

Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disease manifesting both motor and non-motor symptoms. The motor features are generally ascribed to the selective loss of dopamine neurons within the substantia nigra pars compacta. While the precise etiology of PD remains elusive, multiple genetic and environmental elements have emerged as contributing factors. The discovery of MPTP-induced parkinsonism directed intense inquiry towards mitochondrial pathways, with a specific focus on mitochondrial complex I. Consisting of more than 40 subunits, complex I is the first enzyme of the electron transport chain that is required for mitochondrial ATP production. In this review, we present a critical analysis of studies assessing the prevalence and specificity of mitochondrial complex I deficiency in PD. In addition, we take the novel view of incorporating the features of genetically-defined bona fide complex I disorders and the prevalence of nigral involvement in such cases. Through this innovative bi-directional view, we consider both complex I changes in a disease of the substantia nigra and nigral changes in diseases of complex I. We assess the strength of association between nigral cell loss and complex I deficits, as well as the oft under-appreciated heterogeneity of complex I deficiency disorders and the variability of the PD data.
Keywords: Mitochondrial complex I, MPTP, NDUFAF2, Parkinson’s disease, PINK1
1. Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder (de Lau and Breteler, 2006). First formally described by James Parkinson in 1817 as “shaking palsy” (Parkinson, 1817), PD is typically characterized by both motor and non-motor symptoms [reviewed in (Berardelli et al., 2013; Chaudhuri et al., 2006; Titova and Chaudhuri, 2018)]. The motor symptoms are thought to be driven primarily by the severe and near-selective loss of the dopaminergic (DA) neurons of the substantia nigra pars compacta (SNpc) in the ventral midbrain. In late-stage PD, as much as 95% of these neurons may be lost (Burke and O’Malley, 2013; Cheng et al., 2010). In the majority of cases, some surviving DA neurons are characterized by Lewy body inclusions in the cytoplasm and neurites, with α-synuclein as the major protein component of these structures (Dickson et al., 2009). In fact, it is now recognized that Lewy pathology can be found throughout the peripheral and central nervous system (Braak and Del Tredici, 2009). In spite of this widespread dysfunction across multiple neurologic systems, the SNpc clearly manifests an unparalleled degree of neuronal loss in the PD brain (Giguere et al., 2018). Revealing the molecular mechanisms that underlie this selective neurodegeneration has been, perhaps, the greatest mystery in this devastating disease.
Mitochondria are dynamic organelles that play important roles in cellular physiology. Their maintenance, biogenesis, and quality control are vital for maintaining cell health in aging and have emerged as critical areas of investigation in PD [reviewed in (Bose and Beal, 2016; Chen et al., 2019; Park et al., 2018)]. There are two broad aspects of mitochondrial quality control linked to PD. First, the autosomal recessive PD gene products Parkin and PINK1 coordinate one form of mitophagy that enables the selective degradation of aged, dysfunctional, or damaged mitochondria. The role of PINK1/Parkin in mitophagy and mitochondrial dynamics have been well summarized recently in other venues [reviewed in detail in (de Vries and Przedborski, 2013; Pickles et al., 2018; Pickrell and Youle, 2015)]. Mitochondrial DNA (mtDNA) maintenance is another process that contributes to mitochondrial quality. Unlike nuclear DNA, the human mitochondrial genome is not enshrouded by protective histones and is thus prone to increased rates of mtDNA mutagenesis (Alexeyev et al., 2013; van der Wijst and Rots, 2015). The mtDNA encodes subunits of the oxidative phosphorylation pathway (OXPHOS) and a portion of the genes essential for mitochondrial translation (Anderson et al., 1981; Andrews et al., 1999). Mutations in nuclear genes encoding the mitochondrial DNA polymerase gamma (Pol γ) and the mitochondrial DNA helicase Twinkle cause reduced fidelity during mtDNA replication, resulting in increased rates of mtDNA mutation, and thus diminish the mitochondrial quality [reviewed in (Peter and Falkenberg, 2020; Rahman and Copeland, 2019)]. The second link between broad mitochondrial quality control and PD stems from the observation that a subset of these patients develop severe nigral neurodegeneration (Betts-Henderson et al., 2009; Chen et al., 2020; Mehta et al., 2016; Tzoulis et al., 2016). Only a fraction exhibit parkinsonian motor symptoms or Lewy body pathology (Breen et al., 2020; Palin et al., 2013; Reeve et al., 2013; Tzoulis et al., 2013). Thus, failures of mitochondrial quality control, either through loss of function (failed mitophagy) or toxic gain of function (increased mtDNA mutagenesis), have been associated with PD. However, these organelle-wide problems speak to a generalized relationship between mitochondrial dysfunction and PD and do not directly address the primary thesis of this review. This article will instead focus on the specific question: Does mitochondrial complex I activity have a special relationship with the long-term survival of DA neurons of the human substantia nigra (SN)?
Mitochondrial complex I (E.C. 1.6.5.3) (CI), the first enzyme of the electron transport chain (ETC), is a proton-pumping NADH: ubiquinone oxidoreductase. Structural analyses of the holoenzyme have revealed an L-shaped structure that consists of a hydrophilic arm protruding into the mitochondrial matrix and a hydrophobic arm embedded in the inner mitochondrial membrane (Hirst, 2013; Vinothkumar et al., 2014). The NADH-binding site and the prosthetic groups [one flavin mono-nucleotide and eight iron-sulfur (Fe-S) clusters] are located in the matrix arm and are required for the transfer of electrons from NADH to ubiquinone. The ubiquinone-binding center is present at the interface of the matrix and the membrane arms (Berrisford et al., 2016; Wirth et al., 2016) and pharmacologically sensitive to selective inhibition by rotenone. The hydrophobic membrane arm coordinates ubiquinone reduction with proton-pumping, where four protons are translocated per two electrons transferred from NADH to ubiquinone (Galkin et al., 2006; Wikström, 1984). Although CI is conserved from bacteria to humans, the bacterial enzyme consists of only 14 subunits. Called the “core” subunits, they have corresponding orthologs in all eukaryotic CI and are thought to constitute the minimal requirement for its enzymatic activity (Letts and Sazanov, 2015). Mammalian CI is composed of 30 additional “accessory/non-core” subunits, 24 of which are conserved across eukaryotic lineages (Cardol, 2011; Kmita and Zickermann, 2013). These accessory subunits surround the core subunits and are especially concentrated in the membrane arm of CI (Wirth et al., 2016). Although the exact role of the accessory subunits remains poorly understood, they have been hypothesized to regulate enzymatic activity or promote holoenzyme stability (Kmita and Zickermann, 2013; Stroud et al., 2016). Amongst the 44 subunits of mammalian CI, seven core hydrophobic subunits are encoded by the mitochondrial genome and the rest are encoded by the nuclear genome. Thus, eukaryotic CI biogenesis requires the coordination of both the nuclear and mitochondrial genomes (Guerrero-Castillo et al., 2017).
The first indication that mitochondrial dysfunction could be connected to the development of PD came in 1982 at the Santa Clara Valley Medical Center, in San Jose, California. A 42-year-old drug user was admitted to the clinic exhibiting certain motor symptoms associated with atypical PD, such as lead-pipe and cogwheel rigidity, alongside features of catatonic schizophrenia (Langston, 2017; Lewin, 1984). This diagnosis was initially baffling to the clinicians due to the young age of the patient. Soon after, more patients exhibiting similar symptoms were identified and all were linked to the recent use of a new synthetic designer drug. MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) is a by-product of poor MPPP (1-methyl-4-phenyl-4-propionoxy-piperidine) synthesis. A mixture of MPPP contaminated with MPTP was intravenously self-administered by the recreational drug users, leading to the development of a rapid onset akinesia within two weeks of usage (Langston et al., 1983). Similarly, a chemist working on MPTP production for 7 years also developed PD at 37 years of age due to either accidental cutaneous exposure or vapor inhalation (Langston and Ballard, 1983). Later analyses revealed that MPTP is a neurotoxin that is converted to MPP+ (1-methyl-4-phenylpyridinium) by glial monoamine oxidase (Heikkila et al., 1984c). MPP+, a weak mitochondrial CI inhibitor, is released into the extracellular matrix and actively taken up by the DA neurons via the dopamine transporter protein where it is further concentrated into mitochondria via cation transporters to levels sufficient to inhibit CI (Bezard et al., 1999; Gainetdinov et al., 1997; Mizuno et al., 1988, 1987; Przedborski et al., 2004; Ramsay et al., 1986). MPTP-induced inhibition of mitochondrial CI and subsequent DA neuronal loss has been reproduced in mammalian models such as primates and mice (Burns et al., 1983; Heikkila et al., 1984a, 1984b; Kolata, 1983; Mizuno et al., 1987; Tolwani et al., 1999). More recently, a similar model was developed using the selective CI inhibitor, rotenone, albeit with some experimental variability (Betarbet et al., 2000; Cannon et al., 2009; Zeng et al., 2018). These findings inspired a new era of inquiry in PD research towards mitochondrial dysfunction, with a particular focus on CI activity.
2. Mitochondrial complex I activity in brain samples of idiopathic PD patients
The investigation of mitochondrial respiratory chain complexes from idiopathic PD (iPD) brain tissue was first reported by two groups in 1989 (Mizuno et al., 1989; Schapira et al., 1989) and was subsequently followed by various studies by multiple research teams (Table 1, Fig. 1). In these first studies, tissues from neuropathologically-confirmed PD patients were compared to age-matched controls. One of the first analyses investigated the steady-state protein levels of CI subunits in brain tissues by immunoblotting (Table 1A). A decreased abundance of CI subunits in striatal mitochondria was observed in four out of five PD patients (Mizuno et al., 1989). In contrast, one study showing one representative result from a single patient found no difference in the steady-state levels of CI subunits in the SN homogenates (Schapira et al., 1990a). As these studies utilized antisera against the holoenzyme, which is immunoreactive towards multiple CI subunits, the exact assignment of each subunit to a specific immunoreactive band could not be achieved. Interestingly, a recent study also failed to find any changes in multiple OXPHOS subunits, including a CI subunit, detected with a cocktail antibody in prefrontal cortex homogenates of 40 PD compared to 23 controls (Gatt et al., 2016). The lack of a systematic approach to assess the abundance of individual CI subunits in brain tissues of PD patients using antibodies specific to each subunit, alongside comparison to other respiratory complex subunits, limits the ability to understand CI-specific changes from these studies.
Table 1.
A summary of assessments of mitochondrial ETC complexes in different brain regions of PD patients. C: control; PD: Parkinson’s disease; SN: substantia nigra; SNpc: SN pars compacta; SNpr: SN pars reticulata; N.D.: not determined; N.S.: no statistically significant changes observed between PD and control subjects. (A) Abundance of CI subunits tested by SDS-PAGE immunoblotting. (B) Levels of CI assessed by immunostaining using CI antibodies. (C) Enzymatic activities determined spectrophotometrically using tissue homogenates or purified mitochondria. CI: NADH: CoQ1 oxidoreductase activity; CI + III: NADH: cytochrome c oxidoreductase activity; CII + III: succinate: cytochrome c oxidoreductase activity; CII: succinate: ubiquinone oxidoreductase activity; CIII: ubiquinol: cytochrome c oxidoreductase activity; CIV: cytochrome c oxidase activity. CS: citrate synthase activity; “x/CS” refers to activity x (CI, CII, CIII, or CIV) normalized to citrate synthase activity. Activities determined by sensitivity to complex-specific inhibitors such as rotenone or antimycin A are denoted in “blue” font; OCR: oxygen consumption rate; The dipstick assay, refers to a method of assessing CI activity by first immunocapturing the CI holoenzyme followed by assessing the NADH dehydrogenase activity by NBT staining. The studies are grouped based on the presence (light green cells) vs absence (light red cells) of significant difference in CI between control and PD.
| Table 1A. Complex I abundance determined by immunoblotting | |||
|---|---|---|---|
| Study | Source | No. of cases | Results |
| Mizuno et al., 1989 | Striatal and frontal lobe mitochondria | 4 C, 5 PD | Decreased steady-state levels of CI subunits in 4 out of 5 PD patients in striatal mitochondria but no changes in CIII or CIV subunits; N.S. for CI, CIII, CIV subunit levels in frontal lobe mitochondria; |
| Keeney et al., 2006 | Frontal cortex mitochondria | 10 C, 10 PD | 11% increase in 20 kDa ND6 protein, 33% decrease in 8 kDa subunit of CI. The CI antibody recognizes multiple subunits. |
| Schapira et al., 1990a | SN homogenates | 1 C, 1 PD | N.S. for accumulation of CI subunits as detected by anti-CI antisera. |
| Gatt et al., 2016 | Prefrontal cortex homogenates | 23 C, 40 PD | N.S. for CI, CII, CIII, CIV, CV subunit levels normalized to histone H3 levels, assayed using an OXPHOS cocktail antibody. |
| Flones et al., 2018 | N.S. for average assembled complex detected by BN-PAGE immunoblotting analyses. | ||
| Cerebellum homogenates | 2C, 8 PD | ||
| Table 1B. Levels of complex I assessed by immunohistochemical analyses | |||
| Study | Source | No. of cases | Results |
| Hattori et al., 1991 | SN medial, central, lateral neurons | 7 C, 8 PD | CI immunostaining with weak or no staining in 35.6 +/− 17.6% of nigral neurons for PD compared to 13.6 +/− 4.2% in control. CIII, CIV stained intensely. CII had 10–50% weak staining in 3 PD patients. N.S. for CI, CII, CIII, CIV immunostaining in striatal neurons and neuropils. |
| Palin et al., 2013 | Mesencephalic nuclei | 2 C, 6 patients with mutations | Out of 6 patients: 2 with Twinkle mutations, 4 with POLG mutations - 2 exhibit PD. Very low CI immunoreactivity in SN of all 6 patients with three independent CI antibodies. SN neuropil and mesencephalic neuropil had minor loss of CI immunoreactivity. CII immunoreactivity in SN and oculomotor nucleus was more intense in POLG-PD, but N.S. for CIII and CIV immunostaining. |
| Reeve et al., 2013 | SN neurons | 8 C, 5 patients with POLG mutations | All patients had SN neuronal loss, 2/5 patients showed LB pathology, only one patient exhibited parkinsonian symptoms. N.S. for CII, porin immunoreactivity; Neurons with weak CI and CIV immunostaining significantly increased in patients with POLG mutations; CIII, CV immunoreactivity N.D. |
| Grunewald et al., 2016 | SN neurons | 10 C, 10 PD | Decreased CI and CII immunoreactivity quantified relative to porin or GRP75. 49% of PD had combined deficiency in CI and CII immunoreactivity; 25% of patient neurons lacked both CI and CIV immunoreactivity. A decrease in CI and CIV immunoreactivity in controls with an increase in age also noted. |
| Flones et al., 2018 | |||
| principal neurons of red nucleus, basis pontis. | 7C, 17 PD | N.S. for CI immunostaining | |
| Reeve et al., 2017 | SN neurons | 7 C, 3 PD | N.S. for CI and CIV immunostaining in the soma. |
| Table 1C. Enzymatic activities conducted on brain tissue | |||
| Study | Source | No. of cases | Results |
| Schapira et al., 1989; Schapira et al., 1990a | SN homogenates | 9 C, 9 PD | ~39% decrease in CI+III activity; ~31% decrease in CI activity for n=5. N.S for CII+III and CS activity; CIV N.D. |
| Schapira et al., 1990b | |||
| Caudate nucleus, medial and lateral globus pallidus, cerebral cortex, and cerebellum homogenates. | 5 C, 4 PD | N.S. for CI activity, CII+III, CIV, or CS activities | |
| Mann et al., 1992a | |||
| Cerebellum homogenate | 16 C, 16 PD | N.S. for CI activity, CII+III, CIV, and CS activities | |
| Mann et al., 1994 | SN homogenates | 22 C, 17 PD from Mann et al., 1992a + new 7 C, 7 PD | ~35% decrease in CI/CS activity in new PD samples. Together with Mann et al., 1992a: ~38% decrease in CI activity when normalized to protein amount and ~31% decrease in CI/CS activity. Other ETC activities N.D. |
| Janetzky et al., 1994 | |||
| Cortex, putamen homogenates | 3 C, 3 PD | N.S. for absolute CI+III or CI+III/CS activity | |
| Parker et al., 2008 | Frontal cortex: homogenates, crude mitochondrial pellet, and purified mitochondria | 4 C, 5 PD | Statistical difference in CI activity observed only in purified mitochondria: ~50% decrease in CI activity, ~40% decrease in CI+III activitity. N.S. for CII, CIII, and CIV activities. |
| Navarro et al., 2009 | Frontal cortex mitochondria | 9 C, 7 PD | ~59% decrease in CI+III activity; N.S. for CII+III, CIV activities; Decrease in CI-linked OCR; ~49% increase in mitochondrial mass of PD tissue as calculated by CIV activity in mitochondria vs homogenates. |
| Flones et al., 2018 | |||
| Cerebellum homogenate | 7 C, 9 PD | N.S. for CI/CS activity, | |
| Mizuno et al., 1990a | |||
| SN mitochondria | 2 C, 2 PD | N.S. for CI, CII, CIII, or CIV activities. | |
| Cooper et al., 1995 | Posterior putamen homogenates | 12 C, 12 PD | N.S. for CI, CII+III, or CIV activities, before and after CS normalization; CS activity increased by 20%. |
| Keeney et al., 2006 | Frontal cortex mitochondria | 6 C, 10 PD | N.S. for CI activity normalized to mitochondrial protein amounts. For 4 C, 7 PD, when normalized to porin levels by immunoblotting, significant decrease of 30% observed for CI activity. Other ETC activities N.D. |
| Mythri et al., 2011 | Frontal cortex, caudate nucleus, putamen mitochondria | 9 C, 6 PD | N.S. for CI activity; Other ETC activities N.D. |
| Gatt et al., 2016 | N.S. for CI activity assessed by dipstick assay. Other ETC activities not assessed. | ||
| Somatosensory cortex BA1, BA2, BA3 homogenate | 15 C, 15 PD | ||
Fig. 1.
