

Abstract
Introduction:
Macrophage phenotype in the tumor microenvironment correlates with prognosis in non-small cell lung cancer (NSCLC). Immunosuppressive macrophages promote tumor progression, while pro-inflammatory macrophages may drive an anti-tumor immune response. How individual NSCLCs impact macrophage phenotype is a major knowledge gap.
Methods:
To systematically study the impact of lung cancer cells on macrophage phenotypes, we developed an in vitro co-culture model comprised of molecularly and clinically-annotated patient-derived NSCLC lines, human cancer-associated fibroblasts, and murine macrophages. Induced macrophage phenotype was studied through qRT-PCR and validated in vivo using NSCLC xenografts through quantitative immunohistochemistry and clinically with TCGA “matched” patient tumors.
Results:
72 NSCLC cell lines were studied. The most frequent highly induced macrophage-related gene was Arginase-1, reflecting an immunosuppressive M2-like phenotype. This was independent of multiple clinicopathologic factors, which also did not impact M2:M1 ratios in matched TCGA samples. In vivo, xenograft tumors established from high Arginase-1-inducing lines (Arghi) had a significantly elevated density of Arg1+ macrophages. Matched TCGA clinical samples to Arghi NSCLC lines had a significantly higher ratio of M2:M1 macrophages.
Conclusions:
In our in vitro co-culture model, a large panel of patient-derived NSCLC lines most frequently induced high expression Arginase-1 in co-cultured mouse macrophages, independent of major clinicopathologic and oncogenotype-related factors. Arghi cluster-matched TCGA tumors contained a higher ratio of M2:M1 macrophages. Thus, this in vitro model reproducibly characterizes how individual NSCLCs modulate macrophage phenotype, correlates with macrophage polarization in clinical samples, and can serve as an accessible platform for further investigation of macrophage-specific therapeutic strategies.
Keywords: Non-small cell lung cancer, in vitro co-culture model, tumor microenvironment, macrophage phenotype
Introduction
Given the limitations of chemotherapy and oncoprotein-targeted therapy in non-small cell lung cancer (NSCLC), significant effort has been directed towards immunotherapeutic approaches. Remarkable advances in Food and Drug Administration (FDA)-approved immune checkpoint blockade therapies have yielded improvements in objective response and overall survival.1, 2 However, overall long-term benefit from immune checkpoint blockade only occurs in a minority of patients, highlighting a critical need to identify additional mechanisms of immunosuppression including those in the tumor microenvironment (TME), which may impact the efficacy of immunotherapy.3
Macrophages are amongst the most abundant immune cells in the TME and their activity is implicated in tumor progression through multiple mechanisms including cytokine/chemokine production, promotion of chronic inflammation, angiogenesis, response to hypoxia, and immunosuppression.4 In physiologic and malignant processes, macrophages exist on a spectrum between an “M1-like” pro-inflammatory, immunostimulatory phenotype and an “M2-like” anti-inflammatory, angiogenic, immunosuppressive phenotype.4 The type of macrophage present in the NSCLC TME carries clinical significance; the presence of M2-like macrophages in NSCLC stroma is associated with poor prognosis.5, 6 As such, therapeutics that target macrophages are being developed. For example, inhibition of macrophage-derived Arginase-1, an arginine-depleting enzyme that curtails T-cell proliferation, is under preclinical investigation and in early-phase clinical trials.7–9 Thus, it would be important to understand and potentially target the underlying mechanisms generating immunosuppressive macrophage phenotypes, particularly those originating in individual NSCLCs. In fact, studies suggest that oncogenic driver mutations in NSCLC cells contributes to macrophage M2-like phenotype by promoting an immunosuppressive milieu.10, 11 It is likely that distinct NSCLCs differentially impact macrophage phenotype in the TME. While large, molecularly annotated datasets of bulk NSCLC tumors including tumor cells and their TME, as well as new multi-omics and single-cell RNAseq studies of NSCLC are becoming available, what is still needed is a preclinical model system to identify and mechanistically demonstrate specific NSCLC–macrophage connections. Also, such a system would be useful as a preclinical testing platform for potential therapies to overcome such immunosuppressive mechanisms. Thus, we need an accessible and reproducible preclinical model that can be manipulated to dissect how individual NSCLC cell lines generate specific macrophage phenotypes. This model could then serve as a platform to investigate therapeutic strategies for modulating macrophage phenotype. In addition, the most useful preclinical model would be one that could be widely available and easily used in multiple labs.
This led us to explore using a large panel of patient-derived NSCLC cell lines and xenografts that have been clinically annotated, molecularly characterized, and widely distributed to the lung cancer translational research community. We aimed to develop a reproducible and physiologically relevant in vitro co-culture model to investigate tumor cell and TME factors involved in macrophage polarization and to investigate possible therapeutics. While similar co-culture studies have been published, none have interrogated a large panel of patient-derived NSCLC cell lines with a broad spectrum of molecular subtypes on induced macrophage phenotype.12, 13,14 Thus, we established a multicellular co-culture model involving an extensive repository of 72 distinct patient-derived NSCLC cells, human cancer-associated fibroblasts (CAFs), and mouse bone marrow-derived macrophages. We found that each NSCLC cell lines reproducibly induced unique gene expression profiles in co-cultured mouse macrophages, most frequently the immunosuppressive high-Arginase-1 (Arghi) phenotype. Importantly, this macrophage phenotype induced in vitro was also identified in murine macrophages populating the NSCLC xenografts in vivo. Furthermore, patient-derived clinical samples deposited in The Cancer Genome Atlas (TCGA) database, whose tumors were molecularly “matched” by RNA expression and tumor mutations to our NSCLC cell lines which had induced the Arghi macrophage phenotype in the co-culture assay system, also exhibited a higher ratio of immunosuppressive M2 macrophages. Surprisingly, this in vitro model system showed that the induced macrophage phenotypes did not correlate with standard clinical factors, demographics, and oncogenotypes, indicating the possibility of using this system to identify previously unidentified features and mechanisms that did lead to the induced macrophage phenotypes.
Materials and Methods
Patient-derived cell cultures
Patient-derived NSCLC lines and CAFs have been generated at the National Cancer Institute (NCI), the Hamon Center for Therapeutic Oncology Research at UT Southwestern (HCC) or obtained from the American Type Culture Collection (ATCC). These cultures/lines have, in large part, been deposited at the ATCC or are available from the HCC. The HCC4210F CAF line was derived from a 68-year-old white female, former smoker, with lung adenocarcinoma (TTF-1+, Napsin A+) at the time of surgical resection without any prior treatment. Cell lines were maintained in RPMI 1640 (GIBCO, 2.05 mM L-glutamine) supplemented with 5% fetal bovine serum (FBS) (GIBCO). Previously characterized normal human bronchial epithelial-derived cell lines (HBECs) were maintained in Keratinocyte serum-free media (KSFM) supplemented with human recombinant epidermal growth factor and bovine pituitary extract (BPE) at the time of use.15 All cell lines were maintained in a humidified environment in the presence of 5% CO2 at 37°C. All cell lines were DNA fingerprinted (PowerPlex Fusion Kit, Promega) for provenance and found to be mycoplasma-free (Myco Kit, Boca Scientific).
