

Abstract
Bacteria inhabit virtually all environments on earth where they are exposed to numerous endogenous and exogenous DNA damaging agents. To maintain genome integrity and ensure cell survival, bacteria have evolved several DNA repair pathways to correct and repair the different types of DNA damages and non-canonical bases that occur, including strand breaks, nucleotide modifications, cross-links, mismatches, and ribonucleotide incorporations. Recent advances in genome-wide screens, the availability of tens of thousands of whole genome sequences, and advances in structural biology have enabled the rapid discovery and characterization of novel DNA repair pathways and new enzymatic activities across the domain Bacteria. In this Review, we focus on several bacterial excision repair pathways. We review recent advancements in well-known pathways, including advances in base excision repair, nucleotide excision repair, we discuss several new repair processes including the EndoMS mismatch correction pathway, and the MrfAB excision repair system. This Review highlights the impressive breadth of DNA repair capabilities harnessed within the microbial world.
Introduction
Bacteria thrive in diverse environments where they encounter a myriad of endogenous and exogenous causes of DNA damage1. All organisms need to repair large and small base damage sites, DNA strand breaks, and correct misincorporated and noncanonical nucleotides surrounded by millions of properly paired bases1. Maintenance of genetic information through minimizing accumulation of deleterious mutations helps ensure cell survival1–3. DNA damage interferes with accurate DNA replication while also having the potential to block replication fork progression and RNA polymerase during transcription4,5. A block to fork progression could be lethal or lead to genomic instability, including formation of DNA double-strand breaks4,5. Some bacteria use photoreactivation to reverse specific types of UV damage, and direct reversal of alkylation damage, while virtually all organisms use base excision repair (BER) and nucleotide excision repair (NER) to address different types of damaged nucleobases1. Repair of single and double strand DNA breaks is critical, and organisms do so using homologous recombination, nonhomologous end joining, and single strand annealing1,6. Thus, bacteria have evolved a series of repair pathways to address the DNA lesions that occur in vivo.
In this Review, we consider the endogenous and exogenous sources that damage DNA as well as the types of lesions bacteria encounter. Next, we discuss conserved DNA excision repair activities. We discuss bacterial BER, including more recent work describing the BER-dependent repair of DNA crosslinks. We discuss recent advances in our understanding of the mechanism behind NER and we cover three pathways involved in the correction of DNA polymerase errors. Finally, we touch on a recently discovered excision process mediated by a helicase and exonuclease. This review provides a contemporary discussion of the excision-based mechanisms bacteria use to repair the diverse set of lesions they encounter.
Overview of DNA damage sources
Cells must repair a few basic types of lesions including base damage sites (small and bulky), apurinic/apyrimidinic (AP) sites, DNA strand breaks (single and double), cross-links (interstrand, intrastrand, and protein-DNA), ribonucleotide misincorporations and mispaired bases (mismatches)1,7. The relative abundance of each type of lesion depends on each cell’s intrinsic metabolism, the environment occupied, and the frequency with which each type of lesion occurs. In addition, bacteria need to balance the selective pressures required for genome maintenance with the occurrence of mutations in a population that could provide a selective advantage8,9.
Endogenous damage
Endogenous sources of DNA damage result from byproducts of normal cellular metabolism that chemically react with DNA to form lesions (Figure 1)10. Reactive oxygen and nitrogen species chemically altering DNA is the most common source of endogenous lesions1. The primary lesions caused by reactive oxygen species are damaged bases or removal of the base resulting in an AP site10. More than 20 different lesions from oxidative damage can form on intact bases10. These lesions tend to be small and include the very common 8-oxoguanine (8-oxoG), along with base deaminations10. Oxidative DNA damage also results in formamidopyrimidines and AP sites10. AP sites are frequent and are caused by hydrolysis of the N-glycosidic linkage between the nucleobase and the deoxyribose sugar11; hydrolysis occurs spontaneously, by environmental and therapeutic genotoxins, and during BER by DNA glycosylases12. If AP sites are not repaired, the lesion can block progression of DNA replication and transcription13. Interstrand cross-links are a type of lesion that prevents double helix unwinding during DNA replication and transcription, requiring strand breakage or generation of two AP sites to repair14,15.
Figure 1. Examples of endogenous and exogenous sources of DNA damage encountered by bacteria.
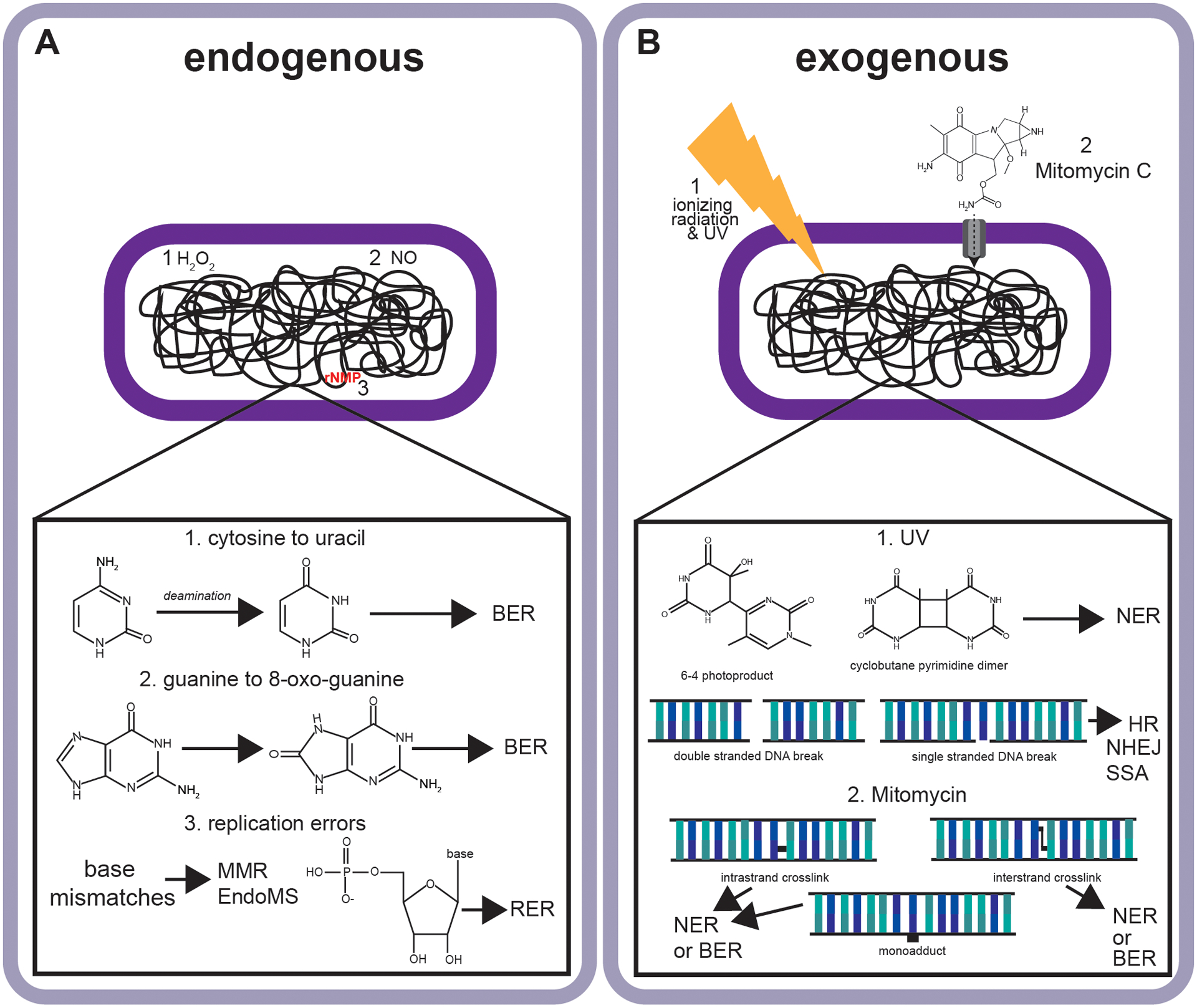
(A) Endogenous sources of damage or errors including base deamination (1), oxidative DNA damage (2), and DNA replication errors including base pairing errors and ribonucleotide errors (3). (B) Examples of exogenous sources of damage including UV cyclobutane pyrimidine dimers (1) and 6–4 photoproducts (1) as well as the uptake of antimicrobial compounds including mitomycin C which damage DNA forming cross-links, and a monoadduct (3). Abbreviations include base excision repair (BER), nucleotide excision repair (NER), homologous recombination (HR), non-homologous end-joining (NHEJ), single strand annealing (SSA), mismatch repair (MMR) and ribonucleotide excision repair (RER).
Other endogenous processes include base-pairing errors and sugar errors, both of which occur during DNA replication16,17. Base-pairing errors occur when the incorrect base is paired during DNA synthesis16. Sugar errors occur when a ribonucleotide is incorporated in place of the cognate deoxyribonucleotide (i.e. AMP in lieu of dAMP)18,19. Both mismatches and sugar errors require correction; however, a distinguishing feature is that the nucleotides themselves are not damaged, rather they are misincorporated requiring removal and correction16,17. Base-pairing errors and sugar errors increase mutation rate16,17. When a base-pairing error occurs, it changes the DNA sequence directly if not corrected before the next round of DNA replication16. Sugar errors can increase mutagenesis when the resynthesis step or removal mechanism is error-prone17,19,20.
Exogenous damage
Exogenous sources of DNA damage are chemicals or radiation that arise from outside the cell, causing direct and indirect damage inside the cell1. Lesions from exogenous sources are specific to the environment inhabited (Figure 1). For bacteria that are plant and animal pathogens or commensals, the exogenous sources of damage also include reactive oxygen and nitrogen species21–23. Pathogenic bacteria taken up by macrophages or plant pathogens and symbionts invading plant tissues are faced with an oxidative burst as a common host defense mechanism23,24. Animal gut commensals and pathogens passing through the animal digestive tract encounter nitrosative stress, oxidative stress, and lesions that can result from the acidic environment in the stomach25.
Bacteria present in nature could be exposed to ultraviolet light (UV), gamma rays and many secondary metabolites produced by soil-dwelling bacteria when present in the same environment26–28. UV exposure causes cyclobutane pyrimidine dimers (CPD) and 6–4 photoproducts, which are considered bulky adducts29. Gamma rays can directly cause single and double strand DNA (ssDNA and dsDNA) breaks while also forming hydroxyl radicals that can produce both types of breaks and base damage sites30,31.
An important group of antibiotic-producing bacteria are from the genus Streptomyces28. Streptomyces produce a glycopeptide called phleomycin that causes DNA breaks, streptozotocin which is capable of alkylating DNA, and mitomycin C (MMC) and azinomycin B (AZB), which can form DNA cross-links and single bulky lesions32–35. Below, we discuss the excision repair pathways responsible for addressing DNA lesions that arise from both endogenous and exogenous sources of DNA damage.
Overview of Base Excision Repair (BER)
Base excision repair (BER) is a process that removes and replaces damaged or non-canonical bases in DNA14,36. The enzymes involved in BER are highly conserved throughout life, underscoring their importance37. First, a DNA glycosylase recognizes a lesion containing a damaged nucleobase. Two types of DNA glycosylases exist, and both release damaged bases but generate different products38. Monofunctional glycosylases cleave the N-glycosidic bond to release the damaged base, causing an AP site (Figure 2A)38. After AP site creation, the DNA backbone is cleaved by an AP endonuclease, leaving a 3’-OH and a 5’ deoxyribose-phosphate (5’-dRP) which is processed by RecJ to create a substrate for DNA resynthesis39. Bifunctional glycosylases also contain lyase activity36. After lesion recognition and N-glycosidic bond cleavage, AP lyase activity of bifunctional glycosylases processes the DNA backbone through β−elimination resulting in a 3’ unsaturated aldehyde and 5’ phosphate36. The 3’-blocking lesion is removed, DNA polymerase fills the gap and DNA ligase seals the nick36.
Figure 2. Advancements in Base Excision Repair.
