

Mitochondrial dysfunction elicits a mitochondrial stress response (MSR) through mitochondrial-nuclear communication and activates ATF4, a master transcriptional regulator of the cellular stress response1. Knowledge of consequences of MSR-triggered ATF4 activation in mitochondrial cardiomyopathy is limited, yet critical for therapeutic approaches.
Mitochondrial phosphatase Ptpmt1 cKO mice (PKO)2 provide a model to investigate in vivo mechanisms of MSR in cardiomyocytes, displaying key features of fetal mitochondrial cardiomyopathy, while evidencing lethality between E16.5–18.52. All mouse protocols were approved by the Institutional Animal Care and Use Committee. Molecular analyses confirmed that loss of PTPMT1 in cardiomyocytes resulted in upregulation of ATF4 and its target genes at E11.5 (Figure A–D), similar to other MSR models. The most well recognized upstream regulator of ATF4 is eIF2α phosphorylation, which increases ATF4 translation. Atf4 is also a direct target of transcriptional repression by HIF1α in cardiomyocytes. Hif1α cKO hearts display increased Atf4 mRNA and protein. mTOR is another upstream regulator of ATF4. We examined these upstream regulators of ATF4 in PKO hearts, and found that phosphorylation of eIF2α was significantly increased in PKO versus control hearts at E11.5, while HIF1α and mTOR phosphorylation were not altered (Figure E), suggesting that eIF2α phosphorylation induced expression of ATF4.
Figure.
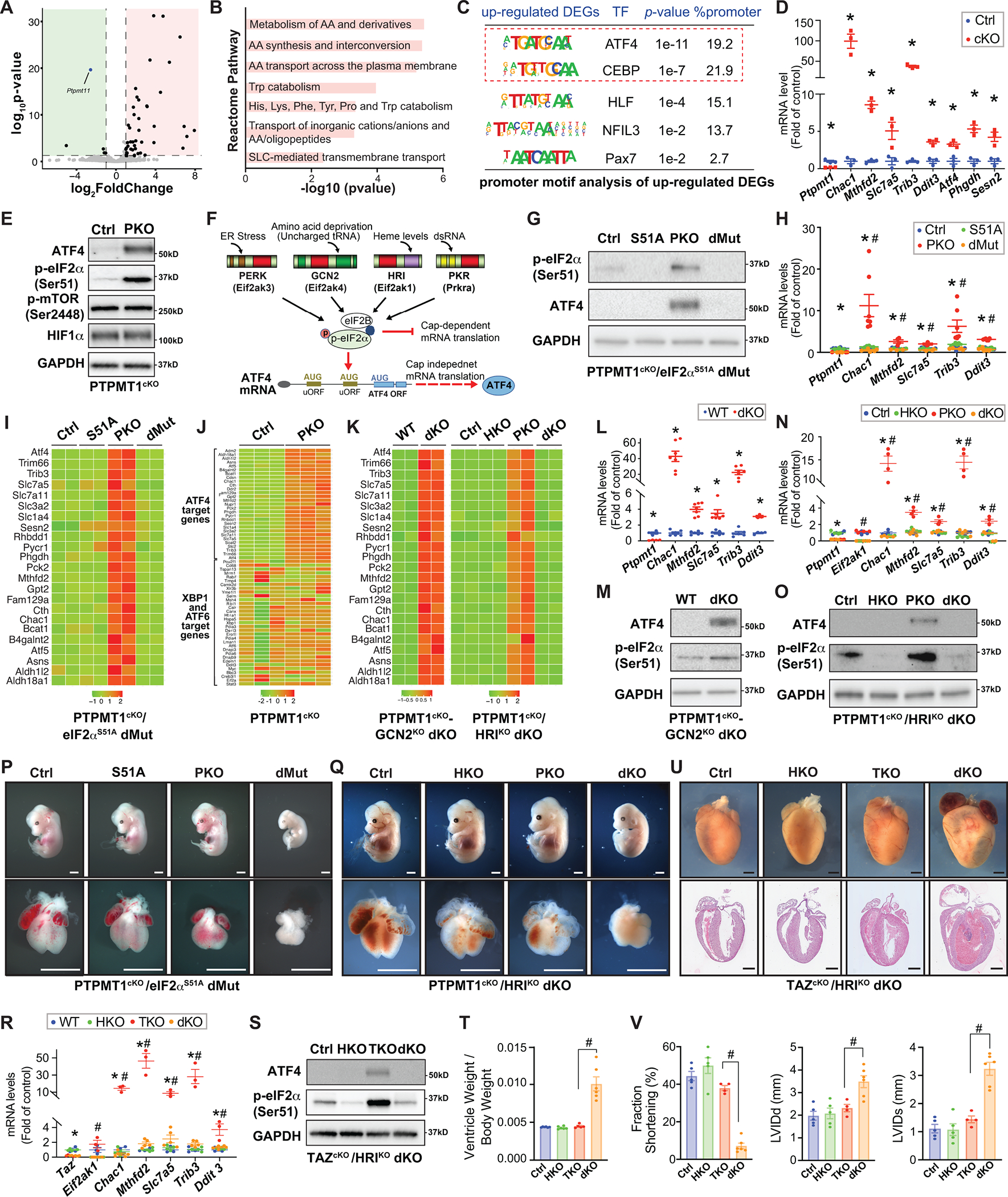
A. Volcano plot obtained from DESeq2 analysis of gene expression in Ptpmt1 cardiomyocyte-specific knockout (Ptpmt1f/f; Xml-Cre+; PKO) versus Cre negative control hearts at E11.5. Genes with adjusted P<0.05 and log2 (fold change) >1 are considered significantly upregulated or downregulated genes in PKO hearts. Green indicates downregulated genes; red indicates upregulated genes. n=3 per group. RNAseq data was deposited to the GEO database (Accession number: GSE201042). B. Functional clustering analysis utilizing the Reactome database revealed that deletion of PTPMT1 in cardiomyocytes significantly upregulates pathways involved in amino acid metabolism, which is classically found to be regulated by ATF4. AA: amino acid; Trp: tryptophan; His: histidine; Lys: lysine; Phe: phenylalanine; SLC: solute carrier superfamily. C. Motif enrichment analysis at promoter regions (±2 kb from transcription start sites) of the genes that were upregulated in PKO hearts revealed that the top enriched transcription factor binding motifs were the ATF4 and its cofactors C/EBP (CCAAT/enhancer-binding protein) or C/EBP homologous protein binding motifs. DEGs: differentially expressed