

Abstract
Methamphetamine (METH) is an addictive illicit drug used worldwide that causes significant damage to blood vessels resulting in cardiovascular dysfunction. Recent studies highlight increased prevalence of cardiovascular disease (CVD) and associated complications including hypertension, vasospasm, left ventricular hypertrophy, and coronary artery disease in younger populations due to METH use. Here we report that METH administration in a mouse model of ‘binge and crash’ decreases cardiovascular function via cystathionine gamma lyase (CSE), hydrogen sulfide (H2S), nitric oxide (NO) (CSE/H2S/NO) dependent pathway. METH significantly reduced H2S and NO bioavailability in plasma and skeletal muscle tissues co-incident with a significant reduction in flow-mediated vasodilation (FMD) and blood flow velocity revealing endothelial dysfunction. METH administration also reduced cardiac ejection fraction (EF) and fractional shortening (FS) associated with increased tissue and perivascular fibrosis. Importantly, METH treatment selectively decreased CSE expression and sulfide bioavailability along with reduced eNOS phosphorylation and NO levels. Exogenous sulfide therapy or endothelial CSE transgenic overexpression corrected cardiovascular and associated pathological responses due to METH implicating a central molecular regulatory pathway for tissue pathology. These findings reveal that therapeutic intervention targeting CSE/H2S bioavailability may be useful in attenuating METH mediated cardiovascular disease.
Keywords: Cystathionine gamma lyase, Hydrogen sulfide, Nitric oxide, Methamphetamine, Endothelial dysfunction, Cardiomyopathy
Graphical abstract
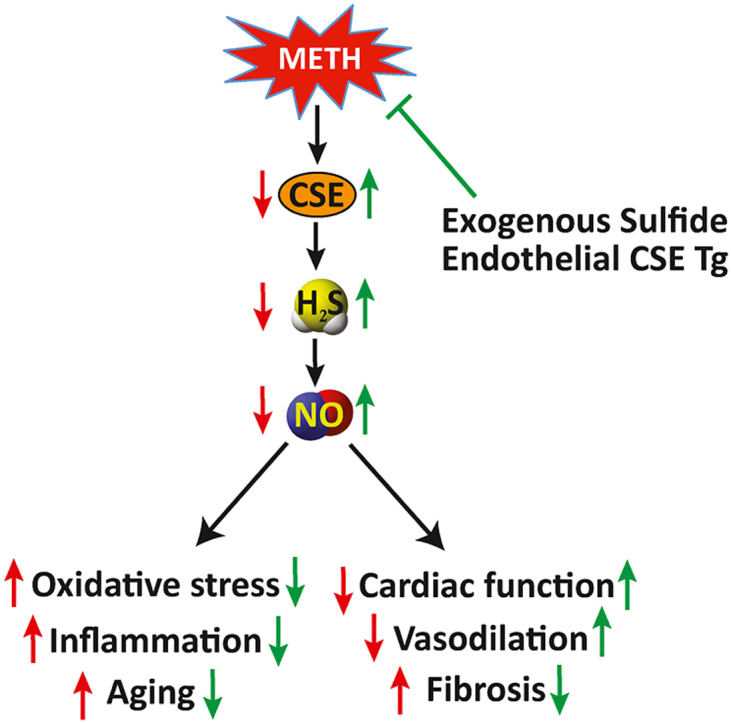
1. Introduction
Methamphetamine (METH) is a widely addictive drug with severe psychological and social risks. Statistics from the National Survey on Drug Use and Health (NSDUH) reveal that an estimated 24.6 million Americans ages 12 or older have used METH in their lifetimes for non-medical reasons [1,2]. Recent studies have revealed increased prevalence of cardiovascular disease (CVD) due to METH use, making cardiovascular complications the second leading cause of death in METH substance users [3]. Importantly, METH use disproportionally increases cardiovascular-related morbidity and mortality in up to three-fourths of its users [4]. However, molecular mechanisms of METH-related cardiovascular pathophysiology remain largely unknown.
The vascular endothelium plays a central role in maintaining cardiovascular homeostasis. Disruption of endothelial function, clinically referred to as endothelial dysfunction, is a key pathophysiological mediator of nearly all cardiovascular disease. Endothelial dysfunction is a hallmark prerequisite of cardiovascular complications including atherosclerosis, hypertension, myocardial infarction, and diabetes. METH can have adverse and potentially fatal effects on arteries and blood vessels, which contribute to increased blood pressure, inflammation and cardiovascular dysfunction including atherosclerosis [[5], [6], [7]]. METH also induces pro-inflammatory signaling responses and increases the production of reactive oxygen species that are detrimental to the cardiovascular system [8]. Thus, METH may lead to cardiovascular disease with endothelial dysfunction as a chief pathological mediator.
Hydrogen sulfide (H2S) and nitric oxide (NO) are gaseous signaling molecules that serve important roles in regulating endothelial and cardiovascular health. Evidence from our group and others has revealed that cystathionine gamma lyase (CSE) and H2S play pivotal regulatory roles in various cardiovascular pathophysiological functions including normal endothelial function and protection against oxidative damage. We and others have also shown that vascular H2S bioavailability directly influences NO bioavailability [[9], [10], [11]]. Also, prolonged exposure of METH can stimulate oxidative and inflammatory events and that are known contributors to endothelial dysfunction thereby accelerating cardiovascular disease. Moreover, while previous studies have been reported examining the relationship between METH and NO, they have primarily focused on neuronal injury with no clear understanding of impacts on vascular function [12]. The objective of this study was to investigate the effect of METH use on H2S and NO production and whether this mediates associated cardiovascular pathology and dysfunction.
2. Materials and methods
2.1. Chemicals and reagents
Chemicals and tissue culture reagents, including Methamphetamine hydrochloride (METH) were obtained from Sigma unless otherwise noted. Anhydrous sodium sulfide was purchased from Alfa‐Aesar Inc. Anti‐CD31 antibody was from BD Biosciences (San Jose, CA, USA), and anti-α-SMA antibody was obtained from Sigma-Aldrich. Vectashield plus DAPI was from Vector Laboratories. All secondary fluorophore‐labeled antibodies were obtained from Jackson Immunoresearch Inc (West Grove, PA, USA).