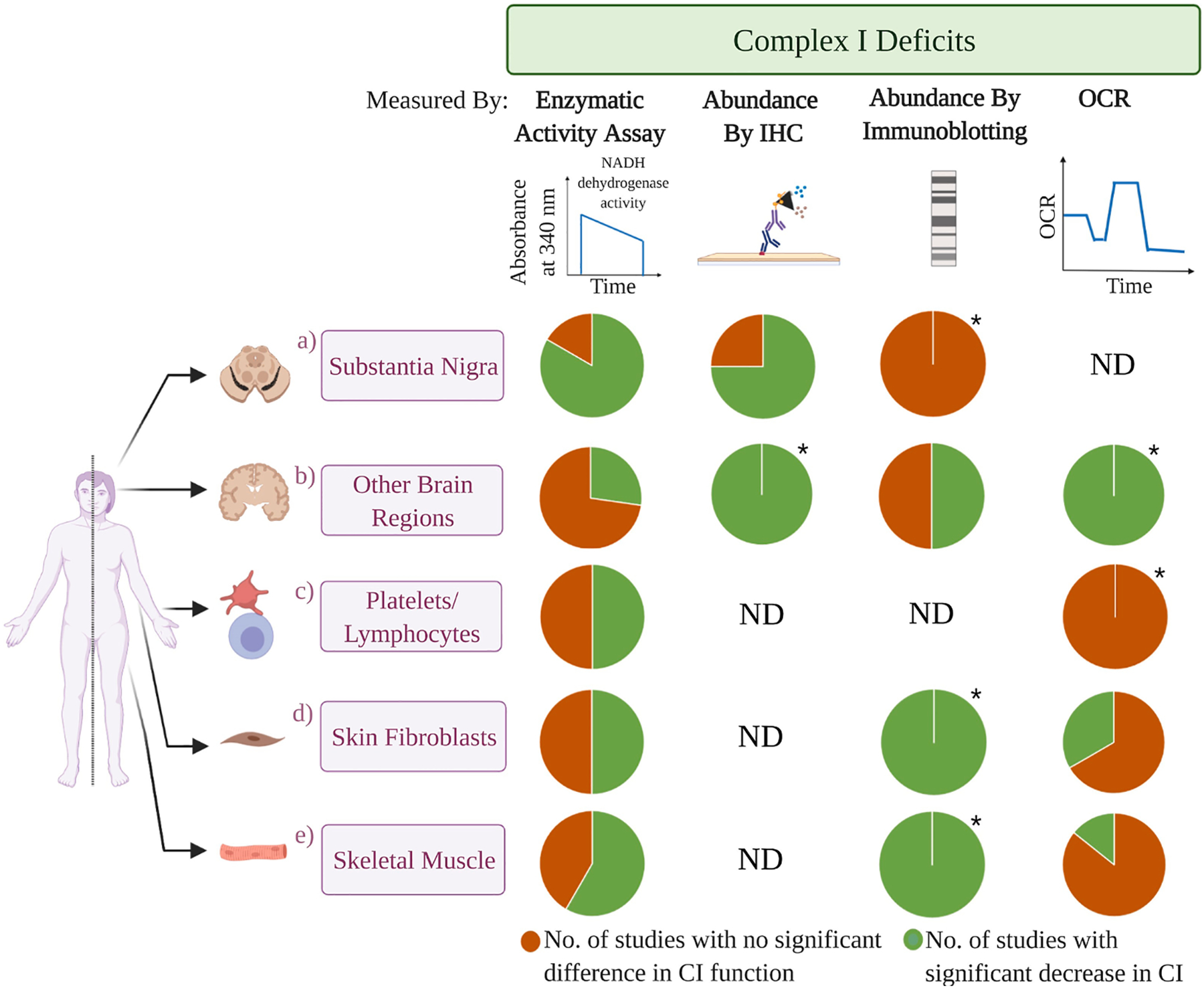
A lack of definitive CI deficiency in a majority of primary tissues from iPD patients. The pie charts represent the approximate percentage of independent studies that report a significant decrease in CI function (green) versus no significant differences (orange) in iPD patient tissues compared to the control subjects, or not determined (ND). Charts are segregated by tissue and assay method. The number of studies (CI-impairment vs no deficit) for a) SN, b) other brain regions, c) platelets/lymphocytes, d) skin fibroblasts, and e) skeletal muscle are as follows. CI enzymatic activities: a) 5 vs 1, b) 3 vs 8, c) 10 vs 10, d) 1 vs 1, e) 7 vs 5; immunohistochemistry: a) 3 vs 1, b) 1 vs 0, c) ND, d) ND, e) ND; SDS-PAGE/BN-PAGE immunoblotting: a) 0 vs 1, b) 2 vs 2, c) ND, d) 1 vs 0, e) 1 vs 0; CI-linked oxygen consumption rate (OCR): a) ND, b) 1 vs 0, c) 0 vs 1, d) 1 vs 2, e) 1 vs 6. The number of subjects tested in each study is highly variable and the details are provided in Tables 1 and 2. “ * ” indicates pie charts generated from only one available study. Created with Biorender.com.
Immunohistochemical staining is another method that has been employed to assess CI levels in the brain with interesting results (Table 1B). An initial study of eight PD patients revealed a 2.6-fold increase in the number of nigral neurons with weak or barely detectable CI immunoreactivity, compared to seven controls (Hattori et al., 1991). On the other hand, the immunohistochemical analyses of other respiratory complexes such as complex III (CIII) and complex IV (CIV) remained unaffected. However, complex II (CII) staining was diminished in three of the eight patients, implying that five of eight PD subjects exhibited a selective decrease in CI abundance in the SN. In contrast, CI immunohistochemical staining was intense in substantia nigra pars reticulata, red nucleus, and oculomotor nucleus, indicating that the decreased CI abundance was unique to the SNpc. One caveat of this study is that while immunohistochemical staining is a valuable method to determine CI localization, it is a poor indicator of absolute quantity. Recently, three additional studies have employed immunohistochemistry to detect the abundance of CI in iPD brain. Decreased immunohistochemical staining of CI subunits NDUFB8 and NDUFA13 was detected in individual SN neurons of 10 iPD patients compared to 10 age-matched controls (Grunewald et al., 2016). In addition, combined CI + CII or CI + CIV deficiencies were also observed in some SN neurons. The second study conducted a broader investigation and observed decreased CI immunohistochemical staining in multiple brain regions of 17 iPD patients, compared to seven controls. CI immunoreactivity was specifically decreased in the SNpc, prefrontal cortex, hippocampus, putamen, pons, Purkinje cells in the cerebellar cortex, and principal neurons of the dentate nucleus, whereas mild CIV decreases were additionally observed in the SNpc alone (Flones et al., 2018). In contrast, a recent analysis reported a lack of significant differences in CI or CIV immunohistochemical staining in the SN neurons of nine PD compared to 10 controls (Reeve et al., 2018). Together, these results indicate that not all iPD cases display isolated CI deficiency in the SN. In addition, decreased CI immunoreactivity is not unique to the SN as it has been observed in multiple brain regions of iPD patients, even though neurodegeneration was selectively observed only in the SN.
Compared to immunohistochemical analyses, the quantitative measure of specific enzymatic activities allows for a more definitive assessment of isolated mitochondrial CI deficiency. This activity is often determined as a function of NADH oxidation in the presence of an electron acceptor, such as decylubiquinone or co-enzyme Q1 (CoQ1) (CI activity) or cytochrome c (CI + III activity), and is ideally calculated as a function of rotenone-sensitive activity (Table 1C). In the first study reporting enzymatic activities from PD patient brain, rotenone-sensitive CI + III activity was decreased by ~39% in the SN averaged across nine PD patients, and rotenone-sensitive CI activity was decreased by ~31% in the SN of five PD patients, whereas the succinate: cytochrome c oxidoreductase (CII + III) activity remained comparable to the non-PD controls (Schapira et al., 1989). The observation of a specific decrease (31–42%) in CI activity of iPD nigral tissue has been reproduced in a total of 47 PD patients across multiple studies conducted by the Schapira and Reichmann groups (Janetzky et al., 1994; Mann et al., 1992a, 1994; Schapira et al., 1990a, 1990b). One outlier, however, is a study of two PD patients that exhibited no significant differences in the activities of any of the respiratory enzymes (Mizuno et al., 1990). It should be noted that there was no clear mention of the use of rotenone in the enzymatic assays to determine CI-specific NADH dehydrogenase activity, potentially undermining the accuracy of the measurements if a CI inhibitor was not used. In conclusion, in the majority of studies, a decrease in rotenone-sensitive CI activity has been observed in the SN of iPD patients.
Similar biochemical studies on other brain regions of PD patient tissue have been conducted to determine if the decreased CI activity is a SN-specific feature (Table 1C). Some studies reported no alterations in the activities of respiratory chain complexes in the frontal lobe, striatum, caudate nucleus, medial globus, lateral globus pallidum, anterior prefrontal cortex, cerebellum, tegmentum, SN pars reticulata, and putamen (Cooper et al., 1995; Flones et al., 2018; Janetzky et al., 1994; Keeney et al., 2006; Mann et al., 1992a; Mizuno et al., 1990; Mythri et al., 2011; Schapira et al., 1990b). In contrast, two separate studies have revealed a significant decrease in rotenone-sensitive CI activity of frontal cortex mitochondrial preparations, with a ~30% deficit in seven PD versus four controls (Keeney et al., 2006), and a ~60% decrease for seven PD versus nine controls (Navarro et al., 2009). In addition to corroborating a decrease in prefrontal cortex tissue, one study reported decreased rotenone-sensitive CI activity in the striatum (eight PD and eight controls) and the cerebellum (nine PD and seven controls) (Flones et al., 2018). In contrast to the traditional enzymatic activity assays described above, one study utilized a unique “dipstick” assay to quantify CI activity in prefrontal and somatosensory cortex tissues. This assay relies upon the immunocapture of CI from tissue homogenates and then tests the ability of the immunocaptured enzyme to oxidize NADH using nitroblue tetrazolium (NBT) as an electron acceptor. However, no decrease in prefrontal cortex CI activity was observed by dipstick assay in 35 PD patients compared to 23 age-matched controls (Gatt et al., 2016). Because this assay normalizes NADH dehydrogenase activity to the abundance of the immunocaptured CI, it may introduce an inherent bias that fails to account for a decrease in holoenzyme abundance in tissue homogenates, as was described above by immunocytochemistry to be affected in PD. Although no change in the accumulation of the CI subunit NDUFB8 (40 iPD vs 23 controls) was observed in the prefrontal cortex by SDS-PAGE immunoblotting, this study still does not fully account for holoenzyme levels. In conclusion, the prevalence of a widespread CI deficiency in non-SN brain tissues remains inconclusive due to mixed results. One possible reason for variations in CI activities between studies is the extent of post-mortem delay (time ranging from death to autopsy and freezing of tissues). An initial study showed a ~9.5% decrease in CI activity per hour of post-mortem interval (Mizuno et al., 1990). Unfortunately, there is a wide variation in post-mortem interval in the various reports ranging from 16–19 h in some studies (Janetzky et al., 1994; Mann et al., 1992a; Schapira et al., 1990a,b) to as high as 120 h in others (Flones et al., 2018).
The majority of biochemical studies using post-mortem tissue analyses support the observation of a selective decrement in CI activity in the SNpc of PD patients (Table 1, Fig. 1). MPTP-driven parkinsonism and the CI defects of the SN may appear to favor the hypothesis of CI deficiency directing the development of PD. However, an important point of consideration while interpreting these results is that significant DA neuronal loss has already occurred in the SN of PD patients at the time of post-mortem analysis (Burke and O’Malley, 2013). Therefore, we cannot rule out that a decrease in CI activity is a consequence of neuronal loss and gliosis in the affected tissue. Furthermore, these two observations do not prove causality - MPTP may cause both CI inhibition and parkinsonism, but that may not be related to the etiology of sporadic/idiopathic PD. Consideration of further evidence is warranted and described below.
3. Mitochondrial complex I deficiency in peripheral tissues of PD patients
The study of post-mortem brain tissues, specifically the SNpc, can provide a focused analysis of mitochondrial respiratory function at the site of the greatest PD neuropathology. However, there are also multiple disadvantages associated with such efforts. Tissues are typically obtained from patients who have died at advanced stages of disease that are characterized by an extraordinarily high degree of neuronal loss with concomitant reactive gliosis in the regions of interest. The biochemical status of CI activity across tissue homogenates in these samples may not reflect the nature of CI in the SN neurons at the initiation of the disease. Thus, it is possible that post-mortem work may not reveal mechanisms of pathogenesis but rather the progression or the consequences of decades of disease. Additionally, patients at advanced stages have already been subjected to prolonged treatments with Levodopa/Carbidopa, dopamine agonists, and/or other medications potentially confounding results. Given that there are no non-invasive technologies that allow for the quantification of CI activity in intact brain, this is as close as we can come to understanding the state of CI activity in the iPD patient brain. To circumvent these limitations, studies have been conducted to determine whether CI deficits in iPD patients would extend into peripheral tissues. A plethora of studies of ETC complexes have been conducted in platelets, lymphocytes, skeletal muscle, and skin fibroblasts from iPD patients and healthy controls (Table 2, Fig. 1).
Table 2.
A summary of biochemical assays conducted on peripheral tissues from PD patients. The different peripheral tissues assayed are (A) platelets, (B) lymphocytes, (C) skeletal muscle, and (D) skin fibroblasts. C: control; SC: spousal control to account for environmental conditions; PD: Parkinson’s disease; PDS: sporadic PD; PDF: patients with first degree relatives with PD; DA: dopaminergic; N.D.: not described; N.S.: no statistically significant difference between PD and control subjects; CI: NADH: CoQ1 oxidoreductase activity; CI + III: NADH: cytochrome c oxidoreductase activity; CII + III: succinate: cytochrome c oxidoreductase activity; CIV: cytochrome c oxidase activity; CS: citrate synthase activity; The color “blue” denotes that the activities were determined with a complex-specific inhibitor (rotenone for CI, antimycin A for CIII); “x/CS” refers to activity x (CI, CII, CIII, or CIV) normalized to citrate synthase activity. L-dopa: Levodopa; OCR: oxygen consumption rate; [3H] DHR: [3H]dihydrorotenone; The symbol (#) denotes that there was no correlation between activity levels and age at onset of disease, severity of disease, duration of disease, and/or treatment. The studies are grouped based on the presence (light green cells) vs absence (light red cells) of significant difference in CI between control and PD.
