

Abstract
Following inflammatory injury in the liver, neutrophils quickly infiltrate the injured tissue to defend against microbes and initiate the repair process; these neutrophils are short lived and rapidly undergo apoptosis. Hepatic stellate cells (HSCs) are the principal precursor cells that transdifferentiate into myofibroblast‐like cells, which produce a large amount of extracellular matrix that promotes repair but can also lead to fibrosis if the injury becomes chronic. The matricellular protein cellular communication network factor 1 (CCN1) acts as a bridging molecule by binding phosphatidylserine in apoptotic cells and integrin αvβ3 in phagocytes, thereby triggering efferocytosis or phagocytic clearance of the apoptotic cells. Here, we show that CCN1 induces liver macrophage efferocytosis of apoptotic neutrophils in carbon tetrachloride (CCl4)‐induced liver injury, leading to the production of activated transforming growth factor (TGF)‐β1, which in turn induces HSC transdifferentiation into myofibroblast‐like cells that promote fibrosis development. Consequently, knock‐in mice expressing a single amino acid substitution in CCN1 rendering it unable to bind αvβ3 or induce efferocytosis are impaired in neutrophil clearance, production of activated TGF‐β1, and HSC transdifferentiation, resulting in greatly diminished liver fibrosis following exposure to CCl4. Conclusion: These results reveal the crucial role of CCN1 in stimulating liver macrophage clearance of apoptotic neutrophils, a process that drives HSC transdifferentiation into myofibroblastic cells and underlies fibrogenesis in chronic liver injury.
We show that CCN1 induces liver macrophage efferocytosis of apoptotic neutrophils in CCl4‐induced liver injury, leading to the production of activated TGF‐β1, which in turn induces HSC transdifferentiation into myofibroblast‐like cells that promote fibrosis development. These results reveal the crucial role of CCN1 in stimulating liver macrophage clearance of apoptotic neutrophils, a process that drives HSC transdifferentiation into myofibroblastic cells and underlies fibrogenesis in chronic liver injury.
INTRODUCTION
Liver fibrosis results from the excessive accumulation of extracellular matrix (ECM) proteins in response to chronic liver injuries, irrespective of the underlying etiology.[ 1 ] Injury and inflammation are followed by rapid infiltration of immune cells and activation of macrophages, leading to the deposition of a provisional ECM, which promotes tissue integrity and cell proliferation.[ 2 ] However, this process can lead to fibrosis if the injury becomes chronic. Extensive analyses using multiple approaches, including lineage tracing and single‐cell RNA sequencing, have determined that hepatic stellate cells (HSCs) are the principal precursor cells that undergo “activation” and transdifferentiate into alpha smooth muscle actin (αSMA)‐expressing myofibroblast‐like fibrogenic cells following liver injury, although portal fibroblasts also play a role in early cholestasis.[ 3 , 4 ] Transforming growth factor‐β1 (TGF‐β1) is the most potent cytokine that induces HSC activation for which Kupffer cells (KCs), the resident macrophages in the liver, are the major source.[ 5 , 6 ] Monocyte‐derived macrophages are also recruited to the liver, particularly following injury.[ 7 ] Time‐specific elimination of macrophages indicated that macrophages can promote fibrogenesis in the early inflammatory phase of injury but stimulate fibrosis resolution in the late phase of healing.[ 8 ] However, significant gaps remain in our understanding of the molecular events that trigger the profibrogenic activities of liver macrophages following liver injury.
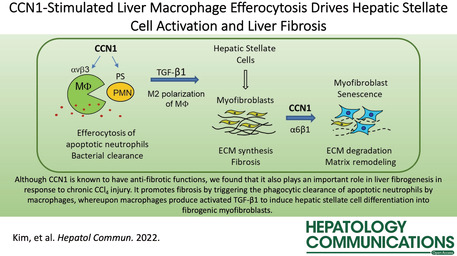
Cellular communication network factor 1 (CCN1) is a 40‐kDa secreted matricellular protein essential for embryonic cardiovascular development.[ 9 , 10 ] In adults, CCN1 plays multiple roles in wound healing and is required for successful injury repair in multiple organs, including the skin, liver, heart, and gut.[ 11 , 12 , 13 , 14 ] In particular, CCN1 induces efferocytosis, the phagocytic clearance of apoptotic cells, by binding phosphatidylserine on apoptotic cells and bridging them to macrophages for engulfment through the engagement of integrin αvβ3, the phagocytic receptor in macrophages.[ 15 ] In the liver, CCN1 can promote biliary regeneration after cholestasis by inducing cholangiocyte proliferation and accelerate fibrosis resolution in chronic inflammatory injuries by inducing senescence in myofibroblasts through integrin α6β1. [ 12 , 16 ] Here, we show that surprisingly, CCN1 can also promote fibrogenesis in the early phase of liver injury repair by triggering the activation of HSCs. This effect is mediated through CCN1‐induced liver macrophage efferocytosis of apoptotic neutrophils, leading to the elevated production of activated TGF‐β1, which in turn induces myofibroblastic differentiation of HSCs. These findings show that liver fibrogenesis is dependent in part on macrophage efferocytosis and that CCN1 plays a key role in this process. Thus, CCN1 can play both profibrotic and antifibrotic roles in the liver at different stages of injury repair through distinct integrins in disparate cell types.
MATERIALS AND METHODS
Mice
Ccn1 D125A/D125A knock‐in mice expressing an αvβ5/αvβ3‐binding defective CCN1 mutant were generated in an svJ129‐C57/BL6J‐mixed background and backcrossed to a C57/BL6J strain more than 8 times.[ 15 ] Male Ccn1 wild type [WT/WT] (C57BL/6J) and Ccn1 D125A/D125A mice 2–3 months of age were maintained in barrier facilities and injected intraperitoneally with carbon tetrachloride (CCl4) (1 ml/kg body weight diluted 1:10 in olive oil) twice weekly for 4 weeks to induce fibrosis. Where indicated, 20 μg of purified recombinant CCN1 protein per mouse was injected intraperitoneally, with buffered saline as control. Fibrosis induction by bile duct ligation (BDL) or feeding of the 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine (DDC) diet was carried out as described.[ 17 , 18 ] All procedures were approved by the University of Illinois Animal Care Committee.
Cell isolation
Neutrophils were isolated from the bone marrow of mice using a Percoll gradient as described.[ 15 , 19 ] Hepatic cells were isolated as described with minor modifications.[ 20 ] Briefly, liver cells liberated by perfusion of livers with elastase, collagenase, and deoxyribonuclease were centrifuged at 30g for 3 min to pellet hepatocytes. The supernatant was centrifuged at 650g for 3 min to pellet nonparenchymal cells. To isolate HSCs, the resuspended pellet was layered on top of 11.5% Nycodenz (weight/volume; Accurate Chemical) and centrifuged at 1500g for 22 min. HSCs form a white band at the Nycodenz mid‐phase. To isolate liver macrophages, the nonparenchymal cell pellet was resuspended and loaded on a 50%/25% Percoll (Amersham) gradient and centrifuged at 1800g for 15 min. The macrophages form a white band in the gradient interphase. Macrophages from uninjured liver showed 98% purity as judged by staining for both F4/80 (Bio‐Rad) and CD68 (Abcam), with 84% galectin‐3‐positive (+) KCs (Figure S1A,B).
