

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a clinically challenging cancer, due to both its late stage at diagnosis and its resistance to chemotherapy. However, recent advances in our understanding of the biology of PDAC have revealed new opportunities for early detection and targeted therapy of PDAC. In this review, we discuss the pathogenesis of PDAC, including molecular alterations in tumor cells, cellular alterations in the tumor microenvironment, and population-level risk factors. We review the current status of surveillance and early detection of PDAC, including populations at high risk and screening approaches. We outline the diagnostic approach to PDAC and highlight key treatment considerations, including how therapeutic approaches change with disease stage and targetable subtypes of PDAC. Recent years have seen significant improvements in our approaches to detect and treat PDAC, but large-scale, coordinated efforts will be needed to maximize the clinical impact for patients and improve overall survival.
Keywords: Pancreatic Cancer, Pancreatic Ductal Adenocarcinoma, Cancer Screening
Pancreatic ductal adenocarcinoma (PDAC) is a deadly disease that is predicted to be the second leading cause of cancer death in the United States by 2030.1 One reason for the dismal prognosis of pancreatic cancer is that 90% of tumors are diagnosed at a late stage after they have spread beyond the pancreas, with systemic metastases in >50%.2,3 This underscores the need for tools to detect PDAC earlier, before it has spread beyond the pancreas, and for therapies that more effectively kill PDAC cells after they have metastasized. In the past decade, basic and translational investigations have significantly improved our understanding of the biological processes that drive pancreatic tumorigenesis, and this understanding is beginning to be leveraged to improve the care of patients with pancreatic cancer. In this review, we discuss the pathogenesis, screening, diagnosis, and treatment of PDAC, focusing on both fundamental concepts and recent advances.
Pathogenesis of Pancreatic Cancer
Pathology of Pancreatic Neoplasia
PDAC arises from noninvasive precancerous lesions that are curable if detected and treated early enough (Figure 1). These precancerous lesions are classified by their size and involvement with the pancreatic ductal system. Most PDACs arise from microscopic pancreatic intraepithelial neoplasia (PanIN), a neoplasm involving pancreatic ducts, which is by definition <5 mm.4 A much smaller proportion of PDACs (<10%) arise from intraductal papillary mucinous neoplasms (IPMNs), macrocystic lesions that involve the pancreatic ductal system.4–6 The least common precancerous neoplasm, mucinous cystic neoplasm, is clinically and pathologically distinct. Mucinous cystic neoplasms do not involve the ductal system and have a characteristic ovarian-type stroma. They are much more common in women and involve the pancreatic body/tail.7 The unique pathological features of each lesion lead to distinct clinical challenges. For example, because of their larger size, IPMNs are commonly detected incidentally on abdominal imaging studies, requiring data-driven approaches to surveillance and intervention for these patients.8 In contrast, PanINs are rarely detected incidentally. Although the pathological features of these precancers are well characterized, the cell of origin of pancreatic cancer remains controversial. The anatomic location of precancers in the ductal system could support a ductal cell of origin, but numerous studies in murine models have shown that acinar cells can give rise to PanINs following pancreatic injury and metaplasia. However, it is challenging to confirm such findings in human samples. This controversy was recently reviewed in detail elsewhere.9
Figure 1.
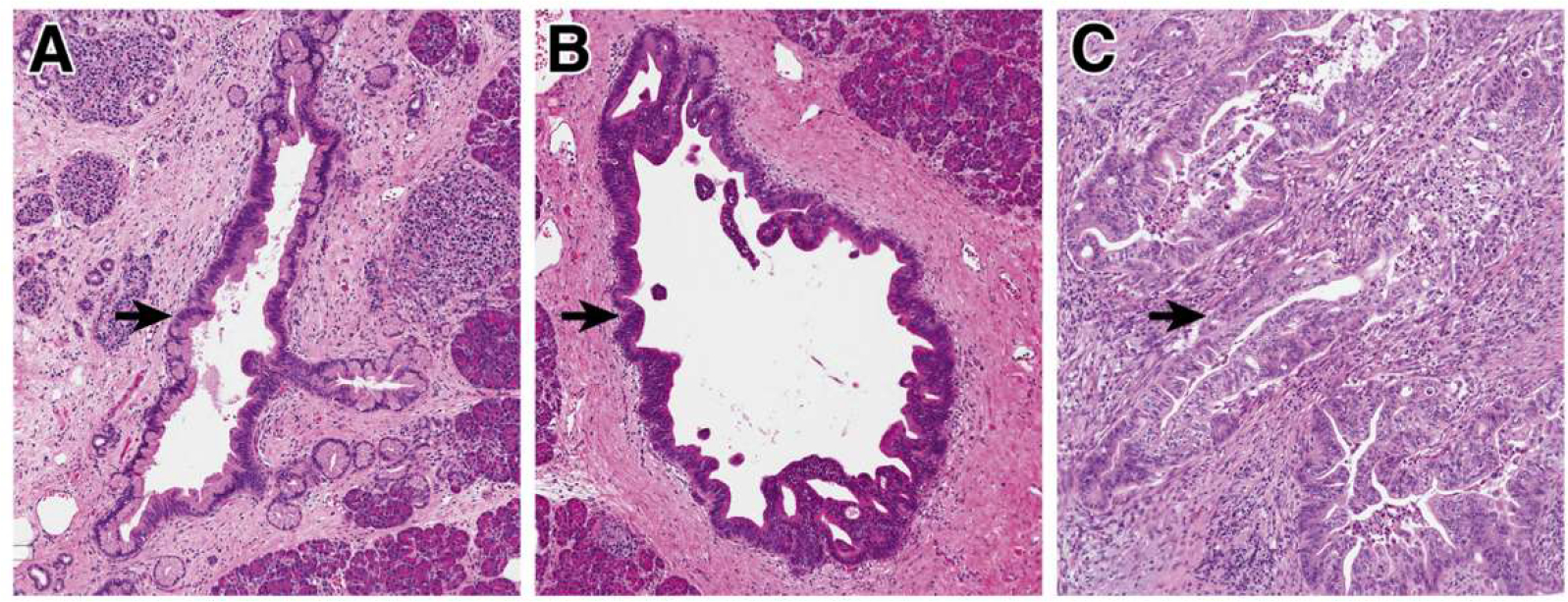
Precancerous neoplasia in the pancreas. (A) Low-grade PanINs involve the pancreatic ducts. They have apical mucin and mild cytologic atypia, with the nuclei still retaining their polarity. (B) High-grade PanIN shows increase architectural and cytologic atypia, which architectural complexity, nuclear pleomorphism, and loss of nuclear polarity. (C) Invasive PDAC shows severe architectural and cytologic atypia with a striking desmoplastic response in the stroma.
Precancerous pancreatic neoplasms are incredibly common and increase in prevalence with age; for example, one autopsy study in older adults identified PanINs in >75% of completely sampled pancreata.10,11 Although these lesions have a risk for progression to PDAC, most will not progress to cancer, and by themselves they have no risk of spread beyond the pancreas.12,13 Morphologically, these lesions are categorized as low-grade or high-grade based on architectural and cytological atypia (Figure 1).4 This 2-tiered grading system (low-grade vs high-grade) is a relatively recent revision of a previously used 3-tiered grading system for pancreatic precancers; the change to a 2-tiered system was driven by increased interobserver reproducibility as well as clinical impact of the revised system.4 Low-grade precancers have basally oriented nuclei and mild to moderate cytologic atypia (Figure 1A), and high-grade precancers have marked architectural alterations (cribriforming, micropapillae, budding), loss of nuclear polarity, and severe cytologic atypia (Figure 1B).4 In IPMNs, the grade of dysplasia is also correlated with the direction of differentiation of the neoplastic cells, with gastric-type IPMNs enriched for low-grade dysplasia and intestinal-type and pancreatobiliary-type more likely to be high-grade.14 When stratified by grade of dysplasia, low-grade precancerous lesions are far more common, whereas high-grade lesions are mostly found in pancreata associated with PDAC, suggesting that high-grade precancers have a higher risk for progression to invasive carcinoma.10,11 Moreover, associated carcinomas are much more frequently associated with high-grade rather than low-grade IPMNs, and recent evolutionary analysis defined high-grade IPMNs as the direct precursor of PDAC.6,15 This evidence highlights that the morphological grade of dysplasia correlates with risk of transformation to invasive carcinoma.
Pathologically, PDAC consists of malignant glands with haphazard architecture embedded in a dense desmoplastic stroma (Figure 1C). This pauci-cellularity significantly complicates the molecular analysis of primary PDAC samples, as most cells in a tissue fragment are likely non-neoplastic; without enrichment for neoplastic cells, often fewer than 10% of cells may be malignant. There are also several morphological variants, including adenosquamous carcinoma and undifferentiated carcinoma with osteoclast-like giant cells, some of which have unique clinical and/or molecular features.16 Most PDACs are not localized at diagnosis, and >50% of patients present with distant metastases.2 The most common site of metastasis is the liver, and increasing evidence suggests that distinct metastatic sites carry different prognostic features, with lung-only metastases having an improved prognosis compared with other sites.17,18 Moreover, autopsy studies suggest that distinct molecular alterations may increase the likelihood of local vs systemic disease progression, with distant metastases more common in patients with somatic mutations in the gene SMAD4.19
Molecular Alterations in Neoplastic Cells
PDAC is caused, at least in part, by somatic mutations in oncogenes and tumor suppressor genes (Figure 2). The most commonly mutated genes in PDAC, the oncogene KRAS and the tumor suppressor genes CDKN2A, TP53, and SMAD4, were originally discovered in the 1980s and 1990s through targeted molecular biology and sequencing approaches.20–23 The first comprehensive look at the PDAC exome came in 2008 with the first whole exome sequencing study of 24 PDACs.24 This study revealed that the PDAC genome landscape is composed of these 4 previously described “mountains” (KRAS, CDKN2A, TP53, SMAD4) as well as a larger number of less frequently mutated “hills.”24 Subsequent large-scale PDAC sequencing studies have refined this landscape in great detail, including efforts by The Cancer Genome Atlas and The International Cancer Genome Consortium.25–28 Several important groups of “hills” have been characterized in these studies, including genes involved in DNA repair, chromatin remodeling, and axon guidance, some of which delineate clinically important groups that respond to specific therapies. Recent efforts have also identified kataegis, a process leading to clustered nucleotide substitutions, in PDAC; this process is likely associated with the activity of APOBEC enzymes.29
Figure 2.