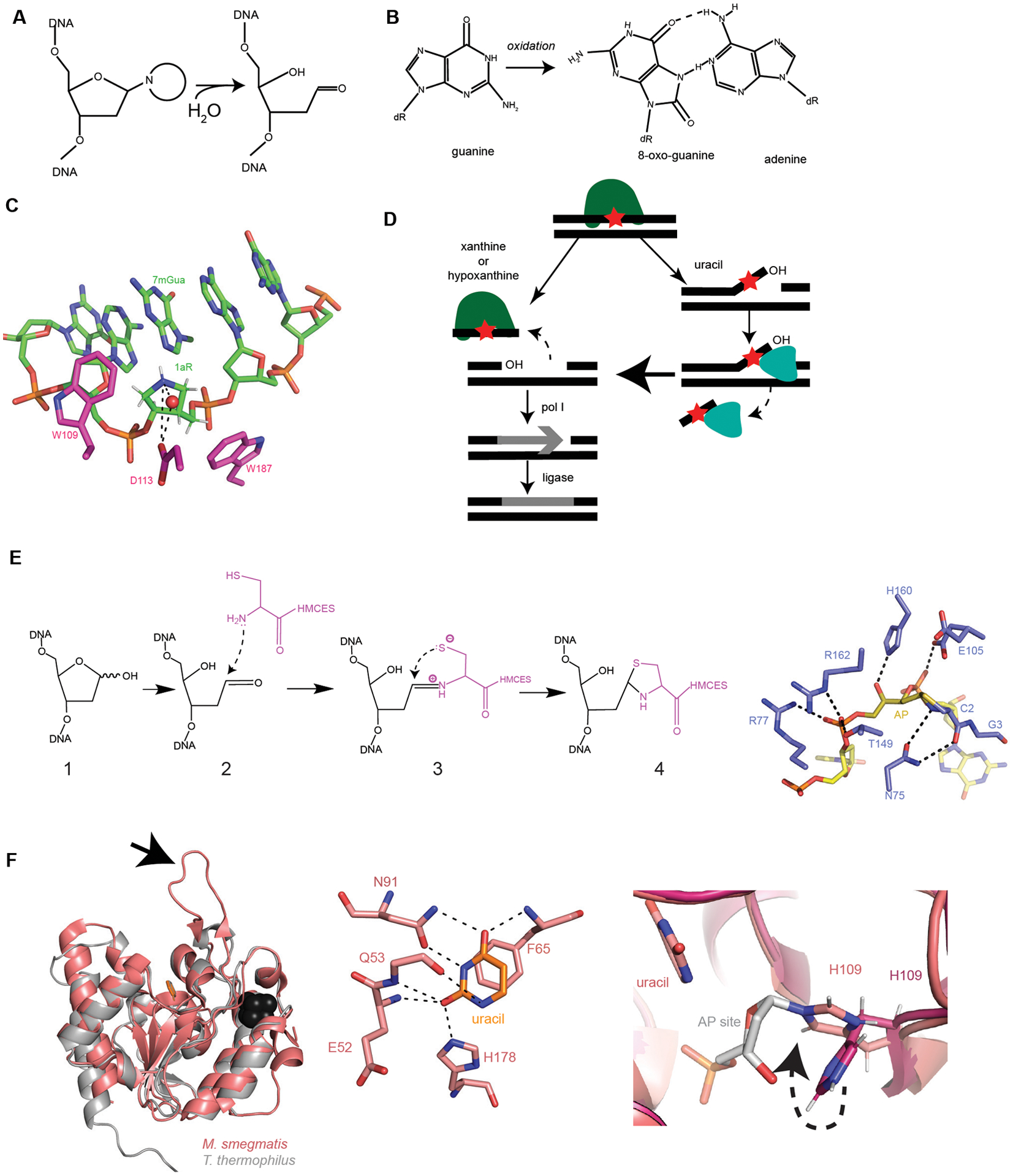
(A) Formation of an AP site through hydrolysis of the N-glycosidic bond (adapted from129). (B) Guanine oxidation into 8-oxo-guanine and mismatched pairing to adenine. (C) AlkD (5kub, shown in pink) protein contacts with depurination intermediate analogue 1aR (1-aza-2’, 4’-dideoxyribose) within DNA (shown in green). Red sphere is a water nucleophile. Dashed black lines are hydrogen bonds. CH/π interactions with W109 and W187 to 1aR (adapted from52). (D) Summary of the EndoV (YwqL)/ExoA repair of deaminated bases (adapted from63). EndoV identifies and incises 3’ of the lesion. If the lesion is xanthine or hypoxanthine, EndoV cuts 5’ of the lesion and pol I fills in the gap. DNA ligase seals the nick to complete repair. If the lesion is uracil or an AP site, the initial endonuclease activity creates a 3’ flap containing the lesion. ExoA uses its 3’ to 5’ exonuclease activity to remove the lesion containing strand, followed by pol I and ligase. (E) Left: Proposed stepwise formation of the thiazolidine linkage between HMCES and DNA based on YedK. 1. AP site within DNA, 2. Nucleophilic attack on AP site, 3. Schiff base intermediate, 4. Thiazolidine link between HMCES and DNA. Right: YedK (6nua, shown in purple) DNA-protein crosslinks with AP site (dark yellow) within DNA (yellow). Critical hydrogen bonds between protein and DNA shown as black dashed lines (adapted from56). Note: E105 is shown with two conformers. (F) Left: Mycobacterium tuberculosis UdgX in salmon pink (6ioa) aligned with a family 4 UDG from Thermus thermophilus in light gray (1ui0), uracil shown in orange and Fe-S cluster in black. Arrow pointing at characteristic protruding loop (adapted from61). Middle: Mycobacterium tuberculosis UdgX (6ioa) interactions with uracil (orange). H178 is the residue responsible for excision of uracil. H178, along with E52, Q53, F65, and N91 hydrogen bond to uracil. Black dashed lines are hydrogen bonds (adapted from60). Right: Mycobacterium tuberculosis apo-UdgX (6ail) in magenta aligned with uracil-bound in salmon (6ajo). H109 changes position within the enzyme (shown with dashed black arrow) when uracil is bound and excised from the DNA backbone to form a covalent bond with the AP site (in gray). H109 serves as the nucleophile instead of water.
BER of Oxidative DNA damage
Guanine is highly susceptible to oxidative damage and is the most common oxidatively damaged base in cells40. The resulting 8-oxo-guanine (8-oxoG) can pair with dC in the anti-conformation or dA when rotation around the N-glycosidic linkage occurs in the syn-conformation (Figure 2B)41. When 8-oxo-dGTP is incorporated into DNA it is highly mutagenic causing G:C to T:A transversions42. Before 8-oxo-dGTP is incorporated into DNA, it can be sanitized by MutT, an 8-oxo-dGTP diphosphatase43. MutT converts 8-oxo-dGTP and 8-oxo-GTP to the corresponding nucleoside monophosphates preventing their incorporation into DNA and RNA43,44. If the 8-oxo-dGTP is incorporated into DNA before removal from the dNTP pools, it becomes a substrate for BER45. The bifunctional formamidopyrimidine-DNA-glycosylase Fgp/MutM recognizes 8-oxo-dG:C base pairs and cleaves the N-glycosidic bond to create an AP site46,47. Then using its lyase activity, MutM cleaves the DNA backbone and heals the gap (by δ elimination) creating 3’ and 5’ phosphates41. Because DNA polymerase requires a 3’ OH, an AP endonuclease such as Exo III or Endo IV is required to remove the remaining 3’ phosphate, allowing for DNA polymerase I to fill the gap41. If mispairing in an 8-oxo-dG:A base pair occurs, monofunctional adenine DNA glycosylase MutY removes the undamaged, mispaired adenine across from the preexisting 8-oxo-dG48. This removal creates an AP site which is then processed to allow for resynthesis generating a substrate for MutM47.
Because 8-oxoG occurs in anti- and syn- conformations, which differ only by orientation of the base attached to the ribose moiety, it can mispair with both cytosine and adenine, respectively41. It was previously unclear whether both anti- or syn-conformations of 8-oxoG could bind to the (MutM/Fpg) required for base excision49. Base-specific excision is the canonical mechanism by which syn-bound 8-oxoG is repaired47. Recently a DNA base-independent excision mechanism was described representing an alternative mechanism for BER49. Importantly, this work suggests that there is no discrimination between DNA lesions and DNA nucleotides within the active site, explaining how 8-oxoG in the anti-conformation could be repaired by MutM/Fgp49.
Non-base flipping DNA glycosylases
Most canonical DNA glycosylases use a base-flipping mechanism for recognition and excision of damaged bases50. In B. cereus, AlkD was found to be the first DNA glycosylase to catalyze BER without flipping the damaged nucleotide from the DNA50. Glycosylases that use a non-base flipping mechanism can provide considerable plasticity in lesion recognition as opposed to canonical base flipping mechanisms where the damaged base is placed in the active site providing a mechanism for lesion specific interactions51. AlkD is specific for cationic lesions and interacts with the sugar-phosphate backbone using CH/π interactions with Trp109 and Trp187 (Figure 2C)52. Asp113 was shown to be critical for orienting the water molecule needed in nucleophilic attack, with mutation dramatically reducing excision (Figure 2C). The interaction between AlkD and a lesion is indirect, and therefore the DNA bends modestly upon lesion binding50,52. For this reason, AlkD can accommodate different positions and sizes of alkyl substituents, including bulky alkylpurine adducts50,52.
AlkD recognizes DNA modifications produced by yatakemycin, a highly toxic DNA alkylating agent within the spirocyclopropylcyclohexadienone family of natural products that produces lesions which do not distort or destabilize DNA53. The stabilization of DNA from yatakemycin lesions occurs through CH-π interactions53. Upon yatakemycin treatment, AlkD widens the minor groove around the lesion by separating the lesion and the modified strand53. AlkD cleaves the N-glycosidic bond connecting the sugar and the base, creating an AP site and leaving the base attached to the YTMA adduct53. AlkC is another non base flipping DNA glycosylase that recognizes 3mG and 3mA substrates54. An Ig-like domain was described in AlkC, which was shown to cause a sharp bend in the DNA and disrupt the base stacking interactions54. As a result, the base is exposed to make direct contact with AlkC, unlike what has been shown in AlkD52,54. Together, AlkD and AlkC provide examples of non-canonical methods for lesion recognition that are likely to be widespread among enzymes that fall into these groups.
Protection of AP sites
AP sites not only occur as a BER intermediate, but they occur spontaneously and are considered a highly abundant lesion in vivo1. Human HMCES and YedK from E. coli possess a SOS-response associated peptidase (SRAP) domain for AP site recognition to block the AP site from use as a template by mutagenic translesion DNA polymerases55,56. These DNA-protein crosslinks are thought to protect DNA from hydrolysis and access by endonucleases and DNA polymerases and are therefore critical for the transient maintenance of genome stability. Additionally, the DNA-protein crosslinks are critical for AP site repair but need to be resolved for replication and transcription to proceed55,57. It was shown previously that covalent HMCES-DNA crosslinks were resolved via ubiquitin-mediated proteolysis55. E. coli YedK has been shown to form a stable thiazolidine link between the ring-opened AP site and the amino-terminal cysteine (Figure 2E, left)56. First, the α-NH2 of Cys2 performs a nucleophilic attack on the AP aldehyde C1’ carbon, resulting in a Schiff base intermediate56. Next, the Cys2 sulfhydryl group attacks C1’ to form the favored thiazolidine linkage (Figure 2E, left). This linkage is resistant to strand cleavage and proteolysis, which differs from the typically unstable, transient cross-links in BER56. Arg77 and Arg162 are essential for DNA binding while His160 forms a hydrogen bond to stabilize the AP site (Figure 2E, right)56. Glu105 and Asn75 are conserved and form hydrogen bonds to the AP site (Figure 2E, right); substitution of these residues results in reduced crosslinking56. In E. coli, the most common model for DNA-protein crosslink repair is through the combined action of NER and homologous recombination,57 with more recent evidence showing that Lon protease helps remove DNA protein-crosslinks indicating a protease dependent mechanism58.
