

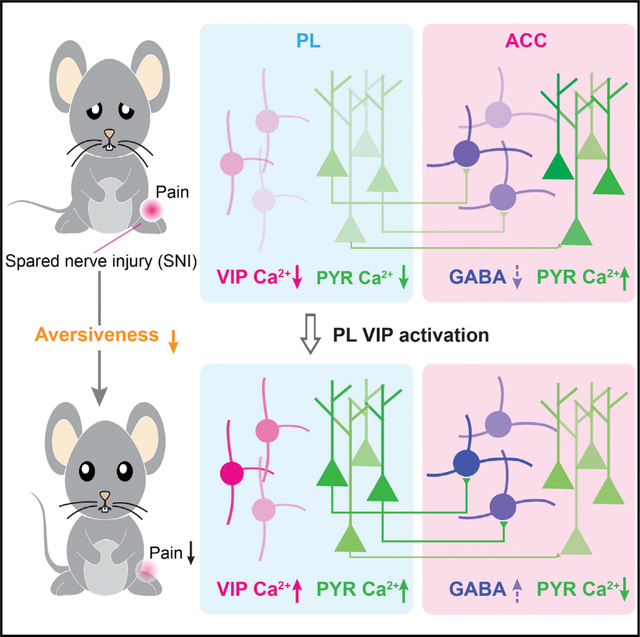
SUMMARY
While dysfunction of the medial prefrontal cortex (mPFC) has been implicated in chronic pain, the underlying neural circuits and the contribution of specific cellular populations remain unclear. Using in vivo Ca2+ imaging, we report that in both male and female mice, peripheral nerve injury-induced neuropathic pain causes a marked reduction of vasoactive intestinal polypeptide (VIP)-expressing interneuron activity in the prelimbic area of the mPFC, which contributes to decreased prefrontal cortical outputs. Moreover, prelimbic glutamatergic projections to GABAergic interneurons in the anterior cingulate cortex (ACC) are diminished, leading to loss of cortical-cortical inhibition and increased pyramidal neuron activity in the ACC. Chemogenetic activation of prelimbic VIP interneurons restores neuronal responses in the mPFC-ACC pathway and attenuates pain-like behaviors in mice. Furthermore, restoration of prelimbic outputs to the ACC reverses nerve injury-induced ACC hyperactivation. These findings reveal mPFC circuit changes associated with neuropathic pain and highlight VIP interneurons as potential therapeutic targets for pain treatment.
In brief
Vasoactive intestinal polypeptide (VIP)-expressing interneurons provide disinhibitory control of cortical circuits. Li et al. show that neuropathic pain reduces VIP neuronal activity in the mouse prefrontal cortex. Chemogenetic activation of VIP interneurons restores prefrontal outputs to the anterior cingulate cortex and ameliorates pain-like behaviors in mice.
Graphical Abstract
INTRODUCTION
Chronic pain is a major cause of disability and disease burden in the states, with an estimated 20.4% of adults in the United States being affected (Dahlhamer et al., 2018). It is increasingly recognized that the neocortical network has a prominent role in pain perception and chronicity (Tan and Kuner, 2021). Neuroimaging studies show that many areas of the cortex, including the anterior cingulate cortex (ACC) and the medial prefrontal cortex (mPFC), are activated in response to acute noxious stimuli in healthy individuals and exhibit differential activity and functional connectivity under chronic pain conditions (Apkarian et al., 2004; Wager et al., 2013). The mPFC is known for its role in providing top-down control of sensory and affective processes. Its projections to the periaqueductal gray (PAG), thalamus, amygdala, and nucleus accumbens (NAc) have been shown to be altered by chronic pain (Cardoso-Cruz et al., 2013; Huang et al., 2019; Ji and Neugebauer, 2014; Lee et al., 2015; Yin et al., 2020). Besides subcortical brain regions, the mPFC projects extensively to other cortices (Gao et al., 2022), including the ACC, a key region processing emotional states associated with pain (Bliss et al., 2016). Whether the neural pathway from the mPFC to the ACC is involved in chronic pain is not known.
Dysfunction in the mPFC is well-documented in rodent models of chronic pain. At cellular levels, chronic pain states have been associated with structural and functional changes in mPFC pyramidal neurons, including changes in dendritic morphology and spine density, decreased excitatory synaptic inputs, impaired excitatory cholinergic modulation, and reduced intrinsic cellular excitability and firing rates (Cheriyan and Sheets, 2018; Dale et al., 2018; Ji and Neugebauer, 2011; Ji et al., 2010; Kelly et al., 2016; Kelly and Martina, 2018; Metz et al., 2009; Mitric et al., 2019; Radzicki et al., 2017; Wang et al., 2015; Zhang et al., 2015). Besides these changes in glutamatergic neurons, alterations to GABAergic interneurons have also been reported. For example, increased excitability of parvalbumin (PV)-expressing interneurons and decreased excitability of somatostatin (SST)-expressing interneurons have been observed in the mPFC of mice with peripheral neuropathic pain (Jones and Sheets, 2020; Zhang et al., 2015). Vasoactive intestinal polypeptide (VIP)-expressing interneurons constitute another major class of GABAergic neurons in the cortex (Tremblay et al., 2016). By inhibiting PV and SST interneurons in the local circuitry, VIP neurons serve key functions to amplify and modulate the activity of local pyramidal cells (Pfeffer et al., 2013; Pi et al., 2013; Silberberg and Markram, 2007). It is unknown, however, whether VIP interneuron responses are altered under chronic pain conditions and contribute to dysfunction of mPFC outputs.
In this study, we examined the Ca2+ activity of pyramidal neurons and VIP interneurons in layer 5 (L5) of the prelimbic cortex (PL; a subregion of the mPFC) in awake mice. Using in vivo Ca2+ imaging, we found that the activity of PL pyramidal neurons, including those projecting to the ACC, was markedly decreased in a mouse model of peripheral nerve injury-induced neuropathic pain. This reduction of PL pyramidal neuron activity was accompanied by a decrease of VIP interneuron responses in local circuits. Activating PL VIP neurons with hM3Dq corrected pyramidal neuron Ca2+ activity, restored PL outputs to the ACC, and mitigated pain-like behaviors in mice. Furthermore, chemogenetic activation of the PL-ACC pathway ameliorated nerve injury-induced pyramidal neuron hyperactivation in the ACC. Our results reveal cell-type-specific changes in PL-ACC circuits that are important for the development of neuropathic pain and underscore the importance of developing brain region- and cell-type-specific targets for chronic pain treatment.
RESULTS
Neuropathic pain deactivates PL outputs to the ACC
To identify prefrontal cortical changes related to neuropathic pain, we used in vivo two-photon Ca2+ imaging to examine somatic activity of pyramidal neurons expressing the genetically encoded Ca2+ indicator GCaMP6s in the PL of awake, head-restrained mice (Cichon et al., 2020). Thy1-GCaMP6s mice were subjected to spared nerve injury (SNI) to induce chronic neuropathic pain (Figure 1A). We first examined the activity of L5 pyramidal neurons, the major output cells of the PL (Cheriyan et al., 2016). Through a glass widow, GCaMP6s-expressing pyramidal neurons in L5 of the PL were visible at a depth of 600–800 μm below the pial surface, with a distance of 400–600 μm to the midline (Figure 1A). When mice were under a quiet resting state, spontaneous Ca2+ transients could be detected in pyramidal somas in the PL contralateral to sham/SNI (Figure 1B). Two weeks after SNI (i.e., the chronic phase of neuropathic pain), we found that the somatic Ca2+ activity in L5 pyramidal neurons was ~60% of that recorded in sham mice, as shown by quantifications of the area under the curve (AUC) (19.17% ± 0.31% versus 32.75% ± 0.80%, p < 0.0001), frequency (3.06 ± 0.08 versus 5.05 ± 0.12, p < 0.0001), peak amplitude (0.75 ± 0.01 versus 1.18 ± 0.03, p < 0.0001), and duration (1.87 ± 0.04 versus 2.79 ± 0.07, p < 0.0001) of Ca2+ transients (Figures 1C–1F). Comparing SNI with sham, a larger fraction (29.54% versus 4.18%) of L5 pyramidal neurons in the PL was inactive (Figure 1C), regardless of the sex of animals (Figure S1). These results indicate that neuropathic pain causes deactivation of PL outputs, consistent with previous electrophysiological findings in vitro (Mitric et al., 2019; Zhang et al., 2015).
Figure 1. Decreased L5 pyramidal somatic Ca2+ activity in the PL after peripheral nerve injury.
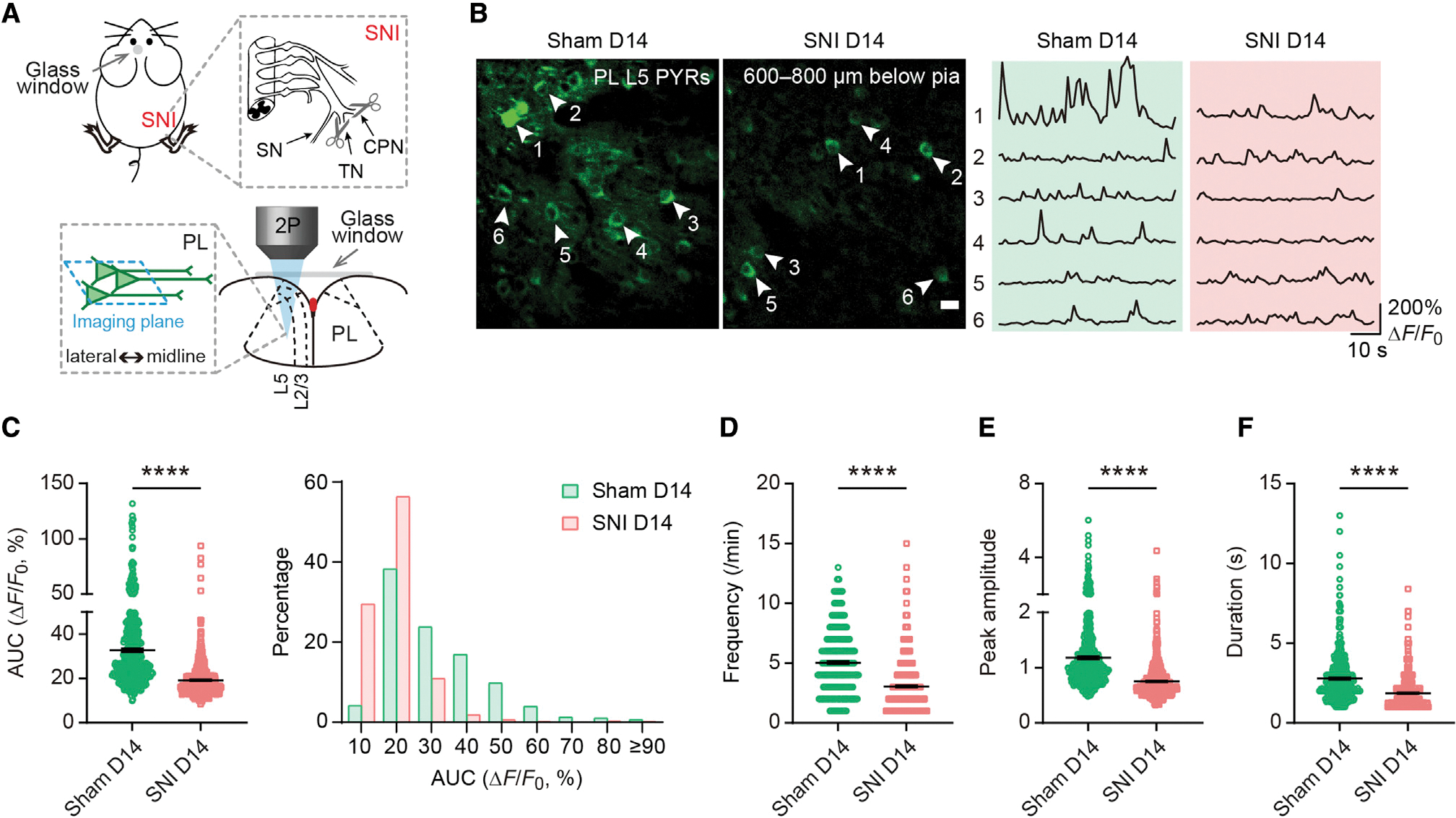
(A) Schematic of spared nerve injury (SNI) and in vivo two-photon (2P) Ca2+ imaging in layer 5 (L5) of the prelimbic cortex (PL) contralateral to SNI. SN, sural nerve; TN, tibial nerve; CPN, common peroneal nerve.
(B) Representative Ca2+ images and fluorescence traces of pyramidal neurons (PYRs) expressing GCaMP6s in L5 of the PL 14 days after surgery. Scale bar, 20 μm.
(C) Left, PYR Ca2+ activity measured by the area under the curve (AUC) 14 days after surgery (p < 0.0001). Right, distribution plot of data shown on the left.
(D–F) The frequency (D), peak amplitude (E), and duration (F) of Ca2+ transients in L5 PYRs 14 days after surgery (p < 0.0001).
In (C)–(F), n = 486 and 633 cells from 5 and 7 mice for sham and SNI, respectively.
Summary data are mean ± SEM. ****p < 0.0001; by Mann-Whitney test.