Mouse bone marrow derived macrophages (BMDMs) isolation and differentiation
Mouse BMDMs were isolated and differentiated using the established Cold Spring Harbor Protocol.16 Mouse L929 cells were grown in T175 flasks with 30 mL of DMEM (Dulbecco’s Modified Eagle’s Medium) (11995040, GIBCO) + 10% FBS (fetal bovine serum). After cells were grown to 100% confluency, media was changed and then cells were cultured for 48 hours. Conditioned media was collected, and new media was added. This collection cycle was repeated 4 times. Collection media (1X PBS + 5% FBS + 1X penicillin/streptomycin), and macrophage media (20% L929 condition media + 20% FBS + 0.5X sodium pyruvate + 1X MEM +1X NEAA (non-essential amino acids) + 1X Glut Max in DMEM without Glutamine) were used. Tibias and femurs were isolated from 6–8-week-old C57BL/6J mice and cleaned in 70% ethanol. Epiphyseal heads were transected to expose the bone marrow-containing medullary cavity. Lumens were flushed with collection media using syringes. Collected bone morrow was spun down (5 min × 1000 rpm at 4°C), supernatant was removed, and the cell pellet resuspended in macrophage media and filtered with a 70-micron filter followed by centrifugation (5 min × 1000 rpm at 4°C). Cells were seeded on petri dishes in 8 mL of macrophage media (3 plates per mouse) or frozen down (90% FBS, 10% DMSO). Fresh macrophage media (4 mL) was added after three days. After an additional two days of culture, macrophages were collected and used for co-culture assays.
Multicellular co-cultures
Multicellular co-cultures were composed of BMDMs, CAFs and NSCLC cells at a 1:10:50 ratio, respectively. We chose these ratios based on immunohistochemical (IHC) quantification of NSCLC patient tumors which reflected the tumor/TME composition.17 NSCLC and CAF cell lines were trypsinized and plated with BMDMs into 6-well plates at 1.5 × 105 total cells/well. Cells were incubated for 40 hours and harvested for real-time quantitative polymerase chain reaction (qRT-PCR) analysis of the mouse expression of macrophage polarization markers (Arginase-1, iNos, Il-6, Il-1b, Ym1, Socs3).6, 7, 18, 19 Macrophages alone were seeded at 1.0 × 105 cells per well. Lipopolysaccharide (LPS) (20 ng/mL, 4-hour stimulation) or IL-4 (40 ng/mL,18-hour stimulation) treatments were used as positive controls for macrophage polarization into M1-like and M2-like phenotypes, respectively. All qPCR data processing was completed in R (see Supplemental File 2: “Co-culture qPCR R code.R”). Primers were designed using NIH nBlast tool against genes of interest specific to Mus musculus (house mouse) transcriptome. To ensure quality control all primer sets were evaluated by: NIH Primer-BLAST to ensure no reactivity with the Homo sapiens transcriptome. Additionally, primer sets were tested with human cell lines to ensure no activity, as well as in polarized macrophages for predicted outcomes.
Xenograft Studies
NSCLC cell lines A427, NCI-H1666, NCI-H2009, NCI-H460, Calu-6, NCI-H1373, and NCI H2073 were grown as subcutaneous xenografts. Tumor cells (1 × 106) suspended in 100 μL of PBS were injected subcutaneously into the right posterior flank of 8-week-old female athymic nude mice. Tumor dimensions and volumes were measured weekly and mice were sacrificed when tumor volumes reached 1000–1500 mm3. Tumors were harvested for quantitative IHC interrogation of macrophage polarization. All mouse experiments were performed under a UTSW Institutional Animal Care and Use Program (IACUC)-approved protocol.
Quantitative IHC Analysis
IHC was performed as previously described.20 Blocking solutions, primary antibodies, secondary antibodies, and relevant information are available in the supplemental methods. Images were captured at 40X magnification using a Vectra Polaris Slide Scanner (AKOYA Biosciences, Delaware, USA). Images were deconvoluted and re-stitched using Phenochart and inForm software (Akoya Biosciences). Reconstituted images underwent multiplex quantitative analysis using HALO software (Akoya Biosciences).
Overview of the discovery approach and macrophage characterization platform
To examine the contribution of specific cancer cell characteristics to macrophage polarity, we utilized archival molecular and clinicopathologic data on our cell line repository, and corroborated these findings with publicly available data from the TCGA (Figure 1, Table 1, Supplemental Table 1, Supplemental Table 2).21 Whole exome and bulk RNA sequencing from NSCLC (n = 72) and SCLC (n = 2) cell lines were used to characterize total mutation burden, copy number variants, and somatic mutations. These data were then used to perform a TCGA “matchup” (Supplemental Table 2, Supplemental Methods). While the individual NSCLC lines were not derived from the TCGA tumor samples, the comparison of mRNA expression and DNA mutation profiles provided quantitative, objective correlation between our NSCLC lines and the TCGA tumor samples. CIBERSORT immune deconvolution software was then used to estimate immune cell populations within the TCGA patient-derived tumor specimens (lung adenocarcinoma (LUAD) n = 490, squamous cell carcinoma (LUSC) n = 490).22 From the CIBERSORT analysis, we used M1-like and M2-like macrophage cell counts for further analyses and then compared the macrophage phenotype counts in the TCGA samples with the induced macrophage phenotypes engendered by the NSCLC lines best matched to the individual TCGA tumors. Additionally, we assessed the contribution of clinicopathologic covariates including pathologic subtype, sex, age, smoking status, clinical stage, anatomic origin of cell line (i.e., primary tumor, metastatic lymph node, distant metastasis) on macrophage polarity in lung cancer co-culture and TCGA samples.
