

Abstract
Purpose:
This article will briefly review the origins and evolution of functional genomics, first describing the experimental technology, and then some of the approaches applied to data analysis and visualization. It will emphasize application of functional genomics to radiation biology, using examples from the author’s work to illustrate several key types of analysis. It concludes with a look at non-coding RNA, alternative reading of the genome, and single-cell transcriptomics, some of the innovative areas that may help to shape future research in radiation biology and oncology.
Conclusions:
Transcriptomic approaches have provided insight into many areas of radiation biology and medicine, and innovations in technology and data analysis approaches promise continued contributions to radiation science in the future.
Keywords: ionizing radiation, gene expression, transcriptomics
Introduction
Exposure to ionizing radiation results in damage to all cellular components, triggering a network of signaling cascades, especially as a response to DNA damage. These signaling cascades coordinate cellular and tissue-level responses including DNA repair, cell cycle arrest, apoptosis, fibrosis, and immune and inflammatory responses. These responses are often directly mediated by post-translational protein modifications that alter protein binding, activity or sub-cellular localization, however, changes in gene expression programs are also recognized as a central component of radiation signaling and response (Amundson 2008). The study of gene expression changes has historically contributed to our understanding of the molecular mechanisms of radiation response, and evolving technologies continue to accelerate such studies.
The study of DNA damage-inducible genes developed out of the SOS response, an error-prone DNA repair system activated in Escherichia coli by exposure to ultraviolet radiation (UV) or alkylating agents (Radman 1975). A set of five coordinately regulated SOS genes were initially cloned in E. coli (Kenyon and Walker 1980), with many more such genes identified since. Six DNA damage responsive genes were also identified in yeast, with early estimates that as many as 80 yeast genes might be DNA damage inducible (Ruby and Szostak 1985).
Several DNA damage-inducible genes, including metallothionein Ila, urokinase-type plasminogen activator, and several keratins, were identified in mammalian cells using hybridization subtraction screening of cDNA libraries (Angel et al. 1986; Rotem et al. 1987; Kartasova et al. 1987). This procedure can only identify very abundant transcripts, however. Soon, it was shown that low-abundance transcripts could be enriched and differential expression could be identified using low-ratio hybridization subtraction screening (Fornace and Mitchell 1986). This approach was used to identify UV induction of metallothioneins I and II, and at least 18 novel sequences (Fornace et al. 1988), many of which were later shown to also respond to ionizing radiation.
In the early 1990s the introduction of RT-PCR-based differential display techniques provided another technical boost to studies of differentially expressed genes (Liang and Pardee 1992). More radiation responsive genes were reported, but because cloning and sequencing was required to determine the identity of individual hits, progress remained slow, with most studies describing only one or a few new radiation-induced genes and following a reductionist approach to their study (Gomez et al. 1996; Yan et al. 1996; Chang-Liu and Woloschak 1997; Goltry et al. 1998; Noel et al. 1998; Okamura et al. 2001). While the earlier focus had been on genes with increased expression following DNA damage, differential display experiments also started to identify genes with decreased abundance after irradiation (Woloschak et al. 1995; Paunesku et al. 2000; Watson et al. 2000; Zhou and Rigaud 2001). As an increasingly complex picture of the transcriptional response to DNA damage began to emerge, it became clear that multiple cellular processes, including apoptosis (Paunesku et al. 2000; Okamura et al. 2001), cell cycle regulation (Gomez et al. 1996; Zhou and Rigaud 2001), and cellular signal transduction pathways (Yan et al. 1996; Watson et al. 2000) could be impacted at the level of mRNA abundance.
Experimental approaches for functional genomics
As the human genome project of the 1990s provided an increasing amount of gene sequence information, a shift in research focus from genomics to functional genomics began. Once all coding sequences had been determined and mapped, the next task would be to understand how the genome was used in a dynamic fashion to achieve cell differentiation and specialization, and to respond to environmental signals and challenges. The introduction of cDNA microarrays was to be revolutionary in many biological fields, including radiation biology.
The first cDNA microarray consisted of 45 arabidopsis genes that were robotically printed onto a glass slide (Schena et al. 1995). Two samples could be labeled with different fluorochromes and the relative expression levels of each gene on the array could be measured simultaneously. Early human cDNA arrays surveyed around 1000 genes, and were used to detect both known and novel genes responding to heat shock and phorbol ester (Schena et al. 1996) or to explore differences related to tumorigenicity (DeRisi et al. 1996).
Despite the technical demands of maintaining and annotating large cDNA libraries and producing consistently printed microarrays, as well as early informatics challenges, the microarray technique was soon applied to the study of the radiation-responsive transcriptome. In the first such study (Amundson et al. 1999), we reported 18 known and 30 new gamma-ray responsive sequences in a human myeloid cell line, and showed different patterns of response to various stress agents in different human cell lines. The radiation responsive genes newly identified in this study included ATF3 and FOSL1, which were both shown to have some level of p53 dependence for their radiation response. This study was followed closely by the initial description of potential blood biomarkers for radiation exposure identified using the same cDNA microarray approach (Amundson et al. 2000).
Around the same time, photolithographic techniques were being applied to construct arrays of short oligonucleotides that did not rely on libraries of cDNA clones (Pease et al. 1994), although they were still limited to known sequences. This approach was commercialized by Affymetrix and widely adopted by many institutional core facilities, making the technology widely accessible.
Long oligonucleotide microarrays, either printed or synthesized in situ, also became commercially available (Ben-Dor et al. 2000; Ramakrishnan et al. 2002). These could be used with two-color hybridization protocols similar to cDNA arrays, but their high degree of standardization and quality control also enabled comparison between samples hybridized to different microarray chips. My group has used the Agilent long oligonucleotide microarray platform to study radiation bystander responses (Ghandhi et al. 2008; Ghandhi et al. 2010; Ghandhi et al. 2011; Ghandhi et al. 2014) and to build on our initial radiation biodosimetry work using human (Paul and Amundson 2008; Paul and Amundson 2011; Broustas et al. 2017; Ghandhi et al. 2019), mouse (Broustas et al. 2018; Mukherjee et al. 2019; Paul et al. 2019; Ghandhi et al. 2020), and non-human primate (Park et al. 2017; Ghandhi et al. 2018) models. Recent reviews (Lacombe et al. 2018; Zhao et al. 2018; Amundson 2021) demonstrate that many laboratories, using various whole-genome transcriptomic techniques, continue to contribute to what is now a considerable body of radiation biodosimetry gene expression work.
More recently, transcriptomic profiling has come full circle, with RNA-Seq, like earlier profiling methods, not requiring a priori knowledge of gene sequence for detection of differential expression (Wilhelm et al. 2008; Nagalakshmi et al. 2008). As sequencing costs have become more competitive with microarrays, RNA-Seq is poised to become the dominant technology for transcriptomic studies and appears to be gradually replacing microarrays (Figure 1). The ability to discover differential expression of previously unidentified genes may be particularly useful in understanding stress responses or disease states, where induced or altered transcripts may not have been represented in the libraries used to define the human genome.
Figure 1.