BER of Uracil
Uracil occurs in DNA when cellular dNTP pools are depleted as a result of ribonucleotide reductase inhibition, or from cytosine deamination1. Uracil incorporated into DNA is repaired using BER and is recognized and excised by uracil DNA glycosylases (UDGs) which exist within six families1. These families of UDGs are characterized by motif A and B sequences for substrate catalysis and stabilization of the uracil-DNA with enzyme complex1. Recently, several studies characterized the structures and activity of UdgX from Mycobacterium smegmatis which has 60% sequence identity with family 4 but possesses a unique, positively charged protruding loop (Figure 2F, left)59. M. smegmatis UdgX is the first uracil DNA glycosylase to form a tight complex with uracil-DNA and several structures of M. smegmatis UdgX were solved in free, uracil-bound, DNA-bound, and ssDNA-bound forms60,61. His178 forms a hydrogen bond to uracil to facilitate N-glycosidic cleavage (Figure 2F, middle) and alanine substitution resulted in a loss of the covalent complex60,61. His109 acts as a nucleophile instead of the typical water molecule creating a covalent linkage to the C1’ of the deoxyribose sugar upon cleavage of the N-glycosidic bond60,61. This linkage is aided by a long-distance hydrogen bond to Gln5360,61 (Figure 2F, right).
The overall model for repair is that MsmUdgX-DNA complex stalls the replication fork and presumably forms gaps that require RecA-mediated repair59. The in vivo relevance of UdgX is unclear. Requiring HR to repair UdgX-DNA seems like an inefficient method to address uracil. More work will be necessary to determine if other processes in addition to HR are used to repair the UdgX-DNA complex.
Alternative repair of deamination
Adenine, guanine, and cytosine are deaminated by hydrolysis or nitric oxide and hydroxylamine62 to produce hypoxanthine, xanthine, and uracil, respectively63. B. subtilis EndoV (YwqL), ExoA and DNA polymerase I constitute an alternative excision repair pathway (Figure 2D)63. EndoV hydrolyzes the second phosphodiester bond 3’ to a lesion with activity on a range of DNA substrates, including hypoxanthine, xanthine, uracil, and AP sites63. EndoV showed preference for hypoxanthine and uracil relative to AP sites, and xanthine, respectively63. DNA polymerase I (polA) and EndoV are epistatic, suggesting that they cooperate although DNA polymerase I is unable to process the product from EndoV. Current work shows that after EndoV cuts at the damaged site, ExoA uses its 3’ to 5’ exonuclease activity to extend the site, followed by DNA polymerase I gap synthesis after lesion excision63. The EndoV, ExoA and Pol I pathway demonstrates the need for bacteria to contain repair pathways that have overlapping functions to address common forming lesions including sites of base deamination.
BER of interstrand cross-links
Interstrand cross-links (ICLs) form a covalent bond between two DNA strands, preventing their separation during replication and transcription64. The most common model for ICL repair involves the combined action of nucleotide excision repair (NER), DNA polymerase I, and homologous recombination15, although other models exist65. Therefore, most current models for ICL repair in bacteria require the participation and coordination of multiple processes15,66.
The availability of genome sequences and functional studies has uncovered a large group of widely conserved DNA glycosylases that repair ICLs14. Glycosylase-dependent ICL repair was identified in Streptomyces sahachiroi, which produces the alkylating agent azinomycin B (AZB) (Figure 3A)67–69. These species contain the gene cluster azi that is responsible for the biosynthesis of AZB and within that gene cluster resides a DNA glycosylase alkZ70. AlkZ is a member of a large family of glycosylases with orthologs present in several thousand diverse bacteria, which also includes human pathogens and environmental species69. The two most well-characterized members from this family are AlkZ from S. sahachiroi and YcaQ from E. coli68,69,71. Like mitomycin C (MMC), AZB is a bifunctional DNA alkylating agent that forms ICLs (Figure 3B)33. Mitomycin C (MMC) forms adducts at the N6 position of adenine or the N2 position of guanine in the GC sequence64, while AZB forms adducts at the N7 position of appropriately placed purines33. Both agents can form a monoadduct where one strand is modified or a diadduct where both strands are modified33,64.
Figure 3. Azinomycin B and nitrogen mustard interstrand crosslink repair.
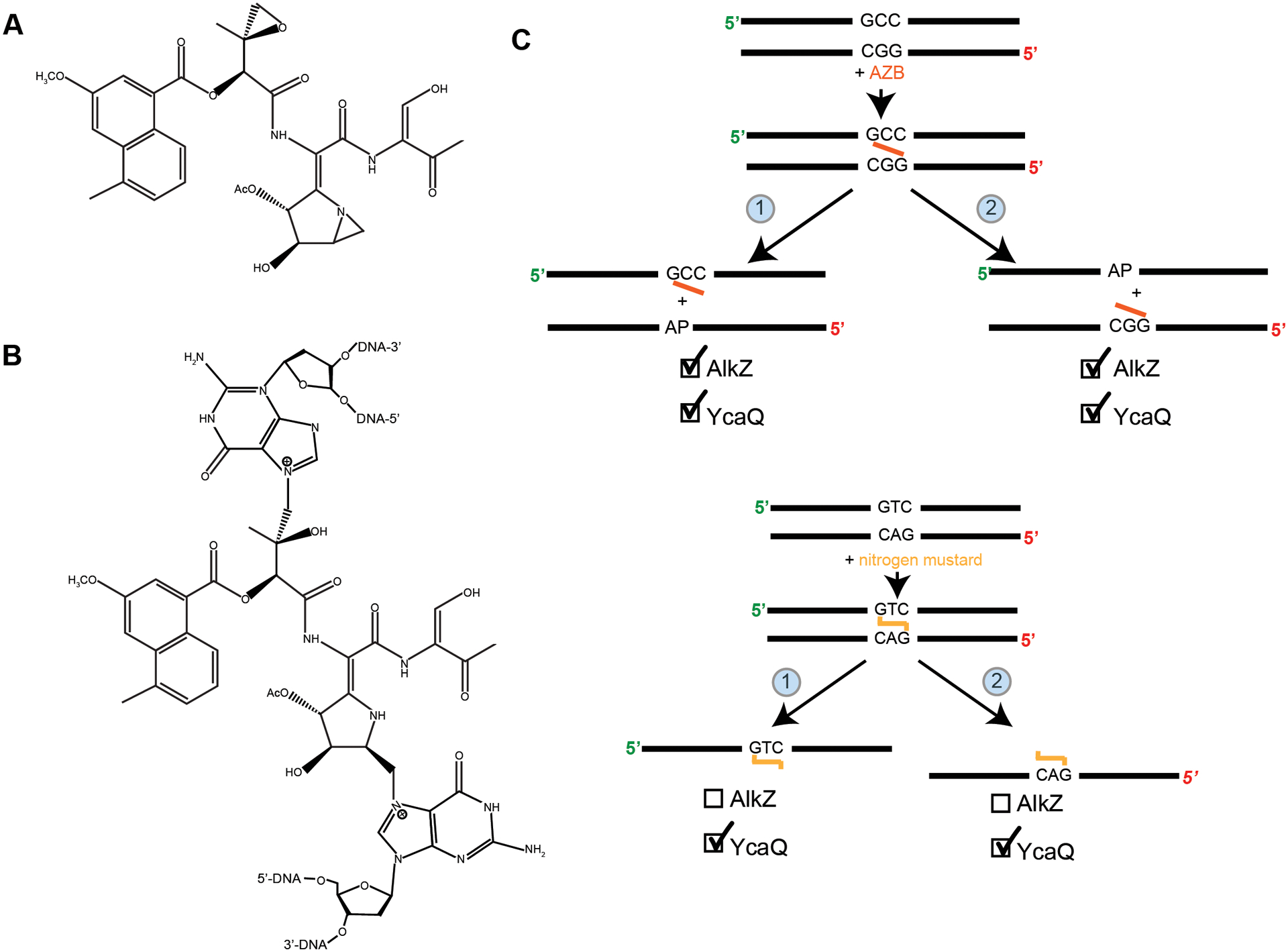
(A) Structure of azinomycin B (AZB) (adapted from71). (B) Structure of interstrand crosslink formed by azinomycin B (adapted from71). (C) Top: AZB asymmetric crosslinked substrate and possible unhooking products. AlkZ was shown to unhook crosslinks from both sides with preference toward the GGC side (product 1). YcaQ was shown to unhook the crosslink from both sides with slight preference toward unhooking the CGG side (product 2). Bottom: Nitrogen mustard symmetric crosslinked substrate and possible unhooking products. YcaQ was able to unhook both sides of the crosslink, while AlkZ showed no activity71.
AlkZ recognizes the ICL and AZB monoadduct68,69. During monoadduct repair, AlkZ binds and cleaves the N-glycosidic bond producing an AP site68,69. During AZB ICL repair, AlkZ unhooks one cross-link generating an AP site followed by unhooking of the second site (Figure 3C, top)71. As with monoadduct repair, the resulting AP sites are processed to complete repair. E. coli YcaQ also unhooks the AZB-generated monoadduct and ICL yielding an AP site that is sensitive to cleavage by an AP endonuclease (Endo IV) (Figure 3C, top)71. As discussed above, some BER glycosylases are highly specific while other glycosylases have more plasticity in lesion recognition. Streptomyces AlkZ is very specific for the monoadduct and ICL produced by AZB71. Perhaps this is not surprising given that the alkZ gene is located within the AZB biosynthesis gene cluster providing a mechanism of survival during AZB production. E. coli YcaQ has more breadth in the type of lesion recognized and excised71. YcaQ was efficient in unhooking ICLs produced by nitrogen mustards, whereas AlkZ was completely ineffective even though ICLs derived from nitrogen mustards react with guanine at the N7 position (Figure 3C, bottom)71. Given these observations, it will be important to learn the lesion specificity among the thousands of other AlkZ family members that remain uncharacterized.
Overview of Nucleotide Excision Repair (NER)
Nucleotide excision repair occurs across all domains of life72. NER repairs bulky DNA adducts, such as CPDs from UV exposure, and interstrand cross-links1. In addition, it can remove non-bulky DNA lesions and ribonucleotides73,74. The breadth of structurally unrelated adducts recognized by NER is an important feature of this pathway and it is still unclear how NER can recognize such a diverse array of nucleobases75. The main steps of NER are the same between prokaryotes and eukaryotes, except bacteria use UvrA, UvrB, and UvrC, while eukaryotes use different proteins that are unrelated to their bacterial counterparts75. In brief, UvrA binds ATP and searches for lesions. UvrA then recruits UvrB ATPase which forms a tight complex on DNA; the interaction timing of UvrA and UvrB is still unclear76,77. Next, UvrC binds and makes two incisions, one 4–5 phosphodiester bonds 3’ to the damage and another 8 phosphodiester bonds 5’ to the damage78,79. Finally, the lesion containing DNA is released by UvrD helicase and DNA polymerase fills the gap, followed by ligation (Figure 4A).
Figure 4. Overview of nucleotide excision repair.