2.2. Mouse model and treatment routes
All animal studies were approved by the LSU Institutional Animal Care and Use Committee (LSU IACUC Protocol# P-08-021) and in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and ARRIVE guidelines. Twelve-week-old male WT (C57BL6/J) and endothelial cell CSE transgenic (ecCSE Tg) male mice [13] were used to study 'Binge' METH administration effects on endovascular function. Mice were randomly assigned to different experimental groups by one investigator and were treated and evaluated by a second blinded investigator. Na2S drinking solutions were made by dissolving the appropriate amount of Na2S with the appropriate volume of tap water for final Na2S concentration (50 μM). Sulfide donor, Na2S 3.9 μg/ml was administered in the drinking water of mice treated with either METH or saline control during the length of the study. Drinking water containing sulfide was changed out every two days and Supplementary Fig. 1 illustrates sulfide levels in drinking water over a 2-day period. Mice were housed at the Louisiana State University Health Sciences Center-Shreveport animal resources facility, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
2.3. ‘Binge and crash’ mouse METH model
Mice were exposed to methamphetamine according to a published protocol that models binge methamphetamine exposure in humans [14]. C57BL/6J male mice received 0–6 mg/kg METH (Methamphetamine HCl, Sigma-Aldrich, St. Louis, MO) through subcutaneous (s.c.) injection five days a week for four weeks. METH was dissolved in a sterile saline (Sigma-Aldrich) solution (0.9% w/v NaCl). Control cohort mice received the same volume of saline alone injection at all-time points for four weeks. METH–treated and saline–control mice were injected subcutaneously with either treatment in a volume of 125 μl (5 ml/kg) with an insulin syringe. Alternating sites of injection were used to avoid any possible damage to tissue and stress to the animal. The dose of METH was escalated over the course of the first cycle, which occurs during the first week of injections (days 1–5) followed by three weeks of repeated cycles of meth injections (days 8–12, 15–19, and 22–26). Mice received 4 injections per day, 2 h apart with doses of METH including 0, 1, 2, 3, 4, 5, and 6 mg/kg subcutaneously as shown in Supplementary Fig. 2. At the end of the 4 weeks, mice were sacrificed by isoflurane overdose and plasma and tissue were collected.
2.4. Echocardiography
Cardiac functional parameters were assessed in mice under isoflurane‐anesthesia with a VisualSonics Vevo 3100 Imaging System (Toronto, ON, Canada) using a 40‐MHz transducer as described [15]. Briefly, 2‐dimensional M‐mode echocardiographic images along the parasternal short axis were recorded by investigators blinded to treatments or genotype to determine LV size and systolic function. Measurements of the LV internal dimensions in diastole and systole (LVID; d and LVID; s, respectively), diastolic thickness of LV posterior wall and diastolic intraventricular septum thickness were recorded. Percent fractional shortening was calculated as [(LVID; d−LVID; s)/LVID; d] × 100. LV mass and volumes were determined from M‐mode measurements using VisualSonics (Toronto, ON, Canada) software.
2.5. Flow mediated dilation (FMD)
Vascular function was determined by mouse FMD as previously reported method [16]. Experimental cohorts of Control (saline), METH (0–6 mg/kg) with or without sulfide drinking water or ecCSE Tg mice, were treated for 4 weeks, and subjected to FMD. Briefly, mice were anesthetized with isoflurane and fur was removed from the hindlimbs. The animals were then placed on a warmed ultrasound table equipped with ECG. A vascular occluder (5 mm diameter, Harvard Apparatus) was placed around the proximal hindlimb to induce transient occlusion of the vessels of the distal hindlimb as an ischemic trigger. The Doppler ultrasound probe (VEVO 3100, VisualSonics) was manually aligned over the femoral artery, distal to the occluder, to take baseline recordings of the vessel for diameter (M mode) and mean velocity (PW mode). The vascular occluder was inflated manually with an air-filled syringe for 1-min, and then deflated. Measurements of diameter and blood flow velocity were recorded for 180s at 30s intervals. The recorded loops were analyzed by Vevo LAB analysis software.
2.6. Measurement of biological pools of H2S
Plasma samples from mouse were analyzed for acid-labile sulfide (ALS), bound sulfane sulfur (BSS), and total sulfide levels using the monobromobimane (MBB) method as we have previously reported [17]. Detection of ALS and BSS was performed with 50 μl plasma processed separately into two sets of 4 mL BD vacutainer tubes. Plasma was added with 100 mM phosphate buffer (pH 2.6, 0.1 mM DTPA) for the ALS reaction, and 100 mM phosphate buffer (pH 2.6, 0.1 mM DTPA) containing 1 mM TCEP for the total sulfide reaction. Following a 30-min incubation on a nutator mixer, to trap the evolved sulfide gas and incubation with 100 mM Tris-HCl buffer (pH 9.5, 0.1 mM DTPA). Determination of individual sulfide metabolites were calculated as previously described [17].
2.7. NO metabolite measurements
NO metabolites (NOx) were measured using an ozone-based chemiluminescent assay (Sievers Nitric Oxide Analyzer 280i, Weddington, NC) as described previously [18]. Plasma and skeletal muscle tissue samples were collected in NO preservation buffer (1.25 mol/L potassium ferricyanide, 56.9 mmol/L N-ethylmaleimide, 6% Nonidet P-40 substitute in PBS). Aliquots of samples were analyzed separately for free nitrite, and nitrosothiol following a reaction with acidic sulfanilamide solution (0.5% v/v) in the dark for 15 min prior to injection into the analyzer.
2.8. Cystathionine γ-lyase (CSE) activity measurement
CSE activity was measured as previously reported [10]. Femoral artery lysates were incubated with 2 mM cystathionine, 0.25 mM pyridoxal 5′-phosphate in 100 mM Tris-HCl buffer (pH 8.3) for 30 min at 37 °C. 20% Trichloroacetic acid was added into reaction mixture. After centrifugation, the supernatant was mixed with 2% ninhydrin reagent and incubated for 5 min at 105 °C then quickly cooled to 4 °C. Samples were then mixed with 97% ethanol and read at 455 nm using a spectrophotometer (Biotek). CSE activity was assessed by cystathionine consumption and enzyme activity expressed as fold change calculated from nanomoles of cystathionine consumed per mg of total protein.
2.9. Western blot analysis
Mouse tissues were homogenized in a solution containing 50 mM Tris buffer (pH 7.4), 2 mM EDTA, 5 mM EGTA, 0.1% SDS, a protease inhibitor cocktail (Roche, Indianapolis, IN), and phosphatase inhibitor cocktail type I and II (Sigma, Saint Louis, MO). Homogenates were centrifuged at 500×g for 15 min and supernatants were collected. Protein concentrations were analyzed using the Bradford protein assay (BioRad, Hercules, CA). Proteins were separated using 10% SDS-PAGE (Bio-Rad, Hercules, CA) and transferred onto PVDF membranes, and incubated with antibodies against CSE catalogue # 12217-1-AP (Fisher Scientific), eNOS catalogue #9572, phospho-eNOS catalogue #9750, and α/β-Tubulin catalogue #2148 (Cell Signaling). Chemiluminescent bands were detected and quantified using NIH Image J software.
