

Abstract
Metastatic breast cancer causes most breast cancer-associated deaths, especially in triple negative breast cancers (TNBC). The metastatic drivers of TNBCs are still poorly understood, and effective treatment non-existent. Here we reveal that the presence of Aurora-A Kinase (AURKA) in the nucleus and metastatic dissemination are molecularly connected through HIF1 (Hypoxia-Inducible Factor-1) signaling. Nuclear AURKA activates transcription of “hypoxia-induced genes” under normoxic conditions (pseudohypoxia) and without upregulation of oxygen-sensitive HIF1A subunit. We uncover that AURKA preferentially binds to HIF1B and co-localizes with the HIF complex on DNA. The mass-spectrometry analysis of the AURKA complex further confirmed the presence of CBP and p300 along with other TFIIB/RNApol II components. Importantly, the expression of multiple HIF-dependent genes induced by nuclear AURKA (N-AURKA), including migration/invasion, survival/death, and stemness, promote early cancer dissemination. These results indicate that nuclear, but not cytoplasmic, AURKA is a novel driver of early metastasis. Analysis of clinical tumor specimens revealed a correlation between N-AURKA presence and decreased patient survival. Our results establish a mechanistic link between two critical pathways in cancer metastasis, identifying nuclear AURKA as a crucial upstream regulator of the HIF1 transcription complex and a target for anti-metastatic therapy.
INTRODUCTION
AURKA is a serine/threonine kinase responsible for centrosome maturation, separation, and spindle formation during mitosis [1]. Normal localization of AURKA is at the centrosomes/cytoplasm [2]. AURKA is also involved in the regulation of migration and invasion [3].
AURKA has recently been reported to localize in the nucleus in various cancers, such as colon [4], lung [5], and breast [6]. Few reports demonstrate the oncogenic potential of nuclear (N-AURKA), but not cytoplasmic, AURKA in cooperation with Ras [7] and Myc [6] oncogenes. AURKA phosphorylates multiple nuclear proteins that promote carcinogenesis, including p53 [8], Akt [9], and NFkB [10], suggesting that these might be targets of nuclear AURKA.
High expression of AURKA was shown to correlate with earlier recurrence of TNBC in patients [11] and indicative of disease progression. The increased AURKA expression was established to promote metastasis by enhancing epithelial to mesenchymal transition [12], stemness [6], and invasion [3]. Although expression of AURKA in the nucleus correlates with poor overall survival [13], the mechanism driving cancer progression associated with N-AURKA is currently unknown.
Hypoxia is a physical state of lower oxygen compared to normal oxygen levels (normoxia). When a cell experiences hypoxia, the hypoxia-inducible factors (HIFs) consisting of alpha/beta subunits (HIF1/2A, HIF1/2B) are stabilized. The HIFA/B heterodimer then translocates into the nucleus, where it binds to DNA on hypoxia-responsive elements (HREs) [14], regulating transcription of genes involved in proliferation, apoptosis, angiogenesis, and metabolism [15]. The association between HIF1 expression, hypoxia genes/signaling, and metastatic breast cancer has been previously documented [15–17].
Here we report, that N-AURKA via binding to HIF complexes activates hypoxia transcriptional programs independent of HIF1/2A stabilization in cells exposed to atmospheric oxygen [18]. Transactivation of hypoxia genes leads to increased migration/invasion and stemness, enabling dissemination and metastases at distant organs without significant delay or dormancy. Inhibiting N-AURKA kinase activity with Alisertib/MLN8237, AURKA-specific inhibitor, significantly reduces metastatic colonization but does not block dissemination. Hence, using nuclear AURKA-targeting compounds in combination with HIF inhibitors might prove beneficial in treating metastases in TNBCs and other cancers. The presence of cells with N-AURKA could serve as a surrogate for metastatic cells and indicate early dissemination.
RESULTS
Nuclear localization of AURKA correlates with disease progression and metastasis
AURKA overexpression in TCGA BC cohort correlates with poorer overall survival (Supplementary Fig. 1A–C), but this dataset does not allow stratifying patients based on subcellular localization of AURKA. To fill this gap, a BC tissue microarray with over 200 patient samples (Supplementary Table S3) was analyzed for AURKA localization and correlated with pathological stage and subtype (Fig. 1A–C). The positivity of normal tissue and ductal carcinoma in-situ (DCIS) for N-AURKA is 0–3%. There is a ten-fold increase in positivity in invasive ductal (IDC), lobular (ILC) carcinomas, and metastases (MIDC-LN) (Fig. 1A, B). The TNBC and HER2+ tumors show up to 12–20% of cells in the tumors with N-AURKA compared to 5% of ER/PR+ cases (Fig. 1C). To evaluate the association between N-AURKA positivity in clinical samples and metastasis, we have procured a set of matched breast tumor-metastases biopsies along with biopsies from patients who had no metastases at the time of biopsy (IRB#WVU011113, Supplementary Fig. 1D, Supplementary Table S4). Spearman’s rank correlation coefficient analysis indicated a strong correlation between N-AURKA positivity and metastatic outcomes (Supplementary Fig. 1E, r = 0.8051, p < 0.0001). Eleven of these biopsies were previously used to produce patient-derived xenograft (PDX) models [19]. Similar to the original biopsy, the low/negative N-AURKA PDXs (PEN_060, PEN_061, PEN_175, PEN_181, PEN_025) had no distant metastases, while N-AURKA high (PEN_014, PEN_056, PEN_76, PEN_116) developed metastases (Supplementary Table S4), indicating that N-AURKA correlates with metastasis. The AURKA localization/expression in BC cell lines shows a correlation between nuclear AURKA positivity and metastatic potential of the cells (Fig. 1D, E, Supplementary Fig. 1F, G).
Fig. 1. Nuclear AURKA positivity correlates with metastasis and more aggressive breast cancer subtypes.
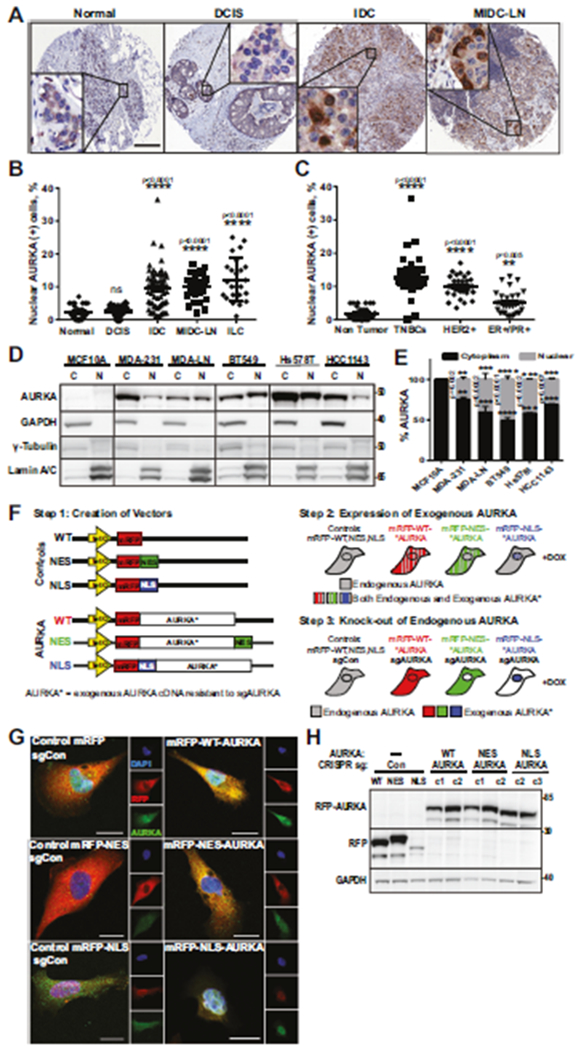
A. Representative images of tissue microarray IHC, (n = 206) stained with AURKA/DAB-brown, hematoxylin-nuclei/blue. Scale bar-300 μm. Insets-x250 enlarged areas. B, C Quantification N-AURKA positive(+) cells as in A, 3 randomly-assigned fields, n = 100 cells/field. B Pathological stages (Normal = 32, DCIS = 24, IDC = 72, MIDC-LN = 32, ILC = 24) and C receptor-based subtypes (Normal = 32, TNBC = 39, HER2+ = 32, ER+/PR+ = 30). D WB analysis of nuclear/cytoplasmic fractionations, as indicated. E Quantification of AURKA in cytoplasm/nucleus, percent-of-total, normalized to controls. F Schematic outline of cell line production. G, H Immunofluorescence and WB analysis of AURKA-sublines produced in F stained with AURKA (green), RFP(red), DAPI-nuclei/blue; clones indicated as c1/c2/c3. Scale bar-10 μm. One-way ANOVA, ±S.E.M, Dunnett’s test, ns non-significant.