Figure 1: Overview of study of induced macrophage phenotypes in NSCLC co-culture system.
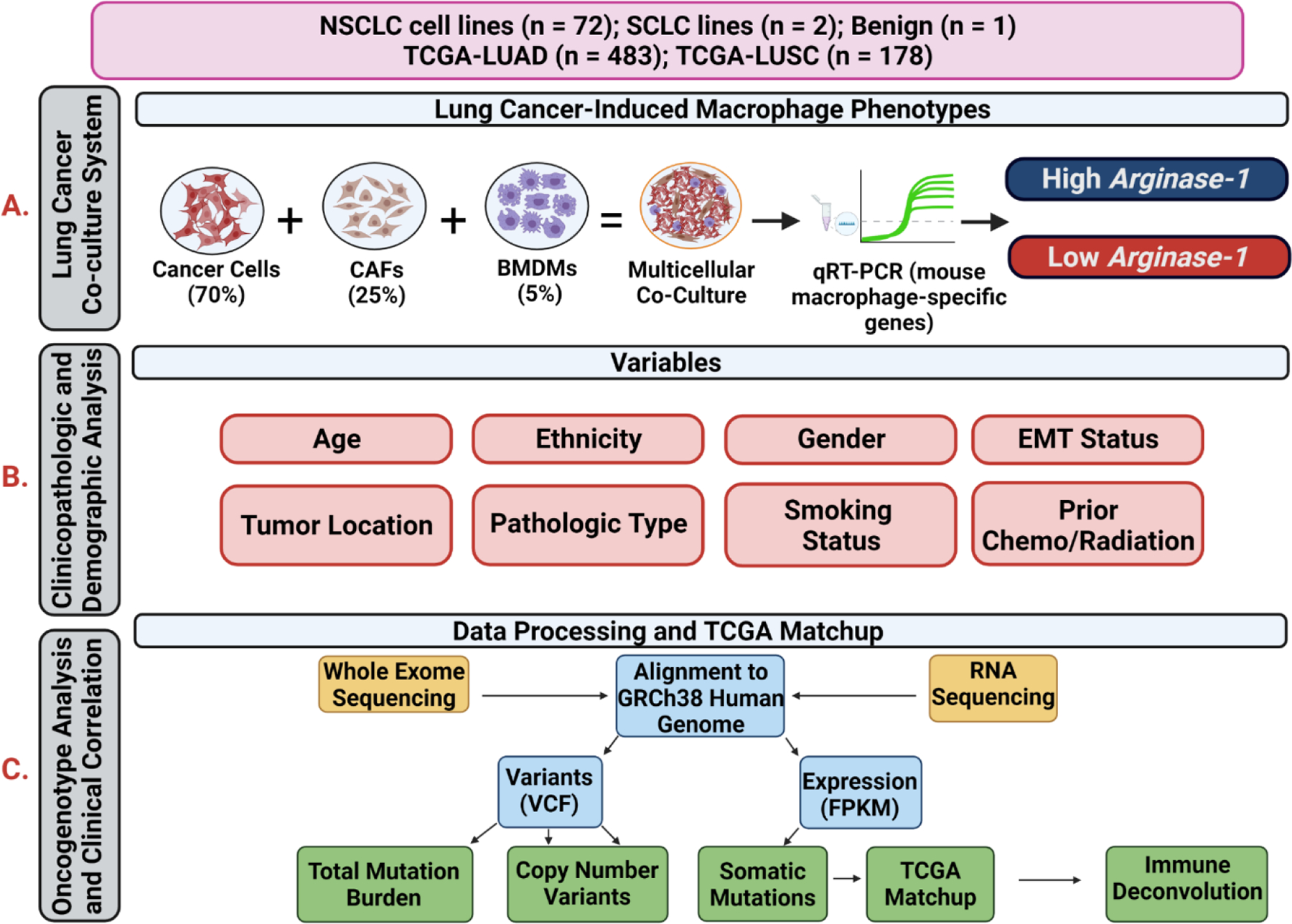
Flowchart showing analytic workflows, integrating molecular and clinicopathologic characteristics, with macrophage phenotypes induced in the lung cancer multicellular co-culture model and the NSCLC cell line information with TCGA datasets. A) Lung Cancer Co-Culture System Assay. We performed co-cultures of NSCLC lines, CAFs, and mouse bone marrow-derived macrophages and assayed by quantitative mRNA expression of mouse macrophage genes by using species-specific primers at 40 hours to determine the induced macrophage phenotype. B) Clinicopathologic and Demographic Analyses. We investigated whether clinicopathologic and demographic data features, or specific mutations or mRNA expression patterns found in the NSCLC lines were correlated with the induced macrophage phenotypes. C) Oncogenotype Analysis and Clinical Correlation. Total mutational burden, copy number variants, and somatic mutation profiling data were abstracted from whole exome and RNAseq analysis from each NSCLC line. These molecular data were “matched” against TCGA molecular data in NSCLC clinical samples by comparing mRNA expression and specific mutations (see Methods) to assign a correlation (ranging from 0.0 to 1.0) between each NSCLC line and TCGA tumor sample. We then performed immune deconvolution using CIBERSORT to estimate immune cell populations in the TCGA bulk tumor data, focusing specifically on M2 and M1 macrophages and tested whether the NSCLC cell line-induced macrophage phenotypes were similar to macrophage phenotypes found in TCGA tumor samples most closely matched to each cell line.
Table 1:
Available clinical and demographic characteristics of patient-derived cell lines with respect to induced macrophage phenotype in the in vitro co-culture model
| Characteristics | Arg hi | Arg low | p-value |
|---|---|---|---|
| Gender | 1.00 | ||
| Male | 15 | 30 | |
| Female | 10 | 20 | |
| Age (Mean ± SD) | 50.36 ± 15.89 | 53.13 ± 13.56 | 0.53 |
| Smoking Status | 0.58 | ||
| Yes | 11 | 16 | |
| No | 2 | 7 | |
| Race | 0.35 | ||
| Caucasian | 18 | 32 | |
| Black | 3 | 3 | |
| Other | 4 | 15 | |
| Tumor Origin | 1.00 | ||
| Primary | 8 | 17 | |
| Lymph node | 6 | 12 | |
| Distant Metastasis | 7 | 16 | |
| Stage | 1.00 | ||
| 1 | 2 | 3 | |
| 2 | 1 | 1 | |
| 3 | 7 | 11 | |
| 4 | 5 | 7 | |
| Prior Chemotherapy | 0.39 | ||
| Yes | 3 | 4 | |
| No | 10 | 28 | |
| Prior Radiation | 0.08 | ||
| Yes | 4 | 6 | |
| No | 9 | 26 | |
| Clinical Response | 0.62 | ||
| Complete/Partial Response | 3 | 2 | |
| Stable/Progressive Disease | 5 | 7 |
Statistical Analysis
All statistical analyses were performed with GraphPad Prism (Version 9) unless otherwise stated. All qPCR data processing was completed using R (R Core Team, Vienna, Austria).
Additional details regarding methods are provided in the supplemental material.
Results
NSCLC cells induce distinct macrophage phenotypes in a multicellular (human tumor cells, CAFs, and mouse macrophages) co-culture model, most frequently high Arginase-1
We established a three component, multicellular, co-culture model of mouse BMDMs, patient-derived NSCLC cells, and patient CAFs to determine the impact of tumor cells on macrophage polarization (Figure 2.A). Since macrophages were derived from mouse bone marrow we utilized a panel of mouse-specific primers for qPCR analysis of expression of macrophage-relevant genes (Arginase-1, Il-1β, Socs3, iNos, Il-6, Ym-1) to assess macrophage phenotype. We interrogated 72 distinct NSCLC cell lines and a patient-derived NSCLC CAF line. We note that inclusion of a CAF line (such as HCC4210F) in the co-culture system was required to identify changes in macrophage phenotype (Figure 2.A and B). Overall, the large panel of NSCLC cell lines most frequently induced increased expression of Arginase-1, Il-1β, and Socs3, compared to macrophages alone. The specific macrophage phenotype induced by each NSCLC cell line was reproducible within assays through multiple technical replicates and stable over time when we repeated testing at different passages (multiple biologic replicates) of any one NSCLC cell line (Supplemental Figure 1.A, Supplemental Methods). To characterize high vs. low expression of any given gene in an unbiased manner between biologic replicates and cell lines, we defined high expression as ≥75-fold change compared to gene expression from macrophages cultured alone and <75-fold change as low expression. With this approach, high expression of Arginase-1 (Arghi), classically associated with the immunosuppressive M2-like phenotype, was the most frequently induced phenotype (32% of lines) (Figure 2.C), followed by high Il-1β, then high Socs3. As elevated expression of ARG by myeloid cells including macrophages has been directly associated with immunosuppression and T-cell dysfunction in NSCLC, we focused on further clinicopathologic and molecular characterization of Arghi vs. Arglow clusters to investigate factors which may be associated with high expression of Arginase-1 in co-cultured macrophages.23
Figure 2: NSCLC cells induce heterogeneous macrophage phenotypes in a multicellular in vitro co-culture model, most frequently, high expression of immunosuppressive Arginase-1.
