

Abstract
Bacillus subtilis is a widely studied Gram-positive bacterium that serves as an important model for understanding processes critical for several areas of biology including biotechnology and human health. B. subtilis has several advantages as a model organism: it is easily grown under laboratory conditions, it has a rapid doubling time, it is relatively inexpensive to maintain, and it is non-pathogenic. Over the last 50 years, advancements in genetic engineering and genetic manipulation have continued to make B. subtilis a genetic workhorse in scientific discovery. In this chapter, we describe methods for traditional gene disruption, use of gene deletion libraries from the Bacillus Genetic Stock Center, allelic exchange, CRISPRi, and CRISPR/Cas9. Additionally, we provide general materials and equipment needed, strengths and limitations, time considerations, and troubleshooting notes to perform each method. Use of the methods outlined in this chapter will allow researchers to create gene insertions, deletions, substitutions, and RNA interference strains through a variety of methods custom to each application.
Keywords: CRISPR/Cas9, genetic manipulation, Bacillus subtilis, genetic engineering, Gram-positive
1. Introduction
The Gram-positive model bacterium Bacillus subtilis is a highly tractable genetic system, and is often more straightforward to manipulate than human pathogens or environmental bacterial isolates from the same phylum. The numerous genetic tools available, the development of genetic competence, and rapid growth rate are a few attributes that have maintained B. subtilis as a model organism for studying a wide variety of conserved biological processes.
B. subtilis can be easily grown under conditions that activate the development of genetic competence for the uptake of exogenous DNA in the laboratory with limited media, tools, and expense [1]. After growing B. subtilis to stationary phase in minimal media, cells induce a genetic program for natural competence allowing for the uptake and integration of extracellular DNA or the uptake, reassembly, and maintenance of plasmids [2]. Growth into stationary phases induces expression of the global competence regulator ComK, which in turn activates expression of the ComK regulon including genes for DNA uptake, and homologous recombination [3–6]. Once B. subtilis cells non-specifically bind DNA, the transforming DNA is fed through a channel where it enters the cytosol as single stranded DNA (ssDNA) [[7] for review [8]]. When incoming DNA has sequence homology with the chromosome, RecA catalyzes homology search and pairing with the B. subtilis chromosome [9]. For comprehensive reviews on the mechanism of natural competence and DNA repair, we direct readers to the following reviews [10,8,11,12]. Harnessing the natural competency of B. subtilis has led to many methods for genetic manipulation, with several new advances occurring within the last 12 years [13–16]. In addition to natural competence, B. subtilis has an efficient homologous recombination system allowing for straightforward and effective genetic alterations to be built using PCR fragments, plasmids, or genomic DNA [1]. For efficient homologous recombination to occur, template DNA should contain approximately 500 bp of homology to the host chromosome. It is important for users to consider the order of gene deletion and the method of integration used when studying mutant alleles in processes that are dependent on or affected by competence and recombination because integration of transforming DNA uses both the genetic competence and homologous recombination machinery.
There are a number of tools readily available to B. subtilis researchers (Figure 1). A deletion library containing erythromycin and kanamycin cassette interruptions of non-essential genes and a library containing CRISPR-interference (CRISPRi) strains of essential genes are housed at the Bacillus Genetic Stock Center (http://www.bgsc.org) (Figure 2). These resources are available for integration of knockout alleles or to knockdown essential genes [14,16]. Once strains are acquired, researchers can extract genomic DNA (gDNA) and use it to transform their strain background of interest [14]. Double and triple knockouts can be achieved using this method (discussed in the next section). CRISPRi strains contain a single guide RNA (sgRNA) for Cas9 targeting to the chromosomal gene of interest, resulting in RNA polymerase stalling and an inhibition of transcription [16]. The level of gene transcription can also be titrated using CRISPRi, reducing expression to exceptionally low levels without completely eliminating gene function [16]. CRISPRi is beneficial for the study of essential genes or genes that require a conditional knockdown in transcription to examine the resulting phenotype [16].
Figure 1.
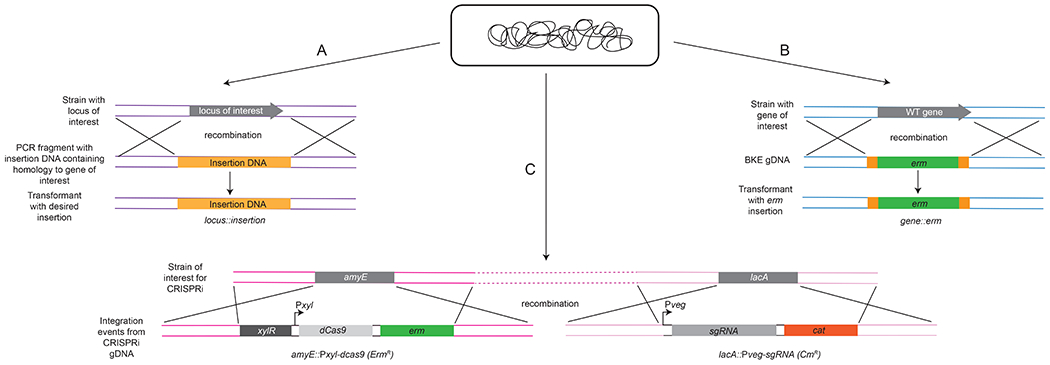
Recombination of DNA into the B. subtilis genome. (A) Recombination of PCR products, (B) Recombination of genomic DNA containing an antibiotic cassette, (C) Recombination of genomic DNA from the CRISPRi library.