N-AURKA drives cancer migration/invasion but does not affect proliferation
To define the impact of N-AURKA on metastasis, cells with nuclear or cytoplasmic AURKA were produced. The cell lines (MDA-MB-231 and BT549) were made using CRISPR/Cas9 to knockout endogenous AURKA and replace with exogenous AURKA targeted to the nucleus (NLS), cytoplasm (NES), or no-localization signal (WT) (Fig. 1F–H, Supplementary Methods). The cells expressing non-targeting sgRNAs and empty vectors were used as controls. In vitro cell proliferation/viability assay and mixed modeling analysis show no statistically significant differences in cell growth or death between NLS- and NES-AURKA cells (Fig. 2A). The morphometric analysis of NLS-AURKA cells shows reduced cell elongation index and nuclei (Fig. 2B–D). The 3D invasion assay in collagen shows that NLS-AURKA cells travel a significantly longer distance and are faster than other cells (Fig. 2E–H). In agreement with previously published reports, the MDA-MB-231 cell viability was not compromised due to attachment loss (Fig. 2I). Interestingly, all cells expressing exogenous AURKA were more resistant to anoikis, but no differences were observed between sublines. The analysis of the number and size of multi-cellular clusters at 48 h post-seeding shows a significant decrease in the clumping/clustering potential of NLS-AURKA cells without effecting cell viability (Fig. 2J, K), suggesting that clumping may not be necessary for the survival of these cells. These results indicate that cells with N-AURKA are highly invasive but do not possess enhanced proliferation or anchorage-independent capabilities.
Fig. 2. N-AURKA drives cancer migration/invasion but does not affect proliferation.
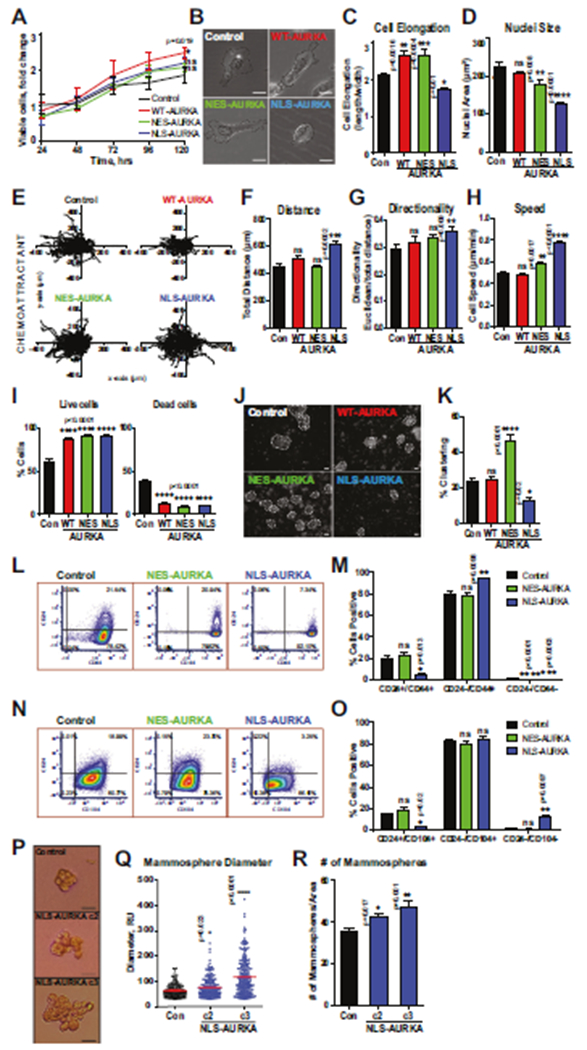
A Cell proliferation/viability assay, mixed-effect model analysis on cell growth. B Representative bright-field images of cells, scale bar-10 μm. C Cell elongation index and D nuclei size. E Individual cell movement tracking-plots toward chemoattractant. Graphs of F total distance, G cell-body directionality, H cell speed. I Quantification of anoikis (live/dead cells). J Representative bright-field images of clusters, scale bar-50 μm, K Quantification of clusters as in J; 3–6 randomly-assigned fields/per clone normalized to area, ≥ 40cells/clone. L–O Representative flow cytometry dot-plots and quantification of CD24/CD44/CD104 positive cells. P Representative images of mammospheres. Scale bar-50 μm. Mammosphere quantification of Q diameter, R # mammospheres/area. One-way ANOVA, ±S.E.M, Dunnett’s test, ns non-significant.
N-AURKA drives cancer cell stemness
The cell-surface antigens, CD24, CD44, and CD104, are used as cancer stem cell (CSC) markers [20, 21]. Similar to previous reports, there is a decrease in the CD24+/CD44+, CD24+/CD104+ cells and a significant increase in CD24−/CD44+, CD24−/CD104− cells in NLS-AURKA cells, showing a shift to more stem-like (Fig. 2L–O). A decrease was observed in the number of stained cells for CD24 and the mean fluorescent intensity (Supplementary Fig. 2A), while CD44 mean fluorescent intensity was increased (Supplementary Fig. 2B, C). Previously, FOXM1 transcription factor was reported to mediate the AURKA driven stemness [22]. The analysis of stem cell markers shows no changes in CD24, CD44, or CD104 upon depletion of FOXM1 (Supplementary Fig. 2D–H), suggesting that this activity of N-AURKA is FOXM1 independent. Similarly, analysis of mammosphere formation shows a significant increase in the number and diameter in NLS-AURKA cells (Fig. 2P–R). Still, this activity was not affected by the depletion of FOXM1 (Supplementary Fig. 2I).
N-AURKA induces hypoxia in xenograft models of breast cancer
To evaluate the effect of N-AURKA on tumor invasion and metastasis in vivo, the cells were injected into the mammary gland of immunocompromised mice (Fig. 3A). There were no significant differences in the growth of mammary tumors between groups (Fig. 3B, Supplementary Fig. 3A, B). Even though tumors were of similar size/volume, there was a significant difference in tumor necrosis and vascularization (Fig. 3C–G). Immunohistochemistry analysis of the tumors showed no significant difference in proliferation (Ki67) (Supplementary Fig. 3C–E). The NLS-AURKA group showed a significant decrease in CD31 staining compared to the control (Fig. 3F, G). To assess oxygenation of tumors, the hypoxia marker-pimonidazole was injected in mice before collection. Tumor analysis shows that over 40% of NLS-AURKA tumors were hypoxic (Fig. 3H, I).