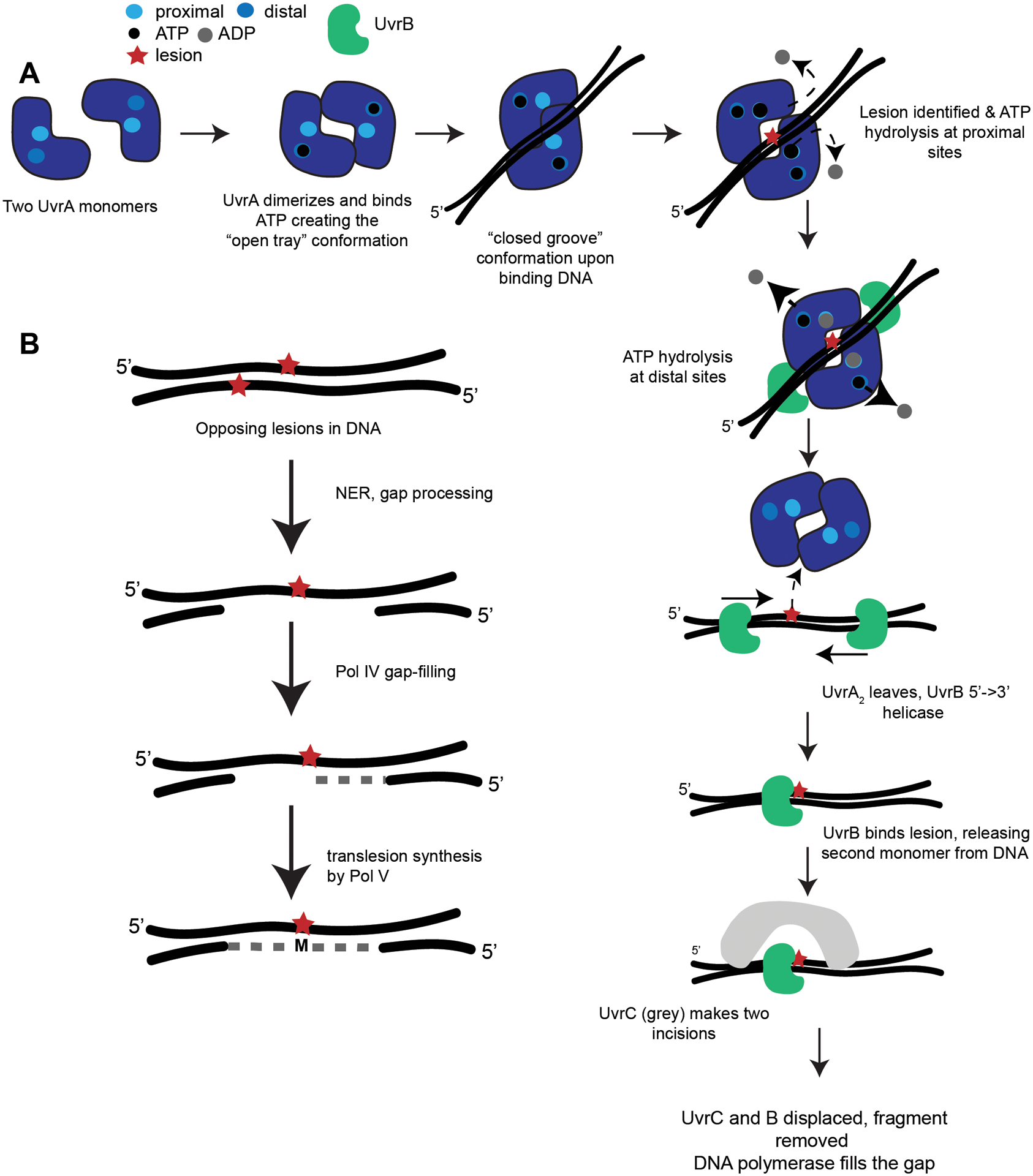
(A) Canonical nucleotide excision repair where UvrA exists as monomers and dimerizes in solution binding ATP in the distal sites. When UvrA2 binds undamaged DNA, it should form a “closed groove” conformation. When UvrA2 encounters a lesion in DNA, ATP is hydrolyzed at proximal sites. The “open tray” should accommodate lesion-bearing DNA. UvrB binds each UvrA monomer and ATP is hydrolyzed at the distal sites. UvrA2 leaves the lesion, and UvrB uses its 5’ to 3’ helicase activity to find the lesion. One UvrB encounters the lesion and clamps down, releasing the second UvrB. UvrC, a dual endonuclease (grey), binds and cleaves the DNA upstream and downstream from the lesion. UvrC and B are displaced and UvrD helicase (or another helicase in organisms lacking UvrD) removes the fragment. Lastly, DNA polymerase and ligase complete repair. See the following review for a detailed mechanistic and structural view of bacterial NER75 (adapted from77,81). (B) Summary of NER-induced mutagenesis (NERiM). When DNA contains opposing lesions, canonical NER performs a 12–13 nucleotide excision and removal of a lesion on one strand. The gap is filled by pol IV or pol II, and subsequent translesion synthesis by pol V occurs. The result is a repaired strand containing a mismatch across from the unrepaired lesion (adapted from97).
Preincision complex
Binding of UvrA to ATP has been shown to promote UvrA2 dimerization and interaction with UvrB80,81. Recent investigations have shown the importance of the UvrA2 dimer ATPase activity and how hydrolysis allows UvrA2 to recognize damaged DNA76. Each monomer of UvrA2 has two ATPase sites, one proximal and one distal to the DNA binding groove76,82. The distal site has high affinity for ATP and performs hydrolysis more quickly with a slow release of ADP76. There appears to be allosteric communication between the distal and proximal sites due to the observation that ATP binding by proximal sites is necessary for ATP hydrolysis, ADP release, and catalytic turnover by distal sites76. This distal hydrolysis creates “mixed” UvrA2 dimers bound to ATP, ADP, or no nucleotide76. From the kinetics, affinity, and stoichiometry experiments, it was shown that the mixed ATP/ADP/apo UvrA2 population is necessary to interact with DNA and initiate NER. Further, ATP hydrolysis by UvrA helps form the UvrB-DNA complex, possibly via the proximal ATPase site82. Recent work found that ATP hydrolysis by UvrB is required to form a UvrB-DNA complex but not required for UvrA to dissociate from the UvrAB-DNA complex82.
The timing of UvrA and B interaction is still not fully resolved. After UvrA dimerization, it is unclear if UvrA recognizes lesions alone or forms a heterotetramer with UvrB prior to lesion recognition83,84. Work by Pakotiparapha et al. showed UvrA2-UvrB2 stoichiometry during lesion recognition in vitro77,85. They also found that the ATPase activity of UvrA is required for recruitment of UvrB77. More recent in vivo single molecule studies suggest that UvrA2 searches in the absence of UvrB, followed by recruitment of UvrB81. Structural characterization of UvrA shows that it forms the “closed groove” or “open tray” configurations (Figure 4A)77. The idea is that UvrA can change conformations between the “open tray” and “closed groove” during the search for damaged DNA77. The “closed groove” appears to only accommodate undamaged DNA while the “open tray” can accommodate lesion-bearing DNA75,77. One possibility is that UvrA toggles between the two conformations as it searches for lesions75. Upon damage recognition, UvrA converts to an “open tray” perhaps stimulating the DNA damage recognition pathway75,77. After formation of the “open tray” UvrA hydrolyzes ATP and leaves the lesion82. Helicase activity of UvrB occurs in the 5’ to 3’ direction on DNA toward the lesion86,87. UvrB kinks the DNA after recognizing the damage to form a stable UvrB-DNA complex, called the pre-incision complex86,88,89. Repair is complete after incision by UvrC and liberation of the lesion-containing segment by UvrD, followed by resynthesis and ligation1.
Additional NER factors
Other proteins have been shown to participate in NER in addition to UvrABC and UvrD90,91. When O6-alkylguanine adducts mispair with thymine they are mutagenic91. Alkyltransferases typically repair these adducts back to guanine92. Alkyltransferase-like (ATL) proteins have high sequence similarity to alkyltransferases but lack the catalytic cysteine residue93. As a result, ATLs can bind O6-alkylguanine adducts but do not transfer alkyl groups, like canonical alkyltransferases94. E. coli YbaZ is in the ATL protein family and works with NER to repair large O6-alkylguanine adducts and other lesions91. Direct interaction between YbaZ and UvrA has been measured, suggesting that YbaZ acts as a “molecular flag” to enhance the efficiency of NER on these lesions91. This provides an important example of a previously uncharacterized gene that enhances the efficiency of the NER pathway for repair of certain lesions. There could be many more protein factors to be discovered that aid in NER repair of specific lesions.
Mutagenesis during NER
An important consideration for NER is that the resynthesis step (gap filling) could be error-prone, leading to lower fidelity repair. In B. subtilis, hexavalent chromium generates reactive oxygen stress, eliciting the SOS response95. At the same time, DNA-protein cross-links were abundant and processed by RecA and NER95. In addition to being involved in processing of UV-induced photodimers96, the Y-family DNA polymerases YqjH and YqjW can act after NER to facilitate low-fidelity DNA synthesis95. This work suggests that NER, homologous recombination, and Y-family DNA polymerases can act together to maintain viability while also resulting in mutagenesis during the resynthesis step95.
In another example, a subpathway for UV-induced lesion repair was identified in E. coli97. This pathway was termed NER-induced mutagenesis (NERiM) for repair of two adjacent lesions on opposing strands (Figure 4B)97. Following UV damage, UvrA and UvrC work to complete canonical NER on lesion-bearing DNA. Next, DNA polymerases Pol II and Pol IV perform gap-filling97. Finally, Pol V is involved in translesion synthesis of the template strand across the opposing lesion resulting in mutagenesis97. Lower fidelity resynthesis allows for DNA replication to bypass a lesion on the template strand. However, overuse of the error-prone pathway has the potential to increase detrimental mutations and therefore decrease cell fitness.
Overview of Mismatch Repair (MMR)
DNA replication errors escape the 3’ to 5’ proofreading activity of replicative polymerases in all organisms. For bacteria, replication errors (mismatches) occur once every 15.5 rounds of replication or approximately one error every 59 million bases replicated16. Although this error rate may seem infrequent, mismatch repair increases the overall fidelity of genome replication to one error per ~909 rounds of replication16. MMR recognizes two major classes of errors, single base mismatches, and insertion and deletion loops (INDELs)1. Replicative DNA polymerases have intrinsic error bias and as a result are more prone to form G-T mismatches instead of unstable C-C mismatches in vivo98. Consequently, the affinity of the mismatch sensing protein MutS for G-T errors is much higher than the affinity of MutS for mismatches that occur less frequently to the point where MutS fails to even bind a C-C mispair99.This suggests that MutS has adapted to recognize the most common errors found in vivo.
Methyl-independent MMR
In bacteria, MMR is either methyl-directed or methylation-independent100,101. The methyl-directed pathway was identified in E. coli and has been well characterized101. DNA adenine methylase (Dam) and hemi-methylation specific endonuclease, MutH, are components of the methylation-dependent mismatch repair pathway present in E. coli and a few closely related bacteria102. In this system, MutH nicks the unmethylated DNA strand representing the newly synthesized strand101.
The vast majority of bacteria lack MutH and Dam methyltransferase102. Considerable evidence in bacteria lacking MutH/Dam has shown that mismatch repair is coupled to the site of DNA replication100,103–106. In bacteria, the replication sliding clamp (β-clamp or DnaN) aids in the recruitment of DNA repair proteins to sites of replication107,108. MutS contains a replication sliding clamp (DnaN) binding motif (QL[SD]LF) that is important for mismatch repair and recruitment of MutS to the site of DNA synthesis serving to confine the search for mismatches to nascent DNA increasing the overall efficiency of mismatch recognition109. Single molecule tracking of MutS in B. subtilis has shown that MutS moves rapidly around the cell but MutS molecules slow and dwell to search the site of DNA synthesis after encountering the replisome106. MutS variants that prevent mismatch recognition have no effect on recruitment to the replisome106,109. However, MutS variants that prevent interaction with the replication sliding clamp reduce or abolish the ability of MutS to dwell at the site of replication impairing the mismatch repair pathway and increasing mutagenesis106,109. Recently, it was shown in the distantly related alpha-proteobacterium Caulobacter crescentus that MutS and MutL localize to the replisome when ectopically expressed in live cells110. Therefore, targeting MMR proteins to the site of DNA replication through interaction with the replication sliding clamp has emerged as a conserved feature in bacteria lacking a methyl-directed signal. One possibility is that targeting of MutS to the replisome is critical to aid in distinguishing the nascent strand from the template in the absence of a methylation signal.
Another hallmark of the methylation-independent mismatch repair pathway is found in MutL homologs that contain a metal-dependent endonuclease active site with the conserved sequence DQHA(X)2E(X)4E111,112. The endonuclease active site was originally identified in the human MutL homolog PMS2 and was shown to cleave DNA using manganese111. The C-terminal structure for B. subtilis MutL with the endonuclease active site revealed a replication sliding clamp (DnaN) binding site located in an extended region of the C-terminal domain112. Mutation of the endonuclease active site or the DnaN binding motif in MutL abolished mismatch repair in vivo demonstrating the importance of MutL-dependent DNA incision and interaction with the sliding clamp112. CryoEM structures of the B. subtilis MutL C-terminal domain and DnaN along with enzymatic assays demonstrated that interaction between MutL and the replication sliding clamp stimulated MutL incision of DNA113. Therefore, in organisms with a methylation-independent pathway, coupling of MutS and MutL to replication through DnaN is critical for both mismatch recognition and stimulating DNA incision.
