2021;37:3048–3050. doi: 10.1093/bioinformatics/btab135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown E.P., Licht A.F., Dugast A.S., Choi I., Bailey-Kellogg C., Alter G., Ackerman M.E. High-throughput, multiplexed IgG subclassing of antigen-specific antibodies from clinical samples. J. Immunol. Methods. 2012;386:117–123. doi: 10.1016/j.jim.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown E.P., Dowell K.G., Boesch A.W., Normandin E., Mahan A.E., Chu T., Barouch D.H., Bailey-Kellogg C., Alter G., Ackerman M.E. Multiplexed Fc array for evaluation of antigen-specific antibody effector profiles. J. Immunol. Methods. 2017;443:33–44. doi: 10.1016/j.jim.2017.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaplonek P., Wang C., Bartsch Y., Fischinger S., Gorman M.J., Bowman K., Kang J., Dayal D., Martin P., Nowak R.P., et al. Early cross-coronavirus reactive signatures of humoral immunity against COVID-19. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abj2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciccullo A., Borghetti A., Zileri Dal Verme L., Tosoni A., Lombardi F., Garcovich M., Biscetti F., Montalto M., Cauda R., Di Giambenedetto S., GEMELLI AGAINST COVID Group Neutrophil-to-lymphocyte ratio and clinical outcome in COVID-19: a report from the Italian front line. Int. J. Antimicrob. Agents. 2020;56 doi: 10.1016/j.ijantimicag.2020.106017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J., Liu Y., Xiang P., Pu L., Xiong H., Li C., Zhang M., Tan J., Xu Y., Song R., et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J. Transl. Med. 2020;18:206–212. doi: 10.1186/s12967-020-02374-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Long L., Zeng X., Zhang X., Xiao W., Guo E., Zhan W., Yang X., Li C., Wu C., Xu T., et al. Short-term outcomes of COVID-19 and risk factors for progression. Eur. Respir. J. 2020;55:2000990. doi: 10.1183/13993003.00990-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin C., Zhou L., Hu Z., Zhang S., Yang S., Tao Y., Xie C., Ma K., Shang K., Wang W., Tian D.S. Dysregulation of immune response in patients with coronavirus 2019 (COVID-19) in wuhan, China. Clin. Infect. Dis. 2020;71:762–768. doi: 10.1093/cid/ciaa248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cai J., Li H., Zhang C., Chen Z., Liu H., Lei F., Qin J.J., Liu Y.M., Zhou F., Song X., et al. The neutrophil-to-lymphocyte ratio determines clinical efficacy of corticosteroid therapy in patients with COVID-19. Cell Metab. 2021;33:258–269.e3. doi: 10.1016/j.cmet.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y., Du X., Chen J., Jin Y., Peng L., Wang H.H.X., Luo M., Chen L., Zhao Y. Neutrophil-to-lymphocyte ratio as an independent risk factor for mortality in hospitalized patients with COVID-19. J. Infect. 2020;81:e6–e12. doi: 10.1016/j.jinf.2020.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andargie T.E., Tsuji N., Seifuddin F., Jang M.K., Yuen P.S., Kong H., Tunc I., Singh K., Charya A., Wilkins K., et al. Cell-free DNA maps COVID-19 tissue injury and risk of death and can cause tissue injury. JCI Insight. 2021;6:147610. doi: 10.1172/jci.insight.147610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinha S., Rosin N.L., Arora R., Labit E., Jaffer A., Cao L., Farias R., Nguyen A.P., de Almeida L.G.N., Dufour A., et al. Dexamethasone modulates immature neutrophils and interferon programming in severe COVID-19. Nat. Med. 2022;28:201–211. doi: 10.1038/s41591-021-01576-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grieshaber-Bouyer R., Radtke F.A., Cunin P., Stifano G., Levescot A., Vijaykumar B., Nelson-Maney N., Blaustein R.B., Monach P.A., Nigrovic P.A., ImmGen Consortium The neutrotime transcriptional signature defines a single continuum of neutrophils across biological compartments. Nat. Commun. 