Fig. 3. N-AURKA induces hypoxia in xenograft models of breast cancer.
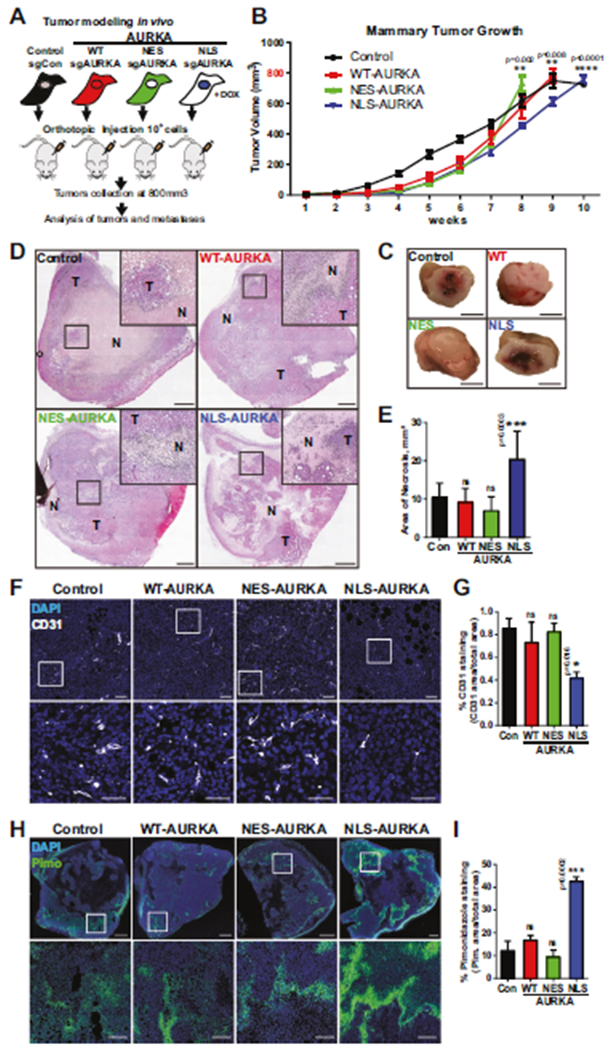
A Experimental design of xenograft study, n = 2 clones/subline, n ≥ 5 mice/group, n = 2 experiments. B Quantification of ultrasound-based tumor volume (mm3) plotted over time. Mixed-effect model analysis. C Representative images of gross tumor pathology, scale bar-5 mm. D Tumor H&E images, inset-necrotic area, scale bar-1 mm. E Quantification of necrosis/area in tumors. F Representative tumor images of F-IHC staining with CD31, scale bar-100 μm(top image), 50 μm(inset). G Quantification of CD31(+) staining area (%) as in F normalized to total area, DAPI/nuclei(blue), CD31(white). H Representative tumor images of F-IHC staining with anti-Pimonidazole (Hypoxyprobe-FITC/green), scale bar-1 mm(top image), 100 μm(inset). I Quantification of Pimonidazole (+) staining area (%) as in H normalized to total area, DAPI/nuclei(blue). One-way ANOVA, ±S.E.M, Dunnett’s test, ns non-significant.
N-AURKA expressing tumors are highly metastatic
The induction of hypoxia often results in metastasis [23]. The analysis of distant organs shows over ten-fold increase in the number and size of metastases in mice bearing NLS-AURKA tumors compared to other groups (Fig. 4A–E, Supplementary Fig. 4A, B). The bone and liver metastases were present in all NLS-AURKA mice (100% penetrance rate) (Fig. 4C), suggesting that NLS-AURKA cells have an increased proficiency to disseminate/grow at multiple distant sites.
Fig. 4. N-AURKA expressing tumors are highly metastatic.

A Representative DAB-IHC images of Lymph Node (LN) metastases stained with anti-RFP-antibody. Scale bar-100 μm. B Representative H&E images of lung and liver metastases; femur/sternum bone metastases stained with human-specific anti-mitochondria antibody. Metastases outlined in black. Scale bar-50 μm(lung), 200 μm(liver), 500 μm(bone). C Quantification of metastatic penetrance, as percent of mice which developed LN, lung, liver, and bone metastases. D Quantification of number of metastases/area of lung, liver, or bone. E Quantification of size of LN and lung metastases. One-way ANOVA, ±S.E.M, Dunnett’s test, ns non-significant.
N-AURKA induces hypoxia signaling via upregulation of HIF-responsive genes
To identify the processes responsible for the increased metastatic potential, the gene expression profiles of cells were generated (GSE154494). The presence of AURKA in the nucleus was associated with a global transcriptional response (Fig. 5A). Principal component analysis of RNA-seq data separated the samples into three groups (Fig. 5B) with a significant number of genes differentially expressed in NLS-AURKA cells (Fig. 5C, Supplementary Fig. 5A, B). The expression of some targets was evaluated by western blotting and qPCR in MDA-MB-231 and BT-549 cells (Fig. 5E, F, Supplementary Fig. 5D–F). The N-AURKA gene-signature was confirmed by qPCR in tumors showing a similar gene expression profile (Supplementary Fig. 5G–I). The Gene Ontology terms analyzed with the REViGO tool show that NLS-AURKA upregulates genes related to differentiation, motility/adhesion, hypoxia/oxygen, and protein phosphorylation (Fig. 5D, Supplementary Fig. 5C). Gene Set Enrichment Analysis (GSEA) showed enrichment for genes involved in mammary stem cell development, EMT, and hypoxia (Fig. 5G). The NLS-AURKA cells show upregulation of genes associated with the hypoxia-response, such as CXCR4, MMP1, ANGPT1, ARNT/HIF1B, MMP14, ITGA6/CD49f, and ARNT2/HIF2B (Fig. 5H). The IPA analysis of RNA-seq identified multiple upstream regulators of hypoxia signaling, including Hypoxia-Inducible Factor-1 alpha (HIF1A), NFκB, and tumor necrosis factor (TNF) to be activated in NLS-AURKA cells (Fig. 5I). The N-AURKA-driven hypoxia gene signature correlates with a worse prognosis and high risk of relapse (Supplementary Fig. 5J, K), suggesting that hypoxia genes identified in our study influence the disease progression.
Fig. 5. RNA-seq profile of N-AURKA cells.