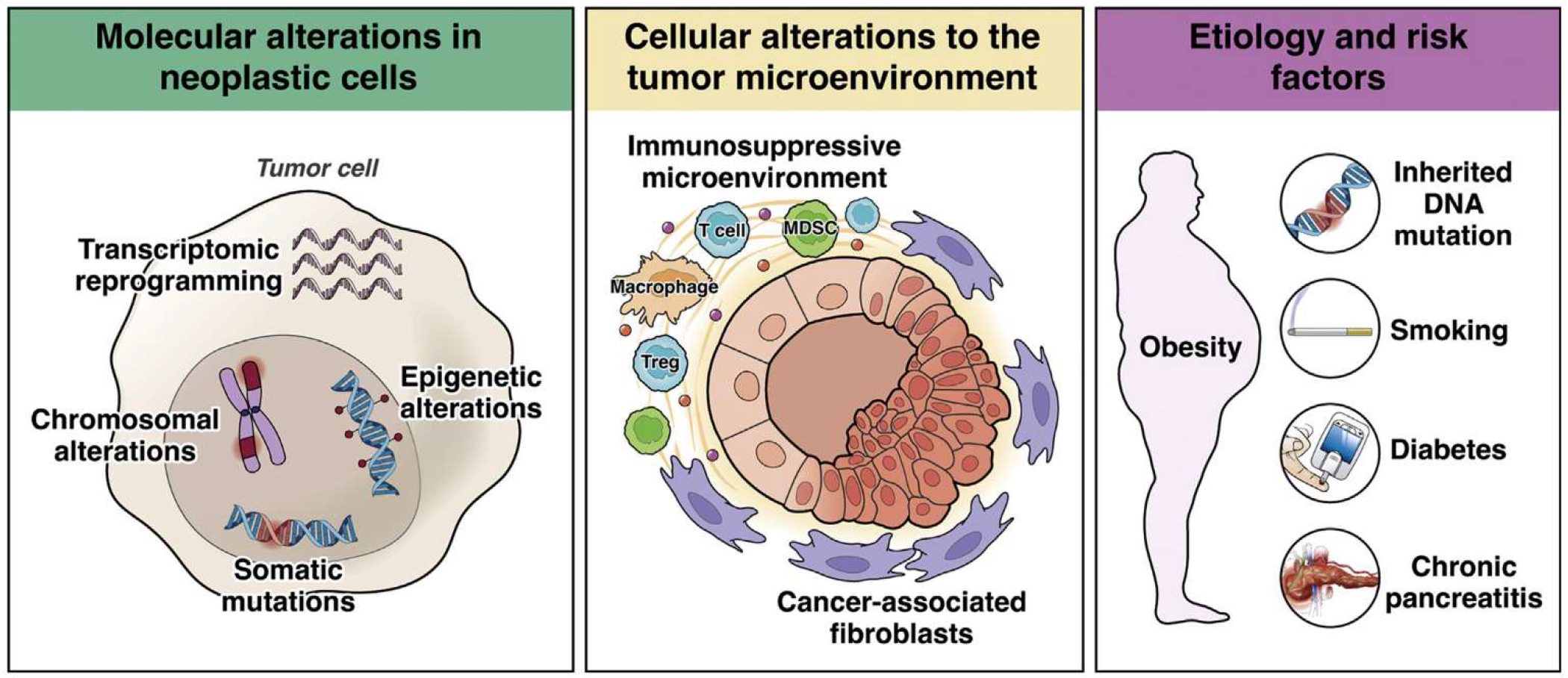
Mechanisms of pancreatic cancer pathogenesis. Molecular contributions to pathogenesis include somatic mutations in driver genes, chromosomal alterations, epigenetic alterations, and transcriptional reprogramming, all occurring in tumor cells. Cellular contributions from the non-neoplastic tumor microenvironment include alterations in cancer-associated fibroblasts and the immune microenvironment. Population-level contributions include inherited DNA mutations, diabetes, obesity, smoking, and chronic pancreatitis.
In addition to small somatic mutations involving coding regions, the expansion of PDAC genomic analysis to include whole genome sequencing has also catalogued large chromosomal alterations, including copy number alterations, chromosomal rearrangements, and chromothripsis. Chromothripsis, a term derived from the Greek for “shattering,” refers to a phenomenon in which 1 or a few chromosomes contain hundreds of clustered genomic rearrangements.29 For the latter, some investigators propose that these alterations are acquired through catastrophic DNA damage events, resulting in punctuated rather than gradual evolution.30 An ongoing challenge in the interpretation of these alterations is the identification of potential driver genes in large altered regions and the distinction of driver alterations from passengers that occur due to PDAC genomic instability. Copy number alterations and chromothripsis have also been reported in precancerous lesions, with a higher prevalence in high-grade lesions, suggesting these chromosomal alterations are a late event in pancreatic tumorigenesis.31
In addition to the identification of genetic drivers, DNA sequencing studies have also revealed other important features of pancreatic tumorigenesis. For example, multiregion sequencing of precancerous lesions, primary tumors, and metastases have allowed evolutionary modeling of various steps in pancreatic tumorigenesis. Studies in IPMNs suggest a period of almost 4 years between the development of high-grade dysplasia and invasive carcinoma.15 Similarly, in PanIN lesions, modeling from sequencing data suggests an interval of approximately 4 years between the common ancestral cell and the founder cell of the associated PDAC.32 Relatedly, modeling from multiregion sequencing of metastases and primary PDACs suggests that many years elapse between tumor initiation and metastasis.33 Intriguingly, these studies show no differences in driver gene mutations between primary and metastatic tumors, suggesting that alterations other than DNA mutations may promote metastasis.33,34 Recent studies in precancerous lesions have revealed surprising genetic heterogeneity, even with respect to well-characterized driver gene mutations.15,35–37 These studies suggest a polyclonal origin for at least a subset of precancerous lesions, convergent evolution in later driver gene mutations, and distinct selective forces at different time points in tumorigenesis (Figure 3). In addition, multifocal neoplasia is also quite common in patients with precancerous pancreatic lesions; co-occurring PanINs and IPMNs/carcinomas are frequently genetically independent (ie, sharing no somatic mutations, which suggests initiation from separate cells), as are IPMNs and their recurrences.6,38–40
Figure 3.
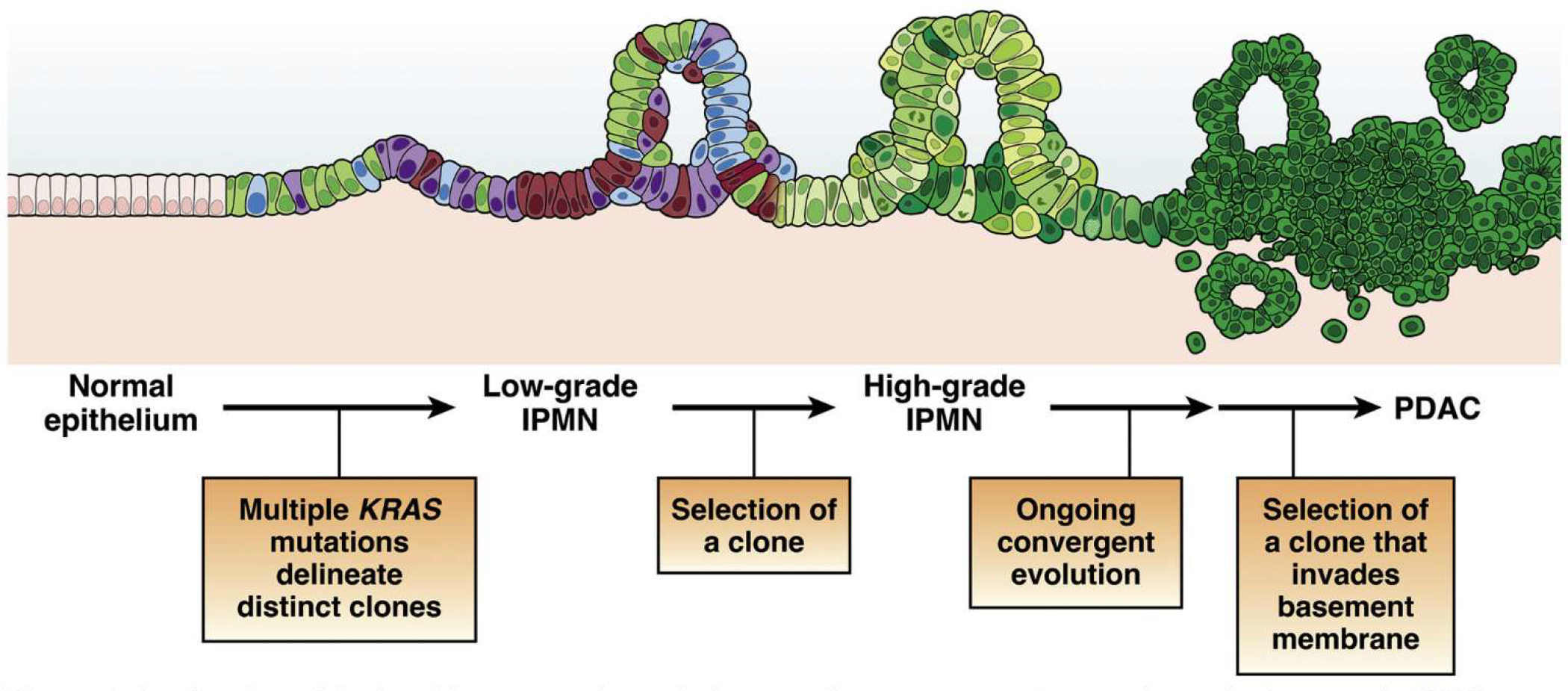
A refined model of multistep tumorigenesis in premalignant pancreatic tumorigenesis. Low-grade IPMNs are characterized by multiple mutations in the initiating oncogene KRAS, suggesting polyclonal evolution of multiple genetically independent precancers. Selection of a clone leads to the development of a high-grade IPMN, which then undergoes ongoing evolution, at times with distinct alterations in the same gene arising in distinct subclones. Selection of an advanced clone that invades the basement membrane leads to the development of invasive PDAC. While this model has been validated in human IPMN samples, its applicability to microscopic human PanINs requires further investigation.
Although alterations in DNA sequence are the most extensively characterized, other types of molecular alterations also contribute to PDAC pathogenesis. Epigenetic modifications to DNA (such as methylation) and histones (such as acetylation and methylation) can heritably modulate chromatin structure and gene expression. Epigenetic inactivation via DNA methylation has been frequently identified for the tumor suppressor gene CDKN2A, but methylation is not a common inactivation mechanism for other PDAC drivers, such as TP53 and SMAD4.41,42 Comprehensive methylation profiling in The Cancer Genome Atlas study identified 2 clusters of PDACs based on the extent of their DNA hypermethylation, and integrated analysis of DNA methylation and messenger RNA expression data identified almost 100 genes that were recurrently silenced by DNA methylation, including ZPF82, PAPR6, and DNAJC15.28 In addition, global epigenomic reprogramming modulates the PDAC genome. Studies in human autopsy samples have shown global alterations in histone states in metastases compared with primary tumors, suggesting that epigenetic alterations may play a role in driving metastasis.43 Such epigenetic alterations also have been reported in precancerous PanIN and IPMN lesions.44–47
Multiple studies have categorized the transcriptomes of human PDACs, with several unique subtyping schemes proposed.26,28,48–51 Integrated analyses of these data suggest that there are likely 2 distinct transcriptional subtypes of PDAC, termed classical and basal, with the basal phenotype enriched in metastases and correlated with a worse prognosis.28,52 Classical tumors are enriched for epithelial and pancreatic lineage transcriptomic programs, whereas basal tumors are enriched for epithelial to mesenchymal transition, cell cycle progression, and transforming growth factor-β signaling.53 Some studies suggest heterogeneity in transcriptional subtype within PDAC tumors, but it is not clear whether basal tumors are inherently more metastatic or the switch to a basal phenotype drives metastasis.52,54 In addition, some studies have identified PDACs with intermediate transcriptomic subtypes or subtypes discordant with different classifiers, but the clinical implications of these intermediate tumors remain to be comprehensively investigated.53,55 Moreover, the timing of acquisition of these transcriptional programs in premalignant pancreatic tumorigenesis is not yet known, highlighting an important area for future research.
