

Significance Statement
Recent studies support a critical role of epigenetics in cyst-associated gene expression and the progression of autosomal dominant polycystic kidney disease (ADPKD), although the underlying molecular mechanisms remain elusive. Here, we show that expression of CDYL, a chromodomain Y-like transcription repressor and crotonyl-CoA hydratase, is suppressed in ADPKD kidneys, accompanied by an increase of histone lysine crotonylation (Kcr). Interestingly, CDYL can form biomolecular condensates, which enhance its catalytic activity on histone Kcr. Through a phase separation–mediated mechanism, overexpression of CDYL reduces histone Kcr, inhibits the expression of cyst-associated genes, and slows cyst growth. Thus, our study establishes a prominent role for CDYL nuclear condensation in regulating histone Kcr, the cyst-associated gene expression program, and ADPKD progression.
Keywords: CDYL, histone crotonylation, phase separation, ADPKD
Visual Abstract

Abstract
Background
Emerging evidence indicates that epigenetic modulation of gene expression plays a key role in the progression of autosomal dominant polycystic kidney disease (ADPKD). However, the molecular basis for how the altered epigenome modulates transcriptional responses, and thereby disease progression in ADPKD, remains largely unknown.
Methods
Kidneys from control and ADPKD mice were examined for the expression of CDYL and histone acylations. CDYL expression and its correlation with disease severity were analyzed in a cohort of patients with ADPKD. Cdyl transgenic mice were crossed with Pkd1 knockout mice to explore CDYL’s role in ADPKD progression. Integrated cistromic and transcriptomic analyses were performed to identify direct CDYL target genes. High-sensitivity mass spectrometry analyses were undertaken to characterize CDYL-regulated histone lysine crotonylations (Kcr). Biochemical analysis and zebrafish models were used for investigating CDYL phase separation.
Results
CDYL was downregulated in ADPKD kidneys, accompanied by an increase of histone Kcr. Genetic overexpression of Cdyl reduced histone Kcr and slowed cyst growth. We identified CDYL-regulated cyst-associated genes, whose downregulation depended on CDYL-mediated suppression of histone Kcr. CDYL assembled nuclear condensates through liquid-liquid phase separation in cultured kidney epithelial cells and in normal kidney tissues. The phase-separating capacity of CDYL was required for efficient suppression of locus-specific histone Kcr, of expression of its target genes, and of cyst growth.
Conclusions
These results elucidate a mechanism by which CDYL nuclear condensation links histone Kcr to transcriptional responses and cystogenesis in ADPKD.
Autosomal dominant polycystic kidney disease (ADPKD) is the most common genetic kidney disease, affecting over 12 million people worldwide. The main clinical manifestation is continuous enlargement of bilateral renal cysts, which leads to a progressive decline of renal function. ADPKD is mainly caused by genetic mutations in the PKD1 or PKD2 genes, which respectively encode polycystin-1 (PC1) or polycystin-2 (PC2).1,2 Defects in these two proteins dysregulate various signaling pathways, leading to activation of a pathologic gene expression program.3,4 Recent studies support a key role for epigenetic changes, such as DNA methylation and histone modifications, in regulating cyst-promoting gene expression.5,6 However, the molecular basis for how the altered epigenome modulates transcriptional responses, and thereby disease progression in ADPKD, remains largely unknown.
Recently, a panel of novel histone post-translational modifications (PTMs) were discovered through high-sensitivity mass spectrometry analysis, and most of them are classified as short-chain lysine acylations, such as crotonylation, propionylation, butyrylation, succinylation, malonylation, lactylation, and β-hydroxybutyrylation.7–11 Although all occurring on the ε-amine group of lysine residues, these newly identified modifications differ from the well-studied lysine acetylation (Kac) in hydrocarbon chain length, charge, or hydrophobicity.12–14 For example, histone lysine crotonylation (Kcr), one of the first-discovered nonacetyl histone acylations, contains an extended hydrocarbon chain and increased rigidity that may lead to enhanced cis effects on the chromatin fiber and gene activation compared with histone Kac.15,16 Histone Kcr exists on a number of core and linker histone lysine residues and is especially abundant at active genomic loci including promoters and enhancers.17,18 This locus-specific histone Kcr is associated with various physiologic and pathologic processes, such as germ cell and endoderm differentiation, acute kidney injury, HIV latency, and cardiac hypertrophy.7,19–22 The level of histone Kcr is largely determined by localized nuclear concentration of crotonyl-CoA, which serves as the acyl group donor for the crotonylation reaction.15,18 CDYL, a chromodomain Y-like transcription corepressor, can recognize histone H3K9 and H3K27 methylation marks and bind to chromatin through its chromodomain.23–26 We have shown previously that CDYL acts as a nuclear crotonyl-CoA hydratase to convert crotonyl-CoA to β-hydroxybutyryl-CoA, and thereby negatively regulates locus-specific histone Kcr.27 The downregulation of histone Kcr by CDYL is intrinsically linked to its transcription repression activity and is functionally implicated in spermatogenesis and stress-induced depressive behaviors in mammals.27,28
Like many other enzymes, the hydratase activity of CDYL relies on its interaction with substrate crotonyl-CoA.27 A common mechanism to increase the rate of enzymatic activity is through liquid-liquid phase separation, which can generate reaction crucibles to compartmentalize and concentrate enzymes and their substrates.29–32 In this study, we found that CDYL formed biomolecular condensates through its intrinsically disordered region (IDR) to enhance its catalytic activity on histone Kcr. We showed that the expression of CDYL was suppressed in ADPKD kidneys, accompanied by an increase of histone Kcr. Overexpression of CDYL reduced histone Kcr, inhibited the expression of cyst-associated genes, and slowed cyst growth through a phase separation–mediated mechanism.
Methods
Human Kidney Specimens
ADPKD kidneys were obtained from patients with nephrectomy or cyst reduction surgery. All patients provided informed consent. Human studies were approved by the Ethics Committee of Shandong Provincial Hospital.
Cell Culture
WT 9-12 cells (Dolichos biflorus agglutinin–positive and immortalized with SV40 virus) were cultured in DMEM/F-12 medium supplemented with 10% FBS and maintained in a 37°C incubator with 5% CO2 as described.33 These cells possess a nucleotide substitution (7877C-T) in PKD1 gene that results in a nonsense mutation (Q2556X).34 The mutation was confirmed by sequencing (Supplemental Figure 1).
Generation of CDYL OE Cell Line
To generate the CDYL OE cell line, CDYL with a TY1 tag was inserted into the pITA vector and transfected into WT 9-12 cells. A stably integrated clone was picked after puromycin selection.
Mice and Drug Treatment
Cdyl transgenic (TG) C57BL/6× CBA mice were described previously.27 Pkd1fl/fl;Cre/Esr1+ C57BL/6 mice were also described previously.35 Pkd1fl/fl;Cre/Esr1+;Cdyl TG mice were generated by crossing Pkd1fl/fl;Cre/Esr1+ mice with Cdyl TG mice and backcrossed to the C57BL/6 background for more than 10 generations. Tamoxifen was injected at postnatal day 10 (P10) (10 mg/kg) to generate the early-onset mouse model. For the late-onset model, tamoxifen was administered at P25 and P28 (250 mg/kg). Cyst area and total area measurement were visualized by hematoxylin and eosin–stained kidney sections and then evaluated by ImageJ software. The cystic index was calculated as the ratio of cyst area to total area. BUN was measured using the QuantiChrom Urea Assay Kit (DIUR; BioAssay Systems). All mouse studies were approved by the Ethical Committee of Tianjin Medical University (permit number SYXK: 2016-0012).
Immunoblotting Analysis
Kidney tissues and WT 9-12 cells were lysed in RIPA buffer with protease inhibitor cocktail and phosphatase inhibitor cocktail. Antibodies were anti-PanKcr (PTM-501; PTM Biolabs), anti-PanKac (PTM-105; PTM Biolabs), anti-PanKpr (PTM-201; PTM Biolabs), anti-PanKbu (PTM-301; PTM Biolabs), anti-PanKbhb (PTM-1201; PTM Biolabs), anti-PanLac (PTM-1401; PTM Biolabs), anti-PanKsu (PTM-401; PTM Biolabs), anti-H3 (ab1791; Abcam), anti-CDYL27 (peptides antigen: IDDRRDQPFDKRLRFSV), anti-H3K18cr (PTM-517; PTM Biolabs), anti-H4K5cr (PTM-521; PTM Biolabs), anti-cherry (26765-1-AP; Proteintech), anti-TY1 (SAB4800032; Sigma), and anti–α-tubulin (10449-1-AP; Proteintech).
Immunofluorescence Staining
Cells were seeded on glass coverslips in six-well plates, fixed with 4% paraformaldehyde for 10 minutes, blocked in 5% BSA for 1 hour, and incubated with the indicated antibodies (anti-TY1 and anti-H3K18cr) overnight at 4°C. After washing three times, cells were incubated with Alexa Fluor 555- or 488-conjugated secondary antibodies for 1 hour at room temperature (RT). The slides were imaged with a fluorescence microscope (DMi8; Leica).
To detect CDYL expression in kidney tissues, kidney specimens were incubated with anti-CDYL antibody in the 5% BSA solution overnight at 4°C. Alexa Fluor 555-conjugated secondary antibody (Thermo Fisher Scientific) was applied after primary antibody incubation. Immunofluorescence slides were examined using a fluorescence microscope (DMi8; Leica) or confocal microscope (LSM 800; Zeiss). For living cells, WT 9-12 cells were grown on 35-mm Nunc glass bottom dishes and transfected with 2 μg of Cherry-CDYL. Immunofluorescence was detected using a fluorescence microscope (DMi8; Leica) or confocal microscope (LSM 800; Zeiss) 24 hours after transfection.
Immunohistochemistry
The human kidney tissues were perfused with 10% formalin overnight and then paraffin-embedded. The kidney sections were blocked with 5% BSA for 1 hour. Tissue sections were incubated with the primary anti-CDYL antibody overnight. Sections were incubated with anti-rabbit HRP secondary antibody at RT for 1 hour. Immunostained sections were evaluated using the H-score method. The staining intensity was graded as low (score one), moderate (score two), or strong (score three). The H-score was assigned by multiplying the percentage of positive cells and immunostaining intensity.
CRISPR-Cas9–Mediated Gene Knockout of CDYL
To generate stable cells expressing Cas9, HEK293T cells were cotransfected with the packaging plasmids (PMD2G and PSPAX2) and lentiCas9-Blast (#52962; Addgene). After transfection for 6–8 hours, the medium was replaced with fresh medium. The lentiviral particles were harvested from HEK293T cells after 48 hours. WT 9-12 cells were then infected with lentiviral particles for 12 hours, and then the medium was replaced with fresh medium for 48 hours. Stably transduced WT 9-12 cells were selected by blasticidin. CDYL knockout (KO) cells were generated by cloning single guide RNAs (sgRNAs) into the sgRNA cloning vector lentiGuide-Puro (#52963; Addgene). Stable WT 9-12 cells expressing Cas9 were infected and then selected with puromycin. The expression of CDYL was detected via immunoblotting analysis. The sgRNA sequences used were as follows: sgRNA forward, 5′-GAATAACTCACTAAATCCAG-3′; and sgRNA reverse, 5′-CTGGATTTAGTGAGTTATTC-3′.