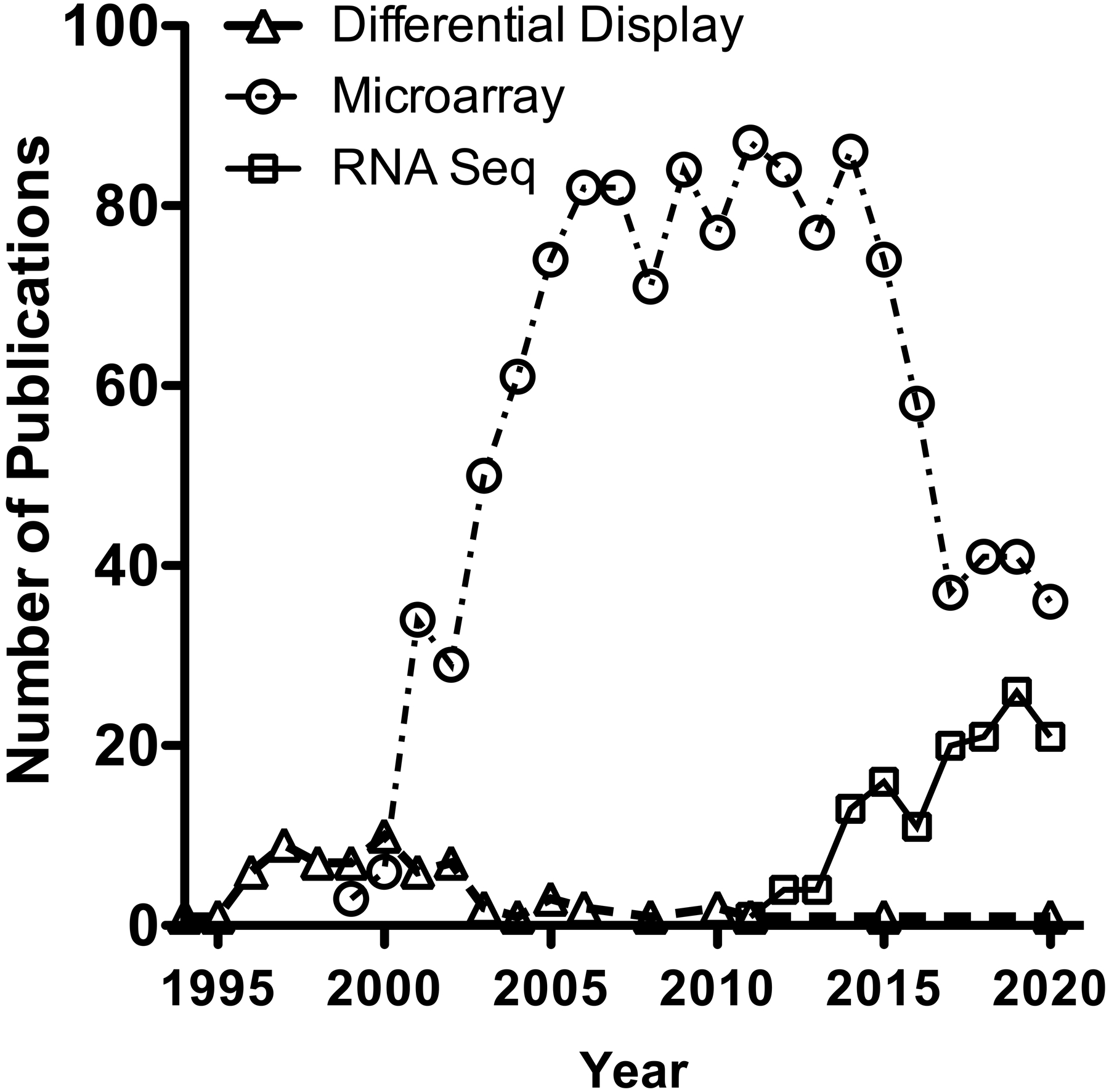
Publications found in Pubmed by searching “ionizing radiation” combined with the terms “differential display”, “microarray”, or “RNA-Seq” plotted by year.
Functional genomics data analysis and visualization
From the early use of printed cDNA microarrays it was almost immediately apparent that the usefulness of whole-genome profiling would be limited mainly by our ability to process and make sense of the huge amounts of data it had now become possible to generate from single samples. Early analyses mainly focused on detecting genes that had significantly different expression levels in different samples. Statistics-based tools, such as DeArray (Chen et al. 1997), provided an improvement over the direct visual or numeric comparison of individual probe intensity between two samples. It was also recognized that performing tens of thousands of simultaneous comparisons could result in many genes achieving apparent statistical significance by chance. Thus, analysis methods incorporating multiple comparison corrections and estimates of the False Discovery Rate (FDR), such as SAM (Significance Analysis of Microarrays) (Tusher et al. 2001) were developed specifically for the identification of genes differentially expressed between groups using microarray data.
Data visualization tools can also be extremely useful for extracting meaningful patterns from large-scale expression data. Many of these are available along with statistical analysis tools, from non-specific statistical tests such as ANOVA or t-tests to multiple comparison adjustment methods, in both commercial and open source platforms. For instance, a huge variety of analysis and visualization tools are available as Bioconductor or other R packages (Carey et al. 2007; Zhang et al. 2009; Huber et al. 2015; McDermaid et al. 2019). User-friendly interfaces are also available, such as BRB Array-Tools (Simon et al. 2007), which provides streamlined access to a curated suite of R packages for microarray data analysis.
Clustering algorithms, such as hierarchical clustering, K-means clustering, and self-organizing maps are often applied to microarray data (Do and Choi 2008; Zhu et al. 2008). These algorithms use different approaches to compare expression patterns of selected genes across all samples in an experiment to group the most similar patterns together. Early studies in yeast found that genes with similar function or shared up-stream regulators could be grouped together in this way (Eisen et al. 1998). Samples can also be clustered as well as genes, for instance to reveal similarities between different tumor types, or to reveal novel sub-types with specific gene expression signatures (Jazaeri et al. 2002). Clustered genes can be displayed as heatmaps, with a row for each gene, a column for each sample, and color intensity representing the relative level of gene expression. Two colors can be used to represent ratios of expression so that both increases and decreases can be clearly visualized, while a single color can be used to represent expression intensities. Figure 2A shows an example of an intensity heatmap visualizing K-means clustering of expression levels of blood genes that significantly responded to an LD50 radiation dose in wild-type mice. Two clusters, representing up-regulated and down-regulated genes, are clearly visible in the wild types, and general ablation of the response can be seen in the two mutant strains. Details of the studies generating this data have been published (Rudqvist et al. 2018).
Figure 2.
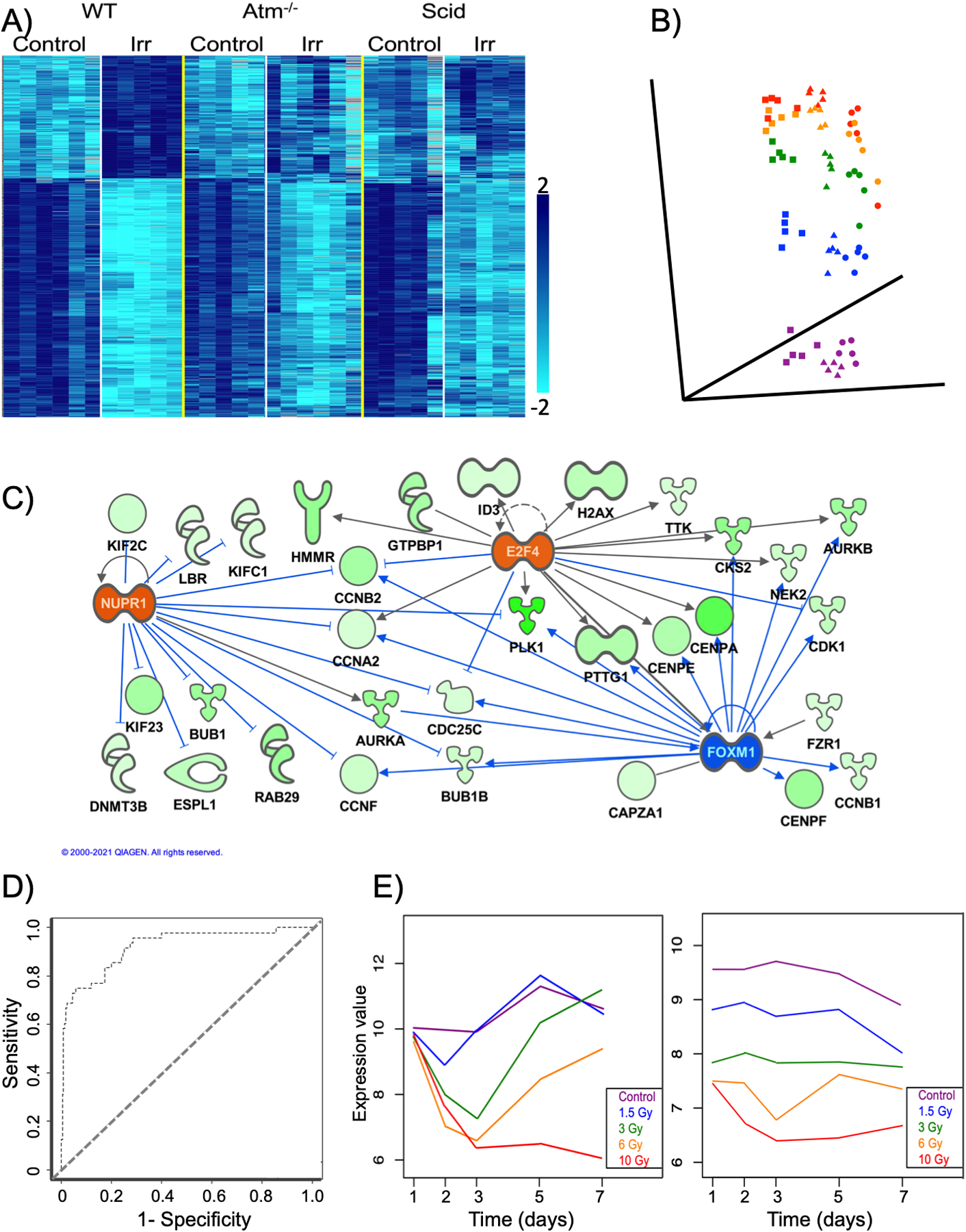
Examples of analysis and visualization tools applied to transcriptomic data.