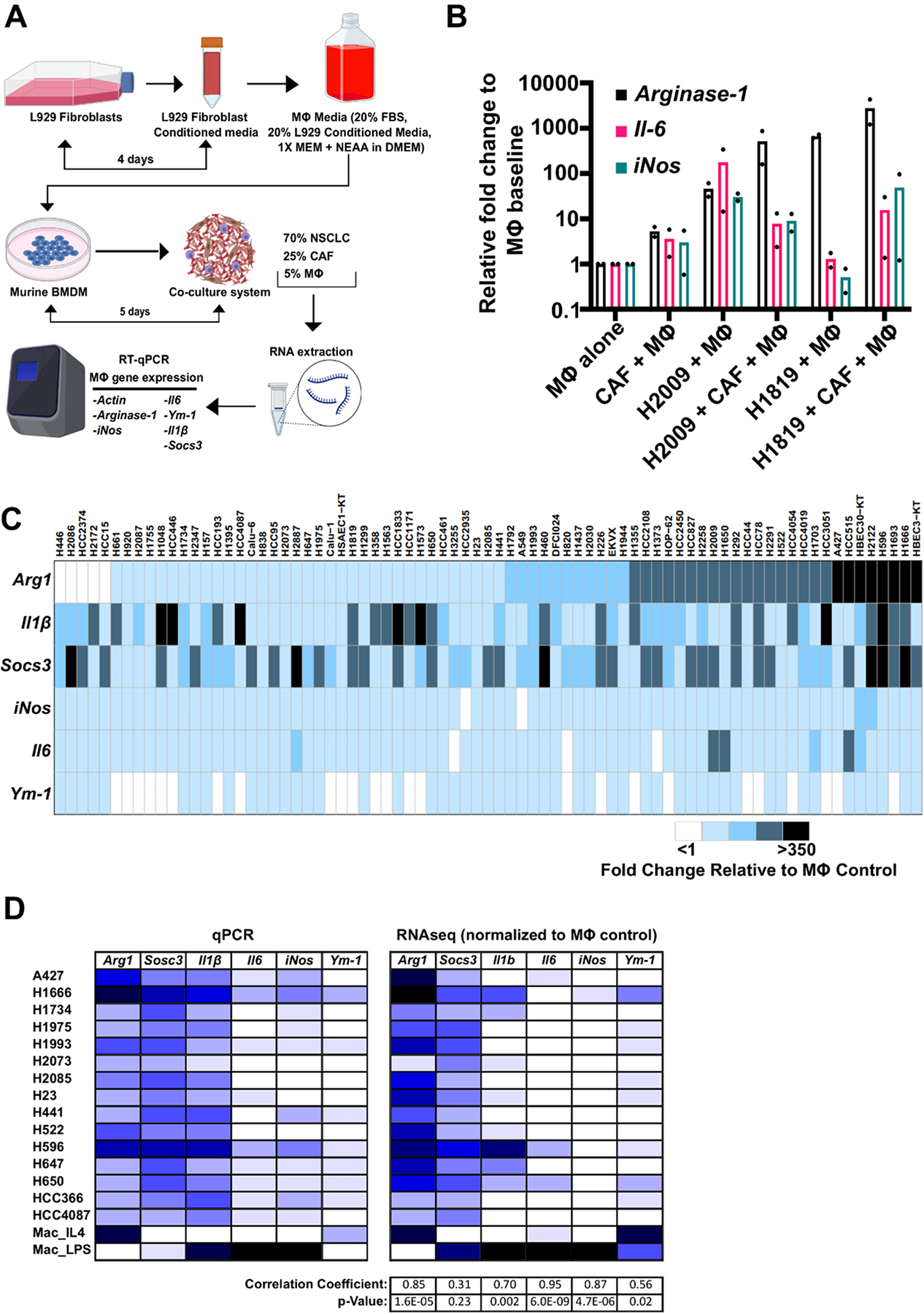
A) Schematic of the NSCLC multicellular co-culture model setup. Co-cultures were comprised of: mouse macrophages (MΦ) isolated and differentiated from mouse bone marrow hematopoietic stem cells (5%) using mouse L929 conditioned media from published methods16, human cancer-associated fibroblasts (CAFs) (25%), and cancer cell lines isolated from NSCLC patients (70%). RNA was extracted from individual co-cultures and qRT-PCR for mouse macrophage-specific genes was conducted. B) Comparison of dual co-cultures and multicellular co-cultures highlight the effects of addition of CAFs to induced macrophage phenotype. qRT-PCR transcriptional analysis of macrophage markers using species specific primers: Arginase-1, Il-6 and iNos. Cultures are dual co-cultures (2 cell types, CAFs and macrophages, or tumor cells and macrophages) or multicellular co-cultures (3 cell types, tumor cells, CAFs, and macrophages) using H2009 and H1819 NSCLC cell lines as examples (results from duplicate assays shown as dots in each bar graph). C) Heatmap of quantitative mRNA expression changes by qRT-PCR analysis of 6 mouse macrophage related genes after NSCLC, CAF, macrophage co-culture compared to culturing the macrophages alone (See Supplemental Materials and Methods). Raw fold change data were normalized to gene expression from macrophages alone. Each experiment included positive controls of macrophages alone treated with IL-4 (strong M2 phenotype induction) or LPS (strong M1 phenotype induction). Each row represents macrophage expression phenotype results average from technical quadruplicates and 3 biologic replicates for the indicated NSCLC cell line. (See Supplemental Figure 1.B for examples of fold changes in macrophage gene expression in the co-cultures). High expression of any given gene was characterized as relative fold change ≥75 from baseline and low expression was characterized as relative fold change <75 from baseline. 24/75 lines (32%) induced high expression of Arginase-1. 72 NSCLC cell lines, 2 SCLC lines and 3 benign individual cell lines were studied. D) qPCR findings of induced macrophage gene expression are conserved in bulk RNAseq analyses. Relative gene expression of 6 macrophage-relevant genes were correlated to expression in bulk RNAseq samples submitted for a large panel of NSCLC co-cultures. Human reads were filtered out and mouse gene expression of each target gene was correlated to our qPCR panel through Pearson correlation coefficient analyses. Arginase-1, Il1b, Il6, and iNos expressions were strongly consistent between qPCR and RNAseq analyses. 15 individual NSCLC cell lines were tested.
To quantitatively investigate macrophage gene expression differences between Arghi vs. Arglow cluster lines, we compared expression of each gene between clusters. Only one macrophage gene, Il-6, was found to be expressed at higher levels in the Arghi compared to Arglow cohort (median fold change 14.5 vs. 5.3, respectively, p<0.0001) (Supplemental Figure 1.B). We note in syngeneic mouse models and human tumors that Rab37-regulated secretion of IL-6 by macrophages is associated with M2-like phenotype, promotion of STAT3-dependent PD-1 expression in CD8+ T-cells, and poor prognosis in NSCLC patients.24 In addition to qRT-PCR measurement of mouse macrophage gene expression, bulk RNAseq was performed on a panel of 15 NSCLC co-cultures, with human reads filtered out, to compare the qRT-PCR and RNAseq results. We found that our qPCR findings of high Arginase-1 expression in macrophages across this panel correlated significantly with macrophage expression patterns observed in our RNAseq samples of the same NSCLC co-culture (r = 0.85, p = 1.6E-5) (Figure 2.D). To further study if the findings from our mouse-macrophage platform could be recapitulated using human macrophages, we used macrophages differentiated from patient umbilical cord blood-derived monocytes co-cultured with 2 Arghi (H1373 and H2009) and 2 Arglow (H647 and H441) cell lines. On flow cytometric analysis, we found that Arghi cell lines similarly polarized co-cultured human macrophages towards an M2-like state (CD68+/CD206+) (Supplemental Figure 1C). Thus, we found through this co-culture model that 72 distinct patient-derived NSCLC cell lines reproducibly (for each cell line model) induce differential expression patterns in co-cultured mouse macrophages, most frequently the immunosuppressive Arghi phenotype.
High expression of Arginase-1+ macrophages is recapitulated in Arghi NSCLC-derived xenografts in vivo
To investigate whether induced macrophage phenotype by Arghi cluster lines was also observed in vivo, we established athymic nude mice subcutaneous xenografts derived from Arghi (5 cell lines) and Arglow lines (6 cell lines) to determine the phenotypes of host mouse macrophages. Tumors were grown to 1,000–1,500 mm3 diameter and harvested for immunohistochemical analysis of the mouse macrophage phenotype in the tumor microenvironment (Figure 3.A, Supplemental Figure 2.A). Median tumor area was not significantly different between cohorts (Supplemental Figure 2.B). We found that compared to NSCLC xenografts established from Arglow cluster lines, those established from Arghi lines had significantly higher median density of total macrophages (characterized as F4/80+) (1245 vs. 414 cells/mm2, p<0.0001) and higher density of Arg1+ macrophages (Arg+ / F4/80+) (142 vs. 44 cells/mm2, p=0.0007) (Figure 3.A). Thus, we observed that xenograft tumors derived from Arghi NSCLC cell lines had significantly higher density of host mouse Arg1+ macrophages in the tumor stroma, providing an independent in vivo confirmation of our in vitro findings.