Recent studies indicate that prefrontal cortical neurons send projections to a variety of brain regions including the ACC (Gao et al., 2022). To examine the effects of neuropathic pain on the PL-ACC pathway, we selectively expressed GCaMP6s in PL neurons projecting to the ACC. Specifically, we injected into the ACC of C57BL/6 mice the retrograde transducing adeno-associated virus (AAVrg) (Tervo et al., 2016) encoding FLP and simultaneously injected into the ipsilateral PL an FLP-dependent GCaMP6s (Figure 2A). Following viral expression, we observed significant retrograde labeling of ACC projecting neurons in the PL, and ~87% of these projection neurons were located in L5 (Figure 2B). Immunostaining of brain slices showed that ~90% of PL-ACC projection neurons were colocalized with CaMKIIα, a marker for excitatory cortical neurons (Figure 2C). Two weeks after sham/SNI, we performed Ca2+ imaging in the PL of these mice (Figure 2D). Consistent with the reduction of L5 pyramidal neuron activity in the PL, the somas of PL-ACC projection neurons exhibited a lower level of spontaneous Ca2+ in SNI mice compared with sham mice (Figure 2E). This reduction of Ca2+ signals in PL-ACC projection neurons was observed in both male and female mice (Figure S2), indicating that PL glutamatergic outputs to the ACC are downregulated under neuropathic pain conditions in a sex-independent manner.
Figure 2. Reduced Ca2+ activity in PL excitatory neurons projecting to the ACC.
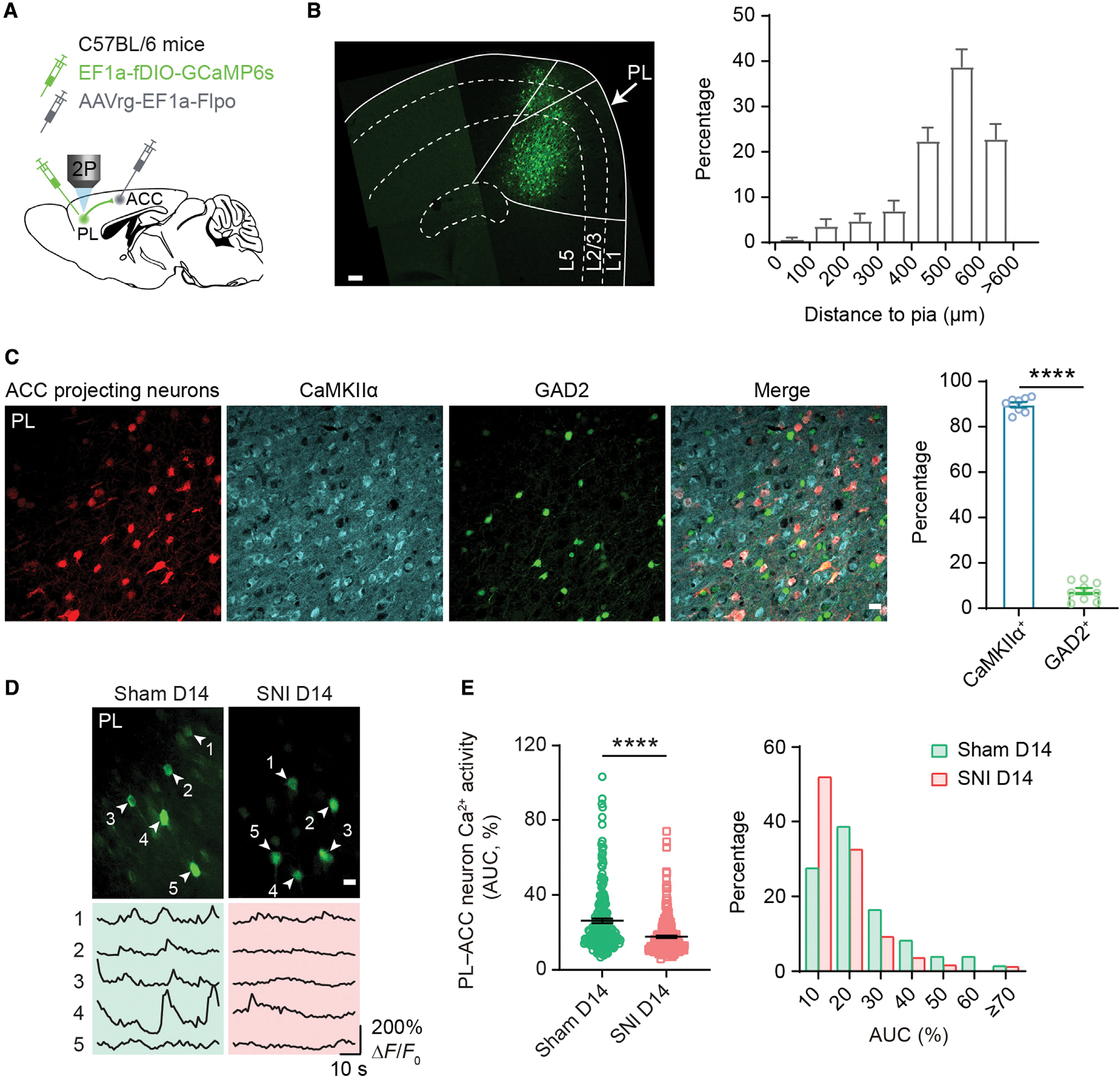
(A) Experimental design for selective expression of GCaMP6s in PL-ACC projection neurons.
(B) Left, a confocal image showing the somas of GCaMP-expressing PL-ACC projection neurons. Scale bar, 100 μm. Right, distribution of GCaMP-expressing somas across layers of the PL. Most ACC projecting neurons are in deeper layers of the PL.
(C) Left, immunofluorescence images of the PL showing ACC projecting neurons (transformed color, anti-GFP+), CaMKIIα+ neurons, and GAD2+ neurons. Scale bar, 20 μm. Right, percentage of PL-ACC projection neurons positive for CaMKIIα (89.72% ± 1.15%) or GAD2 (7.56% ± 1.38%). PL-ACC projection neurons are predominantly colocalized with CaMKIIα (p < 0.0001). n = 8 and 9 sections from 4 mice.
(D) Representative Ca2+ images and fluorescence traces of PL-ACC projection neurons expressing GCaMP6s 14 days after surgery. Scale bar, 20 μm.
(E) Left, average Ca2+ activity of PL-ACC projection neurons 14 days after surgery (p < 0.0001; n = 214 and 249 cells from 7 and 8 mice for sham and SNI, respectively).
Right, distribution plot of data shown on the left. More PL neurons are inactive in SNI than sham mice (51.81% versus 27.54%). AUC, area under the curve.
Summary data are mean ± SEM. ****p < 0.0001; by Mann-Whitney test.
Neuropathic pain decreases PL VIP neuron activity
To understand mechanisms underlying deactivation of the PL-ACC pathway in neuropathic pain, we examined the changes of local inhibitory circuits in the PL during the development of neuropathic pain. In the neocortex, VIP interneurons can selectively inhibit other interneuron types, thereby providing a disinhibitory effect on local pyramidal neurons (Pfeffer et al., 2013; Pi et al., 2013). To measure the impact of neuropathic pain on VIP responses, we used in vivo two-photon Ca2+ imaging to examine the activity of VIP cells expressing GCaMP6s following sham/SNI. In this experiment, VipIRES–Cre mice were injected with a Cre-dependent AAV to induce expression of GCaMP6s specifically in VIP neurons (Figure 3A and S3A–S3C). Comparing sham and SNI groups, there was no difference in the density of VIP neurons in the PL (Figure S3D). Consistent with the reduction of pyramidal neuron activity in mice with neuropathic pain (Figure 1), we found that VIP activity in the PL was markedly decreased 2 weeks after SNI, as evidenced by the measurements of AUC (27.74% ± 0.99% versus 40.04% ± 2.36%, p < 0.0001), frequency (4.21 ± 0.18 versus 5.11 ± 0.23, p = 0.0028), peak amplitude (0.74 ± 0.02 versus 0.87 ± 0.05, p = 0.0019), and duration (2.97 ± 0.18 versus 3.92 ± 0.33, p = 0.0015) of Ca2+ transients (Figures 3C–3F). This decrease in VIP neuron activity occurred as early as 3 days after SNI (Figures 3C–3F). These in vivo Ca2+ recordings reveal a rapid and persistent reduction of VIP interneuron activity during the development of neuropathic pain.
Figure 3. Decreased Ca2+ activity in PL VIP interneurons after peripheral nerve injury.
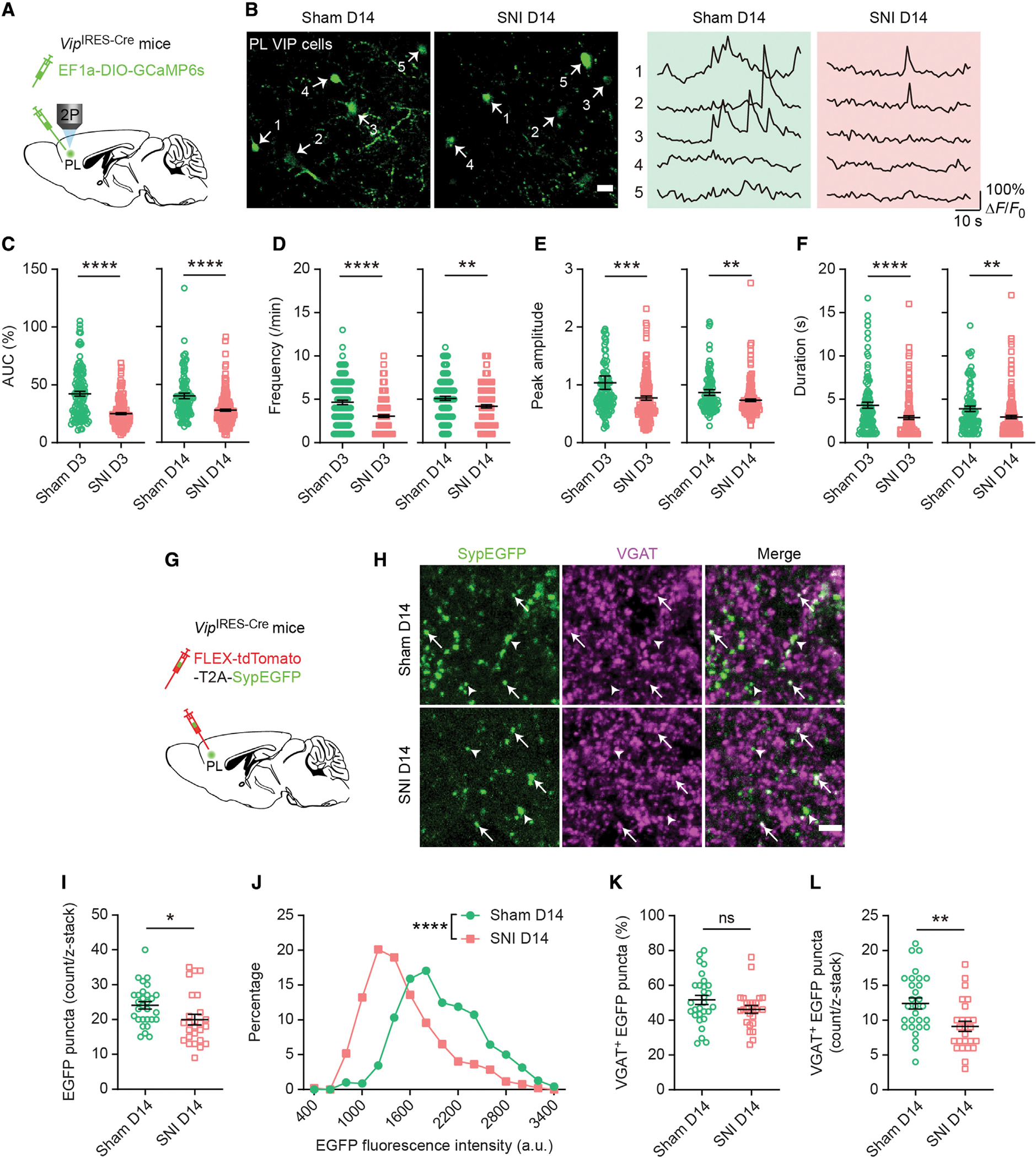
(A) Experimental design to specifically express GCaMP6s in PL VIP interneurons.
(B) Representative Ca2+ images and traces of PL VIP neurons 14 days after sham/SNI. Scale bar, 20 μm.
(C) AUC of VIP Ca2+ transients 3 (p < 0.0001) and 14 days (p < 0.0001) after sham/SNI.
(D) Frequency of VIP Ca2+ transients 3 (p < 0.0001) and 14 days (p = 0.0028) after sham/SNI.
(E) Peak amplitude of VIP Ca2+ transients 3 (p = 0.0001) and 14 days (p = 0.0019) after sham/SNI.
(F) Duration of VIP Ca2+ transients 3 (p < 0.0001) and 14 days (p = 0.0015) after sham/SNI.
In (C)–(F), n = 113, 177, 112, and 182 cells from 6, 12, 6, and 8 mice.
(G) Experimental design to virally express presynaptic (synaptophysin-fused) EGFP in VIP axonal terminals. VGAT was detected by immunofluorescence.
(H) Fluorescence images of the PL 14 days after sham/SNI. Arrows indicate VGAT+ EGFP puncta. Arrowheads indicate VGAT− EGFP puncta. Scale bar, 5 μm.
(I) The count of VIP presynaptic EGFP puncta (per 30 × 30 × 20 μm) in the PL of sham and SNI mice (p = 0.0102).
(J) Distribution of EGFP puncta fluorescence intensity in sham and SNI mice (p < 0.0001).
(K) Percentage of VIP EGFP puncta colocalized with VGAT in sham and SNI mice (p = 0.1469).
(L) The count of VIP VGAT+ EGFP puncta (per 30 × 30 × 20 μm) in the PL of sham and SNI mice (p = 0.0036).
In (I), (K), and (L), n = 29 and 25 sections from 4 and 5 mice for sham and SNI, respectively.