Cellular Alterations to the Tumor Microenvironment
In addition to the extensive molecular alterations described previously in neoplastic cells, interactions with non-neoplastic cells in the tumor microenvironment also play a crucial role in pancreatic tumorigenesis (Figure 2). This topic was recently reviewed in depth by others.56 PDACs exhibit multiple alterations to immune cell subtypes, resulting in an immunosuppressive or “cold” microenvironment. These alterations include increased immunosuppressive regulatory T cells and myeloid-derived suppressor cells, preventing immune-mediated targeting of cancer cells. In addition, tumor-associated macrophages can promote progression of precancerous lesions by secretion of interleukin-6.57 However, these features of the immune microenvironment are not universal across human PDACs, and some investigators have proposed subtyping schemes for the immune microenvironment that include distinct “escape” and “exhausted” phenotypes.58 Immune alterations in PDAC likely influence disease progression and contribute to the lack of response to immunotherapy approaches, such as checkpoint inhibition. Greater mechanistic insights into the signals driving the immunosuppressive microenvironment may reveal novel strategies to enhance therapeutic efficacy, as recently reviewed in depth.59,60
Interactions with nonimmune cells also contribute significantly to the development of PDAC. PDACs have a dense desmoplastic stroma that is produced by cancer-associated fibroblasts (CAFs). CAFs have been studied extensively in vitro, in murine models, and in human tissue samples. Perhaps counterintuitively, CAFs can have both tumor-promoting and tumor-suppressive effects. CAFs can provide metabolic support to PDAC cells, including amino acids and lipids, as well as paracrine signaling to promote growth and survival.61
In addition to direct interactions with tumor cells, CAFs also modulate the immune microenvironment. CAFs may mediate immunosuppression by promoting T cell exclusion, and recent studies suggest that CAFs can present antigens to immune cells, highlighting an additional possible mechanism for immune modulation by CAFs.62,63 Netrin G1 was recently identified as a regulator of both metabolic and immunosuppressive functions of CAFs, suggesting that shared molecular mechanisms underlie distinct CAF functions.64 Despite these seemingly tumor-promoting effects, targeting of CAFs in mice has led to increased tumor growth, and clinical trials of Hedgehog pathway inhibitors that target stromal cells have shown no benefit.65–68 These dual tumor-promoting and tumor-suppressive effects of CAFs may be explained in part by functional heterogeneity of different CAF populations. Recent studies have identified spatially and molecularly distinct subtypes of CAFs, including myofibroblastic and inflammatory CAFs, that likely play distinct roles in pancreatic tumorigenesis.69,70 One recent study showed that inhibition of Hedgehog signaling decreased the proportion of myofibroblastic CAFs and altered the immune cell composition, suggesting intricate crosstalk between different non-neoplastic cell types in the PDAC microenvironment.71 A newly described CAF subtype, antigen-presenting CAFs, express major histocompatibility complex class II and present antigens to T cells, further demonstrating this CAF-immune crosstalk.62
The provision of metabolic support to PDAC cells is not limited to CAFs. Peripheral axons innervating pancreatic tumors release amino acids to support PDAC growth in a nutrient-poor microenvironment.72 Ablation of nerves can slow PDAC development in model systems, highlighting the role of this component of the microenvironment in pancreatic tumorigenesis.73,74
Etiology and Risk Factors
Studies in large patient cohorts have revealed several environmental and chronic disease-related factors that increase the risk of development of PDAC (Figure 2). Cigarette smoking is an established risk factor for pancreatic cancer, with an odds ratio of 1.74 for current smokers compared with never smokers and a population attribute risk of 11% to 32%.75 The mechanisms by which smoking increases the PDAC risk are likely multifactorial and include DNA damage as well as inflammation and fibrosis.76 Similarly, heavy alcohol consumption (>6 drinks per day) increases PDAC risk by 1.6-fold compared with nondrinking controls.
Patients with diabetes mellitus also have an increased risk of PDAC; this increased risk persists among patients with new-onset diabetes as well as those with long-standing diabetes.77 Long-standing diabetes of >3 years increases risk by 1.5- to 2.4-fold.75 Although long-standing diabetes may play a causal role in pancreatic tumorigenesis, it appears that new-onset diabetes (NOD) is an effect rather than a cause of PDAC, and recent studies have focused on this symptom to define a high-risk group for screening.78,79 Approximately 1% or less of patients with NOD develop PDAC within 3 years.80,81 The incidence of PDAC in patients with NOD may increase to as high as 3% in white men after age 50 or range between 1.5% and 6% when a predictive model that considers age at onset, change in weight, and change in blood glucose (Enriching New-Onset Diabetes for Pancreatic Cancer or ENDPAC score82) is used. A large NOD prospective cohort study consisting of subjects 50 years or older is under way to determine the 3-year incidence of PDAC and collect biospecimens as early detection initiative (CPDPC16-01).79
Obesity correlates with increased PDAC risk, although it is challenging to separate cancer risk due solely to obesity from that due to concomitant diabetes mellitus, which is increased in obese patients.77,83 Still, the increased risk associated with obesity persisted even after adjustment for history of diabetes in several studies, suggesting the obesity-associated risk is not solely driven by confounding diabetes.84 Thus, obesity is currently considered an independent risk factor for PDAC (odds ratio 1.72 for body mass index >30 kg/m2).85
Chronic pancreatitis is also associated with increased risk of pancreatic cancer. Like diabetes, a challenge in interpreting co-occurrence of chronic pancreatitis and PDAC is the ability of PDAC to cause chronic pancreatitis via obstruction of the ductal system. Although the risk declines over time after diagnosis of chronic pancreatitis, the elevated risk persists even after long-term follow-up, suggesting that this association is not solely a result of chronic pancreatitis caused by PDAC.86 In addition, smoking and alcohol consumption are risk factors for both chronic pancreatitis and PDAC, further complicating the assessment of specific risk associated with chronic pancreatitis.87
Inherited DNA variants play an important role in PDAC risk, and approximately 10% of PDACs occur in patients with a family history of pancreatic cancer. Increased risk of PDAC is a component of several inherited syndromes. These include familial cancer predisposition syndromes, such as familial atypical mole melanoma syndrome (caused by constitutional CDKN2A mutations) and Peutz-Jeghers syndrome (caused by constitutional STK11 mutations), both of which have strikingly elevated risks of PDAC.88,89 Patients with these syndromes are at increased risk of cancer in the pancreas as well as other organs. Patients with hereditary pancreatitis have an increased cancer risk limited to the pancreas, likely due to the repeated bouts of inflammation and repair. Patients with constitutional mutations in PRSS1, which encodes cationic trypsinogen and is the best characterized hereditary pancreatitis gene, have an astounding cumulative risk of PDAC of approximately 50% by age 75.90 Mutations in PRSS1 result in inappropriate intrapancreatic activation of trypsin, either by enhanced autoactivation or impaired inactivation, leading to pancreatic tissue destruction.91 Outside of these well-characterized inherited syndromes, patients with a family history of pancreatic cancer are at increased risk for PDAC, with the greatest risk among first-degree relatives of patients with PDAC.92 “Familial pancreatic cancer” is defined as families with a pair of affected first-degree relatives.93 Although some cases of familial pancreatic cancer are associated with constitutional alterations in well-characterized cancer predisposition genes (such as BRCA2, ATM), the genetic basis for most familial pancreatic cancer remains unknown.94–96 Pathogenic germline variants in pancreatic cancer susceptibility genes are identified in patients with seemingly sporadic PDAC, in almost 4% of unselected patients with PDAC in one study.97 These results highlight that inherited DNA alterations can increase PDAC risk even in patients without a family history of pancreatic cancer. Taken together, inherited mutations likely play a role in 10% to 15% of pancreatic cancers.
Screening and Early Detection of Pancreatic Cancer
Rationale for Pancreatic Cancer Screening
Although PDAC comprises only 3% of new cancer cases in the United States,98 the prediction that by 2030 it will become the second leading cause of cancer death reflects its high mortality.1 The primary reason for this high mortality is that only a minority (11%) of patients are diagnosed with localized disease, and 52% of PDACs are already metastatic at presentation, with little hope of cure.2,98 This stage distribution implies that any successful strategy in improving PDAC mortality should include screening and early detection of potentially curable cancers. The long-term survival of patients with PDAC is greatly dependent on small tumor size and early disease stage.99
Population-based screening for PDAC is not currently justified due to its relatively low incidence in the United States (3% of new cancers),98 compared with breast (30%), colon (8%), or lung cancer (12%).2 The lifetime risk (age 85) of PDAC for Americans is only 1.7% 98 Enriching the population by targeting subgroups at greater risk for PDAC might improve the diagnostic yield of screening.
Surveillance of High-Risk Individuals
Currently, PDAC surveillance focuses on genetically predisposed individuals. Approximately 10% to 15% of PDAC cases are due to inherited mutations. High-risk individuals (HRIs) with a significantly increased risk for PDAC can be identified and offered surveillance (Table 1). Thresholds for risk over the general population can be set to design screening studies. Currently, an accepted threshold to consider PDAC surveillance is a lifetime risk of 5%. Table 1 summarizes the estimated lifetime risk, relative risk, and recommended age to start surveillance for HRIs supported by the multidisciplinary International Cancer of the Pancreas Consortium in 2019.100 New onset diabetes in an HRI should initiate surveillance regardless of age. The goal of surveillance is to detect and treat stage I PDAC or its high-grade precursor lesions (PanIN or IPMN) to prevent malignant progression.
Table 1.
Candidates for Pancreatic Cancer Screening, Estimated Risk for PDAC, and Recommended Age to Initiate Screening
| Deleterious mutation | Genetic syndrome | Lifetime risk of PDAC (age 70 y) | Relative risk | PDAC in affected blood relative | When to start |
|---|---|---|---|---|---|
|
| |||||
| Group 1 Low to Moderate Risk | |||||
| BRCA1 | Breast-Ovarian Cancer | 1.2% | 2.6 (1.6–4.7) | ≥1 FDR or any 2 | 50–55 a |
| BRCA2 | Breast-Ovarian Cancer | 2%–5% | 3.5 (2.2–12.8) | ≥1 FDR or any 2 | 50–55 ya |
| PALB2 | ? | ? | ≥1 FDR or any 2 | 50–55 ya | |
| ATM | ? | 5.7 | ≥1 FDR or any 2 | 50–55 ya | |
| MLH1, MSH2, MSH6 (DNA mismatch repair) | Lynch | 3.7% | 6.6–8.6 | ≥1 FDR or any 2 | 50–55 ya |
| Unknown | Familial PC | 8%–12% | 6.4 | 2 FDR | 50–55 ya |
| Group 2 High Risk | |||||
| Unknown | Familial PC | 40% | 32 | 3 FDR | 50–55 ya |
| CDKN2A | FAMMM | 10%–17% | 12–34 | None required | 40 y |
| P16 Leiden variant | FAMMM | 17%–58% | 48–80 | None required | 40 y |
| PRSS1 | Hereditary pancreatitis | 25%–40% | 50–80 | None required | 40 yb |
| STK11/LKB1 | Peutz-Jeghers | 30%–60% | 132 | None required | 40 y |
ATM, ataxia telangiectasia mutated; BRCA, breast-related cancer; CDKN2A, cyclin-dependent kinase inhibitor 2A; FAMMM, familial atypical multiple mole melanoma; PALB2, partner and localizer breast cancer 2; PRSS1, cationic trypsinogen gene; STK11, serine threonine kinase 11 gene.
Or start 10 years younger than the youngest affected blood relative. Early age of onset of PDAC <50 years increases risk 9.3-fold.
Or 20 years after first attack of acute pancreatitis.
Nineteen cohort studies (ranging in size from 15–262 HRIs) conducted around the world in the past 2 decades performing endoscopic ultrasound (EUS), magnetic resonance imaging (MRI), and/or computed tomography (CT)-based screening of HRIs report frequent detection of pancreatic lesions. Cystic lesions are commonly identified during screening in HRIs (10.5%–87.5%).101 A solid component within a cyst and main pancreatic duct dilation are detected in 5.8% (95% confidence interval 3%–9%) and 20.2% (95% confidence interval 14%–28%), respectively.102 In addition to conventional imaging, annual glucose testing or assessment of HbA1C is recommended to diagnose NOD in HRIs.100
The risk of PDAC is higher in germline mutation carriers. Hence, genetic testing is recommended for individuals with a PD AC-associated genetic syndrome (Table 1) and at-risk relatives of patients with PDAC. Two large, well-characterized high-risk cohorts with long-term follow-up of more than 350 patients showed significantly greater risk of neoplastic progression to high-grade dysplasia (HGD) or PDAC in mutation carriers compared with familial pancreatic cancer relatives (15% vs 9%, 9% vs 0%).
Five studies have concluded that imaging-based PC screening is cost-effective in appropriately identified HRIs.103–106 A meta-analysis of all prospective studies on surveillance of 7085 HRI estimated that the number needed to screen (NNS) to identify one patient with a high-risk pancreatic lesion (with HGD or PDAC) was 135. Specifically, the NNS is 71 for patients with Peutz-Jegher syndrome and 51 patients with constitutional CDKN2A mutations.101 These low numbers might suggest a high priority for screening in HRIs; however, important data are lacking in determining the NNS to prevent one pancreatic cancer death.
A 2019 systematic review of studies from January 2002 to March 2019 in patients with and without risk factors for PDAC published by the U.S. Preventive Services Task Force concluded that imaging-based screening in groups with a high familial risk can detect PDAC with minimal harm (18 cases of PDAC detected in 1156 familial HRIs). However, the U.S. Preventive Services Task Force recommended against screening for PDAC in asymptomatic adults107 because of the limited evidence to assess the benefits vs harms of surgical intervention, and effect of screening on mortality of screen-detected cancers.