RNA isolation, quantitative reverse‐transcription polymerase chain reaction, and enzyme‐linked immunosorbent assay
Total RNA was purified from liver tissue and cultured cells using GenJET RNA purification kit (Thermo Scientific) following the manufacturer's protocol. RNA was reverse transcribed using MMLV‐Reverse Transcriptase (Promega). Quantitative reverse‐transcription polymerase chain reaction (qRT‐PCR) was performed by mixing complementary DNA and gene‐specific primers (Table S1) with iQ SYBR Green Supermix (Bio‐Rad), and the reaction was carried out in the iCycler Thermal Cycler (Bio‐Rad). PCR specificity was confirmed by agarose gel electrophoresis and melting curve analysis. Cyclophilin E was used as an internal standard. Serum protein levels of total and free active TGF‐β1 were determined using the LEGEND MAX enzyme‐linked immunosorbent assay kit (BioLegend) with the manufacturer's protocol (Table S2).
Histology and immunohistochemistry
Formalin‐fixed paraffin‐embedded liver tissue sections (5 μm) were stained with picrosirius red solution (American MasterTech Scientific), following the manufacturer's protocol. Microphotographs of six random fields (3.4 × 2.5 mm2) from each animal were analyzed using the National Institutes of Health Image J software to quantify the red‐staining fibrotic areas. For immunohistochemistry, epitopes were unmasked by incubation with sodium citrate pH 6.0 and incubated with primary antibodies (Table S3) for αSMA (Abcam), CCN1 (R&D System), lymphocyte antigen 6 complex locus G6D (Ly6G) (BD Biosciences), F4/80(AbDserotec), and myeloperoxidase (MPO) (Invitrogen) overnight. Secondary antibody (GE Healthcare) conjugated with horseradish peroxidase was used with 3,3′‐diaminobenzidine (Sigma) as the chromogen. Samples were counterstained with hematoxylin. Fluorescence microphotographs were acquired using a Leica DM4000B microscope.
Protein assays and western blots
The serum levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) were analyzed using a Beckman Coulter AU480 analyzer at the Biologic Resources facility at the University of Illinois at Chicago (UIC). The MPO assay was performed by using the MPO assay kit (BioVison). Senescence‐associated beta‐galactosidase (SA‐β‐Gal) of HSCs was assayed as described.[ 21 ] Liver collagen was quantified by the amount of hydroxyproline (microgram) per milligram of dried liver tissue as described,[ 22 ] using pure hydroxyproline (Sigma) for standard curves. Western blots were performed with enhanced chemiluminescence using an enhanced chemiluminescence system (GE Heathcare) with standard procedures. Anti‐phosphorylated‐SMAD family member 2 (phospho‐Smad2) (Cell Signaling), anti‐Smad2 (Cell Signaling), anti‐CCN1 (R&D Systems), and anti‐β‐actin (mAbcam8226; Abcam) antibodies were used (Table S4).
Efferocytosis assay
An efferocytosis assay was performed as described.[ 15 ] KCs were plated on a 12‐well culture dish 1 day before the assay and labeled with CellTracker green 5‐chloromethylfluorescein diacetate (Invitrogen). Neutrophils aged for 16 hours and then incubated at 55°C for 2 hours were labeled with 4′,6‐diamidino‐2‐phenylindole (DAPI) and incubated with macrophages for 90 min. WT‐CCN1 or D125A‐CCN1 protein was preincubated with neutrophils for 1 hour before incubation with macrophages where indicated. The efferocytosis index was measured by counting DAPI‐positive macrophages as a percentage of total macrophages.
Adenoviral infection
HSCs were infected with adenovirus expressing β‐Gal (LacZ) or Ccn1 (Clontech Adenovirus expression system) for 48 hours at ~500 plaque‐forming units (PFU)/cell.
Coculture of HSCs and KCs
Primary HSCs and liver macrophages were cocultured in the Transwell system (0.4 μm; Corning). Briefly, HSCs and macrophages were seeded in 12‐well plates and chamber inserts, respectively, and cultured separately overnight to allow cell attachment. The chamber inserts with macrophages were then placed in the 12‐well plates containing HSCs. The cells shared a common medium of Dulbecco's modified Eagle's medium with 10% fetal bovine serum and were incubated at 37°C for 4 days. The HSCs were prepared for immunohistochemical staining and RNA extraction.
Statistical analysis
All experiments were carried out with at least three biological replicates. Two‐tailed Student t tests were used to compare the difference between two groups, and p < 0.05 was considered significant. Statistical analysis was performed with GraphPad Prism9 software.
RESULTS
CCl4 ‐induced liver fibrosis requires CCN1 activities through its integrin αvβ3 binding site
CCN1 exerts distinct cellular functions through direct binding to specific integrin receptors in various cell types.[ 10 ] To uncover the role of CCN1 in liver injury repair mediated through integrins αvβ3/αvβ5, we examined the fibrotic response in Ccn1 D125A/D125A knock‐in mice, which carry a single aspartate to alanine substitution that abrogates CCN1 binding to these integrins.[ 15 , 23 ] Ccn1 WT/WT and Ccn1 D125A/D125A mice were injected twice weekly with the hepatotoxin CCl4 for 4 weeks to induce chronic inflammation and fibrosis.[ 24 ] As expected, Ccn1 WT/WT mice developed substantial fibrosis with central‐to‐central bridging septa as shown by sirius red staining and hydroxyproline content (Figure 1A,B). Surprisingly, Ccn1 D125A/D125A mice did not develop appreciable fibrosis. Serum ALT, AST, and ALP were elevated to comparable levels in both genotypes, indicating that these mice sustained similar extents of liver damage (Figure 1C). Hepatic expression of Ccn1 was induced by approximately 30‐fold within 8 hours of CCl4 exposure and diminished to low levels by 24 to 72 hours, whereas expression of the homologous Ccn2 was also increased but to a lesser extent (Figure S2A,B). CCN1 protein was highly induced around the central vein within 24 hours and became more evenly distributed throughout the liver by 4 weeks of chronic CCl4 injury (Figure S2C,D). Consistent with reduced fibrosis, Ccn1 D125A/D125A livers displayed markedly fewer αSMA‐positive myofibroblastic cells after injury as shown by immunostaining and expression of αSMA and collagen, type I, alpha 1 (Col1a1) (Figure 1D,E). Thus, knock‐in mice expressing CCN1 unable to bind integrins αvβ3/αvβ5 experience reduced liver fibrosis following chronic CCl4 injury, with decreased αSMA‐expressing myofibroblast‐like cells.
FIGURE 1.
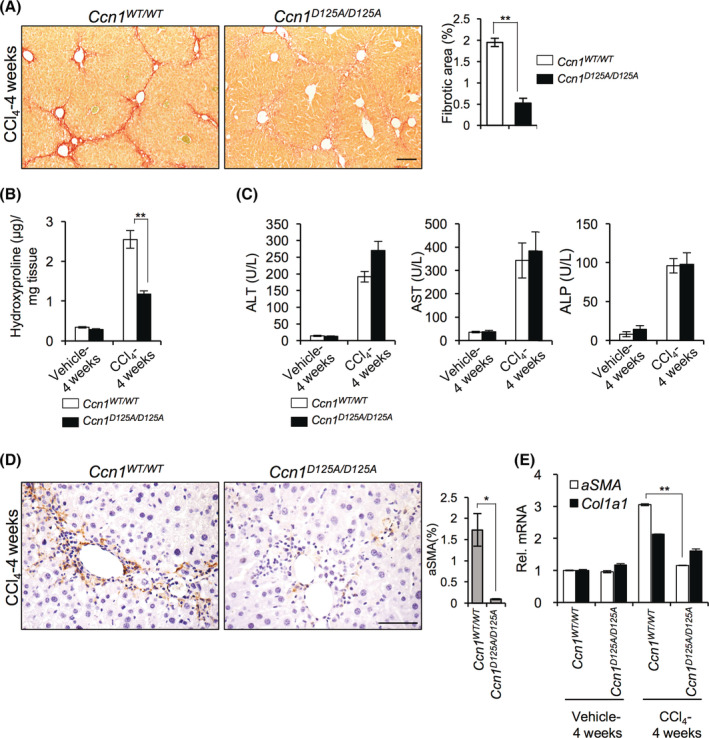
CCN1 is required for liver fibrogenesis in CCl4‐induced injury. Ccn1 WT/WT and Ccn1 D125A/D125A mice were intraperitoneally injected with either vehicle or CCl4 twice weekly for 4 weeks to induce fibrosis (n = 6 each). (A) Liver sections were stained with sirius red for collagen. Fibrotic areas were quantified by Image J analysis of six randomly selected fields. (B) The hydroxyproline content of the livers was determined. (C) Serum ALT, AST, and ALP levels were measured. (D) Liver sections were stained with αSMA antibody (brown) and counterstained with hematoxylin (blue). Percentages of αSMA‐positive cells are shown. (E) Liver αSMA and Col1a1 mRNAs were quantified by quantitative reverse‐transcription polymerase chain reaction. Data are shown as means ± SD. *p < 0.033, **p < 0.002; Student t test. Scale bar, 100 μm. ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CCl4, carbon tetrachloride; Col1a1, collagen type 1 alpha 1; CCN1, cellular communication network factor 1; mRNA, messenger RNA; Rel., relative; WT, wild type; αSMA, alpha smooth muscle actin.