Quantification of Histone PTMs
Histones were acid-extracted with 0.2 mM H2SO4 for 8 hours, followed by precipitation with 25% trichloroacetic acid for 0.5 hours. Purified histones were derivatized with propionic anhydride twice, and then 10 μl fresh propionylation reagent (propionic anhydride/acetonitrile 1:3 [v/v]) was added to 40 μl of the histone samples. NH4OH was further added to the solution to adjust the pH to 8.0. Then, samples were incubated at 37°C for 15 minutes. The histone proteins were obtained by drying out. Next, histones were dissolved in 50 mM NH4HCO3 followed by tryptic digestion (histone/enzyme is 20:1) at 37°C for 16 hours. After digestion, samples were resuspended in 40 μl of 100 mM NH4HCO3 and derivatized twice as described before to derivatize the peptide N terminus.36 The resulting peptides were dissolved in buffer A (0.1% formic acid), injected into a nano–liquid chromatography system (EASY-nLC 1000; Thermo Fisher Scientific), and subjected to separation by reversed-phase chromatography with a 70-minute gradient elution. The gradient comprised an increasing gradient (from 5% to 35%) of HPLC buffer B (0.1% formic acid in acetonitrile) in 50 minutes and then to 80% in 10 minutes, and finally to 90% in 10 minutes. The HPLC eluate was electrosprayed directly into an Orbitrap Q-Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA). A full-scan mass spectrometry spectrum (m/z 300–1100) was acquired. The entire mass range was fragmented at every cycle using windows of 50 m/z. For histone PTMs, data were acquired using data-independent acquisition mode. The relative abundance of each peptide was calculated by the software EpiProfile 2.0 with normalization of the total histone, referred to in previous reports.36,37 The proteomics data were submitted to the ProteomeXchange Consortium via the iProX partner repository with the dataset identifier PXD031029.
Chromatin Immunoprecipitation Sequencing
WT 9-12 cells were fixed with 1% formaldehyde at RT for 10 minutes. Crosslinking was stopped by adding 150 mM of glycine for 5 minutes at RT. Cells were washed with PBS twice and collected using chromatin immunoprecipitation (ChIP) cell lysis buffer (10mM EDTA, 1% SDS, and 50 mM Tris-HCl pH 8.0). Cells were fragmented by sonication (range 200–500 bp). Immunoprecipitation was performed using 2 μg of TY1, PanKcr, or H3K18cr antibody. After elution and reversed crosslinking, samples were treated with RNase A for 30 minutes at 37°C. For ChIP sequencing (ChIP-seq), DNA was purified and sequenced on the Illumina NovaSeq platform. Quality control was performed by FastQC v0.11.9 software. Clean reads were aligned to the human reference genome (hg38) by bowtie2 v2.3.5.1. ChIP-seq peaks were detected by the peak-finding algorithm MACS2 v2.2.7.1. Bigwig files were derived from the output of deepTools v3.4.3. The total signal of ChIP-seq was calculated as reads per million with 10-bp resolution. Annotation of peaks was performed using HOMER. Genomic data were deposited in the Gene Expression Omnibus (GSE183512).
RNA Sequencing and Real-Time Quantitative PCR
Total RNA was isolated from kidney tissues or WT 9-12 cells using TRIzol (15596018; Invitrogen). RNA sequencing (RNA-seq) was performed on the Illumina NovaSeq platform. RNA-seq reads were aligned to the human reference genome (hg38) by HISAT2 v2.1.0. Mapped reads were counted by featureCounts v1.6.0. Differentially expressed genes were calculated by DESeq2. Fold change of less than or equal to −2 or ≥2 and adjusted P value ≤0.05 were considered as significant differential expression. Sequencing data were deposited in the Gene Expression Omnibus (GSE183512). For real-time quantitative PCR (RT-qPCR), 2 μg of RNA was reverse transcribed to cDNA using a cDNA Synthesis Kit (Roche). RT-qPCR was performed with gene-specific primers (Supplemental Table 1).
Protein Expression and Purification
Plasmids containing EGFP- and GST-tagged genes were transformed into Escherichia coli strain BL21. After induction with isopropyl-β-d-thiogalactoside, cells were harvested and then resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 1% Triton X-100, and 100 mM NaCl) containing protease inhibitors. Following sonication and centrifugation, the supernatant was added to glutathione agarose and spun overnight at 4°C. HRV 3C protease (88946; Pierce) and cleavage buffer (50 mM Tris, pH 7.5, and 150 mM NaCl) were added to remove GST tags. Proteins were examined by Coomassie blue staining. Purified proteins were loaded onto a glass slide with PEG 8000, and imaged using an inverted fluorescence microscope (DMi8; Leica).
Fluorescence Recovery after Photobleaching
For purified proteins, droplets of proteins were bleached with a confocal microscope (LSM 800; Zeiss) using a 488-nM laser with a 63×/1.4 oil objective. After droplets were bleached of 80%–90% of the fluorescence, recovery was monitored on a confocal laser scanning microscope every 10 seconds for the indicated periods. For living cells, WT 9-12 cells were grown on 35-mm Nunc glass bottom dishes, transfected with 2 μg of Cherry-CDYL, and analyzed by quantitative fluorescence recovery after photobleaching (FRAP) assays 24 hours after transfection.
In Vitro Kcr Assay
For in vitro enzymatic reactions, 5 mg of native calf thymus histone (CTH) and 1 mM crotonyl-CoA were incubated with various concentrations of recombinant wild-type (WT) and mutant CDYL proteins at 30°C for 1 hour in the reaction buffer (50 mM Tris, pH 8.0, 1 mM DTT, 0.2 mM PMSF, and 10% glycerol). The level of histone crotonylation was analyzed by Western blot.
Zebrafish Studies
Zebrafish experiments were approved by the Institutional Animal Care and Use Committee of Nankai University. Morpholinos (MOs) were obtained from Gene Tools. Sequences of MOs are as follows: a standard control, 5′-CCTCTTACCTCAGTTACAATTTATA-3′; cdyl, 5′-GGCCATCATGTCACGATAATCTTCG-3′; and pkd2, 5′-AGGACGAACGCGACTGGAGCTCATC-3′. For rescue experiments, 200 pg of human CDYL WT mRNA or CDYL mRNA were coinjected with pkd2 MO and cdyl MO into one-cell-stage embryos.
Statistical Analyses
Significance was calculated using the two-tailed unpaired t test to compare two groups of independent samples with GraphPad Prism Software. A P value <0.05 was considered statistically significant. Results are presented as mean±SEM for at least three independent experiments.
Results
Downregulation of CDYL Is Coupled with an Increase of Histone Kcr in ADPKD
To investigate whether histone acylations are involved in ADPKD pathogenesis, we first analyzed the abundance of histone acylations in kidneys from WT and ADPKD mice. We found that histone Kcr was markedly increased and histone β-hydroxybutyrylation (Kbhb) was decreased in ADPKD kidneys, whereas other histone acylations were slightly changed (Figure 1A, Supplemental Figure 2). Because histone acylation levels are tightly controlled by the availability of acyl-CoA, and CDYL can convert crotonyl-CoA to β-hydroxybutyryl-CoA, we hypothesized that the shift of Kcr increase and Kbhb decrease is collectively due to loss of CDYL. To verify this, we analyzed the expression of CDYL in ADPKD mouse kidneys. Consistent with our hypothesis, we found that CDYL protein decreased in kidneys from ADPKD mice compared with normal kidneys (Figure 1B, Supplemental Figure 2A). Immunofluorescence analyses revealed that CDYL expression was markedly decreased in cyst-lining epithelial cells in ADPKD kidneys (Figure 1C, Supplemental Figure 2, B and C). Importantly, we observed that the expression of CDYL was suppressed in kidneys from a cohort of ADPKD patients (Figure 1, D–G, Supplemental Table 2). In addition, we found that CDYL expression correlated positively with eGFR but inversely with serum creatinine and with BUN (Figure 1G), indicating that decreased CDYL expression correlates with increased disease severity. Taken together, these data demonstrate that, accompanied by an increase of histone Kcr, CDYL aberrantly decreased in mouse ADPKD kidneys and its downregulation correlated with disease progression in ADPKD patients.
Figure 1.
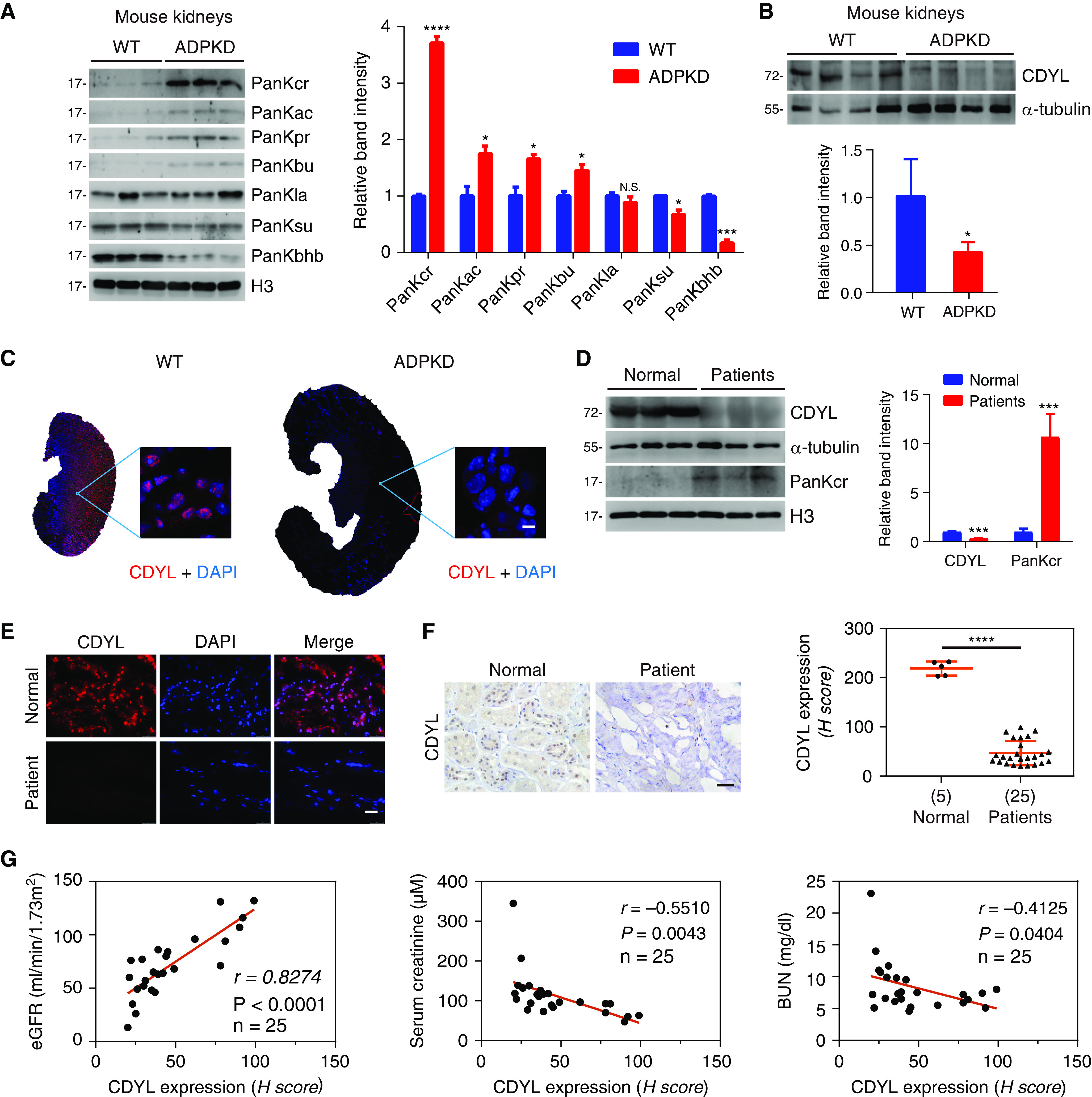
Downregulation of CDYL is accompanied by upregulation of histone Kcr in ADPKD. (A) Western blot analysis of histone lysine acylations in kidneys from WT and early-onset ADPKD mice at P29. Histones were extracted with acid. The antibodies are specific for each type of lysine acylation of protein. Mean data are normalized to H3. Data represent the mean±SEM of three replicates. *P<0.05, ***P<0.001, ****P<0.0001, N.S.: not significant. (B) Western blot analysis of CDYL expression in WT and ADPKD mouse kidneys. α-Tubulin is a protein loading control. Data represent the mean±SEM of five replicates. *P<0.05. (C) Immunofluorescence staining of CDYL in WT and ADPKD mouse kidneys. Scale bar, 5 μm. (D–F) Western blot analysis (D), immunofluorescence staining (E), and immunohistochemistry analysis (F) of CDYL in kidney tissues from healthy people and patients with ADPKD. Data are represented as mean±SEM and were analyzed by two-tailed unpaired t test. ***P<0.001. Scale bar, 25 μm in (E) and 100 μm in (F). Densitometric quantification of CDYL expression in (F) (right). Data are represented as mean±SEM and were analyzed by two-tailed unpaired t test. ****P<0.0001. (G) Correlation between CDYL expression and eGFR (left), serum creatinine (middle), and BUN (right) in ADPKD patients. Pearson’s correlation coefficients are displayed in (G). P values for (G) were determined using linear regression analysis.