2021;12:2856–2921. doi: 10.1038/s41467-021-22973-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R., et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2018;174:1033. doi: 10.1016/j.cell.2018.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Injarabian L., Devin A., Ransac S., Marteyn B.S. Neutrophil metabolic shift during their lifecycle: impact on their survival and activation. Int. J. Mol. Sci. 2019;21:E287. doi: 10.3390/ijms21010287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reyes M., Filbin M.R., Bhattacharyya R.P., Sonny A., Mehta A., Billman K., Kays K.R., Pinilla-Vera M., Benson M.E., Cosimi L.A., et al. Plasma from patients with bacterial sepsis or severe COVID-19 induces suppressive myeloid cell production from hematopoietic progenitors in vitro. Sci. Transl. Med. 2021;13:eabe9599. doi: 10.1126/scitranslmed.abe9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zilionis R., Engblom C., Pfirschke C., Savova V., Zemmour D., Saatcioglu H.D., Krishnan I., Maroni G., Meyerovitz C.V., Kerwin C.M., et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. 2019;50:1317–1334.e10. doi: 10.1016/j.immuni.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Juss J.K., House D., Amour A., Begg M., Herre J., Storisteanu D.M.L., Hoenderdos K., Bradley G., Lennon M., Summers C., et al. Acute respiratory distress syndrome neutrophils have a distinct phenotype and are resistant to phosphoinositide 3-kinase inhibition. Am. J. Respir. Crit. Care Med. 2016;194:961–973. doi: 10.1164/rccm.201509-1818OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spinetti T., Hirzel C., Fux M., Walti L.N., Schober P., Stueber F., Luedi M.M., Schefold J.C. Reduced monocytic human leukocyte antigen-DR expression indicates immunosuppression in critically ill COVID-19 patients. Anesth. Analg. 2020;131:993–999. doi: 10.1213/ANE.0000000000005044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Major L.D., Partridge T.S., Gardner J., Kent S.J., de Rose R., Suhrbier A., Schroder W.A. Induction of SerpinB2 and Th1/Th2 modulation by SerpinB2 during lentiviral infections in vivo. PLoS One. 2013;8 doi: 10.1371/journal.pone.0057343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosa B.A., Ahmed M., Singh D.K., Choreño-Parra J.A., Cole J., Jiménez-Álvarez L.A., Rodríguez-Reyna T.S., Singh B., Gonzalez O., Carrion R., Jr., et al. IFN signaling and neutrophil degranulation transcriptional signatures are induced during SARS-CoV-2 infection. Commun. Biol. 2021;4:290–314. doi: 10.1038/s42003-021-01829-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuo Y., Yalavarthi S., Shi H., Gockman K., Zuo M., Madison J.A., Blair C., Weber A., Barnes B.J., Egeblad M., et al. Neutrophil extracellular traps in COVID-19. JCI Insight. 2020;5:138999. doi: 10.1172/jci.insight.138999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veras F.P., Pontelli M.C., Silva C.M., Toller-Kawahisa J.E., de Lima M., Nascimento D.C., Schneider A.H., Caetité D., Tavares L.A., Paiva I.M., et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020;217:e20201129. doi: 10.1084/jem.20201129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arcanjo A., Logullo J., Menezes C.C.B., de Souza Carvalho Giangiarulo T.C., Dos Reis M.C., de Castro G.M.M., da Silva Fontes Y., Todeschini A.R., Freire-de-Lima L., Decoté-Ricardo D., et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19) Sci. Rep. 2020;10 doi: 10.1038/s41598-020-76781-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mariana J., Kaplan M.R. Neutrophil extracellular traps (NETs): double-edged swords of innate immunity. J. Immunol. 2012;189:2689. doi: 10.4049/jimmunol.1201719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mukhopadhyay S., Sinha S., Mohapatra S.K. Analysis of transcriptomic data sets supports the role of IL-6 in NETosis and immunothrombosis in severe COVID-19. BMC Genom. Data. 2021;22:49. doi: 10.1186/s12863-021-01001-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hamam H.J., Palaniyar N. Post-translational modifications in NETosis and NETs-mediated diseases. Biomolecules. 