Although the diagnostic yield of cross-sectional imaging is high, few studies report on the effect of screening on the morbidity, mortality, effectiveness of treatment, and quality of life of screen-detected PDAC.108 However, data supporting PDAC surveillance continue to accumulate and provide continued hope for positive outcomes from surveillance of HRIs. One meta-analysis of all prospective studies confirmed a stage shift of PDAC diagnosed within formal surveillance programs of HRI, with only 20% of PDAC at stage IV.101 Although limited by the small number of PDACs, there is also evidence that screen-detected PDACs in HRIs are more likely to be resectable (60%–90%)109–112 than sporadic PDAC in the general population (15%). Furthermore, surgical treatment of detected PDAC is associated with a high R0 resection rate and zero mortality.110,111,113,114 PDAC screening also has a positive effect on quality of life, psychological distress, and cancer worry.115,116
Currently, despite advances in surgical and oncologic treatment approaches, the 5-year survival of sporadic PDAC is only 10.8%.98 In contrast, PDAC surveillance programs in Europe and the United States report 5-year survival rates with asymptomatic screen-detected PDAC that are increased compared with the average PDAC (25%109 and 60%,113 respectively). These extended long-term survival rates are not attributable to lead-time bias, but appear to reflect the combined benefits of surgery for earlier stage disease, with adjuvant therapy in patients with better performance status. Overall, we maintain guarded optimism that there is a benefit for surveillance on PDAC survival when performed within formal multidisciplinary programs with state-of-the-art imaging and expertise in genetics, endoscopy, radiology, pancreatic surgery, and pathology.110,111,113,117
Challenges and Future Approaches for HRIs
Current challenges in conventional imaging-based pancreatic cancer screening/surveillance include high cost, invasiveness (at least for EUS), and suboptimal sensitivity and specificity. False positives may lead to unnecessary surgery for findings that do not represent HGD or PDAC114 (Figure 4). False negatives (nonspecific abnormalities such as a mildly dilated pancreatic duct, low-risk cystic lesions) may in turn lead to a delayed recommendation for treatment (Figure 5) or advanced incurable disease. Although PDAC resectability improves in HRI screening, only a handful of potentially curable stage I PDACs have been detected thus far in screening programs. Although screen-detected PDAC resectability is higher, there are still patients in screening who have identification of disease at a later stage than the desired target lesions (high-grade dysplasia or stage I PDAC) (Figure 5). We use well-known conventional cyst imaging features for IPMN progression to PDAC (such as solid mass, marked duct dilation), similar to those used for management of branch-duct IPMN, to recommend operative treatment, but these do not address microscopic PanIN progression.118 The ideal surveillance interval is yet to be known and interval missed cancers still develop,112 particularly in CDKN2A mutation carriers.117
Figure 4.
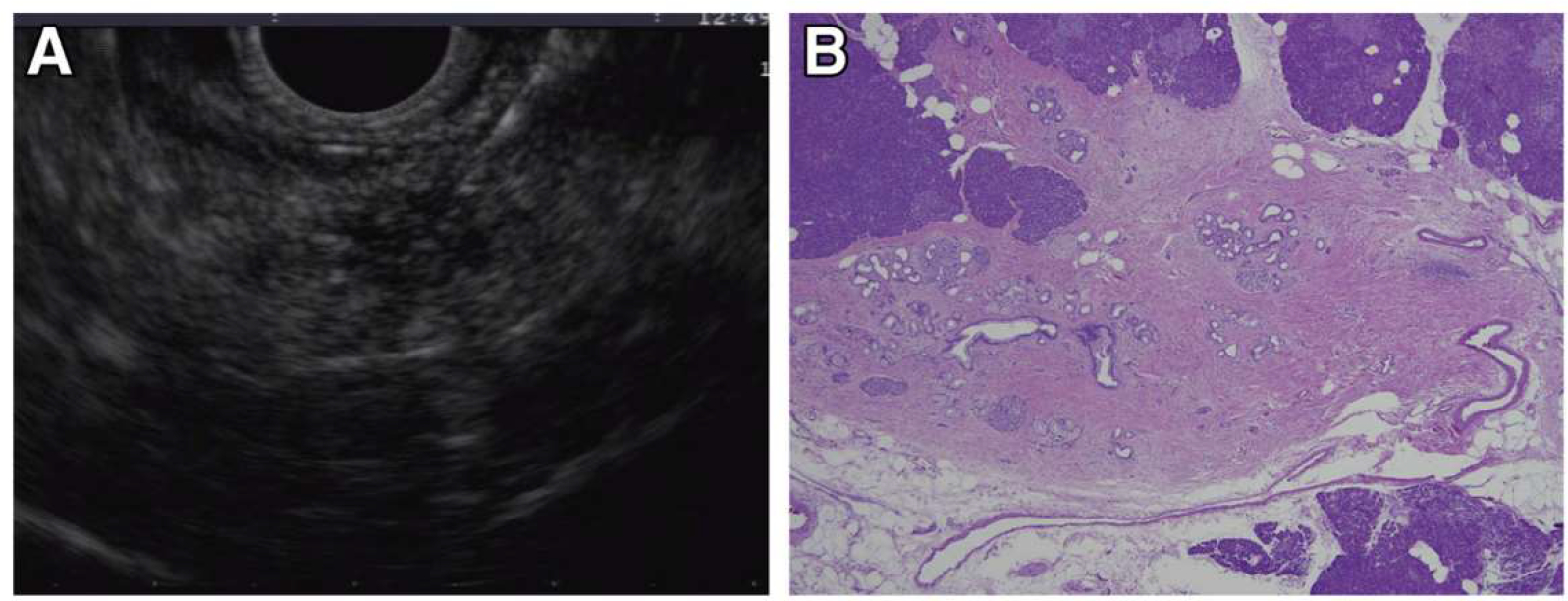
Incident indeterminate solid nodule: low-grade PanIN lesion. EUS and microscopic images from a familial pancreatic cancer relative who developed a 5-mm hypoechoic nodule in the tail (A). EUS-fine needle aspiration performed at 2 separate visits showed atypical cells. EUS-guided tattooing was performed to localize the lesion for surgical resection and histologic evaluation. Distal pancreatectomy with pathology examination of the specimen revealed a focal cluster of multiple low-grade PanIN with adjacent fibrosis (B), corresponding to the location of the preoperative tattoo.
Figure 5.
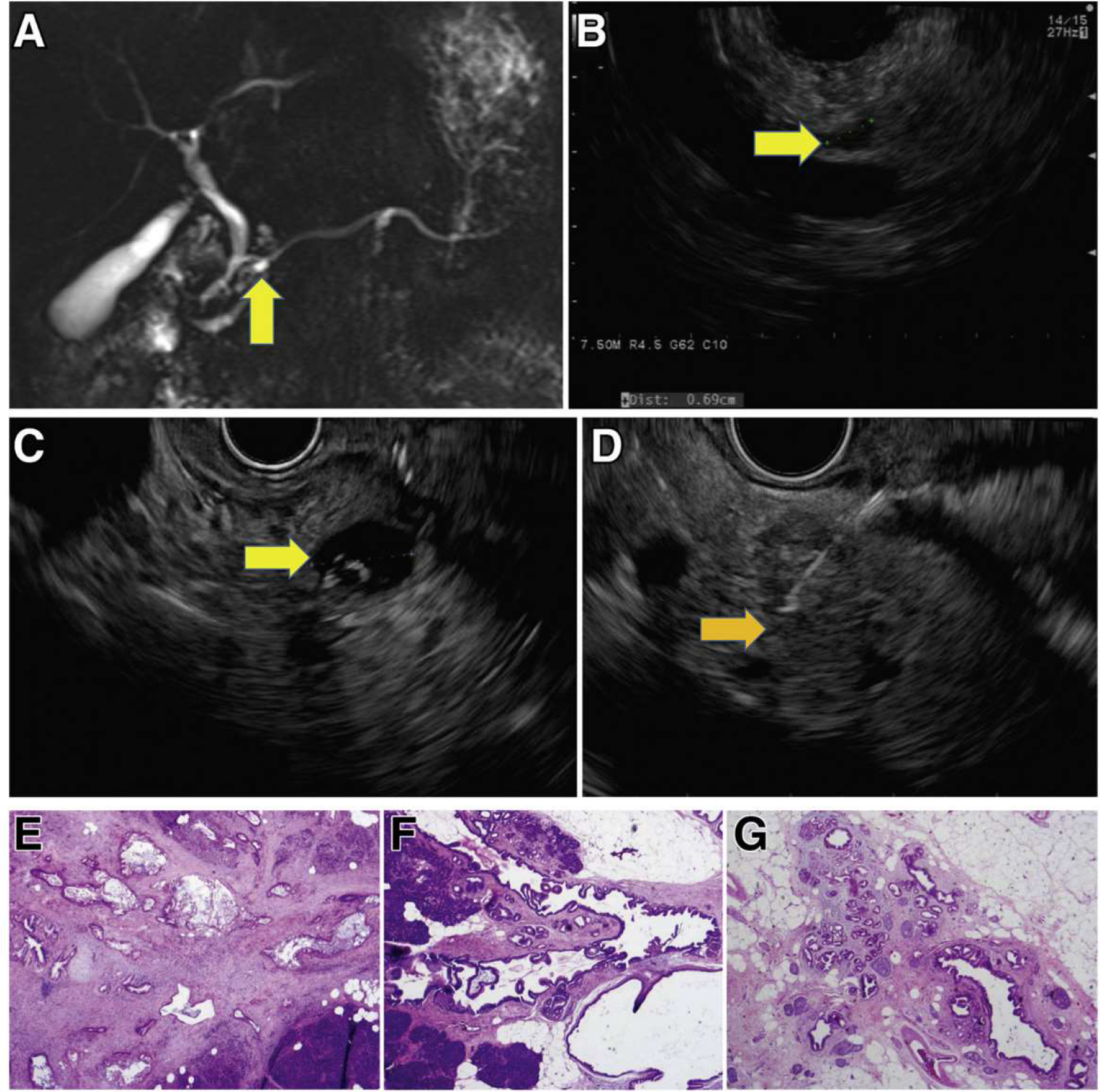
Incident screen-detected PDAC arising in a low-risk BD-IPMN. A familial pancreatic cancer patient with 3 affected FDR, who at baseline screening had subcentimeter cysts in the head (7 mm, yellow arrow) by MRI (A) and EUS (B). Surveillance continued with annual MRI alternating with EUS. Seven years from baseline, EUS showed a mildly dilated main pancreatic duct (4 mm) in the neck of the gland without a mass and 5 low-risk subcentimeter cystic lesions in the head, body, and tail. These changes did not meet international consensus guideline criteria for resection of IPMN. Repeat EUS at the next scheduled visit 1 year later showed the IPMN in the head (C, yellow arrow) associated with an asymptomatic PDAC (orange arrow, D), not visible on CT. Pancreaticoduodenectomy revealed a moderately differentiated T2N1 PDAC (hematoxylin-eosin stained pathology, image E) arising in an IPMN (F), low-grade IPMN, and extensive low-grade PanIN and associated lobulocentric atrophy (G). The patient is alive and well 4 years later.
Early Detection Biomarkers
Biomarkers may help detect microscopic high-grade PanIN precursors or early stage PDAC (Figure 1), clarify ambiguous screen-detected lesions, or prevent interval cancers in patients with a normal pancreas or trivial abnormalities on imaging. We look forward to continued progress with research on the discovery, optimization, and clinical validation of biomarkers in pancreatic juice (such as mutant TP53/SMAD4 DNA119) and blood (such as CA19–9, HbA1C plus other tumor markers,120–122 circulating tumor cells,123 exosomes,123 circulating-tumor DNA [ctDNA],124 or cell-free DNA [cfDNA]124). For the latter, the biggest challenge is the very low levels of peripheral DNA, which affects sensitivity,123 particularly for early-stage disease. A PDAC-specific antibody-based microarray test (IMMray PanCan-d, Immunovia, Marlborough, MA) measuring 8 serum biomarkers plus CA19–9, is being tested to determine the presence of a high-risk signature for PDAC. Although the details of the biomarkers are not publically available, they include proteins in multiple functional classes, including tumor-associated, hormone transport, bone metabolism, complement, coagulation, and protease inhibitor targets. Designed for use in HRIs, it serves to aid risk stratification and identify HRIs needing further clinical evaluation. Although initial results are promising,125 results from the PanFAM-1 longitudinal study in HRIs are pending (NCT03693378). Targeted methylation analysis of circulating cfDNA from pancreatic cancer (as well as multiple other tumor types, for example Galleri, Grail) is another promising approach still undergoing evaluation.126 Cancer-SEEK (Exact Sciences) assays ctDNA for 16 genes and 8 proteins shows promise in detecting cancers across 10 organs, including 7 that have no recommended standard-of-care screening tests, such as PDAC.127 The test by Blue Star Genomics, which received a Breakthrough Device designation from the Food and Drug Administration (FDA) and is undergoing clinical validation studies, involves analysis of 5-hydroxymethylcytosine signatures in cfDNA in various cancer types.128 In addition to these blood-based biomarkers, strategies deploying artificial intelligence129,130 are being developed in parallel to improve risk stratification for neoplastic progression and patient selection for surgery, high priorities for the next decade.