Summary data are mean ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant; by Mann-Whitney test.
Recent studies showed that SST and PV interneurons in the mPFC exhibit differential firing properties in male and female mice under neuropathic pain conditions (Jones and Sheets, 2020). To determine whether there is a sex difference in VIP interneuron responses in neuropathic pain, we compared VIP Ca2+ activity between males and females over the course of neuropathic pain development (Figure S4). We found that the response of VIP neurons was largely similar between male and female mice. In both sham and SNI groups, there were no differences in the AUC (Figures S4A and S4E), peak amplitude (Figures S4C and S4G), and duration (Figures S4D and S4H) of Ca2+ transients between male and female mice. Further analyses showed that within the sham group, the frequency of Ca2+ transients was significantly higher in males than females (Figure S4F). Two weeks after SNI, the frequency of Ca2+ transients was significantly decreased in male mice but not in female mice (Figure S4F). Nevertheless, neuropathic pain causes a decrease in PL VIP neuron activity in both male and female mice.
To further understand the effects of neuropathic pain on VIP interneurons in the PL, we virally expressed presynaptic (synaptophysin-fused) EGFP in these cells (Oh et al., 2014) and subsequently immunostained them for vesicular GABA transporter (VGAT), a marker for inhibitory terminals (Figures 3G and 3H). As expected, synaptophysin-fused EGFP was present in a punctate pattern in the axonal terminals of VIP cells, and a large fraction of them were colocalized with VGAT immunoreactivity (Figure 3H). Two weeks after surgery, we found that the number and fluorescence intensity of VIP presynaptic EGFP puncta were significantly lower in SNI than sham mice (Figures 3I and 3J). While the fraction of EGFP puncta colocalized with VGAT was similar between sham and SNI groups (Figure 3K), the number of VGAT-positive EGFP puncta was markedly deceased after SNI (Figure 3L), regardless of the sex of animals (Figures S4I–S4K). These results suggest a reduction of inhibitory synaptic outputs from PL VIP interneurons in chronic neuropathic pain conditions, which is consistent with the Ca2+ imaging data (Figures 3A–3F).
Transient activation of PL VIP neurons restores PL outputs to the ACC
VIP interneurons can provide disinhibition to pyramidal neurons by inhibiting SST and PV interneurons (Pfeffer et al., 2013; Pi et al., 2013). To test whether the reduction of VIP neuron activity in SNI mice contributes significantly to the reduction of pyramidal neuron activity in the PL, we activated VIP neurons in mice with neuropathic pain using a chemogenetic approach. To increase VIP cell activity in vivo, we specifically infected PL VIP interneurons with a Cre-dependent AAV encoding hM3Dq or mCherry (control) in VipIRES–Cre mice crossed with Thy1-GCaMP6s mice (Figure 4A). Binding of the ligand clozapine N-oxide (CNO) to hM3Dq receptors activates Gq-coupled signaling, thereby increasing neuronal firing in target cells (Urban and Roth, 2015). Two weeks following SNI in mice expressing hM3Dq, we imaged GCaMP6s-expressing pyramidal neurons in the PL before and after CNO injection. Thirty minutes after CNO administration, we found that pyramidal neuron activity in the PL of SNI mice had significantly increased to a level comparable to that of sham mice (p < 0.0001 versus CNO−, p = 0.1058 versus sham) (Figure 4B). In SNI mice expressing mCherry without hM3Dq, administration of CNO had no effect on pyramidal neuron activity in the PL (Figure 4B).
Figure 4. Transient activation of PL VIP interneurons restores PL outputs to the ACC and attenuates pain-like behaviors.
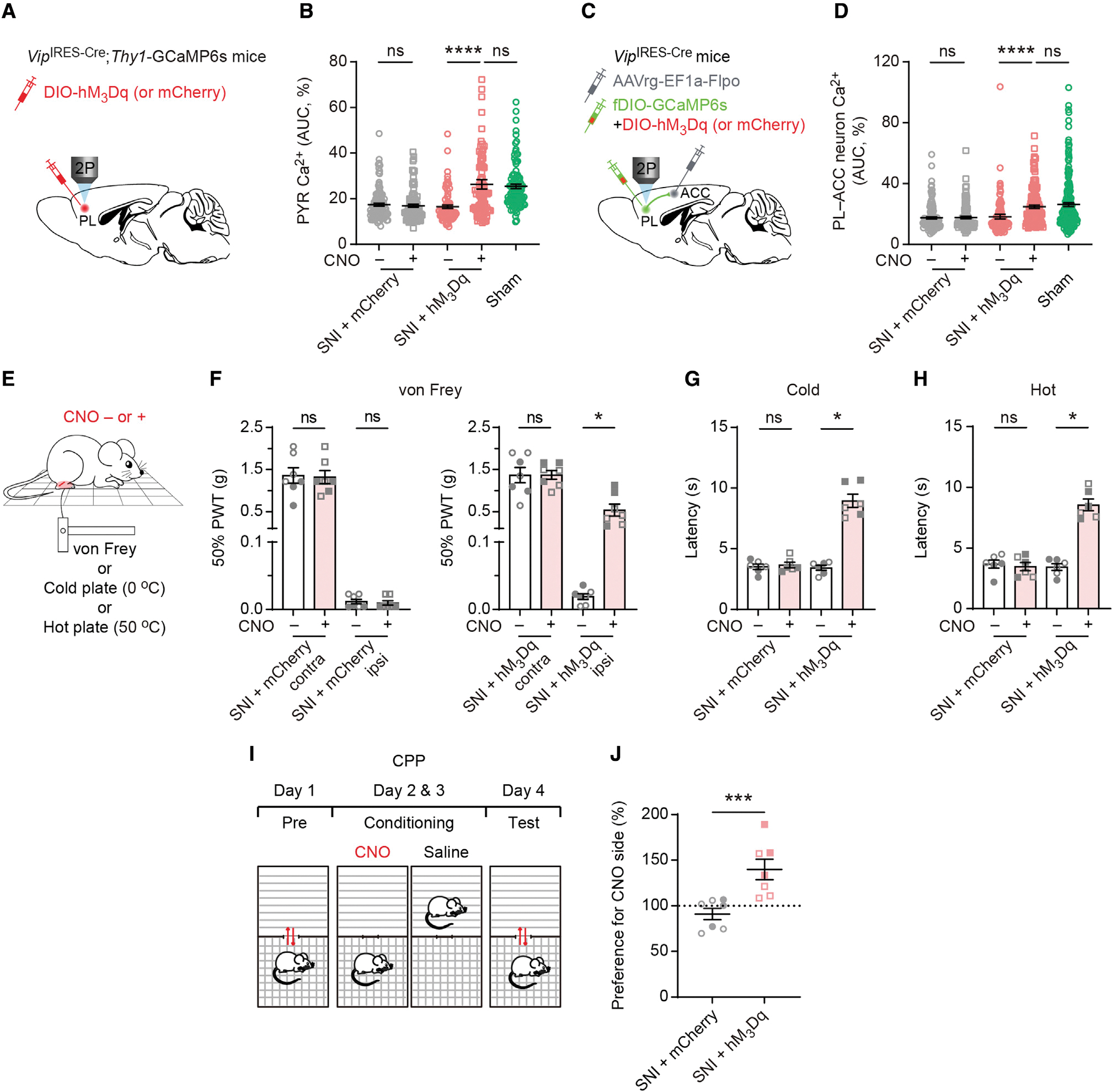
(A) Experimental design for selective expression of hM3Dq or mCherry in PL VIP neurons and in vivo Ca2+ imaging in GCaMP6s-expressing L5 PYRs 14 days post-SNI.
(B) Average Ca2+ activity of PYRs before and after CNO injection in SNI mice expressing mCherry (p = 0.3952; n = 110 cells from 3 mice, 42 and 68 cells from 1 male and 2 females, respectively) or hM3Dq (p < 0.0001; n = 88 cells from 2 mice, 46 and 42 cells from 1 male and 1 female, respectively). Transient activation of VIP neurons in SNI mice reverts pyramidal Ca2+ to the level of sham mice (p = 0.1058).
(C) Experimental design to specifically express hM3Dq or mCherry in PL VIP neurons. In vivo Ca2+ imaging of PL-ACC projection neurons was performed 14 days after SNI.
(D) Average Ca2+ activity of PL-ACC projection neurons before and after CNO injection in SNI mice expressing mCherry (p = 0.6517; n = 126 cells from 3 mice, 74 and 52 cells from 2 males and 1 female, respectively) or hM3Dq (p < 0.0001; n = 144 cells from 4 mice, 70 and 74 cells from 2 males and 2 females, respectively). Acute activation of VIP cells in SNI mice reverts projection neuron activity to the level of sham mice (p = 0.5427).
(E) Schematic of mechanical and thermal tests before and after CNO injection.
(F–H) Measurements of pain sensitivity in SNI mice expressing mCherry or hM3Dq (n = 6–7 mice per group, 3–4 males, 3 females). Acute activation of PL VIP neurons increases the animals’ mechanical (p = 0.0156), cold (p = 0.0313), and hot (p = 0.0313) thresholds in the limb ipsilateral to SNI. CNO injection has no effect on mechanical and thermal thresholds in SNI mice expressing mCherry.
(I) Schematic of conditioned place preference (CPP) tests to assess ongoing pain.
(J) Preference to stay in CNO-paired chamber for SNI mice expressing mCherry or hM3Dq in PL VIP neurons (p = 0.0006; n = 7 mice per group, 4 males, 3 females).
In (F)–(J), filled circles/squares indicate female data, and unfilled circles/squares indicate male data.
Summary data are mean ± SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001; ns, not significant; by Mann-Whitney test for unpaired comparison or Wilcoxon test for paired comparison.
In a separate experiment, we examined the activity of PL-ACC projection neurons while chemogenetically activating VIP interneurons in the PL. Specifically, we injected AAVrg-FLPo into the ACC and simultaneously injected FLP-dependent GCaMP6s and Cre-dependent hM3Dq or mCherry into the PL of VipIRES–Cre mice. This injection scheme allows selective expression of hM3Dq or mCherry in PL VIP neurons and, meanwhile, GCaMP6s in PL neurons projecting to the ACC (Figure 4C). Coinjection of multiple viruses did not cause detectable changes in neuronal density in the injected regions (Figure S5). In mice with neuropathic pain, acute chemogenetic activation of PL VIP neurons significantly increased Ca2+ activity in PL-ACC projection neurons (p < 0.0001) (Figure 4D). Thirty minutes after CNO injection, the level of activity in PL-ACC projection neurons was comparable to that of sham mice (p = 0.5427). These results indicate that activating PL VIP neurons restores PL outputs to the ACC in mice with neuropathic pain.
When we tested pain-like behaviors in VipIRES–Cre mice infected with AAV hM3Dq (Figure 4E), we found that acute activation of PL VIP neurons with a single injection of CNO significantly increased the animals’ mechanical paw withdrawal thresholds in the nerve-injured limb (p = 0.0156) but not the contralateral one (p = 0.8125) (Figure 4F). CNO injection in SNI mice expressing mCherry without hM3Dq had no effect on the animals’ mechanical thresholds (Figure 4F). Similarly, acute activation of PL VIP neurons increased the animals’ thermal thresholds (Figures 4G and 4H).
In addition to nociceptive allodynia, we assessed pain aversiveness in these mice using a conditioned place preference (CPP) test (Figure 4I) (King et al., 2009). In this test, mice were allowed free access to both compartments of the CPP chamber during the pre-conditioning phase. The conditioning phase was performed 2 weeks after SNI, during which one compartment was paired with CNO injection and the other with saline. During the test phase, mice were allowed free access to both compartments without injections (see STAR Methods). Compared with SNI mice expressing mCherry, we found that SNI mice expressing hM3Dq in PL VIP neurons preferred to stay in the compartment associated with CNO treatment (p = 0.0006) (Figure 4J), indicating the relief of ongoing pain after CNO administration to activate PL VIP neurons.
Altogether, these findings demonstrate that in mice with neuropathic pain, acute activation of PL VIP interneurons increases pyramidal neuron activity, including those projecting to the ACC, and attenuates pain-like behaviors.
Continuous activation of PL VIP neurons prevents the development of neuropathic pain
Given the immediate beneficial effects of activating PL VIP interneurons in mice with neuropathic pain, we subsequently asked whether continuous activation of VIP cells after nerve injury could prevent the development of neuropathic pain. To test this possibility, following sham/SNI in mice expressing hM3Dq or mCherry in PL VIP cells, we performed daily CNO injections for 1 week and imaged Ca2+ activity in PL neurons 2 weeks after SNI (Figure 5A). Consistent with previous studies (Alexander et al., 2009), a single injection of CNO activated VIP cells over a period of 4 h (Figure S6). We found that daily CNO treatment for 1 week in SNI mice expressing hM3Dq was sufficient to prevent SNI-induced reduction of VIP neuron activity (p < 0.0001 versus mCherry, p = 0.0699 versus sham) (Figures 5B and 5C). Consistently, SNI-induced reduction of pyramidal neuron activity (p < 0.0001 versus mCherry, p = 0.5503 versus sham) in the PL was also reversed (Figures 5D and 5E). Daily CNO injection in SNI mice expressing mCherry had no effect on pyramidal neuron activity in the PL (p = 0.9992).
Figure 5. Daily activation of PL VIP interneurons following nerve injury prevents the development of neuropathic pain.