Endonuclease Mismatch-Specific Repair
The MutS/MutL MMR pathway is well conserved across all three domains of life,114,115 yet some organisms including Actinobacteria and some archaea lack mutS and mutL116. Instead, these organisms contain a recently described endonuclease mismatch-specific (EndoMS) protein also known as NucS117,118. EndoMS/NucS has been shown to combine with DnaN to form a noncanonical mismatch correction system (Figure 5A)118,117,119,120. EndoMS contains a mismatch-specific RecB-like nuclease domain (Figure 5B) and conserved catalytic domain (Figure 5C)121. Tyr41 and Trp77 exhibit planar stacking with the mismatched base to create the recognition site (Figure 5B). Asn76 forms hydrogen bonds with the base and aids in base discrimination at this site (Figure 5B)121. It was shown that EndoMS prefers G or T mismatches over A or C mismatches. Arg44, Gln78, and Glu103 form critical hydrogen bonds for dsDNA interaction (Figure 5B)121. Within the conserved catalytic domain that resembles type II restriction systems, a critical residue for catalysis Asp165 was mutated to alanine to prevent DNA cleavage (Figure 5C)121. C. glutamicum EndoMS binds and cleaves G/T, G/G and T/T mismatches in vitro120. The absence of EndoMS function in vivo correlates with transition mutations, suggesting a failure to correct DNA replication errors118,120. Further, deletion of the endoMS gene leads to an increase in mutation rate and the mutation rate is synergistic when combined with a replicative DNA polymerase mutant120. EndoMS from several organisms contains the QL[SD]LF DnaN binding motif119,120. DnaN interaction stimulates EndoMS activity on mismatched substrates in vitro, demonstrating a functional interaction. Further, in cells the EndoMS-DnaN interaction is required for mismatch correction along with EndoMS nuclease activity119,120. while over expression of an EndoMS variant that is unable to bind DnaN is still defective in mismatch correction. Together, these results indicate that DnaN targets EndoMS to mismatches while also regulating mismatch-dependent endonuclease activation119,120. Although MutS and EndoMS evolved independently, both require interaction with the DnaN to target their search for mismatches to nascent DNA further underscoring the importance of mismatch recognition near the replisome109,120. One aspect that is unclear is how EndoMS cleavage would be directed to the nascent strand. In vitro reactions show cleavage of both strands 5’ to the mismatch120, which would not occur in vivo. EndoMS/NucS nucleases have also been shown to cleave several other substrates in addition to mismatched bases, including flapped DNA structures and deaminated bases122. Continued investigation of EndoMS/NucS in DNA repair and mismatch correction will be important to fully understand the different ways this family of proteins contributes to genome maintenance.
Figure 5. Structural insights of EndoMS restriction endonuclease activity.
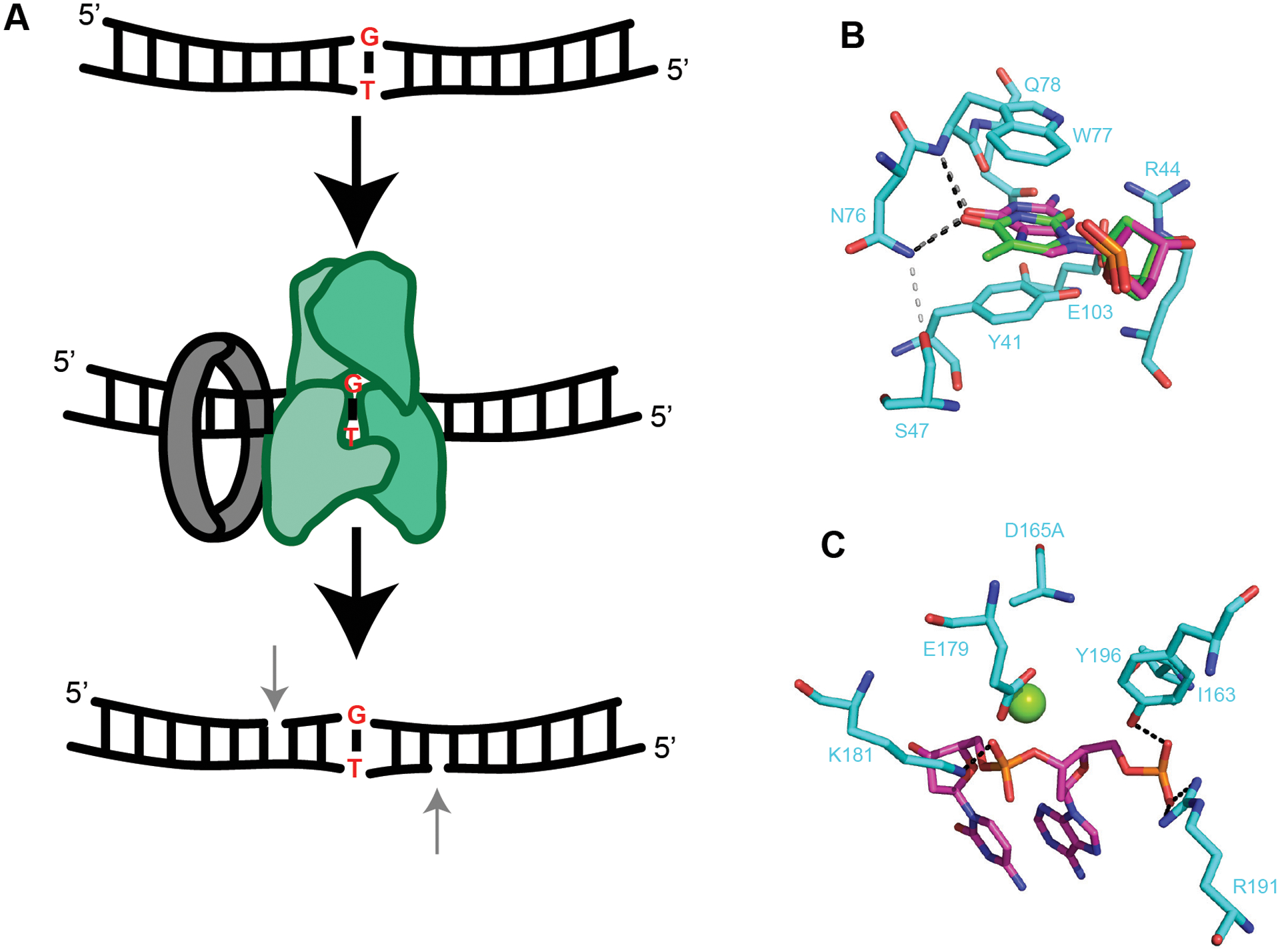
(A) G-T mismatch within duplex DNA (top). EndoMS (green) binds β-clamp (grey) and recognizes the mismatch (middle). In vitro, EndoMS cleaves the third phosphodiester bond 5’ of the mismatch on both sides (bottom)120. In vivo only one 5’ nick should be present on the newly replicated strand. The mechanism of strand discrimination is unknown. (B) Base discrimination site within EndoMS (5gke). dT shown in green and dG shown in dark pink. Critical hydrogen bonds to atoms within dT and dG for base discrimination are shown as black and gray dashed lines, respectively. Light gray dashed line represents a critical hydrogen bond between S47 and N76 (adapted from121). (C) Catalytic site of EndoMS (5gke). Green sphere represents Mg2+. D165 in the structure was substituted to alanine to prevent reaction catalysis. Black dashed lines represent hydrogen bonds (adapted from121).
Genome-wide screens for novel DNA repair activities
Advances in genome-wide screens and homology searches have rapidly enhanced the discovery and characterization of new DNA repair pathways and novel enzymatic activities123. The identification of the large family of ICL repair glycosylases described above highlights the discovery potential of homology searches across sequenced genomes68,69. Recently, the use of random transposon insertions in B. subtilis followed by deep-sequencing (Tn-seq) identified several previously characterized genes including MrfA and MrfB123–125.
MrfA is a member of the highly conserved Superfamily 2 helicases characterized by a C-terminal domain of unknown function (DUF1998)124. This group of helicases are present in a wide range of organisms including Mycobacterium SftH and S. cerevisiae Hrq1126,127. MrfB is a DnaQ-like metal-dependent nuclease active on linear and nicked plasmid DNA124. B. subtilis strains with a mrfA and/or mrfB transposon insertions resulted in a growth sensitivity to MMC, but not phleomycin or MMS123,124. Further, the action of MrfAB was shown to be specific for MMC since mrfA and mrfB deletion mutants failed to sensitize B. subtilis to growth in the presence of another cross-linking agent including psoralen or the bulky guanine monoadduct producing agent 4- nitroquinoline 1 oxide (4-NQO)124. Bacterial two-hybrid interaction data showed that MrfA and MrfB interact and isolation of genomic DNA from MMC treated cells showed that MrfA and MrfB did not repair the ICL, suggesting that these proteins are specific to the MMC monoadduct124. MrfA and B exist together in select species of Proteobacteria and Firmicutes, including B. subtilis and B. cereus124. Overexpression of mrfB from B. cereus and S. pneumoniae in the B. subtilis mrfB deletion background restores growth on MMC to varying degrees124. mrfB from P. aeruginosa, a member of the γ-Proteobacteria phylum, does not restore growth on MMC in the B. subtilis ΔmrfB background, indicating its enzymatic activity differs from that of the B. subtilis protein. It is possible that even in species with MrfA and B functioning together, the MrfB activity has evolved to function differently across phyla124.
The crystal structure for B. subtilis MrfA was solved including the domain of unknown function128 providing novel insight into this group of helicases (Figure 6A). Within the basic groove, kinked DNA is stabilized through hydrogen bonding to residues Asn157-Ser163 and at the 3’ end of DNA with His168 and Lys171128. Residues in the RecA1 and RecA2 domains (Arg148, Arg144, Gly137, Lys108, Arg333, Gly326, and Arg298) bind DNA via the sugar-phosphate backbone, and these interactions are well conserved in MrfA homologs128. F483 forms π-π stacking with the DNA backbone, while Ser486 and Ser489 stabilize the backbone with hydrogen bonds (Figure 6A)128. These interactions are not highly conserved in other homologs, leading authors to suggest that they could be important for helicase activity but are likely not required128. MrfA contains a dual RecA domain with Zn2+ binding in the conserved DUF128. Structural and biochemical characterization of MrfA revealed a novel translocation mechanism (Figure 6B). MrfA is proposed to use a “skipping rope” like mechanism where a molecular “rope” is attached between the winged helix (WH) and oligonucleotide/oligosaccharide binding (OB) domains. In this model the molecular “rope” skips ahead moving along ssDNA, while also preventing backsliding of the helicase128. The Zn2+ binding domain in the DUF contributes to MrfA action by causing a large conformational change induced upon DNA binding128. Given the conservation of MrfA-type helicases involved in repair of MMC-induced lesions, it will be interesting to learn how these helicases load onto DNA and if the translocation and conformational changes observed in MrfA are also conserved in other related helicases.
Figure 6. MrfA involvement in DNA repair.
