

ABSTRACT
Alterations in the epigenome are well known to affect cancer development and progression. Epigenetics is highly influenced by the environment, including diet, which is a source of metabolic substrates that influence the synthesis of cofactors or substrates for chromatin and RNA modifying enzymes. In addition, plants are a common source of bioactives that can directly modify the activity of these enzymes. Here, we review and discuss the impact of diet on epigenetic mechanisms, including chromatin and RNA regulation, and its potential implications for cancer prevention and treatment.
Keywords: histone modifications, RNA modifications, DNA methylation, enhancer, bioactives, obesity, metabolism
Statement of Significance: This review provides the reader with a comprehensive overview of the multiple layers of epigenetic interaction with food relevant for cancer prevention and treatment.
Graphical Abstract
Graphical Abstract.
Introduction
Diet influences the risk of developing cancer (1). Higher consumption of dietary fiber and lower consumption of total sugars are associated with lower risk. Low-carbohydrate and high-protein diets have also been reported to slow tumor growth and prevent cancer initiation. In addition, obesity has been reported to increase cancer risk. Consequently, diet or nutrient modification has been proposed as a complementary strategy to targeting cancer metabolism with pharmacological agents.
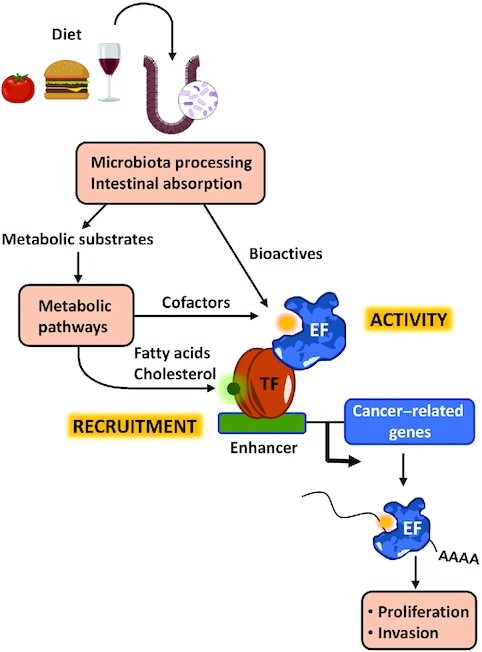
Nutritional epigenetics refers to the influence of diet on gene expression without changing the DNA sequence. Diet modulates epigenetic events, such as DNA, RNA, and histone modifications, by affecting the activity and recruitment to target sites of epigenetic factors (Figure 1). Diet can fuel metabolic processes that generate cofactors needed for the function of epigenetic factors or provide molecules that directly bind and modulate the activity of these factors. In addition, diet can affect the activity of transcription factors impacting the recruitment of epigenetic factors to the genome. Importantly, targeting several epigenetic factors with small synthetic molecules is a current strategy to treat certain cancers. Thus, it is possible that some of the beneficial effects of diets in the onset and progression of cancer are mediated through the modulation of the epigenetic machinery.
FIGURE 1.
Overview of the topics covered by this review. After intake, food is processed by the microbiota in the intestine and nutrients are absorbed. These might be substrates for metabolic reactions or contain bioactives that can modulate directly the activity of EFs. Metabolic processing can generate cofactors needed for the activity of EFs or products such as cholesterol and fatty acids that modulate the activity of TFs that play a role in the recruitment of EFs to chromatin. EF, epigenetic factor; TF, transcription factor.
In this article we will review currently used and promising epigenetic factors as therapeutic targets to treat cancer and the potential effects of dietary products in the regulation of their activity.
Current Status of Knowledge
DNA, RNA, and histone modifications and epigenetic regulators with therapeutic potential
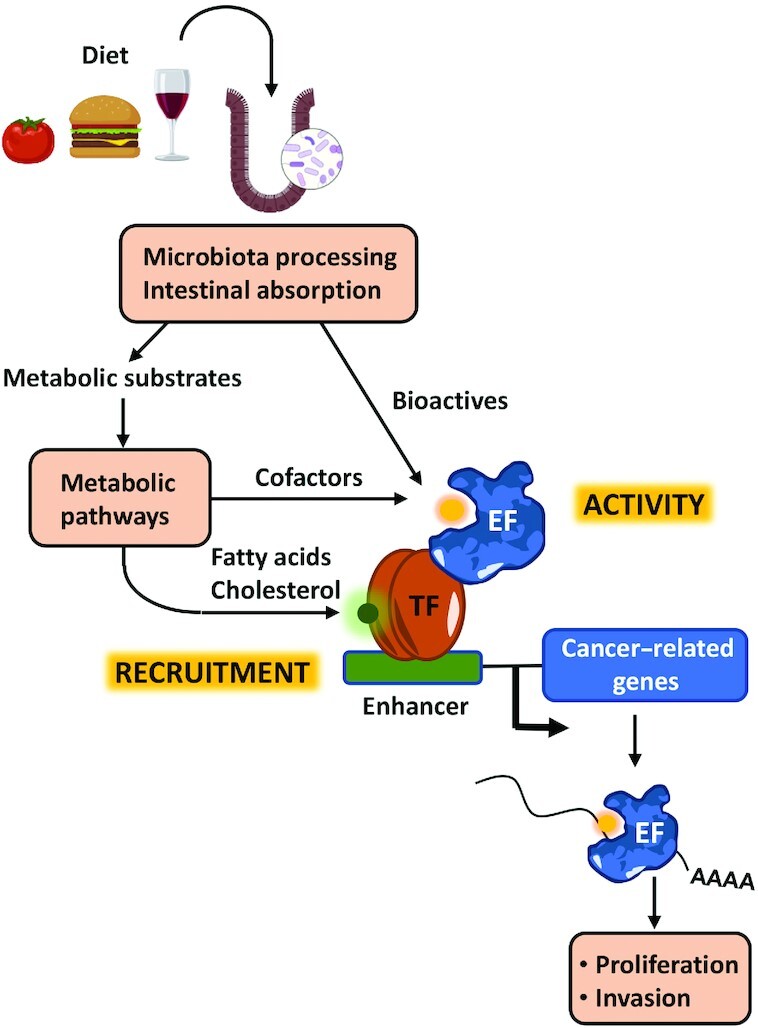
The expression of genes is subject to several layers of epigenetic regulation; among them, modifications of DNA, histones, and RNA play critical roles in this process (Figure 1). DNA in the eukaryotic cell nucleus is wrapped around 2 copies each of the core histones H2A, H2B, H3, and H4 to form chromatin. Chromatin plays important roles in DNA biology, including gene expression regulation. The level of chromatin compaction has important consequences for gene transcription as it influences the accessibility of DNA sequences to transcription factors and other regulatory proteins. Modifications of DNA and histones regulate the level of chromatin compaction either directly or by facilitating the binding of remodeling proteins that recognize modified sites (2). Modifications of RNA add an additional level of gene expression regulation and might influence RNA transport, splicing, stability, and translation, through the binding of specific proteins that recognize the modified sites (Figure 2) (3).
FIGURE 2.
Overview of the many layers of epigenetic regulation that affect gene expression including modifications of DNA, histones, and RNA. DNA and histone modifications control the accessibility of transcription factors and RNA polymerase to chromatin and therefore have a large impact on gene transcription. Some histone modifications might also influence co-transcriptional splicing. mRNA modifications can have an impact in RNA stability, splicing, nuclear export, and translation. Ac, acetylation; Me, methylation.
DNA, RNA, and histone modifications are frequently altered in cancer. These alterations can be due to multiple problems, such as mutations or alterations in the expression of factors involved in these modifications or their improper recruitment to genomic sites. As a result, cancer cells experience important changes in chromatin compaction and accessibility. Among other features, the regulation of accessibility at enhancers is a crucial aspect of transcription regulation and it is often dysregulated in cancer. Enhancers function as nodes that activate gene expression over long distances independently of their orientation with respect to their transcription start sites (4). Enhancers can span large regions, known as super-enhancers (SEs), and drive the expression of genes that control cell identity (5–8). In cancer, SEs have been proposed to be of special importance for tumor dependence (5, 9) and can show de novo demarcation of elements nearby oncogenes to promote cancer progression (6, 8). Examples of de novo activation of SEs have been described in a number of tumor types, including colorectal cancer (10, 11), clear cell renal carcinoma, (12), and adult T-cell leukemia/lymphoma (13).
Accordingly, the development of inhibitors of epigenetic factors able to revert epigenetic alterations in cancer cells holds promise for the treatment of this disease (Table 1). Although many inhibitors that target diverse epigenetic factors have been developed, only a few are currently being used for cancer treatment, including inhibitors of DNA methylation, inhibitors of histone deacetylases (HDACs), and inhibitors of the methyltransferase enhancer of zester 2 (EZH2). Other inhibitors are in clinical trials and/or have shown promising results in preclinical studies.
TABLE 1.
Epigenetic targets with therapeutic potential described in this review1
| Function | Factor | Cofactor/metabolic inhibitor | Consequences of inhibition |
|---|---|---|---|
| DNA methyltransferase | DNMT1 | S-adenosylmethionine | Induction of repetitive elements and tumor suppressors |
| Histone methyltransferase | EZH2 | S-adenosylmethionine | Induction of repetitive elements and tumor suppressors |
| Histone demethylase | LSD1 | Flavin-adenine dinucleotide | |
| Histone deacetylase | HDACs | β-Hydroxybutyrate | Induction of repetitive elements and tumor suppressors |
| Bromodomain-containing protein | BRD4 CREBBP/EP300 |
Silencing of oncogenes associated with SEs | |
| RNA methylase | METTL3 | S-adenosylmethionine | MYC downregulation |
| RNA demethylase | FTO |
2-Oxoglutarate Vitamin C |
MYC downregulation |
BRD, bromodomain; DNMT1, DNA methyltransferase 1; EZH2, Enhancer of zeste 2 polycomb repressive complex 2 subunit; LSD1, lysine specific demethylase 1; HDAC, histone deacetylase; METTL, methyltransferase-like; SE, super-enhancer.