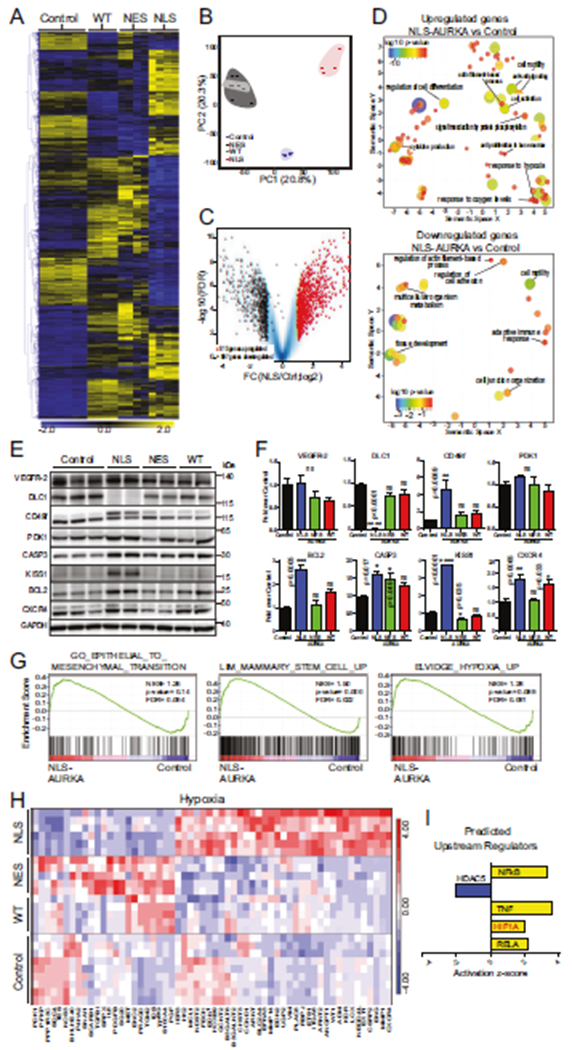
A Heat-map of differentially expressed genes (DEGs, mean values) in cells. Gene expression (n = 3167) is normalized log2 counts/million. B Principal Component Analysis of RNA-Seq libraries as in A. C Volcano-plot analysis of RNAseq data as in A NLS-AURKA vs. control, log2 fold changes of gene expression on the x-axis, FDR statistical significance (−log10 p value) on the y-axis; upregulated (red), downregulated (black) genes in NLS, all genes (blue). Genes with at least a >1.5 fold change and FDR < 0.01 are displayed. D Visualization of Gene Ontology terms for NLS-AURKA vs. control; up/downregulated GOterms (FDR < 0.1) are depicted as circles; the distance indicates the relationship between terms: closer distance means higher similarity. Color and size of circles indicate significance of differential expression of an individual GO term in log10 p value. E WB analysis of selected RNA-seq target proteins as in A. F Quantification of WB in E, fold change over control. G GSEA comparison NLS-AURKA vs. control for selected GOterm gene sets. H Heat-map of HIF-dependent DEGs. I Predicted upstream regulators: activated (yellow), inhibited (blue) using IPA activation z-score. One-way ANOVA, ±S.E.M, Dunnett’s test, ns non-significant.
N-AURKA binds to HIF1A/B and transactivates hypoxia-response genes
To understand how N-AURKA enables the transcription of hypoxia-response genes, protein expression of Hypoxia-Inducible Factors (HIFs) was examined. DMOG (dimethyloxalyl glycine) treatment was used as a positive control, leading to upregulation and stabilization of HIF1/2A. In normoxia, there were no differences in protein levels of HIF1/2A between the sublines (Fig. 6A, B, Supplementary Fig. 6A, B). The DMOG treatment led to increased HIF1A protein independently of AURKA localization. In contrast, HIF2A was not upregulated in DMOG-treated NLS-AURKA cells, suggesting differential action of AURKA on these proteins during hypoxia, but not normoxia. Consistent with RNA-seq results, a significant increase in HIF1/2B proteins was detected (Fig. 6A, B, Supplementary Fig. 6A–E). To determine if N-AURKA interacts with HIFs, we immunoprecipitated AURKA from cell lysates. The AURKA complex contained HIF1A and HIF1B proteins; NLS-AURKA expressing cells preferentially precipitated AURKA in a complex with HIF1B (Fig. 6C, D). Next, the binding of AURKA to hypoxia-response gene promoters was assessed using chromatin immunoprecipitation (ChIP) assay. The CD49f/ITGA6, EPO, and CXCR4 genes were selected as they contain Hypoxia Response Elements (HREs) [24–26]. ChIP for HIF1B was used as the positive control. The qPCR with FOXM1 promoter was used as a positive control for AURKA [22]. ChIP qPCR results show N-AURKA binding to HRE-containing promoters of specific hypoxia genes, also occupied by HIF1B (Fig. 6E, F). Other proteins that interact with N-AURKAon chromatin include hnRNPk, Aurora-B Kinase, Histone H3 and Histone H2B (Fig. 6F). To map the protein interaction network of N-AURKA, we carried out LC-MS/MS analysis of AURKA-IP complex. The identified proteins were clustered based on the cellular compartment and biological function (Fig. 6G). A large subset of proteins was nuclear and classified as DNA/RNA-binding. Notably, the two principal HIF transcription cofactors CBP and P300 were identified within the AURKA complex, along with RNA PolII and multiple proteins involved with the super elongation complex (Fig. 6H, I).
Fig. 6. N-AURKA binds to HIF1A/B and promotes transactivation of hypoxia-response genes.
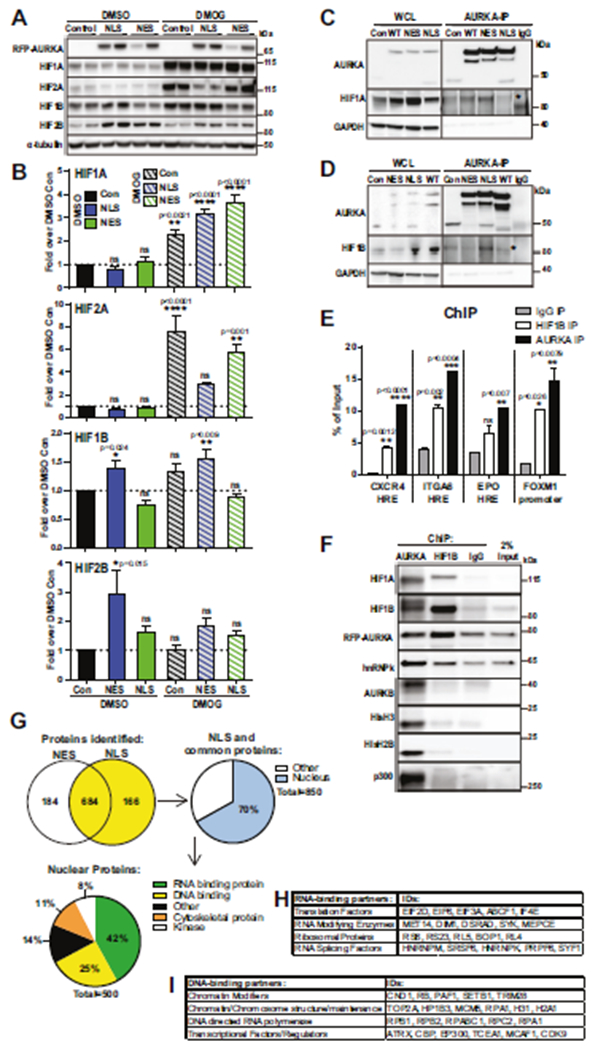
A WB analysis of HIFs in cells with indicated antibodies, DMSO-vehicle or DMOG for 7 h. B Quantification of WB results as in A, fold of change over DMSO-control. C, D Representative images of Immunoprecipitation/WB analysis with indicated antibodies, WCL-whole cell lysate. E Quantification of ChIP qPCR against selected promoter region, normalized to total input. F WB analysis of ChIP (before de-crosslinking). G Mass-spectrometry analysis of AURKA-IP complexes: Venn diagram displaying the numbers of proteins found in the NLS- or NES-AURKA complexes. The 850 proteins/yellow were further filtered, nuclear/blue. Pie-chart showing distribution of N-AURKA binding partners based on functional Panther Gene Ontology terms. Tables showing selected nuclear protein classifications for H RNA-binding and I DNA-binding from PANTHER Gene Ontology. One-way ANOVA, ±S.E.M, Dunnett’s test, ns non-significant.