A) heatmap visualizing K-means clustering of gene expression levels produced using BRB-Array Tools (Simon et al. 2007). Each row represents a gene and each column an individual sample (mouse). Samples are arranged as indicated at the top of the heatmap. WT = wild type; Atm−/− = Atm null; Scid = Prkdcscid; Irr = 24 h post irradiation with an LD50/30 137Cs γ-ray dose. The data are from Rudqvist et al. 2018. The intensity of microarray hybridization, corresponding to the level of gene expression, is represented by the shade of blue as indicated in the key.
B) Multi-dimensional scaling plot created in BRB Array-Tools representing a 72-gene signature of radiation dose (Paul and Amundson 2008) measured in ex vivo irradiated human blood samples at 6 h (squares), 24 h (triangles), or 48 h (circles) after exposure to 0 Gy (purple), 0.5 Gy (blue), 2 Gy (green), 5 Gy (yellow), or 8 Gy (red) 137Cs γ-rays. Data from Paul et al. 2013.
C) Network generated in IPA (Krämer et al. 2014) following upstream regulator analysis of data from Figure 2 of Amundson et al. 2008. Transcription factors colored orange were predicted to be activated by radiation, and blue indicates a transcription factor predicted to be inhibited by radiation. Blue lines indicate the regulatory relationships supporting the prediction. Nodes corresponding to the down-regulated genes (green) have been colored to show the relative radiation response in HL60 cells, with darker color corresponding to a greater magnitude of decrease.
D) ROC curve summarizing performance of a diagonal linear discriminant analysis algorithm built using 20 genes in BRB Array-Tools. Mouse blood was sampled at 1, 2 or 3 days after exposure to doses from 0–10 Gy, and the samples were classified as coming from animals experiencing a survivable exposure (0–6 Gy) or a lethal exposure (10 Gy). The AUC (area under the curve) was 0.92, and the data are from Paul et al. 2019.
E) Two distinct down-regulated gene expression patterns from time-course analysis using MaSigPro (Conesa et al. 2006). Mice were exposed to 0–10 Gy γ-rays and sacrificed for gene expression measurement in blood 1–7 days later (Paul et al. 2019). Relative median expression of all genes in the pattern is plotted as a function of time, with a separate curve for each dose (color coded according to the key). The graph on the left represents 1700 genes, that on the right 98.
Multi-dimensional scaling (MDS) algorithms, such as principal components analysis, can be used to reduce an n-dimensional gene expression space to a 2- or 3-dimensional projection, while maintaining the relative relationships between the samples (Raychaudhuri et al. 2000). In practical terms, this enables visualization of the overall similarity of samples based on the expression of a selected set of genes. The example in Figure 2B illustrates a 72-gene signature of radiation dose (Paul and Amundson 2008) monitored in human blood samples between 6 and 48 hours after radiation exposure (Paul et al. 2013). Each point represents an individual sample, with different colors representing different radiation doses from 0 to 8 Gy, and different shapes representing 6, 24, or 48 hours after irradiation. In this example, it can be seen that the samples cluster by dose, but also separate by the time since exposure, with the effect of dose beginning to diminish after 48 hours compared with the earlier time points.
Network analysis and upstream regulators
Genes that demonstrate patterns of co-expression may also be co-regulated by common upstream pathways. Chromatin Immunoprecipitation (ChIP) assays (Rodríguez-Ubreva and Ballestar 2014) can be used to isolate the DNA sequences bound to a specific transcription factor of interest, such as NFκB or TP53, followed by microarray or sequencing-based identification of the differentially bound genes. Integration of such ChIP-Seq results with expression profiling of the same experimental conditions (Jiang and Mortazavi 2018) can provide insight into mechanisms of transcription regulation following radiation exposure (Janus et al. 2018; Szołtysek et al. 2018; Hafner et al. 2020).
A large amount of non-radiation specific DNA-protein and protein-protein binding data is also publicly available through sources such as the Biomolecular Interaction Network Database (BIND) (Alfarano et al. 2005). In conjunction with gene expression results, such protein-binding information can be used to construct and visualize putative regulatory interaction networks. Free platforms, such as Cytoscape (Shannon et al. 2003), as well as commercial solutions, such as Ingenuity Pathway Analysis (IPA) (Krämer et al. 2014), are available to perform network analyses. We previously used BIND and Cytoscape to build a regulatory network of a strongly down-regulated cluster of TP53-independent radiation responsive genes in the cell lines of the NCI-60 panel. The network analysis indicated E2F4 as a potential upstream regulator of this response, and we were able to confirm response of E2F4 to radiation (Amundson et al. 2008).
A reanalysis of the same NCI-60 gene expression data using IPA is illustrated in Figure 2C. In this case, IPA was first used to predict putative upstream regulators of the genes in the down-regulated cluster. IPA assumes a normal distribution of gene up- or down-regulation for each potential upstream regulator-gene connection, and calculates a z-score (number of standard deviations from the mean) to determine the significant over-representation of “activated” or “inhibited” predictions. For each potential regulator, a z-score of ≥2 is taken as significantly likely to be activated, and a z-score of ≤−2 as significantly inhibited. E2F4 was again predicted to be activated by radiation, but additional transcription factors were also predicted as possible upstream regulators of the down-regulated genes. The strongest predictions were for activation of NUPR1 (z-score 3.5) and inhibition of FOXM1 (z-score −3.7). FOXM1 is a known regulator of cell cycle genes with roles in carcinogenesis (Myatt and Lam 2007), and its down-regulation may enhance radiosensitivity (Nagel et al. 2015; Xiu et al. 2018). Consistent with our prediction from the NCI-60 data, NUPR1 has been shown to be induced by multiple cellular stressors, including ionizing radiation (Gironella et al. 2009). We have also previously reported IPA-predicted activation of NUPR1 by direct irradiation with 123 keV/μm 4He ions, and inhibition in un-irradiated bystanders of the same cells (Ghandhi et al. 2014). This example illustrates the common finding that reanalysis of transcriptomic data using different or updated approaches generally confirms older findings, but also often suggests additional directions for further investigation.
Gene ontology
Gene ontology analyses are also commonly employed to gain insight into the biological functions likely to be affected in an experiment. The Gene Ontology (GO) consortium maintains annotations of biological processes and molecular functions of genes and gene families (The Gene Ontology Consortium 2019). Other databases, such as KEGG (the Kyoto Encyclopedia of Genes and Genomes) (Kanehisa et al. 2017), and Reactome (Jassal et al. 2020), organize genes into pathways, while databases, such as InterPro (Mitchell et al. 2019), SMART (Simple Modular Architecture Research Tool) (Letunic and Bork 2018), or PANTHER (Protein ANalysis THrough Evolutionary Relationships)(Thomas et al. 2003), classify protein functional domains or motifs and associate them to coding genes. Many freely available analysis platforms allow users to query gene lists against multiple such databases in order to look for terms that are overrepresented compared to their expected occurrence in a random gene list of the same length. These tools implement methods to correct for multiple comparison, and include DAVID (the Database for Annotation, Visualization and Integrated Discovery) (Huang et al. 2009), PANTHER Tools (Mi et al. 2019), AmiGO (Carbon et al. 2009), and ToppFun (Transcriptome, ontology, phenotype, proteome, and pharmacome annotations based gene list Functional enrichment analysis)(Chen et al. 2009). Gene Set Enrichment Analysis (GSEA) (Subramanian et al. 2005) is a related approach for revealing enriched biological processes through comparison of experimental results with pre-defined gene lists. An advantage of this approach is that in addition to GO or other annotated categories, any curated gene list can be used for comparison.
Gene expression based classifiers
Numerous algorithms have been applied to develop predictive classifiers based on gene expression. These include such approaches as linear discriminant analysis, the Bayesian compound covariate predictor, and shrunken centroid, nearest centroid and nearest neighbor classifiers (Simon et al. 2007). Artificial intelligence approaches, including random forest classification, support vector machines, radial bias function neural networks, and multilayer perceptron neural networks can also be applied (Pirooznia et al. 2008). Such classification algorithms have been used for gene expression based prediction of the likelihood of cancer metastasis (van’t Veer and Bernards 2008), and to predict radiosensitivity (Torres-Roca et al. 2005; Williams et al. 2011; Williams et al. 2017) or the risk of normal tissue damage (Nuyten and van de Vijver 2008; Lyngholm et al. 2015). They have also been applied in the radiation biodosimetry arena. For example, one early study used Bayesian regression models (West et al. 2001) with leave-one-out cross validation to classify samples from mice as controls or radiation exposed (Dressman et al. 2007). Similarly, a nearest centroid classifier with leave-one-out cross validation was used for non-binary classification of human blood samples irradiated ex vivo as either unexposed, 0.5, 2 or ≥5 Gy (Paul and Amundson 2008). In a different application, independent training and test sets were used with seven classification algorithms (linear discriminant analysis, Bayesian compound covariate predictor, nearest centroid, compound covariate predictor, 1- and 3-nearest neighbors, and support vector machines) to demonstrate improved binary classification of irradiated versus control samples across DNA-repair deficient mouse strains when training sets included all genotypes (Rudqvist et al. 2018).