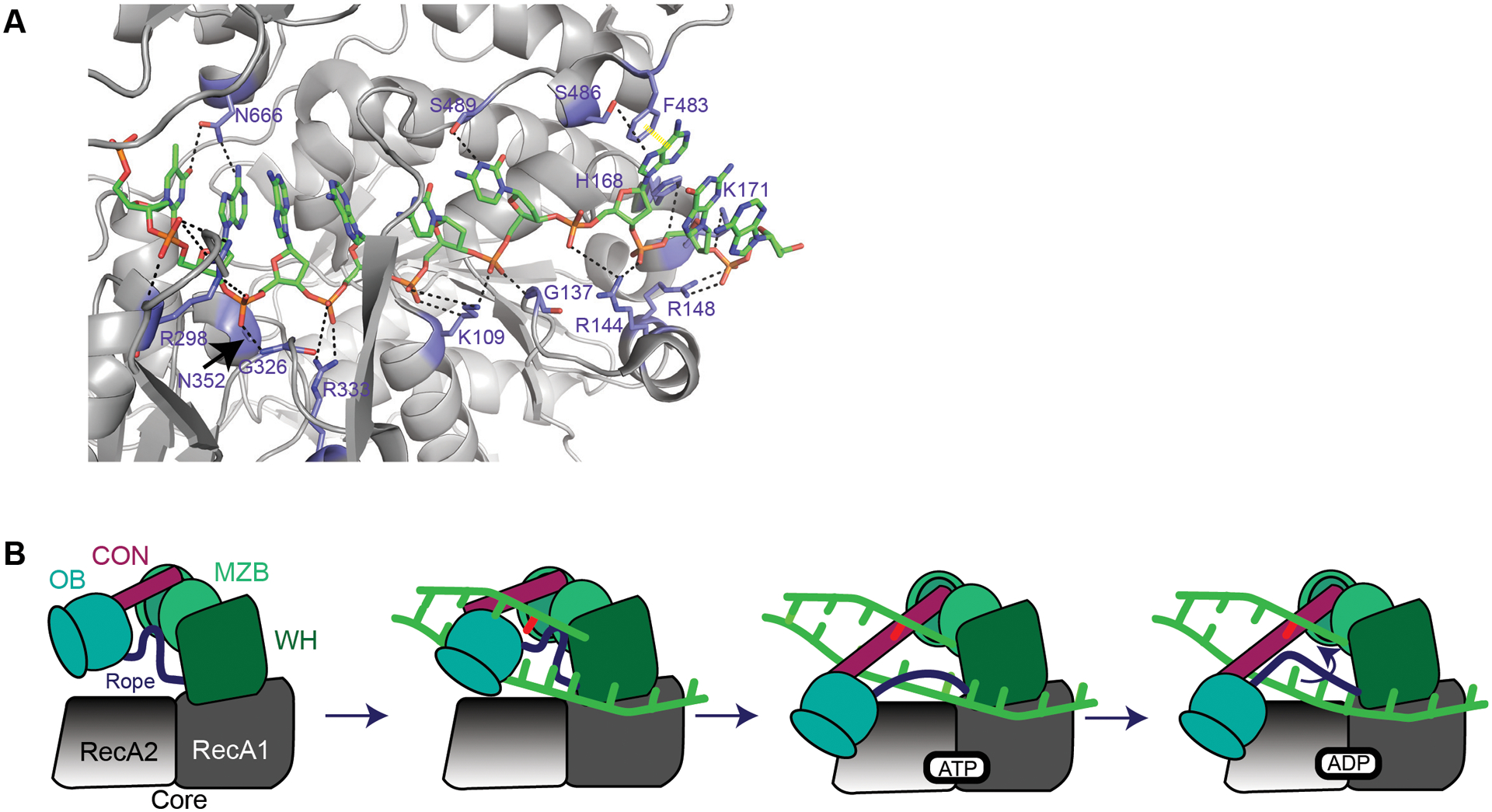
(A) MrfA (6znq) contacts with DNA in the loading strand adapted from128. Note: K108128 is K109. Black dashed lines represent hydrogen bonds. Yellow dashed line represents π-π stacking. A and B are adapted from the following work128. (B) It is unknown how MrfA loads onto DNA or how it identifies a mitomycin C-induced lesion. MrfA contains seven domains: a N-terminal region (NTR, not shown), RecA1 and RecA2 (core domains, grey), a winged helix domain (WHD, green), an oligonucleotide/oligosaccharide binding domain (OB, blue), a connector element (CON, maroon), and a C-terminal DUF1998 that contains a unique MrfA zinc binding domain (MZB, light green)128. DNA shown in bright green with a DNA lesion in red. Upon loading DNA, the OB domain shifts toward RecA2 domain128. The OB domain binds RecA2. As a result, the rope (purple) connecting the OB domain and winged-helix domain undergoes a conformational change. The tightening of this rope across the ssDNA prevents the loading strand from slipping backward128. ATP is bound, causing DNA translocation through an “inchworm”-like mechanism. The mechanism for coupling DNA translocation and unwinding is unknown128. As new ATP is hydrolyzed, DNA is pumped through the protein and products are released.
Conclusions
Because bacteria thrive in such a wide range of habitats, their DNA repair pathways display a broad and unique collection of mechanisms. In this Review we have discussed non-base flipping DNA glycosylases and glycosylases that repair specific types of naturally occurring cross-linking damage. An important future direction will be to study the numerous uncharacterized AlkZ family members to determine their substrate specificity. We have also discussed the MrfAB excision system. Another important future direction will be to determine the mechanism of lesion recognition and to determine if the unstudied enzymes that are part of this family contribute to repair of other types of lesions in the diverse bacteria that use this system. Bacteria have served to drive discovery of novel DNA repair processes and provided new mechanistic insight into established ones; we expect that many new DNA repair strategies will be uncovered in the millions of largely unstudied bacterial species. We look forward to the insights that will be gained in the coming years.
Acknowledgements
The authors wish to thank three anonymous referees for their helpful comments on this work. The authors would also like to acknowledge that due to space limitation many important studies and citations could not be included. This work was funded by the National Institutes of Health grant GM131772 to LAS. KJW was supported by funding from the NIH Cellular Biotechnology Training Grant (T32 GM008353) and a pre-doctoral fellowship from the National Science Foundation (#DEG 1256260).
References
- 1.Friedberg EC et al. DNA Repair and Mutagenesis: Second Edition. (American Society for Microbiology, 2006). [Google Scholar]
- 2.Wang ST et al. The forespore line of gene expression in Bacillus subtilis. J Mol Biol 358, 16–37 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Setlow P I will survive: DNA protection in bacterial spores. Trends Microbiol 15, 172–180 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Park JS, Marr MT & Roberts JWE coli Transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell 109, 757–767 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Goranov AI, Kuester-Schoeck E, Wang JD & Grossman AD Characterization of the global transcriptional responses to different types of DNA damage and disruption of replication in Bacillus subtilis. J Bacteriol 188, 5595–5605 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work characterizes the transcriptional response to mitomycin C. This study also shows that mitomycin C slows replication fork progression in vivo.
- 6.Gupta R, Barkan D, Redelman-Sidi G, Shuman S & Glickman MS Mycobacteria exploit three genetically distinct DNA double-strand break repair pathways. Molecular microbiology 79, 316–330, doi: 10.1111/j.1365-2958.2010.07463.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams JS & Kunkel TA Ribonucleotides in DNA: origins, repair and consequences. DNA Repair (Amst) 19, 27–37, doi: 10.1016/j.dnarep.2014.03.029 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zgur-Bertok D DNA damage repair and bacterial pathogens. PLoS Pathog 9, e1003711, doi: 10.1371/journal.ppat.1003711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matic I Mutation Rate Heterogeneity Increases Odds of Survival in Unpredictable Environments. Mol Cell 75, 421–425, doi: 10.1016/j.molcel.2019.06.029 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Cooke MS, Evans MD, Dizdaroglu M & Lunec J Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17, 1195–1214, doi: 10.1096/fj.02-0752rev (2003). [DOI] [PubMed] [Google Scholar]
- 11.Kow YW & Dare A Detection of abasic sites and oxidative DNA base damage using an ELISA-like assay. Methods 22, 164–169, doi: 10.1006/meth.2000.1057 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Thompson PS & Cortez D New insights into abasic site repair and tolerance. DNA Repair (Amst) 90, 102866, doi: 10.1016/j.dnarep.2020.102866 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sagher D & Strauss B Insertion of nucleotides opposite purinic/apyrimidinic sites in deoxyribonucleic acid during in vitro synthesis: Uniqueness of adenine nucleotides. Biochemistry 22, 4518–4526 (1983). [DOI] [PubMed] [Google Scholar]
- 14.Mullins EA, Rodriguez AA, Bradley NP & Eichman BF Emerging Roles of DNA Glycosylases and the Base Excision Repair Pathway. Trends Biochem Sci 44, 765–781, doi: 10.1016/j.tibs.2019.04.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dronkert ML & Kanaar R Repair of DNA interstrand cross-links. Mutat Res 486, 217–247 (2001). [DOI] [PubMed] [Google Scholar]; This is an excellent comprehensive review covering all aspects of DNA cross-linking agents, and the repair of cross-linking damage in organisms ranging from E. coli to human cells.
- 16.Schroeder JW, Hirst WG, Szewczyk GA & Simmons LA The Effect of Local Sequence Context on Mutational Bias of Genes Encoded on the Leading and Lagging Strands. Curr Biol 26, 692–697, doi: 10.1016/j.cub.2016.01.016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper determines the mutation rate and mutation spectrum caused by a mismatch repair defect in Bacillus subtilis. This work shows that sequence context is the major driver of mutation rate in B. subtilis.
- 17.Schroeder JW, Randall JR, Hirst WG, O’Donnell ME & Simmons LA Mutagenic cost of ribonucleotides in bacterial DNA. Proc Natl Acad Sci U S A 114, 11733–11738, doi: 10.1073/pnas.1710995114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nick McElhinny SA et al. Genome instability due to ribonucleotide incorporation into DNA. Nature chemical biology 6, 774–781, doi: 10.1038/nchembio.424 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yao NY, Schroeder JW, Yurieva O, Simmons LA & O’Donnell ME Cost of rNTP/dNTP pool imbalance at the replication fork. Proceedings of the National Academy of Sciences of the United States of America 110, 12942–12947, doi: 10.1073/pnas.1309506110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vaisman A et al. Investigating the mechanisms of ribonucleotide excision repair in Escherichia coli. Mutat Res Fundam Mol Mech Mutagen 761, 21–33, doi: 10.1016/j.mrfmmm.2014.01.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slauch JM How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol Microbiol 80, 580–583, doi: 10.1111/j.1365-2958.2011.07612.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang FC & Vazquez-Torres A Reactive nitrogen species in host-bacterial interactions. Curr Opin Immunol 60, 96–102, doi: 10.1016/j.coi.2019.05.008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Veen S & Tang CM The BER necessities: the repair of DNA damage in human-adapted bacterial pathogens. Nat Rev Microbiol 13, 83–94, doi: 10.1038/nrmicro3391 (2015). [DOI] [PubMed] [Google Scholar]
- 24.LeVier K, Phillips RW, Grippe VK, Roop RM & Walker GC Similar requirements of a plant symbiont and a mammalian pathogen for prolonged intracellular survival. Science 287, 2492–2493. (2000). [DOI] [PubMed] [Google Scholar]
- 25.Davies BW et al. DNA damage and reactive nitrogen species are barriers to Vibrio cholerae colonization of the infant mouse intestine. PLoS Pathog 7, e1001295 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows how defects in DNA repair pathways prevent passage of V. cholerae through the stomach and a failure to colonize the intestine.
- 26.Cimino GD, Gamper HB, Isaacs ST & Hearst JE Psoralens as photoactive probes of nucleic acid structure and function: organic chemistry, photochemistry, and biochemistry. Annu Rev Biochem 54, 1151–1193, doi: 10.1146/annurev.bi.54.070185.005443 (1985). [DOI] [PubMed] [Google Scholar]
- 27.Barker S, Weinfeld M & Murray D DNA-protein crosslinks: their induction, repair, and biological consequences. Mutat Res 589, 111–135, doi: 10.1016/j.mrrev.2004.11.003 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Procopio RE, Silva IR, Martins MK, Azevedo JL & Araujo JM Antibiotics produced by Streptomyces. Braz J Infect Dis 16, 466–471, doi: 10.1016/j.bjid.2012.08.014 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Barak Y, Cohen-Fix O & Livneh Z Deamination of cytosine-containing pyrimidine photodimers in UV-irradiated DNA. Significance for UV light mutagenesis. J. Biol. Chem 270, 24174–24179 (1995). [DOI] [PubMed] [Google Scholar]
- 30.Prise KM, Pinto M, Newman HC & Michael BD A review of studies of ionizing radiation-induced double-strand break clustering. Radiat. Res. Suppl 156, 572–576 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Ward JF Radiation mutagenesis: the initial DNA lesions responsible. Radiat. Res. Suppl 142, 362–368 (1995). [PubMed] [Google Scholar]
- 32.Hata T et al. Mitomycin, a new antibiotic from Streptomyces. I. J Antibiot (Tokyo) 9, 141–146 (1956). [PubMed] [Google Scholar]
- 33.Armstrong RW, Salvati ME & Nguyen M Novel interstrand cross-links induced by the antitumor antibiotic carzinophilin/azinomycin B. Journal of American Chemical Society 114, 3144–3145 (1992). [Google Scholar]
- 34.Sleigh MJ The mechanism of DNA breakage by phleomycin in vitro. Nucleic Acids Res 3, 891–901 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rizvi RY, Shahabuddin, Rahman A & Hadi SM Effect of alkylation with streptozotocin on the secondary structure of DNA. Biosci Rep 6, 557–564, doi: 10.1007/BF01114952 (1986). [DOI] [PubMed] [Google Scholar]
- 36.Baute J & Depicker A Base excision repair and its role in maintaining genome stability. Crit Rev Biochem Mol Biol 43, 239–276 (2008). [DOI] [PubMed] [Google Scholar]; This is an excellent and comprehensive review of base excision repair across biology. This paper is a tremendous resource detailing lesion types, glycosylases and mechanisms of BER.