DNA methylation
DNA methylation plays important roles in gene silencing and is frequently deregulated in cancer. This mark is catalyzed by DNA methyltransferases (DNMTs), typically at cytosines (5mC), and despite being a relatively stable mark can be reversed by the action of ten-eleven translocation (TET) enzymes that oxidize the methyl group of 5mC to yield 5-hydroxymethylcytosine (5hmC). The DNMT inhibitors (DNMTi) azacytidine (AZA) and decitabine (DAC) are the epigenetic drugs with the longest use in cancer treatment (14). These cytidine analogs, which are currently approved for the treatment of myelodysplastic syndrome and acute myeloid leukemia (AML), are incorporated into DNA and form covalent complexes with DNMTs, depleting the pool of active enzymes in the cell nucleus.
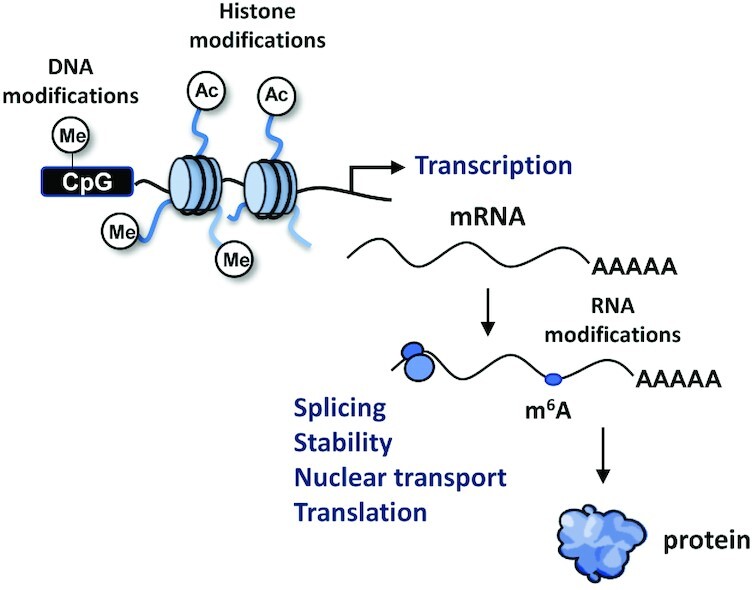
The mechanism of action DNMTi in cancer cells is not completely understood; however, 2 main events may account for their anticancer effects. It has been widely reported that DNMTi cause the induction of tumor suppressor genes, such as cyclin dependent kinase inhibitor 2A (CDKN2A), that are frequently epigenetically silenced by DNA methylation in cancer cells (15). However, recent evidence suggests that other major and more complex effects contribute to the therapeutic action of these inhibitors. Although historically much emphasis has been put on coding genes, these inhibitors have been demonstrated to induce the expression of noncoding repetitive elements typically silenced by DNA methylation, such as endogenous retroviruses (ERVs) (16, 17) (Figure 3A). The transcription of ERVs leads to the accumulation of cytosolic double-stranded RNA (dsRNA) that is sensed as a viral threat and triggers a type I interferon response, leading to apoptosis and expression of immune chemokines in cancer cells.
FIGURE 3.
Epigenetic repressors (A) and activators (B) that are targeted by FDA-approved inhibitors (DNMTs, HDACs, and EZH2) and/or in clinical trials (LSD1, lysine-specific demethylase 1A , BRD) for cancer treatment. Inhibiting enzymes involved in gene repression might have similar molecular effects, leading to the induction of tumor suppressor genes and repetitive sequences such as ERVs. Inhibiting epigenetic activators in cancer cells frequently results in the disruption of expression of oncogenes controlled by super-enhancer regions. BRD, bromodomain; DNMT, DNA methyltransferase; ERV, endogenous retrovirus; EZH2, methyltransferase enhancer of zester 2; HDAC, histone deacetylase.
HDACs
A second type of inhibitors that are currently being used for cancer treatment are HDACs (Figure 3A). Acetylated histones are recognized by proteins that contain bromodomains (BRDs) and will carry out functions involved in gene activation such as chromatin relaxation or recruitment of proteins involved in transcription. Histone acetylation is a typical mark of enhancers, and it is intensively regulated by the action of histone acetyltransferases (HATs) and HDACs. Both inhibitors of HATs and HDACs have shown promising results for cancer treatment. However, only very few inhibitors of proteins with HAT activity have been developed; most notably, inhibitors of the HAT domains of the highly homologous HAT enzymes CREB (cAMP-response element binding protein b)inding protein (CREBBP) and E1A binding protein p300 (EP300) show antiproliferative effects in cancer cell lines (18). A larger number of HDACi have been developed with demonstrated anticancer activity. HDAC inhibitors vorinostat, romidepsin, belinostat, and panobinostat have been FDA approved for the treatment of several hematological malignancies and other HDACi, such as entinostat, are currently undergoing clinical trials (19). Inhibitors of HDACs have been described to induce the expression of repetitive elements with similar consequences to DNMTi (20) (Figure 3A).
EZH2
Tazemetostat, an inhibitor of EZH2, is the most recent epigenetic drug approved by the FDA. EZH2 is the catalytic subunit of the polycomb repressive complex 2 (PRC2) that mediates methylation of histone 3 at lysine 27 (H3K27), a mark involved in gene repression (Figure 3A). EZH2 gain of function is common in cancer, which can be due to EZH2 overexpression, EZH2 activating mutations, or loss of function of the antagonistic remodeling complex SWI/SNF (SWItch/Sucrose Non Fermentable) (21–24). In 2020, the FDA approved the first EZH2 inhibitor, tazemetostat, to treat epithelioid sarcoma with INI1 (also called SMARCB1) deletions and relapsed or refractory follicular lymphomas with EZH2 activating mutations (25, 26). Other tumor types with EZH2 activating mutations might also benefit from these inhibitors in the future. Similar to HDACi and DNMTi, inhibitors of EZH2 might exert their antitumor activity through the induction of the expression of repetitive elements (27, 28).
BRD-containing proteins
BRDs recognize acetylated histone tails and act as effectors of the acetylated signal (Figure 3B). These domains are present in more than 50 human proteins, although the relevance for the functionality of most of these proteins is unknown (29). However, the BRDs of the bromo and extraterminal domain (BET) family of BRD-containing proteins and the HAT enzymes CREBBP/EP300 have been largely studied, and good inhibitors against them have been developed. These inhibitors block the interaction of BET proteins or CREBBP/EP300 with acetylated histones and have shown therapeutic potential in preclinical studies (30–32). Binding of these BRD-containing proteins to large SE regions with very high levels of histone acetylation contributes to maintain high levels of oncogene expression in cancer cells. Accordingly, these inhibitors are particularly effective in reducing the levels of expression of oncogenes such as MYC that are highly expressed in certain tumors (29). More particularly, BET-BRD inhibitors have shown potent antiproliferative effects in cancer cell lines. However, ongoing clinical trials, although encouraging, have not fully met expectations, showing discrepancies between preclinical efficacy and clinical results (33). Future clinical benefit might be improved by the identification of biomarkers to predict sensitivity, the development of highly selective inhibitors to prevent toxicities, and their use as part of combinatorial regimens (34).
LSD1
Histone demethylases are able to remove methyl groups from histone tails contributing to regulate the levels of histone methylation (Figure 3A). Histone demethylases are divided into 2 subgroups: the flavin adenine dinucleotide (FAD)–dependent lysine-specific histone demethylase 1A (LSD1 or KDM1A) and 1B (LSD2 or KDM1B) and the Jumonji C domain (JMJD)–containing protein family (35). LSD1 can remove methylation marks from histone H3 lysine 4 (H3K4), a mark involved in transcriptional activation, working as a transcription co-repressor. Some reports also describe its ability to remove the methylation mark at histone H3 lysine 9 (H3K9) functioning as a coactivator for androgen and estrogen receptors (36). LSD1 is overexpressed in a range of solid tumors and in AML, where it blocks differentiation, promotes proliferation, and has negative effects on prognosis (37–40). In addition, ablating LSD1 genetically or pharmacologically enhances tumor immunogenicity by stimulating endogenous retrovirus expression (41). Several LSD1 inhibitors have been developed and some of them are in clinical trials for the treatment of solid and hematologic malignancies. In light of promising clinical trial results, early in 2021 the FDA granted Orphan Drug designation to the first-in-class LSD1 inhibitor iadademstat (ORY-1001) for the treatment of AML.
m6A RNA modifiers
During recent years, it has become clear that RNA modifications have a direct effect on the regulation of gene expression. This is well described for the methylation of adenosine at position 6 to produce N6-methyladenosine (m6A), a mark catalyzed by the methyltransferase-like (METTL) 3 (METTL3)–METTL14 complex, in which METTL3 is the catalytic subunit (Figure 4) (42). m6A methylation at RNAs can be removed by non-heme–Fe(II)/2-oxoglutarate(2OG)–dependent oxygenases fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5). m6A RNA modifications can be recognized by various m6A binders that participate in the regulation of mRNA splicing, stability, nuclear transport, or translation (43).
FIGURE 4.