For binary classifiers, an algorithm’s performance is commonly illustrated using a receiver operating characteristic (ROC) curve, such as the example in Figure 2D. The ROC curve in the example shows a diagonal linear discriminant analysis using 20 genes selected with a greedy pairs algorithm to discriminate at 1, 2, or 3 days after exposure between mice given a lethal dose of radiation (10 Gy) and mice given survivable doses from 0–6 Gy (Paul et al. 2019). By plotting sensitivity (the true positive rate) against 1-specificity (the false positive rate) it visualizes the trade off between sensitivity and specificity. The point in the extreme upper left corner of the graph represents perfect classification, and the diagonal line represents completely random classification. Classifier performance is reported using the area under the curve (AUC), which would be 1 for perfect classification, and for the case illustrated is 0.92.
Time series analysis
As reflected in the MDS plot in Figure 2B, gene expression in response to radiation or other stresses is a highly dynamic process, and a single microarray or RNA-Seq analysis only provides a snapshot of an instant in time. Time-course data, or more complex experiments, such as a dose response as a function of time, or comparison of multiple irradiation conditions, can become unwieldy to analyze. Pattern clustering approaches, developed for the analysis of time-course data, cluster curves rather than individual points, and can provide insight from time courses or other complex data. One approach developed for transcriptomic data is the Short Time series Expression Miner (STEM) algorithm, which clusters genes into pre-defined patterns based on “units of change” as a function of time and tests significance using a permutation test (Ernst and Bar-Joseph 2006). A Feature Based PAM (Partitioning Around Medoids) Algorithm (FBPA) that incorporates biologically relevant features to summarize gene expression over time has also been developed to cluster gene expression curves without comparison to pre-defined profiles (Sinha and Markatou 2011). In a direct comparison between STEM and FBPA, we were able to extract more biologically relevant clusters from radiation bystander data using FBPA, including identification of a novel methylation pathway involved in the bystander response (Ghandhi et al. 2011). We have also found the maSigPro R package (Conesa et al. 2006) useful for analysis of complex radiation response data sets, including a study monitoring gene expression for two weeks in response to different amounts of internally deposited 137Cs (Ghandhi et al. 2020) and a dose-response study of gene expression covering the first week after external-beam gamma irradiation of mice (Paul et al. 2019). Two of the gene response patterns identified by maSigPro in the latter study are illustrated in Figure 2E. Both represent down-regulated genes, but the genes in these two clusters show very different time and dose relationships. In the first pattern, expression levels decrease during the first few days, and then begin to recover toward control values in a dose dependent manner. Only mice exposed to a lethal 10 Gy dose show no recovery of expression levels. In contrast, the second pattern shows a strong dose dependence, but little time dependence, with gene expression dropping rapidly by the first day after exposure, then remaining at a fairly consistent level throughout the study.
Public accessibility of data
There is an increasing movement of support for open science and open data of all kinds. The wealth of functional genomic data being generated in radiation studies represents a great potential resource for the field. There are a number of repositories available to ensure its continued public availability. Some, such as the Gene Expression Omnibus (Barrett et al. 2009) or ArrayExpress (Brazma et al. 2003) are general transcriptomic repositories, in which it can be difficult to find relevant radiation experiments. Specialized gene expression databases have been developed targeting the radiation community, including Radiation Genes (Chiani et al. 2009) and the NASA GeneLab (Beheshti et al. 2018; Berrios et al. 2021), which integrates radiation datasets from transcriptomic, proteomic, and metabolomic studies. Such efforts, combined with the ongoing development of increasingly powerful analysis tools, which can be applied to existing public datasets, will help ensure that the maximum insight can be derived from all transcriptomic experiments, past and future.
Future directions for radiation functional genomics
Since the initial introduction of cDNA microarrays, functional genomics techniques have expanded to enable an increasingly broad range of studies. Molecular biology has uncovered new levels of regulation by non-coding transcripts and alternatively spliced forms of known genes (Mortazavi et al. 2008). The increasing accessibility of whole-genome RNA-Seq and its ability to detect novel transcripts, as well as third-generation sequencing providing full transcript reads (Cruz-Garcia et al. 2020a), will likely enhance the study of these regulatory layers and their roles in radiation response and disease. Advances in both technology and informatics are also accelerating studies of the heterogeneity of radiation response on the single cell level. These promising directions are discussed briefly below.
Non-coding RNA
When one of the original “growth arrest and DNA-damage inducible” genes, Gadd7, was found to exert growth arrest properties but to lack a protein product (Hollander et al. 1996), it was an intriguing curiosity, but the observation then languished for several decades. Now, however, non-coding RNAs (ncRNA) are understood as key regulatory factors, with roles in diverse processes from chromatin remodeling and gene transcription to protein translation. The importance of ncRNAs is underlined by the finding that their dis-regulation appears to be involved in many pathological states, including cancer (Schmitt and Chang 2013; Choudhari et al. 2020). Although multiple varieties of ncRNA are now known, long non-coding RNAs (lncRNA, such as Gadd7), micro RNAs (miRNA), and circular RNA (circRNA) in particular appear to contribute to the response to radiation, and may be useful as both biomarkers and therapeutic targets (May et al. 2021).
Investigation of miRNA contributions to radiation response was initially enabled by microarray platforms or low-density TaqMan arrays (Goulter et al. 2006). We used the TaqMan low-density array approach to develop miRNA signatures that could classify mouse blood samples as coming from unexposed, low LET exposed or heavy-ion exposed mice with high accuracy (Templin et al. 2011). We also found that accurate classification of human samples from total body irradiation patients was possible using miRNA signatures (Templin et al. 2011). Comparison of miRNA expression in that study with global mRNA expression measured in the same patients (Paul et al. 2011) further identified a set of 37 genes that were downregulated after irradiation, and were also predicted to be targets of consistently upregulated miRNAs. Several biological processes including hematopoiesis and immune response were over-represented among these genes, suggesting a role for miRNA regulation in their response to radiation.
Studies of ncRNA in the context of radiation response continue to expand. Both miRNA and lncRNA have been found to participate in regulating DNA double-strand break repair (Thapar 2018), a critical response to ionizing radiation exposure. Specific lncRNA (Jiang et al. 2017; Hu et al. 2019; Ma et al. 2018), miRNA (Weidhaas et al. 2007; Zhang et al. 2011), and circRNA (Guan et al. 2020; Niu et al. 2020), have been associated with alteration of radio-resistance of human tumors, and may present targets for modification of radiation response. The development of normal tissue damage may also be modulated by ncRNA, with the lncRNA WWC2-AS1 found to be a regulator of radiation-induced fibrosis (Zhou et al. 2019).
The relative stability of ncRNA species, and their presence in serum and in exosomes, also makes them attractive targets for the development of radiation biodosimetry (Jacob et al. 2013; Beer et al. 2017; Aryankalayil et al. 2018; Yadav et al. 2020). miRNA may also provide tissue-specific biomarkers of radiation damage (Khan et al. 2013; Menon et al. 2016; Rogers et al. 2020), or early predictors of death following irradiation (Acharya et al. 2015; Tomasik et al. 2018).
Variant transcripts and translation
Alternative reading of the genome, including the use of alternative transcription start sites, alternative splicing leading to different exon usage, alternative polyadenylation and alternative initiation of translation can all contribute to the functional regulation of the genome, and the broad extent of these alternative processes is being revealed through RNA-Seq (de Klerk and ‘t Hoen 2015). These mechanisms can also play a part in the response to radiation. Ionizing radiation has been shown to induce transcription using alternate promoters in MDM2 (Barak et al. 1994), PPM1D (Rossi et al. 2008), RRM2B and XPC (Forrester et al. 2012), as well as a number of other genes (Sprung et al. 2011). Alternative splicing of genes following irradiation is also being increasingly reported, particularly in the context of radiation biodosimetry (Macaeva et al. 2016; Wahba et al. 2018). The use of radiation-induced exon expression normalized against intragenic control exons of genes including MDM2, PPM1D, and FDXR has been suggested as an attractive approach to provide more robust radiation biodosimetry (Forrester and Sprung 2014; Cruz-Garcia et al. 2020b).
The use of different promoters in irradiated cells has also been linked to different translational profiles for those genes (Barak et al. 1994; Rossi et al. 2008). More broadly, a microarray analysis of polysome-bound mRNA found that radiation exposure had a 10-fold greater impact on gene translation than on transcription (Lü et al. 2006). The translational profiles responding to radiation also appear to be tumor type specific, and to differ between tumor and normal cell lines (Kumaraswamy et al. 2008). RNA-Seq analysis of polysome-bound RNA fractions has revealed a 3-fold enhancement of radiation-induced changes in translation of alternative transcripts compared to their transcription, further emphasizing the interconnection between these levels of functional genomic control (Wahba et al. 2018). Translational control and alternative transcription in response to radiation remain understudied areas worthy of more detailed investigation.