Figure 2.
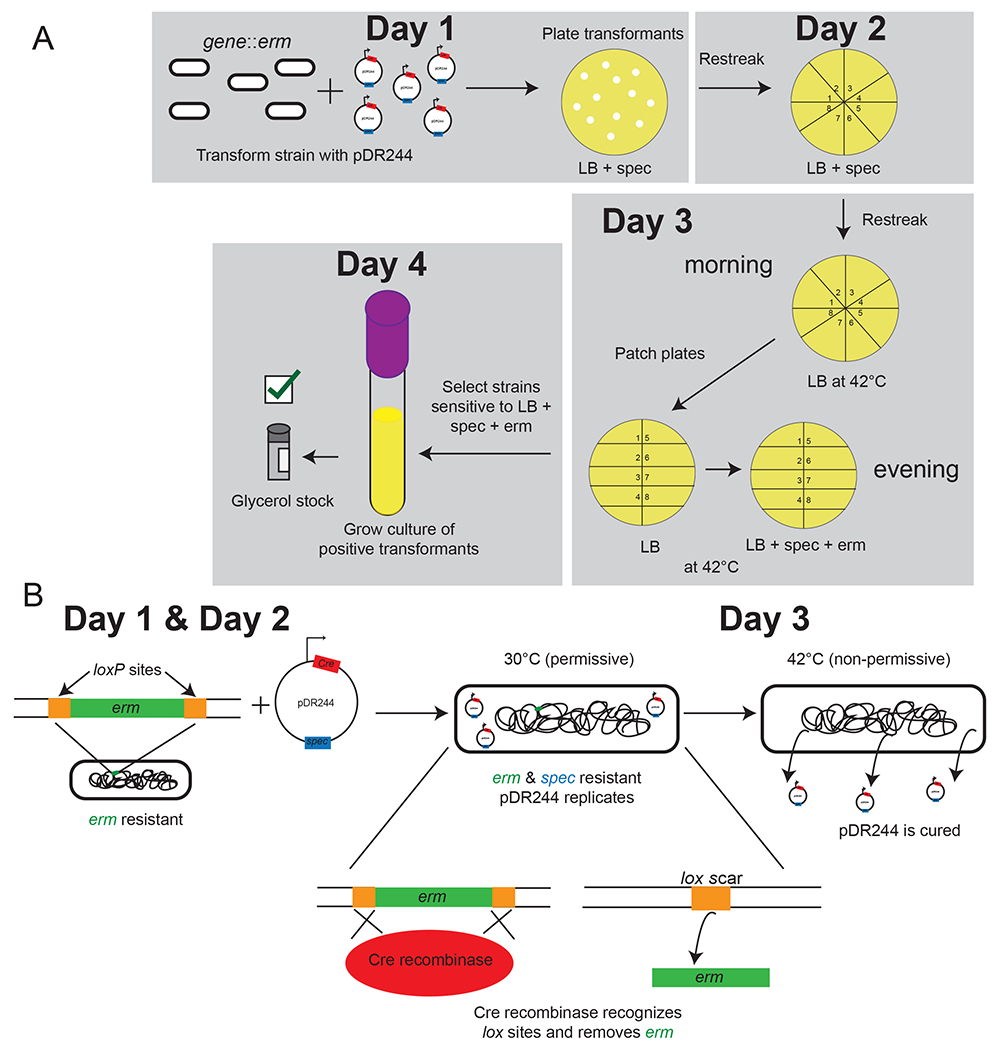
Basic procedure for use of the Bacillus Genetic Stock Center deletion library. (A) Experimental outline of erm cassette removal to create clean deletions using pDR244, (B) pDR244 action inside the cell.
Markerless mutations in B. subtilis can be achieved using several different methods [14,17,13,15]. To create a deletion using the Bacillus gene knockout library, pDR244 aids in the removal of antibiotic insertions. The plasmid pDR244 contains a temperature-sensitive origin of replication and constitutively expresses Cre recombinase [14]. Cre recognizes lox sites flanking an integrated antibiotic cassette in strains from the Bacillus Genetic Stock Center library, allowing for excision of the antibiotic cassettes leaving only a lox scar [14] (Figure 2). A second approach involves the integration and looping out of the pMiniMad plasmid carrying the desired genetic change. The integration vector pMiniMad uses a temperature-sensitive origin of replication to create a markerless change. Cells transformed with pMiniMad are passaged at the non-permissive temperature to encourage a single crossover event in the host genome, conferring erythromycin resistance [15,17]. Once transformants are switched to the permissive temperature, the plasmid excises from the host genome resulting in erythromycin sensitivity [14]. This method also allows for the markerless integration of point mutations, insertions, or deletions. The third and most recent advance in genetic engineering methods is the use of CRISPR/Cas9 in B. subtilis [18,13]. CRISPR/Cas9 allows researchers to make clean substitutions, deletions, and insertions in multiple backgrounds with high efficiency [18,13]. In addition to efficiency, the CRISPR/Cas9 system allows for the insertion or deletion of large fragments on the order of 20 kb [18,13]. Although CRISPR/Cas9 requires more materials than the other methods described above, plasmids engineered for one alteration are easily adapted for the introduction of other genetic manipulations.