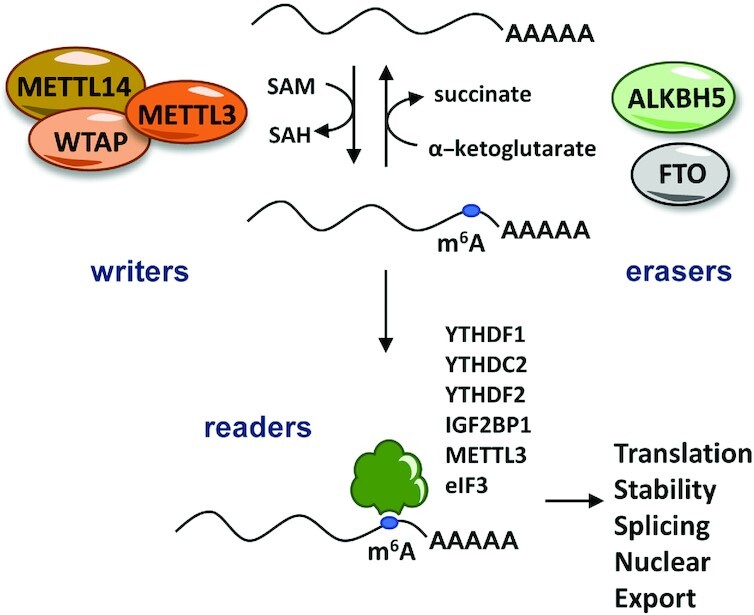
m6A RNA methylation is catalyzed by the writer METTL3-METTL14 complex using SAM as a cofactor. This modification can be removed by the specific RNA demethylases FTO and ALKBH5, which are dependent on α-ketoglutarate as a cofactor. The m6A mark can be recognized by several proteins that play important roles in the regulation of mRNA translation, stability, splicing, and/or nuclear export. ALKBH5, alkB homolog 5; METTL, methyltransferase-like; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine.
Increasing evidence suggests that m6A modifiers are deregulated in cancer and that targeting these enzymes using small molecules could be promising for its treatment (44). METTL3 is upregulated in a number of tumors where it promotes proliferation (45), glycolysis and lipid synthesis (46), and regulates responses to immunotherapies (47). Early compounds targeting this enzyme have recently shown promising preclinical results for treating myeloid leukemias (48). Interestingly, genetic variants in the intron 1 of the FTO gene have been associated with increased risk of obesity and cancer, although the mechanisms are not completely understood (49). Due to its link to obesity, targeting FTO has attracted earliest attention. Several recently developed inhibitors of FTO have been shown to block proliferation, stem cell maintenance, and immune evasion in myeloid leukemias (50, 51) and to impair self-renewal in glioblastoma cells (52). While effects of these inhibitors on body weight remain undescribed, entacapone, an inhibitor that targets both FTO and catechol-O-methyltransferase (COMT), has been reported to reduce body weight and lower fasting blood glucose concentrations in diet-induced obese mice (53). Also, inhibitors of ALKBH5 have been described to enhance the efficacy of cancer immunotherapy in a mouse model of melanoma (54).
Overall, all of the presented scientific evidence shows that targeting epigenetic-related factors is a promising strategy to treat cancer. Interestingly, it has been described that several natural compounds are able to modulate the activity of these factors. Therefore, diets that are enriched or depleted of certain foods might contribute to preventing cancer or aid cancer treatment.
Bioactive natural compounds as epigenetic modulators
Diet supplies molecules that can directly target and modulate the activity of relevant proteins called bioactives. Importantly, a significant number of small-molecule drugs approved by the FDA are closely related to natural products, making the identification of lead compounds from natural products one of the most effective approaches for obtaining useful drugs (55). Natural products show large scaffold diversity and structural complexity, covering a wider area of chemical space compared with typical synthetic small-molecule libraries. However, natural products also present important challenges for drug discovery, such as technical limitations to screening, isolation, characterization, and optimization, which explains the decrease in the use of natural products–based drug discovery programs by the pharmaceutical industry in recent years (56). In addition to serving as lead compounds for drug development, natural products typically show low toxicity and might be interesting to introduce them either as supplements or as part of specific dietetic programs.
Polyphenols are secondary metabolites of plants found in fruits, vegetables, and certain beverages (57). They constitute a large group of bioactive phytochemicals that include several subclasses, such as flavonoids, stilbenes, phenolic acids, and lignans. Many of these compounds have been suggested to have anticancer effects due to their antioxidant, anti-inflammatory, antiproliferative, and chemoprotective properties (57). In addition, other bioactives such as isothiocyanates found in cruciferous vegetables have been reported as cancer chemopreventive agents that target epigenetic factors (58). Bioactive polyphenols have been often identified using phenotypic assays, and deconvolution of their molecular mechanisms of action is not straight forward. Antiproliferative effects in cancer cell lines have been traditionally correlated with the upregulation of the expression of tumor suppressor genes. As induction of tumor suppressors is typically accompanied by changes in histone and DNA modifications, many bioactives have been suggested to directly target the epigenetic machinery involved in these responses, more commonly HDACs and DNMTs (Table 2). Unfortunately, for many natural compounds, direct proof of interaction or inhibition of the catalytic domains of epigenetic enzymes has not been provided for many natural compounds. However, modern chromatography methods and in silico docking assays are starting to provide a more reliable identification of natural compounds able to directly target the epigenetic machinery. Next, we will review described natural compounds able to directly bind and modulate the activity of epigenetic targets.
TABLE 2.
Most prominent natural compounds described to inhibit or activate epigenetic targets1
| Compound | Origin | Modulation | Target | Reference |
|---|---|---|---|---|
| Catechin | Green tea | Inhibitor | DNMT1 | (61) |
| Epicatechin | Green tea | Inhibitor | DNMT1 | (61) |
| Quercetin | Fruits, vegetables, grains | Inhibitor | DNMT1 | (61) |
| Fisetin | Fruits, vegetables | Inhibitor | DNMT1 | (61) |
| Myricetin | Fruits, vegetables, grains | Inhibitor | DNMT1 | (61) |
| EGCG | Green tea | Inhibitor | DNMT1 | (61) |
| Kazinol Q | Formosan plants | Inhibitor | DNMT1 | (63) |
| Curcumin | Turmeric | Inhibitor | DNMT1 | (64) |
| Resveratrol | Red wine | Activator | Sirtuins | (65) |
| Inhibitor | HDAC1, LSD1, BRD4 | (66, 67, 70, 78) | ||
| Emodin | Rhubarb | Inhibitor | HDAC1 | (68) |
| 8-Prenylnaringenin | Hops and beer | Inhibitor | HDAC1 | (69) |
| 6-Prenylnaringenin | Hops and beer | Inhibitor | HDAC1 | (69) |
| Baicalin | Scutellaria baicalensis | Inhibitor | LSD1 | (71) |
| 3-O-acetylpinobanksin | Medicinal plants | Inhibitor | BRD4 | (76) |
| Naringenin | Citrus fruits | Inhibitor | BRD4 | (76) |
| Kaempferol | Green tea, fruits, vegetables | Inhibitor | BRD4 | (76) |
| Amentoflavone | Gingko biloba | Inhibitor | BRD4 | (77) |
| Saikosaponin D | Bupleurum falcatum | Inhibitor | FTO | (80) |
BRD, bromodomain; DNMT1, DNA methyltransferase 1; EGCG, epigallocatechin gallate; FTO: Alpha-Ketoglutarate Dependent Dioxygenase, HDAC, histone deacetylase; LSD1, lysine specific demethylase 1.
Dietary inhibitors of DNMT1
Catechol-containing dietary polyphenols have been suggested to inhibit DNMT activities through 2 potential mechanisms. These natural molecules are excellent substrates for COMT-mediated O-methylation (59), a reaction that generates S-adenosylhomocysteine (SAH), an inhibitor of S-adenosylmethionine (SAM)–dependent enzymes, including DNMT1. Even though this mechanism can contribute to inhibit DNMT1 in vitro, it is unlikely to operate in vivo. First, it has been shown that the ratio of SAH to SAM does not significantly change in tissues after oral administration of catechol-containing polyphenols in mice (60). Second, COMT is mainly localized in the plasma membrane and the cytosol, making it unlikely that the produced SAH molecules reach the nucleus.
More relevant, some catechol-containing dietary polyphenols might act as DNMT1 inhibitors by directly binding and modulating its methyltransferase activity. Catechol-containing dietary polyphenols such as tea catechins [catechin, epicatechin, and epigallocatechin gallate (EGCG)] and bioflavonoids (quercetin, fisetin, and myricetin) have been shown to inhibit the DNA methylation activity of DNMT1 in in vitro assays at varying potencies and efficacies (61). Of them, EGCG, found in green tea, was the more potent and efficacious inhibitor of DNMT1 reported in vitro, with half maximal inhibitory concentration (IC50) values at 0.21–0.47 μM. Moreover, EGCG inhibits the methyltransferase domain of DNMT1 directely and not through its O-methylation and SAH production (61). The reported in vitro inhibition of DNMT1 by other natural compounds such as caffeic acid, chlorogenic acid, and epicatechin appears dependent on the O-methylation of these compounds by COMT and presumably exerted by SAH production (61, 62). Kazinol Q, a compound from formosan plants, was also found to inhibit DNMT1 in vitro independently of SAH production, but at lower efficiency that EGCG (63). Curcumin has been also shown to be a relatively potent inhibitor of DNMT1, presumably through covalent block of the catalytic cysteine of DNMT1 (64).
Dietary modulators of HDACs
Several polyphenols have been described to be activators of HDAC sirtuins, with the most potent activator being resveratrol (3,5,4′-trihydroxystilbene), a polyphenol found in red wine (65). Resveratrol has antioxidant, anti-inflammatory, antiproliferative, and cardioprotective properties that might be exerted, at least in part, through modulation of sirtuins. However, in silico docking analyses suggest that resveratrol has also the ability to inhibit human class I and II HDACs, although with relatively low potency (66, 67).
Additional natural compounds have been described to inhibit HDAC activity in vitro. A recent study screened a library of 131 natural compounds to determine bioactive compounds that inhibit zinc‐dependent HDAC activity. Emodin, an active component of several plants used in traditional Chinese medicine, was identified as an inhibitor of HDAC class I (68). Prenylflavonoids 6-prenylnaringenin (6-PN) and 8-prenylnaringenin (8-PN) can be found in hops and beer in very low concentrations and are also pan-HDAC inhibitors at high concentrations (69).