HIF1A/B is required to mediate N-AURKA-driven invasion
To test the role of HIF1A/B in mediating N-AURKA driven processes, we generated NLS-AURKA and control cell lines with depletion of HIF1A/B (Fig. 7A, B). The 3D invasion/migration analysis shows a significant reduction in invasion of NLS-AURKA/siHIF1 cells as documented by the decrease in speed and distance (Fig. 7C–F). The transactivation of hypoxia-signature genes was also significantly reduced upon depletion of HIF1A/B (Fig. 7G). Interestingly, depletion of HIF1A/B led to a decrease in FOXM1 expression (Fig. 7H). In comparison, depletion of FOXM1 did not change expression levels of HIF1A/B (Fig. 7I, J), suggesting that the N-AURKA/HIF complex is upstream of FOXM1. Supporting this notion, mammosphere formation potential in NLS-AURKA cells was not affected by FOXM1 depletion (Supplementary Fig. 2I).
Fig. 7. HIF1A/B is required for N-AURKA-driven invasion.
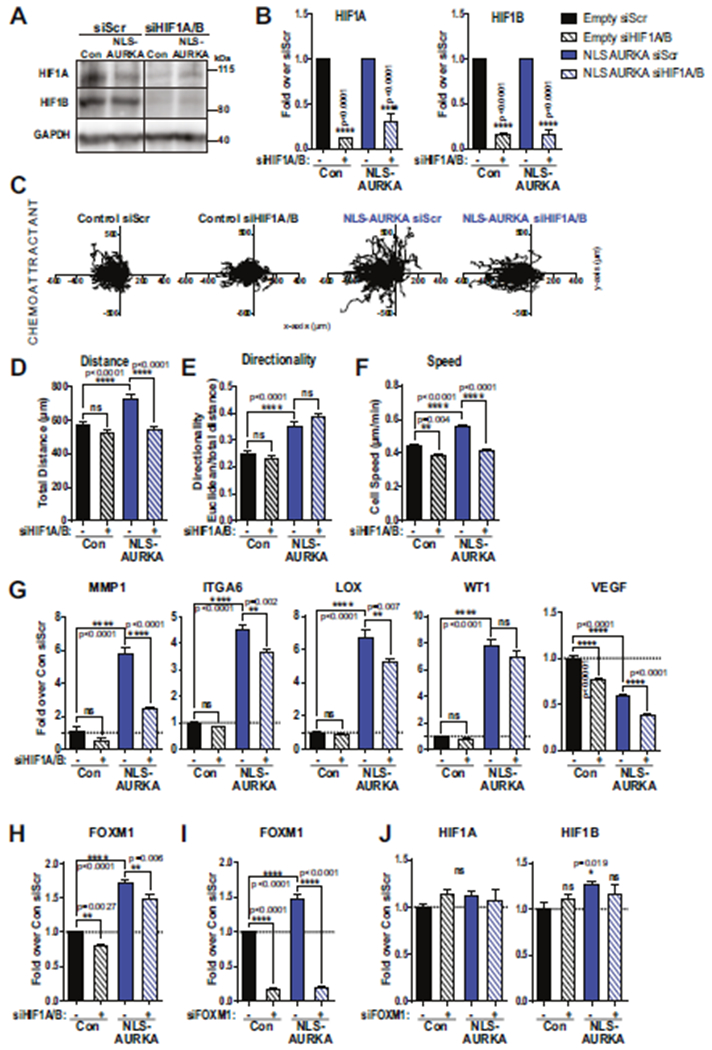
A WB analysis of HIFs in cells as indicated, treated with siRNA: control-scr or anti-HIF1A/B. B Quantification of WB results in A, fold of change over siScr. C Individual cell movement tracking-plots toward chemoattractant. Cell lines as indicated. Graphs of D total distance, E cell-body directionality, F cell speed. G qPCR analysis of select genes in control and NLS-AURKA cells with siScr and siHIF1A/B, fold change over control-siScr. H qPCR analysis of FOXM1 in control and NLS-AURKA cells treated with siScr and siHIF1A/B, fold change over control-siScr. I, J qPCR analysis of FOXM1, HIF1A, and HIF1B in control and NLS-AURKA cells treated with siScr and siFOXM1, fold change over control-siScr. HIF1B: (unpaired t test: con-Scr vs NLS-AURKA-Scr). One-way ANOVA, ±S.E.M. Tukey’s test, ns non-significant.
Inhibition of N-AURKA kinase activity decreases breast cancer metastasis
The orthotopic xenograft study was conducted using control and NLS-AURKA cells treated with AURKA-specific inhibitor, Alisertib/MLN8237 (Fig. 8A, B). The tumor volume was not affected by Alisertib/MLN8237 treatment (Fig. 8C), while metastases in the lungs and liver were significantly decreased (Fig. 8D–F), suggesting that kinase activity might be required to enable metastatic colonization. The metastatic penetrance in the NLS-AURKA group was not affected by the MLN8237 (Fig. 8G) indicating that dissemination of NLS-AURKA cells is kinase-independent. In summary, these findings support the model of N-AURKA-driven activation of hypoxia-response genes via interaction with HIF1 promoting metastasis (Fig. 8H).
Fig. 8. Inhibition of N-AURKA kinase activity decreases breast cancer metastasis.
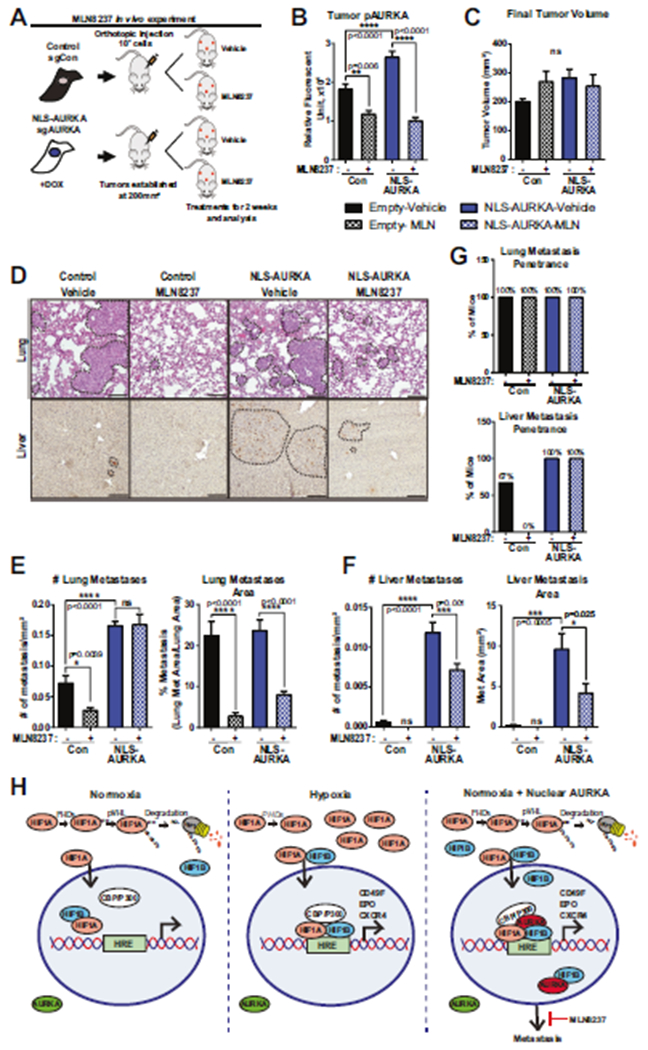
A Experimental design of xenograft study, n = 3 mice/group. B Quantification of Fluorescent-IHC using phospho-AURKA-T288 antibody in tumors. n = 50 mitotic-cells/tumor. C Quantification of final tumor volume/mm3. D Representative H&E images of lung metastases and DAB-IHC images of liver metastases stained with anti-RFP-antibody. Metastases outlined by black line. Scale bar-50 μm. E, F Quantification of metastases: number and area of metastases normalized to total area. G Lung and liver metastasis penetrance in mice treated with vehicle or MLN8237. One-way ANOVA, ±S.E.M. Tukey’s test. H Model of Nuclear-AURKA-HIF1 mediated gene expression. Normoxia: HIF1A is hydroxylated/ubiquitinated by PHDs/VHL resulting in degradation. Hypoxia: low-oxygen stabilizes HIF1A leading to HIF1A/B dimerization/activation, transactivation of hypoxia-response genes. Normoxia+N-AURKA: HIF1A/B activity is increased leading to metastasis. MLN8237 inhibits AURKA activity decreasing metastasis.