2.10. Quantitative PCR
Tissue samples were stored in TRIzol reagent (Thermo Fisher Scientific Inc., Waltham, MA, USA) and RNA was isolated using phenol:chloroform extraction procedure. RNA concentration and purity were evaluated with a NanoDrop 2000 spectrometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). RNA samples with an absorbance ratio OD 260/280 between 1.8 and 2.1 and OD 260/230 between 2 and 2.2 were used for further analysis. Single-stranded cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA), from 1 μg of total RNA in a final volume of 20 μL cDNA was stored at −20 °C for future use. Quantitative PCR reactions were performed using the universal SYBR Green Supermix (Bio-Rad, CA, USA) on a CFX96 thermal cycler with Bio-Rad CFX Manager software (Bio-Rad, Hercules, CA, USA). 50 ng of cDNA were used for each reaction. The mean threshold cycle (Ct) values for each serial dilution was plotted against the logarithm of the cDNA dilution factor. Quntitative PCR primers for genes, including GAPDH, CSE, CBS, eNOS, ICAM-1, VCAM-1, NOX4, gp91phox, p47phox, ATF4, SIRT1 and SIRT6 were used in this study (Supplementary Table 1).
2.11. Masson's trichrome fibrosis staining
Mice paraffin-embedded heart sections were cut in serial 5 μm thin sections, deparaffinized, hydrated, and stained with Masson's Trichrome to assess the extent of fibrotic area [19]. Heart sections were stained and imaged on an Olympus IX43 microscope using bright field mode with a × 20 objective lens in an investigator-blinded manner. Measurements were made of the percent fibrosis area (stained blue with Masson's Trichrome), as a total of the stained myocardium area (μm2) within each microscopic field using the National Institutes of Health (NIH) ImageJ (v1.53r) software (Bethesda, MD) [19]. All quantifications were analyzed in a blinded manner using randomly selected 10–15 high-magnification microscopic fields from each mouse heart section.
2.12. Oxidative stress measurement
Levels of oxidative stress were measured by staining skeletal muscle tissue sections with 5 μM Dihydroethidium (DHE; Sigma-Aldrich, CA USA) as previously reported [20]. Sections were visualized using a Nikon Eclipse Ti-E fluorescence microscope (Nikon Instruments Inc., Melville, NY) and analyzed using Simple PCI software version 6.0 (Compix Inc., Sewickley, PA, USA).
2.13. Statistical analysis
Data were reported as mean ± standard error of the mean (SEM) for all groups. Power calculation analysis at 0.8 level for a minimum 25% difference in various biological and biochemical readouts was performed as we've previously reported [10,16,19]. Statistical analysis was performed with GraphPad Prism using Student's t-test, one-way ANOVA with Tukey post-hoc multiple-comparison tests. A p-value of <0.05 was considered statistically significant.
3. Results
3.1. METH experimental ‘binge and crash’ model causes endothelial cell dysfunction
Arterial vessels are significantly affected by METH use resulting in cardiovascular dysfunction and central nervous system damage [21]. We used a non-invasive flow-mediated dilation (FMD) model in mice to assess femoral artery vascular function at the end of the 4-week ‘binge and crash’ METH treatment protocol. Fig. 1A and B shows reactive dilation blood flow responses between saline control versus METH treated mice, respectively. Panel 1C reports that METH treated mice have significantly blunted flow mediated vasodilation responses with 30 s compared to saline treated control mice. Panel 1D also illustrates that METH treatment significantly blunts mean blood flow velocity recovery at 5 min after occlusion removal. These data show that the binge and crash METH model elicits profound endothelial dysfunction in response to brief tissue ischemia.
Fig. 1.

METH inhibits flow mediated dilation and velocity in femoral artery. Representative doppler images from B-mode and pulse wave velocity of FMD from (A) Saline-treated and (B) METH-treated mice flow mediate dilation of femoral artery. Panel C shows quantitation of percent change in vessel diameter, and (D) blood flow velocity in femoral arteries of control and METH-treated mice, respectively. n = 5 per cohort, ##P < 0.001 vs. saline control.
3.2. METH ‘binge and crash’ decreases hydrogen sulfide and nitric oxide bioavailability
Plasma H2S and NO bioavailability are associated with endothelial health and cardiovascular function [22]. Thus, plasma metabolites of H2S and NO were measured at the end of the 4-week ‘binge and crash’ model of METH treatment. A significant reduction in sulfide metabolites - acid-labile and bound sulfane sulfur pools can be seen in plasma with METH compared to controls (Fig. 2A and B). Similarly, plasma NO metabolites – free nitrite, S-nitrosothiol were decreased in METH treated group (Fig. 2D and E). Importantly, METH caused a 3-fold reduction in overall plasma total sulfide (Fig. 2C) and a two-fold reduction in plasma total NO levels (Fig. 2F). These findings demonstrate that METH treatment elicits a profound reduction in plasma H2S and NO metabolites.
Fig. 2.

METH reduces metabolites of sulfide and NO. Plasma levels of sulfide and NO metabolites were measured in response to METH treatments the following panels illustrate: (A) acid-labile (B) bound sulfane sulfur and (C) total sulfide, (E) free nitrite, (F) s-nitrosothiol, and (G) total NOx, compared to control saline treatment. N = 5–7 per cohort, ###P < 0.0001 vs. saline control; ##P < 0.005 vs. saline control.
3.3. METH ‘binge and crash’ blunts CSE expression and eNOS phosphorylation
H2S is predominantly produced by CSE and CBS enzymes in the vasculature and other tissues, whereas NO bioavailability is enzymatically produced by endothelial nitric oxide synthase (eNOS) [23]. We next examined whether changes in plasma sulfide levels were mediated through enzymatic pathways involving expression of H2S producing enzymes CSE and CBS. METH significantly decreased CSE mRNA expression in gastrocnemius muscle (Fig. 3A); however, no significant changes were observed in mRNA expression of CBS (Fig. 3B) or in eNOS mRNA expression (Fig. 3C). Posttranslational modifications such as phosphorylation of eNOS directly influence NO production and bioavailability [24]. We further examined protein expression of CSE, CBS, p-eNOS and total eNOS (Fig. 3D). Skeletal muscle from METH treated mice showed decreased expressions of CSE protein; yet no change in CBS protein was observed (Fig. 3E and F). Importantly, eNOS total protein was not altered in METH treated skeletal muscle but phospho-eNOS 1177 levels were significantly blunted (Fig. 3G and H). Lastly, a significant decrease in total sulfide and NO levels was also observed in skeletal muscle from METH treated mice (Fig. 3I). These data demonstrate selective reduction of CSE and H2S generation, and eNOS phosphorylation and NO bioavailability that correlates with ischemic limb vascular dysfunction.
Fig. 3.