Overexpression of CDYL Suppresses Histone Kcr and Ameliorates ADPKD Progression
To further explore the role of CDYL in the progression of ADPKD, we generated Pkd1fl/fl;Cre/Esr1+;Cdyl TG C57BL/6 mice by crossing Cdyl TG mice with Pkd1 KO mice. Pkd1 deletion was induced by injection of tamoxifen at P10 and the mice were euthanized 19 days later to collect the kidneys (Figure 2A, Supplemental Table 3). Consistent with previous studies,38,39 loss of Pkd1 resulted in substantial cyst growth and decreased renal function, as assessed by increases of kidney size, kidney weight/body weight ratio, BUN, and serum creatinine in Pkd1 KO mice compared with their WT counterparts (Pkd1−/− versus Pkd1+/+) (Figure 2, B–E, Supplemental Figure 3). We observed no apparent abnormalities in kidney size, kidney weight/body weight ratio, BUN, or serum creatinine in Cdyl TG mice with functional Pkd1 (Pkd1+/+;Cdyl TG versus Pkd1+/+), indicating that overexpression of CDYL alone has little effect on renal development and function. In contrast, overexpression of CDYL in Pkd1−/− mice (Pkd1−/−;Cdyl TG versus Pkd1−/−) inhibited the increase of kidney size, kidney weight/body weight ratio, BUN, serum creatinine, and cystic index caused by Pkd1 deletion (Figure 2, B–G). In addition, CDYL overexpression reversed the upregulation of histone Kcr in Pkd1−/− mice (Pkd1−/−;Cdyl TG versus Pkd1−/−) (Figure 2, G and Figure 2H). Collectively, these results indicate that CDYL overexpression reduces histone Kcr, inhibits cyst growth, and improves renal function in ADPKD.
Figure 2.
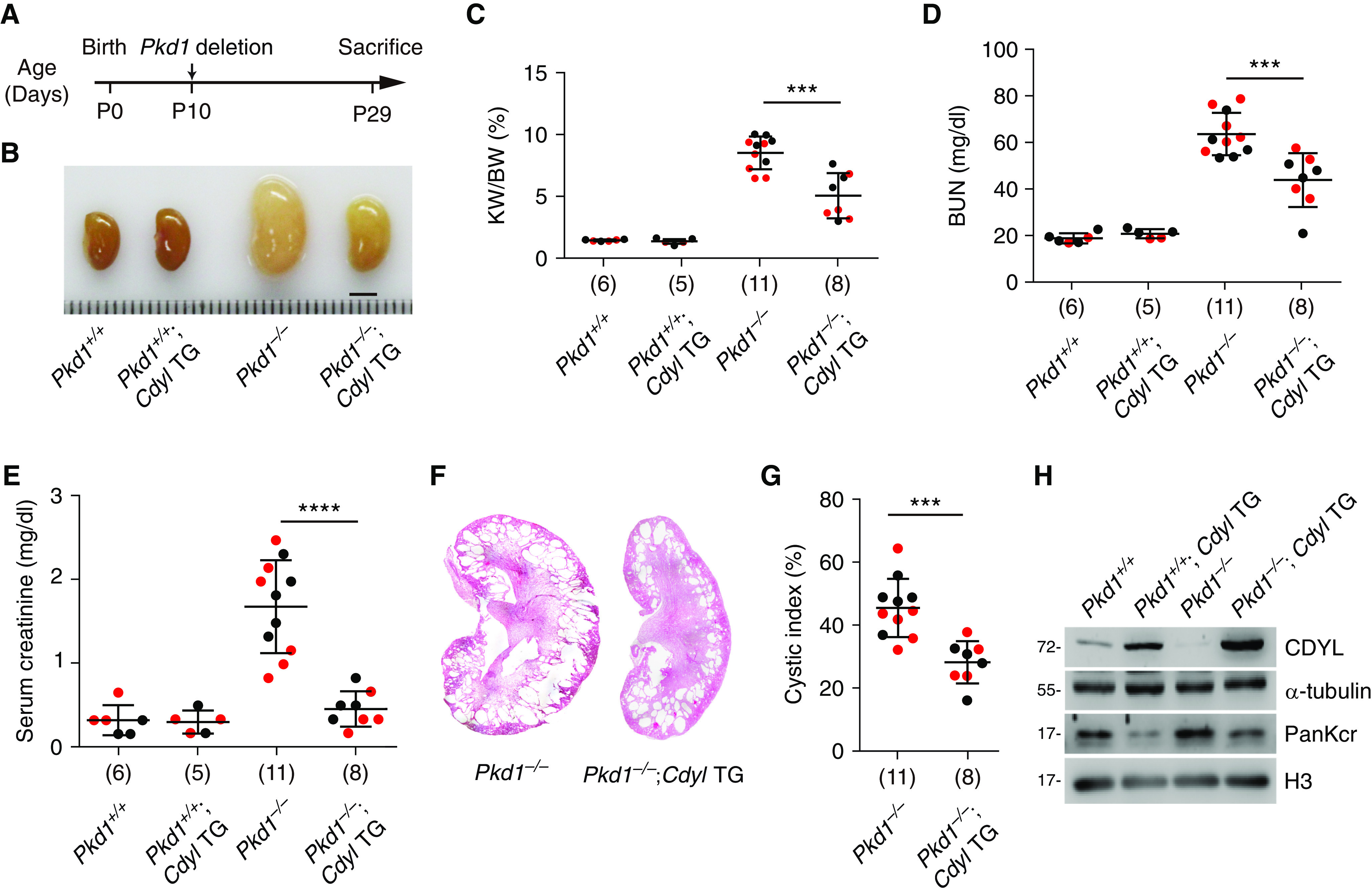
CDYL overexpression slows cyst growth in ADPKD mouse models. (A) Flow chart of Pkd1 deletion induction in Pkd1fl/fl;Cre/Esr1+;Cdyl TG mice. (B) Representative images of P29 kidneys from the indicated groups. Scale bar, 3 mm. (C) Kidney weight/body weight (KW/BW) ratios in the indicated groups. (D and E) BUN (D) and serum creatinine (E) concentrations in the indicated groups. (F) Hematoxylin and eosin (H&E) staining of kidney sections from the indicated groups. (G) Cystic index quantified from H&E images. Data from each mouse are plotted. The number of samples in each group is indicated in parentheses. Red dots represent males and black dots represent females (C–E and G). (H) Western blot analysis of CDYL and histone PanKcr expression level in each of the indicated groups. Data are represented as mean±SEM and were analyzed by two-tailed unpaired t test. ***P<0.001, ****P<0.0001.
Integrated Cistromic and Transcriptomic Analysis Identifies Direct CDYL Target Genes in ADPKD Cells
To investigate the molecular mechanisms underlying the suppression of cyst growth by CDYL overexpression, we generated an engineered ADPKD cell line by stably expressing CDYL with TY1 tag in WT 9-12 human ADPKD cells (CDYL OE). We profiled genome-wide distribution of CDYL through ChIP-seq analysis in both parental WT 9-12 cells (control) and CDYL OE cells. In contrast to an undetectable signal of TY1-CDYL in control cells, we detected 21,717 TY1-CDYL-binding regions in CDYL OE cells (Figure 3A). Genomic distribution analysis of these CDYL-binding regions revealed that 16.82% of them were located within gene promoters, whereas 27.25% and 45.73% were located in intronic and intergenic regions, respectively (Figure 3B). To examine the effects of CDYL-regulated transcriptional output, we performed gene expression analysis by RNA-seq in both control and CDYL OE cells. A total of 517 genes were upregulated and 1232 genes were downregulated in CDYL OE cells compared with control cells (Figure 3, C and D). Because CDYL is widely recognized as a transcriptional repressor, we thus focused on CDYL-downregulated genes in RNA-seq analysis and defined direct CDYL target genes as genes that were both bound by CDYL and repressed by CDYL overexpression. Integrative analysis of CDYL ChIP-seq and RNA-seq data revealed that 476 genes contained CDYL-binding regions and were downregulated in CDYL OE cells (Figure 3E). Gene ontology analysis of these genes showed that CDYL target genes were enriched in previously reported ADPKD-related pathways, such as cell junction organization, cell projection morphogenesis, negative regulation of cell differentiation, and regulation of epithelial cell proliferation (Figure 3F). Representative track files for ChIP-seq and RNA-seq are shown in Figure 3G. We further analyzed the expression of several CDYL direct target genes involved in cyst-associated pathways (Stat5a, Tlr4, and Sema5a)6,40–43 and one profibrotic gene (Col5a1).44,45 As shown in Figure 3H, these target genes were all upregulated upon loss of PKD1 and rescued by CDYL overexpression. Collectively, these results indicate that CDYL ameliorates ADPKD progression by repressing the expression of cyst-associated genes.
Figure 3.