genes. D. qRT-PCR validated the upregulation of classic ATF4 target genes in PKO (red) versus control (Ctrl, blue) hearts at E11.5. n=3–4 per group. E. Western blot analysis of ATF4, phosphorylated eIF2α at Serine 51 (Ser51), phosphorylated mTOR at Serine 2448 (Ser2448), and HIF1α, in PKO and Ctrl hearts at E11.5. n=4 per group. F. Model for eIF2α ‐mediated translational control of ATF4 in response to diverse cellular stress responses. G. Western blot analysis of ATF4 and phosphorylated eIF2α at Ser51 and ATF4 in eIF2αS51A mutant (Ptpmt1f/f; eIF2αm/m; Xml-Cre−) (S51A), PKO (Ptpmt1f/f; eIF2α+/+; Xml-Cre+), PKO/eIF2αS51A double mutant (dMut) (Ptpmt1f/f; eIF2αm/m; Xml-Cre+), and control (Ctrl) (Ptpmt1f/f; eIF2α+/+; Xml-Cre−) hearts at E11.5. n=4 per group. H. qRT-PCR analysis of ATF4 target genes in S51A, PKO, dMut, and Ctrl hearts at E11.5. n=3–4 per group. I. Heatmap representation of transcript levels of selected ATF4 target genes in S51A, PKO, dMut, and Ctrl hearts at E11.5. J. Heatmap representation of transcript levels of selected ER stress-induced genes, including the target genes of ATF4, and XBP1 and ATF6 in PKO and Ctrl hearts at E11.5. K. Heatmap representation of transcript levels of selected ATF4 target genes in PKO/GCN2 double knockout (dKO) (Ptpmt1 f/f-Eif2ak4−/−; Xml-Cre+) and wildtype control (WT) (Ptpmt1 +/+-Eif2ak4+/+; Xml-Cre−) hearts (left), and HRI null (HKO) (Ptpmt1f/f; Eif2ak4−/−; Xml-Cre−), PKO (Ptpmt1f/f; Eif2ak4+/+; Xml-Cre+), PKO/HRI dKO (Ptpmt1f/f; Eif2ak4−/−; Xml-Cre+), and Ctrl (Ptpmt1f/f; Eif2ak4+/+; Xml-Cre−) hearts (right). Note: the genes that encode GCN2 and PTPMT1 are at the same allele, so we could not obtain single knockout littermates. L. qRT-PCR analysis of ATF4 target genes in PKO/GCN2 dKO and WT hearts at E11.5, n=5–7 per group. M. Western blot analysis of ATF4 and phosphorylated eIF2a at Ser51 in PKO/GCN2 dKO and WT hearts. n=4 per group. N. qRT-PCR analysis of ATF4 target genes in HKO, PKO, PKO/HRI dKO, and Ctrl hearts at E11.5, n=3–4 per group. O. Western blot analysis of ATF4 and phosphorylated eIF2a at Serine 51 (Ser51) in HKO, PKO, PKO/HRI dKO, and Ctrl hearts. n=4 per group. P. Whole embryonic (top) and heart (bottom) morphology of S51A, PKO, dMut, and Ctrl hearts at E13.5. Scale bar: 1 mm. Q. Whole embryonic (top) and heart (bottom) morphology of HKO, PKO, PKO/HRI dKO, and Ctrl hearts at E14.5. Scale bar: 1 mm. R. qRT-PCR analysis of ATF4 target genes in HKO (Tazf/Y; Eif2ak4−/−; Xml-Cre−), Tafazzin cardiomyocyte-specific knockout (TKO) (Tazf/Y; Eif2ak4+/+; Xml-Cre+), TKO/HRI dKO (Tazf/Y; Eif2ak4−/−; Xml-Cre+), and Ctrl (Tazf/Y; Eif2ak4+/+; Xml-Cre−) hearts at postnatal day (P) 7. n=3–4 per