The aim of this chapter is to provide an in-depth series of methods that will enable researchers to perform genetic manipulation of several different strains of B. subtilis. We describe five methods for genetic engineering: 1. Traditional gene manipulation, 2. Use of the deletion library, 3. pMiniMad for allelic exchange, 4. CRISPRi, and 5. CRISPR/Cas9. Below, we also provide the resources and protocols for each recombineering method described.
2. Materials:
2.1. Reagents
Luria-Bertani (LB) medium: 10 g NaCl, 10 g tryptone, 5 g yeast extract in 1 L H2O. Autoclave to sterilize.
Luria-Bertani (LB) agar plates: 10 g NaCl, 10 g tryptone, 5 g yeast extract, 15 g agar in 1 L H2O. Autoclave to sterilize and pour into sterile Petri plates once the mixture cools to 60°C. If antibiotics are required, add when the mixture reaches 60°C and mix thoroughly.
LB + starch plates: 1L LB medium, 10 g/L corn starch, 15 g agar. Autoclave to sterilize. Pour ~20 mL into sterile Petri plates once the solution has reached 60°C.
LM medium: LB medium supplemented with 3 mM MgSO4.
PC buffer (10X): 107 g/L potassium hydrate phosphate (anhydrous), 174.2 g/L potassium dihydrate phosphate (anhydrous), 10 g/L trisodium citrate (pentahydrate), up to 1 L H2O. Filter sterilize.
MD medium: 1X PC buffer, 50% w/v glucose, 10 mg/mL L-tryptophan, 2.2 g/mL ferric ammonium citrate, 100 mg/mL potassium aspartate, 1M MgSO4, 10 mg/mL phenylalanine. Store protected from light at 4°C.
Iodide solution: 0.5 g iodine, 5.0 g potassium iodide in 100 mL H2O. Store protected from light at 4°C.
0.5 μg/mL erythromycin for B. subtilis
5 μg/mL chloramphenicol for B. subtilis
100 μg/mL spectinomycin for B. subtilis
100 µg/mL ampicillin for E. coli
Sterile saline: 0.85% NaCl in H2O, or phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; adjust the pH to 7.4 using HCl)
50% glycerol
Molecular grade agarose (to make 0.5-1% final agarose gels for genotyping and purification of amplicons for cloning).
50X TAE buffer: 242 g Tris base, 57.1 ml glacial acetic acid, 100 ml EDTA (pH 8.0), H2O to 1 L.
PCR reagents (Taq polymerase for genotyping, High fidelity polymerase for Sanger Sequencing, dNTPs, buffer, nuclease-free H2O)
MC1061 competent E. coli cells or another competent cell line
BsaI-HF
Cutsmart buffer (NEB)
T4 DNA ligase
Calf intestinal phosphatase (CIP)
T4 polynucleotide kinase (PNK)
TOP10 or equivalent competent E. coli cells
gDNA or PCR amplified DNA for construct creation
pMiniMad
pDR244
pPB41 or pPB105
Genotyping primers are specific to the user and flank the locus of interest
- Optional: genotyping primers flanking amyE and/or lacA to confirm insertion
- amyE: 5′- TTCTTCGCTTGGCTGAAAAT-3′ (forward), 5′- CACCAGGTTTTTGGTTTGCT-3′ (reverse)
- lacA: 5′- TAGACGAAAGCGCCAAGATT-3′ (forward), 5′- CTGGCGTTTTCCGTTTGTAT-3′ (reverse)
- Primers compatible with pPB41 [13]:
- oPEB217: 5′-GAACCTCATTACGAATTCAGCATGC-3′
- oPEB218: 5′-GAATGGCGATTTTCGTTCGTGAATAC-3′
- Primers compatible with CRISPR/Cas9 [13]:
- oPEB232: 5′-GCTGTAGGCATAGGCTTGGTTATG-3′
- oPEB234: 5′-GTATTCACGAACGAAAATCGCCATTCCTAGCAGCACGCCATAGTGACTG-3′
2.2. General Equipment
2 mL cryogenic screw top tubes for glycerol stocks
Round bottom plastic tubes (14 mL) or glass tubes
Wooden sticks or sterile loops for picking colonies
Shaking incubator at 37°C
Shaking incubator at 25°C
Stationary incubator at 37°C
Stationary incubator at 42°C
Heat block at 65°C (see method 3.4)
Thermocycler
Gel electrophoresis apparatus
Sterile spreaders (glass beads, glass or plastic serological pipettes)
PCR tubes
3. Methods:
3.1. Traditional gene manipulation (2-day protocol)
3.1.1. Templates & genotyping primers
B. subtilis can undergo a double crossover event with DNA containing 100 bp to 500 bp (or more) flanking the site of exchange. The template can be plasmid-based, a PCR fragment, or genomic DNA with a selectable marker. For plasmid-based templates, there should be a site of recombination within it (can be the native or ectopic locus). For integration at an ectopic locus, we frequently use the non-essential gene amyE coding for starch utilization [19] or lacA coding for β-galactosidase [20]. Many different integration vectors using the amyE or lacA loci are readily available from the Bacillus Genetic Stock Center (http://www.bgsc.org/_catalogs/Catpart4.pdf). These plasmids and methods described below can be used for a variety of B. subtilis strains including JH642, PY79 and NCIB 3610 with a deletion of a competence inhibitor comI [21]. Other strain backgrounds can also be found at the Bacillus Genetic Stock Center. To create and confirm the desired strain in 2-3 days (after preparing the template), be sure to order genotyping primers specific to the locus of interest before beginning the competency and transformation protocols. Genotyping primers are used to amplify the locus of interest to easily detect insertions or deletions.