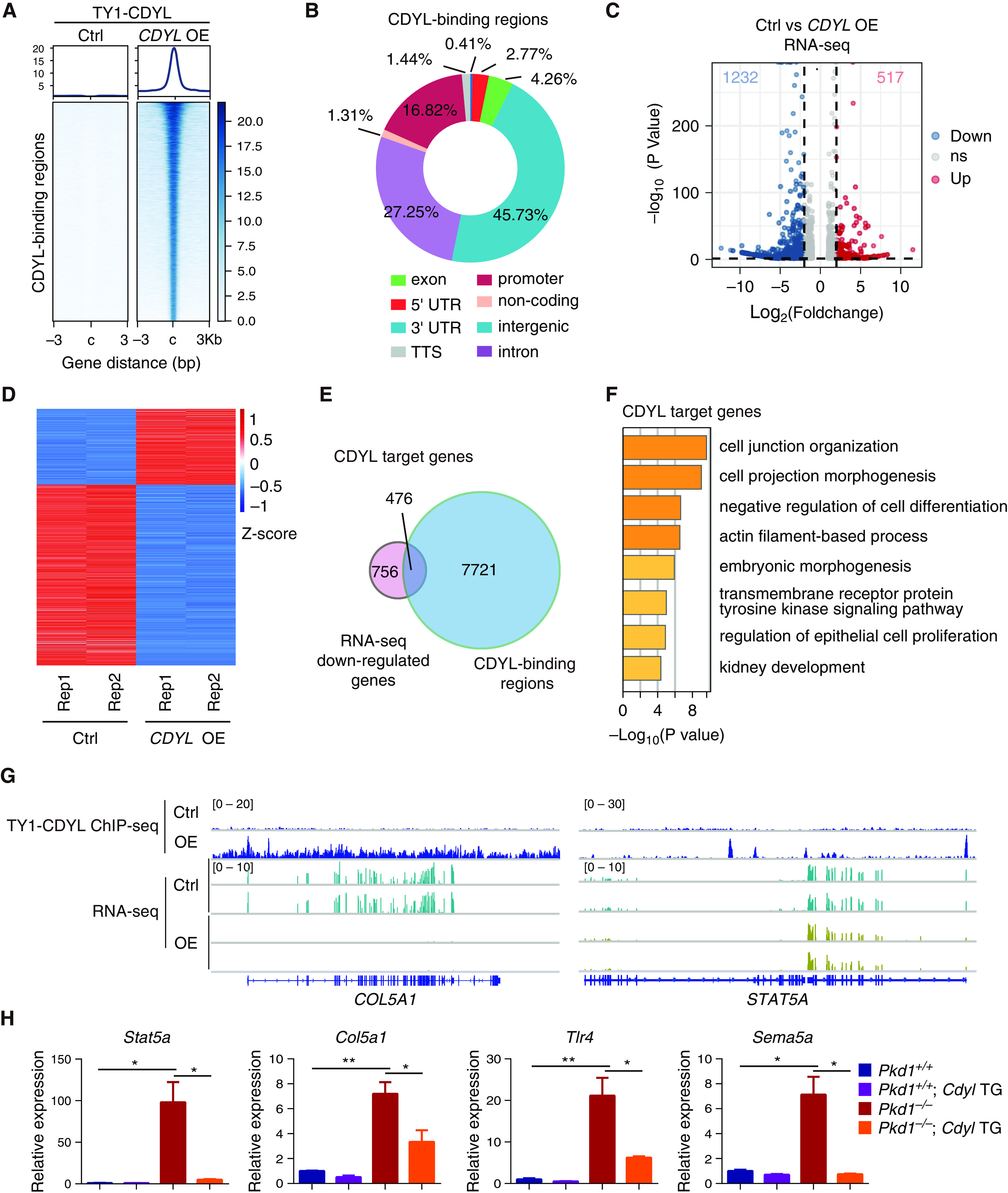
Integrated cistromic and transcriptomic analysis reveals cyst-promoting genes regulated by CDYL in ADPKD cells. (A) Heatmaps showing the occupancy of TY1-CDYL in control and CDYL overexpression (OE) WT 9-12 cells centered on peak summits ±3 kb. The ChIP-seq signal heatmap using a 6-kb window was centered on peak regions. (B) Genomic distribution of CDYL-binding regions in CDYL OE WT 9-12 cells (compared with control cells). (C) Volcano plot showing differentially expressed genes (control versus CDYL OE). (D) Heatmap of differentially expressed genes in control and CDYL OE WT 9-12 cells. (E) Venn diagram showing overlap of CDYL-binding regions and downregulated genes upon CDYL overexpression in CDYL OE WT 9-12 cells. (F) Gene ontology enrichment analysis of CDYL target genes. (G) Gene tracks showing TY1-CDYL ChIP-seq and RNA-seq profiles at the indicated gene loci. (H) RT-qPCR analysis of mRNA level of Stat5a, Col5a1, Tlr4, and Sema5a in each of the indicated groups. Data are represented as mean±SEM and were analyzed by two-sided unpaired t test. *P<0.05, **P<0.01.
CDYL-Mediated Histone Kcr Regulates Cyst-Associated Genes
We have previously shown that CDYL catalyzes the hydroxylation of crotonyl-CoA and thereby depletes the acyl group donor for histone Kcr.27 By neutralizing the positive charge of lysine and weakening the interactions between histones and DNA, histone Kcr contributes to a more open chromatin state and is generally associated with gene activation.15,16 We thus hypothesized that CDYL represses cyst-associated genes by reducing the histone Kcr on these genes. To test this, we examined histone Kcr on CDYL target genes by performing ChIP-seq analyses using an antibody recognizing PanKcr in control and CDYL OE cells. As shown in Figure 4A, CDYL overexpression decreased PanKcr signals on CDYL target genes in CDYL OE cells. Next, we divided CDYL target genes into three groups based on ChIP-seq signals: high, medium, and low (Figure 4B). The intensity of PanKcr ChIP-seq signals was inversely correlated with that of CDYL ChIP-seq signals (Figure 4C). Representative track files are shown in Figure 4D. Furthermore, integrative analysis of PanKcr ChIP-seq data and RNA-seq profiles of control and CDYL OE cells revealed that CDYL overexpression reduced histone Kcr on 351 of 476 direct CDYL target genes, and this CDYL-mediated decrease of histone Kcr positively correlated with the decrease of gene expression (Figure 4E). These results indicate that the enrichment of CDYL on its target genes correlates negatively with both histone Kcr and gene expression. Finally, we performed gene ontology analysis for the 351 CDYL target genes whose histone Kcr was sensitive to CDYL overexpression and found that they were also enriched in cystogenesis-associated pathways (Figure 4F). Thus, we identified cyst-associated genes controlled by CDYL-mediated histone Kcr.
Figure 4.
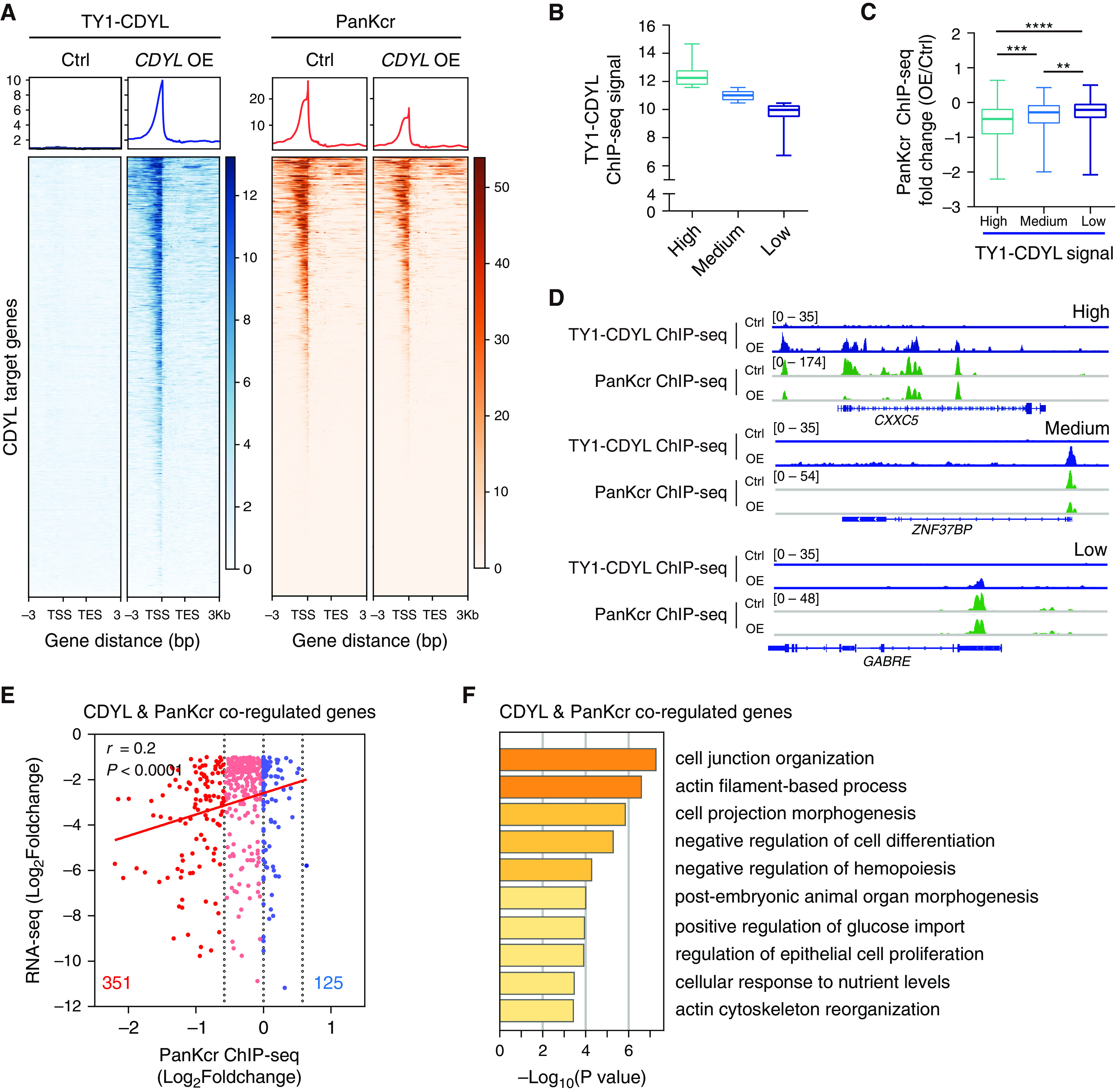
CDYL-mediated histone Kcr modulates the expression of CDYL target genes. (A) Heatmaps showing the occupancy of TY1-CDYL and PanKcr on CDYL target genes in control and CDYL OE WT 9-12 cells. (B and C) PanKcr ChIP-seq signal density in three gene clusters based on TY1-CDYL ChIP-seq signal density on CDYL target genes. (D) Representative integrative genomics viewer tracks showing PanKcr ChIP-seq signal on high, medium, and low TY1-CDYL-binding loci. (E) Pearson’s correlation coefficient of PanKcr ChIP-seq signals and gene expression in CDYL OE WT 9-12 cells. (F) Gene ontology analysis of CDYL and PanKcr coregulated genes.
H3K18 Is the Major Crotonylation Site Targeted by CDYL in ADPKD Cells
Using high-sensitivity mass spectrometry, Tan et al. identified histone Kcr on 28 lysine residues on various histones.7 Because these different modification sites may have different effects on gene expression, we decided to identify the Kcr sites on histones that are targeted by CDYL in ADPKD cells. Because the abundance of histone Kcr is generally low at the basal level,15,18 we chose to knock out CDYL in WT 9-12 cells (CDYL KO) through CRISPR/Cas9-mediated gene editing to increase the overall levels of histone Kcr. We quantified Kcr modifications on different histone residues in CDYL KO versus parental WT 9-12 cells (control) using HPLC tandem mass spectrometry (HPLC-MS/MS)-based proteomics analysis (Figure 5A). We found three lysine sites that were most significantly increased in CDYL KO cells compared with control cells, namely H3K18, H3K23, and H3K79 (Figure 5B). H3K18 crotonylation (H3K18cr) is the dominant site of acyltransferase p300-catalyzed histone crotonylation and is well characterized as an active histone mark for which an antibody is commercially available.17,18 We first validated the increase of H3K18cr in CDYL KO cells (Figure 5C). Consistently, we also observed a marked increase of H3K18cr in ADPKD kidneys compared with normal kidneys (Figure 5D, Supplemental Figure 4). As shown in Figure 5E, H3K18cr was substantially decreased in CDYL OE cells (CDYL OE versus control). Collectively, these findings demonstrate that H3K18cr is one of the major sites controlled by CDYL in ADPKD cells.
Figure 5.