To evaluate whether Ccn1 D125A/D125A mice also show diminished fibrotic response in other models of liver injury, we subjected them to BDL or chronic feeding of DDC to induce cholestatic damage.[ 17 , 25 ] In both injury models, Ccn1 D125A/D125A mice exhibited greatly reduced fibrosis compared to Ccn1 WT/WT mice (Figure S3). Thus, the functions of CCN1 through integrins αvβ3/αvβ5 play key roles in liver fibrogenesis in diverse etiologies, including injuries induced by CCl4, BDL, or DDC diet.
Ccn1 D125A /D125A mice are deficient in injury‐induced HSC activation
HSCs are the principal precursor cells that transdifferentiate into αSMA‐expressing myofibroblast‐like cells following toxic liver injury.[ 3 , 4 ] To test whether CCN1 activates HSCs, we isolated primary HSCs from Ccn1 WT/WT and Ccn1 D125A/D125A mice 48 hours after injection of vehicle or CCl4. Consistent with the finding that HSCs from uninjured mice can be activated when cultured on plastic without the addition of exogenous factors,[ 26 ] Ccn1 WT/WT and Ccn1 D125A/D125A HSCs transdifferentiated into myofibroblastic cells after 7 days in culture, losing their stellate morphology and becoming enlarged with extensive cytoplasmic stress fibers, concomitant with increased αSMA expression (Figure 2A,B). HSCs from CCl4‐injured Ccn1 WT/WT mice showed robust differentiation as early as day 5 in culture that increased further by day 7 with approximately 5‐fold further elevation in αSMA messenger RNA (mRNA) (Figure 2B), indicating that these HSCs were primed for activation. By contrast, HSCs from injured Ccn1 D125A/D125A mice did not show enhanced differentiation, suggesting a deficiency in injury‐induced factors for HSC activation (Figure 2A,B).
FIGURE 2.
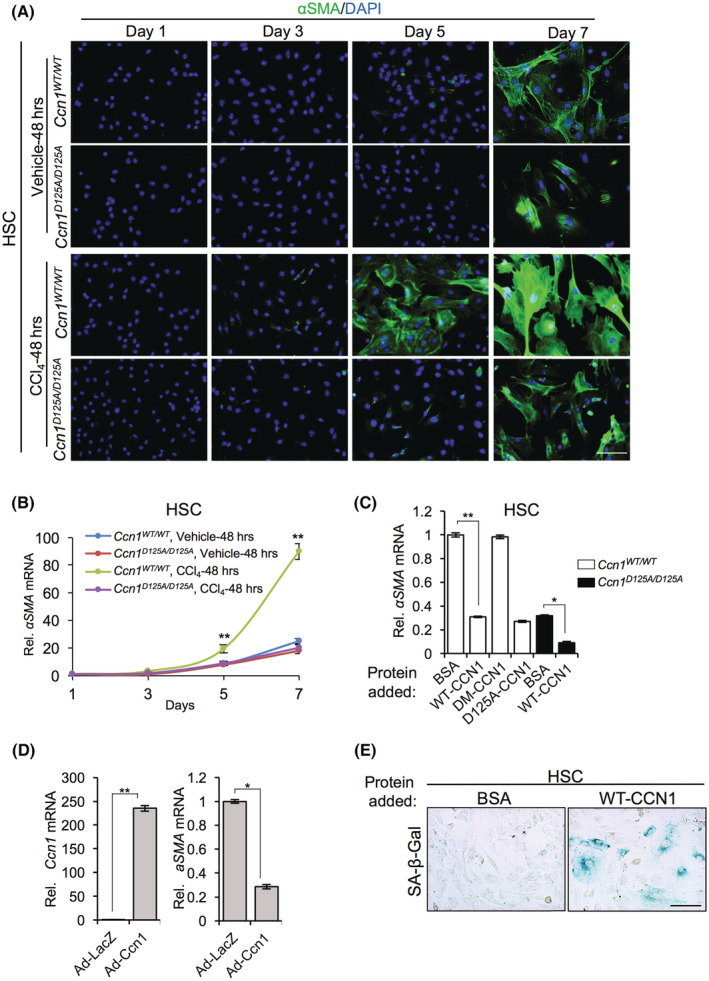
Impaired HSC activation in CCl4‐injured Ccn1 D125A/D125A mice. HSCs isolated from Ccn1 WT/WT and Ccn1 D125A/D125A mice 48 hours after injection with either vehicle or CCl4 were grown in culture. (A) HSCs were stained for αSMA at indicated times. (B) αSMA mRNAs levels were measured by qRT‐PCR. (C) HSCs were incubated with BSA, CCN1, DM‐CCN1, or D125A‐CCN1 proteins (4 μg/ml), as indicated, for 4 days, and αSMA mRNAs were measured by qRT‐PCR. (D) HSCs were transduced with control Ad‐LacZ virus or Ccn1‐overexpressing Ad‐Ccn1 virus, and Ccn1 and αSMA mRNAs were measured by qRT‐PCR after 4 days. (E) HSCs were treated with purified CCN1 protein (2.5 μg/ml) or BSA for 6 days and assayed for SA‐β‐Gal activity. Data represent means ± SD. *p < 0.033, **p < 0.002; Student t test. Scale bar,100 μm. Ad, adenovirus; BSA, bovine serum albumin; CCl4, carbon tetrachloride; CCN1, cellular communication network factor 1; DAPI, 4′,6‐diamidino‐2‐phenylindole; HSC, hepatic stellate cell; LacZ, beta‐galactosidase; mRNA, messenger RNA; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; Rel., relative; SA‐β‐Gal, senescence‐associated beta‐galactosidase; WT, wild type; αSMA, alpha smooth muscle actin.
We tested whether CCN1 can directly induce HSC activation by adding purified CCN1 to HSCs from Ccn1 WT/WT mice. CCN1 did not induce differentiation but instead greatly inhibited αSMA expression (Figure 2C). This inhibitory effect was observed with both CCN1 and D125A‐CCN1 mutant protein unable to bind αvβ3/αvβ5 [ 23 ] but not with DM‐CCN1 mutant protein[ 27 ] unable to bind integrin α6β1 (Figure 2C). HSCs transduced with Ccn1‐expressing adenovirus also blocked αSMA expression (Figure 2D). Because CCN1 can induce senescence in myofibroblasts through integrin α6β1, [ 11 , 16 ] we tested whether it may trigger HSC senescence. Indeed, CCN1‐treated HSCs expressed SA‐β‐Gal and became flattened and enlarged typical of senescent cells (Figure 2E), showing that CCN1 can induce cellular senescence in HSCs. Thus, the effect of CCN1 on HSC activation is likely indirect and potentially mediated through injury‐induced profibrotic factors.