Single-cell transcriptomics
The heterogeneity of the cellular response to radiation exposure has long been recognized. For instance, variation in the intensity of TP53 antibody staining was observed in different cells within a population exposed to the same radiation dose (MacCallum et al. 2001). Early attempts to look at the radiation transcriptional response at a single cell level also indicated a high degree of cell-to-cell variability (Ponnaiya et al. 2007; Ponnaiya et al. 2013) and suggested within-cell correlation of the expression of genes with common upstream regulatory factors (Shang et al. 2019). However, these studies were limited to the measurement of only one or a small number of genes. More recent advances in both sequencing technology and data analysis have enabled studies in which the transcriptome of individual cells within a sample can be studied, further defining the scope of cellular heterogeneity in the response to radiation (Gao et al. 2021). Such studies have also provided more granular molecular insight into heterogeneous radiation responses, for instance, identifying activation of different signaling pathways in subsets of T-lymphocytes (Moreno-Villanueva et al. 2019), and suggesting novel gene expression changes during the development of radiation resistance during tumor treatment (Wu et al. 2019). Reconstructing cell-type specific radiation signaling networks from single-cell sequencing data has also provided insights into the interplay between radiotherapy and immune cell activity (Formenti et al. 2019).
Further technological refinements are enabling true tissue-level systems biology (Moor and Itzkovitz 2017) and deconvolution of the tumor microenvironment (Wang et al. 2021) through retention of spatial information coupled with single-cell resolution transcriptomics (Teves and Won 2020). These spatial transcriptomic techniques have, for example, been applied to bone marrow stem cell niches to study conditions of homeostasis, carcinogenesis, and response to stresses including radiation (Al-Sabah et al. 2020). Spatial and single-cell transcriptomics hold great promise for the development of immunotherapies (Nerurkar et al. 2020; Castellanos-Rueda et al. 2021), and for our understanding of the radiation response of complex tissues, tumors, and interacting cell types. As functional genomic technologies continue to evolve, it seems certain that the field will have an ongoing impact in diverse areas of radiation biology and radiation oncology.
Acknowledgements
The preparation of this manuscript was supported in part by NCI grant no. R01 CA256840 and by the National Institute of Allergy and Infectious Diseases (NIAID) grant no. U19-AI067773 to the Center for High-Throughput Minimally Invasive Radiation Biodosimetry.
Biography
Sally Amundson is an Associate Professor in the Center for Radiological Research at Columbia University Irving Medical Center in New York. She is a member of the National Council on Radiation Protection and Measurements, the Nuclear and Radiation Studies Board of the National Academy of Science, and at the time of writing is serving as Vice President of the Radiation Research Society.
Footnotes
Declaration of interests:
The author reports no conflicts of interest. The author alone is responsible for the content and writing of this paper.
References
- Acharya SS, Fendler W, Watson J, Hamilton A, Pan Y, Gaudiano E, Moskwa P, Bhanja P, Saha S, Guha C et al. 2015. Serum microRNAs are early indicators of survival after radiation-induced hematopoietic injury. Sci Transl Med. 7:287ra69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sabah J, Baccin C, Haas S. 2020. Single-cell and spatial transcriptomics approaches of the bone marrow microenvironment. Curr Opin Oncol. 32:146–153. [DOI] [PubMed] [Google Scholar]
- Alfarano C, Andrade CE, Anthony K, Bahroos N, Bajec M, Bantoft K, Betel D, Bobechko B, Boutilier K, Burgess E et al. 2005. The Biomolecular Interaction Network Database and related tools 2005 update. Nucleic Acids Res. 33:D418–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amundson SA. 2008. Functional Genomics and a New Era in Radiation Biology and Oncology. Bioscience. 58:491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amundson SA. 2021. Transcriptomics for radiation biodosimetry: progress and challenges. Int J Radiat Biol. on line ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amundson SA, Bittner M, Chen Y, Trent J, Meltzer P, Fornace AJJ. 1999. Fluorescent cDNA microarray hybridization reveals complexity and heterogeneity of cellular genotoxic stress responses. Oncogene. 18:3666–3672. [DOI] [PubMed] [Google Scholar]
- Amundson SA, Do KT, Shahab S, Bittner M, Meltzer P, Trent J, Fornace AJJ. 2000. Identification of potential mRNA biomarkers in peripheral blood lymphocytes for human exposure to ionizing radiation. Radiat Res. 154:342–346. [DOI] [PubMed] [Google Scholar]
- Amundson SA, Do KT, Vinikoor LC, Lee RA, Koch-Paiz CA, Ahn J, Reimers M, Chen Y, Scudiero DA, Weinstein JN et al. 2008. Integrating global gene expression and radiation survival parameters across the 60 cell lines of the National Cancer Institute Anticancer Drug Screen. Cancer Res. 68:415–424. [DOI] [PubMed] [Google Scholar]
- Angel P, Pöting A, Mallick U, Rahmsdorf HJ, Schorpp M, Herrlich P. 1986. Induction of metallothionein and other mRNA species by carcinogens and tumor promoters in primary human skin fibroblasts. Mol Cell Biol. 6:1760–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryankalayil MJ, Chopra S, Levin J, Eke I, Makinde A, Das S, Shankavaram U, Vanpouille-Box C, Demaria S, Coleman CN. 2018. Radiation-Induced Long Noncoding RNAs in a Mouse Model after Whole-Body Irradiation. Radiat Res. 189:251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak Y, Gottlieb E, Juven-Gershon T, Oren M. 1994. Regulation of mdm2 expression by p53: alternative promoters produce transcripts with nonidentical translation potential. Genes Dev. 8:1739–1749. [DOI] [PubMed] [Google Scholar]
- Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA et al. 2009. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 37:D885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer L, Nemec L, Wagner T, Ristl R, Altenburger LM, Ankersmit HJ, Mildner M. 2017. Ionizing radiation regulates long non-coding RNAs in human peripheral blood mononuclear cells. J Radiat Res. 58:201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beheshti A, Miller J, Kidane Y, Berrios D, Gebre SG, Costes SV. 2018. NASA GeneLab Project: Bridging Space Radiation Omics with Ground Studies. Radiat Res. 189:553–559. [DOI] [PubMed] [Google Scholar]
- Ben-Dor A, Bruhn L, Friedman N, Nachman I, Schummer M, Yakhini Z. 2000. Tissue classification with gene expression profiles. J Comput Biol. 7:559–583. [DOI] [PubMed] [Google Scholar]
- Berrios DC, Galazka J, Grigorev K, Gebre S, Costes SV. 2021. NASA GeneLab: interfaces for the exploration of space omics data. Nucleic Acids Res. 49:D1515–D1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazma A, Parkinson H, Sarkans U, Shojatalab M, Vilo J, Abeygunawardena N, Holloway E, Kapushesky M, Kemmeren P, Lara GG et al. 2003. ArrayExpress--a public repository for microarray gene expression data at the EBI. Nucleic Acids Res. 31:68–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broustas CG, Harken AD, Garty G, Amundson SA. 2018. Identification of differentially expressed genes and pathways in mice exposed to mixed field neutron/photon radiation. BMC Genomics. 19:504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broustas CG, Xu Y, Harken AD, Chowdhury M, Garty G, Amundson SA. 2017. Impact of Neutron Exposure on Global Gene Expression in a Human Peripheral Blood Model. Radiat Res. 187:433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S, AmiGO H, Web PWG. 2009. AmiGO: online access to ontology and annotation data. Bioinformatics. 25:288–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey VJ, Morgan M, Falcon S, Lazarus R, Gentleman R. 2007. GGtools: analysis of genetics of gene expression in bioconductor. Bioinformatics. 23:522–523. [DOI] [PubMed] [Google Scholar]
- Castellanos-Rueda R, Di Roberto RB, Schlatter FS, Reddy ST. 2021. Leveraging Single-Cell Sequencing for Chimeric Antigen Receptor T Cell Therapies. Trends Biotechnol. [DOI] [PubMed] [Google Scholar]
- Chang-Liu CM, Woloschak GE. 1997. Effect of passage number on cellular response to DNA-damaging agents: cell survival and gene expression. Cancer Lett. 113:77–86. [DOI] [PubMed] [Google Scholar]
- Chen J, Bardes EE, Aronow BJ, Jegga AG. 2009. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37:W305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dougherty ER, Bittner ML. 1997. Ratio-based decisions and the quantitative analysis of cDNA microarray images. J Biomedical Optics. 2:364–374. [DOI] [PubMed] [Google Scholar]