| Table 2A. Assessment of ETC function in platelets | ||||
|---|---|---|---|---|
| Study | Source | No. of cases | Medications for PD | Results |
| Parker et al., 1989 | Mitochondria (Percoll gradient) | 8 C, 10 PD | 8 on L-dopa and other drugs; 2 untreated | ~54% decrease in CI activity. N.S. for CII+III and CIV. |
| Krige et al., 1992 | Mitochondria (differential centrifugation) | 15 C, 25 PD | 21 on L-dopa; 4 untreated | ~16% decrease in CI/CS activity; N.S. for CII+III, CIV, and CS; (#) |
| Yoshino et al., 1992 | Homogenates | 17 C, 20 PD | 17 on L-dopa; 12 on trihexyphendyl; 6 on bromocriptine | ~26% decrease in CI activity; ~19% decrease in CII; N.S. for CIII and CIV; (#) |
| Benecke et al., 1993 | Mitochondria (sucrose gradient ultracentrifugation) | 44 C, 27 PD | 24 on L-dopa/Carbidopa, selegeline/bromocriptine, cabergoline or lisuride; 3 untreated | ~51% statistically significant decrease in piericidinA - sensitive CI activity; ~30% decrease in CIV. N.S. for CIII. 5 early-stage PD measured after one year: CI and CIV activities decreased after one year. N.S. for patients >3 years after diagnosis; (#) |
| Haas et al., 1995 | Mitochondrial (Percoll gradient ultracentrifugation); No medications for at least one month before testing | 1 on trihexylphenidyl; 1 on amantadine; 2 on L-dopa/Carbidopa; 5 on selegiline; Some subjects also received other drugs | ||
| 13C, 13 SC, 13 PD | Compared to age/sex-matched controls (C):~ 32% decrease in CI and ~28% decrease in CII+III activities; N.S. for CI/CS, CII+III/CS and CIV/CS. Compared to SC: ~29% decrease in CI activity, N.S. for CI/CS, CII+III, CIV. | |||
| Gu et al., 1998 | Mitochondira (differential centrifugation) | 8 C, 8 PD | All receiving L-dopa | ~24% decrease in CI/CS; N.S. for CII+III/CS and CIV/CS. 4 PD with worst CI activities chosen to be studied over time: after 1 and 2 months, CI/CS retained similar deficiency. |
| Varghese et al., 2009 | Homogenates | 30 C, 10 PD | 1 under dopamine replacement therapy; others, N.D. | ~50% decrease in NADH: CoQ0 oxidoreductase activity assessed without testing for rotenone sensitivity; N.S. for CII; CIII, CIV: N.D. |
| Bravi et al., 1992 | Mitochondria (differential centrifugation); Medications stopped for minimum 8 h before obtaining platelets | 13 C, 17 PD | 8 on L-dopa/Carbidopa (4 with selegeline), 2 on selegeline (with lisuride or amantadine), 7 untreated. | N.S for CI, CII+IIII, CIV activities, and CI-linked OCR; (#) |
| Mann et al., 1992a | Platelet homogenates | 15 C, 14 PD | 12 on L-dopa. | N.S. for CI, CII+III, CIV activities, before and after CS normalization. |
| Taylor et al., 1994 | Mitochondria (differential centrifugation) | 15 C, 7 PD | All on selegeline and L-dopa; | N.S. for CI, CII+III, CIV activities; (#) |
| Shults et al., 1995 | Mitochondira (differential centrifugation) | 11 PD, 0 C | Three measures: Before medication, 1 month after Carbidopa/L-dopa, and 1 month after Carbiopa/L-dopa + selegiline. | N.S. for CI, CII+III, CIV, and CS activities before or after medications. |
| Blake et al., 1997 | Mitochondria (Percoll gradient centrifugation) | 9 C, 13 PD | 2 untreated; 11 on Carbidopa/L-dopa, with or without selegiline, with or without dopamine agonist or anticholinergic medication - 5 had discontinued drugs 10 days before study. | N.S, for CI, CIII, CIV activities before and after CS normalization; (#) |
| Blandini et al., 1998 | Washed platelets | 16 SC, 16 PD | All receiving L-dopa. | Platelet [3H]DHR binding assay: N.S. for specific binding of [3H]DHR; N.S. for percentage of [3H]DHR binding inhibition by 1 mM MPP+; a correlation was found in % MPP+ inhibition of DHR specific binding and L-dopa dosage; (#) |
| Hanagasi et al., 2009 | Mitochondria (differential centrifugation) | 17C, 17 PDF, 15 PDS | All receiving L-dopa and/or DA drug treatment. | N.S. for CI, CII+III, CIV, before and after CS normalization; (#) |
| Bronstein et al., 2015 | Mitochondria (Percoll gradient ultracentrifugation); No medications at least 12 h before testing | 23 C, 23 PD | N.D. | N.S. for CI and CI+III activities, before and after CS normalization; CIVN.D.; No correlation between CI/CS activity and pesticide exposure. Inverse correlation for CI inhibitors and dithiocarbamates with CI+III activity. |
| Table 2B. Assessment of complex I function in lymphocytes | ||||
| Study | Source | No. of cases | Medications for PD | Results |
| Barroso et al., 1993 | Homogenates | 15 C, 16 PD | Not treated | ~25% decrease CI+III; ~45% decrease in CIV; N.S. for CII+III activity. |
| Shinde et al., 2006 | Homogenates | 30 C, 40 PD | All on L-dopa | ~12% decrease in CI+III, ~26% decrease in CIV, N.S. for CII CII+III, CS activities. |
| Müftüoglu et al., 2003 | Leukocyte mitochondria | 17 C, 10 Parkin PD, 20 iPD | All on L-dopa, selegiline, and/or pergolide. | ~ 62% and ~64% decrease in CI in Parkin PD and iPD, respectively; ~ 60% decrease in CIV activity for iPD, N.S. for CIV activity in Parkin PD; (#) |
| Yoshino et al., 1992 | Homogenates | 17 C, 20 PD | 17 on L-dopa; 12 on trihexyphendyl; 6 on bromocriptine. | N.S. for CI, CIII, and CIV activities; ~13.2% decrease in CII activity; (#) |
| Martin et al., 1996 | Mitochondria (differential centrifugation) | 30 C and 36 PD; ~2.6 years of PD duration | Not treated | N.S. for CI, CI+III, CII, CII+III, CIV, normalized to protein content or CS activity; (#) |
| Ming et al., 2020 | Immortalized lymphocyte mitochondria | 3 C, 3 PDS, 3 Parkin PD | N.D. | N.S. for immunocaptured CI activity |
| Table 2C. Assessment of complex I function in skeletal muscle | ||||
| Study | Source | No. of cases | Medications for PD | Results |
| Bindoff et al., 1989; Bindoff et al., 1991 | Vastus lateralis mitochondria | 7 C, 5 PD | 1 untreated; 1 on sinemet; 1 on sinemet, orphenadrine; 1 on madopar, bromocriptine, diazepam; 1 on sinemet, benzhexol, triazolam. | |
| SDS-PAGE immunoblot using antibody against CI holoenzyme: 2 out of 3 patients show decreased accumulation of subunits | ||||
| Nakagawa-Hattori et al., 1992 | ilipsoas or quadriceps mitochondria. | 6 C, 4 PD | N.D. | ~48% decrease in CI activity. N.S. for CII, CIII, CIV activities |
| Shoffner et al., 1992 | Quadriceps mitochondria | 16 C, 6 PD | All received L-dopa/Carbidopa + combinations of other medications. | 5 out of 6 PD had decreased CI and CI+III activities; One patient had CI, CII+III, CIV deficiency; one patient had decreased activities for CI, CII, CI+III; one patient had decreased CI and CII+III. An association between the severity of CI defect and the level of disability stages observed. No correlation of CI activity with age observed. |
| Cardellach et al., 1993 | Left vastus lateralis mitochondria | 10 C, 8 PD | 6 on L-dopa/Carbidopa; 1 on selegiline; 1 on amantadine. | ~ 25% decrease in CI activity; ~68% decrease in CIV activity; N.S. for CII, CIII, CV activites, and CI-linked OCR; (#) |
| Blin et al., 1994 | Biceps brachii or deltoid muscle mitochondria | 43 C, 27 PD | 6 untreated; 21 on L-dopa. | CI activity: ~71% decrease in treated patients, ~67% decrease in untreated patients. ~34% decrease in CIII activity for only treated patients; N.S. for CII and CIV; (#) |
| Wiedemann et al., 1999 | Mitochondria from saponin-permeabilized M.vastus lateralis fibers | 32 C, 15 PD | N.D. | ~30% decrease in CI+III/CS; N.S. for CII+III/CS, CIV/CS; ~50% increase in CS activity; Flux control coefficient of CI and CIV increased. N.S. for CI-linked respiration. |
| Winkler-Stuck et al., 2005 | Homogenates | 36 C, 19 PD | 15 PD on L-dopa | ~33% and ~30% decrease in CI/CS and CI+III/CS activities, respectively; ~30% decrease for CIV/CS; N.S. for CII+III/CS; Increased CI and CIV flux control coefficients. No decrease in CI-linked respiration. |
| Mann et al., 1992a | Left vastus lateralis mitochondria | 6 C, 9 PD | 5 untreated; 4 on L-dopa. | N.S. for CI, CII+III, CIV activities. N.S. for CI-linked OCR (6 C, 9 PD); No difference in activities in treated vs untreated PD. |
| Anderson et al., 1993 | Quadriceps mitochondria | 6 C, 7 PD | 6 on L-dopa or Carbidopa and other drugs; 1 untreated. | N.S. for CI+III, CII+III or CIV activities, and CI-linked OCR; (#) |
| DiDonato et al., 1993 | Left quadriceps homogenates and mitochondria | 6C, 6 PD (mitochondria), 8 C, 16 PD (homogenates) | 2 untreated; rest on L-dopa. | N.S. for CI, CII, CII+III, or CIV activities in muscle homogenates and mitochondria; (#) |
| Manneschi et al., 1994 | Left quadriceps mitochondria | 53 C, 6 PD out of which 2 PDF | N.D. | N.S for CI/CS, CII+III/CS, CIV/CS activities. |
| Taylor et al., 1994 | Left vastus lateralis mitochondria | 6 C, 3 PD | 2 untreated; 1 on selegeline and L-dopa. | N.S. for CI, CII+III, CIV activities; N.S. for CI-linked OCR (5 C, 3 PD). |
| Table 2D. Assessment of complex I function in skin fibroblasts | ||||
| Study | Source | No. of cases | Medications for PD | Results |
| Wiedemann et al., 1999 | Digitonin-permeabilized skin fibroblasts | 14 C, 14 PD | N.D. | N.S for CI-linked OCR. Increased CI and CIV flux control coefficient. Enzymatic activities N.D. |
| Ambrosi et al., 2014 | Cultured fibroblasts | 7 C, 11 PDS | N.D. | Maximal respiration and rotenone-sensitive respiration were decreased. Enzymatic activities N.D. |
| Winkler-Stuck et al., 2004 | Cultured fibroblasts | 13 C, 18 PD | 15 PD on L-dopa | Increased CI and CIV flux control coefficient; N.S. for CII+III/CS and CIV/CS activity; CI/CS increased by ~ 54%. N.S. for OCR. |
| Carling et al., 2020 | Cultured fibroblasts | 50 C, 100 PDS | N.D. | N.S. for ATP levels, mitochondrial membrane potential; (#) |
| 5 C, 5 PDS | 5 PDS patients selected for most severe ATP defect: Decreased NDUFB8 (CI subunit) and COX2 (CIV subunit) determined by SDS-PAGE immunoblot. | |||
| 6 C, 6 PDS | 75% decrease in immunocaptured CI activity; 37% decrease in CIV activity. | |||
| del Hoyo et al., 2010 | Cultured fibroblasts | 19 C, 20 PD | 18 PD treated with L-dopa, dopamine agonists, selegiline, rasagiline, anticholinergics, and/or amantadine. | N.S. for CI, CII, CIII, CIV, CI+III, CII+III activities normalized to either protein amount or CS activity; ~20% decrease in CV activity normalized to protein or CS activity; |
In 1989, Parker et al., conducted the first analysis of platelet mitochondria from 10 PD patients and eight controls and observed a ~54% decrease in PD platelet CI activity (Parker et al., 1989) (Table 2A). This level of CI deficiency has been subsequently reproduced in platelet homogenates of 10 PD patients from an Indian population (Varghese et al., 2009). However, both studies determined NADH dehydrogenase activity without assessing the sensitivity to a CI inhibitor such as rotenone, which may make these measurements less meaningful. Rotenone-sensitive CI activity can be as high as 80% or as little as <50% of total NADH dehydrogenase activity depending upon cell- or tissue-type (based upon our experience). In subsequent studies conducted on platelet mitochondria and homogenates, conflicting data have been obtained with regards to rotenone-sensitive CI activity. Five studies from four different research groups considering samples from 98 PD patients observed a 16–51% decrease in isolated CI activity or combined with CII and CIV deficiency, with no significant correlation between CI deficiency and age at onset, duration of disease, or severity of disease (Benecke et al., 1993; Gu et al., 1998; Haas et al., 1995; Krige et al., 1992; Yoshino et al., 1992). However, a significant CI defect could not be reproduced in other studies from six research groups, conducted on samples from 89 total PD patients (Blake et al., 1997; Bravi et al., 1992; Bronstein et al., 2015; Hanagasi et al., 2009; Mann et al., 1992b; Taylor et al., 1994). Similar conflicting results have been reported from studies conducted on lymphocytes (Table 2B). While three groups reported a significant decrease in rotenone-sensitive CI or CI + III and CIV activities (Barroso et al., 1993; Muftuoglu et al., 2004; Shinde and Pasupathy, 2006), two other groups observed comparable rotenone-sensitive CI activities in PD and control lymphocytes (Martin et al., 1996; Yoshino et al., 1992). Thus far, none of this work has indicated a consistent decrease in platelet or lymphocyte CI activity.
One possible reason for the discrepancies in the observance and degree of CI deficiency could be the differences in the methods used for collecting platelets and their subsequent processing to yield tissue homogenates or purified mitochondria. Even considering only those studies that used isolated mitochondria for enzymatic analyses, there is still no consistent CI deficiency observed in iPD patient platelets. Three main methods have been utilized for mitochondrial isolation: Percoll gradient, sucrose gradient ultracentrifugation, or differential centrifugation. Parker et al., 1989 conducted plateletpheresis and mitochondrial enrichment via Percoll gradient separation by employing a protocol that required two days of processing (Krige et al., 1992; Parker et al., 1988, 1989), yielding ~54% CI deficiency in iPD platelet mitochondria. Although it is possible that this time-consuming protocol could cause a greater destabilization of mitochondrial complexes resulting in an amplification of a modest CI defect (Parker et al., 1989), another study employing a similar methodology (18 iPD versus 18 controls) showed only a ~24% decrease in rotenone-sensitive CI activity with a concomitant ~20% decrease in CII + III activities in PD platelet mitochondria (Haas et al., 1995). A third study altogether failed to reproduce any CI deficiency in platelet mitochondria from 23 iPD patients versus 23 controls (Bronstein et al., 2015). Mitochondrial purification by other methodologies have likewise yielded inconsistent evidence of CI deficiency in iPD. Mitochondrial isolation from frozen platelets via sucrose gradient ultracentrifugation resulted in a statistically significant 51% decrease in piericidin A-sensitive CI deficiency (Benecke et al., 1993), similar to mitochondrial isolation by Percoll gradient (Parker et al., 1989). On the other hand, a rapid three hour protocol employing differential centrifugation to isolate platelet mitochondria displayed only a modest 16% decrease in rotenone-sensitive CI activity normalized to citrate synthase activity, even though the control activities were comparable to Parker et al., 1989 (Krige et al., 1992). This 16–25% decrease in CI activity with mitochondria isolated by differential centrifugation has been reproduced by the same group (Gu et al., 1998), whereas analyses by another group employing this method has yielded no difference (Hanagasi et al., 2009). Similar conflicting data has been obtained from total platelet homogenates, as well (Mann et al., 1992a; Varghese et al., 2009; Yoshino et al., 1992). These examples highlight the high degree of variability in the measures of platelet CI enzymatic activity from patient samples, even when employing comparable methodologies.
Other sources of variability could be the lack of consistent normalization to control for total mitochondrial content such as with citrate synthase activity or other variable (Table 2), or the frequent utilization of the less accurate NADH: cytochrome c oxidoreductase assay (CI + III) instead of a more consistent employment of the direct rotenone-sensitive NADH: CoQ1 oxidoreductase (CI) activity. In addition, substantial differences in activities amongst the control subjects have also been noted, suggesting a high degree of innate sample-to-sample variability within protocols (Hanagasi et al., 2009; Krige et al., 1992; Mann et al., 1992a; Yoshino et al., 1992).
To determine the onset of a putative CI defect during the development of PD, limited but informative studies have been conducted on platelets obtained from early vs late stage iPD patients. For instance, 18 iPD patients with early stage of PD (~two years post-diagnosis) already displayed a ~23% decrease in rotenone-sensitive CI activity (Haas et al., 1995). A separate study followed five patients with early stage iPD, beginning one year after diagnosis. A decline in platelet CI and CIV activities after one year was observed for patients in the early stages of disease, whereas no correlation between level of CI defect and duration of disease was observed for patients three years after diagnosis (Benecke et al., 1993). Considering the overall discrepancies in iPD platelet CI activity, more studies with larger cohorts is needed to address whether a platelet CI defect is a general feature of iPD.