METH inhibits CSE expression and eNOS phosphorylation. Skeletal muscle tissue was obtained for mRNA and protein analysis from saline control and METH treated mice. qRT-PCR mRNA measurement was performed for (A) CSE (B) CBS and (C) eNOS. Panel D illustrates representative western blot images for CSE, CBS, phospho-eNOS, and total eNOS. Quantitation of protein expression normalized to tubulin is shown for (E) CSE, (F) CBS, (G) eNOS, and (H) p-eNOS (ser1177). Panel I reports skeletal muscle tissue total sulfide and NOx levels between saline control and METH treated mice. n = 4–7 per cohort, ###P < 0.0001 vs. saline control; ##P < 0.001 vs. saline control; #P < 0.05 vs. saline control.
3.4. Endothelial CSE transgenic overexpression or exogenous sulfide rescues METH induced vascular dysfunction
We next examined whether exogenous sulfide or ecCSE Tg mice along with METH treatments could alter endothelial dysfunction. We found a significant recovery of FMD in METH treated mice with either exogenous sulfide therapy or ecCSE Tg overexpression (Fig. 4A). A significant recovery in mean flow velocity (25 and 30% respectively) was also observed after 5 min in METH treated mice with either sulfide therapy or in ecCSE Tg mice, as shown in Fig. 4B. To confirm the effect of either sulfide therapy or ecCSE Tg mice in the vascular system, we measured CSE activity and found a two-fold increased activity in femoral arteries (Fig. 4C). Additionally, we observed an increase in total NOx and sulfide levels in plasma (Fig. 4D and E), and skeletal muscle by three-fold (Fig. 4E and F) compared to METH treated mice. These data reveal that exogenous sulfide therapy or ecCSE Tg overexpression attenuated METH-induced vascular dysfunction.
Fig. 4.

CSE/H2S rescues METH inhibition of vasodilation. Flow mediated vasodilation in response to METH treatment was examined between saline control, METH, METH + Sulfide, or METH + ecCSE transgenic (Tg) cohorts. Panel A shows changes in vessel diameter over different time points after restoration of blood flow. Panel B reports mean blood flow velocity 5 min after dilation responses among cohorts. Panel C illustrates femoral artery CSE activity among the different cohorts. Panels D–G report plasma and tissue Total NOx (D), plasma Total Sulfide (E), and skeletal muscle Total Sulfide (F), and skeletal muscle Total NOx (G), respectively. n = 4–6 per cohort, ##P < 0.001, @@P < 0.001 vs. saline control; *P < 0.05, **P < 0.001, ***P < 0.0001 vs. METH treatment.
3.5. METH ‘binge and crash’ increases skeletal tissue oxidative stress and inflammation
Increased oxidative stress and pro-inflammatory signaling at a cellular level highlight the harmful cascade of events following METH exposure [[25], [26], [27]]. METH-mediated increase in oxidative stress was examined using the fluorescent probe for superoxide, dihydrohydroethidine (DHE) in skeletal muscle using HPLC fluorescent detection, of stained tissue sections from saline or METH treatments, respectively (Fig. 5). There was a significant increase in superoxide levels in skeletal muscle tissues (Fig. 5A) in METH treated mice as shown by abundant DHE fluorescence. Increased DHE fluorescence with METH treatment was attenuated by the administration of exogenous sulfide or in ecCSE Tg mice, respectively (Fig. 5A and B). This corroborates observations from the literature showing that METH increases oxidative stress that may lead to cardiovascular dysfunction [6]. NADPH oxidases (NOX) are major sources of reactive oxygen species in tissues [28]. NOX4 and NOX2 having predominant roles in regulating oxidative stress in the cardiovascular system [29,30]. We observed an insignificant elevation of NOX4 mRNA expression (Fig. 5C); whereas, we found a robust significant increase in NOX2 subunit gp91 and p47 phox expression with METH treatment in skeletal muscles that was remarkably inhibited by either exogenous sulfide or in ecCSE Tg mice (Fig. 5D and E).
Fig. 5.

CSE/H2S reduces METH oxidative stress and inflammation. Oxidative stress and inflammation in response to METH treatment was examined. Plasma (panel A) and skeletal muscle tissue (panel B) superoxide levels in saline control, METH or METH + Sulfide cohorts. Representative skeletal tissue sections from saline control (panel A), METH treatment (panel B), METH + sulfide treatment (panel C), and METH + ecCSETg mice stained with DHE (red) and counterstained with DAPI (blue). Panel E shows image quantitation of DHE staining among the cohorts. Skeletal muscle mRNA levels from saline control, METH, METH + sulfide, and METH + ecCSETg mRNA tissues are shown for ICAM-1 (panel F), VCAM-1 (panel G), NOX4 (panel H), gp91phox (panel I), and p47phox (panel J). n = 5 per cohort, #P < 0.05, ##P < 0.001, ###P < 0.0001 vs. saline treatment; *P < 0.05, **P < 0.001, ***P < 0.0001 vs. METH treatment. Scale bar equals 50 μM. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Chronic METH use can also induce tissue injury involving pro-inflammatory mediators [31,32]. We analyzed for vascular adhesion molecule expression, ICAM-1 and VCAM-1 that critically regulate leukocyte recruitment and atherosclerosis. A concomitant increase in vascular cell adhesion molecule (VCAM-1) and intercellular adhesion molecule (ICAM-1) expression was observed with METH treatment. Importantly, these inflammatory adhesion molecules were significantly reduced by exogenous sulfide or ecCSE Tg (panels 5F and G). Together, these data reveal METH increased oxidative stress and pro-inflammatory phenotypes are corrected by either exogenous sulfide or ecCSE Tg mice.
3.6. METH blunts cardiac function
METH abuse often results in cardiomyopathy, myocardial infarction, and sudden cardiac death [33,34]. Left ventricular function is a crucial echocardiography parameter that corresponds with numerous cardiovascular syndromes resulting in adverse outcomes [35,36]. We next assessed METH-mediated changes in cardiac function using echocardiography on control treated or METH-treated cohorts, as well as METH plus exogenous sulfide or METH treatment of endothelial CSE transgenic mouse cohorts. We observed a 20% decrease in both LVEF (Fig. 6A) and FS (Fig. 6B) in METH treated mice compared to control. This suggests that METH-treated mice developed progressive systolic dysfunction. Notably, cardiac parameters of echocardiography, including LVEF and FS were rectified with exogenous sulfide treatment compared to METH treatment alone (Fig. 6A and B); however, ecCSE Tg mice only significantly improved FS (Fig. 6B). These hemodynamic changes were not accompanied by any significant alterations in diastolic thickness of the interventricular septum and LV posterior wall or increase in LV mass (data not shown) between control and various METH-treatment cohorts. We next evaluated fibrosis deposition in the myocardium following 4 weeks of ‘binge and crash’ METH administration. Heart tissues collected from Control, METH, METH + sulfide and METH + ecCSE Tg mice groups were stained with Masson's Trichrome (Fig. 6C). Image analysis revealed significant upregulation of the percent fibrosis area (Fig. 6D) in METH treatment compared to control. Importantly, exogenous sulfide treatment or ecCSE Tg significantly reduced tissue fibrosis deposition with METH administration (Fig. 6D). NOX2 activation has profound profibrotic effects in the heart in vivo resulting in cardiac dysfunction [37]. Importantly, we observed an increase in gp91phox and p47phox mRNA expression (Fig. 6E and F) similar to skeletal muscle from METH mice (Fig. 5D and E), confirming the association of NOX2 mediated profibrotic phenotype. Importantly, increased cardiac NOX2 expression was significantly abrogated with exogenous sulfide or in ecCSE Tg mice, respectively (Fig. 6E and F).