Figure 3: Macrophage phenotype induced in vitro are observed in vivo and in clinical samples from the TCGA.
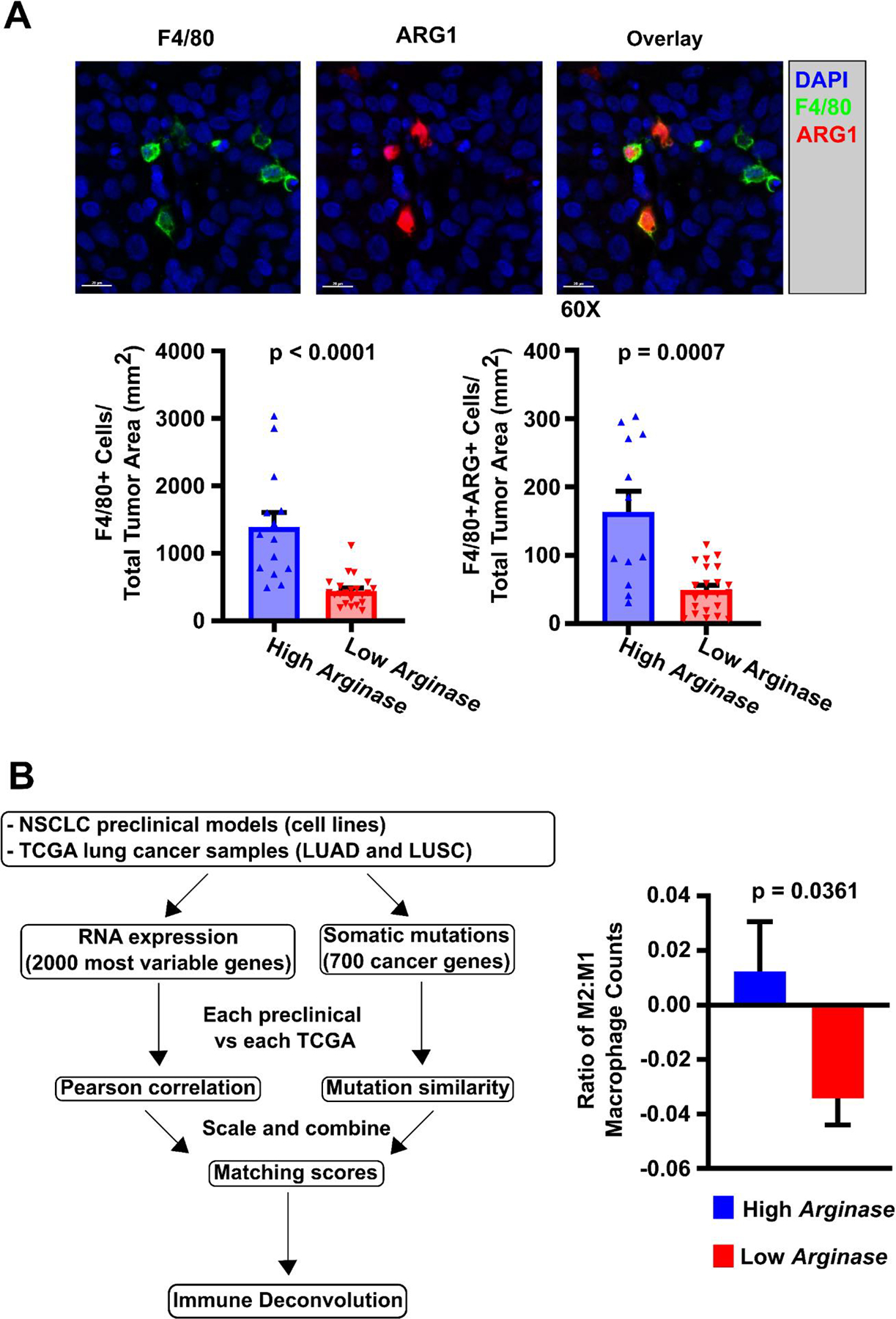
A) Immunohistochemical staining of NSCLC xenografts for macrophage markers. NSCLC xenografts were established from 5 Arghi cluster lines (A427, H1373, H1666, H2009, H522) and 6 Arglow cluster lines (H1993, Calu-6, H460, H647, H2073, H441) using athymic nude mice with subcutaneously injected tumor cells (1×106 cells/mouse) into the right flank to investigate induced macrophage phenotype in vivo (2–7 mice/NSCLC line). As an example, NSCLC xenograft NCI-H2073 was sectioned, immunohistochemically stained, and quantified for macrophage ARG1 expression. F4/80 was used as a co-localizing murine pan-macrophage marker, while staining for human pan-cytokeratin identified epithelial tumor cells. Those xenografts established from Arghi cluster lines had significantly median greater density of total F4/80+ macrophages and ARG+ macrophages. This was consistent with prior qRT-PCR mRNA macrophage expression results from the in vitro co-cultures. Significance was determined using Mann-Whitney U tests between cohorts. B) M2-macrophage phenotype found in NSCLC TCGA data by CIBERSORT analysis correlate with matched NSCLC line-induced macrophage phenotypes. B (left portion): Our NSCLC cell lines were matched to TCGA clinical samples through assessment of RNA expression and mutational profiles to generate a matching score between an individual NSCLC cell line and a “matched” TCGA patient sample (see Supplemental Methods: TCGA Matchup) 36 molecular matches were established. B (right portion): A significantly higher M2:M1 ratio of macrophages was identified through immune deconvolution (CIBERSORT) of TCGA patient data derived from samples that were “matched” (by mRNA expression and DNA mutation profiles) to NSCLC cell lines from the Arghi cluster. Significance was determined using a two-sided T-test. Mean ± SD.
TCGA-deposited clinical NSCLC specimens molecularly matched to co-cultured NSCLC lines exhibit similar tumor-associated macrophage phenotypes
To investigate the external validity of induced macrophage phenotypes in our co-culture system in clinical samples, we investigated RNA expression and mutation data from our patient-derived NSCLC cell lines to “matched” individual patient tumor specimens showing similar mRNA expression and DNA mutation profiles in the TCGA database (Figure 3.B (left), Supplemental Table 2, and Supplemental Methods). We then utilized the bulk RNAseq expression data from the TCGA and CIBERSORT22 to quantify relative M1-like and M2-like macrophages in the TME of these clinical specimens. This approach allowed us to correlate the induced macrophage phenotypes in our NSCLC co-culture system to the ratio of M2:M1 macrophages in clinical samples. We found that the co-culture-induced macrophage expression patterns from any one NSCLC cell line correlated significantly with the macrophage phenotypes (from CIBERSORT RNA expression analyses) found in molecularly matched TCGA patient tumors. Those TCGA specimens matched to Arghi cluster NSCLC cell lines had a significantly higher mean M2:M1 ratio compared to specimens matched to Arglow lines (0.01 vs. −0.03, p=0.0361) (Figure 3.B (right)). These findings indicate that patient-derived NSCLC lines in our co-culture system induce macrophage phenotypes that are not only found for the same NSCLC lines in xenografts in vivo, but also correlate with macrophage phenotypes found in TCGA-derived patient tumor specimens with similar mRNA and mutational profiles. This highlighted that our model has the potential to provide a clinically pertinent platform from which to investigate tumor cell and TME factors that modulate macrophage phenotype.
Clinicopathologic and molecular characteristics of NSCLC cell lines do not correlate with induced macrophage phenotypes.