Another concern in assessing CI defects in iPD patients is the contribution of medications to perceived mitochondrial deficiency. For instance, drugs such as Levodopa/Carbidopa have been implicated in CI deficiency in rat brain mitochondria (Przedborski et al., 1993, 1995). However, no such correlation has been thus far observed between Levodopa dose and platelet respiratory chain activities in PD patients (Benecke et al., 1993; Bravi et al., 1992; Krige et al., 1992). Furthermore, there was no correlation in CI deficiency between untreated and treated patients (Bravi et al., 1992; Krige et al., 1992). Indeed, patients who were never treated or had a history of medications that were stopped one month before the study still displayed a ~25% decrease in platelet CI activity (Haas et al., 1995). There is one thoughtful study that followed 11 patients before medication, one month after Levodopa/Carbidopa, and a second month after treatment with Levodopa/Carbidopa plus the monoamine oxidase inhibitor selegiline (Shults et al., 1995). However, none of the patients displayed respiratory chain defects in platelets and no significant changes were observed before or after medication. In lymphocytes, one study with 16 untreated PD patients reported a ~25% significant decrease in rotenone-sensitive CI + III activity (Barroso et al., 1993), whereas a second study with 36 untreated patients with ~2.6 years of disease duration observed no differences in rotenone-sensitive CI or CI + III activities compared to controls (Martin et al., 1996). These limited studies imply that drug treatment is unlikely to impact respiratory chain function in platelet/lymphocyte mitochondria, and additionally highlight the variable evidence of a CI defect in the first place.
As the pathological consequences of CI inhibitors drove the interest in mitochondrial function in iPD, the primary focus has been on assessing CI enzymatic activity. However, one study employed a different technique for quantifying CI levels. This new assay was based upon the binding capacity of [3H]dihydrorotenone ([3H]DHR), a rotenone analog, to CI in isolated platelets (Blandini et al., 1998). Comparison between 16 iPD and 16 controls revealed no difference in the specific binding of [3H]DHR or in the level of binding competition with 1 mM MPP+. Interestingly, a positive correlation was observed between the percentage of MPP+ competition with [3H]DHR-specific binding and daily Levodopa intake in PD patients, raising the question of Levodopa’s role in changing the MPP+-sensitivity of platelets. Overall, these results indicate an equivalent abundance of the holoenzyme in iPD and control platelets.
Another peripheral tissue that has been extensively targeted for analysis is the skeletal muscle (Table 2C), likely due to the enriched population of mitochondria that can be isolated form this source. The specific muscle tissues utilized for analysis have been the vastus lateralis, iliopsoas, biceps brachii, or deltoid muscle (Bindoff et al., 1989, 1991; Blin et al., 1994; Nakagawa-Hattori et al., 1992; Shoffner et al., 1992). Similar to platelet mitochondria, highly variable information has been obtained from these enzymatic activity assays, although one meta-analysis study reported a significant decrease in muscle CI activity of PD patients across nine studies (Holper et al., 2019). Six research groups observed 26–71% decreases in rotenone-sensitive CI or CI + III activities across a total of 83 PD patients and 148 controls (Bindoff et al., 1989, 1991; Blin et al., 1994; Cardellach et al., 1993; Nakagawa-Hattori et al., 1992; Shoffner et al., 1992; Wiedemann et al., 1999; Winkler-Stuck et al., 2005), whereas four groups found no significant differences in a total of 41 PD subjects and 79 controls (Anderson et al., 1993; DiDonato et al., 1993; Mann et al., 1992a; Manneschi et al., 1994; Taylor et al., 1994). Interestingly, one study that immunoblotted skeletal muscle mitochondria using antisera against the CI holoenzyme showed decreased accumulation of CI subunits in two out of three PD patients (Bindoff et al., 1991). This finding is reminiscent of results from striatal iPD mitochondria (Mizuno et al., 1989), and may suggest a possible frequent decrease in CI subunit levels in iPD tissues. Amongst iPD subjects that did manifest a CI deficiency, a higher incidence of combined CII, CIII, or CIV deficiencies was reported in PD skeletal muscle (Bindoff et al., 1991; Blin et al., 1994; Cardellach et al., 1993; Shoffner et al., 1992). Such degree of combined CI deficiency was not observed in platelets. In one study, CII + III deficiency in skeletal muscle of three patients and CIV deficiency in one patient was reported even in the absence of a CI defect (Manneschi et al., 1994). These disparate findings are perhaps consistent with a general mitochondrial defect in iPD, but importantly, do not endorse a unique loss of CI activity.
Only a limited number of studies have been conducted on patient skin fibroblasts (Table 2D). One methodology employed was the assessment of maximal and rotenone-sensitive oxygen consumption rates as an indirect measure for CI-dependent respiration. Interestingly, the oxygen consumption rates were found to be decreased in PD patient fibroblasts, compared to controls, in two out of three studies (Ambrosi et al., 2014; Wiedemann et al., 1999; Winkler-Stuck et al., 2004). A second methodology measured the flux control coefficients to quantify the relative flux changes in a pathway due to small changes in enzymatic activities. A higher flux control coefficient for CI is indicative of an increased dependence of the flux through ETC on CI activity, implying decreased enzymatic activity. Increased CI and CIV flux coefficients, based on titrations with complex-specific inhibitors, were reported in iPD fibroblasts and skeletal muscle (Wiedemann et al., 1999; Winkler-Stuck et al., 2004, 2005). Very few studies have tested ETC activities on skin fibroblasts of PD patients. In a study of 20 iPD patients, no significant difference was observed in rotenone-sensitive CI or CI + III activities (del Hoyo et al., 2010). In a recent analysis of 100 sporadic PD patient fibroblasts compared to 50 age-matched controls, no overall difference in ATP levels or mitochondrial membrane potential was observed. However, a subset of 12 patients with marked decrease (>2 standard deviation below average) were selected for further testing of specific ETC function. This subset displayed decreased accumulation of CI and CIV subunits by SDS-PAGE immunoblotting (five PD vs five controls), along with decreased CI and CIV activity (six PD vs six controls) (Carling et al., 2020). A caveat of the CI activity assay employed here is that it did not consider rotenone sensitivity. Additionally, although these analyses indicate a possible decrease in CI function in skin fibroblasts, they also imply a coincident CIV defect even in cases selected specifically with a bias towards mitochondrial dysfunction, and thus speak against a unique CI deficiency. In general, the characterization of iPD fibroblasts has been limited by the small number of studies and the lack of direct measurements of rotenone-sensitive NADH dehydrogenase activity. In addition, fibroblasts are traditionally cultured and maintained in concentrations of high glucose, passaged many times and/or immortalized, all of which could introduce inherent metabolic changes in the cell lines that may not reflect the actual status of mitochondrial function in the patient. Thus, we have intentionally focused our attention more heavily on primary tissue analyses from brain, skeletal, and acutely isolated platelets to explore the co-occurrence of CI in PD.
In summary, the presence of a CI-specific deficiency in the peripheral tissues of iPD patients remains inconclusive (Fig. 1). One direction of enquiry currently lacking in the biochemical analyses of patient tissues (brain and peripheral) is the assessment of CI assembly. Keeney et al., 2006 have suggested CI mis-assembly in frontal cortex mitochondria of PD patients, although that conclusion was drawn from the decreased levels of one CI subunit assessed by SDS-PAGE immunoblotting (Keeney et al., 2006). Another study found no differences in CI assembly in the prefrontal cortex (11 PD vs 11 controls), the striatum (six PD vs six controls), and the cerebellum (eight PD vs two controls) (Table 1A) (Flones et al., 2018). A systematic and direct approach to assess the extent of holoenzyme assembly and accumulation of assembly intermediates, using techniques such as one-/two-dimensional Blue-Native PAGE followed by in-gel activities and/or immunoblotting (Schagger, 1995), is required to confirm instances of CI assembly defect. Such methodologies would increase the sensitivity of tissue analyses as even a mild accumulation of assembly intermediates, indicative of an assembly defect, has been previously observed to allow for a normal range of CI activities in certain patient tissues such as fibroblasts (Friederich et al., 2017).
4. Complex I deficiency and mitochondrial encephalomyopathy
Isolated or combined CI deficiency contributes to the majority of genetic OXPHOS disorders and is caused by mutations in either CI subunits or biogenesis factors. CI deficiency can result in a wide range of clinical disorders, the most frequent being Leigh syndrome, leukoencephalopathy, MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like syndrome), hypertrophic cardiomyopathy, and exercise intolerance (Giachin et al., 2016; Pagniez-Mammeri et al., 2012a, 2012b). Pathogenic CI mutations have been identified in seven mitochondrially-encoded subunits, 20 of the 37 nuclear-encoded subunits, and 13 of the 16 biogenesis factors (Fassone and Rahman, 2012; Fiedorczuk and Sazanov, 2018; Formosa et al., 2015; Friederich et al., 2017; Giachin et al., 2016; Koopman et al., 2016; Pagniez-Mammeri et al., 2012a, 2012b; Rodenburg, 2016; Sanchez-Caballero et al., 2016a, 2016b). While CI disorders can have a wide spectrum of symptomology and severity, an important distinction has been noted for patients carrying mitochondrial mutations compared to those carrying nuclear mutations (Distelmaier et al., 2009). The mitochondrial mutations result in a more heterogenous range of symptoms due to the heteroplasmic nature of mtDNA mutations. Such disorders have a wide variability in terms of age of onset, severity of clinical symptoms, and affected tissues/organs. On the other hand, patients with nuclear mutations typically present more consistent clinical phenotypes. The majority of these cases display symptoms within the first year of life with rapid deterioration triggered by respiratory or gastrointestinal infections. However, some mild heterogeneity does exist in CI disorders caused by nuclear mutations as well, such as the lack of correlation between the mutant genes, the manifested clinical phenotypes, or the affected tissues within each patient (Distelmaier et al., 2009; Friederich et al., 2017). The severity of CI deficiency in genetic CI disorders can range widely from 8 to 75%, depending upon the patient, the nature of mutation, and the type of tissues tested within the same patient (Distelmaier et al., 2009, Calvo et al., 2010).
If idiopathic reductions in CI activity play a cardinal role in the development of PD, then it is reasonable to expect that PD neuropathology would consequently be observed in genetic CI disorders. Although certain mtDNA polymorphisms and missense mutations have been associated with increased risk for PD, there is no consistent evidence for mtDNA mutations leading to the development of PD (Chen et al., 2019; Martin-Jimenez et al., 2020). Similarly, the spontaneous loss of SNpc DA neurons is not a universal, or even common, occurrence following pathogenic mutations of nuclear CI genes. The majority of CI disorders are characterized by features of Leigh syndrome or leukoencephalopathy with isolated or combined lesions in the brainstem and the basal ganglia, including both grey and white matter involvement. MELAS is predominantly characterized by grey matter involvement with neuronal loss in the basal ganglia, thalamus, cerebellum, and brain stem (Filosto et al., 2007). Some CI disorders were also reported to have lesions in the cerebellum, spinal cord, corpus callosum, and occasional SN involvement (Fassone and Rahman, 2012; Lebre et al., 2011). Therefore, these disorders where CI activity is reduced patient-wide do not resemble the selective and discrete neuropathology associated with iPD. Moreover, most CI disorders have disease onset within 12 months of birth. The majority of patients do not survive infancy or childhood, with only a limited number of patients surviving into adulthood (Fassone and Rahman, 2012).
Only one nuclear-encoded CI subunit, NDUFV2, has been associated with PD. Located in the matrix arm of CI, NDUFV2 is a highly conserved 24 kDa core subunit that binds a 2Fe-2S cluster and is required for electron relay and/or enzyme stability (Birrell et al., 2013; Sazanov and Hinchliffe, 2006). Furthermore, NDUFV2 is implicated in many other neurological diseases including Leigh syndrome, bipolar disorder, schizophrenia, and encephalomyopathy (Benit et al., 2003; Cameron et al., 2015; Liu et al., 2011; Washizuka et al., 2004, 2006). From a study of 126 PD patients and 112 controls, a homozygous mutation in NDUFV2 leading to an A29V substitution was associated with a 2.4-fold greater risk of developing PD (Hattori et al., 1998). In a second study, the K209R variant was observed in both familial and sporadic PD cases (Nishioka et al., 2010). Unfortunately, neither of the studies included detailed analyses of CI enzymatic activity or assembly to determine the consequence of the NDUFV2 mutations on CI function in patient tissues. Furthermore, reconstruction of the conserved lysine-to-arginine substitution in the unicellular alga Chlamydomonas reinhardtii failed to reveal any relevant defects in respiratory growth, CI activity, or assembly (Subrahmanian et al., 2020). Therefore, the precise biochemical consequence of these mutations on mitochondrial function and the development of PD is unknown.
The contribution of reduced CI activity to the development of parkinsonism has been investigated in mice with a homozygous deletion of Ndufs4 (encoding the 18 kDa CI soluble subunit). Loss of NDUFS4 in mouse embryos bestowed a complete lack of CI activity in mesencephalic neuronal cultures without leading to the expected selective neuronal death of DA neurons in SN or nigral cultures (Choi et al., 2008, 2011). Instead, mice with conditional knockout of Ndufs4 in neurons and glia developed Leigh syndrome-like neuropathology (Quintana et al., 2010). In fact, conditional Ndufs4 knockout in DA neurons of adult mice exhibited decreased dopamine levels in neurons (35% decrease in striatum by 24 months) without significant DA neuronal loss or motor deficits (Choi et al., 2017; Kim et al., 2015). Furthermore, a lack of CI was expected to negate the neurotoxic effects of CI inhibitors. In fact, Ndufs4−/− DA neurons exhibited the same susceptibility to MPP+- and rotenone-induced toxicity, calling into question CI as the common target of their toxicity (Choi et al., 2008, 2011; Kim et al., 2015). Further analyses suggested that rotenone-induced DA neuronal death, observed despite the absence of CI activity, was caused by microtubule depolymerization (Choi et al., 2011). Similarly, MPP+ has also been demonstrated to be an inhibitor of α-ketoglutarate dehydrogenase activity (Mizuno et al., 1988), a microtubule depolymerizing agent (Cappelletti et al., 2001, 2005), and a destabilizer of the D-loop structure in mtDNA (Umeda et al., 2000). Thus, despite the wide acceptance that these toxins inhibit CI activity, there is compelling evidence that their neurotoxicity towards DA neurons may involve targets other than CI. This interpretation is perhaps consistent with the poor correlation between genetic CI deficiencies and the spontaneous degeneration of the SNpc in humans, as described above.
5. The strange case of NDUFAF2
While virtually all cases of CI-related disorders do not support the co-occurrence of selective SNpc neurodegeneration, one rare mitochondrial encephalopathy partly challenges this norm. NDUFAF2 was identified as a potential CI assembly factor, from whole genome subtraction analysis of fungal organisms with the presence or absence of CI encoded by their genomes (Ogilvie et al., 2005). Human NDUFAF2 is a ~20 kDa mitochondrially-localized protein, which is 17% identical and 34% similar to the NDUFA12 soluble subunit of CI (Ogilvie et al., 2005). So far, 12 patients with autosomal recessive NDUFAF2 loss-of-function mutations have been reported (Barghuti et al., 2008; Calvo et al., 2010; Ghaloul-Gonzalez et al., 2016; Herzer et al., 2010; Hoefs et al., 2009; Janssen et al., 2009; Koene et al., 2012; Lesch et al., 2011; Ogilvie et al., 2005). Decreased CI activity in the skeletal muscle or fibroblasts was confirmed in seven patients (Herzer et al., 2010; Janssen et al., 2009; Koene et al., 2012), which is expected as NDUFAF2 has been implicated in both CI assembly (Adjobo-Hermans et al., 2020; Guerrero-Castillo et al., 2017; Ogilvie et al., 2005; Vogel et al., 2007) and activity (Schlehe et al., 2013). In 11 out of the 12 cases, the loss of NDUFAF2 was due to homozygous or hemizygous mutations that lead to the termination of NDUFAF2 synthesis [resulting in the variations: M1L, W3X, Y38X, W74X, I35SfsX17, A73GfsX5, R45X], or homozygous deletions in the NDUFAF2 gene / 450 kb deletion of a region including the NDUFAF2 gene (Barghuti et al., 2008; Calvo et al., 2010; Fassone and Rahman, 2012; Ghaloul-Gonzalez et al., 2016; Herzer et al., 2010; Hoefs et al., 2009; Janssen et al., 2009; Koene et al., 2012; Ogilvie et al., 2005).
Amongst the 11 patients that were studied closely, all exhibited early-onset encephalopathies with a lifespan ranging from nine to 24 months for ten patients and 13 years for one patient (Barghuti et al., 2008; Calvo et al., 2010; Fassone and Rahman, 2012; Ghaloul-Gonzalez et al., 2016; Herzer et al., 2010; Hoefs et al., 2009; Janssen et al., 2009; Koene et al., 2012; Ogilvie et al., 2005). Six patients were characterized with the neuropathology of Leigh syndrome, a disorder that also commonly includes SN involvement (Calvo et al., 2010; Ghaloul-Gonzalez et al., 2016; Herzer et al., 2010; Hoefs et al., 2009). Detailed neuroimaging studies have been reported for three out of six patients with Leigh syndrome. Bilateral symmetrical lesions of the midbrain and/or basal ganglia, characteristic of Leigh Syndrome, were reported for these three patients (Ghaloul-Gonzalez et al., 2016; Herzer et al., 2010; Hoefs et al., 2009). Additionally, two patients exhibited SN involvement, out of which one patient exhibited depigmentation of the SN, typical of DA neuronal loss (Ghaloul-Gonzalez et al., 2016; Herzer et al., 2010). The neuro-imaging of three other patients was distinctive from Leigh syndrome, with initial sparing of cortical and subcortical white matter and the involvement of SN, medial lemniscus, medial longitudinal fasciculus, mamillothalamic tracts, and spinothalamic tracts (Barghuti et al., 2008; Ogilvie et al., 2005). Six patients (three with Leigh syndrome neuropathology and three without) also exhibited motor symptoms (Barghuti et al., 2008; Calvo et al., 2010; Herzer et al., 2010; Hoefs et al., 2009; Ogilvie et al., 2005).