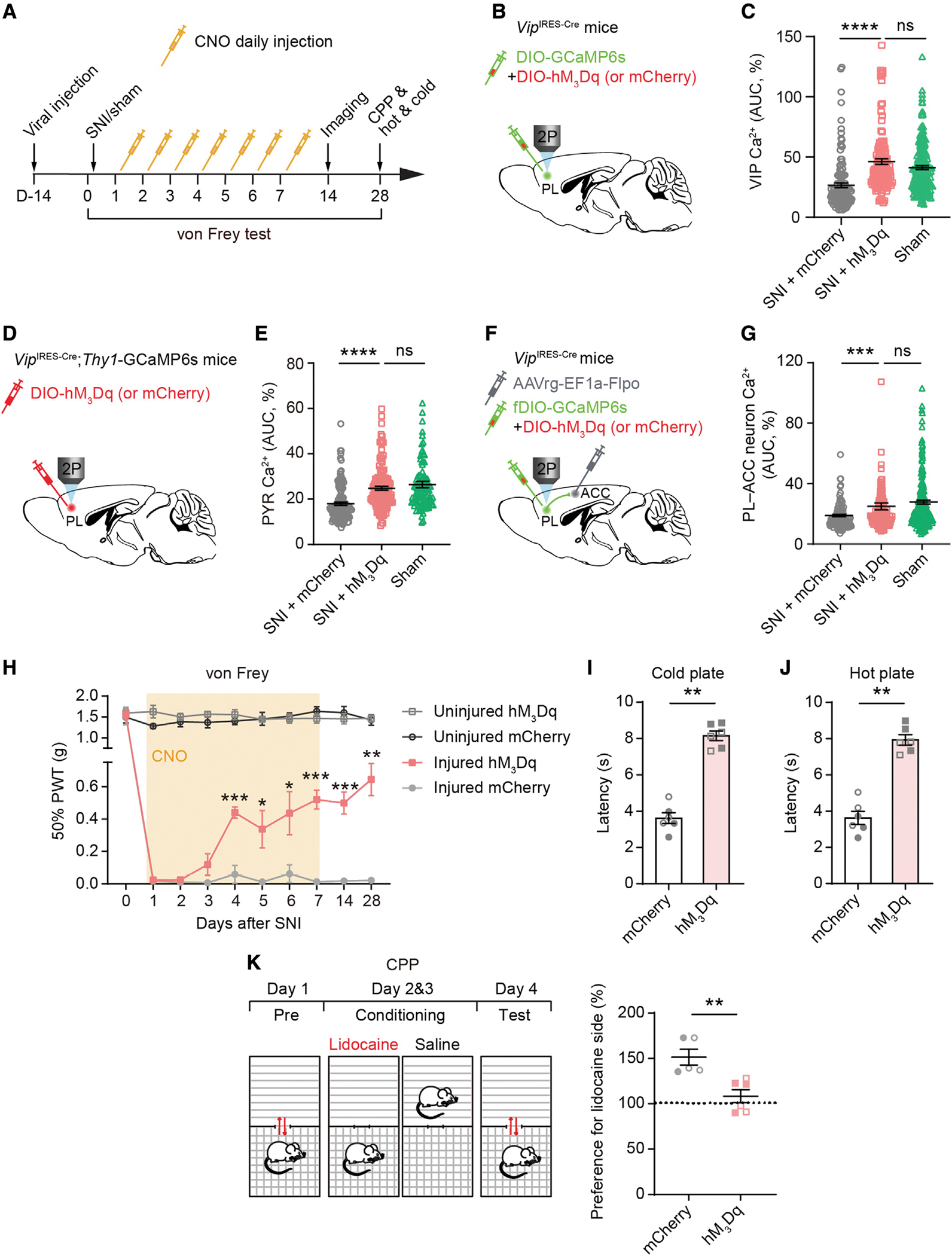
(A) Experimental timeline for viral injection (see design in B, D, and F), sham/SNI surgery, daily CNO application, in vivo Ca2+ imaging, and behavioral tests. For control, SNI mice infected by AAV-mCherry received CNO injections. von Frey tests were performed shortly before daily CNO application during the first week after surgery, as well as on days 14 and 28. Ca2+ imaging was carried out on day 14. CPP and thermal tests were performed 4 weeks post-SNI.
(B) Experimental design to express GCaMP6 and hM3Dq (or mCherry) in PL VIP interneurons contralateral to SNI.
(C) Average Ca2+ activity of PL VIP interneurons. Daily VIP activation in SNI mice restores VIP activity to the level of sham mice (p < 0.0001 versus mCherry, p = 0.0699 versus sham; n = 129, 149, and 225 cells from 3, 4, and 12 mice, with 78, 78, and 102 cells from males and 51, 71, and 123 cells from females, respectively).
(D) Experimental design to express hM3Dq or mCherry in PL VIP interneurons contralateral to SNI.
(E) Average Ca2+ activity of PL PYRs. Daily VIP activation in SNI mice increases pyramidal activity to the level of sham mice (p < 0.0001 versus mCherry, p = 0.5503 versus sham; n = 122, 133, and 108 cells from 3 mice per group, with 75, 79, and 68 cells from males and 47, 54, and 40 cells from females, respectively).
(F) Experimental design to express hM3Dq or mCherry in PL VIP neurons and GCaMP6 in PL-ACC projection neurons.
(G) Average Ca2+ activity of PL-ACC projection neurons. Daily VIP activation in SNI mice expressing hM3Dq increases projection neuron activity to the level of sham mice (p = 0.0004 versus mCherry, p = 0.0955 versus sham; n = 126, 117, and 214 cells from 3, 3, and 7 mice, with 46, 46, and 88 cells from males and 80, 71, and 126 cells from females, respectively).
(H) Daily VIP activation increases mechanical paw withdrawal thresholds of injured limb (injured limb, hM3Dq versus mCherry: p = 0.0003, 0.0369, 0.0372, 0.0003, 0.0008, and 0.0014; n = 6 mice per group, 3 males and 3 females).
(I–J) Daily VIP activation in SNI mice increases paw withdrawal latency on cold (p = 0.0022) or hot plate (p = 0.0022). n = 6 mice (3 males, 3 females) per group.
(K) Left, schematic of CPP tests. Right, preference to stay in lidocaine-paired chamber for SNI mice expressing mCherry or hM3Dq (p = 0.0043; n = 5 and 6 mice, with 3 males and 2 females for mCherry and 3 males and 3 females for hM3Dq, respectively).
In (I)–(K), filled circles/squares indicate female data, and unfilled circles/squares indicate male data.
Summary data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant; by Mann-Whitney test.
To determine whether dysfunction of PL-ACC projections was rescued after continuous activation of VIP interneurons, we specifically infected VIP neurons in the PL with hM3Dq/mCherry and expressed GCaMP6s in PL-ACC projection neurons (Figure 5F). Following the same timeline shown in Figure 5A, we performed daily CNO injection for 1 week and imaged Ca2+ activity in PL-ACC projection neurons 1 week after the last CNO injection (i.e., 2 weeks after SNI). We found that daily CNO treatment for 1 week was sufficient to prevent SNI-induced inhibition of PL-ACC projection neurons. Two weeks after SNI, the level of Ca2+ activity in PL-ACC projection neurons in mice expressing hM3Dq was significantly increased to a level that was comparable to that in sham mice (p = 0.0004 versus mCherry, p = 0.0955 versus sham) (Figure 5G).
When we measured paw withdrawal thresholds over time, we found that VIP activation for 1 week mitigated mechanical and thermal allodynia in SNI mice. One to three weeks after the last CNO injection, the mechanical threshold of the injured limb remained higher in SNI mice with VIP neurons expressing hM3Dq compared with those expressing mCherry (p = 0.0008 and 0.0014 for days 14 and 28 after SNI, respectively) (Figure 5H). Consistently, there was a significant increase of paw withdrawal latency in cold plate (p = 0.0022) and hot plate (p = 0.0022) tests in SNI mice expressing hM3Dq versus mCherry (Figures 5I and 5J). Four weeks after SNI, while mice expressing mCherry preferred to stay in the chamber paired with an analgesic agent, namely lidocaine, mice expressing hM3Dq showed no such preference in the CPP test (Figure 5K), indicating the absence of ongoing pain. Furthermore, SNI mice with daily activation of VIP cells also showed better performance in the novel object recognition and T maze tasks (Figure S7), suggesting the improvement of memory function with the relief of chronic pain symptoms.
Combined, these results showed that daily activation of PL VIP interneurons for 1 week after SNI was sufficient to preserve PL outputs to the ACC and prevent the development of neuropathic pain.
Restoration of PL-ACC pathway reverses ACC hyperactivation
The development of neuropathic pain is associated with increased pyramidal neuron activity in the ACC (Zhao et al., 2018). We next tested the role of the PL-ACC pathway in neuropathic pain-induced ACC hyperactivation. To this end, we used three strategies to excite the PL of SNI mice. In the first set of experiments, we sought to directly activate PL pyramidal neurons by injecting into the PL of Thy1-GCaMP6s mice AAV encoding hM3Dq or mCherry under the control of the glutamatergic cell-specific CaMKIIα promoter (Figure 6A). Two weeks after SNI, we imaged GCaMP6s-expressing pyramidal neurons in the ACC. Consistent with previous reports (Zhao et al., 2018), we observed a robust elevation of L5 pyramidal neuron Ca2+ in the ACC of SNI mice versus sham mice (Figure 6B). Thirty minutes after a single CNO injection to activate PL pyramidal neurons, pyramidal neuron activity in the ACC of SNI mice was decreased to a level comparable to that in sham mice, indicating that acute activation of PL pyramidal neurons corrects pyramidal neuron responses in the ACC.
Figure 6. Restoration of PL-ACC pathway reverses ACC hyperactivation in neuropathic pain mice.
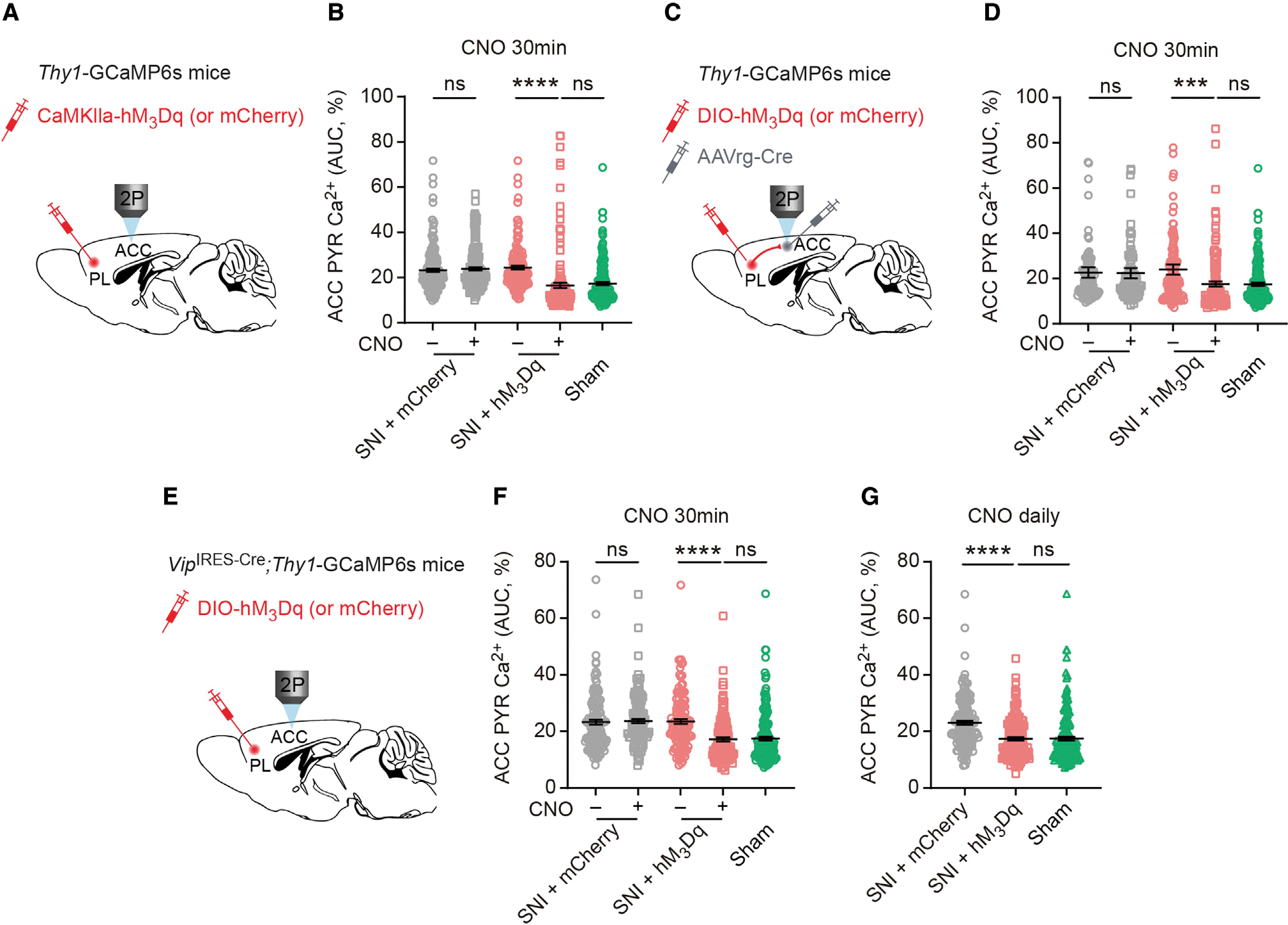
(A) Experimental design to express hM3Dq or mCherry in PL PYRs. Two weeks after SNI, in vivo Ca2+ imaging of ACC PYRs was performed before and 30 min after CNO injection.