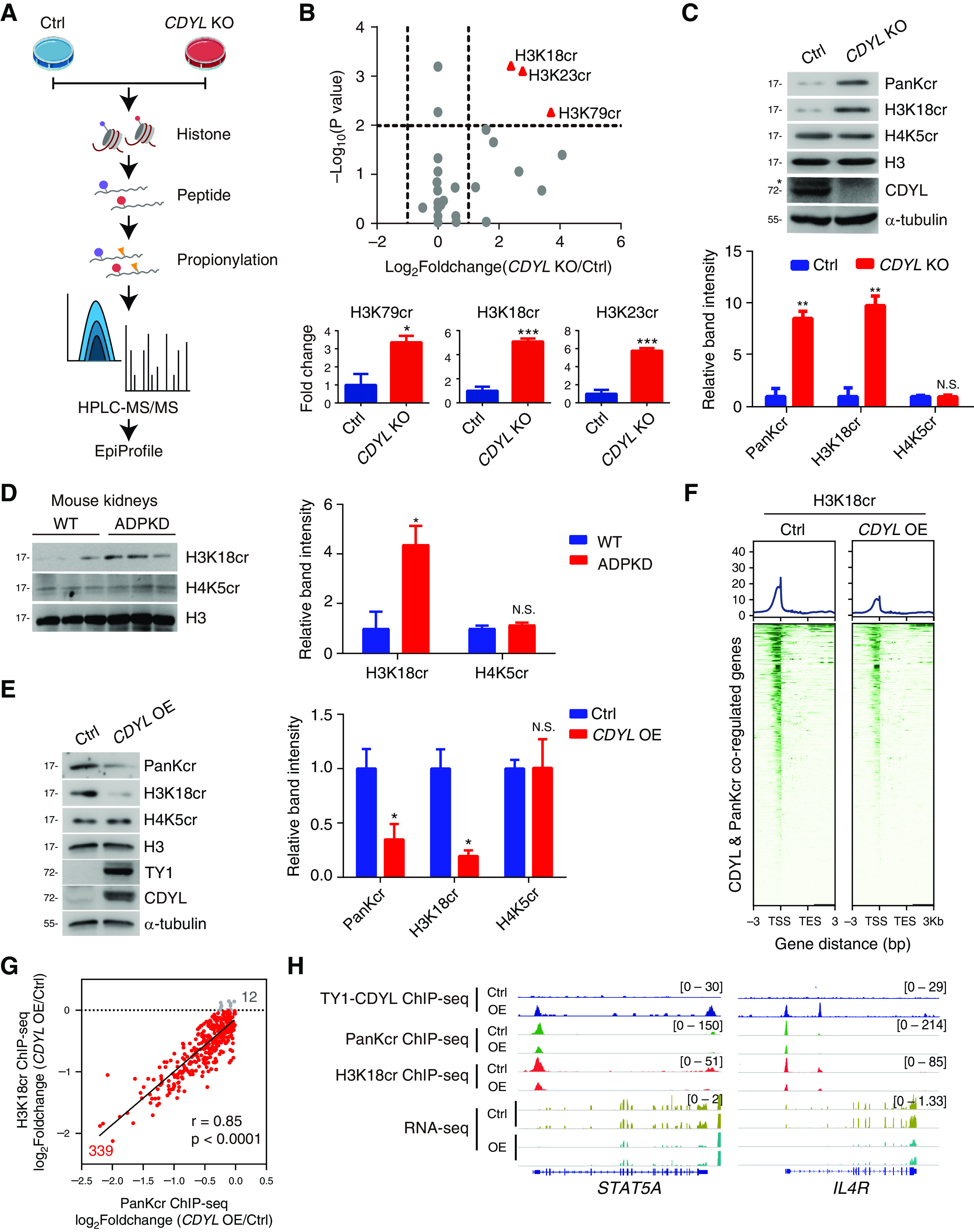
H3K18 is the major crotonylation site controlled by CDYL in ADPKD cells. (A) Experimental workflow of the HPLC-MS/MS assays to quantify histone Kcr in control and CDYL KO WT 9-12 cells. (B) Volcano plot showing histone crotonylation changes versus P values in CDYL OE WT 9-12 cells versus control cells comparisons (top). Bar graphs show relative histone crotonylation in control cells and CDYL OE WT 9-12 cells (bottom). *P<0.05, ***P<0.001. (C) Western blot analysis of histone Kcr in control and CDYL KO WT 9-12 cells (top). *Represents nonspecific band. Averaged histone Kcr levels are shown after normalization to the value for H3 (bottom). Two-tailed unpaired t test. **P<0.01, N.S., not significant. (D) Western blot analysis of histone Kcr in ADPKD mouse kidneys (left). Averaged histone Kcr levels are shown after normalization to the value for H3 (right). Two-tailed unpaired t test. *P<0.05, N.S.: not significant. (E) Western blot analysis of histone Kcr in CDYL OE WT 9-12 cells (left). Quantification of histone Kcr (right). Two-tailed unpaired t test. *P<0.05, N.S., not significant. Two-tailed unpaired t test. (F) Heatmaps showing the occupancy of H3K18cr binding on TY1-CDYL and PanKcr coregulated genes in control cells and CDYL OE WT 9-12 cells. (G) Pearson’s correlation coefficient of H3K18cr ChIP-seq signals and PanKcr ChIP-seq signals. (H) Gene tracks showing representative genes in control and CDYL OE WT 9-12 cells.
Next, we sought to determine whether CDYL repressed H3K18cr on CDYL target genes. To address this, we first explored the effect of CDYL overexpression on the genomic distribution of H3K18cr. We performed ChIP-seq analyses of H3K18cr in both CDYL OE cells and control cells. We found that CDYL overexpression caused a reduction of H3K18cr on the majority (339 of 351) of these CDYL target genes in CDYL OE cells (Figure 5F), and the extent of CDYL-mediated reduction of H3K18cr correlated with the repression of histone PanKcr (Figure 5G). The CDYL-mediated repression of histone PanKcr and H3K18cr on two representative genes is shown in Figure 5H. Collectively, these data indicate that CDYL-mediated reduction of H3K18cr accounts for the majority of the reduction of histone PanKcr on CDYL target genes.
CDYL Undergoes Phase Separation In Vitro and In Vivo
As seen in Figure 6, A and B, immunofluorescence analysis of CDYL showed evident punctate staining in renal cells and mouse kidneys. Further amino acid composition analysis revealed that CDYL contains a predicted IDR between amino acids 52 and 233 (Supplemental Figure 5A). IDR-containing proteins tend to form biomolecular condensates through a liquid-liquid phase separation mechanism, which is crucial for many cellular processes.32,46 By compartmentalizing and concentrating enzymes and their substrates, biomolecular condensates can facilitate enzymatic cascade reaction.29,30 Thus, we hypothesized that CDYL forms phase-separated condensates to promote the hydroxylation reaction on its substrate crotonyl-CoA. To test this, we first overexpressed mCherry-CDYL and performed FRAP assays in WT 9-12 cells. We found that mCherry-CDYL formed nuclear droplets when expressed in WT 9-12 cells and recovered rapidly after photobleaching (Figure 6C). In contrast, mCherry-CDYL with the IDR deleted was distributed diffusely in the nucleus (Supplemental Figure 5B). These data indicate that CDYL protein forms phase-separated condensates in ADPKD cells.
Figure 6.
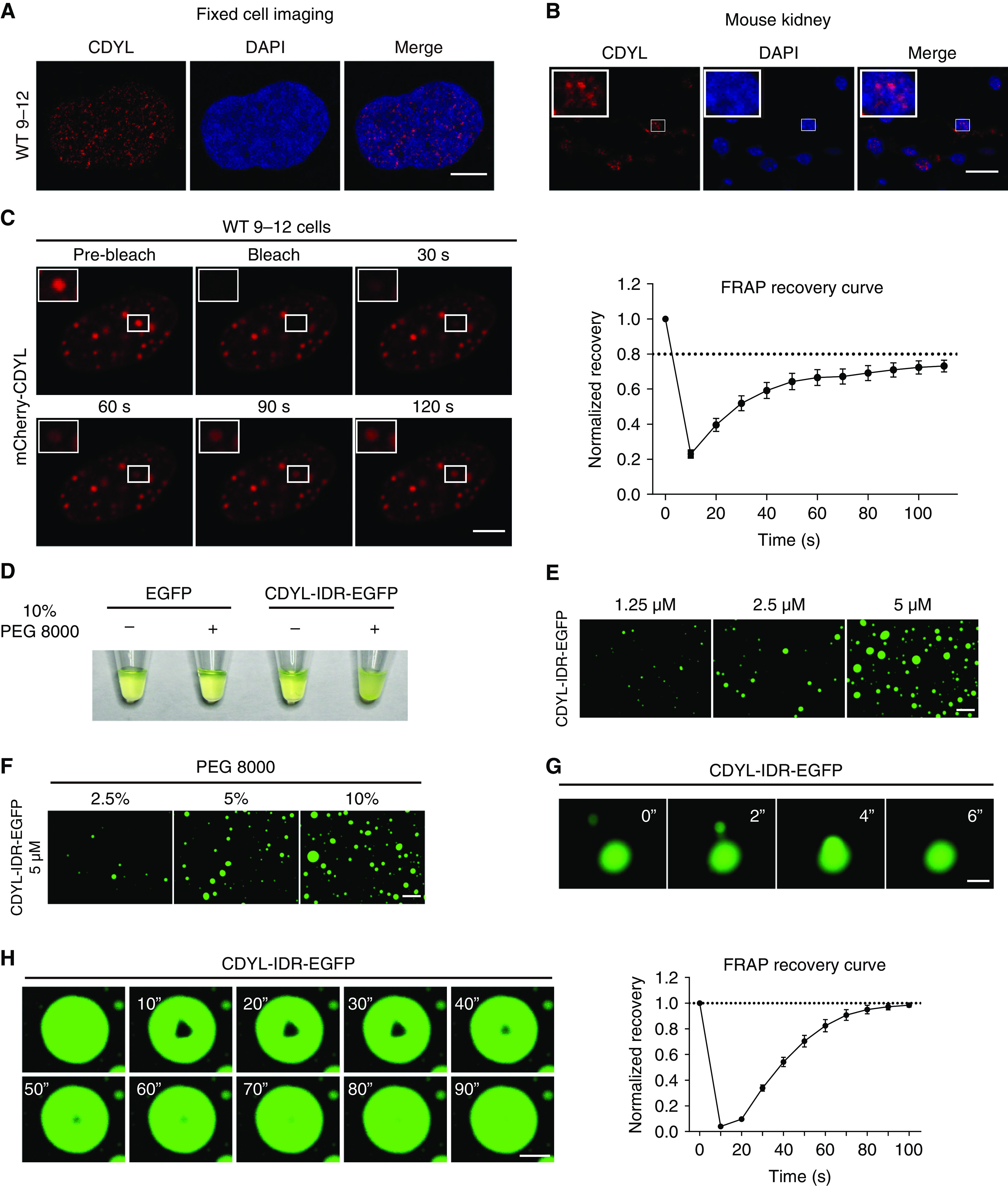
CDYL forms condensates through phase separation in vitro and in vivo. (A) Immunofluorescence imaging of CDYL in WT 9-12 cells. Scale bar, 5 μm. (B) Immunofluorescence imaging of CDYL in normal mouse kidney tissue. Scale bar, 10 μm. (C) FRAP assay of mCherry-CDYL droplets at the indicated time points. Scale bar, 5 μm. Normalized FRAP intensity curve of mCherry-CDYL droplets. Data represent mean±SEM (right, n=7). (D) Visualization of turbidity associated with protein droplet formation. Tubes containing EGFP and CDYL-IDR-EGFP in the absence (−) or presence (+) of PEG 8000 are shown. (E) Representative images of CDYL-IDR-EGFP droplet formation at the indicated concentrations. Scale bar, 50 μm. (F) Representative images of CDYL-IDR-EGFP droplet formation at the indicated PEG 8000 concentrations. Scale bar, 50 μm. (G) Fusion of CDYL-IDR-EGFP droplets over 6 seconds. Scale bar, 5 μm. (H) FRAP assay of CDYL-IDR-EGFP condensates. Scale bar, 5 μm. FRAP recovery curve of CDYL-IDR-EGFP condensates. Data represent mean±SEM (right, n=4).