In spite of the numerous examples of CI-related mutations failing to produce parkinsonian symptoms or selective neurodegeneration of the SNpc, NDUFAF2 loss-of-function is one intriguing example where mitochondrial encephalopathy has been observed with a frequent involvement of SN loss, even in the absence of distinct Leigh syndrome neuropathology, along with broad motor symptomology. One possibility for this unique manifestation of SN injury is that loss of NDUFAF2 affects the mitochondrial respiratory chain in a specific manner that introduces a pathologic gain-of-function (e.g. generation of reactive oxygen species) that mimics mitochondrial dysfunction in iPD. A second possibility is a yet-to-be identified non-CI related function for NDUFAF2,the loss of which is more related to nigral degeneration than the partial decrements in CI activity. Far more work on this enigmatic mitochondrial chaperone is warranted.
6. Role of PINK1 in complex I function
While the majority of PD cases are idiopathic in nature, familial forms of PD (fPD) have been identified with autosomal dominant or autosomal recessive mutations across multiple genes. Most of the established fPD genes are not directly involved in CI function, nor even broadly associated with energy metabolism. Autosomal recessive mutations in the gene encoding the mitochondrial kinase PINK1 (PTEN-induced kinase 1) are causal for early onset fPD (Valente et al., 2004). Interestingly, one notable function of PINK1 involves a partnership with another autosomal recessive fPD gene product, the ubiquitin E3 ligase Parkin, to control the degradation of damaged mitochondria and thus maintain mitochondrial quality control (Narendra et al., 2008, 2012, 2010; Narendra and Youle, 2011). However, in the unique case of PINK1 there are data directly linking its function to CI biology.
In the elucidation of PINK1′s role in mitochondrial function Drosophila, zebrafish, and mice are three major model systems that have been rigorously employed. There are slight variations in each model’s response to loss of PINK1. However, one commonly observed feature of PINK1 loss across these model systems is impaired mitochondrial respiration. While a more generalized mitochondrial defect in Pink1-null mouse brain has also been suggested (Gautier et al., 2008), selective decreases in CI activity were first reported in mouse brain tissue and Drosophila brain-enriched and muscle-enriched mitochondrial homogenates (Morais et al., 2009). Similarly, decreased rotenone-sensitive CI activity was also observed in PINK1-mutant PD patient fibroblasts (Hoepken et al., 2007; Piccoli et al., 2008a, 2008b) and engineered Pink1-null mice fibroblasts transfected with common human PINK1 variants such as G309D, W437X, and K218A (Morais et al., 2009), suggesting that PINK1 function is required for normal mitochondrial CI activity.
The putative selective role of PINK1 in CI function was bolstered by the novel expression of alternative ETC enzymes in Pink1-null flies. The S. cerevisiae alternative NADH dehydrogenase Ndi1p, a monomeric rotenone-insensitive enzyme capable of bypassing the need for CI in animal models, was able to alleviate the pink1 mutant phenotypes (Vilain et al., 2012). Partial rescue was observed for flight incapacity, degeneration of flight muscles, abnormal mitochondrial morphology, and ATP levels. Indeed, pink1 mutants complemented with Ndi1p were capable of normal synaptic transmission at high frequency and the mitochondria at synaptic boutons showed improved membrane polarization. In contrast, expression of C. intestinalis alternative oxidase, a protein which bypasses CIII and CIV function, failed to rescue any of the pink1 phenotypes. These results are consistent with the perspective of a selective role for PINK1 in CI, as first suggested by Morais et al., 2009. It should be noted that PINK1-null zebrafish uniquely exhibit a significant CIII deficiency, in addition to a CI defect (Flinn et al., 2014). However, rescue with alternative ETC enzymes has not been conducted to dissect the role of PINK1-related CI and CIII deficiency in this model system.
The hypothesis for a selective role for PINK1 in CI function was further supported by the identification that knockdown of ND42 in Drosophila cells, encoding the CI soluble subunit NDUFA10, phenocopied pink1 knockdown in Drosophila cells (Pogson et al., 2014). Overexpression of ND42 was able to partially rescue pink1 locomotor defects and mitochondrial integrity, while knockdown of SICILY (encoding a CI chaperone required for NDUFA10 stability) also phenocopied pink1 mutants. Indeed, overexpression of either ND42 or SICILY was capable of fully rescuing the CI enzymatic defect in pink1 Drosophila mutants. A comparative phospho-proteomic analysis of WT and Pink1-null mouse brain and liver tissues revealed a lack of NDUFA10 phosphorylation at Ser250 upon loss of PINK1, implying a PINK1-dependent phosphorylation of NDUFA10 (Morais et al., 2014). The relevance of NDUFA10 phosphorylation was further supported by the capacity of a phosphomimetic S250D variant of NDUFA10, but not the S250A variant, to restore mitochondrial membrane potential and rotenone-sensitive CI activity in Pink1−/− mouse embryonic fibroblasts (MEFs) and Pink1−/− MEFs expressing PD-related PINK1 variants or a kinase-inactive PINK1 variant. Interestingly, conflicting results have been reported regarding NDUFA10 S250A rescue of pink1 Drosophila mutants, wherein one group observed a lack of CI rescue in Pink1−/− mutants by the S250A variant (Morais et al., 2014), whereas another observed WT levels of CI activity with the S250A variant (Pogson et al., 2014). While a role for direct phosphorylation of NDUFA10 by PINK1 kinase activity is indicated by this work, it is yet to be demonstrated. At this stage, we cannot rule out the possibility that NDUFA10 is instead phosphorylated by a different kinase whose activity is dependent on PINK1. Nonetheless, the totality of these data from multiple groups and model systems supports a unique role for the fPD gene product PINK1 in the normal maintenance of CI activity.
The loss of Parkin, encoded by the PARK2 gene, has yielded intriguing results with respect to CI deficiency. Mutations in PARK2 lead to autosomal recessive early-onset parkinsonism characterized by nigral degeneration, in many cases without the development of Lewy bodies (Johansen et al., 2018; Kitada et al., 1998). This manifestation would be ideal for the study of mitochondrial CI as a putative cause of nigral degeneration without an α-synuclein component. However, the few CI analyses conducted on PARK2 patients are limited to peripheral tissues. While one study of immortalized lymphocytes failed to observe any differences in NADH dehydrogenase activity of immunocaptured CI (Ming et al., 2020), another study of 10 PARK2 patients reported a decrease in rotenone-sensitive CI activity in leukocytes (Muftuoglu et al., 2004). In addition, studies from three independent groups on Parkin-deficient patient fibroblasts, encompassing nine patients compared to nine controls, observed a decrease in rotenone-sensitive CI activity (Ferretta et al., 2014; Mortiboys et al., 2008, 2013; Pacelli et al., 2011). This CI defect was reproduced in parkin-deficient zebrafish embryos (Flinn et al., 2009), but could not be recapitulated in PARK2-null mice striatum, midbrain, and cortex mitochondria (Damiano et al., 2014). The inability to reproduce CI deficiency as a consequence of Parkin loss across model systems highlights the necessity of comprehensive studies on CI function in post-mortem nigral tissues and other primary tissues from Parkin-deficient fPD patients to determine if CI deficiency correlates with Parkin function.
While the minimal studies of patient fibroblasts with mutations in other PD-related genes, such as LRRK2 and PARK7 (DJ-1), may imply a CI defect (Di Nottia et al., 2017; Grunewald et al., 2014), direct CI activity measurements from fPD patient tissues are lacking. If CI deficiency is an underlying feature of nigral cell loss, it is reasonable to expect a CI defect in primary tissues such as SN, muscle, and platelets of patients with genetic defects in known PD genes. However, such detailed analyses of rotenone-sensitive CI activity and assembly in post-mortem brain and peripheral tissues from fPD patients is currently lacking in the field. A systematic comparative analysis of CI activities in different primary tissues such as SN neurons, platelets, and skeletal muscle of fPD patients with mutations in each PD-related gene will be essential to determine the contribution of CI function in the etiology of all types of PD. This methodical undertaking needs to be bolstered with uniformly employed considerations including early- vs late-onset PD, disease stage and severity, tissue preparation, mitochondrial isolation, and standardized rotenone-sensitive CI activity assays. Furthermore, a study of CI function and cell survival in patient-derived iPSCs, before and after differentiation into cortical and DA neurons, would provide valuable information regarding the importance of CI dysfunction in DA neuronal loss. Once a consistent correlation between CI deficiency and specific nigral cell loss has been established, future investigations can be directed towards understanding its upstream and/or downstream effects such as changes in mitochondrial biogenesis, CI enzyme or subunit turnover/stability, and post-translational modifications of CI that may contribute towards a bioenergetic deficit and the unique vulnerability of DA neurons in PD.
7. Conclusion
Invaluable discoveries of new mitochondrial pathways in both health and disease have been driven by the seminal observation of parkinsonism caused by human exposure to the mitochondrial CI inhibitor, MPP+ (Langston et al., 1983). What remains enigmatic is whether iPD, or even rare forms of fPD, are caused by selective deficiencies in this enzyme (Fig. 2). While an oft-relied upon tool for inducing experimental parkinsonism, the observation of MPP+-induced nigral neuronal loss in mice lacking CI (Choi et al., 2008) raises questions about the nature of even the oldest established relationship between CI and DA neuron loss. Additionally, environmental exposure to non-CI inhibitors, such as paraquat (Bastias-Candia et al., 2019; Zeng et al., 2018), is also known to cause PD further divorcing CI inhibition from PD epidemiology. The fact that parkinsonism/unique nigral involvement is the exception, not the rule, in isolated CI genetic disorders further clouds the issue. The historical landscape of the PD field consistently endorses an involvement of mitochondrial dysfunction, but only through the prism of broad defects not always confined to or even involving CI. A recent example of this was just reported, where a mutation in a gene encoding a CIII subunit was associated with an autosomal dominant parkinsonism with polyneuropathy (Lin et al., 2020). This further highlights the more generalized link between mitochondrial dysfunction and nigral cell loss, but without a unique or requisite change in CI activity.
Fig. 2.
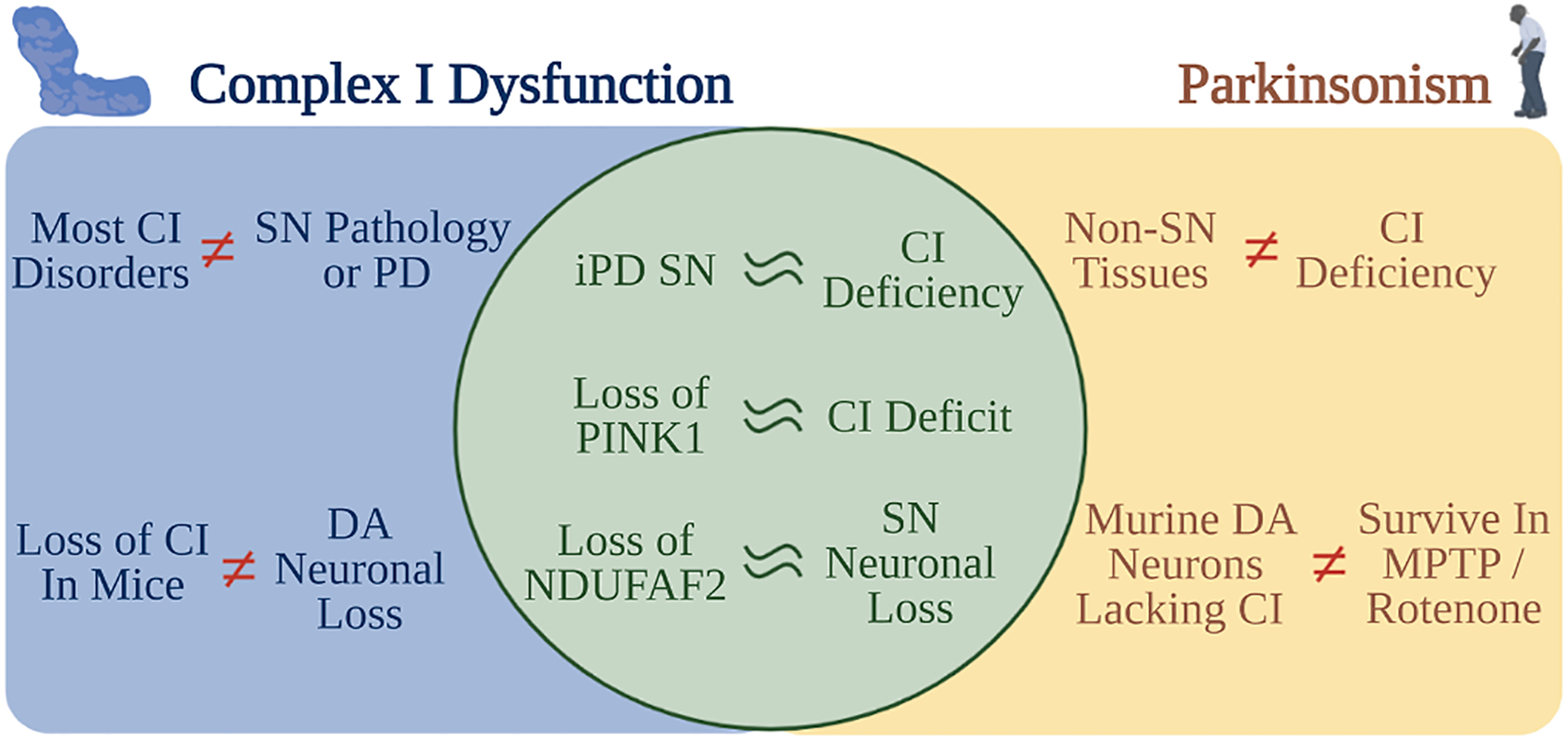
The role of CI activity in selective nigral vulnerability and PD remains elusive. Evidences that support or refute a possible role for CI dysfunction in the etiology of PD as discussed in this manuscript are depicted here. Phenotypes that overlap with CI dysfunction and PD are shown in the central circle. Further empirical evidence is necessary to determine if CI dysfunction occurs upstream or downstream of the pathways that lead to the development of PD, and how important it may be to the manifestation of disease. Created with Biorender.com.
Post-mortem studies have suggested a CI deficiency in the SN of iPD patients, the site of the most severe neuropathology (Figs. 1 and 2). A meta-analysis of these studies has endorsed the correlation between PD and CI deficiency in the SN (Holper et al., 2019). A central question moving forward is whether this association is causal in nature, or a consequence of the severity of the disease in this brain region. If it were causal in nature, it is reasonable to expect that a CI defect would be observed in other tissues, as well. There are conflicting results regarding CI deficiency in other brain regions and peripheral tissues of PD patients (Fig. 1), which may be indicative of a consequence of severe neuronal loss and not pathogenesis. It is also to be noted that subjects with known mutations in CI-related genes typically do not suffer from selective nigral pathology but do display profoundly heterogenous phenotypes and do not always manifest comparable CI deficiency in every tissue. This adds an additional layer of complexity in ascertaining whether CI deficiency could be a driving factor in nigral neurodegeneration. A detailed analysis of ETC complex function in idiopathic, sporadic, and familial PD, compared to CI-deficient patient tissues is required to understand the degree of CI deficiency in PD in comparison to bona fide CI disorders. Specifically, it would be informative to analyze the postmortem SNpc CI activity and degree of neuronal loss in patients with isolated CI disorders. In addition, a systematic study of CI disorder patient-derived iPSCs and DA neuronal cell culture would be beneficial to determine the unique vulnerability of nigral neurons to CI dysfunction. Another avenue for future consideration of the CI hypothesis is to assess whether sub-clinical CI mutations increase the risk for PD. While patients with autosomal recessive CI mutations may fail to display classic parkinsonism, it would be valuable to explore whether heterozygous carriers of these mutations exhibit a selective increased risk for PD, or other neurodegenerative diseases.
8. Thinking beyond Parkinson’s disease
Interestingly, mitochondrial ETC deficits have been observed in other neurodegenerative disorders. Broad mitochondrial dysfunction (Monzio Compagnoni et al., 2020; Wang et al., 2020), including CI deficits (Adav et al., 2019; Holper et al., 2019; Kim et al., 2001; Manczak et al., 2004), have been associated with Alzheimer’s disease. Selective disruption of CI activity has been linked to TDP-43 dependent toxicity implicated in Amyotrophic Lateral Sclerosis (Wang et al., 2019). In a study led by the Heiman lab at MIT, we recently showed that alterations in CI transcript levels were over-represented amongst disease-linked changes in Huntington’s disease (HD) patients and multiple HD mouse models (Lee et al., 2020). Furthermore, cell-type specific profiling revealed these changes to be unique within the neurons most affected in HD, thereby corresponding to tissue-specific changes in CI activity. These findings, and others, may indicate a critical role for intact CI activity in healthy aging of the human brain, whereby genetic or environmental challenges to CI function may broadly affect the risk for adult-onset neurodegenerative diseases. Certainly, the basic and translational value of a full understanding of this large and intricate protein complex continues to grow.