We asked whether there was any correlation between clinicopathologic or demographic factors with induced macrophage phenotypes in our NSCLC models. Prior studies have suggested that multiple clinicopathologic factors may modulate the TME landscape, including macrophage phenotype.21, 22, 25, 26 To date, no large-scale investigation of the impact of these factors across multiple NSCLC tumors or patient-derived cell lines on macrophage phenotype have been performed. Surprisingly, none of the standard clinical or molecular variables correlated significantly with either Arghi or Arglow NSCLC line clusters. These included age, gender, race, smoking status, clinical stage, oncogenotype, total mutation burden, histologic subtype, and epithelial/mesenchymal phenotype (Table 1, Figure 4.A (top), Supplemental Figure 2.C and 2.D).
Figure 4: Total mutation burden and key driver mutations characteristics of the NSCLC cell lines do not correlate with their induced macrophage phenotype in neither co-cultures nor clinical samples.
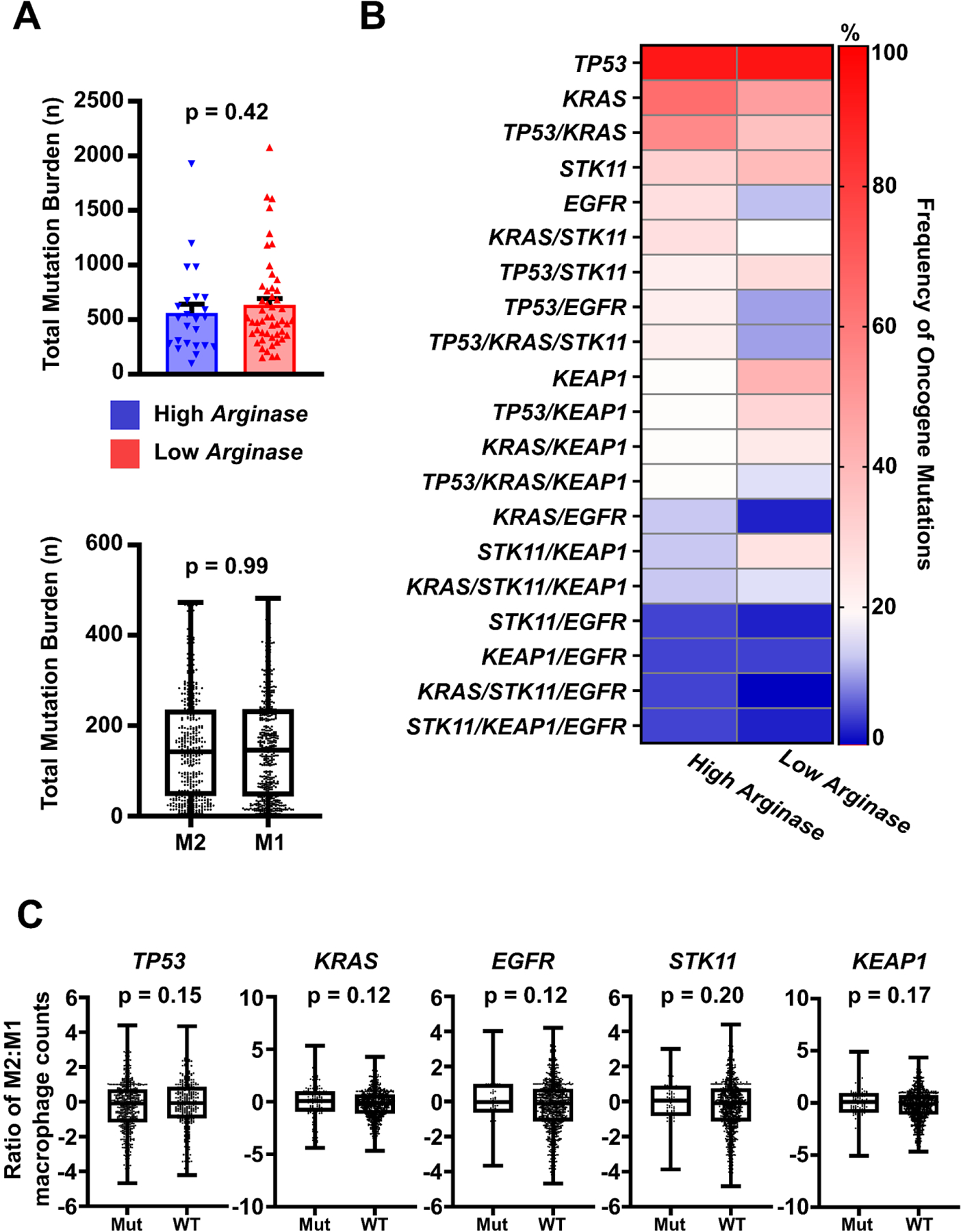
A) Total mutation burden. (Upper) Comparative analyses showed no significant differences in median total mutation burden between Arghi and Arglow cohorts between induced macrophage phenotype between cohorts through Mann-Whitney U tests. (Lower) Total mutational burden was investigated against TCGA-deposited patient samples which contained macrophages with higher M2:M1 (M2) or M1:M2 (M1) ratios on CIBERSORT analysis. No significant differences in total mutation burden was noted on Mann-Whitney U analysis, similar to the findings in our NSCLC-co-culture panel. B) Key Individual Driver Mutations. Frequency of key driver mutations and combinations of mutations were abstracted from cell lines in each cohort. The most commonly mutated genes regardless of cohort included TP53, KRAS, TP53/KRAS, STK11, and EGFR. No significant differences were noted in frequency of oncogene mutations between cohorts for any given single or combination of mutated oncogenes. Significance was determined using two-sided T-tests. Mean ± SD. C) Histology and Key Driver Mutations in TCGA Data and Associated Macrophages. Macrophage M1:M2 ratio determined via immune deconvolution (CIBERSORT) in TCGA NSCLC patient samples were not correlated with mutation status for TP53, KRAS, EGFR, STK11 and KEAP1. M2:M1 ratios between groups were compared through Mann-Whitney U tests. For all cell line analyses, 72 individual NSCLC cell lines were used. Available data from a total of 980 patient lung cancer samples were used for TCGA analyses.
Looking at specific NSCLC oncogenotypes, we found in both Arghi and Arglow cohorts that the most frequently identified gene mutations were TP53 (83, 84%, respectively), KRAS (58, 44%), TP53/KRAS (50, 34%), STK11 (29, 36%), and EGFR (25, 12%), similar to prior studies (Figure 4.B).27, 28 However, no significant differences were identified between cohorts with respect to oncogenotype frequency. We further investigated macrophage Arginase-1 expression quantitatively across our panel of lung cancer lines against presence of key mutant oncogenes individually (TP53, KRAS, STK11, KEAP1) or in combination with one another and did not identify any significant differences (Supplemental Figure 3). Given the absence of significant correlations between NSCLC oncogenotype and macrophage Arginase-1 expression in our in vitro co-culture model, we also asked whether these findings were observed in TCGA-deposited clinical NSCLC samples. We assessed M2:M1 macrophage phenotype ratio in clinical samples using CIBERSORT analysis against the aforementioned demographic, clinical, and molecular characteristics and similarly found no significant differences in the M2:M1 ratio with respect to sex, histologic subtype, age, total mutation burden, or oncogenotype (Figure 4.A (bottom) and 4.C, Supplemental Figure 2.E). Thus, surprisingly, we found that established clinicopathologic, demographic, and molecular characteristics of NSCLC cell lines co-cultured with macrophages were not associated with high or low induction of macrophage Arginase-1 expression, nor were these factors correlated with macrophage phenotypes in the TCGA dataset.