3.1.2. Making B. subtilis competent in a laboratory setting & transformation
We recommend making B. subtilis competent each time a transformation is desired. For some protocols it is standard to freeze stocks of competent Bacillus subtilis using 20% glycerol, however we prefer freshly prepared competent cells because they are often transformed more efficiently.
Streak plates from glycerol or DMSO stocks containing the strain to be manipulated and grow at 30-37°C for 16 hours.
Inoculate 2 mL LM medium (LB supplemented with 3 mM MgSO4) with a single colony in a 14 mL round bottom tube and grow with shaking (200 rpm) for 3 hours at 37°C (until OD600~1). See note 1a.
Using sterile technique, transfer 20 μL of the LM culture to 500 μL MD medium in a test tube and grow for 4 hours at 37°C to reach stationary phase. See note 1b.
Add 1-5 μL of template DNA to turbid MD cultures (up to 100 ng) and let grow for 90 minutes at 37°C. Pre-dry antibiotic plates during this time to help with absorption of liquid.
Transfer 200 μL of turbid MD cells containing DNA onto respective antibiotic containing plates and spread until completely dry using sterile glass beads or glass serological pipettes. See note 2.
3.1.3. Genotyping & storing transformants
After ~16 hours of transformation plate incubation, visible colonies will start to form. For clones with multiple antibiotic resistances, this could take 24-48 hours. See note 1c.
Restreak at least 8 transformations for isolation on fresh antibiotic-containing plates and grow overnight at 30-37°C. Restreaking of single colonies should be done at least twice. Try to pick a variety of colony sizes from different plate locations.
Perform colony PCR on B. subtilis colonies that grew following restreak on antibiotic(s). See notes 3–4.
Grow positive transformants in LB with respective antibiotics for 4-6 hours at 30°C or 37°C for glycerol or DMSO stock preparation followed by storage at −80°C.
Add culture with up to 25% glycerol or 10% DMSO to screw top cryogenic tubes and freeze indefinitely at −80°C.
3.1.4. Considerations
Strengths:
After designing appropriate templates, positive transformants are achieved in 2 days. Media can be made in-house.
Limitations:
Genetic manipulations using genomic DNA (gDNA) as the template need to be distant (~100,000 bp), or the risk of the WT copy recombining and replacing the altered locus is high, requiring further screening to obtain the correct genotype. All templates using this method rely on antibiotic selection to isolate transformants. When using gDNA, perform DNA dilutions to limit the likelihood for transformation and integration of nonlabelled loci by congression (for more information please see [1]).
3.2: Deletion library (available through the Bacillus Genetic Stock Center)
3.2.1. History and mechanism of deletion library strains & pDR244
Koo et al. created a library of erythromycin (BKE) and kanamycin (BKK) single gene interruptions in B. subtilis 168, consisting of 3,968 and 3,970 genes, respectively [14]. These stable, complete libraries allow for creation of multiple mutants with antibiotic markers that can be easily removed by plasmid-borne Cre recombinase driven from pDR244 [14]. Antibiotic cassettes are flanked by lox66 and lox71 sites, which are acted on by Cre recombinase at permissive temperatures to excise the flanked cassette and create a lox scar [14]. Plasmid pDR244 is easily cured from cells following incubation at the non-permissive temperature, resulting in cells without antibiotic resistance and a clean deletion flanked by a lox scar [14]. The double mutant lox72 scar is reduced for Cre binding ability, allowing for multiple mutations to be made in the same background [23]. Additionally, the antibiotic cassettes lack a transcriptional terminator to reduce the likelihood of polar effects on downstream genes in an operon [14].
3.2.2. Competency, transformation, & recovery
If attempting recombination at a single locus from the deletion library, all protocol elements are the same as in section 3.1.2. The DNA template would be gDNA from a strain obtained from the stock center with the desired change. When making multiple mutations dependent on a single antibiotic resistance cassette, integrate constructs sequentially. For example, transform B. subtilis with gDNA for one locus, screen, and make cryogenic stocks for each isolate. Next, remove the antibiotic resistance cassette from the newly created strain using pDR244. After confirmation, make competent cells from the deletion strain and transform with gDNA again and repeat the process. If moving markers from a different genetic background, check all available auxotrophic markers or other genetic markers in the newly created strain.