(B) Average Ca2+ activity of ACC PYRs before and after CNO application in SNI mice expressing mCherry (p = 0.1809; n = 178 cells from 4 mice, 88 and 90 cells from 2 males and 2 females, respectively) or hM3Dq (p < 0.0001; n = 152 cells from 4 mice, 78 and 74 cells from 2 males and 2 females, respectively). Activation of PL PYRs in SNI mice reduces ACC pyramidal activity to the level of sham mice (p = 0.5084).
(C) Experimental design for expressing hM3Dq or mCherry in PL-ACC projection neurons and in vivo Ca2+ imaging in ACC PYRs.
(D) Average Ca2+ activity of ACC PYRs before and after CNO application in SNI mice expressing mCherry (p = 0.5206; n = 114 cells from 3 females) or hM3Dq (p = 0.0006; n = 133 cells from 3 females). Activation of PL-ACC projection neurons in SNI mice reverts ACC neuron activity to the level of sham mice (p = 0.9259).
(E) Experimental design for expressing hM3Dq or mCherry in PL VIP interneurons and in vivo Ca2+ imaging in the ACC.
(F) Average Ca2+ activity of ACC PYRs before and after CNO application in SNI mice expressing mCherry (p = 0.5103; n = 133 cells from 3 males) or hM3Dq (p < 0.0001; n = 127 cells from 3 males). Acute activation of PL VIP neurons in SNI mice reverses ACC PYR hyperactivation (p = 0.9677).
(G) Daily activation of PL VIP neurons during the first week after SNI prevents ACC PYR hyperactivation (p < 0.0001 versus mCherry, p = 0.3635 versus sham; n = 157, 147, and 176 cells from 4 mice per group, with 71, 78, and 84 cells from males and 86, 69, and 92 cells from females, respectively).
Summary data are mean ± SEM. ***p < 0.001, ****p < 0.0001; ns, not significant; by Mann-Whitney test for unpaired comparison or Wilcoxon test for paired comparison.
We next selectively activated PL-ACC projection neurons by injecting AAVrg-Cre and Cre-dependent hM3Dq/mCherry, respectively, into the ACC and PL of Thy1-GCaMP6s mice (Figure 6C). Two weeks after SNI, we found that a single CNO injection to acutely activate PL–ACC projection neurons significantly reduced the activity of L5 pyramidal neurons in the ACC to the sham level (Figure 6D).
Lastly, we tested whether activation of PL VIP neurons could diminish ACC pyramidal neuron hyperactivation associated with neuropathic pain. In this experiment, we injected Cre-dependent hM3Dq/mCherry into the PL of VipIRES–Cre; Thy1-GCaMP6s mice (Figure 6E). In SNI mice expressing hM3Dq in PL VIP neurons, we found that one injection of CNO significantly decreased the activity of L5 pyramidal neurons in the ACC (p < 0.0001 versus CNO−, p = 0.9677 versus sham; Figure 6F). Moreover, daily activation of PL VIP neurons for 1 week was sufficient to prevent SNI-induced ACC hyperactivation. Two weeks after SNI (i.e., 1 week after the last CNO injection), the level of pyramidal neuron activity in the ACC of SNI mice expressing hM3Dq remained comparable to that of sham mice (Figure 6G).
Collectively, these three sets of experiments demonstrate that activation of PL outputs to the ACC prevents ACC pyramidal neuron hyperactivation associated with neuropathic pain.
PL provides feedforward inhibition to ACC local circuits
Finally, we examined the connectivity in the PL-ACC pathway using an AAV-mediated anterograde transsynaptic tracing strategy (Figure 7) (Zingg et al., 2017). Specifically, we injected AAV1-FLPo and FLP-dependent mCherry, respectively, into the PL and ACC of C57BL/6 or GAD2IRES–Cre mice (Figures 7A and 7C). As AAV serotype 1 (AAV1) exhibits anterograde transsynaptic spread properties, this injection scheme enables the selective expression of mCherry in ACC neurons receiving PL projections (ACCPL-ACC neurons). To reveal the identity of these neurons, we simultaneously injected CaMKIIα-EGFP into the ACC of C57BL/6 mice and Cre-dependent EGFP into the ACC of GAD2IRES–Cre mice (Figures 7A and 7C). Three weeks after viral infections, ACC neurons expressing mCherry (ACCPL-ACC neurons) and/or EGFP (glutamatergic neurons or GABAergic neurons) were visualized in brain sections using confocal imaging (Figures 7B and 7D). Quantification showed that ~56% and 35% of ACCPL-ACC neurons were GAD2+ and CaMKIIα+ cells, respectively (Figure 7E). Together with our findings in Figures 2 and 6, these results suggest that the PL primarily sends glutamatergic projections onto GABAergic neurons in the ACC, providing a feedforward inhibition to ACC local circuits.
Figure 7. PL projection neurons mostly innervate GABAergic cells in the ACC.
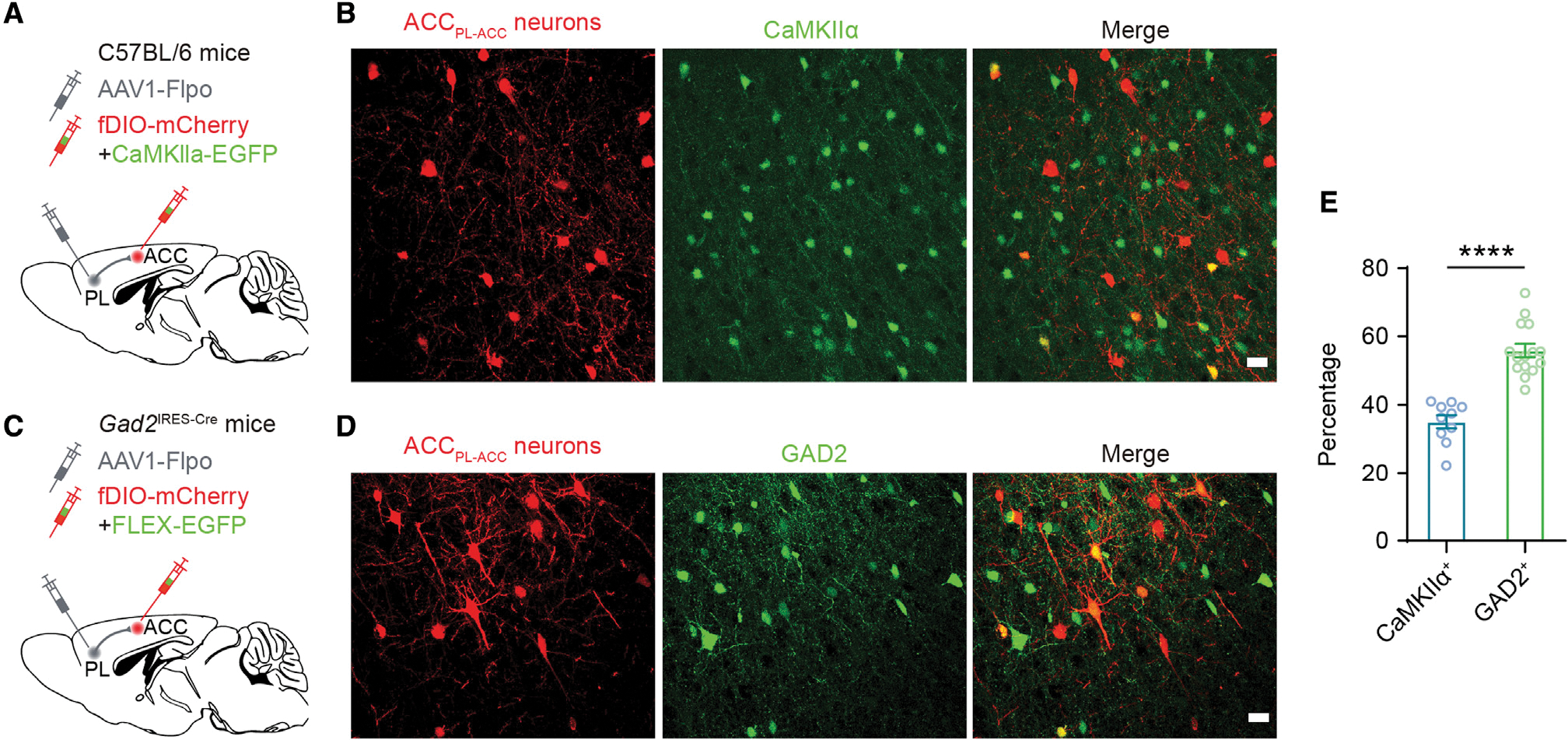
(A) Experimental design for selective expression of mCherry in ACC neurons receiving PL projections (ACCPL-ACC neurons) and EGFP in CaMKIIα+ neurons in the ACC.
(B) Fluorescence images showing ACCPL-ACC neurons and CaMKIIα+ neurons in the ACC. Scale bar, 20 μm.
(C) Experimental design for expressing mCherry in ACCPL-ACC neurons and EGFP in GAD2+ neurons in the ACC.
(D) Fluorescence images showing ACCPL-ACC neurons and GAD2+ neurons in the ACC. Scale bar, 20 μm.
(E) Percentages of ACCPL-ACC neurons expressing CaMKIIα (~35%; n = 15 sections from 5 mice) or GAD2 (~56%; n = 10 sections from 4 mice).
Summary data are mean ± SEM. ****p < 0.0001; by Mann-Whitney test.
DISCUSSION
Following peripheral nerve damage, dysfunction of mPFC contributes to the development of persistent neuropathic pain (Ji et al., 2010; Ott and Nieder, 2019). How prefrontal cortical circuits are reorganized in this chronic pain condition remains unclear. Using virus-based circuit tracing and in vivo two-photon Ca2+ imaging, we found that pyramidal neurons located in L5 of the PL, including PL-ACC projection neurons, were less active in a mouse model of peripheral neuropathic pain. This reduction of PL glutamatergic outputs to the ACC was facilitated by a decrease of VIP interneuron Ca2+ signals in the PL. Chemogenetic activation of PL VIP neurons restored PL outputs to the ACC, reduced ACC pyramidal neuron hyperactivity, and prevented the development of neuropathic pain. These findings reveal maladaptive alterations to the PL-ACC pathway after peripheral nerve injury and suggest that manipulation of PL VIP neuron activity could be an effective strategy for treating chronic neuropathic pain.
Many lines of evidence suggest that deactivation of pyramidal neurons in the mPFC is involved in the development of chronic pain. Using patch clamp in brain slices, previous studies showed that rodents with chronic neuropathic and inflammatory pain exhibit a significant reduction in excitability for L5 pyramidal neurons in prelimbic mPFC (Cheriyan and Sheets, 2018; Kelly et al., 2016; Mitric et al., 2019; Wang et al., 2015; Zhang et al., 2015). Single-unit and population recordings in vivo confirmed that both spontaneous and evoked firing rates of mPFC neurons are suppressed after induction of arthritic or inflammatory pain (Dale et al., 2018; Ji and Neugebauer, 2011; Ji et al., 2010). More recently, in vivo Ca2+ imaging has been employed to understand pain-related cellular and circuit changes in the neocortex (Cichon et al., 2017). A recent study in rats reported that inflammatory pain reduces the overall nociceptive responses of excitatory neurons in prelimbic mPFC (Li et al., 2021a). Consistently, our data showed that the spontaneous Ca2+ activity of L5 pyramidal neurons in the PL is decreased in a mouse model of neuropathic pain. Moreover, spontaneous activity of PL neurons projecting to the ACC is also decreased. Together with previous reports of reduced excitation of mPFC-PAG projections in neuropathic pain (Cheriyan and Sheets, 2018; Huang et al., 2019), these findings imply a generalized reduction of mPFC outputs to its downstream targets, both subcortical and intracortical.
The circuit mechanisms underlying deactivation of mPFC pyramidal neurons remain unclear. Within the mPFC, there is a large diversity of GABAergic interneurons, including PV, SST, and VIP interneurons. These interneurons innervate specific domains of pyramidal neurons and other interneurons, providing a precise spatiotemporal control of mPFC outputs (Tremblay et al., 2016). Specifically, VIP interneurons preferentially inhibit PV and SST cells that provide somatic and dendritic inhibition of neighboring pyramidal neurons (Pfeffer et al., 2013; Pi et al., 2013; Silberberg and Markram, 2007). VIP neurons in the mPFC have been shown to be involved in attention and avoidance behaviors (Lee et al., 2019; Obermayer et al., 2019), but their role in pain perception is not known. It is also unknown whether VIP neurons could change activity in chronic pain conditions. By performing Ca2+ imaging in the PL of awake mice, we showed that VIP neuron activity is downregulated during the development and maintenance of neuropathic pain. This VIP neuron deactivation may lead to an increased inhibitory drive and, consequently, a decreased activation of PL pyramidal neurons. Indeed, it was reported that PV interneurons in the PL are increasingly activated under chronic pain conditions (Zhang et al., 2015). It is worth noting that a small fraction of VIP interneurons in the mouse cortex express acetylcholine and may directly excite local pyramidal neurons through fast cholinergic synaptic transmission (Granger et al., 2020; Obermayer et al., 2019; Tasic et al., 2018). Future studies are needed to better understand how different subclasses of VIP interneurons modulate prefrontal cortical activity during pain perception.
An increasing body of literature suggests sex dimorphism in the pathophysiology of chronic pain (Dawes and Bennett, 2021; Mogil, 2012). Recent studies in experimental animals showed that nerve injury enhances the excitability of PV interneurons in L5 of the PL of male, but not female, mice but reduces the firing rates of SST interneurons in L3 of the PL of female, but not male, mice (Jones and Sheets, 2020). Our Ca2+ imaging data showed that in both male and female mice, nerve injury causes similar alterations to pyramidal neurons and VIP interneurons in L5 of the PL. These results suggest no detectable sex differences in pain-induced maladaptive alterations to somatic Ca2+ in PL neurons. Notably, in the PL of sham mice, we observed a small difference in the frequency of VIP Ca2+ transients between males and females.