Discussion
In this study, we utilized an in vitro multicellular co-culture model of NSCLCs, CAFs, and mouse BMDMs to demonstrate that a large panel (N = 72) of patient-derived NSCLC lines reproducibly induced heterogeneous macrophage gene expression signatures, most frequently the high expression of immunosuppressive Arginase-1. The NSCLC cell lines used in this study are widely available and used by the lung cancer translational research community, and the patient-derived CAFs used will be made freely available.29 Likewise, the methods for harvesting and using mouse bone marrow-derived macrophages are commonly used. The protocol we developed for these assays described in the methods section should be straightforward for other labs to implement and can readily be extended to test the effects of the multiple new lung cancer lines including those from NSCLC and SCLC, as well as patient-derived xenografts.30 We point out that there have been similar co-culture protocols published by other investigators. However, none have been used to interrogate a large panel of NSCLC cell lines as described here.12, 13,14 Differences in induced expression patterns suggest different polarized functions of macrophages in co-culture that serve as a foundational resource for further investigation into how to modulate these phenotypes for potential clinical benefit. Indeed, elevated macrophage Arginase-1 activity is classically associated with an immunosuppressive phenotype that impairs T-cell function and facilitates tumor immune evasion. As expected, this phenotype is associated with poor prognosis.6, 31–33 Therapeutic inhibition of Arginase-1 activity in myeloid cells is under preclinical investigation in several cancers including NSCLC.8, 9, 34
Through xenograft studies, we demonstrated that the Arghi phenotype found in vitro was also recapitulated by the same NSCLC tumors grown in vivo. To further assess the validity and generalizability of the co-culture model to patient tumor specimens, we compared the induced macrophage phenotypes in vitro to those found in molecularly matched NSCLC patient samples in TCGA database. We found that TCGA samples molecularly matched to the NSCLC lines that stimulated high macrophage Arginase-1 expression also had significantly elevated levels of M2-like macrophages. The differences in M2:M1 ratio were modestly but significantly different between cohorts, perhaps reflecting that macrophage phenotypes in the TME are complex and are not completely captured by the traditional M2 (high Arg1) or M1 (high iNos) characterization. To this point, none of our lung cancer lines significantly induced high iNos expression while variably expressing other genetic markers even in Arghi lines, highlighting the principle that macrophages behave on a spectrum between these states. Since our co-culture system characterizes macrophage activity by specific gene signatures a specific biologic context, it serves as a useful model that is not encumbered by this traditional classification while still potentially reflecting clinical validity. In aggregate, these findings reflect the utility of this in vitro co-culture platform to investigate macrophage phenotype in the NSCLC TME.
Previous studies have suggested that gender, ethnicity, smoking status, lung cancer subtype, oncogenotype, and tumor mutation burden may alter the TME landscape.35–38 Therefore, we abstracted these salient characteristics for each cell line studied in the co-culture model and investigated their impact on macrophage phenotype. To our surprise, we did not identify any significant clinicopathologic characteristics that predicted macrophage phenotype in our co-culture system including anatomic origin, sex, smoking status, mutational burden, subtype, or stage. Thus, we were interested in the results of a comparable study of 80 resected NSCLC tumor specimens, where Jackute et al. found that while increased M2 macrophage density was associated with poorer survival compared to M1 macrophage density, no significant differences were identified between M1 or M2 macrophage density with respect to gender, age, histology, or stage.6 The authors did identify more M2 macrophages in poorly differentiated tumors and a higher number of total tumor and stroma-infiltrating M1 and M2 macrophages in samples from smoking vs. non-smoking patients.6 However, in tumor islets, there were no significant differences in M1 and M2 macrophage composition.6
We also found that tumor cell oncogenotype did not correlate with co-culture-induced macrophage phenotype. KRAS, one of the most frequently mutated oncogenes in NSCLC, was identified in nearly 50% of tested NSCLC lines. Prior studies suggest that KRASG12D mutations may be associated with M2-like human macrophage gene signatures including ARGINASE-1.39 A similar observation was noted for the impact of KRAS mutation on M2 vs. M1 macrophage phenotype in pancreatic cancer.40 However, we found KRAS mutations were evenly distributed between Arghi and Arglow cluster lines. Prior patient tumor analyses have demonstrated that EGFR mutations in NSCLC do not distinctly impact infiltration of macrophages, consistent with our findings.41 Cao et al. similarly demonstrated that while M2-like macrophage density was greater in EGFR wild-type tumors, the fraction of M2 to total macrophages was similar between EGFR-mutant vs. wild-type tumors, smokers vs. nonsmokers, and between adenocarcinoma vs. squamous cell carcinoma subtypes.5 Thus, the in vitro data from our co-culture model are consistent with these findings.
Our study has several limitations. We utilized mouse macrophages in the co-culture model to leverage the species specificity and generate mouse-specific primers to evaluate changes in macrophage phenotype. We investigated 4 of our cell lines in a co-culture system with macrophages derived from patient umbilical cord blood-derived macrophages samples with flow cytometry and found that those cell lines that induced high Arginase-1 expression in mouse macrophages similarly polarized human macrophages to an M2-like phenotype. While these findings validate our co-culture system, further investigation of macrophage activity using human-derived cells is warranted. We note that we recapitulated these findings in a smaller panel of NSCLC cell lines. This was substantially more time-consuming and significantly more expensive with respect to financial cost and the need to utilize patient samples, which required consent, monocyte isolation, and a challenging maturation process. As such, we believe that our in vitro co-culture system using mouse macrophages is an accessible, efficient, and biologically consistent initial platform to explore macrophage biology. If meaningful findings are identified, these can be further evaluated in downstream studies. The study of the function of CAFs in the NSCLC TME as they relate to macrophage phenotype is necessary. We have so far studied CAFs from five NSCLC patients and have not found major differences but are continuing to actively investigate the role of CAFs in macrophage polarization in the NSCLC TME. While our studies were performed in 2D co-cultures, organoid-type cultures should be performed. Also, while the xenograft studies were completed on subcutaneous tumors, it will be necessary to study orthotopic models (in the lung and at metastatic sites) as well. Most importantly, the features of individual NSCLC lines which led to the upregulation of Arginase-1 and other genes in this panel need further investigation.
In conclusion, our co-culture system is an accessible, focused, and reproducible model of macrophage activity in the NSCLC TME. We find that a large panel of patient-derived NSCLC lines, when co-cultured with CAFs and mouse macrophages, most frequently induce high expression of immunosuppressive Arginase-1, independent of major clinical, demographic, and molecular oncogenotype-related factors. These phenotypes are recapitulated in xenografts in vivo and are further clinically validated in TCGA-deposited tumor datasets. This co-culture model is an effective tool for early investigation of macrophage biology in the NSCLC TME and an easy-to-use system to investigate genetic expression profiles, clinicopathologic correlates, early validation of planned in vivo studies, and to screen for novel compounds which may target macrophage activity. Thus, our co-culture model serves as a robust, physiologically consistent platform from which to interrogate tumor cell and TME features and novel therapeutics which may affect macrophage phenotype.
Supplementary Material
Acknowledgements
This work was supported by NIH grants R01 CA243577 and U54 CA210181 (to RAB); NIH SPORE P50 CA070907, U54 CA224065, and CPRIT RP160652 (to JDM); the Effie Marie Cain Foundation (to RAB), NCI T32 CA124334 (PI: J Shay) (to JVP), Cancer Center Support Grant P30 CA142543 (LC), and the Burroughs-Wellcome Fund (RC). The results published here are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. The authors thank Novogene Bioinformatics Technology Co. Ltd (Beijing, China) for conducting the RNA sequencing, Simmons Cancer Center Tissue Resource (supported by NCI grant P30 CA142543), McDermott Sequencing, Microarray & Immune Phenotyping Core. We are indebted to Dr. Boning Gao for developing the patient derived CAFs, Elizabeth McMillan for discussion and consultation on biostatistical analyses of this study, Hyunsil Park for the assistance with mouse studies during the COVID-19 pandemic and thank members of the Brekken and Minna laboratories for comments and advice during the development and execution of this project.