To further confirm the phase separation capacity of CDYL protein, we purified recombinant CDYL-IDR-EGFP fusion proteins and investigated their phase separation capacity in vitro (Supplemental Figure 5C). Solutions containing 10% PEG 8000 (a crowding reagent) became turbid after we added purified CDYL-IDR-EGFP fusion proteins, whereas equivalent solutions remained clear when control EGFP proteins were added (Figure 6D). Furthermore, droplet formation assays revealed that CDYL-IDR-EGFP formed liquid droplets in a concentration-dependent manner (Figure 6E). Increasing the concentration of PEG 8000 in the buffer solution also led to larger liquid droplets (Figure 6F). In addition, CDYL-IDR-EGFP droplets exhibited rapid fusion behavior (within seconds) (Figure 6G) and rapid FRAP (Figure 6H). Collectively, these results indicate that CDYL-IDR-EGFP can form phase-separated condensates in vitro and in vivo.
Phase Separation Enhances the Catalytic Activity of CDYL on Histone Kcr
Recent studies have demonstrated that serine (S), phenylalanine (F), lysine (K), and arginine (R) residues are frequently implicated in regulating the phase separation capacity of proteins.47,48 To identify the amino acid residues contributing to the phase-separated property of CDYL, we mutated the above-mentioned residues in the IDR of CDYL to alanine (A) and investigated the resulting mutants’ ability to form phase-separated droplets in vivo (Supplemental Figure 6A). As shown in Figure 7A, similar to IDR deletion, the KR>A mutant exhibited diffuse distribution in cells, whereas the other mutants showed punctate staining similar to WT CDYL, indicating that lysine and arginine in CDYL-IDR are critical for condensate formation. To confirm that K and R residues in the IDR are essential for the phase separation of CDYL, we purified recombinant WT EGFP-CDYL and the KR>A mutant in vitro (Supplemental Figure 6B). In contrast to WT CDYL, the KR>A mutant remained clear when PEG 8000 was added to the solution (Figure 7B) and failed to form droplets (Figure 7C). These data indicate that K and R residues in the IDR are essential for CDYL phase separation.
Figure 7.
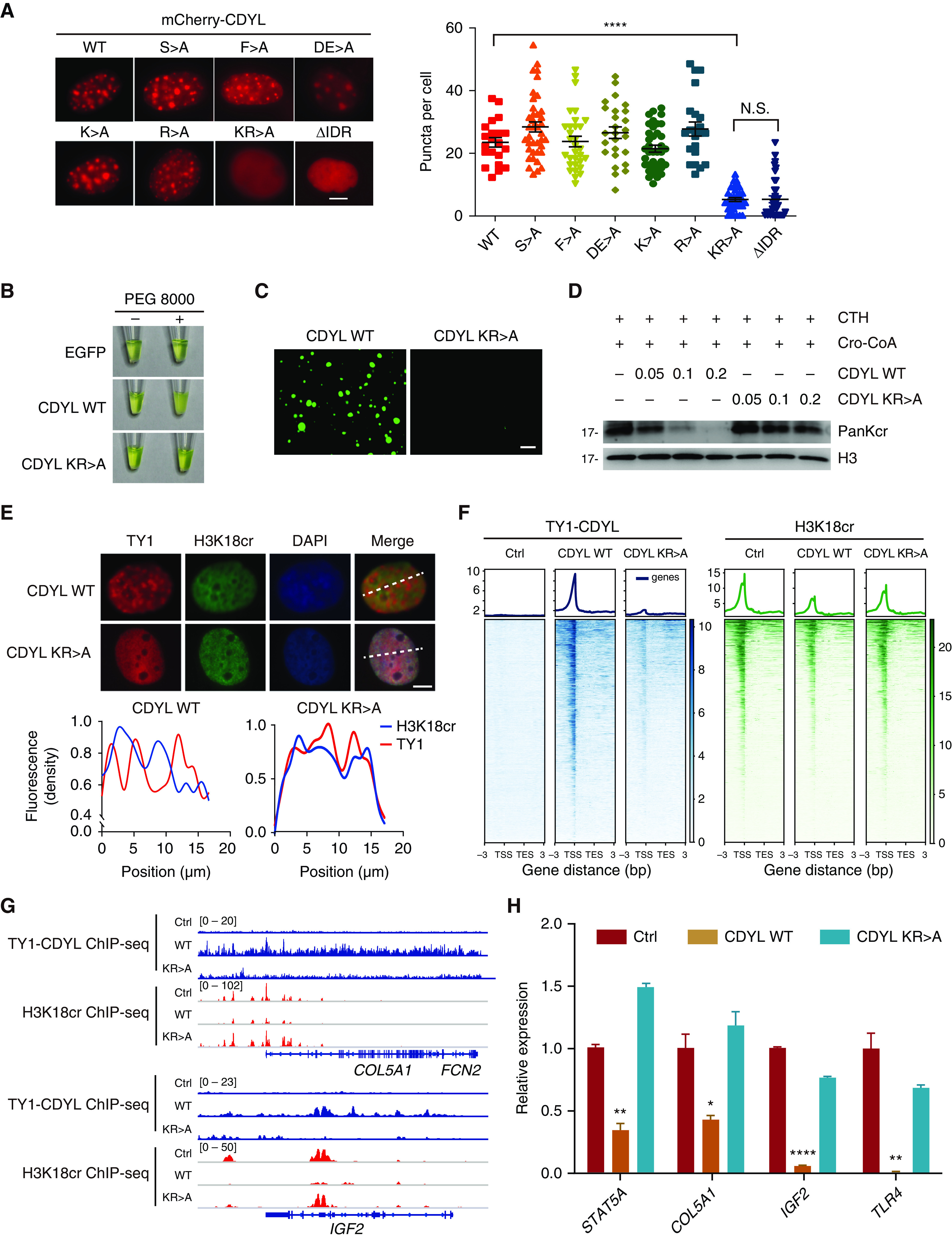
Phase separation increases the catalytic activity of CDYL on histone Kcr. (A) Immunofluorescence imaging of CDYL WT or various mutants in WT 9-12 cells (left). Quantification of CDYL WT or mutants’ puncta (right). Two-tailed unpaired t test. ****P<0.0001, N.S., not significant. Scale bar, 5 μm. (B) Visualization of turbidity associated with protein droplet formation. Tubes containing EGFP, CDYL WT, and CDYL KR>A in the absence (−) or presence (+) of PEG 8000 are shown. (C) Representative images of CDYL WT condensates and CDYL KR>A condensates. Scale bar, 50 μm. (D) In vitro Kcr assays with 0.05, 0.1, or 0.2 μg CDYL WT or KR>A and 50 μM crotonyl-CoA in the presence of 5 μg CTH. Reactions were analyzed by Western blot. (E) Immunofluorescence imaging of TY1-CDYL and H3K18cr in CDYL WT or KR>A WT 9-12 cells (top). Scale bar, 5 μm. Quantitative analysis of TY1-CDYL and H3K18cr immunofluorescence density on the dashed lines (bottom). (F) Heatmaps showing the occupancy of TY1-CDYL (left) and H3K18cr (right) binding on TY1-CDYL target genes in the indicated groups. (G) Gene tracks showing representative genes in the indicated groups. (H) RT-qPCR analysis of relative mRNA expression of STAT5A, COL5A1, IGF2, and TLR4 in each of the indicated groups. Data are represented as mean±SEM and were analyzed by two-tailed unpaired t test, *P<0.05, **P<0.01, and ****P<0.0001.
We next investigated the effects of phase separation on the hydratase activity of CDYL. We incubated purified recombinant WT CDYL or KR>A mutant proteins with crotonyl-CoA in the presence of native CTH. Addition of WT CDYL resulted in a substantial decrease of histone Kcr, whereas inclusion of CDYL KR>A mutant in the reaction had a marginal effect (Figure 7D), indicating that the phase separation capacity is critical for the crotonyl-CoA hydratase activity of CDYL. We further examined whether the formation of CDYL condensates affects histone Kcr in cells. As shown in Figure 7E, H3K18cr staining was abolished in CDYL condensates. As shown in Figure 7F, we then analyzed H3K18cr distribution and target gene expression upon overexpression of CDYL WT or KR>A mutant constructs. We found that KR>A mutation markedly dampened CDYL genomic binding and increased H3K18cr level on CDYL target genes in CDYL KR>A cells compared with CDYL WT cells (Figure 7F). Representative track files for ChIP-seq are shown in Figure 7G. Overexpression of KR>A mutants failed to reduce H3K18cr signals on the majority of CDYL target genes and failed to repress representative CDYL target gene expression, such as STAT5A, COL5A1, IGF2, and TLR4 (Figure 7H). Together, these data indicate that CDYL negatively regulates histone Kcr and target gene expression through a phase separation mechanism.
CDYL Attenuates Cystogenesis through Its Phase Separation Capacity in a Zebrafish ADPKD Model
To test whether phase separation is required for CDYL to inhibit cyst growth in vivo, we used a well-recognized zebrafish ADPKD model, in which MO-induced knockdown of pkd2 causes renal cyst formation and body curvature phenotypes.49 Injection of cdyl MOs alone had no apparent effects on cyst development or body curvature. Coinjection of cdyl and pkd2 MOs led to more severe renal cystogenesis in zebrafish embryos than in embryos coinjected with pkd2 and control MOs (Figure 8, A and B). These data indicate that activation of CDYL-suppressed pathways alone is not sufficient to induce a cystic phenotype, and that knockdown of cdyl can accelerate cyst development and aggravate the cystic kidney phenotype.
Figure 8.