CCN1 regulates TGF‐β1 expression and activation following CCl4 injury
To test whether CCN1 regulates the expression of profibrotic factors following liver damage, we subjected Ccn1 WT/WT and Ccn1 D125A/D125A mice to a single dose of CCl4 to induce acute injury, resulting in similar levels of serum ALT in both genotypes (Figure S4A). The expression of the profibrotic cytokine Tgfb1 was greatly reduced and that of platelet‐derived growth factor subunit A (Pdgfa) and Pdgfb, which can promote fibrosis by enhancing the proliferation of activated HSCs,[ 28 ] was diminished in Ccn1 D125A/D125A mice (Figure 3A,B). Consistent with reduced TGF‐β1 signaling, phosphorylated Smad2 was much reduced in Ccn1 D125A/D125A livers 48 hours after CCl4 injection (Figure 3C). Because TGF‐β1 is synthesized in a latent form and requires activation, we measured the amount of total and active forms of TGF‐β1. Whereas the total TGF‐β1 level in both genotypes without injury was comparable, it rose to 40% higher in Ccn1 WT/WT mice than in Ccn1 D125A/D125A mice after injury (Figure 3D). Most strikingly, the amount of TGF‐β1 in the free active form after CCl4 exposure was >6‐fold lower in Ccn1 D125A/D125A mice (Figure 3D), indicating less efficient activation. Further, liver macrophages isolated from CCl4‐injured Ccn1 D125A/D125A mice expressed lower levels of Tgfb1 and Pdgfa mRNA than Ccn1 WT/WT mice and lower levels of matrix metallopeptidase 9 (Mmp9)‐ and Mmp13‐encoding proteases implicated in the activation of TGF‐β1 (Figure S4B).[ 29 , 30 ] Thus, CCN1 promotes the expression of profibrotic factors, including TGF‐β1, through its αvβ3/αvβ5 binding site in liver injury, which may lead to fibrogenic activation.
FIGURE 3.
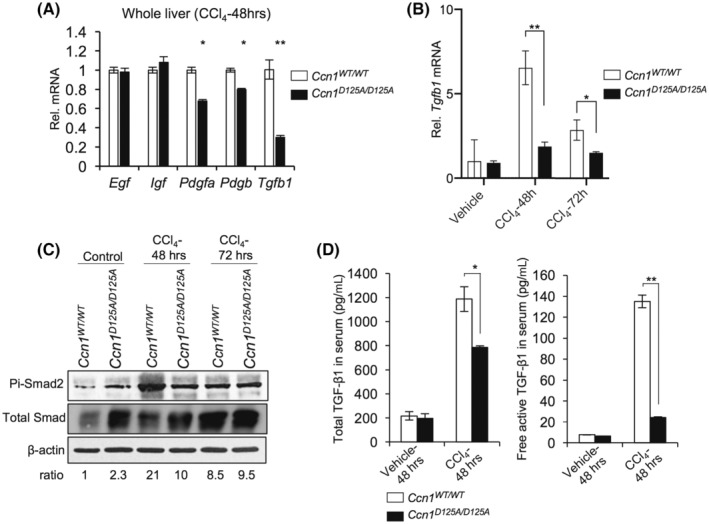
Ccn1 regulates expression of profibrotic factors following CCl4 injury. Livers were isolated from Ccn1 WT/WT and Ccn1 D125A/D125A mice 48 hours after intraperitoneal injection of vehicle or CCl4 (n = 6 each). (A) Expression of indicated genes was assessed by qRT‐PCR. (B) Expression of TGF‐β1 at the indicated time points was assessed by qRT‐PCR. (C) Phosphorylation of Smad‐2 at indicated times was detected by immunoblotting, and its levels were normalized to total Smad. (D) Total (left) or free active (right) TGF‐β1 in serum was quantified by enzyme‐linked immunosorbent assay. Data represent means ± SD. *p < 0.033, **p < 0.002; Student t‐test. CCl4, carbon tetrachloride; CCN1, cellular communication network factor 1; EGF, endothelial growth factor; IGF, insulin‐like growth factor; mRNA, messenger RNA; Pdgfa/b, platelet‐derived growth factor subunit A/B; pi‐Smad2, phosphorylated Smad2; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; Rel., relative; Tgfb1, transforming growth factor‐β1; WT, wild type; αSMA, alpha smooth muscle actin.
CCN1 stimulates liver macrophage efferocytosis and TGF‐β1 expression
Because Ccn1 D125A/D125A macrophages are impaired in TGF‐β1 expression in liver injury, we hypothesized that CCN1 may regulate macrophage functions and gene expression through αv integrins. CCN1 can induce macrophage efferocytosis as a bridging molecule by binding phosphatidylserine, the “eat‐me” signal on apoptotic cells, and activating phagocytosis by binding integrins αvβ3/αvβ5 in macrophages.[ 15 ] Following injury, neutrophils quickly infiltrate the damaged tissue but rapidly undergo apoptosis. To test whether CCN1 induces efferocytosis of apoptotic neutrophils, we isolated liver macrophages from Ccn1 WT/WT or Ccn1 D125A/D125A mice 48 hours after CCl4 injection and incubated them with bone marrow‐derived neutrophils from either Ccn1 WT/WT or Ccn1 D125A/D125A mice made apoptotic by aging and heat treatment (Figure 4A). Ccn1 WT/WT macrophages from CCl4‐injured mice efficiently engulfed apoptotic neutrophils from either genotype, whereas Ccn1 D125A/D125A macrophages were impaired in efferocytosis (Figure 4A,B). Apoptotic neutrophils from Ccn1 D125A/D125A mice were also less efficiently phagocytosed by macrophages of either genotype, and efferocytosis was further reduced when both macrophages and neutrophils were from Ccn1 D125A/D125A mice. However, the addition of purified WT CCN1 protein restored efferocytosis to near wild‐type level (Figure 4B), indicating that Ccn1 D125A/D125A macrophages are capable of efferocytosis but lack functional CCN1.
FIGURE 4.