- 37.Wallace SS Base excision repair: a critical player in many games. DNA Repair (Amst) 19, 14–26, doi: 10.1016/j.dnarep.2014.03.030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Labahn J et al. Structural basis for the excision repair of alkylation-damaged DNA. Cell 86, 321–329, doi: 10.1016/s0092-8674(00)80103-8 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Dianov G & Lindahl T Reconstitution of the DNA base excision-repair pathway. Curr Biol 4, 1069–1076, doi: 10.1016/s0960-9822(00)00245-1 (1994). [DOI] [PubMed] [Google Scholar]
- 40.Neeley WL & Essigmann JM Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem Res Toxicol 19, 491–505, doi: 10.1021/tx0600043 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Gruber CC & Walker GC Incomplete base excision repair contributes to cell death from antibiotics and other stresses. DNA Repair (Amst) 71, 108–117, doi: 10.1016/j.dnarep.2018.08.014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michaels ML, Cruz C, Grollman AP & Miller JH Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc Natl Acad Sci U S A 89, 7022–7025 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maki H & Sekiguchi M MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 355, 273–275 (1992). [DOI] [PubMed] [Google Scholar]
- 44.Fowler RG & Schaaper RM The role of the mutT gene of Escherichia coli in maintaining replication fidelity. FEMS Microbiol Rev 21, 43–54, doi: 10.1111/j.1574-6976.1997.tb00344.x (1997). [DOI] [PubMed] [Google Scholar]
- 45.Lenhart JS, Schroeder JW, Walsh BW & Simmons LA DNA Repair and Genome Maintenance in Bacillus subtilis. Microbiology and molecular biology reviews : MMBR 76, 530–564, doi: 10.1128/MMBR.05020-11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sugahara M et al. Crystal structure of a repair enzyme of oxidatively damaged DNA, MutM (Fpg), from an extreme thermophile, Thermus thermophilus HB8. EMBO J 19, 3857–3869, doi: 10.1093/emboj/19.15.3857 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fromme JC & Verdine GL Structural insights into lesion recognition and repair by the bacterial 8-oxoguanine DNA glycosylase MutM. Nat Struct Biol 9, 544–552, doi: 10.1038/nsb809 (2002). [DOI] [PubMed] [Google Scholar]
- 48.Williams SD & David SS Evidence that MutY is a monofunctional glycosylase capable of forming a covalent Schiff base intermediate with substrate DNA. Nucleic Acids Res 26, 5123–5133, doi: 10.1093/nar/26.22.5123 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kreppel A, Blank ID & Ochsenfeld C Base-Independent DNA Base-Excision Repair of 8-Oxoguanine. J Am Chem Soc 140, 4522–4526, doi: 10.1021/jacs.7b11254 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Mullins EA et al. The DNA glycosylase AlkD uses a non-base-flipping mechanism to excise bulky lesions. Nature 527, 254–258, doi: 10.1038/nature15728 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work solves the crystal structure and uses a modeling approach to demonstrate the first non-base flipping mechanism for a DNA glycosylase.
- 51.Stivers JT Site-specific DNA damage recognition by enzyme-induced base flipping. Prog Nucleic Acid Res Mol Biol 77, 37–65, doi: 10.1016/S0079-6603(04)77002-6 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Parsons ZD, Bland JM, Mullins EA & Eichman BF A Catalytic Role for C-H/pi Interactions in Base Excision Repair by Bacillus cereus DNA Glycosylase AlkD. J Am Chem Soc 138, 11485–11488, doi: 10.1021/jacs.6b07399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mullins EA, Shi R & Eichman BF Toxicity and repair of DNA adducts produced by the natural product yatakemycin. Nat Chem Biol 13, 1002–1008, doi: 10.1038/nchembio.2439 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi R et al. Selective base excision repair of DNA damage by the non-base-flipping DNA glycosylase AlkC. EMBO J 37, 63–74, doi: 10.15252/embj.201797833 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mohni KN et al. HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA. Cell 176, 144–153 e113, doi: 10.1016/j.cell.2018.10.055 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thompson PS, Amidon KM, Mohni KN, Cortez D & Eichman BF Protection of abasic sites during DNA replication by a stable thiazolidine protein-DNA cross-link. Nat Struct Mol Biol 26, 613–618, doi: 10.1038/s41594-019-0255-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work solves the crystal structure of E. coli YedK bound to and protecting an AP site. This work provides a mechanism that is likely to be conserved with the human protein HEMCES.
- 57.Fang Q DNA-protein crosslinks processed by nucleotide excision repair and homologous recombination with base and strand preference in E. coli model system. Mutat Res 741–742, 1–10, doi: 10.1016/j.mrfmmm.2013.02.005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zeinert R et al. A legacy role for DNA binding of Lon protects against genotoxic stress. bioRxiv, doi: 10.1101/317677 (2018). [DOI] [Google Scholar]
- 59.Sang PB, Srinath T, Patil AG, Woo EJ & Varshney U A unique uracil-DNA binding protein of the uracil DNA glycosylase superfamily. Nucleic Acids Res 43, 8452–8463, doi: 10.1093/nar/gkv854 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tu J, Chen R, Yang Y, Cao W & Xie W Suicide inactivation of the uracil DNA glycosylase UdgX by covalent complex formation. Nat Chem Biol 15, 615–622, doi: 10.1038/s41589-019-0290-x (2019). [DOI] [PubMed] [Google Scholar]
- 61.Ahn WC et al. Covalent binding of uracil DNA glycosylase UdgX to abasic DNA upon uracil excision. Nat Chem Biol 15, 607–614, doi: 10.1038/s41589-019-0289-3 (2019). [DOI] [PubMed] [Google Scholar]
- 62.Chatterjee N & Walker GC Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 58, 235–263, doi: 10.1002/em.22087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patlan AG et al. YwqL (EndoV), ExoA and PolA act in a novel alternative excision pathway to repair deaminated DNA bases in Bacillus subtilis. PLoS One 14, e0211653, doi: 10.1371/journal.pone.0211653 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tomasz M et al. Reaction of DNA with chemically or enzymatically activated mitomycin C: isolation and structure of the major covalent adduct. Proc Natl Acad Sci U S A 83, 6702–6706 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cole JM, Acott JD, Courcelle CT & Courcelle J Limited Capacity or Involvement of Excision Repair, Double-Strand Breaks, or Translesion Synthesis for Psoralen Cross-Link Repair in Escherichia coli. Genetics 210, 99–112, doi: 10.1534/genetics.118.301239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cole RS Repair of DNA containing interstrand crosslinks in Escherichia coli: sequential excision and recombination. Proc Natl Acad Sci U S A 70, 1064–1068, doi: 10.1073/pnas.70.4.1064 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Terawaki A & Greenberg J Effect of carzinophillin on bacterial deoxyribonucleic acid: formation of inter-strand cross-links in deoxyribonucleic acid and their disappearance during post-treatment incubation. Nature 209, 481–484, doi: 10.1038/209481a0 (1966). [DOI] [PubMed] [Google Scholar]
- 68.Mullins EA, Warren GM, Bradley NP & Eichman BF Structure of a DNA glycosylase that unhooks interstrand cross-links. Proc Natl Acad Sci U S A 114, 4400–4405, doi: 10.1073/pnas.1703066114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work solves the structure and provides a model for the mechanism of interstrand cross-link repair by DNA glycosylase AlkZ on azinomycin B adducts.
- 69.Wang S et al. Characterization of a novel DNA glycosylase from S. sahachiroi involved in the reduction and repair of azinomycin B induced DNA damage. Nucleic Acids Res 44, 187–197, doi: 10.1093/nar/gkv949 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao Q et al. Characterization of the azinomycin B biosynthetic gene cluster revealing a different iterative type I polyketide synthase for naphthoate biosynthesis. Chem Biol 15, 693–705, doi: 10.1016/j.chembiol.2008.05.021 (2008). [DOI] [PubMed] [Google Scholar]
- 71.Bradley NP, Washburn LA, Christov PP, Watanabe CMH & Eichman BF Escherichia coli YcaQ is a DNA glycosylase that unhooks DNA interstrand crosslinks. Nucleic Acids Res 48, 7005–7017, doi: 10.1093/nar/gkaa346 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sancar A & Reardon JT Nucleotide excision repair in E. coli and man. Adv Protein Chem 69, 43–71, doi: 10.1016/S0065-3233(04)69002-4 (2004). [DOI] [PubMed] [Google Scholar]
- 73.Vaisman A et al. Removal of misincorporated ribonucleotides from prokaryotic genomes: an unexpected role for nucleotide excision repair. PLoS Genet 9, e1003878, doi: 10.1371/journal.pgen.1003878 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows that NER can serve as a back-up pathway to correct misincorporated ribonucleotides in the absence of the canonical RNase HII- mediated ribonucleotide excision repair pathway.
- 74.Van Houten B & Kad N Investigation of bacterial nucleotide excision repair using single-molecule techniques. DNA Repair (Amst) 20, 41–48, doi: 10.1016/j.dnarep.2013.10.012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kraithong T, Hartley S, Jeruzalmi D & Pakotiprapha D A Peek Inside the Machines of Bacterial Nucleotide Excision Repair. Int J Mol Sci 22, doi: 10.3390/ijms22020952 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is an excellent and comprehensive review covering the structural biology and biochemical mechanisms underlying bacterial NER.
- 76.Case BC, Hartley S, Osuga M, Jeruzalmi D & Hingorani MM The ATPase mechanism of UvrA2 reveals the distinct roles of proximal and distal ATPase sites in nucleotide excision repair. Nucleic Acids Res 47, 4136–4152, doi: 10.1093/nar/gkz180 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work details the mechanism of proximal and distal site ATP usage by UvrA during the process of lesion recognition and dissociation from the lesion allowing for UvrB to form the preincision complex.
- 77.Pakotiprapha D, Samuels M, Shen K, Hu JH & Jeruzalmi D Structure and mechanism of the UvrA-UvrB DNA damage sensor. Nat Struct Mol Biol 19, 291–298, doi: 10.1038/nsmb.2240 (2012). [DOI] [PubMed] [Google Scholar]; This manuscript shows that UvrA2 adopts a “closed groove” conformation, which can only accommodate binding of undamaged (native) DNA in the UvrA2B2 lesion recognition stoichiometry.
- 78.Lin JJ & Sancar A Reconstitution of nucleotide excision nuclease with UvrA and UvrB proteins from Escherichia coli and UvrC protein from Bacillus subtilis. J Biol Chem 265, 21337–21341 (1990). [PubMed] [Google Scholar]
- 79.Lin JJ & Sancar A Active site of (A)BC excinuclease. I. Evidence for 5’ incision by UvrC through a catalytic site involving Asp399, Asp438, Asp466, and His538 residues. J Biol Chem 267, 17688–17692 (1992). [PubMed] [Google Scholar]
- 80.Mazur SJ & Grossman L Dimerization of Escherichia coli UvrA and its binding to undamaged and ultraviolet light damaged DNA. Biochemistry 30, 4432–4443 (1991). [DOI] [PubMed] [Google Scholar]
- 81.Stracy M et al. Single-molecule imaging of UvrA and UvrB recruitment to DNA lesions in living Escherichia coli. Nat Commun 7, 12568, doi: 10.1038/ncomms12568 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper examines the single molecule dynamics of UvrA and UvrB in vivo. It shows that lesion recognition takes place in a two-step process with UvrA acting first followed by recruitment of UvrB.