Natural LSD1 inhibitors
Inhibitors of the histone demethylase LSD1 are currently in clinical trials to treat certain cancers (36). Recently, a small number of natural compounds have been reported to inhibit LSD1. Resveratrol (IC50 = 15 μM) (70) and, more prominently, baicalin (IC50 = 3.01 μM) (71) have been shown to inhibit LSD1 in in vitro assays. However, these natural compounds seem rather weak inhibitors compared with the inhibitor ORY-1001 that is currently in clinical trials and has an IC50 under 20 nM (72). In addition, modern chromatography methods have allowed the identification of 6 natural LSD1 inhibitors (including baicalin and wogonoside) present in Scutellaria baicalensis extracts, a plant frequently used in Chinese medicine (73).
Natural HAT inhibitors
Despite the well-known involvement of some enzymes with HAT activity in cancer progression, the development of high-quality chemical probes able to inhibit HATs with good potency and specificity has remained elusive (74). Only recently, a high-quality inhibitor of the HAT-containing enzymes CREBBP/EP300 has been developed and shown to inhibit the growth of several hematological and androgen receptor–positive prostate cancer cell lines (18). In the past, several natural compounds, including curcumin, plumbagin, embelin, EGCG, garcinol (75), anacardic acid, and gossypol, have been claimed to inhibit different enzymes with HAT activity in vitro, but a meticulous recently published work shows that these compounds are poor HAT inhibitors (74). Plumbagin, embelin, and EGCG were found to be nonselective target modulators due to nonspecific thiol reactivity. Although all these compounds showed antiproliferative effects in cancer cell lines, these were found to be nonspecific and likely off-target.
Dietary inhibitors of BRDs
In addition to their therapeutic potential, the good druggability (the likelihood of being able to modulate a target with a small-molecule drug) predictions of BRDs have also contributed to the popularity of screenings against these domains, including natural compounds (29). Among them, flavonoids have emerged as putative modulators of BRDs using molecular docking and dynamic simulation in silico. Rare flavonoids like 3-O-acetylpinobanksin, naringenin diacetate, and kaempferol tetraacetate were found to bind the first BRD of the BET family member BRD4 in silico; however, these interactions were not confirmed in in vitro binding assays (76). Amentoflavone, a biflavonoid from Gingko biloba with reported anticancer properties, was also predicted to bind the first BRD of BRD4 by molecular docking (77). The binding was confirmed in an in vitro AlphaScreen assay in which amentoflavone showed an IC50 of approximately 30 μM, while the IC50 reported for potent BRD4 inhibitors such as JQ1 are in the nanomolar range. A recent report suggests that resveratrol might be a pan-BET inhibitor (78). Isothermal titration calorimetry (ITC) assays show that resveratrol interacts with several BRDs in the BET family, a finding supported by molecular docking data showing binding to the first BRD of BRD4 mimicking the acetyl-lysine interactions. However, resveratrol is likely to be a weak BRD4 inhibitor since the reported Kd in ITC assays for the first BRD of BRD4 were of 6.6 μM while potent BRD4 inhibitors show a Kd in the nanomolar range (32). In addition to potency concerns, none of the published studies addressed the issue of selectivity. BRD inhibitors are well known for showing promiscuity and targeting simultaneously several BRDs, which complicates the interpretation of their biological effects. In addition, BRD inhibitors might also target other domains since inhibitors that target both BRDs and kinases with therapeutically relevant potencies have been described (79).
Natural inhibitors of FTO
Several lines of evidence suggests that RNA-modifying enzymes play a relevant role in cancer. However, the interest in targeting RNA modifying enzymes for therapeutic purposes is relatively novel and potent inhibitors have been developed only recently.
With regard to natural compounds, saikosaponin D and radicicol (from the fungus Diheterospora chlamydosporia) have been described to bind and inhibit FTO (80, 81). Both compounds were predicted to bind FTO by molecular docking analysis and confirmed as FTO inhibitors in in vitro assays. Additionally, saikosaponin D, a triterpenoid saponin compound extracted from Bupleurum falcatum, showed antiproliferative effects in leukemia cell lines while increasing the levels of m6A RNA (81). Since the field of epitranscriptomics is still in its infancy, further research is needed to fully uncover the therapeutic potential of targeting RNA-modifying enzymes.
Overall, many natural compounds have been suggested to target epigenetic factors. However, most studies are based on cellular phenotypic assays that lack specificity. Importantly, in vitro activity assays are not exempt from artifacts. Many assay interference and promiscuous bioactive compounds show nonspecific thiol reactivity, such as the case of EGCG, which has been shown to inhibit multiple targets including EP300 and DNMT1 (74). In addition, some natural compounds are rather weak inhibitors and concentrations needed to effectively inhibit the reported epigenetic targets are far above the levels achieved by dietary consumption. Despite the low concentration of active components that are likely to reach cells in the human body, many clinical studies support the efficacy of dietary plant extracts, most commonly as anti-inflammatory agents (82). There are several potential explanations for these discrepancies, including systemic effects or complex interactions. For example, beneficial effects might be due to mild inhibition of multiple targets, synergistic effects with other compounds found in extracts, or caused by metabolites derived from these compounds. In addition, systemic effects that are not easy to reproduce in vitro, such as effects on the microbiota, cannot be discarded.
Diet and metabolism of epigenetic cofactors
In addition to bioactives, diet supplies a large number of substrates for metabolic reactions. Several metabolites and cofactors play essential roles in mediating the activities of many epigenetic enzymes (Table 1). Examples of such cofactors generated via metabolic process include, SAM, α-ketoglutarate (αKG), acetyl-CoA, and NAD+. Additionally, certain epigenetic enzymes are modulated by dietary nutrients such as vitamin C. Thus, epigenetic enzymes can act as sensors of the nutritional status, modulating accordingly the marks on the epigenome. This section will focus on the role of cofactors in the regulation of epigenetic enzymes, the metabolic pathways involved in their production, and the impact of nutrition on such pathways.
Cofactors for histone acetylation
Acetyl-CoA is the donor of acetyl groups for acetylation, which is promoted by acetyl-CoA and inhibited by its product, CoA. Therefore, the ratio of acetyl-CoA to CoA is likely to have an impact in the acetylation of histones and it is influenced by the rate of glycolysis and nutrient abundance. During glycolysis, glucose is converted into 2 pyruvate molecules that can be used for lactate production or transported into the mitochondria to generate acetyl-CoA that will enter the Krebs cycle for energy production (Figure 5). Cancer cells, even in oxygenated conditions, show high rates of glucose uptake and preferential production of lactate, a trend known as the Warburg effect (83). This results in changes in metabolite concentrations such as acetyl-CoA that are used as substrates or cofactors for enzymes that carry out post-translational modifications, including histone acetylation (84).
FIGURE 5.
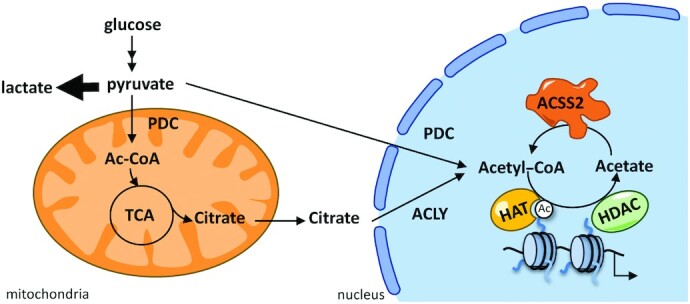
Sources of acetyl-CoA in the nucleus. Acetyl-CoA can be generated in the nucleus from pyruvate by the PDC, from citrate by ACLY, and from acetate by ACSS2. ACLY, ATP-citrate lyase; ACSS2, acyl-CoA synthetase short-chain family member 2; HAT, histone acetyltransferase; HDAC, histone deacetylase; PDC, pyruvate dehydrogenase complex.
Acetyl-CoA can be generated in multiple cellular compartments; however, it needs to reach the nucleus to have an impact on histone acetylation. Therefore, both concentrations of acetyl-CoA and the compartmentalization of its production are relevant physiological parameters for histone acetylation (85). Acetyl-CoA produced in the mitochondria cannot pass through mitochondrial membranes. Thus, acetyl-CoA relevant for histone acetylation is likely to be the one produced in the cytosol and nucleus from mitochondria-exported citrate by ATP-citrate lyase (ACLY), and from acetate by acyl-CoA synthetase short-chain family member 2 (ACSS2) (Figure 5). Both enzymes are upregulated in multiple cancer types, and likely contribute to their malignancy (86–88). In addition, a recent study reported a source of acetyl-CoA for histone acetylation by nuclear glycogenolysis (89).
Nutrition, including diets that play a role in cancer risk and progression such as high-fat diets (HFDs), fasting, and caloric restriction, can impact histone acetylation through multiple mechanisms. An HFD, which increases cancer risk and aggressiveness, results in a decrease in the acetyl-CoA:CoA ratio that correlates with reduced histone acetylation in the white adipose tissue of mice (90). In addition to providing substrates that fuel certain metabolic pathways affecting the acetyl-CoA pool, an HFD might also have an impact in the expression and subcellular localization of ACLY or ACSS2, as these genes are targets of the sterol regulatory element binding transcription factors (SREBPs) that regulate lipid homeostasis (91, 92). Also, other short-chain acyl modifications of histone lysine residues different from acetylation have been identified, including propionylation, butyrylation, hydroxybutyrylation, crotonylation, malonylation, succinylation, and glutarylation. These less-known histone modifications are also regulated by HATs and HDACs and are also influenced by glucose and fatty acid availability (14). Despite the great complexity of acetyl-CoA–related metabolism and histone acylation, targeting metabolic regulators or even dietary interventions that modulate acetyl-CoA availability and likely impact histone acetylation are emerging as potential strategies for cancer therapy (93, 94).