Fig. 6.

METH blunts cardiac function and CSE/H2S levels. Measurement of cardiac function and histological changes in response to METH treatment was examined between saline control, METH, METH + Sulfide, or METH + ecCSE transgenic (Tg) cohorts. Panel A reports ejection fraction %, and panel B reports fractional shortening (%), panels C shows fibrosis in heart sections by Masson's trichrome staining, which is quantified in panel D, panel E reports total sulfide levels among the various cohorts, and panel F and G, shows expressions of gp91 and p47phox respectively. N = 4–6 per cohort, #P < 0.05 vs. saline control; *P < 0.05, **P < 0.001, ***P < 0.0001 vs. METH treatment.
3.7. CSE/sulfide corrects pro-aging effects of METH
Prolonged METH use alters molecular signaling that accelerates systemic inflammation and cellular aging making people look years older than their natural age [38,39]. Moreover, advanced vascular aging promotes cardiovascular disease (CVD), ultimately contributing to death [40]. Aging and subsequent complications includes artery stiffening and endothelial dysfunction, which are responsible for the development of CVD. In addition, the transcription factor ATF4 is known to regulate CSE gene expression [41]. Therefore, we checked for changes in age-related genes, Sirtuins (Sirt), including ATF4 and CSE in the skeletal muscle and heart tissues of METH treated mice. METH treatment significantly decreased genes related to sulfide and redox regulation, and anti-aging genes (Fig. 7). Notably, these gene defects were rectified with exogenous sulfide or endogenous CSE overexpression. A significant increase in CSE and ATF4 expressions (Fig. 7A, B and E, F in skeletal muscle and heart, respectively) were observed with exogenous sulfide therapy; however, ecCSE Tg mice increased CSE mRNA but not ATF4 mRNA in skeletal muscle, which was elevated in the heart tissues (Fig. 7F). Likewise, expressions of Sirt1 and Sirt6 (Fig. 7C and D and 7G, H, in skeletal muscle and heart, respectively) were also elevated by exogenous sulfide treatment and in ecCSE Tg mice. This suggests that the pro-aging effects of METH can be effectively attenuated by exogenous sulfide therapy or in ecCSE Tg mice.
Fig. 7.

CSE/H2S reduces METH changes in skeletal muscle aging associated genes. Skeletal muscle and heart gene expressions between saline control, METH, METH + sulfide, and METH + ecCSETg mice is shown for CSE (panel A and E), ATF4 (panel B and F), SIRT1 (panel C and G) and SIRT6 (panel D and H). n = 5 per cohort, #P < 0.05, @@P < 0.001 vs. saline control; *P < 0.05, **P < 0.001 vs. METH treatment.
4. Discussion
Binge and crash cycles of METH use are frequently reported in substance abusers suffering from METH-associated disorders [19,[42], [43], [44], [45]]. The magnitude of risks due to recreational substance usage, and early onset of atherosclerotic cardiovascular disease due to METH use is still under appreciated, despite increasing incidences among young predominantly male substance users [46,47]. Therefore, our study exposed male mice to the ‘binge and crash’ METH model, in a similar manner to human METH users, which developed profound endothelial dysfunction leading to cardiovascular impairment. In this model, mice are administered an increasing dose of METH followed by an abrupt reduction of METH intake [43]. The model provides a useful tool for identifying molecular mechanisms involved in METH-mediated disorders, including both neurological and cardiovascular dysfunctions.
Cystathionine γ-lyase (CSE) is a major enzyme producing H2S in the vascular system that serves critical roles in endothelial function and cardiovascular health [10,35]. Previous studies from our group and others have revealed CSE/H2S regulation of NO bioavailability via NOS-dependent and independent pathways that regulate ischemic vascular growth and remodeling [9,10,16]. Importantly, findings here demonstrate that METH treatment impairs CSE mRNA and protein expression in skeletal muscle tissues resulting in a cascade of molecular pathophysiological events that are similar to CSE genetic deficient mice. The fact that METH selectively decreases CSE expression and bioavailable sulfide leading to decreased eNOS phosphorylation and a reduction in NO levels further confirms this important reciprocal relationship between these gasotransmitters for cardiovascular health while identifying a potential therapeutic target. While we currently don't know how METH selectively reduces CSE expression, future studies are planned to reveal these mechanisms.
METH can induce cardiomyopathy that leads to a reduction in LVEF, reduced cardiac function and increased fibrosis [19]. Importantly, we found that either exogenous sulfide therapy or transgenic overexpression of endothelial cell CSE substantially ameliorated METH induced cardiac dysfunction and fibrosis. Similarly, METH-mediated FMD and blood flow velocity impairments were also rectified with endothelial CSE overexpression or exogenous sulfide treatment. These observations tie together the importance of endothelial CSE and H2S production in the vasculature that regulates cardiovascular function, vascular tone, and blood flow.
METH use is associated with enhanced oxidative stress; however, specific molecular changes underlying the cardiovascular complications have not been understood. Significantly elevated DHE fluorescence in tissues revealed an increase in oxidative stress due to METH treatment. Our observations substantiate that METH-mediated increase in oxidative stress is associated with cardiovascular dysfunction [6]. NOX2 and its catalytic subunits p47 and gp91phox increases oxidative stress that can play regulatory roles in METH-induced endothelium dysfunction and blood-brain barrier disruption [28,[48], [49], [50]]. Importantly, we found that sulfide bioavailability and endothelial CSE expression are crucial in preventing METH-mediated increase in NOX2 expression. Chronic METH use can induce pro-inflammatory cytokines (e.g. TNF-α and IFN-γ) contributing to increased ICAM-1 and VCAM-1 expression exacerbating atherosclerosis and neuroinflammation [31,32]. Our findings also demonstrate that exogenous sulfide or ecCSE Tg interventions critically attenuate increased inflammatory cell adhesion molecule expression. Together, these findings show that H2S bioavailability or endogenous endothelial CSE expression prevents oxidative stress and inflammation, and protects endothelial function [51].