2019;9:E369. doi: 10.3390/biom9080369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kimura Y., Nakai Y., Shin J., Hara M., Takeda Y., Kubo S., Jeremiah S.S., Ino Y., Akiyama T., Moriyama K., et al. Identification of serum prognostic biomarkers of severe COVID-19 using a quantitative proteomic approach. Sci. Rep. 2021;11:20638. doi: 10.1038/s41598-021-98253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu P., Chen D., Ding W., Wu P., Hou H., Bai Y., Zhou Y., Li K., Xiang S., Liu P., et al. The trans-omics landscape of COVID-19. Nat. Commun. 2021;12:4543. doi: 10.1038/s41467-021-24482-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bauer W., Weber M., Diehl-Wiesenecker E., Galtung N., Prpic M., Somasundaram R., Tauber R., Schwenk J.M., Micke P., Kappert K. Plasma proteome fingerprints reveal distinctiveness and clinical outcome of SARS-CoV-2 infection. Viruses. 2021;13:2456. doi: 10.3390/v13122456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krishnan S., Nordqvist H., Ambikan A.T., Gupta S., Sperk M., Svensson-Akusjärvi S., Mikaeloff F., Benfeitas R., Saccon E., Ponnan S.M., et al. Metabolic perturbation associated with COVID-19 disease severity and SARS-CoV-2 replication. Mol. Cell. Proteomics. 2021;20:100159. doi: 10.1016/j.mcpro.2021.100159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lacy P. Mechanisms of degranulation in neutrophils. Allergy Asthma Clin. Immunol. 2006;2:98–108. doi: 10.1186/1710-1492-2-3-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aarts C.E.M., Hiemstra I.H., Tool A.T.J., van den Berg T.K., Mul E., van Bruggen R., Kuijpers T.W. Neutrophils as suppressors of T cell proliferation: does age matter? Front. Immunol. 2019;10:2144. doi: 10.3389/fimmu.2019.02144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Darcy C.J., Minigo G., Piera K.A., Davis J.S., McNeil Y.R., Chen Y., Volkheimer A.D., Weinberg J.B., Anstey N.M., Woodberry T. Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit. Care. 2014;18:R163. doi: 10.1186/cc14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vonwirth V., Bülbül Y., Werner A., Echchannaoui H., Windschmitt J., Habermeier A., Ioannidis S., Shin N., Conradi R., Bros M., et al. Inhibition of arginase 1 liberates potent T cell immunostimulatory activity of human neutrophil granulocytes. Front. Immunol. 2020;11 doi: 10.3389/fimmu.2020.617699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaltenmeier C., Yazdani H.O., Morder K., Geller D.A., Simmons R.L., Tohme S. Neutrophil extracellular traps promote T cell exhaustion in the tumor microenvironment. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.785222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kobayashi S.D., Malachowa N., DeLeo F.R. Neutrophils and bacterial immune evasion. J. Innate Immun. 2018;10:432–441. doi: 10.1159/000487756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y., Schneider A.M., Mehta A., Sade-Feldman M., Kays K.R., Gentili M., Charland N.C., Gonye A.L., Gushterova I., Khanna H.K., et al. SARS-CoV-2 viremia is associated with distinct proteomic pathways and predicts COVID-19 outcomes. J. Clin. Invest. 2021;131:148635. doi: 10.1172/JCI148635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chung A.W., Alter G. Systems serology: profiling vaccine induced humoral immunity against HIV. Retrovirology. 2017;14:57. doi: 10.1186/s12977-017-0380-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karsten C.B., Mehta N., Shin S.A., Diefenbach T.J., Slein M.D., Karpinski W., Irvine E.B., Broge T., Suscovich T.J., Alter G. A versatile high-throughput assay to characterize antibody-mediated neutrophil phagocytosis. J. Immunol. Methods. 2019;471:46–56. doi: 10.1016/j.jim.2019.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swystun L.L., Liaw P.C. The role of leukocytes in thrombosis. Blood. 2016;128:753–762. doi: 10.1182/blood-2016-05-718114. [DOI] [PubMed] [Google Scholar]