Our study provides evidence that impaired VIP neuron activity is involved in the development of neuropathic pain. Given the reduction of VIP Ca2+ activity in SNI mice, we acutely manipulated the activity of VIP neurons and demonstrated that VIP cell activation was sufficient to disinhibit PL pyramidal neurons. Importantly, daily activation of VIP cells for 1 week following SNI diminished PL pyramidal neuron hypoactivity, mitigated mechanical and thermal allodynia, and abolished the ongoing pain. These findings suggest that the initiation of chronic neuropathic pain could be prevented by early treatments targeting the abnormal activity in the pain pathways. This is consistent with our previous finding that continuous activation of SST interneurons in the somatosensory cortex for 1 week following SNI prevents the development of mechanical allodynia in mice (Cichon et al., 2017). However, it was also reported that peripheral nerve blockade for 1 week in a rat SNI model has no effect on the development of allodynia (Suter et al., 2003). This discrepancy is possibly because, besides peripheral discharge from damaged axons, ectopic activity arising within dorsal root ganglia also contributes significantly to the ascending pain flow (Wall and Devor, 1983).
The mPFC projects extensively to the ACC (Gao et al., 2022), but little is known about this cortical-cortical pathway and its function in chronic pain. In this study, we used both anterograde and retrograde viral labeling approaches to specifically identify PL neurons projecting to the ACC, as well as ACC neurons receiving projections from the PL. Our results showed that the vast majority of PL-ACC projection neurons are glutamatergic. While these excitatory PL neurons project onto both glutamatergic and GABAergic cells in the ACC, a larger fraction of them are inhibitory, implying an overall feedforward inhibition from the PL to the ACC. Indeed, chemogenetic activation of PL projection neurons potently reduced the pyramidal neuronal activity in the ACC. These results suggest a model in which peripheral nerve injury preferentially diminishes PL glutamatergic projections onto ACC GABAergic cells, ultimately increasing ACC pyramidal neuron activity. In support, direct activation of PL-ACC projection neurons reverses ACC pyramidal neuron hyperactivity in neuropathic pain. Our findings reveal a cortical-cortical pathway by which the mPFC modulates ACC circuits, and maladaptive changes to this pathway may contribute to ACC hyperactivation in chronic pain.
As a key component in central pain pathways, the ACC - receives direct projections from the medial thalamic nuclei and is particularly involved in processing affective aspects of pain (Hogrefe et al., 2022; Yang et al., 2006; Zhu et al., 2021). Cellular dysfunctions in the ACC, such as enhanced excitatory synaptic transmission, increased intrinsic cellular excitability, and decreased inhibitory postsynaptic currents, have been extensively documented in animal pain models (Blom et al., 2014; Koga et al., 2015; Santello and Nevian, 2015; Xu et al., 2008). With neuropathic pain, both spontaneous and stimuli-evoked activity is increased in ACC pyramidal neurons, the degree of which is correlated with pain sensitivity in mice (Zhao et al., 2018). Previous studies have shown that targeting local circuits in the ACC can relieve pain hypersensitivity and aversion in animals with chronic pain conditions (Juarez-Salinas et al., 2019; Sellmeijer et al., 2018; Zhang et al., 2017; Zhao et al., 2006; Zhou et al., 2018). Built on these studies, our data showed that in mice with neuropathic pain, activation of PL-ACC projections is sufficient to attenuate mechanical and thermal allodynia and ameliorate negative affects associated with neuropathic pain.
Limitations of the study
Our study in mice revealed that alterations to prefrontal cortical circuits associated with neuropathic pain involve the deactivation of PL projections to the ACC, in the virtue of VIP interneurons. However, a limitation of our study is that we were not able to determine whether the firing rates of VIP cells are indeed altered by neuropathic pain, as calcium signals are not equivalent to neuronal spikes. In addition, our study demonstrated that chemogenetic activation of PL VIP interneurons restores neuronal responses in PL-ACC circuits and ameliorates pain-like behaviors in mice. However, it is possible that activation of VIP cells may also disinhibit PL projections to PAG, a key midbrain structure involved in central pain integration. It is likely that the beneficial effects of prefrontal activation in alleviating pain are synergistically mediated by its cortical-cortical and cortical-subcortical projections.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Guang Yang (gy2268@cumc.columbia.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Summary data reported in this paper are provided in Table S1.
No original code has been generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J (Stock No: 000664), VipIRES–Cre (Stock No: 010908) and Gad2IRES-Cre (stock No: 010802) mice were purchased from the Jackson Laboratory. Transgenic mice expressing GCaMP6 slow in pyramidal neurons, Thy1.2-GCaMP6s founder line 3, have been described in the previous publication (Cichon et al., 2020). To image cortical pyramidal neuron activity while chemogenetically manipulating VIP interneurons, VipIRES-Cre mice were crossed with Thy1-GCaMP6s mice. Mice were group-housed in temperature- and humidity-controlled rooms on a 12-h light-dark cycle and were assigned to different treatment groups using simple random sampling. Specifically, mice were assigned random numbers generated by the Excel program that allocated them to one of the treatment groups. Two-to three-month-old mice of both sexes were used for all the experiments. All animal procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at Columbia University as consistent with National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals.
METHOD DETAILS
Spared nerve injury
Spared nerve injury (SNI) was performed by ligation and transection of the tibial and common peroneal branches of the sciatic nerve, leaving the sural nerve intact (Cichon et al., 2018; Shields et al., 2003). Briefly, mice were deeply anesthetized with an intraperitoneal (i.p.) injection of 100 mg/kg ketamine and 15 mg/kg xylazine. Using sterile techniques, a small incision was made in the right thigh to expose the sciatic nerve. The tibial and common peroneal branches of the sciatic nerve were ligated and transected, leaving the sural nerve intact. Muscle and skin were closed and sutured in two layers. For sham surgery, the sciatic nerve was exposed but not ligated or cut. Throughout the surgical procedure and recovery, the animal’s body temperature was maintained at ~37°C. The experimenters were blinded to treatment groups during subsequent Ca2+ imaging, behavioral and immunohistochemistry studies.
Surgical preparation for imaging awake, head-restrained mice
In vivo Ca2+ imaging was carried out in the PL and the ACC of awake, head-restrained mice as previously described (Li et al., 2021b; Yang et al., 2013; Zhao et al., 2018). Specifically, mice were deeply anesthetized with an i.p. injection of 100 mg/kg ketamine and 15 mg/kg xylazine. After shaving the fur, the skull surface was exposed with a midline scalp incision and the periosteum tissue was removed. A head bar was subsequently glued to the animal’s skull to reduce motion artifact during imaging. A small skull region (~1 mm in diameter) over the PL (anterior-posterior (AP) +2.68 mm, medial-lateral (ML) 0.5 mm) or ACC (AP +0.75 mm, ML 0.5 mm) was removed with precaution to avoid damaging the dura matter. A round glass coverslip (approximately the same size as the bone being removed) was glued onto the skull. Dental acrylic cement was applied to the surrounding area to further secure the glass window. Throughout the surgical procedure and recovery, the animal’s body temperature was kept at ~37°C.
Upon awakening, mice with head bars were habituated three times (10 min each time) to the imaging platform to minimize potential stress related to head restraining and imaging. Imaging experiments were performed 24 h after window implantation, free from anesthetic effects.
In vivo Ca2+ imaging and data analysis
The genetically encoded Ca2+ indicator GCaMP6s was used for Ca2+ recording. To image VIP neurons, Cre-dependent GCaMP6s was expressed with recombinant adeno-associated virus (AAV) under the synapsin promoter (AAV9-Syn-FLEx-GCaMP6s; 100845; Addgene). 0.2 μL of AAV was slowly injected (Picospritzer III; 15 p.s.i., 10 ms, 0.5 Hz) over 10–15 min into the left PL of VipIRES–Cre mice using a glass microelectrode around the coordinates of AP +2.68 mm, ML 0.5 mm, subpial (SP) 0.85 mm. For Ca2+ imaging of PL neurons projecting to the ACC, 0.1 μL of retrograde AAV encoding FLP (AAVrg-EF1a-FLPo; 55637; Addgene) was injected into the left ACC (AP +0.75 mm, ML 0.5 mm, SP 1 mm) and 0.1 μL of FLP-dependent GCaMP6s (AAV8-EF1a-fDIO-GCaMP6s; 105714; Addgene) was injected into the left PL of C57BL/6 mice. Thy1-GCaMP6s mice were utilized for imaging pyramidal neurons in the PL and the ACC in combination with chemogenetic manipulation.
All mice infected with AAV were prepared for head-fixation and imaging 2–3 weeks following AAV injections. In vivo Ca2+ imaging was performed with a Scientifica two-photon system equipped with a Ti:Sapphire laser (Chameleon Vision-S, Coherent) tuned to 920 nm. All experiments were performed using a 25× objective (1.05 N.A.) immersed in an artificial cerebrospinal fluid solution, with a digital zoom of 1×. All images were acquired at a frame rate of ~1.69 Hz (2-ms pixel dwell time) and a resolution of 512 × 512 pixels. Image acquisition was performed using ScanImage software and analyzed post hoc using NIH ImageJ software.
During quiet resting, motion artifact due to respiration and heartbeat was typically less than 2 μm as detected in our cortical measurements. Vertical movements were infrequent and minimized by habituation, the head bar attached to the animal’s skull, and a custom-built imaging platform. If the animal struggled on the imaging platform, imaging time points from those segments were excluded from data analysis.
Regions of interest (ROIs) corresponding to visually identifiable somas were selected for quantification as previously described (Cichon et al., 2017). L5 neurons were identified based on their somatic distance to the midline. The fluorescence time course of each cell was measured by averaging all pixels within the ROIs covering the somas. A background value was first subtracted from GCaMP6 fluorescence values. All Ca2+ transients were calculated as ΔF/F0, where ΔF/F0 is (F−F0)/F0 and F0 is the baseline value defined as averaged fluorescence over a 6-s period of lowest fluorescence signal during the recording period. The average integrated Ca2+ activity was quantified as the area under the curve (AUC) of ΔF/F0 over 1 min. The threshold for a Ca2+ event was >3 × s.d. of the baseline fluorescence of GCaMP6s. The peak amplitude was the average of peak ΔF/F0 of each Ca2+ transient over 1 min. The frequency was the number of Ca2+ transients per min. The duration was the average of the full width of each Ca2+ transient over 1 min recording.
Chemogenetic manipulation
To activate VIP neurons in vivo, 0.2 μL of Cre-dependent AAV encoding hM3Dq (AAV9-hSyn-DIO-hM3Dq-mCherry; 44361; Addgene) or mCherry (AAV9-hSyn-DIO-mCherry; 50459; Addgene) was injected into the left PL (AP +2.68 mm, ML 0.5 mm, SP 0.85 mm) of VipIRES-Cre mice or VipIRES-Cre; Thy1-GCaMP6s mice. For activation of pyramidal neurons in the PL, 0.2 μL of AAV encoding hM3Dq or mCherry under a CaMKIIα promotor (AAV8-CaMKIIα-hM3Dq-mCherry, AAV1-CaMKIIα-mCherry; 50476, 114469; Addgene) was delivered to the left PL. To specifically manipulate PL–ACC projection neurons, 0.1 μL of retrograde AAV encoding Cre (AAVrg-Pgk-Cre; 24593; Addgene) was injected into the left ACC (AP +0.75 mm, ML 0.5 mm, SP 1 mm) and 0.1 μL of Cre-dependent hM3Dq or mCherry was injected into the left PL of Thy1-GCaMP6s mice.
The right hindlimb contralateral to the AAV injection site was subjected to sham or SNI surgery. For acute activation experiments, SNI mice expressing hM3Dq or mCherry were administrated 5 mg/kg Clozapine N-oxide (CNO, 1 mg/mL in saline, i.p.) 30 min before imaging and behavioral experiments. For daily activation experiments, SNI mice were injected 5 mg/kg CNO daily, at 8 am each day, for a total of 7 days after SNI. Following chemogenetic manipulation, Ca2+ recordings and behavioral assessments were performed in different sets of animals.
von Frey tests
We measured the animals’ paw withdrawal thresholds using an up/down method as previously described with minor modifications (Dixon, 1980). The minor modification to the present up/down method versus Dixon/Chapman is that we started with the smallest filament for testing rather than the median (middle) filament for withdrawal assessment. In brief, animals were individually placed in clear acrylic boxes (10 cm × 7 cm × 7 cm) placed over a mesh table and habituated for 30 min prior to testing procedures. A series of von Frey fibers (0.008, 0.02, 0.07, 0.16, 0.4, 0.6, 1.0, 1.4, 2.0, and 4.0 g) were presented in a consecutive ascending order. In the absence of paw withdrawal response, the next stronger stimulus was applied; in the event of paw withdrawal, the next weaker stimulus was chosen. After the response threshold was first crossed, six data points were counted, at which time the two responses straddling the threshold were retrospectively designated as the first two responses of the series of 6. 50% response threshold was calculated as: 50% g threshold = 10(Xf + κδ − 4), where Xf is the value (in log units) of the final von Frey fiber used; κ is the tabular value for the pattern of positive/negative responses (Chaplan et al., 1994); and δ is the mean difference (in log units) between stimuli (here, 0.2699).