- Chiani F, Iannone C, Negri R, Paoletti D, D’Antonio M, De Meo PD, Castrignano T. 2009. Radiation Genes: a database devoted to microarrays screenings revealing transcriptome alterations induced by ionizing radiation in mammalian cells. Database (Oxford). 2009:bap007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhari R, Sedano MJ, Harrison AL, Subramani R, Lin KY, Ramos EI, Lakshmanaswamy R, Gadad SS. 2020. Long noncoding RNAs in cancer: From discovery to therapeutic targets. Adv Clin Chem. 95:105–147. [DOI] [PubMed] [Google Scholar]
- Conesa A, Nueda MJ, Ferrer A, Talón M. 2006. maSigPro: a method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics. 22:1096–1102. [DOI] [PubMed] [Google Scholar]
- Cruz-Garcia L, O’Brien G, Sipos B, Mayes S, Love MI, Turner DJ, Badie C. 2020a. Generation of a Transcriptional Radiation Exposure Signature in Human Blood Using Long-Read Nanopore Sequencing. Radiat Res. 193:143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Garcia L, O’Brien G, Sipos B, Mayes S, Tichý A, Sirák I, Davídková M, Marková M, Turner DJ, Badie C. 2020b. In Vivo Validation of Alternative FDXR Transcripts in Human Blood in Response to Ionizing Radiation. Int J Mol Sci. 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Klerk E, ‘t Hoen PA. 2015. Alternative mRNA transcription, processing, and translation: insights from RNA sequencing. Trends Genet. 31:128–139. [DOI] [PubMed] [Google Scholar]
- DeRisi J, Penland L, Brown PO, Bittner ML, Meltzer PS, Ray M, Chen Y, Su YA, Trent JM. 1996. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet. 14:457–460. [DOI] [PubMed] [Google Scholar]
- Do JH, Choi DK. 2008. Clustering approaches to identifying gene expression patterns from DNA microarray data. Mol Cells. 25:279–288. [PubMed] [Google Scholar]
- Dressman HK, Muramoto GG, Chao NJ, Meadows S, Marshall D, Ginsburg GS, Nevins JR, Chute JP. 2007. Gene expression signatures that predict radiation exposure in mice and humans. PLoS Med. 4:e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 95:14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Bar-Joseph Z. 2006. STEM: a tool for the analysis of short time series gene expression data. BMC Bioinformatics. 7:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formenti SC, Hawtin RE, Dixit N, Evensen E, Lee P, Goldberg JD, Li X, Vanpouille-Box C, Schaue D, McBride WH et al. 2019. Baseline T cell dysfunction by single cell network profiling in metastatic breast cancer patients. J Immunother Cancer. 7:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornace AJ, Mitchell JB. 1986. Induction of B2 RNA polymerase III transcription by heat shock: enrichment for heat shock induced sequences in rodent cells by hybridization subtraction. Nucleic Acids Res. 14:5793–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornace AJ Jr, Alamo IJ, Hollander MC. 1988. DNA damage-inducible transcripts in mammalian cells. Proc Natl Acad Sci U S A. 85:8800–8804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester HB, Li J, Hovan D, Ivashkevich AN, Sprung CN. 2012. DNA repair genes: alternative transcription and gene expression at the exon level in response to the DNA damaging agent, ionizing radiation. PLoS One. 7:e53358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester HB, Sprung CN. 2014. Intragenic controls utilizing radiation-induced alternative transcript regions improves gene expression biodosimetry. Radiat Res. 181:314–323. [DOI] [PubMed] [Google Scholar]
- Gao Y, Duan Q, Wu N, Xu B. 2021. A heterogeneous cellular response to ionizing radiation revealed by single cell transcriptome sequencing. Am J Cancer Res. 11:513–529. [PMC free article] [PubMed] [Google Scholar]
- Ghandhi SA, Ming L, Ivanov VN, Hei TK, Amundson SA. 2010. Regulation of early signaling and gene expression in the alpha-particle and bystander response of IMR-90 human fibroblasts. BMC Med Genomics. 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandhi SA, Ponnaiya B, Panigrahi SK, Hopkins KM, Cui Q, Hei TK, Amundson SA, Lieberman HB. 2014. RAD9 deficiency enhances radiation induced bystander DNA damage and transcriptomal response. Radiat Oncol. 9:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandhi SA, Shuryak I, Morton SR, Amundson SA, Brenner DJ. 2019. New Approaches for Quantitative Reconstruction of Radiation Dose in Human Blood Cells. Sci Rep. 9:18441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandhi SA, Sima C, Weber WM, Melo DR, Rudqvist N, Morton SR, Turner HC, Amundson SA. 2020. Dose and Dose-Rate Effects in a Mouse Model of Internal Exposure to 137Cs. Part 1: Global Transcriptomic Responses in Blood. Radiat Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandhi SA, Sinha A, Markatou M, Amundson SA. 2011. Time-series clustering of gene expression in irradiated and bystander fibroblasts: an application of FBPA clustering. BMC Genomics. 12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandhi SA, Turner HC, Shuryak I, Dugan GO, Bourland JD, Olson JD, Tooze JA, Morton SR, Batinic-Haberle I, Cline JM et al. 2018. Whole thorax irradiation of non-human primates induces persistent nuclear damage and gene expression changes in peripheral blood cells. PLoS One. 13:e0191402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandhi SA, Yaghoubian B, Amundson SA. 2008. Global gene expression analyses of bystander and alpha particle irradiated normal human lung fibroblasts: synchronous and differential responses. BMC Med Genomics. 1:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gironella M, Malicet C, Cano C, Sandi MJ, Hamidi T, Tauil RM, Baston M, Valaco P, Moreno S, Lopez F et al. 2009. p8/nupr1 regulates DNA-repair activity after double-strand gamma irradiation-induced DNA damage. J Cell Physiol. 221:594–602. [DOI] [PubMed] [Google Scholar]
- Goltry KL, Epperly MW, Greenberger JS. 1998. Induction of serum amyloid A inflammatory response genes in irradiated bone marrow cells. Radiat Res. 149:570–578. [PubMed] [Google Scholar]
- Gomez LA, Strasberg Rieber M, Rieber M. 1996. PCR-mediated differential display and cloning of a melanocyte gene decreased in malignant melanoma and up-regulated with sensitization to DNA damage. DNA Cell Biol. 15:423–427. [DOI] [PubMed] [Google Scholar]
- Goulter AB, Harmer DW, Clark KL. 2006. Evaluation of low density array technology for quantitative parallel measurement of multiple genes in human tissue. BMC Genomics. 7:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Cao Z, Du J, Liu T, Wang T. 2020. Circular RNA circPITX1 knockdown inhibits glycolysis to enhance radiosensitivity of glioma cells by miR-329–3p/NEK2 axis. Cancer Cell Int. 20:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner A, Kublo L, Tsabar M, Lahav G, Stewart-Ornstein J. 2020. Identification of universal and cell-type specific p53 DNA binding. BMC Mol Cell Biol. 21:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander MC, Alamo I, Fornace AJ. 1996. A novel DNA damage-inducible transcript, gadd7, inhibits cell growth, but lacks a protein product. Nucleic Acids Res. 24:1589–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Ding D, Zhang J, Cui J. 2019. Knockdown of lncRNA HOTAIR sensitizes breast cancer cells to ionizing radiation through activating miR-218. Biosci Rep. 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4:44–57. [DOI] [PubMed] [Google Scholar]
- Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, Bravo HC, Davis S, Gatto L, Girke T et al. 2015. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods. 12:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob NK, Cooley JV, Yee TN, Jacob J, Alder H, Wickramasinghe P, Maclean KH, Chakravarti A. 2013. Identification of sensitive serum microRNA biomarkers for radiation biodosimetry. PLoS One. 8:e57603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janus P, Szołtysek K, Zając G, Stokowy T, Walaszczyk A, Widłak W, Wojtaś B, Gielniewski B, Iwanaszko M, Braun R et al. 2018. Pro-inflammatory cytokine and high doses of ionizing radiation have similar effects on the expression of NF-kappaB-dependent genes. Cell Signal. 46:23–31. [DOI] [PubMed] [Google Scholar]
- Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R et al. 2020. The reactome pathway knowledgebase. Nucleic Acids Res. 48:D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazaeri AA, Yee CJ, Sotiriou C, Brantley KR, Boyd J, Liu ET. 2002. Gene expression profiles of BRCA1-linked, BRCA2-linked, and sporadic ovarian cancers. J Natl Cancer Inst. 94:990–1000. [DOI] [PubMed] [Google Scholar]