3.2.3. Use of pDR244 to create a loxP-flanked clean deletion (4-day protocol)
Streak cells onto plates from glycerol or DMSO stocks containing the strain to be engineered and grow at 30-37°C for 16 hours (day 1).
Inoculate 2 mL LM media (LB supplemented with 3 mM MgSO4) with a single colony in a 14 mL round bottom tube and grow with shaking (200 rpm) for 3 hours at 37°C (until OD600~1). See note 1a.
Transfer 20 μL of the LM culture to 500 μL MD medium in a test tube and grow for 4 hours at 37°C to reach stationary phase. See note 1b.
Add 1-2 μL of pDR244 (up to 100 ng total) to 500 µl of cells from MD tubes and let grow for 90 minutes at 37°C for transformation. Pre-dry antibiotic plates containing spectinomycin (selection for pDR244) and the antibiotic within your gene interruption. See note 2.
Transfer 200 μL of transformation reaction onto plates containing spectinomycin and spread until completely dry using sterile glass beads or glass serological pipettes.
3.2.4. Screening & excising the antibiotic resistance cassette
The next day (day 3), streak at least 8 transformants for isolation on spectinomycin plates and incubate at 37°C for 16 hours. See note 1c.
Streak a single line onto a LB plate and grow at 42°C for 12 hours.
Streak a single line from the LB plate onto a new LB plate, and a plate containing spectinomycin+antibiotic (erythromycin if using BKE strain or kanamycin if using BKK strain), and a third plate containing antibiotic but lacking spectinomycin (in that order) and grow at 42°C overnight. See note 5.
The next day (day 4), assess growth on plates. Transformants containing clean deletions will have grown on LB only. If there is still growth on spectinomycin+antibiotic (erm or kan), then pDR244 was not cured. If there is growth on antibiotic only plates, the cassette remains interrupting the gene and has not recombined out. See notes 3–4.
Grow culture & cryogenic stock as in method 1.
3.2.5. Considerations
Strengths:
Gene interruption strains are available from the Bacillus Genetic Stock Center at the Ohio State University. Once your order is placed, Dr. Zeigler will next-day ship your strains via UPS.
Limitations:
The step-wise nature of making deletions takes >2 days.
3.3. pMiniMAD (6-day protocol)
3.3.1. History and mechanism of pMiniMAD
The pMiniMAD (also known as pMiniMAD2) plasmid was developed by Patrick & Kearns in 2008 for allelic replacement [15]. The plasmid pMiniMAD contains a temperature sensitive origin of replication (ColE1) and an ampicillin and erythromycin cassette [15]. In E. coli, the plasmid replicates at the permissive temperature and can be selected for using ampicillin resistance. In B. subtilis at a non-permissive temperature, a single crossover event will occur flanking the site of your choice and confer erythromycin resistance. B. subtilis transformants are subsequently grown without erythromycin at the permissive temperature, causing the plasmid to replicate and excise from the genome [15]. The result is markerless allelic replacement in your desired B. subtilis strain. An E. coli strain containing the pMiniMAD plasmid is available at the Bacillus Genetic Stock Center (strain ECE765). Around-the-world PCR amplification [24] of the plasmid can be done using only one primer set, allowing for easy ligation of your desired gene to create the full plasmid [15].
3.3.2. Competency & transformation
The protocol for making competent cells and transformation is the same as in section 3.1.2 (steps 1-5). See note 1.
After plating transformants on erythromycin (section 3.1.2, step 5), grow at 37°C overnight (day 1). See note 2.
3.2.3. Recovery & screening
Pick 4 colonies from transformation plates and use to inoculate 3 mL each of LB. Incubate for 10 hours at 25°C (day 2).
Inoculate 30 μL of each culture (may not be visibly turbid, this is OK to continue with) into 3 mL of fresh LB and incubate at 25°C overnight.
The next day (day 3), repeat steps 1 & 2.
Inoculate 30 μL of cultures again into 3 mL each of fresh LB. Grow at 37°C for ~3 hours (day 4).
Once the cultures reach OD600 1.1-1.3, perform 10-fold serial dilutions in sterile saline (10−4, 10−5, 10−6). Spread 200 μL of each dilution onto the respective LB plate. At the end of the day, let the plates grow overnight at 37°C.
On day 5, identify which serial dilution for each culture results in the best isolation of individual colonies per plate (approximately 20-80). From that dilution plate, select at least 12 transformants and restreak for single colonies on LB and LB+erm. Grow overnight at 37°C.
The next day (day 6), assess which colonies were sensitive to erythromycin. Perform colony PCR on these colonies to determine if the desired genetic changes is complete. See note 3 and 6.
Grow transformants in LB at 37°C for glycerol or DMSO stocks. Sanger Sequence the PCR product of the region of interest from each strain to ensuring the nucleotide substitution or other genetic change is correct.