Financial support:
This work was supported by NIH grants R01 CA243577 and U54 CA210181 (to RAB); NIH SPORE P50 CA070907, U54 CA224065, and CPRIT RP160652 (to JDM); the Effie Marie Cain Foundation (to RAB), NCI T32 CA124334 (PI: J Shay) (to JVP) Cancer Center Support Grant P30 CA142543 (LC), and the UTSW TARDIS Physician-Scientist Fellowship through Burroughs-Wellcome Fund (RC).
Abbreviations:
- NSCLC
non-small cell lung cancer
- LUAD
lung adenocarcinoma
- LUSC
Lung Squamous Cell Carcinoma
- BMDMs
bone marrow-derived macrophages
- CAFs
cancer-associated fibroblasts
- TME
tumor microenvironment
- TCGA
The Cancer Genome Atlas
- IHC
immunohistochemistry
- HCC
Hamon Center for Therapeutic Oncology Research
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: JDM receives licensing fees from the NIH and UTSW for distribution of human tumor cell lines. The other authors have no potential conflicts of interest to declare.
Literature Cited
- 1.Ruiz-Cordero R, Devine WP. Targeted Therapy and Checkpoint Immunotherapy in Lung Cancer. Surg Pathol Clin 2020;13:17–33. [DOI] [PubMed] [Google Scholar]
- 2.Zhang R, Zhu J, Liu Y, et al. Efficacy of immune checkpoint inhibitors in the treatment of non-small cell lung cancer patients with different genes mutation: A meta-analysis. Medicine 2021;100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haslam A, Prasad V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA network open 2019;2:e192535–e192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poh AR, Ernst M. Targeting Macrophages in Cancer: From Bench to Bedside. Front Oncol 2018;8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao L, Che X, Qiu X, et al. M2 macrophage infiltration into tumor islets leads to poor prognosis in non-small-cell lung cancer. Cancer Manag Res 2019;11:6125–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jackute J, Zemaitis M, Pranys D, et al. Distribution of M1 and M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC Immunol 2018;19:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arlauckas SP, Garren SB, Garris CS, et al. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics 2018;8:5842–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao J, Du Z, Li Z, et al. 6-Gingerol as an arginase inhibitor prevents urethane-induced lung carcinogenesis by reprogramming tumor supporting M2 macrophages to M1 phenotype. Food Funct 2018;9:4611–4620. [DOI] [PubMed] [Google Scholar]
- 9.Javle MM, Bridgewater JA, Gbolahan OB, et al. A phase I/II study of safety and efficacy of the arginase inhibitor INCB001158 plus chemotherapy in patients (Pts) with advanced biliary tract cancers. Journal of Clinical Oncology 2021;39:311–311. [Google Scholar]
- 10.Yan K, Wang Y, Lu Y, et al. Coexpressed Genes That Promote the Infiltration of M2 Macrophages in Melanoma Can Evaluate the Prognosis and Immunotherapy Outcome. Journal of Immunology Research 2021;2021:6664791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cavnar MJ, Zeng S, Kim TS, et al. KIT oncogene inhibition drives intratumoral macrophage M2 polarization. Journal of Experimental Medicine 2013;210:2873–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Müller-Quernheim UC, Potthast L, Müller-Quernheim J, et al. Tumor-cell co-culture induced alternative activation of macrophages is modulated by interferons in vitro. Journal of Interferon & Cytokine Research 2012;32:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuen J, Darowski D, Kluge T, et al. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PloS one 2017;12:e0182039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X-q, Kiefl R, Roskopf C, et al. Interactions among lung cancer cells, fibroblasts, and macrophages in 3D co-cultures and the impact on MMP-1 and VEGF expression. PLoS One 2016;11:e0156268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramirez RD, Sheridan S, Girard L, et al. Immortalization of Human Bronchial Epithelial Cells in the Absence of Viral Oncoproteins. Cancer Research 2004;64:9027–9034. [DOI] [PubMed] [Google Scholar]
- 16.Weischenfeldt J, Porse B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protoc 2008;2008:pdb prot5080. [DOI] [PubMed] [Google Scholar]
- 17.Banat GA, Tretyn A, Pullamsetti SS, et al. Immune and Inflammatory Cell Composition of Human Lung Cancer Stroma. PloS one 2015;10:e0139073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xuefeng X, Hou M-X, Yang Z-W, et al. Epithelial–mesenchymal transition and metastasis of colon cancer cells induced by the FAK pathway in cancer-associated fibroblasts. 2020;48:0300060520931242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray PJ. Macrophage Polarization. Annu Rev Physiol 2017;79:541–566. [DOI] [PubMed] [Google Scholar]
- 20.Sorrelle N, Ganguly D, Dominguez ATA, et al. Improved Multiplex Immunohistochemistry for Immune Microenvironment Evaluation of Mouse Formalin-Fixed, Paraffin-Embedded Tissues. J Immunol 2019;202:292–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gazdar AF, Minna JD. NCI series of cell lines: an historical perspective. J Cell Biochem Suppl 1996;24:1–11. [DOI] [PubMed] [Google Scholar]
- 22.Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37:773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miret JJ, Kirschmeier P, Koyama S, et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a NSCLC mouse model by activating anti-tumor immunity. Journal for immunotherapy of cancer 2019;7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuo I-Y, Yang Y-E, Yang P-S, et al. Converged Rab37/IL-6 trafficking and STAT3/PD-1 transcription axes elicit an immunosuppressive lung tumor microenvironment. Theranostics 2021;11:7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petty AJ, Yang Y. Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy 2017;9:289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMillan EA, Ryu MJ, Diep CH, et al. Chemistry-First Approach for Nomination of Personalized Treatment in Lung Cancer. Cell 2018;173:864–878.e829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dearden S, Stevens J, Wu Y-L, et al. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). 2013;24:2371–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chevallier M, Borgeaud M, Addeo A, et al. Oncogenic driver mutations in non-small cell lung cancer: Past, present and future. 2021;12:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mulshine JL, Ujhazy P, Antman M, et al. From clinical specimens to human cancer preclinical models—a journey the NCI‐cell line database—25 years later. Journal of cellular biochemistry 2020;121:3986–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun H, Cao S, Mashl RJ, et al. Comprehensive characterization of 536 patient-derived xenograft models prioritizes candidates for targeted treatment. Nature communications 2021;12:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hao NB, Lu MH, Fan YH, et al. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol 2012;2012:948098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sumitomo R, Hirai T, Fujita M, et al. M2 tumor-associated macrophages promote tumor progression in non-small-cell lung cancer. Exp Ther Med 2019;18:4490–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Byers LA, Diao L, Wang J, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013;19:279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steggerda SM, Bennett MK, Chen J, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. 2017;5:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ye Y, Jing Y, Li L, et al. Sex-associated molecular differences for cancer immunotherapy. Nat Commun 2020;11:1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abdou Y, Attwood K, Cheng TD, et al. Racial differences in CD8(+) T cell infiltration in breast tumors from Black and White women. Breast Cancer Res 2020;22:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Busch SE, Hanke ML, Kargl J, et al. Lung Cancer Subtypes Generate Unique Immune Responses. J Immunol 2016;197:4493–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Danaher P, Warren S, Lu R, et al. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA). Journal for ImmunoTherapy of Cancer 2018;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim BS, Clinton J, Wang Q, et al. Targeting ST2 expressing activated regulatory T cells in Kras-mutant lung cancer. Oncoimmunology 2020;9:1682380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vayrynen SA, Zhang J, Yuan C, et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin Cancer Res 2021;27:1069–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim DW, Min HS, Lee KH, et al. High tumour islet macrophage infiltration correlates with improved patient survival but not with EGFR mutations, gene copy number or protein expression in resected non-small cell lung cancer. Br J Cancer 2008;98:1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gazdar AF, Girard L, Lockwood WW, et al. Lung Cancer Cell Lines as Tools for Biomedical Discovery and Research. JNCI: Journal of the National Cancer Institute 2010;102:1310–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.