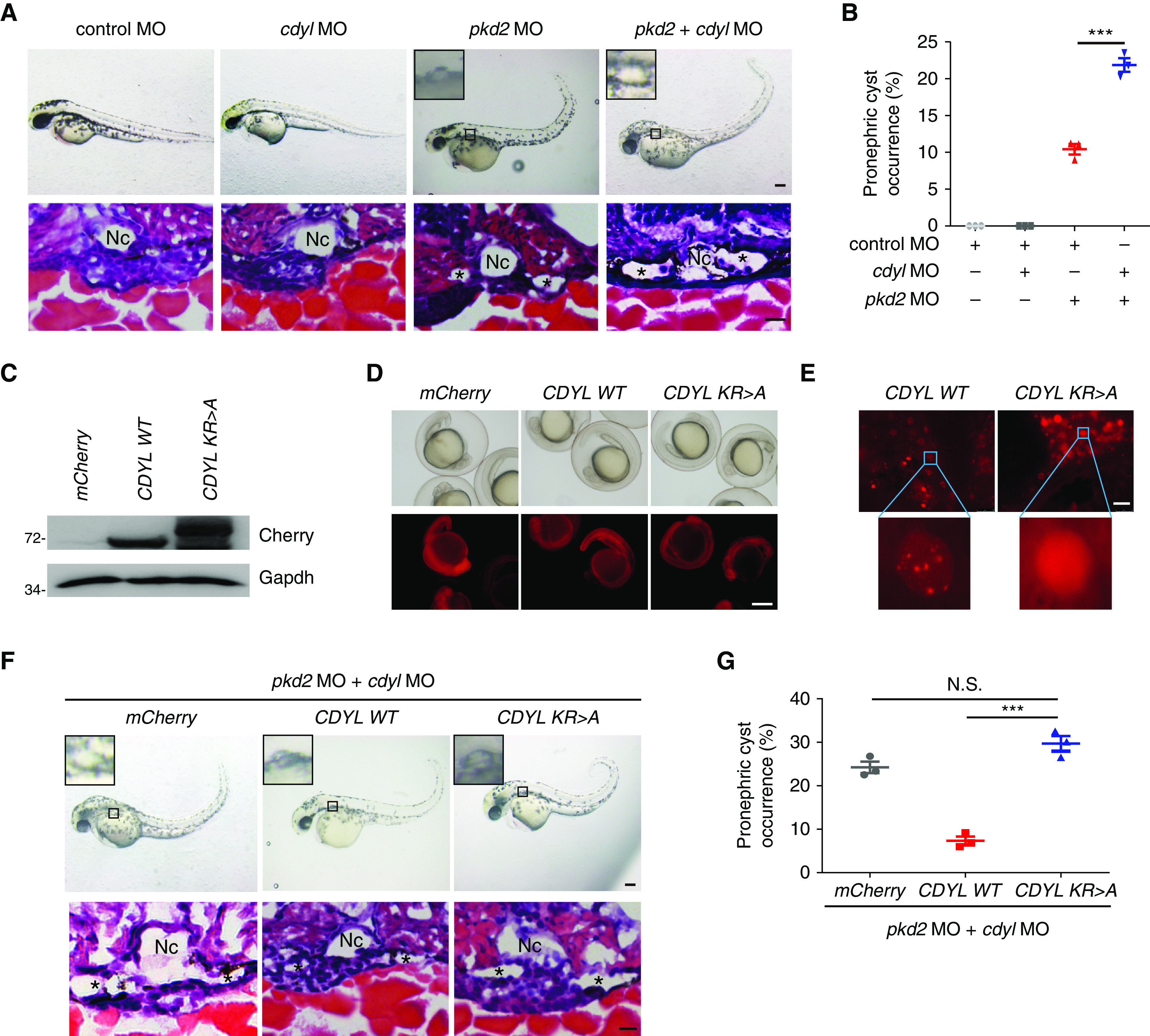
CDYL slows cyst growth through its phase separation capacity in a zebrafish ADPKD model. (A) Representative pictures of zebrafish WT embryos injected with the indicated MOs. Images were taken at 2 dpf. Notochord was indicated by “Nc.” Asterisks (*) indicate dilated pronephric tubules. Scale bar, 200 μm (top), 10 μm (bottom). (B) Quantitative analysis of pronephric cyst occurrence in zebrafish embryos: control MO injection alone (n=70, 68, and 72), cdyl MO (n=65, 75, and 70), pkd2 MO (n=63, 71, and 78), and coinjection (n=69, 69, and 76). (C) Western blot analysis of exogenous mCherry-CDYL levels in zebrafish embryos from each group. (D and E) Live-cell images showing mCherry-CDYL expression in zebrafish embryos microinjected with human mCherry-CDYL mRNA (WT and KR>A). Scale bar, 300 μm in (D) and 25 μm in (E). (F) Representative images of zebrafish embryos microinjected with the indicated MOs and mRNAs. Images were taken at 2 dpf. Scale bar, 200 μm. (G) Quantitative analysis of pronephric cyst occurrence in zebrafish embryos: mCherry mRNA (n=63, 79, or 65), CDYL WT mRNA (n=69, 65, or 72), and CDYL KR>A mRNA (n=68, 63, or 65). Quantification data (n>60) are represented as mean±SEM of three replicates in (B) and (G) and were analyzed by two-tailed unpaired t test, ***P<0.001, N.S., not significant.
We finally sought to determine whether CDYL condensates modulate cystogenesis. To address this, we injected human WT CDYL or KR>A mutant mRNA into pkd2/cdyl double KO morphants. The expression of exogenous mCherry-CDYL (WT and KR>A) was examined by Western blot analysis and fluorescence imaging (Figure 8, C and D). In contrast to KR>A mutant proteins, WT CDYL proteins formed droplets in zebrafish tissues (Figure 8E). Injection of human WT CDYL mRNA clearly inhibited kidney cystogenesis, whereas injection of KR>A mutant CDYL mRNA failed to rescue renal cyst formation (Figure 8, F and G). Together, these genetic studies indicate that CDYL attenuates cystogenesis through a phase separation mechanism in vivo.
Discussion
Epigenetic control of gene expression plays key roles in regulating cellular plasticity and cell fate transitions in development and various diseases.50,51 During cystogenesis, renal tubular epithelial cells undergo profound changes of cellular states. The proliferation of fully differentiated tubular cells ceases before birth in the normal kidney. Defects in PC1 and PC2 alter multiple signaling pathways, activate a pathologic gene expression program, lock tubular cells in an abnormal differentiated state, and promote uncontrolled cell proliferation.3,52 The epigenetic changes, and how they affect transcriptional responses and cystogenesis, in ADPKD remain poorly understood. In this study, we discover that downregulation of the crotonyl-CoA hydratase CDYL is accompanied by an elevation of histone Kcr in ADPKD cells. The mechanism underlying CDYL downregulation and the link with primary genetic/molecular defects in ADPKD are currently unknown. We did not observe a change of its mRNA level (Supplemental Figure 7), thereby excluding the possibility of regulation via transcription or mRNA stability. We suspect that CDYL downregulation is due to dysregulation of protein stability. PTMs of proteins including phosphorylation and ubiquitination are crucial for controlling protein stability.53 Further studies are needed to validate the above hypothesis. Functionally, overexpression of CDYL suppresses histone Kcr and slows cyst growth in an early-onset ADPKD mouse model. Because this is a developmental model, further analysis with a long-lived and adult-onset model is needed to more closely mimic human ADPKD progression. Through high-sensitivity mass spectrometry analysis, we identify H3K18cr as the major histone Kcr site regulated by CDYL. Overexpression of CDYL reduces histone Kcr levels and suppresses target gene expression controlled by CDYL-mediated histone Kcr. A variety of histone acylations have been discovered in recent years, but their regulation by the metabolism of the different acyl-CoA forms, the molecular mechanisms underlying their modulation of gene expression, and their biologic and pathologic functions remain largely unknown.12,54 This study establishes a key role for CDYL-mediated histone Kcr in connecting chromatin dynamics and gene transcription in ADPKD, illustrating a context-dependent role for epigenetic regulation in metabolic disease.
Altered cellular metabolism has recently been recognized as a prominent feature of ADPKD, including increased glycolysis, pentose phosphate pathway, and glutamine metabolism, as well as defective fatty acid β-oxidation and altered mitochondrial function.55–57 In the past decade, targeted treatment regimens for metabolic abnormalities have shown good therapeutic prospects for ADPKD, including dietary restrictions, 2-deoxy-glucose treatment, methionine restriction, and a ketogenic diet.58–63 Given the profound influence of these pharmacologic and dietary interventions on cellular metabolism, the levels of acyl-CoAs and their accompanying histone acylations may alter. Through modulation of chromatin architecture, histone acylations may reprogram the transcriptome and account for the beneficial effects of these treatments. Future studies are needed to test this hypothesis using chemicals that target metabolic changes, together with genome-wide epigenetic and transcriptomic analysis. Furthermore, it is worth identifying the key intermediate metabolites during ADPKD progression, as well as under these diet- and pharmacology-based therapeutic interventions, which would help to clarify the role of metabolism-epigenetics crosstalk and to develop safer and more effective therapeutic strategies for this disease. In this study, we also observed a marked decrease of histone β-hydroxybutyrylation (Kbhb) in ADPKD kidneys. β-hydroxybutyrate (BHB), a natural ketone body during ketogenic conditions, functions as a substrate for β-hydroxybutyrylation of histones. Histone Kbhb marks are dramatically induced in response to elevation of BHB levels in cultured cells and in liver and kidney tissues, which turns on genes involved in ketogenesis-responsive metabolic pathways.11 A ketogenic diet or oral administration of BHB strongly inhibits cyst development in PKD animal models.63 Further research is therefore needed to explore whether the therapeutic effects of a ketogenic diet rely on changes of histone Kbhb and the gene expression program regulated by this modification.
A particularly interesting discovery in our study is that CDYL forms nuclear condensates through phase separation in kidney cells. Importantly, this phase-separating capacity is required for CDYL-mediated crotonyl-CoA hydration, which suppresses histone Kcr, cyst-associated gene expression, and cyst growth. Formation of biomolecular condensates concentrates enzymes and substrates, enhances their interaction, and therefore promotes catalysis.29–31 Our ChIP-seq results show that the genome-wide distribution of CDYL is mostly concentrated in transcription regulatory regions, including promoters and enhancers. This locus-specific genomic binding and condensate assembly of CDYL may establish a domain-specific microenvironment, which could compartmentalize crotonyl-CoA into a dense phase to enhance the hydration reaction, and thus reduce local histone Kcr. Recent studies support the existence of such subnuclear metabolic niches or microdomains, which would enable site-specific recruitment of metabolic enzymes to efficiently modulate metabolite levels, and thus accurately modify chromatin and control gene expression.54 Indeed, metabolic enzymes involved in the synthesis of acyl-CoA, such as ACLY, ACSS2, or α-KGDH, are able to localize in the nucleus and interact with chromatin to produce acyl-CoA at specific genomic loci.64 In contrast to these acyl-CoA synthetases, CDYL functions as a crotonyl-CoA hydratase to reduce acyl-CoA. Thus, the locus-specific modulation of histone Kcr by CDYL via nuclear condensation provides new evidence in support of this subnuclear metabolic niche model for precise regulation of chromatin modifications and gene transcription.
Disclosures
All authors have nothing to disclose.
Funding
This work was supported by the National Natural Science Foundation of China (grants 82070689 and 81770658 to Y. Chen; 81861148027 to J. Liang; and 21874100 to K. Zhang), the National Key Research and Development Program of China (grant 2017YFA0504102 to Y. Chen), and the Tianjin Science and Technology Committee (grant 19JCJQJC63800 to Y. Chen).
Supplementary Material
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
See related editorial, “Decorating Histones in Polycystic Kidney Disease,” on pages 1629–1630.
Author Contributions
Y. Chen, J. Liang, K. Zhang, and L. Zhang conceptualized the study; Y. Chen was responsible for project administration and supervision; L. Yang was responsible for visualization; P. Cao, X. Cao, L. Dang, and T. Zhang were responsible for investigation; X. Cao, L. Dang, T. Gong, S. Tian, L. Yang, S. Yu, and T. Zhang were responsible for formal analysis; X. Cao and L. Dang were responsible for software; X. Cao, L. Dang, T. Gong, T. Liu, Y. Sun, S. Tian, H. Xiong, and T. Zhang were responsible for validation; Y. Chen, Y. Li, J. Liang, and K. Zhang were responsible for resources; L. Dang was responsible for methodology; Y. Chen, and L. Zhang wrote the original draft; and Y. Chen, J. Liang, and K. Zhang reviewed and edited the manuscript.
Data Sharing Statement
The ChIP-seq and RNA-seq data have been deposited in the Gene Expression Omnibus (GSE183512). Further information and requests for resources and reagents should be directed to Yupeng Chen (ychen@tmu.edu.cn).
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2021111425/-/DCSupplemental.
Supplemental Table 1. Primers for RT-qPCR.
Supplemental Table 2. Characteristics of ADPKD patients.
Supplemental Table 3. Characteristics of ADPKD mice.
Supplemental Figure 1. Genetic analysis reveals homozygous mutation of PKD1 in WT 9-12 cells.
Supplemental Figure 2. Downregulation of CDYL is accompanied by upregulation of histone Kcr in early-onset and late-onset ADPKD mouse model.
Supplemental Figure 3. Body weight of WT and ADPKD mice in the indicated groups.
Supplemental Figure 4. Upregulation of H3K18cr in early-onset and late-onset ADPKD mouse model.
Supplemental Figure 5. Intrinsic disorder of CDYL.