Diagnosis of Pancreatic Cancer
Clinical Presentation
Most patients with pancreatic cancer do not develop symptoms until the advanced stage of the disease. When patients develop symptoms, they are often vague and nonspecific, leading to a delay in diagnosis that may extend several months.131 The most commonly reported symptoms are fatigue, weight loss, anorexia, and abdominal or back pain. Tumors that arise in the pancreatic head and neck may be associated with pruritis and jaundice from biliary obstruction, whereas tumors in the body and tail of the gland more commonly present with pain. The development of NOD in the setting of any of the previously listed symptoms should prompt the patient’s physician to consider pancreatic cancer in the differential diagnosis. A thorough family history of cancer should be obtained to assess whether the patient may be a member of a familial pancreatic cancer kindred, or may carry a pathogenic germline mutation, especially if there are close family members with breast, colon, or skin (melanoma) cancers. Less common presenting symptoms include steatorrhea due to malignant obstruction of the main pancreatic duct, acute pancreatitis, gastric outlet obstruction, and venous thromboembolism.
Diagnostic Evaluation
After a complete history and physical examination, patients should undergo lab work to include a complete blood count, electrolyte panel, and liver function tests, the latter of which can help assess if an obstructive biliary process is present. A serum amylase is obtained for patients who present with epigastric and/or back pain to evaluate for the presence of acute pancreatitis. The tumor marker sialylated Lewisa blood group antigen CA 19–9 is a commonly used serum biomarker for PDAC, with a sensitivity and specificity of 80% to 90% in patients presenting with symptomatic disease.132 CA 19–9 levels are related to tumor size, presence of metastasis, and patient prognosis.132 Serial monitoring of CA 19–9 is useful to track response to systemic therapy in patients who present with elevated levels. Ten percent of the population do not express Lewis antigens,133 and in this situation the CA 19–9 level is deemed uninformative. CA 19–9 also can be elevated in the setting of benign pancreaticobiliary disease and biliary obstruction. Thus, in patients with PDAC with biliary obstruction who undergo biliary stent placement, CA 19–9 levels should be re-checked to more accurately assess true tumor burden before the initiation of therapy. In patients with localized, resectable disease and high CA 19–9 levels,134 consideration should be given to a neoadjuvant first approach, although National Comprehensive Cancer Network (NCCN) guidelines do not recommend a specific CA 19–9 cutoff level. Although CA 19–9 alone lacks adequate sensitivity for the early detection of pancreatic cancer, its use in combination with other biomarkers may provide added benefit in a blood-based early detection blood test in the future.122,135
Imaging
The imaging test of choice for the evaluation of a patient with a suspected pancreatic adenocarcinoma is a thin-cut, pancreatic protocol CT scan of the abdomen, providing a sensitivity in the detection of pancreatic cancer ranging from 76% to 96%.136 CT, compared with MRI, provides better anatomic detail of the relationship of the tumor to the surrounding vasculature, important in tumor staging. Pancreatic cancer typically appears as hypodense compared with the surrounding pancreatic parenchyma, reflecting poor penetration of contrast into these tumors, which possess high interstitial pressures due to extensive desmoplasia.137 Standard guidelines regarding the technical aspects of the performance of the pancreatic cancer CT protocol are included in NCCN guidelines, and include submillimeter slice thickness, proper timing of arterial and venous imaging, and 3-dimentional reconstruction for full vascular assessment.138 MRI is reserved for use in patients with contraindications to CT scanning, significant renal insufficiency, or a severe contrast allergy. MRI may also be a useful adjunct in patients to determine the etiology of indeterminate liver lesions. Positron emission tomography scans are not routinely used in the diagnostic evaluation of patients with PDAC, but may be helpful to detect occult metastatic disease, particularly in the setting of a high CA 19–9 disproportionately elevated based on tumor staging. Endoscopic ultrasound is used primarily for tissue acquisition to confirm the diagnosis of pancreatic cancer, but may be useful for diagnosis of small, isodense lesions.
Staging
PDAC is formally staged using a tumor nodes metastasis (TNM) system based on the eighth edition of the American Joint Committee on Cancer Staging Manual, with TNM staging used to provide prognostic information regarding patient outcomes. In practicality, clinicians use a 4-category staging system to classify tumors and guide treatment decisions: resectable, borderline resectable, locally advanced, and metastatic disease. NCCN guidelines (shown in Supplementary Table 1) define resectable disease as lacking arterial and venous involvement, with no distant spread.139 Tumors with venous contact >180 degrees, venous contour irregularity, or thrombosis are considered borderline resectable as long as the venous segment is reconstructible. Unreconstructable involvement of the superior mesenteric vein or portal vein is categorized as locally advanced.138 Arterial contact <180 degrees is considered borderline resectable, whereas contact >180 degrees is considered locally advanced. Resectability status should be determined in the context of a multidisciplinary team evaluation.
Treatment of Pancreatic Cancer
Pancreatic cancer has a very poor prognosis with a median survival of approximately 10 to 12 months with treatment and 5 to 6 months without treatment due to the advanced stage of diagnosis for many patients and the low percentage of patients eligible for surgical resection. The current standard of care for first-line therapy is FOLFIRONOX (a combination of 5-fluorouracil [5-FU], leucovorin, irinotecan, and oxaliplatin) or gemcitabine plus albumin-bound (nab) paclitaxel. Other combinations to date have not shown significant survival benefits over these treatments and/or result in treatment-limiting toxicities. However, current research on the molecular and tumor microenvironmental changes driving pancreatic cancer development, progression, and metastasis is uncovering distinct targetable pathways in certain patient subpopulations. By identifying these populations early in diagnosis, it may be possible to personalize therapy for some patients and improve their treatment outcome. As we learn more about these PDAC driving pathways, it should be possible to develop new drugs and combinations that further improve treatment outcomes for most patients.
Targetable Subtypes of Pancreatic Cancer
Many of the subtypes of PDAC susceptible to targeted therapies are associated with specific genetic alterations. Approximately 10% to 15% of PDACs are thought to be associated with inherited mutations, and all PDACs have somatic mutations in distinct combinations of driver genes. These mutations affect a range of cellular processes, including DNA repair, cellular proliferation pathways, and transcriptional activation of genes involved in cancer progression.
BRCA1/BRCA2 mutations.
BRCA1 (breast cancer type 1 susceptibility protein) and BRCA2 genes, which are involved in repairing double-strand DNA breaks via homologous DNA repair (HDR), are the most extensively characterized mutations. Mutations in these 2 genes were initially implicated as risk factors in breast and ovarian cancer and were later linked to pancreatic cancer. Mutations of other genes involved in the homologous DNA repair pathway, such as PALB2 and ATM, have also been implicated in PDAC, as well as what is referred to as a “BRCAness” HDR impaired phenotype. Mutations in BRCA1/2 are found in 5% to 10% of PDACs and occur in both familial and nonfamilial cases. In the Ashkenazi Jewish population, up to 20% patients may harbor these mutations.140
Breast and ovarian cancers with BRCA1, BRCA2, and/or related HDR mutations are susceptible to drugs that cause double-stranded DNA breaks, such as platinum salts and topoisomerase inhibitors. Recent retrospective141,142 and prospective143 studies examining the use of cisplatin in patients with PDAC with altered BRCA1/2 have shown clinical benefit, although superiority over standard of care treatments and optimal dosing and combination therapies are still being investigated.
PARP inhibitors such as niraparib and olaparib have also shown promise as treatment for tumors with BRCA mutations. These inhibitors act by preventing single-strand break repair, resulting in double-stranded breaks that are unable to be repaired by HDR deficient tumors, causing cell cycle arrest and apoptosis. Olaparib was shown to increase progression-free survival when used as a maintenance therapy for patients with pancreatic cancer who responded to first-line platinum-based therapy, leading to its FDA approval in 2019.144 Although these therapies show promise, they are also plagued by high cost and development of drug resistance. The development of optimal combination therapies for PDAC with the “BRCAness” phenotype should be pursued and may prove to be of great benefit to this subset of patients.140,145
Microsatellite instability.
Another distinct pancreatic cancer subtype is characterized by microsatellite instability (MSI). These tumors are defective in DNA mismatch repair, and can occur in inherited disorders such as Lynch Syndrome. These patients with PDAC (1% of cases) have been found to differ from patients with microsatellite stable tumors by being less responsive to 5-FU and gemcitabine and more responsive to FOLFIRINOX.146,147
In many cancers, these MSI tumors are more responsive to immune-based therapies and have a favorable prognosis.148 Since 2017, pembrolizumab, an inhibitor of the immune checkpoint protein PD-1 (programmed cell death protein-1) has been approved for mismatch-repair deficient cancers at all tumor sites. It acts as an immune checkpoint blockade preventing the binding of PD-1 to PD-L1 (programmed death ligand-1), which is selectively expressed on the surface of cancer cells but not normal cells. PD-1/PD-L1 binding suppresses the proliferation of antigen-specific T cells and causes apoptosis of regulatory T cells. Therefore, blocking of this interaction increases the patient’s innate immune response to the tumor.
PDAC, however, has proven to be more difficult to treat with immune-based therapies due to large quantities of immune-suppressor cells and characteristics of the stromal tissue. Pembrolizumab has shown some success in patients with MSI-high PDAC but not MSI-low tumors. Numerous phase I/II clinical trials have sought to increase the efficacy of PD-1/PD-L1 inhibitors in MSI-low PDACs by combining them with chemotherapy, radiotherapy, other immunotherapies, and vaccines to increase their efficacy. The COMBAT trial (NCT02826486) recently demonstrated that immunomodulation with a CXCR4 antagonist and pembrolizumab may improve clinical response to chemotherapy.149 There is also evidence suggesting that the CD40 agonist selicrelumab may modulate the tumor microenvironment in pancreatic cancer making it more immunologically active.150 Ghidini et al151 recently published a review of the current immune-based treatment approaches for patients with PDAC with regard to MSI. Although significant increases in overall survival have yet to be observed from these combinations, measurable changes in tumor immunogenicity indicate that further research in this area may lead to significantly improved immune therapies for pancreatic cancer.151
KRAS mutations.
In contrast to the preceding mutations and phenotypes that only affect a small subset of PDACs, mutations in KRAS (Kirsten rat sarcoma viral oncogene homolog) are found in 90% to 95% of PDACs and may be a driving force in pancreatic tumorigenesis. KRAS is a membrane-bound GTPase that signals cellular growth through the MAPK and PI3K pathways in its GTP form. The most prevalent KRAS oncogenic mutations have been shown to disrupt the balance of GTP- and GDP-bound KRAS.
Although mutant KRAS has been pursued as a therapeutic target for many years, its high picomolar binding affinity for GTP/GDP have made small molecule inhibition efforts of the nucleotide binding pocket difficult. However, in 2013, Ostrem et al152 published crystallographic evidence of an unknown allosteric binding pocket of KRASG12C, a common KRAS mutation in many cancers. In 2018, Janes et al153 published the discovery of a small molecular compound, ARS-1620, that is specific to this binding pocket and acts to sequester KRAS in its inactive GDP bound form. This proof-of-concept study spurred renewed interest in selective inhibition of mutated KRAS. Just 4 years later, 2 next-generation KRASG12C inhibitors have advanced to clinical use. In 2021 Sotorasib (AMG510) was FDA approved154 and Adagrasib (MRTX849) received FDA breakthrough therapy designation.155 A timely review of the clinical progress of KRASG12C inhibitors was recently published in January 2022.156
Although the advancement of KRASG12C inhibitors is impactful for treatment of certain cancers, such as lung cancer, this mutation is relatively rare in pancreatic cancer, which primarily has G12D and G12V mutations. Other innovative KRAS inhibition approaches, including Pan KRAS inhibitors that target the guanine exchange factor SOS1 (BI 1701963),157 a number of SHP2 inhibitors, and combination therapies including KRAS-specific vaccines158 may benefit patients with pancreatic cancer in the future. A promising G12D-specific inhibitor, MRTX1133, is also poised to undergo clinical testing in 2022.159 Active work in this area is focusing on the development of inhibitors to mutations relevant to PDAC and combination therapies to combat compensatory signaling pathways.