Thermal tests
Cold allodynia was tested using a custom-made cold plate assembled with a thermoelectric Peltier cold plate (10 cm × 8 cm) (CP-031; TE Technologies, USA) and a temperature controller (TC-720; TE Technologies, USA). Hot allodynia was assessed on a hot plate (Ugo Basile 7280; Italy). Before testing, mice were individually placed in a clear acrylic container (10 cm × 7 cm × 20 cm) placed on the plate at room temperature and habituated for 30 min. During testing, the temperature was set at 0°C for cold plate and 50°C for hot plate. A stopwatch was used to record the latency of paw withdrawal response which was determined by hind paw lifting coupled with flinching. For each animal, paw withdrawal latency was measured 3 times with a 30-min interval.
Conditioned place preference
The conditioned place preference (CPP) test was performed in a two-compartment CPP chamber (Ugo Basile; Italy). The chamber (32 cm × 15 cm × 25 cm) consists of two compartments of the same size connected through a removable door (4 cm wide × 6 cm high). Each compartment has distinct visual and textured cues (e.g., wall patterns, floor patterns, and texture). The CPP test was performed 2 weeks after SNI. In the pre-conditioning phase (day 1), mice were placed in the CPP chamber with the door removed, and recorded for 20 min per session, a total of 2 sessions. The time mice spent in each compartment was recorded. The animals showing strong unconditioned preference (>75% of time in one compartment) were excluded. During conditioning (days 2 and 3), mice were injected with saline (0.1 mL per 20 g body weight, i.p.) in the morning and ~30 min later, confined to the paired compartment for 30 min; about 6 h later, mice were injected with CNO (1 mg/mL, 0.1 mL per 20 g body weight, i.p.) and ~30 min later, placed in the other compartment for 30 min. For CPP tests using lidocaine (0.2% in 50 μL saline), the same conditioning procedures were used except that lidocaine or saline was injected into the nerve injury site and mice were immediately confined to the paired compartment. Compartment assignments were counterbalanced among all the test mice. Test was carried out on day 4. Mice were allowed free access to both compartments of CPP chamber for 20 min. The time spent in each compartment was recorded for each animal. The change of preference was calculated as the ratio of time spent in the drug-paired compartment between the test and pre-conditioning phase: The preference for treatment side = test/preconditioning × 100%.
Novel object recognition test
Novel object recognition test was performed in an opaque box (40 cm × 40 cm × 35 cm). Mice were first habituated to the testing environment for 10 min with no objects in the box. Twenty-four hours after the habituation, mice were placed in a testing environment with two identical sample objects (the familiar object) placed in neighboring corners, for 10 min. Six hours later, mice were returned to the testing environment for 10 min, with one of the sample objects replaced by a novel object. Behavior was videotaped and analyzed with ANY-maze software (Stoelting, Wood Dale, IL). Measures of interaction were taken as the amount of time the animal spent with its head and nose oriented toward and within 2 cm of the object. The discrimination ratio was calculated as the time spent exploring the novel object divided by the total time spent with both novel and familiar objects.
T maze test
T maze test was conducted in a chamber consisting of three arms of equal length (Stoelting, Wood Dale, IL). During the training session, mice were placed in the designated start arm and allowed access to two of three arms for 2 min. A removable door was used to prevent mice from accessing the third arm. Six hours later, mice were returned to the start location in the T maze and allowed to explore all three arms for 2 min. Behavior was videotaped and analyzed with ANY-maze software. Novel arm preference was calculated as the time spent in the novel arm divided by the time spent in both novel and familiar arms.
Cell-type specific circuit dissection
To label ACC neurons receiving monosynaptic projections from the PL (ACCPL-ACC neurons), 0.1 μL of AAV1-EF1a-FLPo (55,637; Addgene) was injected into the PL (AP +2.68 mm, ML 0.5 mm, SP 0.85 mm) to allow the virus to spread anterogradely to postsynaptic cells to express FLP. Simultaneously, 0.1 μL mixture of FLP-dependent mCherry (AAV1-EF1a-fDIO-mCherry; 114471; Addgene) and AAV5-CaMKIIα-EGFP (50469; Addgene) (dilute 20x, 1:1) was injected into the ACC (AP +0.75 mm, ML 0.5 mm, SP 1 mm) to visualize ACCPL-ACC neurons and CaMKIIα+ neurons in C57BL/6 mice. To visualize GABAergic neurons, Cre-dependent EGFP (AAV1-CAG-FLEx-EGFP; 51502; Addgene) was injected into the ACC of Gad2IRES-Cre mice. Three weeks following AAV expression, mice were perfused, and brain slices were obtained for analyzing mCherry and EGFP signals.
Immunohistochemistry and confocal imaging
Mice were deeply anesthetized and transcardially perfused with a phosphate-buffered solution (PBS) followed by 4% paraformaldehyde (PFA). The brains were carefully removed, post-fixed in 4% PFA at 4°C overnight and then incubated in 20% and 30% (w/v) sucrose, each for 24 h to allow sufficient dehydration. Coronal sections in 20 μm thickness were prepared using a cryostat (Microm HM505E). For immunofluorescence, floating sections were permeabilized and blocked in 0.1% Triton X-100 with 10% goat serum in PBS for 1 h at room temperature, and then incubated overnight at 4°C with primary antibodies: rabbit anti-GFP (1:500, ab6556, Abcam), mouse anti-GFP (1:500, SAB2702197, Sigma Aldrich), mouse anti-CaMKIIα (1:300, 50049S, CST), mouse anti-GAD65 (1:100, ab26113, Abcam), mouse anti-VGAT (1:100, 131011, Synaptic Systems), rabbit anti-VIP (1:500, 20077, Immunostar), mouse anti-NeuN (1:500, MAB377, Sigma Aldrich). The sections were washed 3 times with PBS and incubated with fluorophore-conjugated secondary antibodies (1:400, Invitrogen) for 2 h at room temperature. After washing, the sections were mounted in medium (010001; SouthernBiotech) for confocal imaging.
Confocal imaging was performed using a Nikon Ti laser scanning confocal system using a 10× or 20× objective. Images were captured at 1024 × 1024 pixels with a resolution of 0.622 μm/pixel for visualizing somas and 0.207 μm/pixel for visualizing axonal terminals. Z-stacks of images (20 μm thick) were collected at 2.5-μm step sizes for somas and 0.7-μm step sizes for axons and projected at a maximum intensity to create the final multi-channel images, which were then analyzed using ImageJ software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Summary data are presented as mean ± S.E.M. Sample sizes were chosen to ensure adequate power with the statistical analyses while minimizing the number of animals used. No animals that were successfully imaged or measured were excluded from the analysis. Statistical tests were determined after data collection using the data normality test. Because normality was rejected, we performed nonparametric statistical tests. To compare two groups, we used Mann-Whitney U test (or Wilcoxon matched-pairs signed rank test) for unpaired (or paired) comparison. To compare multiple matched groups, we used Friedman test followed by Dunn’s multiple comparisons test. Significant levels were set at p ≤ 0.05. All statistical analyses were performed using GraphPad Prism software.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit polyclonal anti-GFP | Abcam | Cat#ab6556; RRID:AB_305564 |
| Mouse monoclonal anti-GFP | Sigma Aldrich | Cat#SAB2702197 |
| Mouse monoclonal anti- CaMKIIα | Cell Signaling Technology | Cat#50049S; RRID:AB_2721906 |
| Mouse monoclonal anti-GAD65 | Abcam | Cat#ab26113; RRID:AB_448989 |
| Mouse monoclonal anti-VGAT | Synaptic Systems | Cat#131011; RRID:AB_887872 |
| Rabbit polyclonal anti-VIP | ImmunoStar | Cat#20077; RRID:AB_572270 |
| Mouse monoclonal anti-NeuN | Millipore | Cat#MAB377; RRID:AB_2298772 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| AAVrg-EF1a-FLPo | Addgene | Cat#55637-AAVrg |
| AAV8-EF1a-fDIO-GCaMP6s | Addgene | Cat#105714-AAV8 |
| AAV9-Syn-FLEX-GCaMP6s | Addgene | Cat#100845-AAV9 |
| AAV1-phSyn1(S)-FLEX-tdTomato-T2A-SypEGFP | Addgene | Cat# 51509-AAV1 |
| AAV9-hSyn-DIO-hM3Dq-mCherry | Addgene | Cat#44361 -AAV9 |
| AAV9-hSyn-DIO-mCherry | Addgene | Cat#50459-AAV9 |
| AAV8-CaMKIIα-hM3Dq-mCherry | Addgene | Cat#50476-AAV8 |
| AAV1-CaMKIIα-mCherry | Addgene | Cat#114469-AAV1 |
| AAVrg-Pgk-Cre | Addgene | Cat#24593-AAVrg |
| AAV1-EF1a-FLPo | Addgene | Cat#55637-AAV1 |
| AAV1-EF1a-fDIO-mCherry | Addgene | Cat#114471-AAV1 |
| AAV5-CaMKIIα-EGFP | Addgene | Cat#50469-AAV5 |
| AAV1-CAG-FLEX-EGFP | Addgene | Cat#51502-AAV1 |
|
| ||
| Chemicals | ||
|
| ||
| Clozapine N-oxide | Tocris | Cat#4936 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | JAX | Stock No: 000664; RRID:IMSR_JAX:000664 |
| Mouse: VipIRES-Cre: Viptm1(cre)Zih/J | JAX | Stock No: 010908; RRID:IMSR_JAX:010908 |
| Mouse: Gad2IRES-Cre: Gad2tm2(cre)Zih/J | JAX | stock No: 010802; RRID:IMSR_JAX:010802 |
| Mouse: Thy1-GCaMP6s: Thy1.2-GCaMP6s founder lines 3 | Cichon et al., 2020 | N/A |
| Mouse: VipIRES-Cre; Thy1-GCaMP6s | Crossed line by original lines listed above | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ Fiji | NIH | https://imagej.net/software/fiji/downloads |
| GraphPad Prism 9.0 | Graphpad Software Inc. | https://www.graphpad.com/ |
| ANY-maze software | Stoelting Co. | https://www.any-maze.com/ |
Highlights.
Neuropathic pain reduces VIP neuronal activity in the medial prefrontal cortex (mPFC)
Neuropathic pain deactivates mPFC outputs to the anterior cingulate cortex (ACC)
Activation of VIP interneurons restores mPFC-ACC outputs and relieves pain in mice
Restoration of mPFC-ACC outputs reverses neuropathic pain-induced ACC hyperactivation
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health R01AA027108 (G.Y.) and R35GM131765 (G.Y.) and a Columbia University Medical Center Target of Opportunity award to the Department of Anesthesiology.
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflict of interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111333.