Cofactors for histone deacetylation
The mammalian NAD+-dependent HDACs SIRT1, SIRT2, and SIRT7 contribute to regulate gene expression depending on NAD+ abundance. NAD+ levels are high in situations of energy deficiency, such as exercise, caloric restriction, or fasting, leading to sirtuin activation. On the contrary, NAD+ is depleted when energy is in excess, increasing the NAD+:NADH ratio and inhibiting sirtuin activity (95). This suggests a direct link between the nutritional status and epigenetic control through sirtuins; however, the diversity of sirtuins’ histone and nonhistone targets complicates the comprehension of their molecular mechanisms of action.
Classical HDACs, despite their independence from cofactors for catalytic activity, are linked to cellular metabolism since SCFAs β-hydroxybutyrate and lactate are inhibitors of their activity. In animals, the synthesis of β-hydroxybutyrate and other ketone bodies takes place mainly in the mitochondria of liver cells and provides an energy source to the heart and brain during starvation, fasting, or intense exercise conditions. In addition, β-hydroxybutyrate is derived from the fermentation of dietary fiber by the gut microbiota, providing the preferred source of fuel for colonocytes in the large bowel, as well as playing important epigenetic roles in the colon epithelia. By inhibiting HDACs, β-hydroxybutyrate induces the expression of genes involved in ketogenesis and transcription factors that regulate cell-cycle genes in intestinal epithelial progenitors (96, 97).
Several studies have demonstrated that β-hydroxybutyrate is able to inhibit the growth of different types of tumor cells, presumably through its HDAC inhibitor properties (98). In agreement, diets rich in fiber are associated with a lower risk of developing colorectal cancer. Ketogenic diets and intermittent fasting are popular diets that can significantly increase the concentrations of circulating β-hydroxybutyrate. While their long-term benefits in the general population remain controversial, ketogenic diets might be beneficial for cancer patients and are currently under evaluation as adjuvant therapy to conventional radiation and chemotherapies (99). Despite the reported anti-tumor effects of caloric restriction, a well-designed ketogenic diet would be preferred in a range of cancer patients, particularly those with cachexia (100).
Lactate is produced under conditions of high glycolysis, such as the Warburg effect, making it an important metabolite in cancer cells. Despite this, the effects of lactate on epigenetic regulation remain obscure. Lactate has been described to induce histone acetylation and changes in gene expression similar to well-known HDAC inhibitors in colon cancer cells (101). However, in vitro assays suggest that lactate is a very weak HDAC inhibitor compared with β-hydroxybutyrate (102). More recently it has been shown that lactate accumulation induces lactate-derived lactylation of histone lysine residues and stimulates gene transcription (102). The relevance of this modification for cancer treatment remains to be determined.
SAM and the methylation of DNA, RNA, and histones
SAM is the primary methyl group donor for methylation reactions. SAM is primarily generated from methionine in the one-carbon metabolism pathway involving the folate and methionine cycles (Figure 6). Methionine adenosyltransferases (MATs) are the key enzymes that generate SAM from methionine. SAM donates its methyl group to become SAH. The one-carbon metabolism has been suggested to be an integrator of cellular nutrient status by cycling carbon units from amino acid inputs to generate diverse outputs, including nucleotides, proteins, and lipids, reducing power and substrates for methylation (1).
FIGURE 6.
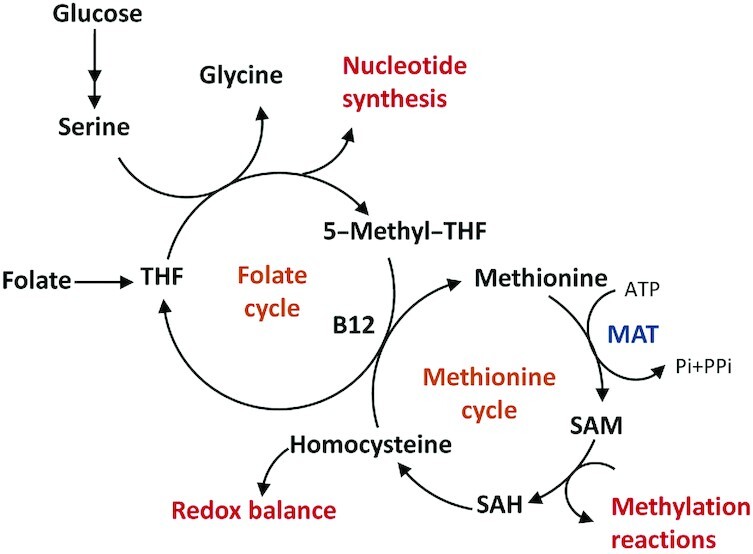
Simplified one-carbon metabolism involved in SAM production. The folate cycle begins with the conversion of dietary folate to THF. THF accepts methyl groups from serine and be further reduced to 5-methyl-THF, which delivers one-carbon units into methionine cycle as is converted back to THF to complete the folate cycle. In the methionine cycle, homocysteine is re-methylated using a one-carbon unit from methyl-THF to form methionine. Methionine is converted to SAM by MAT and SAM then acts as a substrate used by a diverse group of methyltransferases. The product of these methylation reactions is SAH, which can be hydrolyzed into homocysteine, completing the cycle. B12, vitamin B-12; MAT, methyl-adenosyl transferase; THF, tetrahydrofolate; PPi, pyrophosphate; Pi, inorganic phosphate; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine.
Alterations of the one-carbon pathway play a role in cancer and might provide a link to cellular epigenetic status through the synthesis of SAM (1). Although most of these pathway reactions take place in the cytosol, or the mitochondria, MAT isoforms MATIIα and MATIIβ have been detected in the nucleus and defects in their activity have been correlated with a decrease in histone methylation (103). In addition, MATIIα and MATIIβ interact with several methyltransferases and transcription factors and are recruited to the promoter of several genes to repress transcription by affecting histone methylation (104, 105).
The one-carbon metabolism can be influenced by diet in multiple ways (Figure 6). Folate cannot be synthesized de novo by humans and needs to be supplied with diet, mostly from leafy green vegetables. Vitamin B-12, an important cofactor for the one-carbon cycle, is produced solely by bacteria through aerobic or anaerobic pathways. Given the important functions of the one-carbon metabolism, methionine, folate, or vitamin B-12 deprivation have been reported to have detrimental consequences for health (106). In addition, various studies have correlated folate and vitamin B-12 intake with alterations in DNA methylation and cancer risk. For example, decreased folate intake is associated with colorectal cancer, breast cancer, and global DNA hypomethylation (107, 108).
Despite the detrimental consequences of impairing the one-carbon metabolism, targeting enzymes in this pathway with small molecules has been a successful strategy to treat cancer (109). Alternatively, diets that restrict the intake of folate, vitamin B-12, or amino acids such as methionine, serine, or glycine can contribute to cancer treatment and prevention through the modulation of one-carbon metabolism. Eventually, such beneficial effects might be exerted, at least partially, by regulating the activity of SAM-dependent epigenetic enzymes. Importantly, human dietary behaviors, such as fruit and vegetable consumption versus carbohydrate-rich diets, induce fluctuations in methionine concentrations in serum with the potential to modulate histone methylation (110). In mice, a methionine deprivation diet caused a global decrease in histone H3K4 methylation and subsequent changes in gene expression in the liver (110, 111). A decrease in histone, DNA, and RNA methylation were also observed in vitro in colon cancer cells cultured under methionine starvation conditions (110, 112). In these cells, methionine restriction caused a reduction in intracellular SAM concentrations, decreasing the levels of H3K4 methylation, and affecting the expression of important cancer-associated genes, such as MYC (110). Although methionine is the dominant methyl provider for SAM production, serine- and glycine-deprived diets have also been reported to have anti-tumoral effects in mice (113). In colon cancer cells, it has been shown that serine, a major influencer on the growth and metabolism in these cells, facilitates the methylation of DNA and RNA from methionine-derived SAM by providing de novo ATP synthesis (112).
Cofactors for DNA, RNA, and histone demethylation
The abundance of cofactors that modulate the activity of DNA, RNA, and histone demethylases is also influenced by diet and plays a role in cancer progression. JMJD-containing histone lysine demethylases, TET enzymes, and RNA demethylates are 2-oxoglutarate-dependent dioxygenases (2OGDDs) that use the TCA cycle intermediate 2OG (also known as αKG) to remove methyl groups from histones, DNA, or RNA. These hydroxylation reactions also require Fe2+ as a cofactor, O2 as a co-substrate, and ascorbic acid (vitamin C) as a reductase of Fe(III) to Fe(II) to restore enzyme activity. Importantly, the activity of 2OGDDs can be competitively inhibited by the 2OG analogs fumarate, succinate, and the R enantiomer of 2-hydroxyglutarate (R-2HG). Therefore, 2OGDDs have the potential to sense oxygen, reactive oxygen species, iron availability, vitamins, and specific metabolites. Each 2OGDD has different affinity for their cofactors and competitive inhibitors establishing complex relations between metabolism and gene expression regulation (114).
Some cancers are caused by mutations in the genes encoding fumarate hydratase (FH), succinate dehydrogenase (SDH), and NADP-dependent isocitrate dehydrogenase isoforms (IDH1/2), which lead to the accumulation of the 2OGDDs inhibitors fumarate, succinate, and R-2HG, respectively, leading to profound effects on DNA and histone methylation that contribute to malignancy (115–117). Interestingly, these epigenetic effects can be reversed by increasing the intracellular concentrations of 2OG, which might be modulated by diet (115, 118, 119). Since 2OG supplementation has antiproliferative effects in cultured cancer cells, the possibility of using 2OG or its precursors in specific dietetic plans as anticancer agents to counteract oncogenic epigenetic processes is under consideration.