- Jiang H, Hu X, Zhang H, Li W. 2017. Down-regulation of LncRNA TUG1 enhances radiosensitivity in bladder cancer via suppressing HMGB1 expression. Radiat Oncol. 12:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Mortazavi A. 2018. Integrating ChIP-seq with other functional genomics data. Brief Funct Genomics. 17:104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. 2017. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45:D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartasova T, Cornelissen BJ, Belt P, van de Putte P. 1987. Effects of UV, 4-NQO and TPA on gene expression in cultured human epidermal keratinocytes. Nucleic Acids Res. 15:5945–5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ, Walker GC. 1980. DNA-damaging agents stimulate gene expression at specific loci in Escherichia coli. Proc Natl Acad Sci U S A. 77:2819–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SY, Tariq MA, Perrott JP, Brumbaugh CD, Kim HJ, Shabbir MI, Ramesh GT, Pourmand N. 2013. Distinctive microRNA expression signatures in proton-irradiated mice. Mol Cell Biochem. 382:225–235. [DOI] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J, Tugendreich S. 2014. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 30:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaraswamy S, Chinnaiyan P, Shankavaram UT, Lu X, Camphausen K, Tofilon PJ. 2008. Radiation-induced gene translation profiles reveal tumor type and cancer-specific components. Cancer Res. 68:3819–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacombe J, Sima C, Amundson SA, Zenhausern F. 2018. Candidate gene biodosimetry markers of exposure to external ionizing radiation in human blood: A systematic review. PLoS One. 13:e0198851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P. 2018. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46:D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang P, Pardee AB. 1992. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 257:967–971. [DOI] [PubMed] [Google Scholar]
- Lü X, de la Peña L, Barker C, Camphausen K, Tofilon PJ. 2006. Radiation-induced changes in gene expression involve recruitment of existing messenger RNAs to and away from polysomes. Cancer Res. 66:1052–1061. [DOI] [PubMed] [Google Scholar]
- Lyngholm CD, Overgaard J, Christiansen PM, Alsner J. Validation of a gene expression profile predictive of the risk of radiation-induced fibrosis in women treated with breast conserving therapy.[letter]. Acta Oncol 2015;54(9):1665–1668. [DOI] [PubMed] [Google Scholar]
- Ma X, Zhou J, Liu J, Wu G, Yu Y, Zhu H, Liu J. 2018. LncRNA ANCR promotes proliferation and radiation resistance of nasopharyngeal carcinoma by inhibiting PTEN expression. Onco Targets Ther. 11:8399–8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaeva E, Saeys Y, Tabury K, Janssen A, Michaux A, Benotmane MA, De Vos WH, Baatout S, Quintens R. 2016. Radiation-induced alternative transcription and splicing events and their applicability to practical biodosimetry. Sci Rep. 6:19251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCallum DE, Hall PA, Wright EG. 2001. The Trp53 pathway is induced in vivo by low doses of gamma radiation. Radiat Res. 156:324–327. [DOI] [PubMed] [Google Scholar]
- May JM, Bylicky M, Chopra S, Coleman CN, Aryankalayil MJ. 2021. Long and short non-coding RNA and radiation response: a review. Transl Res. in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermaid A, Monier B, Zhao J, Liu B, Ma Q. 2019. Interpretation of differential gene expression results of RNA-seq data: review and integration. Brief Bioinform. 20:2044–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon N, Rogers CJ, Lukaszewicz AI, Axtelle J, Yadav M, Song F, Chakravarti A, Jacob NK. 2016. Detection of Acute Radiation Sickness: A Feasibility Study in Non-Human Primates Circulating miRNAs for Triage in Radiological Events. PLoS One. 11:e0167333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. 2019. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47:D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AL, Attwood TK, Babbitt PC, Blum M, Bork P, Bridge A, Brown SD, Chang HY, El-Gebali S, Fraser MI et al. 2019. InterPro in 2019: improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 47:D351–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moor AE, Itzkovitz S. 2017. Spatial transcriptomics: paving the way for tissue-level systems biology. Curr Opin Biotechnol. 46:126–133. [DOI] [PubMed] [Google Scholar]
- Moreno-Villanueva M, Zhang Y, Feiveson A, Mistretta B, Pan Y, Chatterjee S, Wu W, Clanton R, Nelman-Gonzalez M, Krieger S et al. 2019. Single-Cell RNA-Sequencing Identifies Activation of TP53 and STAT1 Pathways in Human T Lymphocyte Subpopulations in Response to Ex Vivo Radiation Exposure. Int J Mol Sci. 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 5:621–628. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Laiakis EC, Fornace AJ, Amundson SA. 2019. Impact of inflammatory signaling on radiation biodosimetry: mouse model of inflammatory bowel disease. BMC Genomics. 20:329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myatt SS, Lam EW. 2007. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 7:847–859. [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320:1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel R, Stigter-van Walsum M, Buijze M, van den Berg J, van der Meulen IH, Hodzic J, Piersma SR, Pham TV, Jiménez CR, van Beusechem VW et al. 2015. Genome-wide siRNA Screen Identifies the Radiosensitizing Effect of Downregulation of MASTL and FOXM1 in NSCLC. Mol Cancer Ther. 14:1434–1444. [DOI] [PubMed] [Google Scholar]
- Nerurkar SN, Goh D, Cheung CCL, Nga PQY, Lim JCT, Yeong JPS. 2020. Transcriptional Spatial Profiling of Cancer Tissues in the Era of Immunotherapy: The Potential and Promise. Cancers (Basel). 12:E2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu H, Zhang L, Chen YH, Yuan BY, Wu ZF, Cheng JC, Lin Q, Zeng ZC. 2020. Circular RNA TUBD1 Acts as the miR-146a-5p Sponge to Affect the Viability and Pro-Inflammatory Cytokine Production of LX-2 Cells through the TLR4 Pathway. Radiat Res. 193:383–393. [DOI] [PubMed] [Google Scholar]
- Noel F, Gumin GJ, Raju U, Tofilon PJ. 1998. Increased expression of prohormone convertase-2 in the irradiated rat brain. FASEB J. 12:1725–1730. [DOI] [PubMed] [Google Scholar]
- Nuyten DS, van de Vijver MJ. 2008. Using microarray analysis as a prognostic and predictive tool in oncology: focus on breast cancer and normal tissue toxicity. Semin Radiat Oncol. 18:105–114. [DOI] [PubMed] [Google Scholar]
- Okamura S, Arakawa H, Tanaka T, Nakanishi H, Ng CC, Taya Y, Monden M, Nakamura Y. 2001. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol Cell. 8:85–94. [DOI] [PubMed] [Google Scholar]
- Park JG, Paul S, Briones N, Zeng J, Gillis K, Wallstrom G, LaBaer J, Amundson SA. 2017. Developing Human Radiation Biodosimetry Models: Testing Cross-Species Conversion Approaches Using an Ex Vivo Model System. Radiat Res. 187:708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Amundson SA. 2008. Development of gene expression signatures for practical radiation biodosimetry. Int J Radiat Oncol Biol Phys. 71:1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Amundson SA. 2011. Gene expression signatures of radiation exposure in peripheral white blood cells of smokers and non-smokers. Int J Radiat Biol. 87:791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Barker CA, Turner HC, McLane A, Wolden SL, Amundson SA. 2011. Prediction of in vivo radiation dose status in radiotherapy patients using ex vivo and in vivo gene expression signatures. Radiat Res. 175:257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Kleiman NJ, Amundson SA. 2019. Transcriptomic responses in mouse blood during the first week after in vivo gamma irradiation. Sci Rep. 9:18364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Smilenov LB, Amundson SA. 2013. Widespread Decreased Expression of Immune Function Genes in Human Peripheral Blood Following Radiation Exposure. Radiat Res. 180:575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paunesku T, Chang-Liu CM, Shearin-Jones P, Watson C, Milton J, Oryhon J, Salbego D, Milosavljevic A, Woloschak GE. 2000. Identification of genes regulated by UV/salicylic acid. Int J Radiat Biol. 76:189–198. [DOI] [PubMed] [Google Scholar]
- Pease AC, Solas D, Sullivan EJ, Cronin MT, Holmes CP, Fodor SP. 1994. Light-generated oligonucleotide arrays for rapid DNA sequence analysis. Proc Natl Acad Sci U S A. 91:5022–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia M, Yang JY, Yang MQ, Deng Y. 2008. A comparative study of different machine learning methods on microarray gene expression data. BMC Genomics. 9 Suppl 1:S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnaiya B, Amundson SA, Ghandhi SA, Smilenov LB, Geard CR, Buonanno M, Brenner DJ. 2013. Single-cell responses to ionizing radiation. Radiat Environ Biophys. 52:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnaiya B, Jenkins-Baker G, Randers-Pherson G, Geard CR. 2007. Quantifying a bystander response following microbeam irradiation using single-cell RT-PCR analyses. Exp Hematol. 35:64–68. [DOI] [PubMed] [Google Scholar]
- Radman M. 1975. SOS repair hypothesis: phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic Life Sci. 5A:355–367. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R, Dorris D, Lublinsky A, Nguyen A, Domanus M, Prokhorova A, Gieser L, Touma E, Lockner R, Tata M et al. 2002. An assessment of Motorola CodeLink microarray performance for gene expression profiling applications. Nucleic Acids Res. 30:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Stuart JM, Altman RB. 2000. Principal components analysis to summarize microarray experiments: application to sporulation time series. Pac Symp Biocomput. 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Ubreva J, Ballestar E. 2014. Chromatin immunoprecipitation. Methods Mol Biol. 1094:309–318. [DOI] [PubMed] [Google Scholar]