group. S. Western blot analysis of ATF4 and phosphorylated eIF2α at Serine 51 (Ser51) in HKO, TKO, TKO/HRI dKO, and Ctrl hearts at P7. n=4 per group. T. Cardiac ventricular weight (VW) to body weight (BW) ratio for HKO, TKO, TKO/HRI dKO, and Ctrl hearts at P7. n= 5–7 per group. U. Whole-mount (top) and H&E stained sections (bottom) for HKO, TKO, TKO/HRI dKO, and Ctrl hearts at P8. Scale bar: 1 mm. V. Echocardiographic measurements of left ventricular percentage of fractional shortening (% FS) (left), end-diastolic LV internal diameter (LVIDd) (middle), and end-systolic LV internal diameter (LVIDs) (right) for HKO, TKO, TKO/HRI dKO, and Ctrl mice at P7. n = 4–6 mice per group. GAPDH was used as a loading control for western blots. qRT-PCR data were normalized to corresponding 18S levels, and levels in mutants are expressed as the fold-change versus Ctrl. Data are represented as the mean ± SEM. *P < 0.05 PKO, TKO, or dMut, or dKO vs. Ctrl, or as indicated; #P < 0.05 dMut or dKO vs. PKO or TKO, or as indicated, by 2-tailed Student’s t test (two group comparison) or two-way ANOVA (four group comparison).
Phosphorylation of eIF2α at serine 51 integrates signals from diverse cellular stress responses (Figure F)1. We generated a “phosphorylation-resistant” eIF2α mutant allele1, in which the serine 51 phosphorylation site was mutated to alanine (eIF2αS51A), and crossed this allele into PKO mice to generate PKO/eIF2αS51A double mutant mice (dMut). Absence of eIF2α phosphorylation in eIF2αS51A and dMut hearts was validated (Figure G). ATF4 protein and downstream targets were significantly decreased to baseline levels in dMut hearts (Figure G–I), confirming that eIF2α phosphorylation was essential for MSR-triggered ATF4 activation in PKO hearts.
Four eIF2α kinases are activated by distinct forms of stress (Figure F)1. RNA-seq analysis from PKOs indicated increased expression of ATF4 targets, but no increase in XBP1- or ATF6 (Figure J), the latter negating the likelihood that a PERK-mediated stress response was involved1. Next, we generated null alleles for GCN2 or HRI, and generated PKO/GCN2 and PKO/HRI double knockout mice (dKO). Loss of HRI abolished activation of eIF2α-ATF4 signaling in PKOs, while eIF2α-ATF4 remained activated in PKO/GCN2 dKO hearts (Figure K–O). Thus, MSR induced eIF2α-ATF4 signaling was dependent on HRI, while other eIF2α kinases were not involved. Our results are consistent with recent in vitro findings in HeLa cells3,4, providing the first demonstration of the in vivo relevance of this pathway in mitochondrial cardiomyopathy. It is not clear whether activation of HRI depends on the amount of heme in cardiomyocytes.