Vitamin C is a cofactor for 2OGDDs that exists predominantly as an ascorbate anion under physiological pH conditions. Ascorbate has been described to induce the removal of DNA methylation by enhancing TET enzymes and promoting conversion of 5mC to 5hmC (120), and to impact the levels of histone methylation through activation of JMJD-containing histone demethylases (121). Vitamin C is an essential dietary micronutrient for humans and its deficiency is rare in the general population, due to its abundance in certain fruits and vegetables. However, genetic variation in ascorbate transporters and certain intestinal diseases might influence its absorption. Vitamin C serum concentrations might be also lowered by unhealthy habits such as smoking and alcohol consumption (122). Importantly, vitamin C deficiency is frequently observed in patients with cancer and correlates with shorter survival (123, 124). In addition, loss of 5hmC is also common in cancer, mainly due to alterations in TET enzymes or in enzymes that produce 2OG analogs (125). Therefore, supplementation with vitamin C might have positive outcomes in cancer treatment, as suggested by several studies. Treatment of IDH-mutant AML cells with vitamin C reduced proliferation and increased differentiation in correlation with changes in DNA methylation (126). Vitamin C might be also used in combination with other epigenetic drugs to boost their effects. For example, vitamin C has been shown to improve the antiproliferative effects of inhibitors of DNA methylation and enhance the expression of ERVs in several cancer cell lines (123). Moreover, normalization of plasma vitamin C by oral supplementation in myeloid patients treated with the DNA methylation inhibitor 5-azacytidine improved the ratio of global 5hmC:5mC (127). In addition to dietary supplementation of vitamin C, pharmacological doses of vitamin C can be injected intravenously to achieve higher and lasting plasma concentrations, boosting the benefits of vitamin C as an adjuvant for existing chemotherapies to improve their therapeutic potential.
As described in this section, most experimental data have been reported in cellular or animal models. The impact of particular diets on cofactor metabolism and how this translates into changes in the activity of epigenetic enzymes is not completely understood and will need to be further investigated and tested in clinical trials.
Conclusions and Future Perspectives
Inhibitors that target different epigenetic factors are currently used for cancer treatment or hold promise for future treatment. In a similar way, diet can have an important epigenetic impact, affecting gene expression at multiple levels, and might influence the risk and prognosis of different types of cancer. Diet can affect the metabolism of cofactors needed for the proper function of epigenetic enzymes. In addition, several natural compounds have been described to modulate the activity of such enzymes. Moreover, fat, cholesterol, and dietary fiber have been demonstrated to influence cancer risk and can impact in the activity of transcription factors that recruit epigenetic regulators to chromatin as depicted in Figure 1. Thus, the relation between diet, DNA, RNA, histone modifications, and gene expression and cancer is complex.
Most studies testing the anticancer effects of natural compounds or cofactor metabolism have been conducted in vitro or in animal models under a precise control of food intake or dietary exposures. However, the effects of food in humans might be more difficult to assess due to the complexity of diets, accuracy of methods to evaluate them, and the multifactorial character of human nutrition (influenced by physical activity, microbiota, and genetic background, among others). From the molecular point of view, understanding how food intake translates into the accumulation of compounds in the cell nucleus at a level able to inhibit the epigenetic machinery is challenging. Moreover, bioactives might be able to bind several targets while cofactors can affect different types of activities with different outcomes (for example, SAM availability might affect DNA, histone, and RNA methylation). While it is known that healthy diets help to prevent cancer and that those have an epigenetic impact, it is clear that a deeper understanding of nutrition and the molecular action of nutritional biomolecules will be needed to define specific and personalized diets that impact epigenetics for cancer prevention and to aid cancer treatment.
ACKNOWLEDGEMENTS
The authors’ responsibilities were as follows—MJB and PC: conducted the literature search and wrote the manuscript; HWL and ARdM: performed the critical revision of the article; MJB and ARdM: conceptually designed the manuscript; and all authors: read and approved the final manuscript.
Notes
Research at Ana Ramírez de Molina Laboratory is funded by the Spanish Ministry of Science (Plan Nacional I+D+i PID2019-110183RB-C21), the Regional Government of Community of Madrid (P2018/BAA-4343-ALIBIRD2020-CM, Y2020/BIO-6350 and REACT EU Program FACINGLCOVID-CM project, Comunidad de Madrid and The European Regional Development Fund. ERDF. European Union), and the Ramón Areces Foundation (CIVP19A5937).
Author disclosures: The authors report no conflicts of interest.
Abbreviations used: ACLY, ATP-citrate lyase; ACSS2, acyl-CoA synthetase short-chain family member 2; ALKBH5, alkB homolog 5; AML, acute myeloid leukemia; BET, bromo and extraterminal domain; BRD, bromodomain; COMT, catechol-O-methyltransferase; DNMT, DNA methyltransferase; DNMTi, DNMT inhibitor; EGCG, epigallocatechin gallate; ERV, endogenous retrovirus; EZH2, Enhancer of zeste 2 polycomb repressive complex 2 subunit ; H3K4, histone H3 lysine 4; HAT, histone acetyltransferase; HDAC, histone deacetylase; HDACi, HDAC inhibitor; HFD, high-fat diet; IC50, half maximal inhibitory concentration; ITC, isothermal titration calorimetry; JMJD, Jumonji C domain; m6A, N6-methyladenosine; MAT, methionine adenosyltransferase; METTL, methyltransferase-like; PN, prenylnaringenin; R-2HG, 2-hydroxyglutarate; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SE, super-enhancer; TET, ten-eleven translocation; αKG, α-ketoglutarate; 2OG, 2-oxoglutarate; 2OGDD, 2-oxoglutarate-dependent dioxygenase; 5hmC, 5-hydroxymethylcytosine; 5mC, 5 methylcytosine.
Contributor Information
Maria J Barrero, Molecular Oncology Group, IMDEA Food, CEI UAM+CSIC, Madrid, Spain; Models and Mechanisms Unit, Institute of Rare Diseases Research (IIER), Instituto de Salud Carlos III (ISCIII), Madrid, Spain.
Paloma Cejas, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA; Center for Functional Cancer Epigenetics, Dana-Farber Cancer Institute, Boston, MA, USA; Translational Oncology Laboratory, Hospital La Paz Institute for Health Research, Madrid, Spain.
Henry W Long, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA; Center for Functional Cancer Epigenetics, Dana-Farber Cancer Institute, Boston, MA, USA.
Ana Ramirez de Molina, Molecular Oncology Group, IMDEA Food, CEI UAM+CSIC, Madrid, Spain.
References
- 1. Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13(8):572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang T, Cooper S, Brockdorff N. The interplay of histone modifications—writers that read. EMBO Rep. 2015;16(11):1467–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20(10):608–24. [DOI] [PubMed] [Google Scholar]
- 4. Banerji J, Rusconi S, Schaffner W. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27(2):299–308. [DOI] [PubMed] [Google Scholar]
- 5. Cejas P, Drier Y, Dreijerink KMA, Brosens LAA, Deshpande V, Epstein CBet al. . Enhancer signatures stratify and predict outcomes of non-functional pancreatic neuroendocrine tumors. Nat Med. 2019;25:1260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AAet al. . Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sabari BR, Dall'Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas Ket al. . Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018;361(6400):eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MHet al. . Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CRet al. . Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cejas P, Li L, O'Neill NK, Duarte M, Rao P, Bowden Met al. . Chromatin immunoprecipitation from fixed clinical tissues reveals tumor-specific enhancer profiles. Nat Med. 2016;22:685–91. [DOI] [PubMed] [Google Scholar]
- 11. Cohen AJ, Saiakhova A, Corradin O, Luppino JM, Lovrenert K, Bartels CFet al. . Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat Commun. 2017;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yao X, Tan J, Lim KJ, Koh J, Ooi WF, Li Zet al. . VHL deficiency drives enhancer activation of oncogenes in clear cell renal cell carcinoma. Cancer Discov. 2017;7(11):1284–305. [DOI] [PubMed] [Google Scholar]
- 13. Wong RWJ, Ngoc PCT, Leong WZ, Yam AWY, Zhang T, Asamitsu Ket al. . Enhancer profiling identifies critical cancer genes and characterizes cell identity in adult T-cell leukemia. Blood. 2017;130:2326–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mehdipour P, Murphy T, De Carvalho DD. The role of DNA-demethylating agents in cancer therapy. Pharmacol Ther. 2020;205:107416. [DOI] [PubMed] [Google Scholar]
- 15. Bender CM, Pao MM, Jones PA. Inhibition of DNA methylation by 5-Aza-2′-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res. 1998;58:95–101. [PubMed] [Google Scholar]
- 16. Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman Bet al. . Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162:974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SYet al. . DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell. 2015;162:961–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino ELet al. . Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017;550:128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McClure JJ, Li X, Chou CJ. Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv Cancer Res. 2018;138:183–211. [DOI] [PubMed] [Google Scholar]
- 20. Brocks D, Schmidt CR, Daskalakis M, Jang HS, Shah NM, Li Det al. . DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat Genet. 2017;49(7):1052–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AVet al. . Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21(3):231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GSet al. . EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–12. [DOI] [PubMed] [Google Scholar]