Acknowledgements
We thank Adamantios Mamais, Rebecca Wallings, Mary Herrick, Lauren Laboissonniere, Aimee Aylward, and Dwindy Gerbier for the critical reading of the manuscript.
Funding
The authors would like to acknowledge the financial support of the NIH grants (NS065013 and NS110188), the Michael J. Fox Foundation, and the Parkinson’s Foundation for the authorship of this article.
Abbreviations:
- CI
Complex I (NADH: ubiquinone oxidoreductase)
- CII
complex II (succinate: ubiquinone oxidoreductase)
- CIII
complex III (ubiquinol: cytochrome c oxidoreductase)
- CIV
complex IV (cytochrome c oxidase)
- CI+III activity
NADH: cytochrome c oxidoreductase activity
- CII+III activity
succinate: cytochrome c oxidoreductase activity
- DA
dopaminergic
- [3H]DHR
[3H]dihydrorotenone
- ETC
electron transport chain
- Fe-S cluster
iron-sulfur cluster
- fPD
familial Parkinson’s disease
- iPD
idiopathic Parkinson’s disease
- MELAS
Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like syndrome
- mtDNA
mitochondrial DNA
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MPP+
1-methyl-4-phenylpyridinium
- OXPHOS
oxidative phosphorylation
- PD
Parkinson’s disease
- PINK1
PTEN-induced kinase 1
- SN
substantia nigra
- SNpc
SN pars compacta
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Adav SS, Park JE, Sze SK, 2019. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol Brain. 12, 8. 10.1186/s13041-019-0430-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adjobo-Hermans MJW, de Haas R, Willems P, Wojtala A, van Emst-de Vries SE, Wagenaars JA, van den Brand M, Rodenburg RJ, Smeitink JAM, Nijtmans LG, Sazanov LA, Wieckowski MR, Koopman WJH, 2020. NDUFS4 deletion triggers loss of NDUFA12 in Ndufs4(−/−) mice and Leigh syndrome patients: A stabilizing role for NDUFAF2. Biochim. Biophys. Acta, Bioenerg 1861, 148213. [DOI] [PubMed] [Google Scholar]
- Alexeyev M, Shokolenko I, Wilson G, LeDoux S, 2013. The Maintenance of Mitochondrial DNA Integrity-Critical Analysis and Update. Cold Spring Harbor Perspectives in Biology. 10.1101/cshperspect.a012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosi G, Ghezzi C, Sepe S, Milanese C, Payan-Gomez C, Bombardieri CR, Armentero MT, Zangaglia R, Pacchetti C, Mastroberardino PG, Blandini F, 2014. Bioenergetic and proteolytic defects in fibroblasts from patients with sporadic Parkinson’s disease. Biochim. Biophys. Acta 1842, 1385–1394. [DOI] [PubMed] [Google Scholar]
- Anderson JJ, Bravi D, Ferrari R, Davis TL, Baronti F, Chase TN, Dagani F, 1993. No evidence for altered muscle mitochondrial function in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 56, 477–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG, 1981. Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N, 1999. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet 23, 147. 10.1038/13779. [DOI] [PubMed] [Google Scholar]
- Barghuti F, Elian K, Gomori JM, Shaag A, Edvardson S, Saada A, Elpeleg O, 2008. The unique neuroradiology of complex I deficiency due to NDUFA12L defect. Mol. Genet. Metab 94, 78–82. [DOI] [PubMed] [Google Scholar]
- Barroso N, Campos Y, Huertas R, Esteban J, Molina JA, Alonso A, Gutierrez-Rivas E, Arenas J, 1993. Respiratory chain enzyme activities in lymphocytes from untreated patients with Parkinson disease. Clin. Chem 39, 667–669. [PubMed] [Google Scholar]
- Bastias-Candia S, Zolezzi JM, Inestrosa NC, 2019. Revisiting the paraquat-induced sporadic Parkinson’s disease-like model. Mol. Neurobiol 56, 1044–1055. [DOI] [PubMed] [Google Scholar]
- Benecke R, Strumper P, Weiss H, 1993. Electron transfer complexes I and IV of platelets are abnormal in Parkinson’s disease but normal in Parkinson-plus syndromes. Brain 116 (Pt 6), 1451–1463. [DOI] [PubMed] [Google Scholar]
- Benit P, Beugnot R, Chretien D, Giurgea I, De Lonlay-Debeney P, Issartel JP, Corral-Debrinski M, Kerscher S, Rustin P, Rotig A, Munnich A, 2003. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum. Mutat 21, 582–586. 10.1002/humu.10225. [DOI] [PubMed] [Google Scholar]
- Berardelli A, Wenning GK, Antonini A, Berg D, Bloem BR, Bonifati V, Brooks D, Burn DJ, Colosimo C, Fanciulli A, Ferreira J, Gasser T, Grandas F, Kanovsky P, Kostic V, Kulisevsky J, Oertel W, Poewe W, Reese JP, Relja M, Ruzicka E, Schrag A, Seppi K, Taba P, Vidailhet M, 2013. EFNS/MDS-ES/ENS [corrected] recommendations for the diagnosis of Parkinson’s disease. Eur. J. Neurol 20, 16–34. [DOI] [PubMed] [Google Scholar]
- Berrisford JM, Baradaran R, Sazanov LA, 2016. Structure of bacterial respiratory complex I. BBA 1857, 892–901. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT, 2000. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci 3, 1301–1306. 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Betts-Henderson J, Jaros E, Krishnan KJ, Perry RH, Reeve AK, Schaefer AM, Taylor RW, Turnbull DM, 2009. Alpha-synuclein pathology and Parkinsonism associated with POLG1 mutations and multiple mitochondrial DNA deletions. Neuropathol. Appl. Neurobiol 35, 120–124. 10.1111/j.1365-2990.2008.00981.x. [DOI] [PubMed] [Google Scholar]
- Bezard E, Gross CE, Fournier MC, Dovero S, Bloch B, Jaber M, 1999. Absence of MPTP-induced neuronal death in mice lacking the dopamine transporter. Exp. Neurol 155, 268–273. [DOI] [PubMed] [Google Scholar]
- Bindoff LA, Birch-Machin M, Cartlidge NE, Parker WD Jr., Turnbull DM, 1989. Mitochondrial function in Parkinson’s disease. Lancet 2, 49. [DOI] [PubMed] [Google Scholar]
- Bindoff LA, Birch-Machin MA, Cartlidge NE, Parker WD Jr., Turnbull DM, 1991. Respiratory chain abnormalities in skeletal muscle from patients with Parkinson’s disease. J. Neurol. Sci 104, 203–208. [DOI] [PubMed] [Google Scholar]
- Birrell JA, Morina K, Bridges HR, Friedrich T, Hirst J, 2013. Investigating the function of [2Fe-2S] cluster N1a, the off-pathway cluster in complex I, by manipulating its reduction potential. Biochem. J 456, 139–146. 10.1042/BJ20130606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake CI, Spitz E, Leehey M, Hoffer BJ, Boyson SJ, 1997. Platelet mitochondrial respiratory chain function in Parkinson’s disease. Mov. Disord 12, 3–8. [DOI] [PubMed] [Google Scholar]
- Blandini F, Nappi G, Greenamyre JT, 1998. Quantitative study of mitochondrial complex I in platelets of parkinsonian patients. Mov. Disord 13, 11–15. [DOI] [PubMed] [Google Scholar]
- Blin O, Desnuelle C, Rascol O, Borg M, Peyro Saint Paul H, Azulay JP, Bille F, Figarella D, Coulom F, Pellissier JF, et al. , 1994. Mitochondrial respiratory failure in skeletal muscle from patients with Parkinson’s disease and multiple system atrophy. J. Neurol. Sci 125, 95–101. 10.1016/0022-510x(94)90248-8. [DOI] [PubMed] [Google Scholar]
- Bose A, Beal MF, 2016. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem 139 (Suppl 1), 216–231. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, 2009. Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv. Anat. Embryol. Cell Biol 201, 1–119. [PubMed] [Google Scholar]
- Bravi D, Anderson JJ, Dagani F, Davis TL, Ferrari R, Gillespie M, Chase TN, 1992. Effect of aging and dopaminomimetic therapy on mitochondrial respiratory function in Parkinson’s disease. Mov. Disord 7, 228–231. [DOI] [PubMed] [Google Scholar]
- Breen DP, Munoz DG, Lang AE, 2020. Twinkle-associated familial parkinsonism with Lewy pathology: cause or predisposition? Neurology 95, 644–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronstein JM, Paul K, Yang L, Haas RH, Shults CW, Le T, Ritz B, 2015. Platelet mitochondrial activity and pesticide exposure in early Parkinson’s disease. Mov. Disord 30, 862–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE, O’Malley K, 2013. Axon degeneration in Parkinson’s disease. Exp. Neurol 246, 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ, 1983. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc. Natl. Acad. Sci. U.S.A 80, 4546–4550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Tucker EJ, Compton AG, Kirby DM, Crawford G, Burtt NP, Rivas M, Guiducci C, Bruno DL, Goldberger OA, Redman MC, Wiltshire E, Wilson CJ, Altshuler D, Gabriel SB, Daly MJ, Thorburn DR, Mootha VK, 2010. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet 42, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron JM, MacKay N, Feigenbaum A, Tarnopolsky M, Blaser S, Robinson BH, Schulze A, 2015. Exome sequencing identifies complex I NDUFV2 mutations as a novel cause of Leigh syndrome. Eur. J. Paediatr. Neurol [DOI] [PubMed] [Google Scholar]
- Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT, 2009. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol. Dis 34, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappelletti G, Pedrotti B, Maggioni MG, Maci R, 2001. Microtubule assembly is directly affected by MPP(+)in vitro. Cell Biol. Int 25, 981–984. [DOI] [PubMed] [Google Scholar]
- Cappelletti G, Surrey T, Maci R, 2005. The parkinsonism producing neurotoxin MPP + affects microtubule dynamics by acting as a destabilising factor. FEBS Lett. 579, 4781–4786. [DOI] [PubMed] [Google Scholar]
- Cardellach F, Marti MJ, Fernandez-Sola J, Marin C, Hoek JB, Tolosa E, Urbano-Marquez A, 1993. Mitochondrial respiratory chain activity in skeletal muscle from patients with Parkinson’s disease. Neurology 43, 2258–2262. 10.1212/wnl.43.11.2258. [DOI] [PubMed] [Google Scholar]
- Cardol P, 2011. Mitochondrial NADH:ubiquinone oxidoreductase (complex I) in eukaryotes: a highly conserved subunit composition highlighted by mining of protein databases. Biochim. Biophys. Acta 1807, 1390–1397. [DOI] [PubMed] [Google Scholar]
- Carling PJ, Mortiboys H, Green C, Mihaylov S, Sandor C, Schwartzentruber A, Taylor R, Wei W, Hastings C, Wong S, Lo C, Evetts S, Clemmens H, Wyles M, Willcox S, Payne T, Hughes R, Ferraiuolo L, Webber C, Hide W, Wade-Martins R, Talbot K, Hu MT, Bandmann O, 2020. Deep phenotyping of peripheral tissue facilitates mechanistic disease stratification in sporadic Parkinson’s disease. Prog. Neurobiol 187, 101772. [DOI] [PubMed] [Google Scholar]
- Chaudhuri KR, Healy DG, Schapira AH, National Institute for Clinical, E., 2006. Nonmotor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 5, 235–245. 10.1016/S1474-4422(06)70373-8. [DOI] [PubMed] [Google Scholar]
- Chen C, Turnbull DM, Reeve AK, 2019. Mitochondrial dysfunction in Parkinson’s disease-cause or consequence? Biology (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Vincent AE, Blain AP, Smith AL, Turnbull DM, Reeve AK, 2020. Investigation of mitochondrial biogenesis defects in single substantia nigra neurons using post-mortem human tissues. Neurobiol. Dis 134, 104631. [DOI] [PubMed] [Google Scholar]
- Cheng HC, Ulane CM, Burke RE, 2010. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol 67, 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WS, Kruse SE, Palmiter RD, Xia Z, 2008. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc. Natl. Acad. Sci. U.S.A 105, 15136–15141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WS, Palmiter RD, Xia Z, 2011. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. J. Cell Biol 192, 873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WS, Kim HW, Tronche F, Palmiter RD, Storm DR, Xia Z, 2017. Conditional deletion of Ndufs4 in dopaminergic neurons promotes Parkinson’s disease-like non-motor symptoms without loss of dopamine neurons. Sci. Rep 7, 44989. 10.1038/srep44989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JM, Daniel SE, Marsden CD, Schapira AH, 1995. L-dihydroxyphenylalanine and complex I deficiency in Parkinson’s disease brain. Mov. Disord 10, 295–297. [DOI] [PubMed] [Google Scholar]
- Damiano M, Gautier CA, Bulteau AL, Ferrando-Miguel R, Gouarne C, Paoli MG, Pruss R, Auchere F, L’Hermitte-Stead C, Bouillaud F, Brice A, Corti O, Lombes A, 2014. Tissue- and cell-specific mitochondrial defect in Parkin-deficient mice. PLoS ONE 9, e99898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM, 2006. Epidemiology of Parkinson’s disease. Lancet Neurol. 5, 525–535. [DOI] [PubMed] [Google Scholar]
- de Vries RL, Przedborski S, 2013. Mitophagy and Parkinson’s disease: be eaten to stay healthy. Mol. Cell. Neurosci 55, 37–43. [DOI] [PubMed] [Google Scholar]
- del Hoyo P, Garcia-Redondo A, de Bustos F, Molina JA, Sayed Y, Alonso-Navarro H, Caballero L, Arenas J, Agundez JA, Jimenez-Jimenez FJ, 2010. Oxidative stress in skin fibroblasts cultures from patients with Parkinson’s disease. BMC Neurol. 10, 95. 10.1186/1471-2377-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nottia M, Masciullo M, Verrigni D, Petrillo S, Modoni A, Rizzo V, Di Giuda D, Rizza T, Niceta M, Torraco A, Bianchi M, Santoro M, Bentivoglio AR, Bertini E, Piemonte F, Carrozzo R, Silvestri G, 2017. DJ-1 modulates mitochondrial response to oxidative stress: clues from a novel diagnosis of PARK7. Clin. Genet 92, 18–25. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I, 2009. Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol. 8, 1150–1157. [DOI] [PubMed] [Google Scholar]
- DiDonato S, Zeviani M, Giovannini P, Savarese N, Rimoldi M, Mariotti C, Girotti F, Caraceni T, 1993. Respiratory chain and mitochondrial DNA in muscle and brain in Parkinson’s disease patients. Neurology 43, 2262–2268. 10.1212/wnl.43.11.2262. [DOI] [PubMed] [Google Scholar]
- Distelmaier F, Koopman WJH, van den Heuvel LP, Rodenburg RJ, Mayatepek E, Willems PHGM, Smeitink JAM, 2009. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain 132, 833–842. 10.1093/brain/awp058. [DOI] [PubMed] [Google Scholar]
- Fassone E, Rahman S, 2012. Complex I deficiency: clinical features, biochemistry and molecular genetics. J. Med. Genet 49, 578–590. [DOI] [PubMed] [Google Scholar]
- Ferretta A, Gaballo A, Tanzarella P, Piccoli C, Capitanio N, Nico B, Annese T, Di Paola M, Dell’aquila C, De Mari M, Ferranini E, Bonifati V, Pacelli C, Cocco T, 2014. Effect of resveratrol on mitochondrial function: implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta 1842, 902–915. [DOI] [PubMed] [Google Scholar]
- Fiedorczuk K, Sazanov LA, 2018. Mammalian mitochondrial complex I structure and disease-causing mutations. Trends Cell Biol. 28, 835–867. [DOI] [PubMed] [Google Scholar]
- Filosto M, Tomelleri G, Tonin P, Scarpelli M, Vattemi G, Rizzuto N, Padovani A, Simonati A, 2007. Neuropathology of mitochondrial diseases. Biosci. Rep 27, 23–30. [DOI] [PubMed] [Google Scholar]
- Flinn L, Mortiboys H, Volkmann K, Koster RW, Ingham PW, Bandmann O, 2009. Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 132, 1613–1623. [DOI] [PubMed] [Google Scholar]
- Flones IH, Fernandez-Vizarra E, Lykouri M, Brakedal B, Skeie GO, Miletic H, Lilleng PK, Alves G, Tysnes OB, Haugarvoll K, Dolle C, Zeviani M, Tzoulis C, 2018. Neuronal complex I deficiency occurs throughout the Parkinson’s disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol. 135, 409–425. [DOI] [PubMed] [Google Scholar]
- Formosa LE, Mimaki M, Frazier AE, McKenzie M, Stait TL, Thorburn DR, Stroud DA, Ryan MT, 2015. Characterization of mitochondrial FOXRED1 in the assembly of respiratory chain complex I. Hum. Mol. Genet 24, 2952–2965. [DOI] [PubMed] [Google Scholar]