- 66.Stacey H.D., Golubeva D., Posca A., Ang J.C., Novakowski K.E., Zahoor M.A., Kaushic C., Cairns E., Bowdish D.M.E., Mullarkey C.E., Miller M.S. IgA potentiates NETosis in response to viral infection. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2101497118. e2101497118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anthony R.M., Nimmerjahn F. The role of differential IgG glycosylation in the interaction of antibodies with FcγRs in vivo. Curr. Opin. Organ Transplant. 2011;16:7–14. doi: 10.1097/MOT.0b013e328342538f. [DOI] [PubMed] [Google Scholar]
- 68.Chakraborty S., Gonzalez J.C., Sievers B.L., Mallajosyula V., Chakraborty S., Dubey M., Ashraf U., Cheng B.Y.L., Kathale N., Tran K.Q.T., et al. Early non-neutralizing, afucosylated antibody responses are associated with COVID-19 severity. Sci. Transl. Med. 2022;14:eabm7853. doi: 10.1126/scitranslmed.abm7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mayadas T.N., Cullere X., Lowell C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014;9:181–218. doi: 10.1146/annurev-pathol-020712-164023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chakraborty S., Gonzalez J., Edwards K., Mallajosyula V., Buzzanco A.S., Sherwood R., Buffone C., Kathale N., Providenza S., Xie M.M., et al. Proinflammatory IgG Fc structures in patients with severe COVID-19. Nat. Immunol. 2021;22:67–73. doi: 10.1038/s41590-020-00828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Najder K., Rugi M., Lebel M., Schröder J., Oster L., Schimmelpfennig S., Sargin S., Pethő Z., Bulk E., Schwab A. Role of the intracellular sodium homeostasis in chemotaxis of activated murine neutrophils. Front. Immunol. 2020;11:2124. doi: 10.3389/fimmu.2020.02124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Santoni K., Pericat D., Gorse L., Buyck J., Pinilla M., Prouvensier L., Bagayoko S., Hessel A., Leon-Icaza S.A., Bellard E., et al. Caspase-1-driven neutrophil pyroptosis and its role in host susceptibility to Pseudomonas aeruginosa. PLoS Pathog. 2022;18 doi: 10.1371/journal.ppat.1010305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Naegelen I., Beaume N., Plançon S., Schenten V., Tschirhart E.J., Bréchard S. Regulation of neutrophil degranulation and cytokine secretion: a novel model approach based on linear fitting. J. Immunol. Res. 2015;2015:817038. doi: 10.1155/2015/817038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Borsutzky S., Cazac B.B., Roes J., Guzmán C.A. TGF-beta receptor signaling is critical for mucosal IgA responses. J. Immunol. 2004;173:3305–3309. doi: 10.4049/jimmunol.173.5.3305. [DOI] [PubMed] [Google Scholar]
- 75.Witkowski M., Tizian C., Ferreira-Gomes M., Niemeyer D., Jones T.C., Heinrich F., Frischbutter S., Angermair S., Hohnstein T., Mattiola I., et al. Untimely TGFβ responses in COVID-19 limit antiviral functions of NK cells. Nature. 2021;600:295–301. doi: 10.1038/s41586-021-04142-6. [DOI] [PubMed] [Google Scholar]
- 76.Donnelly R.P., Kotenko S.V. Interferon-lambda: a new addition to an old family. J. Interferon Cytokine Res. 2010;30:555–564. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lim A., Radujkovic A., Weigand M.A., Merle U. Soluble receptor for advanced glycation end products (sRAGE) as a biomarker of COVID-19 disease severity and indicator of the need for mechanical ventilation, ARDS and mortality. Ann. Intensive Care. 2021;11:1–13. doi: 10.1186/s13613-021-00836-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haljasmägi L., Salumets A., Rumm A.P., Jürgenson M., Krassohhina E., Remm A., Sein H., Kareinen L., Vapalahti O., Sironen T., et al. Longitudinal proteomic profiling reveals increased early inflammation and sustained apoptosis proteins in severe COVID-19. Sci. Rep. 2020;10:20533. doi: 10.1038/s41598-020-77525-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Su Y., Chen D., Yuan D., Lausted C., Choi J., Dai C.L., Voillet V., Duvvuri V.R., Scherler K., Troisch P., et al. Multi-Omics resolves a sharp disease-state shift between mild and moderate COVID-19. Cell. 2020;183:1479–1495.e20. doi: 10.1016/j.cell.2020.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shu T., Ning W., Wu D., Xu J., Han Q., Huang M., Zou X., Yang Q., Yuan Y., Bie Y., et al. Plasma proteomics identify biomarkers and pathogenesis of COVID-19. Immunity. 2020;53:1108–1122.e5. doi: 10.1016/j.immuni.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Y., Fear D.J., Willis-Owen S.A.G., Cookson W.O., Moffatt M.F. Global gene regulation during activation of immunoglobulin class switching in human B cells. Sci. Rep. 2016;6:37988. doi: 10.1038/srep37988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abraham E., Gyetko M.R., Kuhn K., Arcaroli J., Strassheim D., Park J.S., Shetty S., Idell S. Urokinase-type plasminogen activator potentiates lipopolysaccharide-induced neutrophil activation. J. Immunol. 2003;170:5644–5651. doi: 10.4049/jimmunol.170.11.5644. [DOI] [PubMed] [Google Scholar]
- 83.Wendisch D., Dietrich O., Mari T., von Stillfried S., Ibarra I.L., Mittermaier M., Mache C., Chua R.L., Knoll R., Timm S., et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell. 2021;184:6243–6261.e27. doi: 10.1016/j.cell.2021.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Russell M.W., Moldoveanu Z., Ogra P.L., Mestecky J. Mucosal immunity in COVID-19: a neglected but critical aspect of SARS-CoV-2 infection. Front. Immunol. 2020;11:611337. doi: 10.3389/fimmu.2020.611337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Masso-Silva J.A., Moshensky A., Lam M.T.Y., Odish M., Patel A., Xu L., Hansen E., Trescott S., Nguyen C., Kim R., et al. Increased peripheral blood neutrophil activation phenotypes and NETosis in critically ill COVID-19 patients: a case series and review of the literature. Clin. Infect. Dis. 2021 doi: 10.1093/cid/ciab437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Skendros P., Mitsios A., Chrysanthopoulou A., Mastellos D.C., Metallidis S., Rafailidis P., Ntinopoulou M., Sertaridou E., Tsironidou V., Tsigalou C., et al. Complement and tissue factor–enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Invest. 2020;130:6151–6157. doi: 10.1172/JCI141374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mutua V., Gershwin L.J. A review of neutrophil extracellular traps (NETs) in disease: potential anti-NETs therapeutics. Clin. Rev. Allergy Immunol. 2021;61:194–211. doi: 10.1007/s12016-020-08804-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kaneko N., Kuo H.H., Boucau J., Farmer J.R., Allard-Chamard H., Mahajan V.S., Piechocka-Trocha A., Lefteri K., Osborn M., Bals J., et al. The loss of bcl-6 expressing T follicular helper cells and the absence of germinal centers in COVID-19. SSRN. 2020 doi: 10.1016/j.cell.2020.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brito C.A., Paiva J.G., Pimentel F.N., Guimarães R.S., Moreira M.R. COVID-19 in patients with rheumatological diseases treated with anti-TNF. Ann. Rheum. Dis. 2021;80:e62. doi: 10.1136/annrheumdis-2020-218171. [DOI] [PubMed] [Google Scholar]