- 23. Chan-Penebre E, Armstrong K, Drew A, Grassian AR, Feldman I, Knutson SKet al. . Selective killing of SMARCA2- and SMARCA4-deficient small cell carcinoma of the ovary, hypercalcemic type cells by inhibition of EZH2: in vitro and in vivo preclinical models. Mol Cancer Ther. 2017;16(5):850–60. [DOI] [PubMed] [Google Scholar]
- 24. Harms PW, Hristov AC, Kim DS, Anens T, Quist MJ, Siddiqui Jet al. . Activating mutations of the oncogene EZH2 in cutaneous melanoma revealed by next generation sequencing. Hum Pathol Case Rep. 2014;1:21–8. [Google Scholar]
- 25. First EZH2 inhibitor approved-for rare sarcoma. Cancer Discov. 2020;10:333–4. [DOI] [PubMed] [Google Scholar]
- 26. Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline Set al. . Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020;21(11):1433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deblois G, Madani Tonekaboni SA, Grillo G, Martinez C, Kao YI, Tai Fet al. . Epigenetic switch-induced viral mimicry evasion in chemotherapy resistant breast cancer. Cancer Discov. 2020;10(9):1312–29. [DOI] [PubMed] [Google Scholar]
- 28. Arbuckle JH, Gardina PJ, Gordon DN, Hickman HD, Yewdell JW, Pierson TCet al. . Inhibitors of the histone methyltransferases EZH2/1 induce a potent antiviral state and suppress infection by diverse viral pathogens. Mbio. 2017;8(4):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discovery. 2014;13(5):337–56. [DOI] [PubMed] [Google Scholar]
- 30. Garcia-Carpizo V, Ruiz-Llorente S, Sarmentero J, González-Corpas A, Barrero MJ. CREBBP/EP300 bromodomain inhibition affects the proliferation of AR positive breast cancer cell lines. Mol Cancer Res. 2019;17(3):720–30. [DOI] [PubMed] [Google Scholar]
- 31. Garcia-Carpizo V, Ruiz-Llorente S, Sarmentero J, Graña-Castro O, Pisano DG, Barrero MJ. CREBBP/EP300 bromodomains are critical to sustain the GATA1/MYC regulatory axis in proliferation. Epigenet Chromatin. 2018;11:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov Oet al. . Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun Y, Han J, Wang Z, Li X, Sun Y, Hu Z. Safety and efficacy of bromodomain and extra-terminal inhibitors for the treatment of hematological malignancies and solid tumors: a systematic study of clinical trials. Front Pharmacol. 2021;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shorstova T, Foulkes WD, Witcher M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br J Cancer. 2021;124(9):1478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sainathan S, Paul S, Ramalingam S, Baranda J, Anant S, Dhar A. Histone demethylases in cancer. Curr Pharmacol Rep. 2015;1(4):234–44. [Google Scholar]
- 36. Yang G-J, Lei P-M, Wong S-Y, Ma D-L, Leung C-H. Pharmacological inhibition of LSD1 for cancer treatment. Molecules. 2018;23(12):3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schenk T, Chen WC, Göllner S, Howell L, Jin L, Hebestreit Ket al. . Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18(4):605–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schulte JH, Lim S, Schramm A, Friedrichs N, Koster J, Versteeg Ret al. . Lysine-specific demethylase 1 is strongly expressed in poorly differentiated neuroblastoma: implications for therapy. Cancer Res. 2009;69(5):2065–71. [DOI] [PubMed] [Google Scholar]
- 39. Lim S, Janzer A, Becker A, Zimmer A, Schüle R, Buettner Ret al. . Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis. 2010;31(3):512–20. [DOI] [PubMed] [Google Scholar]
- 40. Kahl P, Gullotti L, Heukamp LC, Wolf S, Friedrichs N, Vorreuther Ret al. . Androgen receptor coactivators lysine-specific histone demethylase 1 and four and a half LIM domain protein 2 predict risk of prostate cancer recurrence. Cancer Res. 2006;66(23):11341–47. [DOI] [PubMed] [Google Scholar]
- 41. Bailey CP, Figueroa M, Gangadharan A, Yang Y, Romero MM, Kennis BAet al. . Pharmacologic inhibition of lysine-specific demethylase 1 as a therapeutic and immune-sensitization strategy in pediatric high-grade glioma. Neuro-oncol. 2020;22(9):1302–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang Let al. . A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dai X-Y, Shi L, Li Z, Yang H-Y, Wei J-F, Ding Q. Main N6-Methyladenosine readers: YTH family proteins in cancers. Front Oncol. 2021;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20(6):303–22. [DOI] [PubMed] [Google Scholar]
- 45. Zeng C, Huang W, Li Y, Weng H. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol. 2020;13(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Han X, Wang L, Han Q. Advances in the role of m6A RNA modification in cancer metabolic reprogramming. Cell Biosci. 2020;10:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, Yuan J, Rana TM. m6 A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 2020;39:(20):e104514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga Get al. . Small molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:(7860):597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lan N, Lu Y, Zhang Y, Pu S, Xi H, Nie XFTO—a common genetic basis for obesity and cancer et al. FTO—a common genetic basis for obesity and cancer . Front Genet. 2020;11:559138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Su R, Dong L, Li Y, Gao M, Han L, Wunderlich Met al. . Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 2020;38(1):79–96, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu Het al. . Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. 2019;35(4):677–91, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huff S, Tiwari SK, Gonzalez GM, Wang Y, Rana TM. M6A-RNA demethylase FTO inhibitors impair self-renewal in glioblastoma stem cells. ACS Chem Biol. 2021;16(2):324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Peng S, Xiao W, Ju D, Sun B, Hou N, Liu Qet al. . Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Sci Translat Med. 2019;11:7116. [DOI] [PubMed] [Google Scholar]
- 54. Li N, Kang Y, Wang L, Huff S, Tang R, Hui Het al. . ALKBH5 regulates anti–PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci. 2020;117(33):20159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Harvey AL, Edrada-Ebel R, Quinn RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discovery. 2015;14(2):111–29. [DOI] [PubMed] [Google Scholar]
- 56. Atanasov AG, Zotchev SB, Dirsch VM, Orhan IE, Banach M, Rollinger JMet al. . Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discovery. 2021;20(3):200–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fraga CG, Croft KD, Kennedy DO, Tomás-Barberán FA. The effects of polyphenols and other bioactives on human health. Food Function. 2019;10(2):514–28. [DOI] [PubMed] [Google Scholar]
- 58. Hudlikar R, Wang L, Wu R, Li S, Peter R, Shannar Aet al. . Epigenetics/epigenomics and prevention of early stages of cancer by isothiocyanates. Cancer Prev Res. 2021;14(2):151–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhu, BT, Ezell EL, Liehr JG. Catechol-O-methyltransferase-catalyzed rapid O-methylation of mutagenic flavonoids. Metabolic inactivation as a possible reason for their lack of carcinogenicity in vivo. J Biol Chem. 1994;269:292–99. [PubMed] [Google Scholar]
- 60. Fang M, Chen D, Yang CS. Dietary polyphenols may affect DNA methylation. J Nutr. 2007;137(1 Suppl):223S–8S. [DOI] [PubMed] [Google Scholar]
- 61. Won JL, Shim JY, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol. 2005;68:1018–30. [DOI] [PubMed] [Google Scholar]
- 62. Lee WJ, Zhu BT. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis. 2006;27(2):269–77. [DOI] [PubMed] [Google Scholar]
- 63. Weng J-R, Lai I-L, Yang H-C, Lin C-N, Bai L-Y. Identification of kazinol q, a natural product from formosan plants, as an inhibitor of DNA methyltransferase. Phytother Res. 2014;28(1):49–54. [DOI] [PubMed] [Google Scholar]
- 64. Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu S, Yu Jet al. . Curcumin is a potent DNA hypomethylation agent. Bioorg Med Chem Lett. 2009;19(3):706–9. [DOI] [PubMed] [Google Scholar]
- 65. Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JGet al. . Small molecule activators of sirtuins extend saccharomyces cerevisiae lifespan. Nature. 2003;425(6954):191–6. [DOI] [PubMed] [Google Scholar]
- 66. Venturelli S, Berger A, Böcker A, Busch C, Weiland T, Noor Set al. . Resveratrol as a pan-HDAC inhibitor alters the acetylation status of Jistone proteins in human-derived hepatoblastoma cells. PLoS One. 2013;8(8):e73097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dayangaç-Erden D, Bora G, Ayhan P, Kocaefe Ç, Dalkara S, Yelekçi Ket al. . Histone deacetylase inhibition activity and molecular docking of (e )-resveratrol: its therapeutic potential in spinal muscular atrophy. Chem Biol Drug Des. 2009;73:355–64. [DOI] [PubMed] [Google Scholar]
- 68. Godoy LD, Lucas JE, Bender AJ, Romanick SS, Ferguson BS. Targeting the epigenome: screening bioactive compounds that regulate histone deacetylase activity. Mol Nutr Food Res. 2017;61:1600744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Venturelli S, Niessner H, Sinnberg T, Berger A, Burkard M, Urmann Cet al. . 6- and 8-Prenylnaringenin, novel natural histone deacetylase inhibitors found in hops, exert antitumor activity on melanoma cells. Cell Physiol Biochem. 2018;51(2):543–56. [DOI] [PubMed] [Google Scholar]
- 70. Abdulla A, Zhao X, Yang F. Natural polyphenols inhibit lysine-specific demethylase-1 in vitro. J Biochem Pharmacol Res. 2013;1:56–63. [PMC free article] [PubMed] [Google Scholar]
- 71. Zheng YC, Shen DD, Ren M, Liu XQ, Wang ZR, Liu Yet al. . Baicalin, a natural LSD1 inhibitor. Bioorganic Chem. 2016;69:129–31. [DOI] [PubMed] [Google Scholar]
- 72. Fang Y, Liao G, Yu B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J Hematol Oncol. 2019;12:(1):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Han C, Wang S, Li Z, Chen C, Hou J, Xu Det al. . Bioactivity-guided cut countercurrent chromatography for isolation of lysine-specific demethylase 1 inhibitors from Scutellaria baicalensis Georgi. Anal Chim Acta. 2018;1016:59–68. [DOI] [PubMed] [Google Scholar]
- 74. Dahlin JL, Nelson KM, Strasser JM, Barsyte-Lovejoy D, Szewczyk MM, Organ Set al. . Assay interference and off-target liabilities of reported histone acetyltransferase inhibitors. Nat Commun. 2017;8(1):1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Balasubramanyam K, Altaf M, Varier RA, Swaminathan V, Ravindran A, Sadhale PPet al. . Polyisoprenylated benzophenone, garcinol, a natural histone acetyltransferase inhibitor, represses chromatin transcription and alters global gene expression. J Biol Chem. 2004;279(32):33716–26. [DOI] [PubMed] [Google Scholar]