DISCUSSION
AURKA has recently been found in the nucleus in multiple cancers; [4–6, 27] however, the role of nuclear AURKA (N-AURKA) in metastasis is currently unknown. Here, we report that N-AURKA promotes metastasis. The AURKA subcellular distribution in BC-TMA shows accumulation in the nucleus correlating with more aggressive subtypes, invasive stage, and metastasis (Fig. 1A–C, Supplementary Fig. 1E). The analysis of TCGA data indicates a strong correlation between levels of AURKA mRNA and poor outcomes (Supplementary Fig. 1A–C). The NLS-AURKA induced hypoxia gene-signature correlates with decreased overall and relapse-free survival (Supplementary Fig. 5J, K). In human breast cancer cell lines, the localization of AURKA in the nucleus closely correlates with the metastatic proficiency of the cells (Fig. 1D, E, Supplementary Fig. 1F, G).
The critical role of N-AURKA in metastasis was documented by the enhanced metastatic burden in orthotopic mouse models (Fig.4). The metastatic penetrance of NLS-AURKA cells is 100% in the lungs, lymph node, liver, and bone (Fig. 4C) without the need for multiple rounds of selection [28, 29]. NLS-AURKA upregulates genes that promote bone metastasis, including ADAMST1, FGF5, FST, CXCR4, IL-11, MMP1 [28]. Note that tumor cells with cytoplasmic AURKA, found in non-transformed cells, have limited to no metastatic capacity (Figs. 2A and 4).
The increased metastatic capabilities are supported by changes in morphology, migration, and stemness. Cells with nuclear AURKA are round and small (Fig. 2B–D), enabling fast movement throughout a constricted environment [30, 31], as in the invasion assay (Fig. 2E–H). The upregulation of cytoskeleton (RHOJ, ARHGAP22, ARHGAP24, ELMO1, DEF6), adhesion (CADM2, SELL, ITGA6), matrix-degrading (ADAMTS5, ADAMTS1, MMP1, MMP14), and tight junction (CLDN4, CLDN11, CLDN12, CLDN23) proteins enables metastatic proficiency of N-AURKA cells (Fig. 5). The decrease in extracellular matrix proteins (LAMB1, LAMC2) might explain the decline in cluster formation (Fig. 2J, K). Interestingly, clustering disability did not affect the sensitivity to anoikis (Fig. 2I) or metastatic proficiency (Fig. 4), suggesting uncoupling of these processes.
The analysis of stem cell markers showed an increase in CD24−/CD44+ and CD24−/CD104+ cells (Fig. 2L, O) and a reduction in mean fluorescent intensity of CD24 (Supplementary Fig. 2A–C). These findings agree with a recent study showing that N-AURKA promotes breast cancer stemness [6]. The NLS-AURKA cells produced more and larger mammospheres, indicating an increase in stemness (Fig. 2P–R). The depletion of FOXM1 was insufficient to change the stem cell markers or mammosphere formation (Supplementary Fig. 2D–I). This activity of N-AURKA might be mediated by HIF1A/B, which in turn might upregulate FOXM1 (Fig. 7H–J). These findings differ from previously published reports, using either an overexpression system [22] or NES-AURKA inducible system [6]. Neither of those modeling approaches allows analysis of nuclear AURKA-specific function directly.
The RNAseq analysis show changes in expression of hypoxia, transcription, and metastasis-related pathways (Fig. 5, Supplementary Fig. 5). It is well-established that hypoxia is an inducer of metastasis [32, 33]. Our findings show that NLS-AURKA interacts with HIF1 and is a potent inducer of the hypoxia-response genes in vitro and in vivo (Fig. 6, Supplementary Fig. 6). Hypoxia signaling is primarily driven by hypoxia-inducible factors (HIFs). The activation/expression of HIF1A at diagnosis is predictive of early relapse and metastasis [34–37].
The NLS-AURKA-ChIP complex analysis and qPCR support the notion that AURKA is binding to HRE-containing promoters with HIF1A/B (Fig. 6E–I). These changes are reported under normoxic conditions and could be further induced under hypoxia. A similar phenotype was observed in other breast cancer models [38]. N-AURKA gene-signature changes were associated with metabolic reprogramming (HK1/2), angiogenesis (ADM, ANGPT1, EDN1, FECH), invasion (CXCR4, LOX, MMP1, MMP14, ITGA6, CSRP2), and proliferation (ENG, WT1, CCND1). Depletion of HIF1A/B in NLS-AURKA cells led to a reduction in migration/invasion and expression of the key hypoxia signature genes, suggesting that this metastasis-driving activity of AURKA is HIF1 dependent (Fig. 7A–G). Note that not all genes connected to hypoxia were upregulated. Some canonical HIF1A-induced genes PFKP, SNAI1, and NOS3 were downregulated, suggesting the involvement of additional factors affecting transcription (Fig. 5I).
The mass-spectrometry analysis of the AURKA-complex identified p300/CBP and RNAPII complex proteins, supporting the role of N-AURKA in transcription (Fig. 6G–I). These findings are in agreement with the previous reports showing AURKA binding to the MYC promoter [6]. In concordance with this report, we show that nuclear AURKA binds to chromatin complexes that contain hnRNPk (Fig. 6F, H). The global ChIP-seq analysis of AURKA has not been reported; therefore, many of the potential targets are still unknown.
Previously it was shown that transcription-related activities of nuclear AURKA are kinase-independent. In our study, inhibition of N-AURKA leads to a significant decrease in metastatic burden (Fig. 8). Interestingly, the kinase activity was not required for dissemination but was critical for colonization of distant organs. Combining AURKA inhibitors with HIF-targeting compounds might prove to be even more beneficial in treating metastatic cancer [6, 19]. A better understanding of the nuclear AURKA functions may enable discovery of new biomarkers for metastatic disease and therapeutic targets.
MATERIALS AND METHODS
Cell Culture, plasmids, and reagents
Cell lines MDA-MB-231, BT-549, HCC1143, Hs578t, MCF7, MCF10A, SK-BR-3, and BT-474 were purchased from and authenticated by American Type Culture Collection. MDA-MB-231LN (PerkinElmer) were grown based on manufacturer’s recommendations, low passage cells were used in the study. The sgRNAs, siRNAs, primers for site-directed mutagenesis is outlined in Supplementary Table S1. Details about lentivirus constructs/production/infection and reagents are outlined in Supplementary Methods.
Western blotting (WB)
Cells/tumors were either lysed in Laemmli or M-PER lysis buffer (ThermoFisher) and processed as previously described [39]. Proteins are normalized to loading controls (GAPDH or tubulin), n = 2 biological/clones, n = 2–3 experiments. Antibodies and dilutions are listed in Supplementary Table S2.
Immunofluorescent cell analysis
Immunofluorescence was performed as previously described [40]. Images were captured using a Zeiss/LSM-510 confocal microscope with 63X Plan-Apochromat/NA1.4, oil objective. 3D-confocal images were processed using Zen and ImageJ/NIH software. Images inside each data set were collected with the same microscopy/image capture settings. Antibodies used are listed in Supplementary Table S2.