- Friederich MW, Erdogan AJ, Coughlin CR 2nd, Elos MT, Jiang H, O’Rourke CP, Lovell MA, Wartchow E, Gowan K, Chatfield KC, Chick WS, Spector EB, Van Hove JLK, Riemer J, 2017. Mutations in the accessory subunit NDUFB10 result in isolated complex I deficiency and illustrate the critical role of intermembrane space import for complex I holoenzyme assembly. Hum. Mol. Genet 26, 702–716. 10.1093/hmg/ddw431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Fumagalli F, Jones SR, Caron MG, 1997. Dopamine transporter is required for in vivo MPTP neurotoxicity: evidence from mice lacking the transporter. J. Neurochem 69, 1322–1325. 10.1046/j.1471-4159.1997.69031322.x. [DOI] [PubMed] [Google Scholar]
- Galkin A, Dröse S, Brandt U, 2006. The proton pumping stoichiometry of purified mitochondrial complex I reconstituted into proteoliposomes. Biochim. Biophys. Acta 1757, 1575–1581. [DOI] [PubMed] [Google Scholar]
- Gatt AP, Duncan OF, Attems J, Francis PT, Ballard CG, Bateman JM, 2016. Dementia in Parkinson’s disease is associated with enhanced mitochondrial complex I deficiency. Mov. Disord 31, 352–359. [DOI] [PubMed] [Google Scholar]
- Gautier CA, Kitada T, Shen J, 2008. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. U.S.A 105, 11364–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaloul-Gonzalez Lina, Goldstein Amy, Vockley Walsh, Catherine Dobrowolski, Steven Biery, Amy Irani, Afifa Ibarra, Jordan Morton, D Holmes Mohsen, Al-Walid Vockley, Jerry, 2016. Mitochondrial respiratory chain disorders in the Old Order Amish population. Molecular Genetics and Metabolism 118 (4), 296–303. 10.1016/j.ymgme.2016.06.005. [DOI] [PubMed] [Google Scholar]
- Giachin G, Bouverot R, Acajjaoui S, Pantalone S, Soler-Lopez M, 2016. Dynamics of human mitochondrial complex I assembly: implications for neurodegenerative diseases. Front. Mol. Biosci 3, 43. 10.3389/fmolb.2016.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giguere N, Burke Nanni S, Trudeau LE, 2018. On cell loss and selective vulnerability of neuronal populations in Parkinson’s disease. Front. Neurol 9, 455. 10.3389/fneur.2018.00455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald A, Arns B, Meier B, Brockmann K, Tadic V, Klein C, 2014. Does uncoupling protein 2 expression qualify as marker of disease status in LRRK2-associated Parkinson’s disease? Antioxid. Redox Signal 20, 1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald A, Rygiel KA, Hepplewhite PD, Morris CM, Picard M, Turnbull DM, 2016. Mitochondrial DNA depletion in respiratory chain-deficient Parkinson disease neurons. Ann. Neurol 79, 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Cooper JM, Taanman JW, Schapira AH, 1998. Mitochondrial DNA transmission of the mitochondrial defect in Parkinson’s disease. Ann. Neurol 44, 177–186. [DOI] [PubMed] [Google Scholar]
- Guerrero-Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, Nijtmans L, 2017. The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. 25, 128–139. [DOI] [PubMed] [Google Scholar]
- Haas RH, Nasirian F, Nakano K, Ward D, Pay M, Hill R, Shults CW, 1995. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol 37, 714–722. [DOI] [PubMed] [Google Scholar]
- Hanagasi HA, Serdaroglu P, Ozansoy M, Basak N, Tasli H, Emre M, 2009. Mitochondrial pathology in muscle of a patient with a novel parkin mutation. Int. J. Neurosci 119, 1572–1583. [DOI] [PubMed] [Google Scholar]
- Hattori N, Tanaka M, Ozawa T, Mizuno Y, 1991. Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Ann. Neurol 30, 563–571. [DOI] [PubMed] [Google Scholar]
- Hattori N, Yoshino H, Tanaka M, Suzuki H, Mizuno Y, 1998. Genotype in the 24-kDa subunit gene (NDUFV2) of mitochondrial complex I and susceptibility to Parkinson disease. Genomics 49, 52–58. [DOI] [PubMed] [Google Scholar]
- Heikkila RE, Cabbat FS, Manzino L, Duvoisin RC, 1984a. Effects of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine on neostriatal dopamine in mice. Neuropharmacology 23, 711–713. [DOI] [PubMed] [Google Scholar]
- Heikkila RE, Hess A, Duvoisin RC, 1984b. Dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine in mice. Science 224, 1451–1453. 10.1126/science.6610213. [DOI] [PubMed] [Google Scholar]
- Heikkila RE, Manzino L, Cabbat FS, Duvoisin RC, 1984c. Protection against the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine by monoamine oxidase inhibitors. Nature 311, 467–469. [DOI] [PubMed] [Google Scholar]
- Herzer M, Koch J, Prokisch H, Rodenburg R, Rauscher C, Radauer W, Forstner R, Pilz P, Rolinski B, Freisinger P, Mayr JA, Sperl W, 2010. Leigh disease with brainstem involvement in complex I deficiency due to assembly factor NDUFAF2 defect. Neuropediatrics 41, 30–34. [DOI] [PubMed] [Google Scholar]
- Hirst J, 2013. Mitochondrial complex I. Annu. Rev. Biochem 82, 551–575. [DOI] [PubMed] [Google Scholar]
- Hoefs SJ, Dieteren CE, Rodenburg RJ, Naess K, Bruhn H, Wibom R, Wagena E, Willems PH, Smeitink JA, Nijtmans LG, van den Heuvel LP, 2009. Baculovirus complementation restores a novel NDUFAF2 mutation causing complex I deficiency. Hum. Mutat 30, E728–E736. [DOI] [PubMed] [Google Scholar]
- Hoepken HH, Gispert S, Morales B, Wingerter O, Del Turco D, Mulsch A, Nussbaum RL, Muller K, Drose S, Brandt U, Deller T, Wirth B, Kudin AP, Kunz WS, Auburger G, 2007. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis 25, 401–411. [DOI] [PubMed] [Google Scholar]
- Holper L, Ben-Shachar D, Mann JJ, 2019. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 44, 837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetzky B, Hauck S, Youdim MB, Riederer P, Jellinger K, Pantucek F, Zochling R, Boissl KW, Reichmann H, 1994. Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson’s disease. Neurosci. Lett 169, 126–128. [DOI] [PubMed] [Google Scholar]
- Janssen RJ, Distelmaier F, Smeets R, Wijnhoven T, Ostergaard E, Jaspers NG, Raams A, Kemp S, Rodenburg RJ, Willems PH, van den Heuvel LP, Smeitink JA, Nijtmans LG, 2009. Contiguous gene deletion of ELOVL7, ERCC8 and NDUFAF2 in a patient with a fatal multisystem disorder. Hum. Mol. Genet 18, 3365–3374. [DOI] [PubMed] [Google Scholar]
- Johansen KK, Torp SH, Farrer MJ, Gustavsson EK, Aasly JO, 2018. A case of Parkinson’s disease with no lewy body pathology due to a homozygous exon deletion in Parkin. Case Rep. Neurol. Med 2018, 6838965. 10.1155/2018/6838965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney PM, Xie J, Capaldi RA, Bennett JP Jr., 2006. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci 26, 5256–5264. 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HW, Choi WS, Sorscher N, Park HJ, Tronche F, Palmiter RD, Xia Z, 2015. Genetic reduction of mitochondrial complex I function does not lead to loss of dopamine neurons in vivo. Neurobiol. Aging 36, 2617–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Vlkolinsky R, Cairns N, Fountoulakis M, Lubec G, 2001. The reduction of NADH ubiquinone oxidoreductase 24- and 75-kDa subunits in brains of patients with Down syndrome and Alzheimer’s disease. Life Sci. 68, 2741–2750. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N, 1998. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Kmita K, Zickermann V, 2013. Accessory subunits of mitochondrial complex I. Biochem. Soc. Trans 41, 1272–1279. 10.1042/BST20130091. [DOI] [PubMed] [Google Scholar]
- Koene S, Rodenburg RJ, van der Knaap MS, Willemsen MAAP, Sperl W, Laugel V, Ostergaard E, Tarnopolsky M, Martin MA, Nesbitt V, Fletcher J, Edvardson S, Procaccio V, Slama A, van den Heuvel LPWJ, Smeitink JAM, 2012. Natural disease course and genotype-phenotype correlations in Complex I deficiency caused by nuclear gene defects: what we learned from 130 cases. J. Inherit. Metab. Dis 35, 737–747. 10.1007/s10545-012-9492-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolata G, 1983. Monkey model of Parkinson’s disease. Science 220, 705. 10.1126/science.6403987. [DOI] [PubMed] [Google Scholar]
- Koopman WJ, Beyrath J, Fung CW, Koene S, Rodenburg RJ, Willems PH, Smeitink JA, 2016. Mitochondrial disorders in children: toward development of small-molecule treatment strategies. EMBO Mol. Med 8, 311–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krige D, Carroll MT, Cooper JM, Marsden CD, Schapira AH, 1992. Platelet mitochondrial function in Parkinson’s disease. The Royal Kings and Queens Parkinson Disease Research Group. Ann. Neurol 32, 782–788. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I, 1983. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard PA Jr., 1983. Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med 309, 310. 10.1056/nejm198308043090511. [DOI] [PubMed] [Google Scholar]
- Langston JW, 2017. The MPTP Story. J. Parkinsons Dis 7, S11–S19. 10.3233/JPD-179006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebre AS, Rio M, Faivre d’Arcier L, Vernerey D, Landrieu P, Slama A, Jardel C, Laforet P, Rodriguez D, Dorison N, Galanaud D, Chabrol B, Paquis-Flucklinger V, Grevent D, Edvardson S, Steffann J, Funalot B, Villeneuve N, Valayannopoulos V, de Lonlay P, Desguerre I, Brunelle F, Bonnefont JP, Rotig A, Munnich A, Boddaert N, 2011. A common pattern of brain MRI imaging in mitochondrial diseases with complex I deficiency. J. Med. Genet 48, 16–23. [DOI] [PubMed] [Google Scholar]
- Lee H, Fenster RJ, Pineda SS, Gibbs WS, Mohammadi S, Davila-Velderrain J, Garcia FJ, Therrien M, Novis HS, Gao F, Wilkinson H, Vogt T, Kellis M, LaVoie MJ, Heiman M, 2020. Cell type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron 107, 891-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch K-P, Selch S, Renner TJ, Jacob C, Nguyen TT, Hahn T, Romanos M, Walitza S, Shoichet S, Dempfle A, Heine M, Boreatti-Hümmer A, Romanos J, Gross-Lesch S, Zerlaut H, Wultsch T, Heinzel S, Fassnacht M, Fallgatter A, Allolio B, Schäfer H, Warnke A, Reif A, Ropers H-H, Ullmann R, 2011. Genome-wide copy number variation analysis in attention-deficit/hyperactivity disorder: association with neuropeptide Y gene dosage in an extended pedigree. Molecular Psychiatry 16, 491–503. 10.1038/mp.2010.29. [DOI] [PubMed] [Google Scholar]
- Letts JA, Sazanov LA, 2015. Gaining mass: the structure of respiratory complex I — from bacterial towards mitochondrial versions. Curr. Opin. Struct. Biol 33, 135–145. [DOI] [PubMed] [Google Scholar]
- Lewin R, 1984. Trail of ironies to Parkinson’s disease. Science 224, 1083–1085. 10.1126/science.6426059. [DOI] [PubMed] [Google Scholar]
- Lin CH, Tsai PI, Lin HY, Hattori N, Funayama M, Jeon B, Sato K, Abe K, Mukai Y, Takahashi Y, Li Y, Nishioka K, Yoshino H, Daida K, Chen ML, Cheng J, Huang CY, Tzeng SR, Wu YS, Lai HJ, Tsai HH, Yen RF, Lee NC, Lo WC, Hung YC, Chan CC, Ke YC, Chao CC, Hsieh ST, Farrer M, Wu RM, 2020. Mitochondrial UQCRC1 mutations cause autosomal dominant parkinsonism with polyneuropathy. Brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HY, Liao PC, Chuang KT, Kao MC, 2011. Mitochondrial targeting of human NADH dehydrogenase (ubiquinone) flavoprotein 2 (NDUFV2) and its association with early-onset hypertrophic cardiomyopathy and encephalopathy. J. Biomed. Sci 18, 29. 10.1186/1423-0127-18-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Park BS, Jung Y, Reddy PH, 2004. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 5, 147–162. 10.1385/NMM:5:2:147. [DOI] [PubMed] [Google Scholar]
- Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD, 1992a. Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson’s disease. Brain 115 (Pt 2), 333–342. [DOI] [PubMed] [Google Scholar]
- Mann VM, Cooper JM, Schapira AH, 1992b. Quantitation of a mitochondrial DNA deletion in Parkinson’s disease. FEBS Lett. 299, 218–222. [DOI] [PubMed] [Google Scholar]
- Mann VM, Cooper JM, Daniel SE, Srai K, Jenner P, Marsden CD, Schapira AH, 1994. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann. Neurol 36, 876–881. [DOI] [PubMed] [Google Scholar]
- Manneschi L, Dotti MT, Battisti C, de Stefano N, Federico A, 1994. Muscle respiratory chain enzyme activities in parkinson’s disease and in multisystem extrapyramidal disorders with parkinsonism as the main clinical feature. Arch. Gerontol. Geriatr 19 (Suppl 1), 155–161. [DOI] [PubMed] [Google Scholar]
- Martin-Jimenez R, Lurette O, Hebert-Chatelain E, 2020. Damage in Mitochondrial DNA Associated with Parkinson’s Disease. DNA Cell Biol. 39, 1421–1430. [DOI] [PubMed] [Google Scholar]
- Martin MA, Molina JA, Jimenez-Jimenez FJ, Benito-Leon J, Orti-Pareja M, Campos Y, Arenas J, 1996. Respiratory-chain enzyme activities in isolated mitochondria of lymphocytes from untreated Parkinson’s disease patients. Grupo-Centro de Trastornos del Movimiento. Neurology 46, 1343–1346. 10.1212/wnl.46.5.1343. [DOI] [PubMed] [Google Scholar]
- Mehta SH, Dickson DW, Morgan JC, Singleton AB, Majounie E, Sethi KD, 2016. Juvenile onset Parkinsonism with “pure nigral” degeneration and POLG1 mutation. Parkinsonism Relat Disord. 30, 83–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming F, Tan J, Qin L, Zhang H, Tang J, Tan X, Wang C, 2020. The PARK2 mutation associated with Parkinson’s disease enhances the vulnerability of peripheral blood lymphocytes to paraquat. Biomed Res. Int 2020, 4658109. 10.1155/2020/4658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Suzuki K, Sone N, Saitoh T, 1988. Inhibition of mitochondrial respiration by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in mouse brain in vivo. Neurosci. Lett 91, 349–353. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Suzuki K, Sone N, Saitoh T, 1987. Inhibition of ATP synthesis by 1-methyl-4-phenylpyridinium ion (MPP+) in isolated mitochondria from mouse brains. Neurosci. Lett 81 (1–2), 204–208. 10.1016/0304-3940(87)90366-1. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y, 1989. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun 163, 1450–1455. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Suzuki K, Ohta S, 1990. Postmortem changes in mitochondrial respiratory enzymes in brain and a preliminary observation in Parkinson’s disease. J. Neurol. Sci 96, 49–57. [DOI] [PubMed] [Google Scholar]
- Monzio Compagnoni G, Di Fonzo A, Corti S, Comi GP, Bresolin N, Masliah E, 2020. The role of mitochondria in neurodegenerative diseases: the lesson from Alzheimer’s disease and Parkinson’s disease. Mol. Neurobiol 57, 2959–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais VA, Verstreken P, Roethig A, Smet J, Snellinx A, Vanbrabant M, Haddad D, Frezza C, Mandemakers W, Vogt-Weisenhorn D, Van Coster R, Wurst W, Scorrano L, De Strooper B, 2009. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med 1, 99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais VA, Haddad D, Craessaerts K, De Bock PJ, Swerts J, Vilain S, Aerts L, Overbergh L, Grunewald A, Seibler P, Klein C, Gevaert K, Verstreken P, De Strooper B, 2014. PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 344, 203–207. [DOI] [PubMed] [Google Scholar]
- Mortiboys H, Thomas KJ, Koopman WJ, Klaffke S, Abou-Sleiman P, Olpin S, Wood NW, Willems PH, Smeitink JA, Cookson MR, Bandmann O, 2008. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol 64, 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortiboys H, Aasly J, Bandmann O, 2013. Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson’s disease. Brain. 136, 3038–3050. [DOI] [PubMed] [Google Scholar]