Endothelial cell senescence aggravates vascular dysfunction, inflammation, and oxidative stress, and directly contributes to cardiovascular aging. Sirtuins are critical regulators of vascular senescence that also modulate eNOS/NO signaling and prevent oxidative stress; whereas pro-inflammatory stimuli accelerate pro-aging process [52]. We found that METH treatment reduced sirtuin 1 and 6 levels that are associated with accelerated cellular senescence and vascular dysfunction. Although skeletal muscle tissues were used to demonstrate changes in METH-mediated H2S/NO levels, enzyme expressions, and anti-aging genes, the corresponding results can be extended to the cardiovascular system due to metabolic and structural similarities [53]. Moreover, increasing evidence demonstrate the mutual crosstalk between skeletal muscle and the cardiovascular system with respect to biochemical interplay on the failing heart [[54], [55], [56]]. Additionally, our data shows distinctive phenotypic alterations due to METH treatment on cardiovascular function (FMD and echo) and large vessel biochemical differences (e.g. CSE enzyme activity) that demonstrate METH-mediated cardiovascular damage. However, future studies will be necessary to understand discrete effects of METH on cardiac and vascular (macro versus microvascular) responses and dysfunction. Exogenous H2S therapy and endothelial CSE transgenic overexpression inhibits METH-mediated pro-aging signaling and restores cardiovascular functions consistent with previous studies demonstrating beneficial effects of exogenous H2S for aging and age-associated diseases [57,58]. These observations highlight the potential clinical utility of augmenting H2S bioavailability or increasing CSE expression levels as therapeutic approaches for METH cardiovascular dysfunction and have broader implications beyond that of METH acceleration of aging. Lastly, exogenous sulfide intervention may be ideally suited as a therapeutic approach as it was found to increase ATF4 expression, which can directly augment CSE mRNA expression [59].
In summary, our results show that METH mediated decrease in CSE expression and activity, and subsequent reduction in H2S/NO bioavailability contributes to cardiovascular dysfunction leading to oxidative stress and pro-inflammatory signaling in mice. Future studies are needed in human subjects that engage in chronic METH substance abuse to fully understand the importance of these pathways for cardiovascular pathology and possible clinical intervention.
Acknowledgements
This work was supported by an Institutional Development Award (IDeA) from the National Institutes of General Medical Sciences of the NIH under grant number P20GM121307 and HL149264 to C.G.K; NIH HL098435, HL133497, and HL141155 to A.W.O; HL131844 to M.D.W.; HL122354 and HL145753 to M.S.B.; and an NIH COBRE Pilot Grant to G.K.K.
G.K.K. and C.G.K. have a provisional patent for the use of hydrogen sulfide compounds and restoration of CSE in treating METH use mediated cardiovascular diseases.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102480.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Supplementary Figure 1. The figure shows the stability and amount of sulfide in drinking water over two days.
Supplementary Figure 2. The figure illustrates showing METH dosage and treatment over time.
Supplementary Table 1. qRT PCR Primers used in the study.
Data availability
Data will be made available on request.
References
- 1.Health NSoDUa . 2014. Results from the 2013 National Survey on Drug Use and Health: Summary of National Findings. [Google Scholar]
- 2.SAMHSA. Behavioral . 2015. Health Trends in the United States: Results from the 2014 National Survey on Drug Use and Health. [Google Scholar]
- 3.Darke S., Duflou J., Kaye S. Prevalence and nature of cardiovascular disease in methamphetamine-related death: a national study. Drug Alcohol Depend. 2017;179:174–179. doi: 10.1016/j.drugalcdep.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Won S., Hong R.A., Shohet R.V., Seto T.B., Parikh N.I. Methamphetamine-associated cardiomyopathy. Clin. Cardiol. 2013;36:737–742. doi: 10.1002/clc.22195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tar M.T., Martinez L.R., Nosanchuk J.D., Davies K.P. The effect of methamphetamine on an animal model of erectile function. Andrology. 2014;2:531–536. doi: 10.1111/j.2047-2927.2014.00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lord K.C., Shenouda S.K., McIlwain E., Charalampidis D., Lucchesi P.A., Varner K.J. Oxidative stress contributes to methamphetamine-induced left ventricular dysfunction. Cardiovasc. Res. 2010;87:111–118. doi: 10.1093/cvr/cvq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaye S., McKetin R., Duflou J., Darke S. Methamphetamine and cardiovascular pathology: a review of the evidence. Addiction. 2007;102:1204–1211. doi: 10.1111/j.1360-0443.2007.01874.x. [DOI] [PubMed] [Google Scholar]
- 8.McDonnell-Dowling K., Kelly J.P. The role of oxidative stress in methamphetamine-induced toxicity and sources of variation in the design of animal studies. Curr. Neuropharmacol. 2017;15:300–314. doi: 10.2174/1570159X14666160428110329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bir S.C., Kolluru G.K., McCarthy P., Shen X., Pardue S., Pattillo C.B., Kevil C.G. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1alpha and vascular endothelial growth factor-dependent angiogenesis. J. Am. Heart Assoc. 2012;1 doi: 10.1161/JAHA.112.004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolluru G.K., Bir S.C., Yuan S., Shen X., Pardue S., Wang R., Kevil C.G. Cystathionine gamma-lyase regulates arteriogenesis through NO-dependent monocyte recruitment. Cardiovasc. Res. 2015;107:590–600. doi: 10.1093/cvr/cvv198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondo K., Bhushan S., King A.L., Prabhu S.D., Hamid T., Koenig S., Murohara T., Predmore B.L., Gojon G., Sr., Gojon G., Jr., Wang R., Karusula N., Nicholson C.K., Calvert J.W., Lefer D.J. H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;127:1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaVoie M.J., Hastings T.G. Peroxynitrite- and nitrite-induced oxidation of dopamine: implications for nitric oxide in dopaminergic cell loss. J. Neurochem. 1999;73:2546–2554. doi: 10.1046/j.1471-4159.1999.0732546.x. [DOI] [PubMed] [Google Scholar]