ARID1A mutations.
Another commonly mutated gene across a range of cancers is AR1D1A (AT-rich interaction domain 1A), a subunit of the chromatin remodeling complex SWI/SNF. In PDAC, ARID 1A deficiency correlates with poorly differentiated tumors and is a poor prognostic indicator. ARID1A is functionally complex, having both tumor suppressor and oncogenic properties across a number of cellular processes, including telomere maintenance, DNA damage repair, and transcriptional control.160 Recent studies have found that loss of ARID1A function correlates with the development of MSI and is associated with high mutational burden, high PD-L1 expression, and an increase in tumor infiltrating lymphocytes, which are all predictive of sensitivity to immune checkpoint blockade therapy.161 Indeed, increased progression-free and overall survival has been observed retrospectively in patients with ARID1A-deficient tumors that have undergone immunotherapy.162,163 Clinical and mouse model data also indicate increased sensitivity to PARP inhibition.164 Preclinical studies also have implicated that EZH2, HDAC6, and PI3K/AKT/mTOR inhibitors may also have increased efficacy against ARIDlA-deficient tumors.161 The exact role of ARID 1A specifically in PDAC and the use of its mutational status as an indicator for targeted therapy are still actively being investigated.
Treatment Based on Disease Stage
Other promising targeted PDAC therapies are based on disease stage. Localized tumors are categorized as resectable, borderline resectable, or locally advanced. The standard of care for resectable tumors, which lack of distant metastases and have limited vascular invasion, is partial surgical removal of the affected region of the pancreas followed by adjuvant chemotherapy. Partial removal includes pancreaticoduodenectomy surgery, also known as Whipple procedure, to remove the head of the pancreas or distal pancreatectomy to remove the tail of the organ. A total pancreatectomy is performed when the entire pancreas is affected. In addition, neoadjuvant treatment is sometimes used in this patient group. Randomized data comparing outcomes in up-front surgery vs neoadjuvant treatment followed by surgery in resectable patients is not yet available.
Neoadjuvant chemotherapy and/or radiotherapy before surgery is used in locally advanced and in most borderline resectable tumors to improve margin-negative resection rates and to potentially induce tumor downstaging. Improvements in surgical techniques have led to increased use of venous and arterial reconstruction to render some locally advanced tumors resectable. Tumor tissue acquisition pretreatment (via biopsy) and at surgery offers a unique opportunity to observe the intratumoral changes in the tumor and TME after various treatment regimens.165,160 A recent analysis by Farren et al167 that examined gene expression and spatial distribution of numerous immunologically relevant cells and proteins showed distinct differences in resected tumors from untreated pancreatic tumors vs tumors treated with neoadjuvant therapy. As a retrospective study, the relevance of these changes to patient outcome and response to further adjuvant therapy are not addressed; however, the results indicate further research in this area is needed.167
Adjuvant therapy typically occurs within 4 to 8 weeks of resection once the patient has recovered from surgery. Selection of the proper treatment regimen for each patient may vary based on the patient’s health status and cancer stage. Recent progress in the identification of genetic biomarkers, the use of ctDNA as a disease state indicator, and improved imaging methods are beginning to allow for improved ability to optimize adjuvant therapy selection. In a recent review, Turpin et al168 gives a comprehensive overview of the current state of adjuvant therapy selection. This is an active area of research that may eventually allow for the personalization of adjuvant therapy for every patient.
The treatment of nonresectable and metastatic disease varies greatly depending on treatment response, the existence of any of the aforementioned genetic mutations or phenotypes, and the overall health of the patient. In most cases, the standard first-line treatment, as with localized disease, is FOLFIRINOX or gemcitabine plus albumin-bound (nab) paclitaxel. The select subgroups noted previously may benefit from alternative targeted treatments at this stage. Second-line treatment for patients capable of more combination therapy would be the alternate treatment, that is, fluoropyrimidine-based treated patients would receive gemcitabine plus albumin-bound (nab) paclitaxel and gemcitabine-treated patients would get FOLFIRONOX or a modified 5-FU-based regimen such as FOLFIRI (5-FU, folic acid, and irinotecan) or FOLFOX (leucovorin, 5-FU, oxaliplatin). Single-agent treatments may be given to patients with a lower performance status.
Recent research is also showing that maintenance therapy may be beneficial to select patient groups. Olaparib was approved as a maintenance therapy in 2019 for patients with BRCA1 or BRCA2 mutations and metastatic disease.144 Clinical trials examining a combination of 5-FU and leucovorin or gemcitabine as PDAC maintenance therapy have also shown encouraging results.169,170 Many clinical trials are now incorporating maintenance therapy for pancreatic cancer minimal residual disease detected by ctDNA to improve long-term prognosis. As more targetable subtypes of pancreatic cancer are discovered, the treatment of both localized and metastatic disease should improve.
Future Treatment Considerations
To date, the few FDA-approved chemotherapy and targeted therapies have only increased the 5-year overall survival of pancreatic cancer patients from about 2% a decade ago to 11% in 2022. However, an increase in our understanding of the biology of subtypes of PDAC have led to more targeted approaches. New clinical trial designs that incorporate drug lead-in, neoadjuvant testing of investigational agents, and platform studies that allow rapid testing of combinations are facilitating progress. Over the next decade, we should expect to see improved clinical outcomes for a greater number of patients who will be treated with patient-specific drug combinations.
Conclusions
An improved understanding of the biologic processes driving pancreatic tumorigenesis is taking place. With the advent of increased use of genetic testing, individuals at elevated risk of pancreatic cancer are being identified and entered into screening programs, and targeted therapies for subtypes of patients with pancreatic cancer are coming into clinical use. Although these efforts show promise, large-scale, coordinated efforts will be needed to maximize the clinical impact for patients and improve overall survival.
Supplementary Material
Abbreviations:
- CAF
cancer-associated fibroblast
- cfDNA
cell-free DNA
- CT
computed tomography
- ctDNA
circulating-tumor DNA
- EUS
endoscopic ultrasound
- FDA
Food and Drug Administration
- HDR
homologous DNA repair
- HGD
high-grade dysplasia
- HRI
high-risk individual
- IPMN
intraductal papillary mucinous neoplasm
- MRI
magnetic resonance imaging
- MSI
microsatellite instability
- NCCN
National Comprehensive Cancer Network
- NNS
number needed to screen
- NOD
new-onset diabetes
- PanIN
pancreatic intraepithelial neoplasia
- PD-1
programmed cell death protein-1
- PDAC
pancreatic ductal adenocarcinoma
- PD-L1
programmed death ligand-1
Footnotes
Conflict of interest
These authors disclose the following: Elizabeth M. Jaffee is the Dana and Albert “Cubby” Professor of Oncology. She is a paid consultant for Adaptive Biotech, CSTONE, Achilles, DragonFly, Candel Therapeutics, NextCure, STIMIT, and Genocea. She receives funding from Lustgarten Foundation, AstraZeneca, Genentech, and Bristol Myer Squibb. She is the Chief Medical Advisor for Lustgarten and SAB advisor to the Parker Institute for Cancer Immunotherapy (PICI) and for the Break Through Cancer Institute. She is a founding member of Abmeta. Diane M. Simeone is the Laura and Isaac Perlmutter Professor of Surgery. She serves in an advisory capacity for Merck, Bluestar Genomics, and Interpace, and receives funding from the Pancreatic Cancer Action Network, Cyteir Therapeutics, Tempus, Micronoma and Novartis. Marcia Irene Canto is a Professor of Medicine and Oncology. She is a consultant for Castle Biosciences, BlueStar Genomics, and Pentax Medical Corporation. She receives royalties from UpToDate. She receives funding for research from Pentax Medical Corporation and Endogastric Solutions. The remaining author discloses no conflicts.
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at https://doi.org/10.1053/j.gastro.2022.03.056.
References
- 1.Rahib L, Smith B, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913–2921. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Fuchs HE, et al. Cancer Statistics, 2021. CA Cancer J Clin 2021;71:7–33. [DOI] [PubMed] [Google Scholar]
- 3.Kommalapati A, Telia SH, Goyal G, et al. Contemporary management of localized resectable pancreatic cancer. Cancers (Basel) 2018;10:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basturk O, Hong SM, Wood LD, et al. A revised classification system and recommendations from the baltimore consensus meeting for neoplastic precursor lesions in the pancreas. Am J Surg Pathol 2015;39:1730–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winter JM, Cameron JL, Campbell KA, et al. 1423 pancreaticoduodenectomies for pancreatic cancer: a single-institution experience. J Gastrointest Surg 2006;10:1199–1210; discussion 1210–1191. [DOI] [PubMed] [Google Scholar]
- 6.Felsenstein M, Noe M, Masica DL, et al. IPMNs with cooccurring invasive cancers: neighbours but not always relatives. Gut 2018;67:1652–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zamboni G, Scarpa A, Bogina G, et al. Mucinous cystic tumors of the pancreas: clinicopathological features, prognosis, and relationship to other mucinous cystic tumors. Am J Surg Pathol 1999;23:410–422. [DOI] [PubMed] [Google Scholar]
- 8.Laffan TA, Horton KM, Klein AP, et al. Prevalence of unsuspected pancreatic cysts on MDCT. AJR Am J Roentgenol 2008;191:802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grimont A, Leach SD, Chandwani R. Uncertain beginnings: acinar and ductal cell plasticity in the development of pancreatic cancer. Cell Mol Gastroenterol Hepatol 2021;13:369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsuda Y, Furukawa T, Yachina S, et al. The prevalence and clinicopathological characteristics of high-grade pancreatic intraepithelial neoplasia: autopsy study evaluating the entire pancreatic parenchyma. Pancreas 2017;46:658–664. [DOI] [PubMed] [Google Scholar]
- 11.Andea A, Sarkar F, Adsay VN. Clinicopathological correlates of pancreatic intraepithelial neoplasia: a comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod Pathol 2003;16:996–1006. [DOI] [PubMed] [Google Scholar]
- 12.Rezaee N, Barbon C, Zaki A, et al. Intraductal papillary mucinous neoplasm (IPMN) with high-grade dysplasia is a risk factor for the subsequent development of pancreatic ductal adenocarcinoma. HPB (Oxford) 2016;18:236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oyama H, Tada M, Takagi K, et al. Long-term risk of malignancy in branch-duct intraductal papillary mucinous neoplasms. Gastroenterology 2020;158:226–237.e225. [DOI] [PubMed] [Google Scholar]
- 14.Furukawa T, Kloppel G, Adsay NV, et al. Classification of types of intraductal papillary-mucinous neoplasm of the pancreas: a consensus study. Virchows Archiv 2005;447:794–799. [DOI] [PubMed] [Google Scholar]
- 15.Noë M, Niknafs N, Fischer CG, et al. Genomic characterization of malignant progression in neoplastic pancreatic cysts. Nat Commun 2020;11:4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luchini C, Capelli P, Scarpa A. Pancreatic ductal adenocarcinoma and its variants. Surg Pathol Clin 2016;9:547–560. [DOI] [PubMed] [Google Scholar]
- 17.Groot VP, Gemenetzis G, Blair AB, et al. Implications of the pattern of disease recurrence on survival following pancreatectomy for pancreatic ductal adenocarcinoma. Ann Surg Oncol 2018;25:2475–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wangjam T, Zhang Z, Zhou XC, et al. Resected pancreatic ductal adenocarcinomas with recurrence limited in lung have a significantly better prognosis than those with other recurrence patterns. Oncotarget 2015;6:36903–36910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.lacobuzio-Donahue CA, Fu B, Yachida S, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol 2009;27:1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Almoguera C, Shibata D, Forrester K, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988;53:549–554. [DOI] [PubMed] [Google Scholar]
- 21.Scarpa A, Capelli P, Mukai K, et al. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am J Pathol 1993;142:1534–1543. [PMC free article] [PubMed] [Google Scholar]