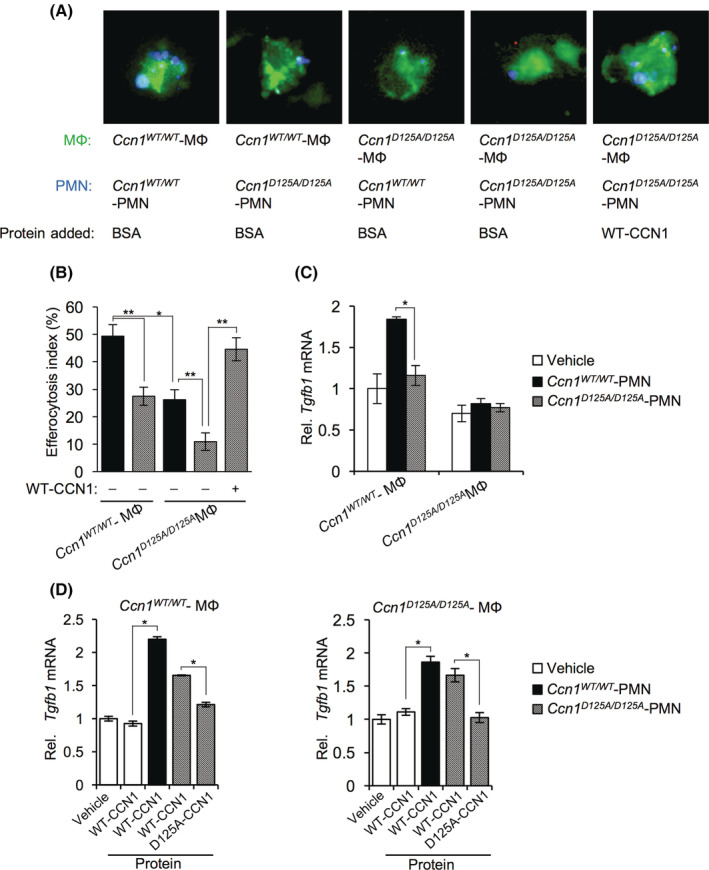
CCN1 stimulates liver macrophage efferocytosis and enhances Tgfb1 expression. Liver macrophages and neutrophils (PMN) were isolated from Ccn1 WT/WT and Ccn1 D125A/D125A mice 48 hours after CCl4 injury. (A) Apoptotic neutrophils (PMN; stained blue with DAPI) were pretreated with BSA or CCN1 protein (4 μg/ml) for 60 min and incubated with macrophages (stained with CellTracker Green) for 90 min. The field of view in each panel of Figure 4A is 35 × 35 μm. The width of each cell is approximately 20 μm. (B) The number of DAPI‐positive macrophages was counted and expressed as a percentage of total macrophages. (C) The expression of Tgfb1 was assessed by qRT‐PCR. The vehicle samples contained no PMN. (D) Apoptotic neutrophils from Ccn1 WT/WT and Ccn1 D125A/D125A were pretreated with the indicated proteins for 60 min before incubation with macrophages from Ccn1 WT/WT (left panel) and Ccn1 D125A/D125A (right panel) mice, and the expression of Tgfb1 was assessed by qRT‐PCR. Data represent means ± SD. *p < 0.033, **p < 0.002; Student t test. BSA, bovine serum albumin; CCl4, carbon tetrachloride; CCN1, cellular communication network factor 1; DAPI, 4′,6‐diamidino‐2‐phenylindole; MФ, macrophage; mRNA, messenger RNA; PMN, polymorphonuclear; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; Rel., relative; Tgfb1, transforming growth factor‐β1; WT, wild type; αSMA, alpha smooth muscle actin.
Phagocytosis can trigger the polarization of macrophages to M2‐like cells that express cytokines, such as TGF‐β1.[ 31 , 32 ] Tgfb1 mRNA was elevated in Ccn1 WT/WT macrophages in the presence of WT apoptotic neutrophils (polymorphonuclear [PMN]) but not in Ccn1 D125A/D125A macrophages with Ccn1 WT/WT or Ccn1 D125A/D125A neutrophils (Figure 4C). The addition of CCN1 alone to macrophages did not stimulate Tgfb1 expression but did so only in the presence of apoptotic neutrophils to allow efferocytosis (Figure 4D). The addition of the D125A‐CCN1 mutant protein, which does not induce efferocytosis, did not affect Tgfb1 expression (Figure 4D). These data show that CCN1 stimulates efferocytosis of apoptotic neutrophils by liver macrophages, after which Tgfb1 expression is increased. Whereas the liver macrophages employed here were isolated from uninjured livers and were mostly galectin‐3+ KCs (Figure S1), it is noteworthy that CCN1 can also strongly induce efferocytosis in monocyte‐derived macrophages.[ 15 ]
The notion that CCN1 is critical for efferocytosis predicts that clearance of apoptotic cells would be impaired in Ccn1 D125A/D125A livers after injury. Indeed, more apoptotic cells and apoptotic neutrophils were accumulated in Ccn1 D125A/D125A livers compared to Ccn1 WT/WT mice after CCl4 injury as judged by cleaved caspase‐3 and costaining with Ly6G, whereas the levels of macrophages were similar (Figure 5A). MPO activity confirmed the abundance of neutrophils in Ccn1 D125A/D125A livers (Figure 5B). Remarkably, injection of CCl4‐injured Ccn1 D125A/D125A mice with CCN1 protein after CCl4 exposure resulted in greatly enhanced neutrophil clearance (Figure S5A), showing the potency of CCN1 in inducing efferocytosis.
FIGURE 5.
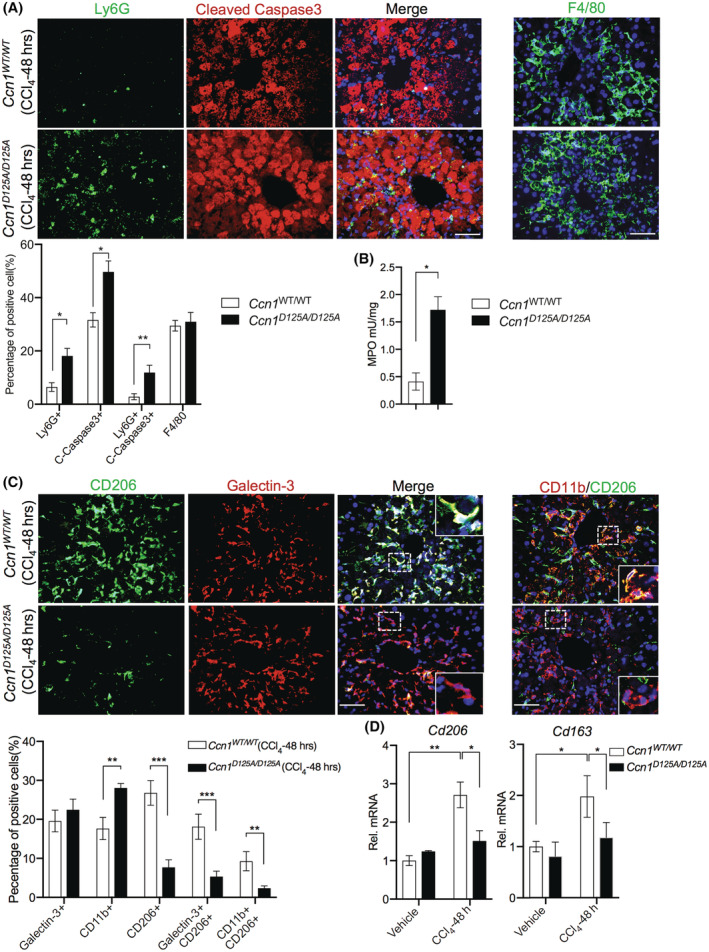
CCN1 regulates neutrophil clearance in CCl4‐induced liver injury. (A) Liver sections from Ccn1 WT/WT and Ccn1 D125A/D125A mice 48 hours after CCl4 injection were costained for Ly6G (neutrophils) and cleaved caspase 3 (apoptotic cells) as indicated and counterstained with DAPI (blue) (n = 6 each). The percentages of positive cells were quantified. (B) An MPO assay was performed with liver tissues from mice 48 hours after CCl4 injection. (C) The livers were stained for F4/80 (macrophages), and the number of F4/80‐positive cells was quantified. (D) The liver sections were also costained with CD206 and galectin‐3 or CD206 and CD11b. The percentages of positive cells were quantified. (E) The expression of cd206 and cd163 was assessed by qRT‐PCR. The percentages of positive cells were quantified and shown. Data represent means ± SD. *p < 0.033, **p < 0.002, ***p < 0.001; Student t‐test. Scale bar, 100 μm. CCl4, carbon tetrachloride; CCN1, cellular communication network factor 1; DAPI, 4′,6‐diamidino‐2‐phenylindole; Ly6G, lymphocyte antigen 6 complex locus G6D; MPO, myeloperoxidase; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; Rel., relative; mRNA, messenger RNA; Tgfb1, transforming growth factor‐β1; WT, wild type.