- Muftuoglu M, Elibol B, Dalmizrak O, Ercan A, Kulaksiz G, Ogus H, Dalkara T, Ozer N, 2004. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov. Disord 19, 544–548. 10.1002/mds.10695. [DOI] [PubMed] [Google Scholar]
- Mythri RB, Venkateshappa C, Harish G, Mahadevan A, Muthane UB, Yasha TC, Srinivas Bharath MM, Shankar SK, 2011. Evaluation of markers of oxidative stress, antioxidant function and astrocytic proliferation in the striatum and frontal cortex of Parkinson’s disease brains. Neurochem. Res 36, 1452–1463. [DOI] [PubMed] [Google Scholar]
- Nakagawa-Hattori Y, Yoshino H, Kondo T, Mizuno Y, Horai S, 1992. Is Parkinson’s disease a mitochondrial disorder? J. Neurol. Sci 107, 29–33. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ, 2008. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol 183, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Walker JE, Youle R, 2012. Mitochondrial quality control mediated by PINK1 and Parkin: links to Parkinsonism. Cold Spring Harb. Perspect. Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ, 2010. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Youle RJ, 2011. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal 14, 1929–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Boveris A, Bandez MJ, Sanchez-Pino MJ, Gomez C, Muntane G, Ferrer I, 2009. Human brain cortex: mitochondrial oxidative damage and adaptive response in Parkinson disease and in dementia with Lewy bodies. Free Radic. Biol. Med 46, 1574–1580. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Vilarino-Guell C, Cobb SA, Kachergus JM, Ross OA, Hentati E, Hentati F, Farrer MJ, 2010. Genetic variation of the mitochondrial complex I subunit NDUFV2 and Parkinson’s disease. Parkinsonism Relat. Disord 16, 686–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie I, Kennaway NG, Shoubridge EA, 2005. A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J. Clin. Invest 115, 2784–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacelli C, De Rasmo D, Signorile A, Grattagliano I, di Tullio G, D’Orazio A, Nico B, Comi GP, Ronchi D, Ferranini E, Pirolo D, Seibel P, Schubert S, Gaballo A, Villani G, Cocco T, 2011. Mitochondrial defect and PGC-1alpha dysfunction in parkin-associated familial Parkinson’s disease. Biochim. Biophys. Acta 1812, 1041–1053. [DOI] [PubMed] [Google Scholar]
- Pagniez-Mammeri H, Loublier S, Legrand A, Benit P, Rustin P, Slama A, 2012a. Mitochondrial complex I deficiency of nuclear origin I. Structural genes. Mol Genet Metab. 105, 163–172. [DOI] [PubMed] [Google Scholar]
- Pagniez-Mammeri H, Rak M, Legrand A, Benit P, Rustin P, Slama A, 2012b. Mitochondrial complex I deficiency of nuclear origin II. Non-structural genes. Mol Genet Metab. 105, 173–179. [DOI] [PubMed] [Google Scholar]
- Palin EJ, Paetau A, Suomalainen A, 2013. Mesencephalic complex I deficiency does not correlate with parkinsonism in mitochondrial DNA maintenance disorders. Brain 136, 2379–2392. [DOI] [PubMed] [Google Scholar]
- Park JS, Davis RL, Sue CM, 2018. Mitochondrial dysfunction in Parkinson’s disease: new mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep 18, 21. 10.1007/s11910-018-0829-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker WD Jr., Frerman F, Haas R, Parks JK, 1988. Myxothiazol resistance in human mitochondria. Biochim. Biophys. Acta 936, 133–138. [DOI] [PubMed] [Google Scholar]
- Parker WD Jr., Boyson SJ, Parks JK, 1989. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann. Neurol 26, 719–723. [DOI] [PubMed] [Google Scholar]
- Parkinson J, 1817. An essay on the shaking palsy. 1817. Whittingham and Rowland for Sherwood, Needly and Jones, London; J Neuropsychiatry Clin Neurosci 14 (2), 223–236. 10.1176/jnp.14.2.223. [DOI] [PubMed] [Google Scholar]
- Peter B, Falkenberg M, 2020. TWINKLE and Other Human Mitochondrial DNA Helicases: Structure, Function and Disease. Genes (Basel) 11. 10.3390/genes11040408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli C, Ripoli M, Quarato G, Scrima R, D’Aprile A, Boffoli D, Margaglione M, Criscuolo C, De Michele G, Sardanelli A, Papa S, Capitanio N, 2008a. Coexistence of mutations in PINK1 and mitochondrial DNA in early onset parkinsonism. J. Med. Genet 45, 596–602. [DOI] [PubMed] [Google Scholar]
- Piccoli C, Sardanelli A, Scrima R, Ripoli M, Quarato G, D’Aprile A, Bellomo F, Scacco S, De Michele G, Filla A, Iuso A, Boffoli D, Capitanio N, Papa S, 2008b. Mitochondrial respiratory dysfunction in familiar parkinsonism associated with PINK1 mutation. Neurochem. Res 33, 2565–2574. [DOI] [PubMed] [Google Scholar]
- Pickles S, Vigie P, Youle RJ, 2018. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol 28, R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ, 2015. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogson JH, Ivatt RM, Sanchez-Martinez A, Tufi R, Wilson E, Mortiboys H, Whitworth AJ, 2014. The complex I subunit NDUFA10 selectively rescues Drosophila pink1 mutants through a mechanism independent of mitophagy. PLoS Genet. 10, e1004815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Muthane U, Jiang H, Ferreira M, Naini AB, Fahn S, 1993. Chronic levodopa administration alters cerebral mitochondrial respiratory chain activity. Ann. Neurol 34, 715–723. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Fahn S, 1995. Antiparkinsonian therapies and brain mitochondrial complex I activity. Mov. Disord 10, 312–317. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Tieu K, Perier C, Vila M, 2004. MPTP as a mitochondrial neurotoxic model of Parkinson’s disease. J. Bioenerg. Biomembr 36, 375–379. 10.1023/B:JOBB.0000041771.66775.d5. [DOI] [PubMed] [Google Scholar]
- Quintana A, Kruse SE, Kapur RP, Sanz E, Palmiter RD, 2010. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. U.S.A 107, 10996–11001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S, Copeland WC, 2019. POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol 15, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay RR, Salach JI, Dadgar J, Singer TP, 1986. Inhibition of mitochondrial NADH dehydrogenase by pyridine derivatives and its possible relation to experimental and idiopathic parkinsonism. Biochem. Biophys. Res. Commun 135, 269–275. [DOI] [PubMed] [Google Scholar]
- Reeve A, Meagher M, Lax N, Simcox E, Hepplewhite P, Jaros E, Turnbull D, 2013. The impact of pathogenic mitochondrial DNA mutations on substantia nigra neurons. J. Neurosci 33, 10790–10801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AK, Grady JP, Cosgrave EM, Bennison E, Chen C, Hepplewhite PD, Morris CM, 2018. Mitochondrial dysfunction within the synapses of substantia nigra neurons in Parkinson’s disease. NPJ Parkinsons Dis. 4, 9. 10.1038/s41531-018-0044-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenburg RJ, 2016. Mitochondrial complex I-linked disease. Biochim. Biophys. Acta 1857, 938–945. [DOI] [PubMed] [Google Scholar]
- Sanchez-Caballero L, Guerrero-Castillo S, Nijtmans L, 2016a. Unraveling the complexity of mitochondrial complex I assembly: a dynamic process. Biochim. Biophys. Acta 1857, 980–990. [DOI] [PubMed] [Google Scholar]
- Sanchez-Caballero L, Ruzzenente B, Bianchi L, Assouline Z, Barcia G, Metodiev MD, Rio M, Funalot B, van den Brand MA, Guerrero-Castillo S, Molenaar JP, Koolen D, Brandt U, Rodenburg RJ, Nijtmans LG, Rotig A, 2016b. Mutations in complex I assembly factor TMEM126B result in muscle weakness and isolated complex I deficiency. Am. J. Hum. Genet 99, 208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazanov LA, Hinchliffe P, 2006. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 311, 1430–1436. [DOI] [PubMed] [Google Scholar]
- Schagger H, 1995. Quantification of oxidative phosphorylation enzymes after blue native electrophoresis and two-dimensional resolution: normal complex I protein amounts in Parkinson’s disease conflict with reduced catalytic activities. Electrophoresis 16, 763–770. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD, 1989. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1, 1269. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD, 1990a. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem 54, 823–827. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Mann VM, Cooper JM, Dexter D, Daniel SE, Jenner P, Clark JB, Marsden CD, 1990b. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J. Neurochem 55, 2142–2145. [DOI] [PubMed] [Google Scholar]
- Schlehe JS, Journel MS, Taylor KP, Amodeo KD, LaVoie MJ, 2013. The mitochondrial disease associated protein Ndufaf2 is dispensable for Complex-1 assembly but critical for the regulation of oxidative stress. Neurobiol. Dis 58, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde S, Pasupathy K, 2006. Respiratory-chain enzyme activities in isolated mitochondria of lymphocytes from patients with Parkinson’s disease: preliminary study. Neurol. India 54, 390–393. [DOI] [PubMed] [Google Scholar]
- Shoffner JM, Wallace DC, Watts RL, Juncos JL, Torroni A, 1992. Mitochondrial oxidative phosphorylation defects in Parkinson’s disease. Ann. Neurol 32, 113–114. [DOI] [PubMed] [Google Scholar]
- Shults CW, Nasirian F, Ward DM, Nakano K, Pay M, Hill LR, Haas RH, 1995. Carbidopa/levodopa and selegiline do not affect platelet mitochondrial function in early parkinsonism. Neurology 45, 344–348. [DOI] [PubMed] [Google Scholar]
- Stroud DA, Surgenor EE, Formosa LE, Reljic B, Frazier AE, Dibley MG, Osellame LD, Stait T, Beilharz TH, Thorburn DR, Salim A, Ryan MT, 2016. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 538, 123–126. [DOI] [PubMed] [Google Scholar]
- Subrahmanian N, Castonguay AD, Fatnes TA, Hamel PP, 2020. Chlamydomonas reinhardtii as a plant model system to study mitochondrial complex I dysfunction. Plant Direct. 4, e00200 10.1002/pld3.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DJ, Krige D, Barnes PR, Kemp GJ, Carroll MT, Mann VM, Cooper JM, Marsden CD, Schapira AH, 1994. A 31P magnetic resonance spectroscopy study of mitochondrial function in skeletal muscle of patients with Parkinson’s disease. J. Neurol. Sci 125, 77–81. [DOI] [PubMed] [Google Scholar]
- Titova N, Chaudhuri KR, 2018. Non-motor Parkinson disease: new concepts and personalised management. Med. J. Aust 208, 404–409. [DOI] [PubMed] [Google Scholar]
- Tolwani RJ, Jakowec MW, Petzinger GM, Green S, Waggie K, 1999. Experimental models of Parkinson’s disease: insights from many models. Lab. Anim. Sci 49, 363–371. [PubMed] [Google Scholar]
- Tzoulis C, Tran GT, Schwarzlmuller T, Specht K, Haugarvoll K, Balafkan N, Lilleng PK, Miletic H, Biermann M, Bindoff LA, 2013. Severe nigrostriatal degeneration without clinical parkinsonism in patients with polymerase gamma mutations. Brain 136, 2393–2404. [DOI] [PubMed] [Google Scholar]
- Tzoulis C, Schwarzlmuller T, Biermann M, Haugarvoll K, Bindoff LA, 2016. Mitochondrial DNA homeostasis is essential for nigrostriatal integrity. Mitochondrion 28, 33–37. [DOI] [PubMed] [Google Scholar]
- Umeda S, Muta T, Ohsato T, Takamatsu C, Hamasaki N, Kang D, 2000. The D-loop structure of human mtDNA is destabilized directly by 1-methyl-4-phenylpyridinium ion (MPP+), a parkinsonism-causing toxin. Eur. J. Biochem 267, 200–206. 10.1046/j.1432-1327.2000.00990.x. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW, 2004. Hereditary earlyonset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- van der Wijst MGP, Rots MG, 2015. Mitochondrial epigenetics: an overlooked layer of regulation? Trends Genet. 31, 353–356. [DOI] [PubMed] [Google Scholar]
- Varghese M, Pandey M, Samanta A, Gangopadhyay PK, Mohanakumar KP, 2009. Reduced NADH coenzyme Q dehydrogenase activity in platelets of Parkinson’s disease, but not Parkinson plus patients, from an Indian population. J. Neurol. Sci 279, 39–42. [DOI] [PubMed] [Google Scholar]
- Vilain S, Esposito G, Haddad D, Schaap O, Dobreva MP, Vos M, Van Meensel S, Morais VA, De Strooper B, Verstreken P, 2012. The yeast complex I equivalent NADH dehydrogenase rescues pink1 mutants. PLoS Genet. 8, e1002456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar KR, Zhu J, Hirst J, 2014. Architecture of mammalian respiratory complex I. Nature 515, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel RO, van den Brand MA, Rodenburg RJ, van den Heuvel LP, Tsuneoka M, Smeitink JA, Nijtmans LG, 2007. Investigation of the complex I assembly chaperones B17.2L and NDUFAF1 in a cohort of CI deficient patients. Mol. Genet. Metab 91, 176–182. [DOI] [PubMed] [Google Scholar]
- Wang P, Deng J, Dong J, Liu J, Bigio EH, Mesulam M, Wang T, Sun L, Wang L, Lee AY, McGee WA, Chen X, Fushimi K, Zhu L, Wu JY, 2019. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 15, e1007947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Zhao F, Ma X, Perry G, Zhu X, 2020. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: recent advances. Mol. Neurodegener 15, 30. 10.1186/s13024-020-00376-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washizuka S, Iwamoto K, Kazuno AA, Kakiuchi C, Mori K, Kametani M, Yamada K, Kunugi H, Tajima O, Akiyama T, Nanko S, Yoshikawa T, Kato T, 2004. Association of mitochondrial complex I subunit gene NDUFV2 at 18p11 with bipolar disorder in Japanese and the National Institute of Mental Health pedigrees. Biol. Psychiatry 56, 483–489. [DOI] [PubMed] [Google Scholar]
- Washizuka S, Kametani M, Sasaki T, Tochigi M, Umekage T, Kohda K, Kato T, 2006. Association of mitochondrial complex I subunit gene NDUFV2 at 18p11 with schizophrenia in the Japanese population. Am. J. Med. Genet. B Neuropsychiatr. Genet 141B, 301–304. [DOI] [PubMed] [Google Scholar]
- Wiedemann FR, Winkler K, Lins H, Wallesch CW, Kunz WS, 1999. Detection of respiratory chain defects in cultivated skin fibroblasts and skeletal muscle of patients with Parkinson’s disease. Ann. N. Y. Acad. Sci 893, 426–429. [DOI] [PubMed] [Google Scholar]
- Wikström M, 1984. Two protons are pumped from the mitochondrial matrix per electron transferred between NADH and ubiquinone. FEBS Lett. 169, 300–304. [DOI] [PubMed] [Google Scholar]
- Winkler-Stuck K, Wiedemann FR, Wallesch CW, Kunz WS, 2004. Effect of coenzyme Q10 on the mitochondrial function of skin fibroblasts from Parkinson patients. J. Neurol. Sci 220, 41–48. [DOI] [PubMed] [Google Scholar]
- Winkler-Stuck K, Kirches E, Mawrin C, Dietzmann K, Lins H, Wallesch CW, Kunz WS, Wiedemann FR, 2005. Re-evaluation of the dysfunction of mitochondrial respiratory chain in skeletal muscle of patients with Parkinson’s disease. J. Neural. Transm. (Vienna) 112, 499–518. 10.1007/s00702-004-0195-y. [DOI] [PubMed] [Google Scholar]
- Wirth C, Brandt U, Hunte C, Zickermann V, 2016. Structure and function of mitochondrial complex I. Biochim. Biophys. Acta 1857, 902–914. [DOI] [PubMed] [Google Scholar]
- Yoshino H, Nakagawa-Hattori Y, Kondo T, Mizuno Y, 1992. Mitochondrial complex I and II activities of lymphocytes and platelets in Parkinson’s disease. J. Neural Transm. Park. Dis. Dement. Sect 4, 27–34. 10.1007/BF02257619. [DOI] [PubMed] [Google Scholar]
- Zeng XS, Geng WS, Jia JJ, 2018. Neurotoxin-induced animal models of Parkinson disease: pathogenic mechanism and assessment. Asn Neuro 10. [DOI] [PMC free article] [PubMed] [Google Scholar]