- 13.Watts M., Kolluru G.K., Dherange P., Pardue S., Si M., Shen X., Trosclair K., Glawe J., Al-Yafeai Z., Iqbal M., Pearson B.H., Hamilton K.A., Orr A.W., Glasscock E., Kevil C.G., Dominic P. Decreased bioavailability of hydrogen sulfide links vascular endothelium and atrial remodeling in atrial fibrillation. Redox Biol. 2021;38 doi: 10.1016/j.redox.2020.101817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kesby J.P., Chang A., Markou A., Semenova S. Modeling human methamphetamine use patterns in mice: chronic and binge methamphetamine exposure, reward function and neurochemistry. Addiction Biol. 2018;23:206–218. doi: 10.1111/adb.12502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abdullah C.S., Alam S., Aishwarya R., Miriyala S., Panchatcharam M., Bhuiyan M.A.N., Peretik J.M., Orr A.W., James J., Osinska H., Robbins J., Lorenz J.N., Bhuiyan M.S. Cardiac dysfunction in the Sigma 1 receptor knockout mouse associated with impaired mitochondrial dynamics and bioenergetics. J. Am. Heart Assoc. 2018;7 doi: 10.1161/JAHA.118.009775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pardue S., Kolluru G.K., Shen X., Lewis S.E., Saffle C.B., Kelley E.E., Kevil C.G. Hydrogen sulfide stimulates xanthine oxidoreductase conversion to nitrite reductase and formation of NO. Redox Biol. 2020;34 doi: 10.1016/j.redox.2020.101447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen X., Kolluru G.K., Yuan S., Kevil C.G. Measurement of H2S in vivo and in vitro by the monobromobimane method. Methods Enzymol. 2015;554:31–45. doi: 10.1016/bs.mie.2014.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolluru G.K., Yuan S., Shen X., Kevil C.G. H2S regulation of nitric oxide metabolism. Methods Enzymol. 2015;554:271–297. doi: 10.1016/bs.mie.2014.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdullah C.S., Aishwarya R., Alam S., Morshed M., Remex N.S., Nitu S., Kolluru G.K., Traylor J., Miriyala S., Panchatcharam M., Hartman B., King J., Bhuiyan M.A.N., Chandran S., Woolard M.D., Yu X., Goeders N.E., Dominic P., Arnold C.L., Stokes K., Kevil C.G., Orr A.W., Bhuiyan M.S. Methamphetamine induces cardiomyopathy by Sigmar1 inhibition-dependent impairment of mitochondrial dynamics and function. Commun. Biol. 2020;3 doi: 10.1038/s42003-020-01408-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pattillo C.B., Pardue S., Shen X., Fang K., Langston W., Jourd'heuil D., Kavanagh T.J., Patel R.P., Kevil C.G. ICAM-1 cytoplasmic tail regulates endothelial glutathione synthesis through a NOX4/PI3-kinase-dependent pathway. Free Radic. Biol. Med. 2010;49:1119–1128. doi: 10.1016/j.freeradbiomed.2010.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polesskaya O., Silva J., Sanfilippo C., Desrosiers T., Sun A., Shen J., Feng C., Polesskiy A., Deane R., Zlokovic B., Kasischke K., Dewhurst S. Methamphetamine causes sustained depression in cerebral blood flow. Brain Res. 2011;1373:91–100. doi: 10.1016/j.brainres.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun H.-J., Wu Z.-Y., Nie X.-W., Bian J.-S. Role of endothelial dysfunction in cardiovascular diseases: the link between inflammation and hydrogen sulfide. Front. Pharmacol. 2020;10:1568. doi: 10.3389/fphar.2019.01568. 1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolluru G.K., Shen X., Kevil C.G. A tale of two gases: NO and H2S, foes or friends for life? Redox Biol. 2013;1:313–318. doi: 10.1016/j.redox.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rafikov R., Fonseca F.V., Kumar S., Pardo D., Darragh C., Elms S., Fulton D., Black S.M. eNOS activation and NO function: structural motifs responsible for the posttranslational control of endothelial nitric oxide synthase activity. J. Endocrinol. 2011;210:271–284. doi: 10.1530/JOE-11-0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lord K.C., Shenouda S.K., McIlwain E., Charalampidis D., Lucchesi P.A., Varner K.J. Oxidative stress contributes to methamphetamine-induced left ventricular dysfunction. Cardiovasc. Res. 2010;87:111–118. doi: 10.1093/cvr/cvq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nazari A., Zahmatkesh M., Mortaz E., Hosseinzadeh S. Effect of methamphetamine exposure on the plasma levels of endothelial-derived microparticles. Drug Alcohol Depend. 2018;186:219–225. doi: 10.1016/j.drugalcdep.2018.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Kevil C.G., Goeders N.E., Woolard M.D., Bhuiyan M.S., Dominic P., Kolluru G.K., Arnold C.L., Traylor J.G., Orr A.W. Methamphetamine use and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2019;39:1739–1746. doi: 10.1161/ATVBAHA.119.312461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panday A., Sahoo M.K., Osorio D., Batra S. NADPH oxidases: an overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015;12:5–23. doi: 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuroda J., Ago T., Matsushima S., Zhai P., Schneider M.D., Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. U. S. A. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lassegue B., San Martin A., Griendling K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao B., Li L., Zhu P., Zhang M., Hou L., Sun Y., Liu X., Peng X., Gu Y. Chronic administration of methamphetamine promotes atherosclerosis formation in ApoE-/- knockout mice fed normal diet. Atherosclerosis. 2015;243:268–277. doi: 10.1016/j.atherosclerosis.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Goncalves J., Baptista S., Martins T., Milhazes N., Borges F., Ribeiro C.F., Malva J.O., Silva A.P. Methamphetamine-induced neuroinflammation and neuronal dysfunction in the mice hippocampus: preventive effect of indomethacin. Eur. J. Neurosci. 2010;31:315–326. doi: 10.1111/j.1460-9568.2009.07059.x. [DOI] [PubMed] [Google Scholar]
- 33.Kaye S., Darke S., Duflou J., McKetin R. Methamphetamine-related fatalities in Australia: demographics, circumstances, toxicology and major organ pathology. Addiction. 2008;103:1353–1360. doi: 10.1111/j.1360-0443.2008.02231.x. [DOI] [PubMed] [Google Scholar]
- 34.Kevil C.G., Goeders N.E., Woolard M.D., Bhuiyan M.S., Dominic P., Kolluru G.K., Arnold C.L., Traylor J.G., Orr A.W. Methamphetamine use and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2019;39:1739–1746. doi: 10.1161/ATVBAHA.119.312461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao S., Ho D., Vatner D.E., Vatner S.F. Echocardiography in mice. Curr. Protoc. Mol. Biol. 2011;1:71–83. doi: 10.1002/9780470942390.mo100130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamaguchi M., Tsuruda T., Watanabe Y., Onitsuka H., Furukawa K., Ideguchi T., Kawagoe J., Ishikawa T., Kato J., Takenaga M., Kitamura K. Reduced fractional shortening of right ventricular outflow tract is associated with adverse outcomes in patients with left ventricular dysfunction. Cardiovasc. Ultrasound. 2013;11:19. doi: 10.1186/1476-7120-11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murdoch C.E., Chaubey S., Zeng L., Yu B., Ivetic A., Walker S.J., Vanhoutte D., Heymans S., Grieve D.J., Cave A.C., Brewer A.C., Zhang M., Shah A.M. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J. Am. Coll. Cardiol. 2014;63:2734–2741. doi: 10.1016/j.jacc.2014.02.572. [DOI] [PubMed] [Google Scholar]