- Rogers CJ, Lukaszewicz AI, Yamada-Hanff J, Micewicz ED, Ratikan JA, Starbird MA, Miller TA, Nguyen C, Lee JT, Olafsen T et al. 2020. Identification of miRNA signatures associated with radiation-induced late lung injury in mice. PLoS One. 15:e0232411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Demidov ON, Anderson CW, Appella E, Mazur SJ. 2008. Induction of PPM1D following DNA-damaging treatments through a conserved p53 response element coincides with a shift in the use of transcription initiation sites. Nucleic Acids Res. 36:7168–7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem N, Axelrod JH, Miskin R. 1987. Induction of urokinase-type plasminogen activator by UV light in human fetal fibroblasts is mediated through a UV-induced secreted protein. Mol Cell Biol. 7:622–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby SW, Szostak JW. 1985. Specific Saccharomyces cerevisiae genes are expressed in response to DNA-damaging agents. Mol Cell Biol. 5:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudqvist N, Laiakis EC, Ghandhi SA, Kumar S, Knotts JD, Chowdhury M, Fornace AJ, Amundson SA. 2018. Global Gene Expression Response in Mouse Models of DNA Repair Deficiency after Gamma Irradiation. Radiat Res. 189:337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. 1995. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 270:467–470. [DOI] [PubMed] [Google Scholar]
- Schena M, Shalon D, Heller R, Chai A, Brown PO, Davis RW. 1996. Parallel human genome analysis: microarray-based expression monitoring of 1000 genes. Proc Natl Acad Sci U S A. 93:10614–10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AM, Chang HY. 2013. Gene regulation: Long RNAs wire up cancer growth. Nature. 500:536–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J, Welch D, Buonanno M, Ponnaiya B, Garty G, Olsen T, Amundson SA, Lin Q. 2019. An Integrated Preprocessing Approach for Exploring Single-Cell Gene Expression in Rare Cells. Sci Rep. 9:19758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Lam A, Li M-C, Ngan M, Menenzes S, Zhao Y. 2007. Analysis of gene expression data using BRB-Array Tools. Cancer Informatics. 2:11–17. [PMC free article] [PubMed] [Google Scholar]
- Sinha A, Markatou M. 2011. A platform for processing expression of short time series (PESTS). BMC Bioinformatics. 12:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprung CN, Li J, Hovan D, McKay MJ, Forrester HB. 2011. Alternative transcript initiation and splicing as a response to DNA damage. PLoS One. 6:e25758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szołtysek K, Janus P, Zając G, Stokowy T, Walaszczyk A, Widłak W, Wojtaś B, Gielniewski B, Cockell S, Perkins ND et al. 2018. RRAD, IL4I1, CDKN1A, and SERPINE1 genes are potentially co-regulated by NF-κB and p53 transcription factors in cells exposed to high doses of ionizing radiation. BMC Genomics. 19:813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templin T, Amundson SA, Brenner DJ, Smilenov LB. 2011. Whole mouse blood microRNA as biomarkers for exposure to γ-rays and (56)Fe ion. Int J Radiat Biol. 87:653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves JM, Won KJ. 2020. Mapping Cellular Coordinates through Advances in Spatial Transcriptomics Technology. Mol Cells. 43:591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapar R. 2018. Regulation of DNA Double-Strand Break Repair by Non-Coding RNAs. Molecules. 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. 2019. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47:D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. 2003. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 13:2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasik B, Chałubińska-Fendler J, Chowdhury D, Fendler W. 2018. Potential of serum microRNAs as biomarkers of radiation injury and tools for individualization of radiotherapy. Transl Res. 201:71–83. [DOI] [PubMed] [Google Scholar]
- Torres-Roca JF, Eschrich S, Zhao H, Bloom G, Sung J, McCarthy S, Cantor AB, Scuto A, Li C, Zhang S et al. 2005. Prediction of radiation sensitivity using a gene expression classifier. Cancer Res. 65:7169–7176. [DOI] [PubMed] [Google Scholar]
- Tusher VG, Tibshirani R, Chu G. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 98:5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van’t Veer LJ, Bernards R. 2008. Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature. 452:564–570. [DOI] [PubMed] [Google Scholar]
- Wahba A, Ryan MC, Shankavaram UT, Camphausen K, Tofilon PJ. 2018. Radiation-induced alternative transcripts as detected in total and polysome-bound mRNA. Oncotarget. 9:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Li X, Wang R, Ding Z. 2021. Spatial transcriptomics and proteomics technologies for deconvoluting the tumor microenvironment. Biotechnol J. e2100041. [DOI] [PubMed] [Google Scholar]
- Watson CA, Chang-Liu CM, Woloschak GE. 2000. Modulation of calmodulin by UV and X-rays in primary human endothelial cell cultures. Int J Radiat Biol. 76:1455–1461. [DOI] [PubMed] [Google Scholar]
- Weidhaas JB, Babar I, Nallur SM, Trang P, Roush S, Boehm M, Gillespie E, Slack FJ. 2007. MicroRNAs as potential agents to alter resistance to cytotoxic anticancer therapy. Cancer Res. 67:11111–11116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West M, Blanchette C, Dressman H, Huang E, Ishida S, Spang R, Zuzan H, Olson JA, Marks JR, Nevins JR. 2001. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc Natl Acad Sci U S A. 98:11462–11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, Penkett CJ, Rogers J, Bähler J. 2008. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 453:1239–1243. [DOI] [PubMed] [Google Scholar]
- Williams NL, Dan T, Zaorsky NG, Garg S, Den RB. 2017. The Role of Genomic Techniques in Predicting Response to Radiation Therapy. Oncology (Williston Park). 31:562–570. [PubMed] [Google Scholar]
- Williams PD, Owens CR, Dziegielewski J, Moskaluk CA, Read PW, Larner JM, Story MD, Brock WA, Amundson SA, Lee JK et al. 2011. Cyclophilin B expression is associated with in vitro radioresistance and clinical outcome after radiotherapy. Neoplasia. 13:1122–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woloschak GE, Paunesku T, Chang-Liu CM, Grdina DJ. 1995. Expression of thymidine kinase messenger RNA and a related transcript is modulated by radioprotector WR1065. Cancer Res. 55:4788–4792. [PubMed] [Google Scholar]
- Wu H, Yu J, Kong D, Xu Y, Zhang Z, Shui J, Li Z, Luo H, Wang K. 2019. Population and single-cell transcriptome analyses reveal diverse transcriptional changes associated with radioresistance in esophageal squamous cell carcinoma. Int J Oncol. 55:1237–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiu G, Sui X, Wang Y, Zhang Z. 2018. FOXM1 regulates radiosensitivity of lung cancer cell partly by upregulating KIF20A. Eur J Pharmacol. 833:79–85. [DOI] [PubMed] [Google Scholar]
- Yadav M, Bhayana S, Liu J, Lu L, Huang J, Ma Y, Qamri Z, Mo X, Jacob DS, Parasa ST et al. 2020. Two-miRNA-based finger-stick assay for estimation of absorbed ionizing radiation dose. Sci Transl Med. 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Khanna KK, Lavin MF. 1996. Induction of inositol 1,4,5 trisphosphate receptor genes by ionizing radiation. Int J Radiat Biol. 69:539–546. [DOI] [PubMed] [Google Scholar]
- Zhang C, Kang C, Wang P, Cao Y, Lv Z, Yu S, Wang G, Zhang A, Jia Z, Han L et al. 2011. MicroRNA-221 and −222 regulate radiation sensitivity by targeting the PTEN pathway. Int J Radiat Oncol Biol Phys. 80:240–248. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Szustakowski J, Schinke M. 2009. Bioinformatics analysis of microarray data. Methods Mol Biol. 573:259–284. [DOI] [PubMed] [Google Scholar]
- Zhao JZL, Mucaki EJ, Rogan PK. 2018. Predicting ionizing radiation exposure using biochemically-inspired genomic machine learning. F1000Res. 7:233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JM, Liang R, Zhu SY, Wang H, Zou M, Zou WJ, Nie SL. 2019. LncRNA WWC2-AS1 functions AS a novel competing endogenous RNA in the regulation of FGF2 expression by sponging miR-16 in radiation-induced intestinal fibrosis. BMC Cancer. 19:647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou PK, Rigaud O. 2001. Down-regulation of the human CDC16 gene after exposure to ionizing radiation: a possible role in the radioadaptive response. Radiat Res. 155:43–49. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Wang Z, Miller DJ, Clarke R, Xuan J, Hoffman EP, Wang Y. 2008. A ground truth based comparative study on clustering of gene expression data. Front Biosci. 13:3839–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]