- 82.Kraithong T et al. Real-time investigation of the roles of ATP hydrolysis by UvrA and UvrB during DNA damage recognition in nucleotide excision repair. DNA Repair (Amst) 97, 103024, doi: 10.1016/j.dnarep.2020.103024 (2021). [DOI] [PubMed] [Google Scholar]
- 83.Orren DK & Sancar A The (A)BC excinuclease of Escherichia coli has only the UvrB and UvrC subunits in the incision complex. Proc Natl Acad Sci U S A 86, 5237–5241 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Verhoeven EE, Wyman C, Moolenaar GF & Goosen N The presence of two UvrB subunits in the UvrAB complex ensures damage detection in both DNA strands. EMBO J 21, 4196–4205, doi: 10.1093/emboj/cdf396 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pakotiprapha D & Jeruzalmi D Small-angle X-ray scattering reveals architecture and A(2)B(2) stoichiometry of the UvrA-UvrB DNA damage sensor. Proteins 81, 132–139, doi: 10.1002/prot.24170 (2013). [DOI] [PubMed] [Google Scholar]
- 86.Lee SJ, Sung RJ & Verdine GL Mechanism of DNA Lesion Homing and Recognition by the Uvr Nucleotide Excision Repair System. Research (Wash D C) 2019, 5641746, doi: 10.34133/2019/5641746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Oh EY & Grossman L Helicase properties of the Escherichia coli UvrAB protein complex. Proc Natl Acad Sci U S A 84, 3638–3642, doi: 10.1073/pnas.84.11.3638 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shi Q, Thresher R, Sancar A & Griffith J Electron microscopic study of (A)BC excinuclease. DNA is sharply bent in the UvrB-DNA complex. J Mol Biol 226, 425–432, doi: 10.1016/0022-2836(92)90957-l (1992). [DOI] [PubMed] [Google Scholar]
- 89.Delagoutte E, Fuchs RP & Bertrand-Burggraf E The isomerization of the UvrB-DNA preincision complex couples the UvrB and UvrC activities. J Mol Biol 320, 73–84, doi: 10.1016/S0022-2836(02)00401-1 (2002). [DOI] [PubMed] [Google Scholar]
- 90.Perera AV, Mendenhall JB, Courcelle CT & Courcelle J Cho Endonuclease Functions during DNA Interstrand Cross-Link Repair in Escherichia coli. J Bacteriol 198, 3099–3108, doi: 10.1128/JB.00509-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mazon G, Philippin G, Cadet J, Gasparutto D & Fuchs RP The alkyltransferase-like ybaZ gene product enhances nucleotide excision repair of O(6)-alkylguanine adducts in E. coli. DNA Repair (Amst) 8, 697–703, doi: 10.1016/j.dnarep.2009.01.022 (2009). [DOI] [PubMed] [Google Scholar]
- 92.Mielecki D, Wrzesinski M & Grzesiuk E Inducible repair of alkylated DNA in microorganisms. Mutat Res Rev Mutat Res 763, 294–305, doi: 10.1016/j.mrrev.2014.12.001 (2015). [DOI] [PubMed] [Google Scholar]
- 93.Margison GP et al. Alkyltransferase-like proteins. DNA Repair (Amst) 6, 1222–1228, doi: 10.1016/j.dnarep.2007.03.014 (2007). [DOI] [PubMed] [Google Scholar]
- 94.Pearson SJ, Ferguson J, Santibanez-Koref M & Margison GP Inhibition of O6-methylguanine-DNA methyltransferase by an alkyltransferase-like protein from Escherichia coli. Nucleic Acids Res 33, 3837–3844, doi: 10.1093/nar/gki696 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Santos-Escobar F, Leyva-Sanchez HC, Ramirez-Ramirez N, Obregon-Herrera A & Pedraza-Reyes M Roles of Bacillus subtilis RecA, Nucleotide Excision Repair, and Translesion Synthesis Polymerases in Counteracting Cr(VI)-Promoted DNA Damage. J Bacteriol 201, doi: 10.1128/JB.00073-19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rivas-Castillo AM, Yasbin RE, Robleto E, Nicholson WL & Pedraza-Reyes M Role of the Y-family DNA polymerases YqjH and YqjW in protecting sporulating Bacillus subtilis cells from DNA damage. Current microbiology 60, 263–267, doi: 10.1007/s00284-009-9535-3 (2010). [DOI] [PubMed] [Google Scholar]
- 97.Janel-Bintz R, Napolitano RL, Isogawa A, Fujii S & Fuchs RP Processing closely spaced lesions during Nucleotide Excision Repair triggers mutagenesis in E. coli. PLoS Genet 13, e1006881, doi: 10.1371/journal.pgen.1006881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Johnson SJ & Beese LS Structures of mismatch replication errors observed in a DNA polymerase. Cell 116, 803–816, doi: 10.1016/s0092-8674(04)00252-1 (2004). [DOI] [PubMed] [Google Scholar]
- 99.Su SS, Lahue RS, Au KG & Modrich P Mispair specificity of methyl-directed DNA mismatch correction in vitro. J Biol Chem 263, 6829–6835 (1988). [PubMed] [Google Scholar]; This paper biochemically reconstitutes methyl-directed mismatch repair showing that for E. coli strand discrimination is signaled by methylation state.
- 100.Lenhart JS, Pillon MC, Guarne A, Biteen JS & Simmons LA Mismatch repair in Gram-positive bacteria. Res Microbiol 167, 4–12, doi: 10.1016/j.resmic.2015.08.006 (2015). [DOI] [PubMed] [Google Scholar]
- 101.Modrich P Methyl-directed DNA mismatch correction. J. Biol. Chem 264, 6597–6600 (1989). [PubMed] [Google Scholar]
- 102.Putnam CD Evolution of the methyl directed mismatch repair system in Escherichia coli. DNA Repair (Amst) 38, 32–41, doi: 10.1016/j.dnarep.2015.11.016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Klocko AD et al. Mismatch repair causes the dynamic release of an essential DNA polymerase from the replication fork. Mol Microbiol 82, 648–663 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lenhart JS, Pillon MC, Guarne A & Simmons LA Trapping and visualizing intermediate steps in the mismatch repair pathway in vivo. Molecular microbiology 90, 680–698, doi: 10.1111/mmi.12389 (2013). [DOI] [PubMed] [Google Scholar]
- 105.Lenhart JS, Sharma A, Hingorani MM & Simmons LA DnaN clamp zones provide a platform for spatiotemporal coupling of mismatch detection to DNA replication. Molecular microbiology 87, 553–568, doi: 10.1111/mmi.12115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liao Y, Schroeder JW, Gao B, Simmons LA & Biteen JS Single-molecule motions and interactions in live cells reveal target search dynamics in mismatch repair. Proc Natl Acad Sci U S A 112, E6898–6906, doi: 10.1073/pnas.1507386112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This manuscript uses a single-molecule approach showing that MutS is recruited to search nascent DNA for mismatches at the replisome. This paper also shows that MutS tracks with replication forks in synchronized cells.
- 107.Lopez de Saro FJ & O’Donnell M Interaction of the beta sliding clamp with MutS, ligase, and DNA polymerase I. Proc. Natl. Acad. Sci. U.S.A 98, 8376–8380 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dalrymple BP, Kongsuwan K, Wijffels G, Dixon NE & Jennings PA A universal protein-protein interaction motif in the eubacterial DNA replication and repair systems. Proc. Natl. Acad. Sci. U.S.A 98, 11627–11632 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Simmons LA, Davies BW, Grossman AD & Walker GC Beta clamp directs localization of mismatch repair in Bacillus subtilis. Mol Cell 29, 291–301 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chai T, Terrettaz C & Collier J Spatial coupling between DNA replication and mismatch repair in Caulobacter crescentus. Nucleic Acids Res 49, 3308–3321, doi: 10.1093/nar/gkab112 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kadyrov FA, Dzantiev L, Constantin N & Modrich P Endonucleolytic function of MutLalpha in human mismatch repair. Cell 126, 297–308 (2006). [DOI] [PubMed] [Google Scholar]; This paper identifies the endonuclease active site in the human MutL homolog PMS2. This study also demonstrates that the site is identical in organisms throughout biology including several bacterial MutL homologs.
- 112.Pillon MC et al. Structure of the endonuclease domain of MutL: unlicensed to cut. Mol Cell 39, 145–151 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pillon MC et al. The sliding clamp tethers the endonuclease domain of MutL to DNA. Nucleic Acids Res 43, 10746–10759, doi: 10.1093/nar/gkv918 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Culligan KM, Meyer-Gauen G, Lyons-Weiler J & Hays JB Evolutionary origin, diversification and specialization of eukaryotic MutS homolog mismatch repair proteins. Nucleic Acids Res 28, 463–471 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sachadyn P Conservation and diversity of MutS proteins. Mutat Res 694, 20–30, doi: 10.1016/j.mrfmmm.2010.08.009 (2010). [DOI] [PubMed] [Google Scholar]
- 116.Cole ST et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544, doi: 10.1038/31159 (1998). [DOI] [PubMed] [Google Scholar]
- 117.Ishino S et al. Identification of a mismatch-specific endonuclease in hyperthermophilic Archaea. Nucleic Acids Res 44, 2977–2986, doi: 10.1093/nar/gkw153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Castaneda-Garcia A et al. A non-canonical mismatch repair pathway in prokaryotes. Nat Commun 8, 14246, doi: 10.1038/ncomms14246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work shows that EndMS/NucS functions in mismatch correction in Mycobacterium, and shows conservation among other bacteria lacking the canonical mismatch repair proteins MutS and MutL.
- 119.Ishino S et al. Activation of the mismatch-specific endonuclease EndoMS/NucS by the replication clamp is required for high fidelity DNA replication. Nucleic Acids Res 46, 6206–6217, doi: 10.1093/nar/gky460 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Takemoto N, Numata I, Su’etsugu M & Miyoshi-Akiyama T Bacterial EndoMS/NucS acts as a clamp-mediated mismatch endonuclease to prevent asymmetric accumulation of replication errors. Nucleic Acids Res 46, 6152–6165, doi: 10.1093/nar/gky481 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nakae S et al. Structure of the EndoMS-DNA Complex as Mismatch Restriction Endonuclease. Structure 24, 1960–1971, doi: 10.1016/j.str.2016.09.005 (2016). [DOI] [PubMed] [Google Scholar]
- 122.Zhang L, Jiang D, Wu M, Yang Z & Oger PM New Insights Into DNA Repair Revealed by NucS Endonucleases From Hyperthermophilic Archaea. Front Microbiol 11, 1263, doi: 10.3389/fmicb.2020.01263 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Burby PE, Simmons ZW, Schroeder JW & Simmons LA Discovery of a dual protease mechanism that promotes DNA damage checkpoint recovery. PLoS Genet 14, e1007512, doi: 10.1371/journal.pgen.1007512 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Burby PE & Simmons LA A bacterial DNA repair pathway specific to a natural antibiotic. Mol Microbiol 111, 338–353, doi: 10.1111/mmi.14158 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper discovers a mitomycin C repair pathway in Bacillus subtilis mediated by newly characterized helicase MrfA and exonuclease MrfB.
- 125.Burby PE, Simmons ZW & Simmons LA DdcA antagonizes a bacterial DNA damage checkpoint. Mol Microbiol 111, 237–253, doi: 10.1111/mmi.14151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yakovleva L & Shuman S Mycobacterium smegmatis SftH exemplifies a distinctive clade of superfamily II DNA-dependent ATPases with 3’ to 5’ translocase and helicase activities. Nucleic Acids Res 40, 7465–7475, doi: 10.1093/nar/gks417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bochman ML, Paeschke K, Chan A & Zakian VA Hrq1, a homolog of the human RecQ4 helicase, acts catalytically and structurally to promote genome integrity. Cell Rep 6, 346–356, doi: 10.1016/j.celrep.2013.12.037 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roske JJ, Liu S, Loll B, Neu U & Wahl MC A skipping rope translocation mechanism in a widespread family of DNA repair helicases. Nucleic Acids Res 49, 504–518, doi: 10.1093/nar/gkaa1174 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work solves the crystal structure for Bacillus subtilis MrfA helicase demonstrating a novel translocation mechanism.
- 129.Amidon KM & Eichman BF Structural biology of DNA abasic site protection by SRAP proteins. DNA Repair (Amst) 94, 102903, doi: 10.1016/j.dnarep.2020.102903 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]