The macrophages in CCl4‐injured livers consisted of both resident KCs (galectin‐3+) and monocyte‐derived cells (CD11b+; Figure 5C). However, both types of macrophages expressed M2 marker genes in Ccn1 WT/WT livers to a much greater extent than in Ccn1 D125A/D125A livers, as indicated by CD206 staining and expression of Cd206 and Cd163 (Figure 5C,D). Similar findings were also observed in liver macrophages isolated from these strains of mice (Figure S5B). These findings suggest that both resident and monocyte‐derived macrophages participate in efferocytosis following injury, thereby undergoing M2 phenotypic change. Thus, CCN1 induces the removal of apoptotic neutrophils by macrophages after liver injuries, and this efferocytosis process drives the production of fibrogenic cytokines, including TGF‐β1 (Figures 3 and 4).
CCN1‐induced liver macrophage efferocytosis leads to HSC activation
The above findings suggest that CCN1 stimulated macrophage efferocytosis of apoptotic cells after liver injury, resulting in increased activated TGF‐β1, which in turn induced HSC transdifferentiation into fibrogenic myofibroblastic cells. To test this hypothesis, we isolated macrophages from uninjured Ccn1 WT/WT mice and cultured them in the presence or absence of apoptotic neutrophils, with or without the addition of CCN1 for 4 hours (Figure 6A). When either CCN1 or apoptotic neutrophils were added to Ccn1 WT/WT macrophages from uninjured mice, there was little effect on Tgfb1 expression, but when both were added together, allowing efferocytosis, a large increase in Tgfb1 expression was observed (Figure 6A). Concomitantly, the expression of CD206 was increased, indicating an M2 switch.
FIGURE 6.
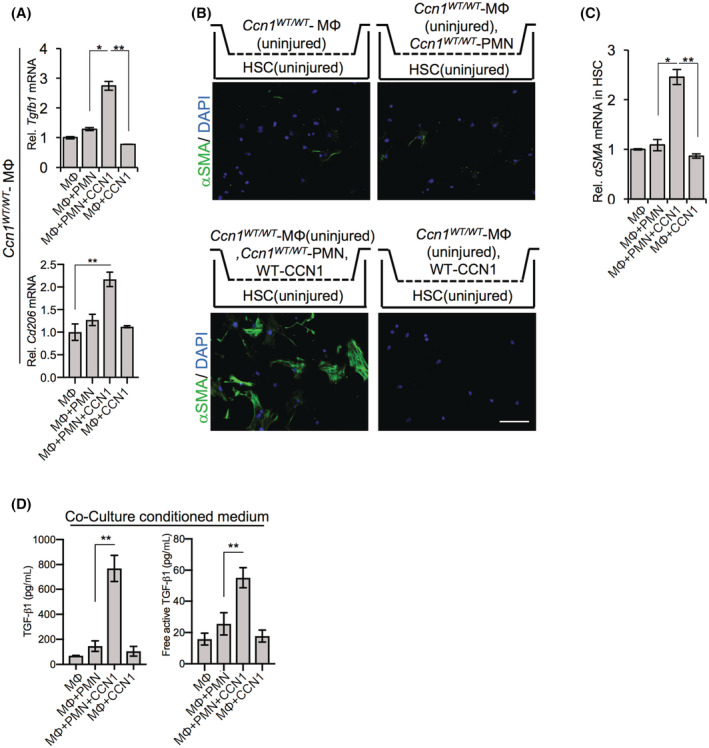
Liver macrophage efferocytosis leads to myofibroblast differentiation of HSCs. Coculture of liver macrophages with HSCs. (A) HSCs isolated from uninjured Ccn1 D125A/D125A mice were seeded on the bottom chamber of a Transwell and cocultured with macrophages in the top chamber isolated from Ccn1 WT/WT and Ccn1 D125A/D125A mice 48 hours after injection with CCl4. After 5 days of coculture, HSCs were stained with anti‐αSMA antibody. (B) αSMA mRNA levels were quantified by qRT‐PCR. (C) Anti‐TGF‐β1 antibody or control IgG was added to co‐cultures of HSCs and macrophages above. HSCs were stained with anti‐αSMA antibody after 5 days of culture. (D) αSMA mRNA levels of HSCs were evaluated by qRT‐PCR. Apoptotic neutrophils (PMN) from Ccn1 WT/WT mice were pretreated with or without CCN1 protein for 60 min before incubation with macrophages from uninjured Ccn1 WT/WT liver for 4 hours. (E) Tgfb1 and Cd206 expression of the above macrophages was assessed by qRT‐PCR. (F) Total (upper) or free active (lower) TGF‐β1 in the indicated conditional medium were quantified by enzyme‐linked immunosorbent assay after culturing for 5 days. Macrophages isolated from uninjured Ccn1 WT/WT liver were incubated with apoptotic neutrophils and/or CCN1 protein in a Transwell insert (upper chamber) for 4 hours before being cocultured with HSCs from uninjured Ccn1 D125A/D125A mice in the lower chamber. (G) After 5 days of coculture, the HSCs were stained with anti‐αSMA antibodies. Only cocultures with macrophages and neutrophils and CCN1 showed myofibroblastic differentiation. (H) αSMA mRNA from above was measured. Data represent means ± SD.*p < 0.033, **p < 0.002; Student t test. Scale bar, 100 μm. CCl4, carbon tetrachloride; CCN1, cellular communication network factor 1; HSC, hepatic stellate cell; MФ, macrophage; mRNA, messenger RNA; PMN, polymorphonuclear; qRT‐PCR, quantitative reverse‐transcription polymerase chain reaction; Rel., relative; Tgfb1, transforming growth factor‐β1; WT, wild type; αSMA, alpha smooth muscle actin.
We then cocultured liver macrophages and HSCs in a Transwell system in which these cells were grown in separate chambers but shared the culture media and exchanged soluble factors. HSCs from uninjured Ccn1 WT/WT mice were seeded in the bottom chamber and cocultured with liver macrophages isolated from uninjured Ccn1 WT/WT mice in the top chamber. In such cocultures, macrophages did not induce myofibroblastic differentiation of HSCs by themselves, even in the presence of either apoptotic neutrophils or CCN1 protein (Figure 6B,C). However, in the presence of both apoptotic neutrophils and CCN1, there was a significant induction of HSC myofibroblastic differentiation (Figure 6B,C), suggesting that CCN1‐induced efferocytosis drives HSC activation. Correspondingly, high levels of total and free active TGF‐β1 were found only in conditioned media of cocultures that included both CCN1 protein and apoptotic neutrophils (Figure 6D), consistent with the induction of HSC activation by TGF‐β1. Whereas the liver macrophages from uninjured liver used in these experiments are mostly resident KCs (Figure S1A), a substantial number of monocyte‐derived macrophages are recruited following injury (Figure 5C; Figure S5B).[ 33 ] We found that bone marrow‐derived macrophages also express TGF‐β1 and M2 marker genes following exposure to CCN1 and apoptotic neutrophils, thereby acquire the ability to induce HSC activation (Figure S6A–C). These results indicate that CCN1 stimulates liver macrophage efferocytosis of apoptotic neutrophils to induce the production of activated TGF‐β1, which triggers HSC transdifferentiation into myofibroblastic cells.