Immunohistochemistry (IHC)
The detailed protocol for processing and analysis of Fluorescent-IHC and DAB-IHC is outlined in the Supplementary Methods.
Tissue microarray and patient data
Breast cancer survival data were evaluated using the breast cancer gene chip data of the online databases Kaplan-Meier Plotter [41, 42], the best cutoff values were selected according to the defined high or low expression of AURKA, HIF1A/B, N-AURKA-signature genes. High-density breast cancer TMA BR2082 (US Biomax Supplementary Tables S3 and S4) were collected with full donor consent under the approved IRB protocol. The detailed protocol is outlined in the Supplementary Methods.
Nuclear/cytoplasmic fractionation
The detailed protocol is outlined in the Supplementary Methods.
Cell viability analysis
Guava/Viacount (Luminex) reagent and Guava Eascyte-HT Flow Cytometer (Millipore) were used for analysis of cell viability per manufacturer′s instructions. 1 × 105 cells/well in a 12-well plate were collected 24–120 h post-plating. Parameters for the flow cytometer were set using positive (live) and negative (dead) cells/controls. n = 2 experiments with n = 2 biological, n = 2 technical replicates.
Cell elongation and nuclear size analysis
1 × 104 attached cells/well were stained with Hoechst33342 (ThermoFisher) and bright-field images captured using Zeiss-Axio Imager-Z2 microscope, 20x Plan-Apochromat/0.80NA objective. Cell elongation was measured as a function of cell length divided by cell width in ImageJ software. 50 cells/condition quantified in 3–4 random fields with n = 2 biological/clones, n = 2 technical replicates.
Live-cell imaging of individual cell invasion/migration
Protocol and quantification methods were previously reported [43]. 2.5 × 105 cells were imaged with confocal microscopy, 2 μm/steps. n = 3 experiments with n = 2 biological, n = 2 technical replicates, and ≥50cells traced/per condition.
Anoikis assay
The detailed protocol is outlined in Supplementary Methods.
Flow cytometry analysis
The staining, collection and gating protocols outlined in Supplementary Methods. Antibodies used are listed in Supplementary Table S2.
Mammosphere assay
Mammosphere assay was done as described [6]. At day 10, mammospheres were imaged on ECHO Rebel microscope (VWR) at 4× and 10×. Analysis of sphere’s diameter and number was done in ImageJ. n = 2 experiments, n = 2 biological/clones, n = 3 technical replicates per group with 100 cells counted in 6–10 random field/condition.
Orthotopic xenograft models of breast cancer
Orthotopic injections, bioluminescent imaging, and ultrasound of xenografts were performed as described [44]. All procedures were approved by the WVU Institutional Animal Care and Use Committee and followed guidelines established by the NIH. Details for the orthotopic experiments are outlined in the Supplementary Methods.
RNA sequencing and bioinformatics
Total RNA was isolated from cells (2 clones/construct) with RNeasy Mini-kit (Qiagen). The RNA sequencing data were generated by the WVU Genomics core facility and deposited to GEO with accession number GSE154494. The detailed protocol is outlined in the Supplementary Methods.
Quantitative RT-PCR
RNA (RNeasy Mini-Kit) was used for cDNA generation with MaximaH-Minus First-Strand cDNA-Synthesis kit (ThermoFisher). Custom Taqman-Array in 96-well format (AppliedBiosciences #4391524) was created based on RNA sequencing. The detailed protocol is outlined in the Supplementary Methods. Primer assays and sequences are listed in Supplementary Table S5.
Mass spectrometry
The 6 × 106 cells (MDA-MB-231-NES/or NLS-AURKA) were used (2 clones combined/cell line). The detailed protocol is outlined in Supplementary Methods.
Immunoprecipitation (IP) assay
The detailed protocol for immunoprecipitation of AURKA is outlined in Supplementary Methods.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) was performed using ChIP-grade antibodies against human AURKA (BethylLabs, #IHC-00062) and the SimpleChIP Enzymatic ChromatinIP Kit (CellSignaling) according to manufacturer’s instructions. The detailed protocol is outlined in Supplementary Methods. The primers used for ChIP qPCR analysis are listed in Supplementary Table S6.
Statistical analysis
Statistical comparisons were made using two-tailed Student’s t test, one-way or two-way analysis of variance (ANOVA). P ≥ 0.05 was considered to be significant (*). All groups were compared to empty-vector control/s unless mentioned otherwise. Experimental values were reported as the means with ±S.E.M (standard error of mean), p values are reported as adjusted, and calculations of statistical significance were made using Prism7 software (Graphpad Software). All experimental data sets reported here were collected from multiple independent experiments with multiple technical and biological replicas. The analyses were performed by staff on numbered, de-identified samples.
Supplementary Material
ACKNOWLEDGEMENTS
We thank WVU HSC Office of Graduate Research and Education for outstanding administrative support, WVU Tissue Bank, Genetic Tumor Modeling Core, and Animal Models & Imaging Facilities, supported by the WVUCI and NIH grants P20 RR016440, P30 RR032138/GM103488, and S10RR026378. Flow Cytometry Facility was supported by NIH grants P30GM103488, P30RR032138, and RCP1101809. Bioinformatics Core by NIH grants P20GM103434 and U54 GM-104942. This work was supported by a grant from NIH-NCI CA148671 to ENP. Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number CA148671 (to ENP). Small animal imaging and cell image analysis were performed in the West Virginia University Animal Models & Imaging Facility, supported by NIH grants P20RR016440, P30GM103488, U54GM104942, P20GM103434, and S10RR026378. The flow cytometry analysis was done in WVU Flow Cytometry & Single Cell Core Facility, supported by NIH grants GM121322, GM104942, GM103434, and OD016165. The genetic tumor modeling (GTM) core facility provided animal models supported by NIH grant GM121322. The WVU Bioinformatics Core Facility supported by NIH grants P20GM103434 and U54 GM-104942 assisted with RNA-Seq data analysis and interpretation.
Footnotes
Disclaimers
The authors declare the following: (1) This manuscript contains original work only, (2) All authors have directly participated in the planning, execution, and analysis of this study, (3) All have approved the submitted version of this manuscript, (4) The described data has not been published nor submitted elsewhere. The RNA-seq data have been deposited to the public database GSE154494.
COMPETING INTERESTS
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41388-021-01969-1.