- 22.Caldas C, Hahn SA, da Cost LT, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet 1994;8:27–32. [DOI] [PubMed] [Google Scholar]
- 23.Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996;271:350–353. [DOI] [PubMed] [Google Scholar]
- 24.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 27.Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017;32:185–203.e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020;578:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Notta F, Chang-Seng-Yue M, Lemire M, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016;538:378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hata T, Suenaga M, Marchionni L, et al. Genome-wide somatic copy number alterations and mutations in high-grade pancreatic intraepithelial neoplasia. Am J Pathol 2018;188:1723–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makohon-Moore AP, Matsukuma K, Zhang M, et al. Precancerous neoplastic cells can move through the pancreatic ductal system. Nature 2018;561:201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010;467:1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Makohon-Moore AP, Zhang M, Reiter JG, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet 2017;49:358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuboki Y, Fischer CG, Beleva Guthrie V, et al. Single-cell sequencing defines genetic heterogeneity in pancreatic cancer precursor lesions. J Pathol 2019;247:347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fischer CG, Beleva Guthrie V, Braxton AM, et al. Intraductal papillary mucinous neoplasms arise from multiple independent clones, each with distinct mutations. Gastroenterology 2019;157:1123–1137.e1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fujikura K, Hosoda W, Felsenstein M, et al. Multiregion whole-exome sequencing of intraductal papillary mucinous neoplasms reveals frequent somatic KLF4 mutations predominantly in low-grade regions. Gut 2020;70:928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hosoda W, Chianchiano P, Griffin JF, et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. J Pathol 2017;242:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pea A, Yu J, Rezaee N, et al. Targeted DNA sequencing reveals patterns of local progression in the pancreatic remnant following resection of intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg 2017;266:133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Omori Y, Ono Y, Tanino M, et al. Pathways of progression from intraductal papillary mucinous neoplasm to pancreatic ductal adenocarcinoma based on molecular features. Gastroenterology 2019;156:647–661.e642. [DOI] [PubMed] [Google Scholar]
- 41.Schutte M, Hruban RH, Geradts J, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res 1997;57:3126–3130. [PubMed] [Google Scholar]
- 42.Vincent A, Omura N, Hong SM, et al. Genome-wide analysis of promoter methylation associated with gene expression profile in pancreatic adenocarcinoma. Clin Cancer Res 2011;17:4341–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDonald OG, Li X, Saunders T, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet 2017;49:367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukushima N, Sato N, Ueki T, et al. Aberrant methylation of preproenkephalin and p16 genes in pancreatic intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am J Pathol 2002;160:1573–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato N, Fukushima N, Hruban RH, et al. CpG island methylation profile of pancreatic intraepithelial neoplasia. Mod Pathol 2008;21:238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sato N, Ueki T, Fukushima N, et al. Aberrant methylation of CpG islands in intraductal papillary mucinous neoplasms of the pancreas. Gastroenterology 2002;123:365–372. [DOI] [PubMed] [Google Scholar]
- 47.Fujikura K, Alruwaii Zl, Haffner M, et al. Downregulation of 5-hydroxymethylcytosine is an early event in pancreatic tumorigenesis. J Pathol 2021;254:279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47:1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011;17:500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Puleo F, Nicolle R, Blum Y, et al. Stratification of pancreatic ductal adenocarcinomas based on tumor and microenvironment features. Gastroenterology 2018;155:1999–2013.e1993. [DOI] [PubMed] [Google Scholar]
- 51.Chan-Seng-Yue M, Kim JC, Wilson GW, et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat Genet 2020;52:231–240. [DOI] [PubMed] [Google Scholar]
- 52.Connor AA, Denroche RE, Jang GH, et al. Integration of genomic and transcriptional features in pancreatic cancer reveals increased cell cycle progression in metastases. Cancer Cell 2019;35:267–282.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raghavan S, Winter PS, Navia AW, et al. Microenvironment drives cell state, plasticity, and drug response in pancreatic cancer. Cell 2021;184:6119–6137.e6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hayashi A, Fan J, Chen R, et al. A unifying paradigm for transcriptional heterogeneity and squamous features in pancreatic ductal adenocarcinoma. Nature Cancer 2020;1:59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Topham JT, Karasinka JM, Lee MKC, et al. Subtype-discordant pancreatic ductal adenocarcinoma tumors show intermediate clinical and molecular characteristics. Clin Cancer Res 2021;27:150–157. [DOI] [PubMed] [Google Scholar]
- 56.Herting CJ, Karpovsky I, Lesinski GB. The tumor microenvironment in pancreatic ductal adenocarcinoma: current perspectives and future directions. Cancer Metastasis Rev 2021;40:675–689. [DOI] [PubMed] [Google Scholar]
- 57.Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011;19:456–469. [DOI] [PubMed] [Google Scholar]
- 58.Karamitopoulou E Tumour microenvironment of pancreatic cancer: immune landscape is dictated by molecular and histopathological features. Br J Cancer 2019;121:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ho WJ, Jaffee EM, Zheng L. The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat Rev Clin Oncol 2020;17:527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heumann T, Azad N. Next-generation immunotherapy for pancreatic ductal adenocarcinoma: navigating pathways of immune resistance. Cancer Metastasis Rev 2021;40:837–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Helms E, Onate MK, Sherman MH. Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer Discov 2020;10:648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elyada E, Bolisetty M, Laise P, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov 2019;9:1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 2013;110:20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Francescone R, Vendramini-Costa DB, Franco-Barraza J, et al. Netrin G1 promotes pancreatic tumorigenesis through cancer-associated fibroblast-driven nutritional support and immunosuppression. Cancer Discov 2021;11:446–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014;25:735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee JJ, Perera RM, Wang H, et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci U S A 2014;111:E3091–E3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Jesus-Acosta A, Sugar EA, O’Dwyer PJ, et al. Phase 2 study of vismodegib, a hedgehog inhibitor, combined with gemcitabine and nab-paclitaxel in patients with untreated metastatic pancreatic adenocarcinoma. Br J Cancer 2020;122:498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Catenacci DV, Junttila MR, Karrison T, et al. Randomized phase Ib/ll study of gemcitabine plus placebo or vismodegib, a Hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol 2015;33:4284–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ohlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214:579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Biffi G, Oni TE, Spielman B, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 2019;9:282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Steele NG, Biffi G, Kemp SB, et al. Inhibition of Hedgehog signaling alters fibroblast composition in pancreatic cancer. Clin Cancer Res 2021;27:2023–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Banh RS, Biancur DE, Yamamoto K, et al. Neurons release serine to support mRNA translation in pancreatic cancer. Cell 2020;183:1202–1218.e1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saloman JL, Albers KM, Li D, et al. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc Natl Acad Sci U S A 2016;113:3078–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Renz BW, Takahashi R, Tanaka T, et al. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell 2018;33:75–90.e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klein AP. Pancreatic cancer epidemiology: understanding the role of lifestyle and inherited risk factors. Nat Rev Gastroenterol Hepatol 2021;18:493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Momi N, Kaur S, Ponnusamy MP, et al. Interplay between smoking-induced genotoxicity and altered signaling in pancreatic carcinogenesis. Carcinogenesis 2012;33:1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hassan MM, Bondy ML, Wolff RA, et al. Risk factors for pancreatic cancer: case-control study. Am J Gastroenterol 2007;102:2696–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sah RP, Nagpal SJ, Mukhopadhyay D, et al. New insights into pancreatic cancer-induced paraneoplastic diabetes. Nat Rev Gastroenterol Hepatol 2013;10:423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maitra A, Sharma A, Brand RE, et al. A prospective study to establish a new-onset diabetes cohort: From the Consortium for the Study of Chronic Pancreatitis, Diabetes, and Pancreatic Cancer. Pancreas 2018;47:1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chari ST, Leibson CL, Rabe KG, et al. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology 2005;129:504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen W, Butler RK, Lustigova E, et al. Validation of the enriching new-onset diabetes for pancreatic cancer model in a diverse and integrated healthcare setting. Dig Dis Sci 2021;66:78–87. [DOI] [PubMed] [Google Scholar]
- 82.Sharma A, Kandlakunta H, Nagpal SJS, et al. Model to determine risk of pancreatic cancer in patients with new-onset diabetes. Gastroenterology 2018;155:730–739.e733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bracci PM. Obesity and pancreatic cancer: overview of epidemiologic evidence and biologic mechanisms. Mol Carcinog 2012;51:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Berrington de Gonzalez A, Sweetland S, Spencer E. A meta-analysis of obesity and the risk of pancreatic cancer. Br J Cancer 2003;89:519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Michaud DS, Giovannucci E, Willett WC, et al. Physical activity, obesity, height, and the risk of pancreatic cancer. JAMA 2001;286:921–929. [DOI] [PubMed] [Google Scholar]
- 86.Kirkegard J, Mortensen FV, Cronin-Fenton D. Chronic pancreatitis and pancreatic cancer risk: a systematic review and meta-analysis. Am J Gastroenterol 2017;112:1366–1372. [DOI] [PubMed] [Google Scholar]
- 87.Weissman S, Takakura K, Eibl G, et al. The diverse involvement of cigarette smoking in pancreatic cancer development and prognosis. Pancreas 2020;49:612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Giardiello FM, Welsh SB, Hamilton SR, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med 1987;316:1511–1514. [DOI] [PubMed] [Google Scholar]
- 89.Goldstein AM, Fraser MC, Struewing JP, et al. Increased risk of pancreatic cancer in melanoma-prone kindreds with p16INK4 mutations. N Engl J Med 1995;333:970–974. [DOI] [PubMed] [Google Scholar]
- 90.Rebours V, Boutron-Ruault MC, Schnee M, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am J Gastroenterol 2008;103:111–119. [DOI] [PubMed] [Google Scholar]
- 91.Teich N, Rosendahl J, Tóth M, et al. Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum Mutat 2006;27:721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Petersen GM. Familial pancreatic adenocarcinoma. Hematol Oncol Clin North Am 2015;29:641–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Klein AP. Identifying people at a high risk of developing pancreatic cancer. Nat Rev Cancer 2013;13:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Roberts NJ, Norris AL, Peterson GM, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov 2016;6:166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roberts NJ, Jiao T, Yu J, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov 2012;2:41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Couch FJ, Johnson MR, Rabe KG, et al. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol Biomarkers Prev 2007;16:342–346. [DOI] [PubMed] [Google Scholar]
- 97.Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol 2017;35:3382–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.National Cancer Institute. Cancer stat facts: pancreatic cancer. 2021. Available at: https://seer.cancer.gov/statfacts/html/pancreas.html. Accessed May 28, 2021.