DISCUSSION
This study has revealed a surprising role for the matricellular protein CCN1 in liver fibrogenesis. CCN1 acts as a bridging molecule to stimulate the phagocytic clearance of apoptotic neutrophils by macrophages through integrin αvβ3, leading to the elevated production of activated TGF‐β1, which induces the differentiation of HSCs into myofibroblast‐like cells critical for liver fibrogenesis. Ccn1 D125A/D125A knock‐in mice expressing a point mutation rendering CCN1 unable to bind integrin αvβ3 are deficient for neutrophil clearance, resulting in reduced expression of activated TGF‐β1, diminished HSC activation, and markedly reduced fibrosis after chronic liver injuries from CCl4 or cholestasis. These results underscore the key role of CCN1 in accelerating neutrophil clearance and the important contribution of CCN1 in macrophage efferocytosis in liver fibrogenesis. These findings provide molecular details to the observations that macrophages are profibrotic in the early phase of injury response and that macrophage engulfment of apoptotic bodies promotes the expression of profibrotic cytokines.[ 8 , 34 ]
CCN1 is quickly released from platelet α‐granules following tissue damage.[ 35 ] It is also encoded by an immediate early gene that is rapidly activated in various cell types by a variety of cellular stimuli.[ 10 ] Thus, CCN1 is promptly available at the sites of injury where it plays a surprisingly broad spectrum of roles in repair (Figure 7). In addition to acting as a bridging molecule to stimulate macrophage clearance of apoptotic cells,[ 15 ] CCN1 also functions as a powerful opsonin to stimulate the phagocytic removal of gram‐positive and gram‐negative bacteria.[ 36 ] Thus, CCN1 participates in bacterial clearance during injury, which is also a key function of neutrophils. However, as neutrophils are short lived, CCN1 helps to remove the apoptotic neutrophils by inducing efferocytosis. CCN1‐induced efferocytosis leads to polarization of macrophages and TGF‐β1 expression, activation of HSCs, and ECM production to promote tissue integrity during injury repair (Figure 7). In addition, CCN1 can also stimulate the proliferation of specific cell types, including cholangiocytes, to promote regeneration.[ 12 ]
FIGURE 7.
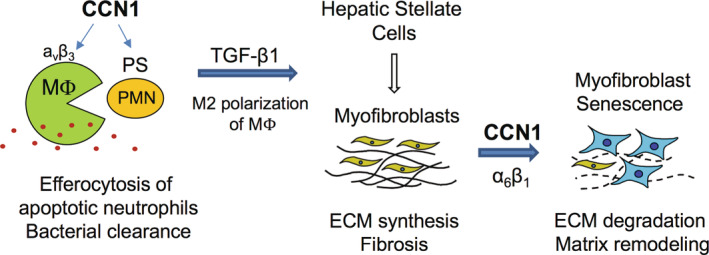
Schematic model of profibrotic and antifibrotic functions of CCN1 in liver injury. In the initial stages of injury, CCN1 acts as an opsonin for bacterial clearance[ 36 ] and induces the clearance of apoptotic neutrophils (PMN) by stimulating their phagocytosis through the engagement of integrin αvβ3 in macrophages.[ 15 ] Efferocytosis leads to the expression of activated TGF‐β1, which in turn induces HSC transdifferentiation to myofibroblastic cells that promote fibrosis. In later stages of injury repair, CCN1 accumulates and binds to integrin α6β1 in myofibroblasts to trigger cellular senescence, after which the senescent myofibroblasts express matrix‐degrading enzymes to promote fibrosis resolution.[ 11 , 16 ] CCN1, cellular communication network factor 1; ECM, extracellular matrix; HSC, hepatic stellate cell; MФ, macrophage; PMN, polymorphonuclear; PS, phosphatidylserine; TGF‐β1, transforming growth factor‐β1.
Paradoxically, CCN1 also has an antifibrotic function that occurs in later stages of tissue repair. CCN1 can trigger myofibroblast senescence through direct binding to integrin α6β1, after which the senescent myofibroblasts secrete matrix‐degrading enzymes and down‐regulate collagen as part of the senescence‐associated secretory phenotype.[ 11 , 16 , 37 ] Thus, the profibrotic function of CCN1 occurs early in the injury response when CCl4 toxicity induces cell death and a high level of neutrophil infiltration in the pericentral region, [38 ] and CCN1 acts by binding integrin αvβ3 in macrophages.[ 15 ] CCN1 is accumulated within 48 hours of CCl4 exposure in the central vein region (Figure S2C) where it can induce efferocytosis. Myofibroblasts are rare in the early phase of injury. By contrast, the antifibrotic function of CCN1 occurs late in the injury response when the myofibroblasts level is high, [39 ] and CCN1 induces their senescence by binding integrin α6β1. [ 11 ] Interestingly, HSCs express a much higher level of integrin α6 as they transdifferentiate into myofibroblasts (Figure S7), facilitating CCN1 binding to evoke senescence. Thus, whereas CCN1 is profibrotic by stimulating efferocytosis in the early injury response where it is antifibrotic by inducing myofibroblast senescence in the late stages of injury response (Figure 7).
Similarly, multiple subsets of macrophages exist in the liver that can play both profibrotic and antifibrotic roles during injury repair.[ 40 , 41 ] In the early phase of injury, circulating
lymphocyte antigen 6 complex, locus C (Ly6Chi) monocytes are recruited to the liver and differentiate into macrophages that play a proinflammatory and profibrotic role in the early stages of injury repair.[ 33 ] Following phagocytosis of cell debris, these cells phenotypically switch to Ly6clo macrophages and promote fibrosis resolution by elevating the expression of MMPs.[ 42 ] Interestingly, the expression of Tgfb1 in Ly6Chi monocytes changes very little as they transition to Ly6clo monocytes.[ 42 ] Here, we found that liver macrophages, which are comprised of both resident KCs and recruited monocyte‐derived macrophages (Figure 5C), express a high level of Tgfb1 in response to efferocytosis capable of inducing HSC transdifferentiation into myofibroblasts (Figures 3 and 6; Figure S6). Indeed, the profibrogenic roles of both KCs and monocyte‐derived macrophages have been observed.[ 7 ] Ccn1 D125A/D125A mice are deficient in neutrophil clearance, expression of activated TGF‐β1, myofibroblastic differentiation of HSCs, and fibrotic change after CCl4 exposure, underscoring the role of CCN1‐induced efferocytosis as a driver for fibrogenesis.
Neutrophils can also play diverse roles in liver fibrosis, including activation of HSCs, through the production of reactive oxygen species (ROS), MPO, and interleukin‐17, thus enhancing fibrosis.[ 43 , 44 , 45 ] On the other hand, neutrophils can also promote fibrosis resolution by the expression of MMPs[ 46 ] as well as the polarization of macrophages to the proresolving phenotype through ROS and microRNA‐223.[ 47 , 48 ] High levels of neutrophils in the early phase of injury correlate with fibrogenesis,[ 38 ] yet high neutrophils in later stages promote resolution.[ 46 ] Thus, neutrophils can also exert profibrotic and antifibrotic effects in liver fibrosis. Interestingly, the metabolic functions of neutrophils may be distinct from those of the apoptotic bodies that they leave behind. Consistent with this interpretation, fibrosis was enhanced when neutrophils were depleted by anti‐Ly6G monoclonal antibodies during the recovery phase of CCl4 injury, possibly because the cell debris from neutrophil death fueled liver macrophage phagocytosis and consequent HSC activation.[ 48 ]
Ccn1 D125A/D125A knock‐in mice develop minimal fibrosis in response to chronic CCl4 injury and cholestasis, suggesting that potential therapeutic interventions blocking CCN1 functions in efferocytosis might ameliorate fibrosis. However, because CCN1 performs multiple roles in liver injury repair, albeit acting through distinct integrins and following disparate cell types (Figure 7), assessing whether blockade of CCN1 activities may have adverse consequences warrants further investigation.
CONFLICT OF INTEREST
Nothing to report.
Supporting information
Appendix S1
Kim K‐H, Cheng N, Lau LF. Cellular communication network factor 1‐stimulated liver macrophage efferocytosis drives hepatic stellate cell activation and liver fibrosis. Hepatol Commun. 2022;6:2798–2811. 10.1002/hep4.2057
Ki‐Hyun Kim and Naiyuan Cheng contributed equally to this work.