3.3.4. Considerations
Strengths:
Creation of a construct without antibiotic resistance. pMiniMAD is easily changed and available through the Bacillus Genetic Stock Center.
Limitations:
Takes 6 days and requires constant passaging and screening of many colonies. This does not work for essential genes or can be challenging for gene disruptions that cause a severe growth phenotype.
3.4: CRISPRi (2-day protocol)
3.4.1. History of CRISPRi and its application in B. subtilis
Peters et al., 2016 created a library of essential gene knockdowns in B. subtilis using CRISPR interference [16]. To perform CRISPRi, constructs have two alterations: lacA::Pxyl-dCas9 (ermR) and amyE::Pveg-sgRNA(gene) (cmR).The sgRNAs target the non-template strand [16]. Pveg drives expression of the sgRNA to the gene of interest during vegetative growth. dCas9 is induced with xylose and upon induction, binds the sgRNA, and blocks RNA polymerase from transcribing [16]. This method is important to study B. subtilis because it provides a conditional knockout or knockdown of essential genes or genes that cause a severe growth phenotype when disrupted or depleted.
3.4.2. Competency, transformation, & screening
Streak plates with glycerol or DMSO stocks containing the strain to be manipulated and grow at 30-37°C for 16 hours.
Inoculate 2 mL LM medium (LB supplemented with 3 mM MgSO4) with a single colony in a 14 mL round bottom tube and grow with shaking (200 rpm) for 3 hours at 37°C (until OD600~1). See note 1a.
Transfer 20 μL of the LM culture to 500 μL MD medium in a test tube and grow for 4 hours at 37°C to reach stationary phase. See note 1b.
Add 1-5 μL your template DNA to 500 µl of cells from MD tubes (up to 100 ng) and let grow for 90 minutes at 37°C & pre-dry antibiotic plates.
Transfer 200 μL of transformation reaction onto LB+cm+erm plates and spread until completely dry using sterile glass beads, or sterile glass serological pipettes. See note 2.
After ~16 hours of transformation plate incubation, visible colonies will start to form. For constructs with multiple antibiotic resistances, this could take 24 hours or longer.
Restreak at least 8 transformations for isolation on fresh antibiotic-containing plates and grow overnight at 30-37°C. Try to pick a variety of colony sizes from different plate locations.
Perform colony PCR on isolates that grew following restreak on antibiotics at both the amyE and lacA loci. See note 7.
Grow positive transformants in LB with chloramphenicol and erythromycin for 4-6 hours at 37°C for subsequent glycerol or DMSO stock preparation.
Add culture with up to 25% glycerol or 10% DMSO to cryogenic screw-top tubes and freeze indefinitely at −80°C.
3.4.3. Use of newly constructed strains
To test interference of your desired gene, we recommend titrating xylose at several different concentrations (or percentages) on LB plates. Start in 10-fold increments and then fine-tune within the increments based on your desired result. Alternatively, perform qPCR to measure transcript levels quantitatively.
Be sure to keep both erythromycin and chloramphenicol in the media when you use the CRISPRi strains.
3.4.4. Considerations
Strengths:
This protocol is fast and includes essential genes. The researcher can fine tune the level of transcript by carefully modulating gene expression.
Limitations:
The desired titration amount is up to the researcher and is determined experimentally. Starting concentrations are usually 0.5% or 1% xylose (w/v). In addition, there are two antibiotic cassettes and integrations for a single CRISPRi strain.
3.5: CRISPR/Cas9 (2-day protocol)
3.5.1. History of CRISPR/Cas9 manipulation of genes in B. subtilis
With the recent application of CRISPR/Cas9 to create mutations in many organisms, an efficient protocol was established for manipulation of B. subtilis by Burby and Simmons [18,13]. CRISPR/Cas9 allows the user to make deletions, fusions, and point mutations in multiple backgrounds. The plasmids required for this procedure are available at the Bacillus Genetic Stock Center [18,13]. The editing plasmids pPB41 (SpecR & AmpR) or pPB105 (CmR & AmpR) are modified to insert a proto-spacer sequence to target the locus of interest [18,13]. The second modification of pPB41 serves as the editing DNA template for introduction into the genome [18,13]. After constructing both pieces of pPB41 using standard molecular biology methods, the final editing plasmid is assembled. In short, the plasmid backbone containing antibiotic resistance, editing template, Cas9, and protospacer sequences are ligated using Gibson Assembly [22] to create one final plasmid. The resulting plasmid can be used to transform B. subtilis to introduce the desired genetic change and is subsequently cured from cells with ease [13].
3.5.2. Protocol for creating plasmid with proto-spacer for CRISPR/Cas9 alteration
Digest pPB41 or pPB105 with restriction endonuclease BsaI.
Construct a phosphorylated proto-spacer for insertion into pPB41 or pPB105.
Ligate plasmid and proto-spacer together, transform E. coli, and isolate plasmid.
3.5.3. Protocol for creating editing plasmid
Use oPEB217 and oPEB218 with Q5 DNA polymerase to linearize pPB41. Gel extract and purify PCR product.