- 90.Kridin K., Schonmann Y., Damiani G., Peretz A., Onn E., Bitan D.T., Cohen A.D. Tumor necrosis factor inhibitors are associated with a decreased risk of COVID-19-associated hospitalization in patients with psoriasis-A population-based cohort study. Dermatol. Ther. 2021;34 doi: 10.1111/dth.15003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brenner E.J., Ungaro R.C., Gearry R.B., Kaplan G.G., Kissous-Hunt M., Lewis J.D., Ng S.C., Rahier J.F., Reinisch W., Ruemmele F.M., et al. Corticosteroids, but not TNF antagonists, are associated with adverse COVID-19 outcomes in patients with inflammatory bowel diseases: results from an international registry. Gastroenterology. 2020:481–491.e3. doi: 10.1053/j.gastro.2020.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Uhlen M., Karlsson M.J., Zhong W., Tebani A., Pou C., Mikes J., Lakshmikanth T., Forsström B., Edfors F., Odeberg J., et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science. 2019;366:eaax9198. doi: 10.1126/science.aax9198. [DOI] [PubMed] [Google Scholar]
- 93.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Korotkevich G., Sukhov V., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2016 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- 97.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Picelli S., Björklund Å.K., Faridani O.R., Sagasser S., Winberg G., Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat. Methods. 2013;10:1096–1098. doi: 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 99.Villani A.-C., Satija R., Reynolds G., Sarkizova S., Shekhar K., Fletcher J., Griesbeck M., Butler A., Zheng S., Lazo S., et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. 2017;356:eaah4573. doi: 10.1126/science.aah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Guan W.-J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S.C., et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020;382:1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Puram S.V., Tirosh I., Parikh A.S., Patel A.P., Yizhak K., Gillespie S., Rodman C., Luo C.L., Mroz E.A., Emerick K.S., et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171:1611–1624.e24. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kim J., Kwiatkowski D., McConkey D.J., Meeks J.J., Freeman S.S., Bellmunt J., Getz G., Lerner S.P. The cancer genome atlas expression subtypes stratify response to checkpoint inhibition in advanced urothelial cancer and identify a subset of patients with high survival probability. Eur. Urol. 2019;75:961–964. doi: 10.1016/j.eururo.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 103.Freeman S.S., Sade-Feldman M., Kim J., Stewart C., Gonye A.L.K., Ravi A., Arniella M.B., Gushterova I., LaSalle T.J., Blaum E.M., et al. Combined tumor and immune signals from genomes or transcriptomes predict outcomes of checkpoint inhibition in melanoma. Cell Rep. Med. 2022;3 doi: 10.1016/j.xcrm.2021.100500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ramilowski J.A., Goldberg T., Harshbarger J., Kloppmann E., Kloppman E., Lizio M., Satagopam V.P., Itoh M., Kawaji H., Carninci P., et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2015;6:7866. doi: 10.1038/ncomms8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw RNA sequencing data reported in this study cannot be deposited in a public repository because these data were collected at the beginning of the COVID-19 pandemic, and as such, a waiver of informed consent was approved by the Massachusetts General Hospital governing institutional review board, in compliance with the Code of Federal Regulation (45CFR 46, 2018 Common Rule). To protect the identity of individual subjects, public posting of raw sequencing data from the patients has not been approved; therefore, raw data is not provided. However, the read count matrix and TPM matrix used in this study are available in GEO under accession number GSE212041 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE212041). All code required to run the analyses in this manuscript is deposited in Zenodo (https://doi.org/10.5281/zenodo.7030528) based on the associated Github repository (https://github.com/lasalletj/COVID_Neutrophils). Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.