REFERENCES
- 1. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18:151–66. [DOI] [PubMed] [Google Scholar]
- 2. Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol. 2007;127:514–25. [DOI] [PubMed] [Google Scholar]
- 3. Kisseleva T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology. 2017;65:1039–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang W, He H, Wang T, Su N, Zhang F, Jiang K, et al. Single‐cell transcriptomic analysis reveals a hepatic stellate cell‐activation roadmap and myofibroblast origin during liver fibrosis in mice. Hepatology. 2021;74:2774–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411. [DOI] [PubMed] [Google Scholar]
- 6. Dooley S, ten Dijke P. TGF‐beta in progression of liver disease. Cell Tissue Res. 2012;347:245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases‐diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. 2021;18:45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jun JI, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov. 2011;10:945–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lau LF. CCN1/CYR61: the very model of a modern matricellular protein. Cell Mol Life Sci. 2011;68:3149–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jun JI, Lau LF. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol. 2010;12:676–85. Erratum in: Nat Cell Biol. 2010;12:1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim KH, Chen CC, Alpini G, Lau LF. CCN1 induces hepatic ductular reaction through integrin αvβ5‐mediated activation of NF‐κB. J Clin Invest. 2015;125:1886–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feng T, Meng J, Kou S, Jiang Z, Huang X, Lu Z, et al. CCN1‐induced cellular senescence promotes heart regeneration. Circulation. 2019;139:2495–8. [DOI] [PubMed] [Google Scholar]
- 14. Choi JS, Kim KH, Lau LF. The matricellular protein CCN1 promotes mucosal healing in murine colitis through IL‐6. Mucosal Immunol. 2015;8:1285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jun JI, Kim KH, Lau LF. The matricellular protein CCN1 mediates neutrophil efferocytosis in cutaneous wound healing. Nat Commun. 2015;6:7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim KH, Chen CC, Monzon RI, Lau LF. The matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol. 2013;33:2078–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kountouras J, Billing BH, Scheuer PJ. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br J Exp Pathol. 1984;65:305–11. [PMC free article] [PubMed] [Google Scholar]
- 18. Jelnes P, Santoni‐Rugiu E, Rasmussen M, Friis SL, Nielsen JH, Tygstrup N, et al. Remarkable heterogeneity displayed by oval cells in rat and mouse models of stem cell‐mediated liver regeneration. Hepatology. 2007;45:1462–70. [DOI] [PubMed] [Google Scholar]
- 19. Boxio R, Bossenmeyer‐Pourie C, Steinckwich N, Dournon C, Nusse O. Mouse bone marrow contains large numbers of functionally competent neutrophils. J Leukoc Biol. 2004;75:604–11. [DOI] [PubMed] [Google Scholar]
- 20. Weiskirchen R, Gressner AM. Isolation and culture of hepatic stellate cells. Methods Mol Med. 2005;117:99–113. [DOI] [PubMed] [Google Scholar]
- 21. Debacq‐Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence‐associated beta‐galactosidase (SA‐betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4:1798–806. [DOI] [PubMed] [Google Scholar]
- 22. Kliment CR, Englert JM, Crum LP, Oury TD. A novel method for accurate collagen and biochemical assessment of pulmonary tissue utilizing one animal. Int J Clin Exp Pathol. 2011;4:349–55. [PMC free article] [PubMed] [Google Scholar]
- 23. Chen N, Leu S‐J, Todorovic V, Lam SC‐T, Lau LF. Identification of a novel integrin alphavbeta3 binding site in CCN1 (CYR61) critical for pro‐angiogenic activities in vascular endothelial cells. J Biol Chem. 2004;279:44166–76. [DOI] [PubMed] [Google Scholar]
- 24. Constandinou C, Henderson N, Iredale JP. Modeling liver fibrosis in rodents. Methods Mol Med. 2005;117:237–50. [DOI] [PubMed] [Google Scholar]
- 25. Preisegger KH, Factor VM, Fuchsbichler A, Stumptner C, Denk H, Thorgeirsson SS. Atypical ductular proliferation and its inhibition by transforming growth factor beta1 in the 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine mouse model for chronic alcoholic liver disease. Lab Invest. 1999;79:103–9. [PubMed] [Google Scholar]
- 26. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leu S‐J, Liu Y, Chen N, Chen CC, Lam SC, Lau LF. Identification of a novel integrin α6β1 binding site in the angiogenic Inducer CCN1 (CYR61). J Biol Chem. 2003;278:33801–8. [DOI] [PubMed] [Google Scholar]
- 28. Borkham‐Kamphorst E, Weiskirchen R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev. 2016;28:53–61. [DOI] [PubMed] [Google Scholar]
- 29. Yu Q, Stamenkovic I. Cell surface‐localized matrix metalloproteinase‐9 proteolytically activates TGF‐beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–76. [PMC free article] [PubMed] [Google Scholar]
- 30. Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1‐MMP‐dependent activation of TGF‐beta1. J Cell Biol. 2002;157:493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF‐beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huynh ML, Fadok VA, Henson PM. Phosphatidylserine‐dependent ingestion of apoptotic cells promotes TGF‐beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology. 2009;50:261–74. [DOI] [PubMed] [Google Scholar]
- 34. Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology. 2003;38:1188–98. [DOI] [PubMed] [Google Scholar]
- 35. Hviid CV, Samulin Erdem J, Drechsler S, Weixelbaumer K, Ahmed MS, Attramadal H, et al. The matricellular “cysteine‐rich protein 61” is released from activated platelets and increased in the circulation during experimentally induced sepsis. Shock. 2014;41:233–40. [DOI] [PubMed] [Google Scholar]
- 36. Jun JI, Lau LF. CCN1 is an opsonin for bacterial clearance and a direct activator of Toll‐like receptor signaling. Nat Commun. 2020;11:1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jun JI, Lau LF. Resolution of organ fibrosis. J Clin Invest. 2018;128:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Louis H, Van Laethem JL, Wu W, Quertinmont E, Degraef C, Van den Berg K, et al. Interleukin‐10 controls neutrophilic infiltration, hepatocyte proliferation, and liver fibrosis induced by carbon tetrachloride in mice. Hepatology. 1998;28:1607–15. [DOI] [PubMed] [Google Scholar]
- 39. Georges PC, Hui JJ, Gombos Z, McCormick ME, Wang AY, Uemura M, et al. Increased stiffness of the rat liver precedes matrix deposition: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1147–54. [DOI] [PubMed] [Google Scholar]
- 40. Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60:1090–6. [DOI] [PubMed] [Google Scholar]
- 42. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly‐6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109:E3186–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pulli B, Ali M, Iwamoto Y, Zeller MW, Schob S, Linnoila JJ, et al. Myeloperoxidase‐hepatocyte‐stellate cell cross talk promotes hepatocyte injury and fibrosis in experimental nonalcoholic steatohepatitis. Antioxid Redox Signal. 2015;23:1255–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou Z, Xu MJ, Cai Y, Wang W, Jiang JX, Varga ZV, et al. Neutrophil‐hepatic stellate cell interactions promote fibrosis in experimental steatohepatitis. Cell Mol Gastroenterol Hepatol. 2018;5:399–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fabre T, Molina MF, Soucy G, Goulet JP, Willems B, Villeneuve JP, et al. Type 3 cytokines IL‐17A and IL‐22 drive TGF‐beta‐dependent liver fibrosis. Sci Immunol. 2018;3:eaar7754. [DOI] [PubMed] [Google Scholar]
- 46. Saijou E, Enomoto Y, Matsuda M, Yuet‐Yin Kok C, Akira S, Tanaka M, et al. Neutrophils alleviate fibrosis in the CCl4‐induced mouse chronic liver injury model. Hepatol Commun. 2018;2:703–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang W, Tao Y, Wu Y, Zhao X, Ye W, Zhao D, et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat Commun. 2019;10:1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Calvente CJ, Tameda M, Johnson CD, Del Pilar H, Lin YC, Adronikou N, et al. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA‐223. J Clin Invest. 2019;129:4091–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1