- 76. Dhananjayan K. Molecular docking study characterization of rare flavonoids at the nac-binding site of the first bromodomain of BRD4 (BRD4 BD1). J Cancer Res. 2015;2015:1–15. [Google Scholar]
- 77. Prieto-Martínez F, Medina-Franco J. Flavonoids as putative epi-modulators: insight into their binding mode with BRD4 bromodomains using molecular docking and dynamics. Biomolecules. 2018;8(3):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dutra LA, Heidenreich D, da Silva GDB, Chin CM, Knapp S, dos Santos JL. Dietary compound resveratrol is a pan-BET bromodomain inhibitor. Nutrients. 2017;9(11):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ciceri P, Müller S, O'Mahony A, Fedorov O, Filippakopoulos P, Hunt JPet al. . Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat Chem Biol. 2014;10(4):305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sun K, Du Y, Hou Y, Zhao M, Li J, Du Yet al. . Saikosaponin D exhibits anti-leukemic activity by targeting FTO/m 6 a signaling. Theranostics. 2021;11(12):5831–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wang R, Han Z, Liu B, Zhou B, Wang N, Jiang Qet al. . Identification of natural compound radicicol as a potent FTO inhibitor. Mol Pharmaceutics. 2018;15(9):4092–98. [DOI] [PubMed] [Google Scholar]
- 82. Barros Silva Soares de Souza EP, Faria RX, Rocha LM. Clinical trials studies of plant extracts with anti-inflammatory activity. J Appl Pharmaceut Sci. 2016;6:224–32. [Google Scholar]
- 83. Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–37. [DOI] [PubMed] [Google Scholar]
- 84. Cluntun AA, Huang H, Dai L, Liu X, Zhao Y, Locasale JW. The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab. 2015;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wellen KE, Snyder NW. Should we consider subcellular compartmentalization of metabolites, and if so, how do we measure them? Curr Opin Clin Nutr Metab Care. 2019;22(5):347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura Met al. . ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008;68(20):8547–54. [DOI] [PubMed] [Google Scholar]
- 87. Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AKet al. . Acetate dependence of tumors. Cell. 2014;159(7):1591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam ISet al. . Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27(1):57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sun RC, Dukhande VV, Zhou Z, Young LEA, Emanuelle S, Brainson CFet al. . Nuclear glycogenolysis modulates histone acetylation in human non-small cell lung cancers. Cell Metab. 2019;30(5):903–16, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Carrer A, Parris JLD, Trefely S, Henry RA, Montgomery DC, Torres Aet al. . Impact of a high-fat diet on tissue Acyl-CoA and histone acetylation levels. J Biol Chem. 2017;292:3312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Luong A, Hannah VC, Brown MS, Goldstein JL. Molecular characterization of human acetyl-CoA synthetase, an enzyme regulated by sterol regulatory element-binding proteins. J Biol Chem. 2000;275(34):26458–66. [DOI] [PubMed] [Google Scholar]
- 92. Sato R, Okamoto A, Inoue J, Miyamoto W, Sakai Y, Emoto Net al. . Transcriptional regulation of the ATP citrate-lyase gene by sterol regulatory element-binding proteins. J Biol Chem. 2000;275(17):12497–502. [DOI] [PubMed] [Google Scholar]
- 93. Vernieri C, Casola S, Foiani M, Pietrantonio F, de Braud F, Longo V. Targeting cancer metabolism: dietary and pharmacological interventions. Cancer Discov. 2016;6(12):1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Montesdeoca N, López M, Ariza X, Herrero L, Makowski K. Inhibitors of lipogenic enzymes as a potential therapy against cancer. FASEB J. 2020;34(9):11355–81. [DOI] [PubMed] [Google Scholar]
- 95. Anderson KA, Madsen AS, Olsen CA, Hirschey MD. Metabolic control by sirtuins and other enzymes that sense NAD+, NADH, or their ratio. Biochim Biophys Acta Bioenergetics. 2017;1858(12):991–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Camarero N, Nadal A, Barrero MJ, Haro D, Marrero PF. Histone deacetylase inhibitors stimulate mitochondrial HMG-CoA synthase gene expression via a promoter proximal Sp1 site. Nucleic Acids Res. 2003;31(6):1693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJet al. . The colonic crypt protects stem cells from microbiota-derived metabolites. Cell. 2016;165:1708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chen J, Zhao KN, Vitetta L. Effects of intestinal microbial-elaborated butyrate on oncogenic signaling pathways. Nutrients. 2019;11:(5):1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Allen BG, Bhatia SK, Anderson CM, Eichenberger-Gilmore JM, Sibenaller ZA, Mapuskar KAet al. . Ketogenic diets as an adjuvant cancer therapy: history and potential mechanism. Redox Biol. 2014;2:963–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Weber DD, Aminazdeh-Gohari S, Kofler B. Ketogenic diet in cancer therapy. Aging. 2018;10:(2):164–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Latham T, MacKay L, Sproul D, Karim M, Culley J, Harrison DJet al. . Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 2012;40(11):4794–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Yet al. . Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Hayashi T, Teruya T, Chaleckis R, Morigasaki S, Yanagida M. S-adenosylmethionine synthetase is required for cell growth, maintenance of G0 phase, and termination of quiescence in fission yeast. Iscience. 2018;5:38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kera Y, Katoh Y, Ohta M, Matsumoto M, Takano-Yamamoto T, Igarashi K. Methionine adenosyltransferase II-dependent histone H3K9 methylation at the COX-2 gene locus. J Biol Chem. 2013;288(19):13592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Katoh Y, Ikura T, Hoshikawa Y, Tashiro S, Ito T, Ohta Met al. . Methionine adenosyltransferase II serves as a transcriptional corepressor of MAF oncoprotein. Mol Cell. 2011;41(5):554–66. [DOI] [PubMed] [Google Scholar]
- 106. Lyon P, Strippoli V, Fang B, Cimmino L. B vitamins and one-carbon metabolism: implications in human health and disease. Nutrients. 2020;12(9):1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Christensen BC, Kelsey KT, Zheng S, Andres Houseman E, Marsit CJ, Wrensch MRet al. . Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 2010;6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Pufulete M, Al-Ghnaniem R, Leather AJM, Appleby P, Gout S, Terry Cet al. . Folate status, genomic DNA hypomethylation, and risk of colorectal adenoma and cancer: a case control study. Gastroenterology. 2003;124(5):1240–8. [DOI] [PubMed] [Google Scholar]
- 109. Cuthbertson CR, Arabzada Z, Bankhead A, Kyani A, Neamati N. A review of small-molecule inhibitors of one-carbon enzymes: SHMT2 and MTHFD2 in the spotlight. ACS Pharmacol Translat Sci. 2021;4(2):624–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks Det al. . Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 2015;22(5):861–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Dai Z, Mentch SJ, Gao X, Nichenametla SN, Locasale JW. Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width. Nat Commun. 2018;9(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Maddocks ODK, Labuschagne CF, Adams PD, Vousden KH. Serine metabolism supports the methionine cycle and DNA/RNA methylation through de novo ATP synthesis in cancer cells. Mol Cell. 2016;61(2):210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Maddocks ODK, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb Eet al. . Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature. 2013;493(7433):542–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Losman JA, Koivunen P, Kaelin WG. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat Rev Cancer. 2020;20(12):710–26. [DOI] [PubMed] [Google Scholar]
- 115. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SHet al. . Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi Cet al. . SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23(6):739–52. [DOI] [PubMed] [Google Scholar]
- 117. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu Het al. . Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Tran TQ, Lowman XH, Kong M. Molecular pathways: metabolic control of histone methylation and gene expression in cancer. Clin Cancer Res. 2017;23(15):4004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu Xet al. . Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol. 2016;18(10)1090–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, Mahapatra Set al. . Vitamin C induces TET-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature. 2013;500(7461):222–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wang T, Chen K, Zeng X, Yang J, Wu Y, Shi Xet al. . The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell. 2011;9(6):575–87. [DOI] [PubMed] [Google Scholar]
- 122. Camarena V, Wang G. The epigenetic role of vitamin C in health and disease. Cell Mol Life Sci. 2016;73(8):1645–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Liu M, Ohtani H, Zhou W, Ørskov AD, Charlet J, Zhang YWet al. . Vitamin C increases viral mimicry induced by 5-aza-2’-deoxycytidine. Proc Natl Acad Sci. 2016;113(37):10238–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Mayland CR, Bennett MI, Allan K. Vitamin C deficiency in cancer patients. Palliat Med. 2005;19:17–20. [DOI] [PubMed] [Google Scholar]
- 125. Kroeze LI, van der Reijden BA, Jansen JH. 5-Hydroxymethylcytosine: an epigenetic mark frequently deregulated in cancer. Biochimica Biophys Acta Rev Cancer. 2015;1855(2):144–54. [DOI] [PubMed] [Google Scholar]
- 126. Mingay M, Chaturvedi A, Bilenky M, Cao Q, Jackson L, Hui Tet al. . Vitamin C-induced epigenomic remodelling in IDH1 mutant acute myeloid leukaemia. Leukemia. 2018;32:11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Gillberg L, Ørskov AD, Nasif A, Ohtani H, Madaj Z, Hansen JWet al. . Oral vitamin C supplementation to patients with myeloid cancer on azacitidine treatment: normalization of plasma vitamin c induces epigenetic changes. Clin Epigenet. 2019;11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]