Whether the eIF2α pathway was adaptive or maladaptive remained to be addressed. Although global suppression of protein synthesis by eIF2α phosphorylation conserves energy, and increasing ATF4 translation allows cells to survive periods of stress, persistent ATF4 and suppression of protein synthesis may also be detrimental. Homozygous eIF2αS51A mutants die within 18 hours after birth1. However, constitutive activation of eIF2α by deleting both eIF2α phosphatases Ppp1r15a and Ppp1r15b results in embryonic lethality1. Thus far, in vivo physiological consequences of eIF2α phosphorylation in response to stress have not been addressed. We analyzed morphology and survival of PKO/eIF2αS51A dMut mice. PKO/eIF2αS51A dMut mice died at E13.5, while PKO mice survived at this stage with abnormal heart morphology, demonstrating that blocking eIF2α phosphorylation negatively impacted survival (Figure P).
Although HRI null mice were viable1 and displayed normal cardiac development and function at baseline, the role of HRI activation in mitochondrial cardiomyopathy remained to be addressed. We found that PKO/HRI dKOs died between E14.5-E16.5, while PKO littermates survived with abnormal cardiac morphology (Figure Q). Thus, although milder than effects of the eIF2α S51A mutant, deletion of HRI was also detrimental to survival. Thus HRI-eIF2α activation was protective for fetal mitochondrial cardiomyopathy
To determine the role of HRI in adult mitochondrial cardiomyopathy, we crossed HRI knockout mice with Tafazzin cKO (TKO) mice. TKO mice display mitochondrial dysfunction at 2 months and dilated cardiomyopathy at 4 months, but survive more than one year with impaired cardiac function5. Western blot and qPCR analysis confirmed that eIF2α-ATF4 signaling was activated in TKO hearts but abolished in TKO/HRI dKO hearts (Figure R–S). TKO/HRI dKO mice died between postnatal day (P) 7–10 with enlarged hearts, compared to TKO mice that survived more than one year5. We also observed increased ventricular weight to body weight ratios in dKOs (Figure T–U). Echocardiographic analysis revealed severe cardiac dysfunction in dKOs (Figure V). Thus, MSR triggered HRI-eIF2α was also protective for adult mitochondrial cardiomyopathy.
Overall we demonstrated that an HRI-eIF2α pathway mediated mitochondrial-nuclear communication and MSR-triggered ATF4 activation in both embryonic and adult heart. Our results uncovered a protective role for MSR triggered HRI-eIF2α-ATF4 signaling in both fetal and adult mitochondrial cardiomyopathy. Therefore, intention to inhibit the HRI-eIF2α-ATF4 pathway in mitochondrial cardiomyopathy could be detrimental, rather than beneficial, for cardiac dysfunction.
The data, analytical methods, and study materials that support the findings of this study will be available to other researchers from the corresponding authors on reasonable request. RNAseq data was deposited to the GEO database (Accession number: GSE201042).
Sources of Funding
XF is supported by NIH grants. SME is supported by NIH grants and the Foundation Leducq (16 CVD 03).
Footnotes
Disclosures
Jun Zhao is currently employed by Guardant Health, US. This work is not related to her employment at Guardant Health.
Reference:
- 1.Costa-Mattioli M and Walter P. The integrated stress response: From mechanism to disease. Science. 2020. Apr 24;368(6489):eaat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen Z, Zhu S, Wang H, Wang L, Zhang J, Gu Y, Tan C, Dhanani M, Wever E, Wang X, Xie B, Wang S, Huang L, van Kampen AHC, Liu J, Han Z, Patel HH, Vaz FM, Fang X, Chen J and Ouyang K. PTPMT1 Is Required for Embryonic Cardiac Cardiolipin Biosynthesis to Regulate Mitochondrial Morphogenesis and Heart Development. Circulation. 2021;144:403–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fessler E, Eckl EM, Schmitt S, Mancilla IA, Meyer-Bender MF, Hanf M, Philippou-Massier J, Krebs S, Zischka H and Jae LT. A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature. 2020;579:433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, Wiita AP, Xu K, Correia MA and Kampmann M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature. 2020;579:427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu S, Chen Z, Zhu M, Shen Y, Leon LJ, Chi L, Spinozzi S, Tan C, Gu Y, Nguyen A, Zhou Y, Feng W, Vaz FM, Wang X, Gustafsson AB, Evans SM, Kunfu O and Fang X. Cardiolipin Remodeling Defects Impair Mitochondrial Architecture and Function in a Murine Model of Barth Syndrome Cardiomyopathy. Circ Heart Fail. 2021;14:e008289. [DOI] [PMC free article] [PubMed] [Google Scholar]