REFERENCES
- 1.Schumacher JM, Ashcroft N, Donovan PJ, Golden A. A highly conserved centrosomal kinase, AIR-1, is required for accurate cell cycle progression and segregation of developmental factors in Caenorhabditis elegans embryos. Development. 1998;125:4391–402. [DOI] [PubMed] [Google Scholar]
- 2.Giet R, McLean D, Descamps S, Lee MJ, Raff JW, Prigent C, et al. Drosophila Aurora A kinase is required to localize D-TACC to centrosomes and to regulate astral microtubules. J Cell Biol. 2002;156:437–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang LH, Xiang J, Yan M, Zhang Y, Zhao Y, Yue CF, et al. The mitotic kinase Aurora-A induces mammary cell migration and breast cancer metastasis by activating the Cofilin-F-actin pathway. Cancer Res. 2010;70:9118–28. [DOI] [PubMed] [Google Scholar]
- 4.Baba Y, Nosho K, Shima K, Irahara N, Kure S, Toyoda S, et al. Aurora-A expression is independently associated with chromosomal instability in colorectal cancer. Neoplasia. 2009;11:418–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu HT, Ma L, Qi FJ, Liu Y, Yu JH, Dai SD, et al. Expression of serine threonine kinase 15 is associated with poor differentiation in lung squamous cell carcinoma and adenocarcinoma. Pathol Int. 2006;56:375–80. [DOI] [PubMed] [Google Scholar]
- 6.Zheng F, Yue C, Li G, He B, Cheng W, Wang X, et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat Commun. 2016;7:10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tatsuka M, Sato S, Kanda A, Miki T, Kamata N, Kitajima S, et al. Oncogenic role of nuclear accumulated Aurora-A. Mol Carcinog. 2009;48:810–20. [DOI] [PubMed] [Google Scholar]
- 8.Mao JH, Wu D, Perez-Losada J, Jiang T, Li Q, Neve RM, et al. Crosstalk between Aurora-A and p53: frequent deletion or downregulation of Aurora-A in tumors from p53 null mice. Cancer Cell. 2007;11:161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang H, He L, Kruk P, Nicosia SV, Cheng JQ. Aurora-A induces cell survival and chemoresistance by activation of Akt through a p53-dependent manner in ovarian cancer cells. Int J Cancer. 2006;119:2304–12. [DOI] [PubMed] [Google Scholar]
- 10.Briassouli P, Chan F, Savage K, Reis-Filho JS, Linardopoulos S. Aurora-A regulation of nuclear factor-kappaB signaling by phosphorylation of IkappaBalpha. Cancer Res. 2007;67:1689–95. [DOI] [PubMed] [Google Scholar]
- 11.Xu J, Wu X, Zhou WH, Liu AW, Wu JB, Deng JY, et al. Aurora-A identifies early recurrence and poor prognosis and promises a potential therapeutic target in triple negative breast cancer. PLoS One. 2013;8:e56919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Assoro AB, Liu T, Quatraro C, Amato A, Opyrchal M, Leontovich A, et al. The mitotic kinase Aurora-a promotes distant metastases by inducing epithelial-to-mesenchymal transition in ERalpha(+) breast cancer cells. Oncogene. 2014;33:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nadler Y, Camp RL, Schwartz C, Rimm DL, Kluger HM, Kluger Y. Expression of Aurora A (but not Aurora B) is predictive of survival in breast cancer. Clin Cancer Res. 2008;14:4455–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996;271:17771–8. [DOI] [PubMed] [Google Scholar]
- 15.Semenza GL. The hypoxic tumor microenvironment: a driving force for breast cancer progression. Biochim Biophys Acta. 2016;1863:382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Bock K, Mazzone M, Carmeliet P. Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or not? Nat Rev Clin Oncol. 2011;8:393–404. [DOI] [PubMed] [Google Scholar]
- 17.Zhang H, Wong CC, Wei H, Gilkes DM, Korangath P, Chaturvedi P, et al. HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene. 2012;31:1757–70. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.Hayashi Y, Yokota A, Harada H, Huang G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1alpha in cancer. Cancer Sci. 2019;110:1510–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozyreva VK, Kiseleva AA, Ice RJ, Jones BC, Loskutov YV, Matalkah F, et al. Combination of Eribulin and Aurora A inhibitor MLN8237 prevents metastatic colonization and induces cytotoxic autophagy in breast cancer. Mol Cancer Ther. 2016;15:1809–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bierie B, Pierce SE, Kroeger C, Stover DG, Pattabiraman DR, Thiru P, et al. Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc Natl Acad Sci USA. 2017;114:E2337–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang N, Wang C, Wang Z, Zona S, Lin SX, Wang X, et al. FOXM1 recruits nuclear Aurora kinase A to participate in a positive feedback loop essential for the self-renewal of breast cancer stem cells. Oncogene 2017;36:3428–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood 2012;119:5782–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melchionna R, Romani M, Ambrosino V, D’Arcangelo D, Cencioni C, Porcelli D, et al. Role of HIF-1alpha in proton-mediated CXCR4 down-regulation in endothelial cells. Cardiovasc Res. 2010;86:293–301. [DOI] [PubMed] [Google Scholar]
- 25.Orlando IMC, Lafleur VN, Storti F, Spielmann P, Crowther L, Santambrogio S, et al. Distal and proximal hypoxia response elements cooperate to regulate organ-specific erythropoietin gene expression. Haematologica. 2020;105:2774–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks DL, Schwab LP, Krutilina R, Parke DN, Sethuraman A, Hoogewijs D, et al. ITGA6 is directly regulated by hypoxia-inducible factors and enriches for cancer stem cell activity and invasion in metastatic breast cancer models. Mol Cancer. 2016;15:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanaka E, Hashimoto Y, Ito T, Okumura T, Kan T, Watanabe G, et al. The clinical significance of Aurora-A/STK15/BTAK expression in human esophageal squamous cell carcinoma. Clin Cancer Res. 2005;11:1827–34. [DOI] [PubMed] [Google Scholar]
- 28.Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–49. [DOI] [PubMed] [Google Scholar]
- 29.Andrzejewski S, Klimcakova E, Johnson RM, Tabaries S, Annis MG, McGuirk S, et al. PGC-1alpha promotes breast cancer metastasis and confers bioenergetic flexibility against metabolic drugs. Cell Metab. 2017;26:778–87 e5. [DOI] [PubMed] [Google Scholar]
- 30.Friedl P, Wolf K, Lammerding J. Nuclear mechanics during cell migration. Curr Opin Cell Biol. 2011;23:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pankova K, Rosel D, Novotny M, Brabek J. The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell Mol Life Sci. 2010;67:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science 2016;352:175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen A, Sceneay J, Godde N, Kinwel T, Ham S, Thompson EW, et al. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene. 2018;37:4214–25. [DOI] [PubMed] [Google Scholar]
- 34.Bos R, van der Groep P, Greijer AE, Shvarts A, Meijer S, Pinedo HM, et al. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer. 2003;97:1573–81. [DOI] [PubMed] [Google Scholar]
- 35.Gruber G, Greiner RH, Hlushchuk R, Aebersold DM, Altermatt HJ, Berclaz G, et al. Hypoxia-inducible factor 1 alpha in high-risk breast cancer: an independent prognostic parameter? Breast Cancer Res. 2004;6:R191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Generali D, Berruti A, Brizzi MP, Campo L, Bonardi S, Wigfield S, et al. Hypoxia-inducible factor-1alpha expression predicts a poor response to primary chemoendocrine therapy and disease-free survival in primary human breast cancer. Clin Cancer Res. 2006;12:4562–8. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, He YF, Sun QK, Wang Y, Han XH, Peng DF, et al. Hypoxia-inducible factor 1alpha in breast cancer prognosis. Clin Chim Acta. 2014;428:32–7. [DOI] [PubMed] [Google Scholar]
- 38.Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG, et al. PDK1-dependent metabolic reprogramming dictates metastatic potential in breast cancer. Cell Metab. 2015;22:577–89. [DOI] [PubMed] [Google Scholar]
- 39.Pugacheva EN, Golemis EA. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat Cell Biol. 2005;7:937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129:1351–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725–31. [DOI] [PubMed] [Google Scholar]
- 42.Nagy A, Lanczky A, Menyhart O, Gyorffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018;8:9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones BC, Kelley LC, Loskutov YV, Marinak KM, Kozyreva VK, Smolkin MB, et al. Dual targeting of mesenchymal and amoeboid motility hinders metastatic behavior. Mol Cancer Res. 2017;15:670–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ice RJ, McLaughlin SL, Livengood RH, Culp MV, Eddy ER, Ivanov AV, et al. NEDD9 depletion destabilizes Aurora A kinase and heightens the efficacy of Aurora A inhibitors: implications for treatment of metastatic solid tumors. Cancer Res. 2013;73:3168–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.