Use oPEB232 and oPEB234 with Q5 DNA polymerase to PCR amplify CRISPR/Cas9 from plasmid created in step 1. Gel extract and purify PCR product.
PCR amplify editing template, gel extract and purify.
Assemble full editing plasmid using Gibson Assembly.
3.5.4. Transformation of plasmids and screening of transformants
Make desired B. subtilis strain competent using Method 3.1.2.
After incubation in MD culture, add 200-600 ng of editing plasmid DNA and incubate for 60-90 minutes at 37°C.
Plate 200 μL transformants on LB+spec and incubate at 30°C overnight.
Restreak single colonies on LB+spec for purity.
Cure isolates of plasmid by restreaking on LB for single colonies and incubating overnight at 45°C.
Screen isolates for plasmid loss by restreaking single colonies onto LB and LB+spec and incubate again overnight at 45°C. There should be no growth on LB+spec plates if cells are cured of the plasmid.
Use genotyping primers to confirm alteration of interest.
3.5.5. Considerations
Strengths:
It is possible to introduce multiple mutations of your own design in many different backgrounds with high efficiency (80-100% positive clones). This method lacks antibiotic cassettes or remnants of vector DNA used during cloning.
Limitations:
Mutations are contingent upon a proto-spacer adjacent motif (PAM) sequence, NGG in B. subtilis, near to the desired locus and requires more reagents than the other methods.
Acknowledgements
Work in the Simmons lab is funded by grants from the National Institutes of Health (R35GM131772) and the National Science Foundation (MCB 1714539). KJW was funded in part by an NIH Cellular Biotechnology Training Grant (T32 GM008353), a pre-doctoral fellowship from the National Science Foundation (#DEG 1256260), and a Rackham Merit Fellowship from the University of Michigan. We would like to thank the Bacillus Genetic Stock Center and the director, Dr. Daniel Zeigler for his continued service to the Bacillus scientific community.
4. Notes
On competency and transformation: a. Glass or plastic tubes both work well for incubation of cells for transformation; be sure the culture takes up 1/10 or less of the container volume for proper aeration. Shaking incubation can be performed in a test tube rack in a warm room, water bath, or a rolling rack within an incubator. b. Prepare MD tube for each transformation. c. If initial transformation of B. subtilis with DNA is unsuccessful or for strains that grow more slowly due to other genetic changes, increasing the incubation in LM media could be extended from 4 hours to ~6 hours, and incubation with gDNA could be increased from 90 minutes to 3 hours.
On plating transformations: Pre-drying plates before spreading transformation mix is important to help the liquid absorb into the plate for growth of single transformant colonies. Any remaining transformation culture can be spread onto another plate (as is or diluted with MD medium to make 1:2, 1:5, and/or 1:10 dilutions).
On confirmation of transformants: Use genotyping primers to confirm correct amplicon size of transformants. It is recommended to submit samples for Sanger Sequencing when making substitutions or piecing together constructs using Gibson Assembly [22]. If using the amyE locus, add up to 1 mL iodide solution to starch plate “patch” restreaks. If the gene is disrupted, and your construct was successfully inserted, you will no longer see a starch clearing or “halo” around the patch. Please note that iodide treatment is lethal to the cells on the starch plate.
On ensuring a double crossover event with exogenous DNA (method 1): Homology between the desired B. subtilis locus and amplicon DNA will help increase likelihood of recombination. If homology is sufficient and transformation is unsuccessful, titrating the amount of amplicon DNA in the transformation could lead to the desired recombination event.
Notes on curing an antibiotic resistance marker using pDR244 (method 2): It is not necessary to go back to the original LB streak when streaking plates containing antibiotics to assess if pDR244 has been cured. Simply collect some bacteria from the first plate and continue streaking a single line on all plates. Due to the high cell density of bacteria from the original plate during patch plating, there are still enough cells to streak on the last plate of the series.
On increasing the number of markerless desired clones (method 3): After transformation and integration of pMiniMad, passaging >4 cultures through room temperature incubations and screening >12 colonies from each could allow the researcher to find additional clones.
On CRISPRi (method 4): As the guide RNA is driven by a promoter inserted at amyE, streak a single line onto LB plates containing 10 g/L starch to ensure this locus has been disrupted. Streak WT as a control and incubate with the other antibiotic plates. Use genotyping primers to confirm correct amplicon size of transformants. To determine if the amyE locus is disrupted, add up to 1 mL iodide solution to starch plate streaks. You will no longer see a starch clearing or “halo” if the insertion at amyE occurred.