- 38.Astarita G., Avanesian A., Grimaldi B., Realini N., Justinova Z., Panlilio L.V., Basit A., Goldberg S.R., Piomelli D. Methamphetamine accelerates cellular senescence through stimulation of de novo ceramide biosynthesis. PLoS One. 2015;10 doi: 10.1371/journal.pone.0116961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farazdaghi E., Nait-Ali A. International Conference on Bio-Engineering for Smart Technologies (BioSMART) 2016. Face aging predictive model due to methamphetamine addiction; pp. 1–4. 2016. [Google Scholar]
- 40.Donato A.J., Machin D.R., Lesniewski L.A. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ. Res. 2018;123:825–848. doi: 10.1161/CIRCRESAHA.118.312563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dickhout J.G., Carlisle R.E., Jerome D.E., Mohammed-Ali Z., Jiang H., Yang G., Mani S., Garg S.K., Banerjee R., Kaufman R.J., Maclean K.N., Wang R., Austin R.C. Integrated stress response modulates cellular redox state via induction of cystathionine gamma-lyase: cross-talk between integrated stress response and thiol metabolism. J. Biol. Chem. 2012;287:7603–7614. doi: 10.1074/jbc.M111.304576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng W.S., Garfein R.S., Semple S.J., Strathdee S.A., Zians J.K., Patterson T.L. Binge use and sex and drug use behaviors among HIV(–), heterosexual methamphetamine users in san diego. Subst. Use Misuse. 2010;45:116–133. doi: 10.3109/10826080902869620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cusick M. Mens rea and methamphetamine: high time for a modern doctrine acknowledging the neuroscience of addiction. Fordham Law Rev. 2017;85:2417–2449. [PubMed] [Google Scholar]
- 44.Shabani S., Houlton S.K., Hellmuth L., Mojica E., Mootz J.R.K., Zhu Z., Reed C., Phillips T.J. A mouse model for binge-level methamphetamine use. Front. Neurosci. 2016;10 doi: 10.3389/fnins.2016.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuczenski R., Segal D.S., Melega W.P., Lacan G., McCunney S.J. Human methamphetamine pharmacokinetics simulated in the rat: behavioral and neurochemical effects of a 72-h binge. Neuropsychopharmacology. 2009;34:2430–2441. doi: 10.1038/npp.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mahtta D., Ramsey D., Krittanawong C., Al Rifai M., Khurram N., Samad Z., Jneid H., Ballantyne C., Petersen L.A., Virani S.S. Recreational substance use among patients with premature atherosclerotic cardiovascular disease. Heart. 2021;107:650–656. doi: 10.1136/heartjnl-2020-318119. [DOI] [PubMed] [Google Scholar]
- 47.Scott M.L., Murnane K.S., Orr A.W. Young at heart? Drugs of abuse cause early-onset cardiovascular disease in the young. Heart. 2021;107:604–606. doi: 10.1136/heartjnl-2020-318856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rastogi R., Geng X., Li F., Ding Y. NOX activation by subunit interaction and underlying mechanisms in disease. Front. Cell. Neurosci. 2017;10 doi: 10.3389/fncel.2016.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park M., Hennig B., Toborek M. Methamphetamine alters occludin expression via NADPH oxidase-induced oxidative insult and intact caveolae. J. Cell Mol. Med. 2012;16:362–375. doi: 10.1111/j.1582-4934.2011.01320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sajja R.K., Rahman S., Cucullo L. Drugs of abuse and blood-brain barrier endothelial dysfunction: a focus on the role of oxidative stress. J. Cerebr. Blood Flow Metabol. 2016;36:539–554. doi: 10.1177/0271678X15616978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zanardo R.C., Brancaleone V., Distrutti E., Fiorucci S., Cirino G., Wallace J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. Faseb. J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- 52.Jia G., Aroor A.R., Jia C., Sowers J.R. Endothelial cell senescence in aging-related vascular dysfunction. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2019;1865:1802–1809. doi: 10.1016/j.bbadis.2018.08.008. [DOI] [PubMed] [Google Scholar]
- 53.Bekfani T., Elsaied M.B., Derlien S., Nisser J., Westermann M., Nietzsche S., Hamadanchi A., Fröb E., Westphal J., Haase D., Kretzschmar T., Schlattmann P., Smolenski U.C., Lichtenauer M., Wernly B., Jirak P., Lehmann G., Möbius-Winkler S., Schulze P.C. Skeletal muscle function, structure, and metabolism in patients with heart failure with reduced ejection fraction and heart failure with preserved ejection fraction. Circulation: Heart Fail. 2020 Dec;13(12) doi: 10.1161/CIRCHEARTFAILURE.120.007198. [DOI] [PubMed] [Google Scholar]
- 54.Loncar G., Fülster S., von Haehling S., Popovic V. Metabolism and the heart: an overview of muscle, fat, and bone metabolism in heart failure. Int. J. Cardiol. 2013;162:77–85. doi: 10.1016/j.ijcard.2011.09.079. [DOI] [PubMed] [Google Scholar]
- 55.Seiler M., Bowen T.S., Rolim N., Dieterlen M.T., Werner S., Hoshi T., Fischer T., Mangner N., Linke A., Schuler G., Halle M., Wisloff U., Adams V. Skeletal muscle alterations are exacerbated in heart failure with reduced compared with preserved ejection fraction: mediated by circulating cytokines? Circ. Heart Fail. 2016 Sept;9(9) doi: 10.1161/CIRCHEARTFAILURE.116.003027. [DOI] [PubMed] [Google Scholar]
- 56.Keng B.M.H., Gao F., Teo L.L.Y., Lim W.S., Tan R.S., Ruan W., Ewe S.H., Koh W.P., Koh A.S. Associations between skeletal muscle and myocardium in aging: a syndrome of "Cardio-Sarcopenia". J. Am. Geriatr. Soc. 2019;67:2568–2573. doi: 10.1111/jgs.16132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang G., Zhao K., Ju Y., Mani S., Cao Q., Puukila S., Khaper N., Wu L., Wang R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxidants Redox Signal. 2013;18:1906–1919. doi: 10.1089/ars.2012.4645. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Y., Tang Z.-H., Ren Z., Qu S.-L., Liu M.-H., Liu L.-S., Jiang Z.-S. Hydrogen sulfide, the next potent preventive and therapeutic agent in aging and age-associated diseases. Mol. Cell Biol. 2013;33:1104–1113. doi: 10.1128/MCB.01215-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dickhout J.G., Carlisle R.E., Jerome D.E., Mohammed-Ali Z., Jiang H., Yang G., Mani S., Garg S.K., Banerjee R., Kaufman R.J., Maclean K.N., Wang R., Austin R.C. Integrated stress response modulates cellular redox state via induction of cystathionine γ-lyase: cross-talk between integrated stress response and thiol metabolism. J. Biol. Chem. 2012;287:7603–7614. doi: 10.1074/jbc.M111.304576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.