Supplemental Figure 6. CDYL WT and KR>A mutant fusion proteins.
Supplemental Figure 7. Cdyl mRNA in ADPKD mice.
References
- 1.Cornec-Le Gall E, Alam A, Perrone RD: Autosomal dominant polycystic kidney disease. Lancet 393: 919–935, 2019 [DOI] [PubMed] [Google Scholar]
- 2.Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE: Polycystic kidney disease. Nat Rev Dis Primers 4: 50, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris PC, Torres VE: Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest 124: 2315–2324, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapin HC, Caplan MJ: The cell biology of polycystic kidney disease. J Cell Biol 191: 701–710, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li X: Epigenetics and cell cycle regulation in cystogenesis. Cell Signal 68: 109509, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X: Polycystic Kidney Disease, eBOOK Ed., Brisbane, Codon Publications, 2015 [PubMed] [Google Scholar]
- 7.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, et al. : Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146: 1016–1028, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Sprung R, Tang Y, Ball H, Sangras B, Kim SC, et al. : Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics 6: 812–819, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, et al. : Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics 11: 100–107, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. : Metabolic regulation of gene expression by histone lactylation. Nature 574: 575–580, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie Z, Zhang D, Chung D, Tang Z, Huang H, Dai L, et al. : Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol Cell 62: 194–206, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabari BR, Zhang D, Allis CD, Zhao Y: Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol 18: 90–101, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rousseaux S, Khochbin S: Histone acylation beyond acetylation: Terra incognita in chromatin biology. Cell J 17: 1–6, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dutta A, Abmayr SM, Workman JL: Diverse activities of histone acylations connect metabolism to chromatin function. Mol Cell 63: 547–552, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ntorla A, Burgoyne JR: The regulation and function of histone crotonylation. Front Cell Dev Biol 9: 624914, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li K, Wang Z: Histone crotonylation-centric gene regulation. Epigenetics Chromatin 14: 10, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Sabari BR, Panchenko T, Wen H, Zhao D, Guan H, et al. : Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Mol Cell 62: 181–193, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sabari BR, Tang Z, Huang H, Yong-Gonzalez V, Molina H, Kong HE, et al. : Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol Cell 58: 203–215, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang Y, Xu X, Ding J, Yang L, Doan MT, Karmaus PWF, et al. : Histone crotonylation promotes mesoendodermal commitment of human embryonic stem cells. Cell Stem Cell 28: 748–763, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang X, Chen XF, Sun X, Xu P, Zhao X, Tong Y, et al. : Short-chain enoyl-CoA hydratase mediates histone crotonylation and contributes to cardiac homeostasis. Circulation 143: 1066–1069, 2021 [DOI] [PubMed] [Google Scholar]
- 21.Ruiz-Andres O, Sanchez-Niño MD, Cannata-Ortiz P, Ruiz-Ortega M, Egido J, Ortiz A, et al. : Histone lysine crotonylation during acute kidney injury in mice. Dis Model Mech 9: 633–645, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang G, Nguyen D, Archin NM, Yukl SA, Méndez-Lagares G, Tang Y, et al. : HIV latency is reversed by ACSS2-driven histone crotonylation. J Clin Invest 128: 1190–1198, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Yang X, Gui B, Xie G, Zhang D, Shang Y, et al. : Corepressor protein CDYL functions as a molecular bridge between polycomb repressor complex 2 and repressive chromatin mark trimethylated histone lysine 27. J Biol Chem 286: 42414–42425, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franz H, Mosch K, Soeroes S, Urlaub H, Fischle W: Multimerization and H3K9me3 binding are required for CDYL1b heterochromatin association. J Biol Chem 284: 35049–35059, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caron C, Pivot-Pajot C, van Grunsven LA, Col E, Lestrat C, Rousseaux S, et al. : Cdyl: A new transcriptional co-repressor. EMBO Rep 4: 877–882, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mulligan P, Westbrook TF, Ottinger M, Pavlova N, Chang B, Macia E, et al. : CDYL bridges REST and histone methyltransferases for gene repression and suppression of cellular transformation. Mol Cell 32: 718–726, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu S, Yu H, Liu Y, Liu X, Zhang Y, Bu C, et al. : Chromodomain protein CDYL acts as a crotonyl-CoA hydratase to regulate histone crotonylation and spermatogenesis. Mol Cell 67: 853–866, 2017 [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Li M, Fan M, Song Y, Yu H, Zhi X, et al. : Chromodomain Y-like protein-mediated histone crotonylation regulates stress-induced depressive behaviors. Biol Psychiatry 85: 635–649, 2019 [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Narlikar GJ, Kutateladze TG: Enzymatic reactions inside biological condensates. J Mol Biol 433: 166624, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Flynn BG, Mittag T: The role of liquid-liquid phase separation in regulating enzyme activity. Curr Opin Cell Biol 69: 70–79, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kojima T, Takayama S: Membraneless compartmentalization facilitates enzymatic cascade reactions and reduces substrate inhibition. ACS Appl Mater Interfaces 10: 32782–32791, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shin Y, Brangwynne CP: Liquid phase condensation in cell physiology and disease. Science 357: eaaf4382, 2017 [DOI] [PubMed] [Google Scholar]
- 33.Loghman-Adham M, Nauli SM, Soto CE, Kariuki B, Zhou J: Immortalized epithelial cells from human autosomal dominant polycystic kidney cysts. Am J Physiol Renal Physiol 285: F397–F412, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Nauli SM, Rossetti S, Kolb RJ, Alenghat FJ, Consugar MB, Harris PC, et al. : Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J Am Soc Nephrol 17: 1015–1025, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Yang Y, Chen M, Zhou J, Lv J, Song S, Fu L, et al. : Interactions between macrophages and cyst-lining epithelial cells promote kidney cyst growth in Pkd1-deficient mice. J Am Soc Nephrol 29: 2310–2325, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo Z, Pan F, Peng L, Tian S, Jiao J, Liao L, et al. : Systematic proteome and lysine succinylome analysis reveals enhanced cell migration by hyposuccinylation in esophageal squamous cell carcinoma. Mol Cell Proteomics 20: 100053, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuan ZF, Sidoli S, Marchione DM, Simithy J, Janssen KA, Szurgot MR, et al. : EpiProfile 2.0: A computational platform for processing epi-proteomics mass spectrometry data. J Proteome Res 17: 2533–2541, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Y, Liu Z, Cao X, Lu Y, Mi Z, He C, et al. : Activation of P-TEFb by cAMP-PKA signaling in autosomal dominant polycystic kidney disease. Sci Adv 5: eaaw3593, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu Y, Sun Y, Liu Z, Lu Y, Zhu X, Lan B, et al. : Activation of NRF2 ameliorates oxidative stress and cystogenesis in autosomal dominant polycystic kidney disease. Sci Transl Med 12: eaba3613, 2020 [DOI] [PubMed] [Google Scholar]
- 40.Fragiadaki M, Lannoy M, Themanns M, Maurer B, Leonhard WN, Peters DJ, et al. : STAT5 drives abnormal proliferation in autosomal dominant polycystic kidney disease. Kidney Int 91: 575–586, 2017 [DOI] [PubMed] [Google Scholar]
- 41.Lakhia R, Yheskel M, Flaten A, Ramalingam H, Aboudehen K, Ferrè S, et al. : Interstitial microRNA miR-214 attenuates inflammation and polycystic kidney disease progression. JCI Insight 5: e133785, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vanden Heuvel GB: CD14 : A candidate biomarker for the prognosis of polycystic kidney disease. Kidney Int 78: 537–538, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sadanandam A, Rosenbaugh EG, Singh S, Varney M, Singh RK: Semaphorin 5A promotes angiogenesis by increasing endothelial cell proliferation, migration, and decreasing apoptosis. Microvasc Res 79: 1–9, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lei G-S, Kline HL, Lee C-H, Wilkes DS, Zhang C: Regulation of collagen V expression and epithelial-mesenchymal transition by mir-185 AND mir-186 DURING idiopathic pulmonary fibrosis. Am J Pathol 186: 2310–2316, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nosalski R, Siedlinski M, Denby L, McGinnigle E, Nowak M, Cat AND, et al. : T-cell-derived mirna-214 mediates perivascular fibrosis in hypertension. Circ Res 126: 988–1003, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Banani SF, Lee HO, Hyman AA, Rosen MK: Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18: 285–298, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alberti S, Gladfelter A, Mittag T: Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 176: 419–434, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bianchi G, Longhi S, Grandori R, Brocca S: Relevance of electrostatic charges in compactness, aggregation, and phase separation of intrinsically disordered proteins. Int J Mol Sci 21: 6208, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao Y, Semanchik N, Lee SH, Somlo S, Barbano PE, Coifman R, et al. : Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc Natl Acad Sci U S A 106: 21819–21824, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moris N, Pina C, Arias AM: Transition states and cell fate decisions in epigenetic landscapes. Nat Rev Genet 17: 693–703, 2016 [DOI] [PubMed] [Google Scholar]
- 51.Wang H, Yang Y, Liu J, Qian L: Direct cell reprogramming: approaches, mechanisms and progress. Nat Rev Mol Cell Biol 22: 410–424, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Calvet JP: Polycystic kidney disease: primary extracellular matrix abnormality or defective cellular differentiation?. Kidney Int 43: 101–108, 1993 [DOI] [PubMed] [Google Scholar]
- 53.Baker PJ, De Nardo D, Moghaddas F, Tran LS, Bachem A, Nguyen T, et al. : Posttranslational modification as a critical determinant of cytoplasmic innate immune recognition. Physiol Rev 97: 1165–1209, 2017 [DOI] [PubMed] [Google Scholar]
- 54.Nitsch S, Zorro Shahidian L, Schneider R: Histone acylations and chromatin dynamics: concepts, challenges, and links to metabolism. EMBO Rep 22: e52774, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Padovano V, Podrini C, Boletta A, Caplan MJ: Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat Rev Nephrol 14: 678–687, 2018 [DOI] [PubMed] [Google Scholar]
- 56.Podrini C, Cassina L, Boletta A: Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell Signal 67: 109495, 2020 [DOI] [PubMed] [Google Scholar]
- 57.Menezes LF, Germino GG: The pathobiology of polycystic kidney disease from a metabolic viewpoint. Nat Rev Nephrol 15: 735–749, 2019 [DOI] [PubMed] [Google Scholar]
- 58.Warner G, Hein KZ, Nin V, Edwards M, Chini CC, Hopp K, et al. : Food restriction ameliorates the development of polycystic kidney disease. J Am Soc Nephrol 27: 1437–1447, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kipp KR, Rezaei M, Lin L, Dewey EC, Weimbs T: A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am J Physiol Renal Physiol 310: F726–F731, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chiaravalli M, Rowe I, Mannella V, Quilici G, Canu T, Bianchi V, et al. : 2-deoxy-d-glucose ameliorates PKD progression. J Am Soc Nephrol 27: 1958–1969, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rowe I, Chiaravalli M, Mannella V, Ulisse V, Quilici G, Pema M, et al. : Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat Med 19: 488–493, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramalingam H, Kashyap S, Cobo-Stark P, Flaten A, Chang CME, Hajarnis S, et al. : A methionine-Mettl3-N-6-methyladenosine axis promotes polycystic kidney disease. Cell Metab 33: 1234–1247, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Torres JA, Kruger SL, Broderick C, Amarlkhagva T, Agrawal S, Dodam JR, et al. : Ketosis ameliorates renal cyst growth in polycystic kidney disease. Cell Metab 30: 1007–1023, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boukouris AE, Zervopoulos SD, Michelakis ED: Metabolic enzymes moonlighting in the nucleus: Metabolic regulation of gene transcription. Trends Biochem Sci 41: 712–730, 2016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.