REFERENCES
- Alexander GM, Rogan SC, Abbas AI, Armbruster BN, Pei Y, Allen JA, Nonneman RJ, Hartmann J, Moy SS, Nicolelis MA, et al. (2009). Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39. 10.1016/j.neuron.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, and Gitelman DR (2004). Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J. Neurosci. 24, 10410–10415. 10.1523/JNEUROSCI.2541-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL, Kaang BK, and Zhuo M (2016). Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat. Rev. Neurosci. 17, 485–496. 10.1038/nrn.2016.68. [DOI] [PubMed] [Google Scholar]
- Blom SM, Pfister JP, Santello M, Senn W, and Nevian T (2014). Nerve injury-induced neuropathic pain causes disinhibition of the anterior cingulate cortex. J. Neurosci. 34, 5754–5764. 10.1523/JNEUROSCI.3667-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso-Cruz H, Sousa M, Vieira JB, Lima D, and Galhardo V (2013). Prefrontal cortex and mediodorsal thalamus reduced connectivity is associated with spatial working memory impairment in rats with inflammatory pain. Pain 154, 2397–2406. 10.1016/j.pain.2013.07.020. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, and Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cheriyan J, Kaushik MK, Ferreira AN, and Sheets PL (2016). Specific targeting of the basolateral amygdala to projectionally defined pyramidal neurons in prelimbic and infralimbic cortex. eNeuro 3, ENEURO.0002, 16.2016. 10.1523/ENEURO.0002-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheriyan J, and Sheets PL (2018). Altered excitability and local connectivity of mPFC-PAG neurons in a mouse model of neuropathic pain. J. Neurosci. 38, 4829–4839. 10.1523/JNEUROSCI.2731-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichon J, Blanck TJJ, Gan WB, and Yang G (2017). Activation of cortical somatostatin interneurons prevents the development of neuropathic pain. Nat. Neurosci. 20, 1122–1132. 10.1038/nn.4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichon J, Magrané J, Shtridler E, Chen C, Sun L, Yang G, and Gan WB (2020). Imaging neuronal activity in the central and peripheral nervous systems using new Thy1.2-GCaMP6 transgenic mouse lines. J. Neurosci. Methods 334, 108535. 10.1016/j.jneumeth.2019.108535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichon J, Sun L, and Yang G (2018). Spared nerve injury model of neuropathic pain in mice. Bio. Protoc. 8, e2777. 10.21769/bioprotoc.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlhamer J, Lucas J, Zelaya C, Nahin R, Mackey S, DeBar L, Kerns R, Von Korff M, Porter L, and Helmick C (2018). Prevalence of chronic pain and high-impact chronic pain among adults - United States, 2016. MMWR Morb. Mortal. Wkly. Rep. 67, 1001–1006. 10.15585/mmwr.mm6736a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale J, Zhou H, Zhang Q, Martinez E, Hu S, Liu K, Urien L, Chen Z, and Wang J (2018). Scaling up cortical control inhibits pain. Cell Rep. 23, 1301–1313. 10.1016/j.celrep.2018.03.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawes JM, and Bennett DL (2021). Addressing the gender pain gap. Neuron 109, 2641–2642. 10.1016/j.neuron.2021.08.006. [DOI] [PubMed] [Google Scholar]
- Dixon WJ (1980). Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 20, 441–462. 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Gao L, Liu S, Gou L, Hu Y, Liu Y, Deng L, Ma D, Wang H, Yang Q, Chen Z, et al. (2022). Single-neuron projectome of mouse prefrontal cortex. Nat. Neurosci. 25, 515–529. 10.1038/s41593-022-01041-5. [DOI] [PubMed] [Google Scholar]
- Granger AJ, Wang W, Robertson K, El-Rifai M, Zanello AF, Bistrong K, Saunders A, Chow BW, Nuñez V, Turrero García M, et al. (2020). Cortical ChAT(+) neurons co-transmit acetylcholine and GABA in a target- and brain-region-specific manner. Elife 9, e57749. 10.7554/eLife.57749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogrefe N, Blom SM, Valentinova K, Ntamati NR, Jonker LJE, Nevian NE, and Nevian T (2022). Long-lasting, pathway-specific impairment of a novel form of spike-timing-dependent long-term depression by neuropathic pain in the anterior cingulate cortex. J. Neurosci. 42, 2166–2179. 10.1523/JNEUROSCI.0326-21.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Gadotti VM, Chen L, Souza IA, Huang S, Wang D, Ramakrishnan C, Deisseroth K, Zhang Z, and Zamponi GW (2019). A neuronal circuit for activating descending modulation of neuropathic pain. Nat. Neurosci. 22, 1659–1668. 10.1038/s41593-019-0481-5. [DOI] [PubMed] [Google Scholar]
- Ji G, and Neugebauer V (2011). Pain-related deactivation of medial prefrontal cortical neurons involves mGluR1 and GABA(A) receptors. J. Neurophysiol. 106, 2642–2652. 10.1152/jn.00461.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, and Neugebauer V (2014). CB1 augments mGluR5 function in medial prefrontal cortical neurons to inhibit amygdala hyperactivity in an arthritis pain model. Eur. J. Neurosci. 39, 455–466. 10.1111/ejn.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Sun H, Fu Y, Li Z, Pais-Vieira M, Galhardo V, and Neugebauer V (2010). Cognitive impairment in pain through amygdala-driven prefrontal cortical deactivation. J. Neurosci. 30, 5451–5464. 10.1523/JNEUROSCI.0225-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AF, and Sheets PL (2020). Sex-specific disruption of distinct mPFC inhibitory neurons in spared-nerve injury model of neuropathic pain. Cell Rep. 31, 107729. 10.1016/j.celrep.2020.107729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juarez-Salinas DL, Braz JM, Etlin A, Gee S, Sohal V, and Basbaum AI (2019). GABAergic cell transplants in the anterior cingulate cortex reduce neuropathic pain aversiveness. Brain 142, 2655–2669. 10.1093/brain/awz203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CJ, Huang M, Meltzer H, and Martina M (2016). Reduced glutamatergic currents and dendritic branching of layer 5 pyramidal cells contribute to medial prefrontal cortex deactivation in a rat model of neuropathic pain. Front. Cell. Neurosci. 10, 133. 10.3389/fncel.2016.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly CJ, and Martina M (2018). Circuit-selective properties of glutamatergic inputs to the rat prelimbic cortex and their alterations in neuropathic pain. Brain Struct. Funct. 223, 2627–2639. 10.1007/s00429-018-1648-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, and Porreca F (2009). Unmasking the tonic-aversive state in neuropathic pain. Nat. Neurosci. 12, 1364–1366. 10.1038/nn.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga K, Descalzi G, Chen T, Ko HG, Lu J, Li S, Son J, Kim T, Kwak C, Huganir RL, et al. (2015). Coexistence of two forms of LTP in ACC provides a synaptic mechanism for the interactions between anxiety and chronic pain. Neuron 85, 377–389. 10.1016/j.neuron.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AT, Cunniff MM, See JZ, Wilke SA, Luongo FJ, Ellwood IT, Ponnavolu S, and Sohal VS (2019). VIP interneurons contribute to avoidance behavior by regulating information flow across hippocampal-prefrontal networks. Neuron 102, 1223–1234.e4. 10.1016/j.neuron.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Manders TR, Eberle SE, Su C, D’Amour J, Yang R, Lin HY, Deisseroth K, Froemke RC, and Wang J (2015). Activation of corticostriatal circuitry relieves chronic neuropathic pain. J. Neurosci. 35, 5247–5259. 10.1523/JNEUROSCI.3494-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li A, Liu Y, Zhang Q, Friesner I, Jee HJ, Chen ZS, and Wang J (2021a). Disrupted population coding in the prefrontal cortex underlies pain aversion. Cell Rep. 37, 109978. 10.1016/j.celrep.2021.109978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Cabrera-Garcia D, Salling MC, Au E, Yang G, and Harrison NL (2021b). Alcohol reduces the activity of somatostatin interneurons in the mouse prefrontal cortex: a neural basis for its disinhibitory effect? Neuropharmacology 188, 108501. 10.1016/j.neuropharm.2021.108501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz AE, Yau HJ, Centeno MV, Apkarian AV, and Martina M (2009). Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc. Natl. Acad. Sci. USA 106, 2423–2428. 10.1073/pnas.0809897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrić M, Seewald A, Moschetti G, Sacerdote P, Ferraguti F, Kummer KK, and Kress M (2019). Layer- and subregion-specific electrophysiological and morphological changes of the medial prefrontal cortex in a mouse model of neuropathic pain. Sci. Rep. 9, 9479. 10.1038/s41598-019-45677-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS (2012). Sex differences in pain and pain inhibition: multiple explanations of a controversial phenomenon. Nat. Rev. Neurosci. 13, 859–866. 10.1038/nrn3360. [DOI] [PubMed] [Google Scholar]
- Obermayer J, Luchicchi A, Heistek TS, de Kloet SF, Terra H, Bruinsma B, Mnie-Filali O, Kortleven C, Galakhova AA, Khalil AJ, et al. (2019). Prefrontal cortical ChAT-VIP interneurons provide local excitation by cholinergic synaptic transmission and control attention. Nat. Commun. 10, 5280. 10.1038/s41467-019-13244-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SW, Harris JA, Ng L, Winslow B, Cain N, Mihalas S, Wang Q, Lau C, Kuan L, Henry AM, et al. (2014). A mesoscale connectome of the mouse brain. Nature 508, 207–214. 10.1038/nature13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott T, and Nieder A (2019). Dopamine and cognitive control in prefrontal cortex. Trends Cogn. Sci. 23, 213–234. 10.1016/j.tics.2018.12.006. [DOI] [PubMed] [Google Scholar]
- Pfeffer CK, Xue M, He M, Huang ZJ, and Scanziani M (2013). Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat. Neurosci. 16, 1068–1076. 10.1038/nn.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pi HJ, Hangya B, Kvitsiani D, Sanders JI, Huang ZJ, and Kepecs A (2013). Cortical interneurons that specialize in disinhibitory control. Nature 503, 521–524. 10.1038/nature12676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzicki D, Pollema-Mays SL, Sanz-Clemente A, and Martina M (2017). Loss of M1 receptor dependent cholinergic excitation contributes to mPFC deactivation in neuropathic pain. J. Neurosci. 37, 2292–2304. 10.1523/JNEUROSCI.1553-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, and Nevian T (2015). Dysfunction of cortical dendritic integration in neuropathic pain reversed by serotoninergic neuromodulation. Neuron 86, 233–246. 10.1016/j.neuron.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Sellmeijer J, Mathis V, Hugel S, Li XH, Song Q, Chen QY, Barthas F, Lutz PE, Karatas M, Luthi A, et al. (2018). Hyperactivity of anterior cingulate cortex areas 24a/24b drives chronic pain-induced anxiodepressive-like consequences. J. Neurosci. 38, 3102–3115. 10.1523/JNEUR-OSCI.3195-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields SD, Eckert WA 3rd, and Basbaum AI (2003). Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J. Pain 4, 465–470. 10.1067/s1526-5900(03)00781-8. [DOI] [PubMed] [Google Scholar]
- Silberberg G, and Markram H (2007). Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron 53, 735–746. 10.1016/j.neuron.2007.02.012. [DOI] [PubMed] [Google Scholar]
- Suter MR, Papaloïzos M, Berde CB, Woolf CJ, Gilliard N, Spahn DR, and Decosterd I (2003). Development of neuropathic pain in the rat spared nerve injury model is not prevented by a peripheral nerve block. Anesthesiology 99, 1402–1408. 10.1097/00000542-200312000-00025. [DOI] [PubMed] [Google Scholar]
- Tan LL, and Kuner R (2021). Neocortical circuits in pain and pain relief. Nat. Rev. Neurosci. 22, 458–471. 10.1038/s41583-021-00468-2. [DOI] [PubMed] [Google Scholar]
- Tasic B, Yao Z, Graybuck LT, Smith KA, Nguyen TN, Bertagnolli D, Goldy J, Garren E, Economo MN, Viswanathan S, et al. (2018). Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78. 10.1038/s41586-018-0654-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervo DGR, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D, et al. (2016). A designer AAV variant permits efficient retrograde Access to projection neurons. Neuron 92, 372–382. 10.1016/j.neuron.2016.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay R, Lee S, and Rudy B (2016). GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91, 260–292. 10.1016/j.neuron.2016.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban DJ, and Roth BL (2015). DREADDs (designer receptors exclusively activated by designer drugs): chemogenetic tools with therapeutic utility. Annu. Rev. Pharmacol. Toxicol. 55, 399–417. 10.1146/annurev-pharmtox-010814-124803. [DOI] [PubMed] [Google Scholar]
- Wager TD, Atlas LY, Lindquist MA, Roy M, Woo CW, and Kross E (2013). An fMRI-based neurologic signature of physical pain. N. Engl. J. Med. 368, 1388–1397. 10.1056/NEJMoa1204471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall PD, and Devor M (1983). Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain 17, 321–339. 10.1016/0304-3959(83)90164-1. [DOI] [PubMed] [Google Scholar]
- Wang GQ, Cen C, Li C, Cao S, Wang N, Zhou Z, Liu XM, Xu Y, Tian NX, Zhang Y, et al. (2015). Deactivation of excitatory neurons in the prelimbic cortex via Cdk5 promotes pain sensation and anxiety. Nat. Commun. 6, 7660. 10.1038/ncomms8660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Wu LJ, Wang H, Zhang X, Vadakkan KI, Kim SS, Steenland HW, and Zhuo M (2008). Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J. Neurosci. 28, 7445–7453. 10.1523/JNEUROSCI.1812-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Chang PC, Gooden F, and Gan WB (2013). Transcranial two-photon imaging of synaptic structures in the cortex of awake head-restrained mice. Methods Mol. Biol. 1010, 35–43. 10.1007/978-1-62703-411-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JW, Shih HC, and Shyu BC (2006). Intracortical circuits in rat anterior cingulate cortex are activated by nociceptive inputs mediated by medial thalamus. J. Neurophysiol. 96, 3409–3422. 10.1152/jn.00623.2006. [DOI] [PubMed] [Google Scholar]
- Yin JB, Liang SH, Li F, Zhao WJ, Bai Y, Sun Y, Wu ZY, Ding T, Sun Y, Liu HX, et al. (2020). dmPFC-vlPAG projection neurons contribute to pain threshold maintenance and antianxiety behaviors. J. Clin. Invest. 130, 6555–6570. 10.1172/JCI127607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Manders T, Tong AP, Yang R, Garg A, Martinez E, Zhou H, Dale J, Goyal A, Urien L, et al. (2017). Chronic pain induces generalized enhancement of aversion. Elife 6, e25302. 10.7554/eLife.25302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gadotti VM, Chen L, Souza IA, Stemkowski PL, and Zamponi GW (2015). Role of prelimbic GABAergic circuits in sensory and emotional aspects of neuropathic pain. Cell Rep. 12, 752–759. 10.1016/j.celrep.2015.07.001. [DOI] [PubMed] [Google Scholar]
- Zhao MG, Ko SW, Wu LJ, Toyoda H, Xu H, Quan J, Li J, Jia Y, Ren M, Xu ZC, and Zhuo M (2006). Enhanced presynaptic neurotransmitter release in the anterior cingulate cortex of mice with chronic pain. J. Neurosci. 26, 8923–8930. 10.1523/JNEUROSCI.2103-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Zhou H, Huang L, Xie Z, Wang J, Gan WB, and Yang G (2018). Neuropathic pain causes pyramidal neuronal hyperactivity in the anterior cingulate cortex. Front. Cell. Neurosci. 12, 107. 10.3389/fncel.2018.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Zhang Q, Martinez E, Dale J, Hu S, Zhang E, Liu K, Huang D, Yang G, Chen Z, and Wang J (2018). Ketamine reduces aversion in rodent pain models by suppressing hyperactivity of the anterior cingulate cortex. Nat. Commun. 9, 3751. 10.1038/s41467-018-06295-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Tang HD, Dong WY, Kang F, Liu A, Mao Y, Xie W, Zhang X, Cao P, Zhou W, et al. (2021). Distinct thalamocortical circuits underlie allodynia induced by tissue injury and by depression-like states. Nat. Neurosci. 24, 542–553. 10.1038/s41593-021-00811-x. [DOI] [PubMed] [Google Scholar]
- Zingg B, Chou XL, Zhang ZG, Mesik L, Liang F, Tao HW, and Zhang LI (2017). AAV-mediated anterograde transsynaptic tagging: mapping corticocollicular input-defined neural pathways for defense behaviors. Neuron 93, 33–47. 10.1016/j.neuron.2016.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary data reported in this paper are provided in Table S1.
No original code has been generated in this study.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.