5. References
- 1.Hardwood CR, Cutting SM (1990) Molecular Biological Methods for Bacillus. John Wiley & Sons, Chichester: Pages 1–35 [Google Scholar]
- 2.Dubnau D (1991) Genetic competence in Bacillus subtilis. Microbiol Rev 55 (3):395–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van Sinderen D, Luttinger A, Kong L, Dubnau D, Venema G, Hamoen L (1995) comK encodes the competence transcription factor, the key regulatory protein for competence development in Bacillus subtilis. Molecular microbiology 15 (3):455–462 [DOI] [PubMed] [Google Scholar]
- 4.van Sinderen D, ten Berge A, Hayema BJ, Hamoen L, Venema G (1994) Molecular cloning and sequence of comK, a gene required for genetic competence in Bacillus subtilis. Molecular microbiology 11 (4):695–703 [DOI] [PubMed] [Google Scholar]
- 5.Hamoen LW, Haijema B, Bijlsma JJ, Venema G, Lovett CM (2001) The Bacillus subtilis competence transcription factor, ComK, overrides LexA-imposed transcriptional inhibition without physically displacing LexA. J Biol Chem 276 (46):42901–42907 [DOI] [PubMed] [Google Scholar]
- 6.Ogura M, Yamaguchi H, Kobayashi K, Ogasawara N, Fujita Y, Tanaka T (2002) Whole-genome analysis of genes regulated by the Bacillus subtilis competence transcription factor ComK. J Bacteriol 184 (9):2344–2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hahn J, Maier B, Haijema BJ, Sheetz M, Dubnau D (2005) Transformation proteins and DNA uptake localize to the cell poles in Bacillus subtilis. Cell 122 (1):59–71. doi: 10.1016/j.cell.2005.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubnau D, Blokesch M (2019) Mechanisms of DNA Uptake by Naturally Competent Bacteria. Annu Rev Genet 53:217–237. doi: 10.1146/annurev-genet-112618-043641 [DOI] [PubMed] [Google Scholar]
- 9.Yadav T, Carrasco B, Myers AR, George NP, Keck JL, Alonso JC (2012) Genetic recombination in Bacillus subtilis: a division of labor between two single-strand DNA-binding proteins. Nucleic Acids Res 40 (12):5546–5559. doi: 10.1093/nar/gks173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dubnau D, Provvedi R (2000) Internalizing DNA. Res Microbiol 151 (6):475–480. doi: 10.1016/s0923-2508(00)00166-2 [DOI] [PubMed] [Google Scholar]
- 11.Lenhart JS, Schroeder JW, Walsh BW, Simmons LA (2012) DNA Repair and Genome Maintenance in Bacillus subtilis. Microbiology and molecular biology reviews : MMBR 76 (3):530–564. doi: 10.1128/MMBR.05020-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ayora S, Carrasco B, Cardenas PP, Cesar CE, Canas C, Yadav T, Marchisone C, Alonso JC (2011) Double-strand break repair in bacteria: a view from Bacillus subtilis. FEMS Microbiol Rev [DOI] [PubMed] [Google Scholar]
- 13.Burby PE, Simmons LA (2017) CRISPR/Cas9 Editing of the Bacillus subtilis Genome. Bio Protoc 7 (8). doi: 10.21769/BioProtoc.2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koo BM, Kritikos G, Farelli JD, Todor H, Tong K, Kimsey H, Wapinski I, Galardini M, Cabal A, Peters JM, Hachmann AB, Rudner DZ, Allen KN, Typas A, Gross CA (2017) Construction and Analysis of Two Genome-Scale Deletion Libraries for Bacillus subtilis. Cell Syst 4 (3):291–305 e297. doi: 10.1016/j.cels.2016.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patrick JE, Kearns DB (2008) MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol Microbiol 70 (5):1166–1179 [DOI] [PubMed] [Google Scholar]
- 16.Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CHS, Koo BM, Marta E, Shiver AL, Whitehead EH, Weissman JS, Brown ED, Qi LS, Huang KC, Gross CA (2016) A Comprehensive, CRISPR-based Functional Analysis of Essential Genes in Bacteria. Cell 165 (6):1493–1506. doi: 10.1016/j.cell.2016.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cozy LM, Kearns DB (2010) Gene position in a long operon governs motility development in Bacillus subtilis. Mol Microbiol 76 (2):273–285. doi: 10.1111/j.1365-2958.2010.07112.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burby PE, Simmons LA (2016) MutS2 promotes homologous recombination in Bacillus subtilis. J Bacteriol. doi: 10.1128/JB.00682-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimotsu H, Henner DJ (1986) Construction of a single-copy integration vector and its use in analysis of regulation of the trp operon of Bacillus subtilis. Gene 43 (1-2):85–94. doi: 10.1016/0378-1119(86)90011-9 [DOI] [PubMed] [Google Scholar]
- 20.Hartl B, Wehrl W, Wiegert T, Homuth G, Schumann W (2001) Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J Bacteriol 183 (8):2696–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Konkol MA, Blair KM, Kearns DB (2013) Plasmid-encoded ComI inhibits competence in the ancestral 3610 strain of Bacillus subtilis. J Bacteriol 195 (18):4085–4093. doi: 10.1128/JB.00696-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6 (5):343–345. doi: 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 23.Yan X, Yu HJ, Hong Q, Li SP (2008) Cre/lox system and PCR-based genome engineering in Bacillus subtilis. Appl Environ Microbiol 74 (17):5556–5562. doi: 10.1128/AEM.01156-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, Naismith JH (2008) An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 8:91. doi: 10.1186/1472-6750-8-91 [DOI] [PMC free article] [PubMed] [Google Scholar]