- 99.Hur C, Tramontano AC, Dowling EC, et al. Early pancreatic ductal adenocarcinoma survival is dependent on size: positive implications for future targeted screening. Pancreas 2016;45:1062–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Goggins M, Overbeek KA, Brand R, et al. Management of patients with increased risk for familial pancreatic cancer: updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 2020;69:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Corral JE, Mareth KF, Riegert-Johnson DL, et al. Diagnostic yield from screening asymptomatic individuals at high risk for pancreatic cancer: a meta-analysis of cohort studies. Clin Gastroenterol Hepatol 2019;17:41–53. [DOI] [PubMed] [Google Scholar]
- 102.Signoretti M, Bruno MJ, Zerboni G, et al. Results of surveillance in individuals at high-risk of pancreatic cancer: a systematic review and meta-analysis. United European Gastroenterol J 2018;6:489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Corral JE, Das A, Bruno MJ, et al. Cost-effectiveness of pancreatic cancer surveillance in high-risk individuals: an economic analysis. Pancreas 2019;48:526–536. [DOI] [PubMed] [Google Scholar]
- 104.Bruenderman E, Martin RC 2nd. A cost analysis of a pancreatic cancer screening protocol in high-risk populations. Am J Surg 2015;210:409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Joergensen MT, Gerdes AM, Sorensen J, et al. Is screening for pancreatic cancer in high-risk groups cost-effective? Experience from a Danish national screening program. Pancreatology 2016;16:584–592. [DOI] [PubMed] [Google Scholar]
- 106.Rulyak SJ, Kimmey MB, Veenstra DL, et al. Cost-effectiveness of pancreatic cancer screening in familial pancreatic cancer kindreds. Gastrointest Endosc 2003;57:23–29. [DOI] [PubMed] [Google Scholar]
- 107.U.S. Preventive Services Task force. Screening for pancreatic cancer: US Preventive Services Task Force Reaffirmation Recommendation Statement. JAMA 2019;322:438–444. [DOI] [PubMed] [Google Scholar]
- 108.Henrikson NB, Bowles EJA, Blasi PR, et al. Screening for pancreatic cancer: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA 2019;322:445–454. [DOI] [PubMed] [Google Scholar]
- 109.Vasen H, Ibrahim I, Ponce CG, et al. Benefit of surveillance for pancreatic cancer in high-risk individuals: outcome of long-term prospective follow-up studies from three European expert centers. J Clin Oncol 2016;34:2010–2019. [DOI] [PubMed] [Google Scholar]
- 110.Canto Ml, Almario JA, Schulick RD, et al. Risk of neoplastic progression in individuals at high risk for pancreatic cancer undergoing long-term surveillance. Gastroenterology 2018;155:740–751.e742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Overbeek KA, Levink IJM, Koopmann BDM, et al. Long-term yield of pancreatic cancer surveillance in high-risk individuals. Gut 2022;71:1152–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Overbeek KA, Goggins MG, Dbouk M, et al. Timeline of development of pancreatic cancer and implications for successful early detection in high-risk individuals. Gastroenterology 2022;162:772–785.e2. [DOI] [PubMed] [Google Scholar]
- 113.Canto Ml, Kerdisirichairat T, Yeo CJ, et al. Surgical outcomes after pancreatic resection of screening-detected lesions in individuals at high risk for developing pancreatic cancer. J Gastrointest Surg 2020;24:1101–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Paiella S, Salvia R, De Pastena M, et al. Screening/surveillance programs for pancreatic cancer in familial high-risk individuals: a systematic review and proportion meta-analysis of screening results. Pancreatology 2018;18:420–428. [DOI] [PubMed] [Google Scholar]
- 115.Harinck F, Nagtegaal T, Kluijt I, et al. Feasibility of a pancreatic cancer surveillance program from a psychological point of view. Genet Med 2011;13:1015–1024. [DOI] [PubMed] [Google Scholar]
- 116.O’Neill RS, Meiser B, Emmanuel S, et al. Long-term positive psychological outcomes in an Australian pancreatic cancer screening program. Familial Cancer 2020;19:23–35. [DOI] [PubMed] [Google Scholar]
- 117.Vasen HF, Wasser M, van Mil A, et al. Magnetic resonance imaging surveillance detects early-stage pancreatic cancer in carriers of a p16-Leiden mutation. Gastroenterology 2011;140:850–856. [DOI] [PubMed] [Google Scholar]
- 118.Dbouk M, Brewer Gutierrez Ol, Lennon AM, et al. Guidelines on management of pancreatic cysts detected in high-risk individuals: an evaluation of the 2017 Fukuoka guidelines and the 2020 International Cancer of the Pancreas Screening (CAPS) consortium statements. Pancreatology 2021;21:613–621. [DOI] [PubMed] [Google Scholar]
- 119.Suenaga M, Yu J, Shindo K, et al. Pancreatic juice mutation concentrations can help predict the grade of dysplasia in patients undergoing pancreatic surveillance. Clin Cancer Res 2018;24:2963–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fahrmann JF, Schmidt CM, Mao K, et al. Lead-time trajectory of CA19–9 as an anchor marker for pancreatic cancer early detection. Gastroenterology 2021;160:1373–1383.e1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Abe T, Koi C, Kohi S, et al. Gene variants that affect levels of circulating tumor markers increase identification of patients with pancreatic cancer. Clin Gastroenterol Hepatol 2020;18:1161–1169.e1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim J, Bamlet WR, Oberg AL, et al. Detection of early pancreatic ductal adenocarcinoma with thrombospondin-2 and CA19–9 blood markers. Sci Transl Med 2017;9:eaah5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhu Y, Yang T, Wu Q, et al. Diagnostic performance of various liquid biopsy methods in detecting colorectal cancer a meta-analysis. Cancer Med 2020;9:5699–5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Grunvald MW, Jacobson RA, Kuzel TM, et al. Current status of circulating tumor DNA liquid biopsy in pancreatic cancer. Int J Mol Sci 2020;21:7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mellby LD, Nyberg AP, Johansen JS, et al. Serum biomarker signature-based liquid biopsy for diagnosis of early-stage pancreatic cancer. J Clin Oncol 2018;36:2887–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu MC, Oxnard GR, Klein EA, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol 2020;31:745–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018;359:926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Song CX, Yin S, Ma L, et al. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res 2017;27:1231–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kenner B, Chari ST, Kelsen D, et al. Artificial intelligence and early detection of pancreatic cancer: 2020 summative review. Pancreas 2021;50:251–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chu LC, Park S, Kawamoto S, et al. Application of deep learning to pancreatic cancer detection: lessons learned from our initial experience. J Am Coll Radiol 2019;16:1338–1342. [DOI] [PubMed] [Google Scholar]
- 131.Raptis DA, Fessas C, Belasyse-Smith P, et al. Clinical presentation and waiting time targets do not affect prognosis in patients with pancreatic cancer. Surgeon 2010;8:239–246. [DOI] [PubMed] [Google Scholar]
- 132.Steinberg W The clinical utility of the CA 19–9 tumor-associated antigen. Am J Gastroenterol 1990;85:350–355. [PubMed] [Google Scholar]
- 133.Scara S, Bottoni P, Scatena R. CA 19–9: biochemical and clinical aspects. Adv Exp Med Biol 2015;867:247–260. [DOI] [PubMed] [Google Scholar]
- 134.Maithel SK, Maloney S, Winston C, et al. Preoperative CA 19–9 and the yield of staging laparoscopy in patients with radiographically resectable pancreatic adenocarcinoma. Ann Surg Oncol 2008;15:3512–3520. [DOI] [PubMed] [Google Scholar]
- 135.Kaur S, Smith LM, Patel A, et al. A combination of MUC5AC and CA19–9 improves the diagnosis of pancreatic cancer: a multicenter study. Am J Gastroenterol 2017;112:172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chu LC, Goggins MG, Fishman EK. Diagnosis and detection of pancreatic cancer. Cancer J 2017;23:333–342. [DOI] [PubMed] [Google Scholar]
- 137.Provenzano PP, Hingorani SR. Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br J Cancer 2013;108:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Al-Hawary MM, Francis IR, Chari ST, et al. Pancreatic ductal adenocarcinoma radiology reporting template: consensus statement of the society of abdominal radiology and the american pancreatic association. Gastroenterology 2014;146:291–304.e291. [DOI] [PubMed] [Google Scholar]
- 139.Tempero MA, Malafa MP, Al-Hawary M, et al. Pancreatic adenocarcinoma, version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Cane Netw 2021;19:439–457. [DOI] [PubMed] [Google Scholar]
- 140.Wong W, Raufi AG, Safyan RA, et al. BRCA mutations in pancreas cancer: spectrum, current management, challenges and future prospects. Cancer Manag Res 2020;12:2731–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wattenberg MM, Asch D, Yu S, et al. Platinum response characteristics of patients with pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or PALB2 mutation. Br J Cancer 2020;122:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer 2014;111:1132–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.O’Reilly EM, Lee JW, Zalupski M, et al. Randomized, multicenter, Phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J Clin Oncol 2020;38:1378–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med 2019;381:317–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kasi A, Al-Jumayli M, Park R, et al. Update on the role of poly (ADP-ribose) polymerase inhibitors in the DNA repair-deficient pancreatic cancers: a narrative review. J Pancreat Cancer 2020;6:107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cloyd JM, Katz MHG, Wang H, et al. Clinical and genetic implications of DNA mismatch repair deficiency in patients with pancreatic ductal adenocarcinoma. JAMA Surg 2017;152:1086–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Riazy M, Kalloger SE, Sheffield BS, et al. Mismatch repair status may predict response to adjuvant chemotherapy in resectable pancreatic ductal adenocarcinoma. Mod Pathol 2015;28:1383–1389. [DOI] [PubMed] [Google Scholar]
- 148.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bockorny B, Semenisty V, Macarulla T, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med 2020;26:878–885. [DOI] [PubMed] [Google Scholar]
- 150.Byrne KT, Betts CB, Mick R, et al. Neoadjuvant selicrelumab, an agonist CD40 antibody, induces changes in the tumor microenvironment in patients with resectable pancreatic cancer. Clin Cancer Res 2021;27:4574–4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ghidini M, Lampis A, Mirchev MB, et al. Immune-based therapies and the role of microsatellite instability in pancreatic cancer. Genes 2020;12:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ostrem JM, Peters U, Sos ML, et al. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013;503:548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Janes MR, Zhang J, Li LS, et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 2018;172:578–589.e517. [DOI] [PubMed] [Google Scholar]
- 154.Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019;575:217–223. [DOI] [PubMed] [Google Scholar]
- 155.Fell JB, Fischer JP, Baer BR, et al. Identification of the clinical development candidate MRTX849, a Covalent KRAS(G12C) inhibitor for the treatment of cancer. J Med Chem 2020;63:6679–6693. [DOI] [PubMed] [Google Scholar]
- 156.Kwan AK, Piazza GA, Keeton AB, et al. The path to the clinic: a comprehensive review on direct KRAS(G12C) inhibitors. J Exp Clin Cancer Res 2022;41:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hofmann MH, Gmachl M, Ramharter J, et al. BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov 2021;11:142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Krishnan T, Roberts-Thomson R, Broadbridge V, et al. Targeting mutated KRAS genes to treat solid tumours. Mol Diagn Ther 2022;26:39–49. [DOI] [PubMed] [Google Scholar]
- 159.Wang X, Allen S, Blake JF, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRAS(G12D) inhibitor. J Med Chem 2022;65:3123–3133. [DOI] [PubMed] [Google Scholar]
- 160.Ferri-Borgogno S, Barui S, McGee AM, et al. Paradoxical role of AT-rich interactive domain 1A in restraining pancreatic carcinogenesis. Cancers (Basel) 2020;12:2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wang L, Qu J, Zhou N, et al. Effect and biomarker of immune checkpoint blockade therapy for ARID1A deficiency cancers. Biomed Pharmacother 2020;130:110626. [DOI] [PubMed] [Google Scholar]
- 162.Jiang T, Chen X, Su C, et al. Pan-cancer analysis of ARID1A alterations as biomarkers for immunotherapy outcomes. J Cancer 2020;11:776–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Okamura R, Kato S, Lee S, et al. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J Immunother Cancer 2020;8:e000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Park Y, Chui MH, Rahmato YS, et al. Loss of ARID1A in tumor cells renders selective vulnerability to combined ionizing radiation and PARP inhibitor therapy. Clin Cancer Res 2019;25:5584–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Yu Y, Zheng P, Chen Y, et al. Advances and challenges of neoadjuvant therapy in pancreatic cancer. Asia Pac J Clin Oncol 2020;17:425–434. [DOI] [PubMed] [Google Scholar]
- 166.Muller PC, Frey MC, Ruzza CM, et al. Neoadjuvant chemotherapy in pancreatic cancer: an appraisal of the current high-level evidence. Pharmacology 2021;106:143–153. [DOI] [PubMed] [Google Scholar]
- 167.Farren MR, Sayegh L, Ware MB, et al. Immunologic alterations in the pancreatic cancer microenvironment of patients treated with neoadjuvant chemotherapy and radiotherapy. JCI Insight 2020;5:e130362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Turpin A, El Amrani M, Bachet JB, et al. Adjuvant pancreatic cancer management: towards new perspectives in 2021. Cancers (Basel) 2020;12:3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Dahan L, Williet N, Le Malicot K, et al. Randomized phase II trial evaluating two sequential treatments in first line of metastatic pancreatic cancer: results of the PANOPTIMOX-PRODIGE 35 trial. J Clin Oncol 2021;39:3242–3250. [DOI] [PubMed] [Google Scholar]
- 170.Petrioli R, Torre P, Pesola G, et al. Gemcitabine plus nab-paclitaxel followed by maintenance treatment with gemcitabine alone as first-line treatment for older adults with locally advanced or metastatic pancreatic cancer. J Geriatr Oncol 2020;11:647–651. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.