

Abstract
Malignant rhabdoid tumors (MRTs) are rare and highly aggressive pediatric cancers with no standard of care. MRTs are characterized by loss of SMARCB1, which results in upregulated expression of enhancer of zeste homolog 2 (EZH2), which is responsible for the methylation of lysine 27 of histone H3 (H3K27me3), leading to the repression of gene expression. Although previous reports suggest EZH2 as an effective therapeutic target, the functions of EZH1, the other homolog of EZH, in MRT remain unknown. Here, we show that EZH1, as well as EZH2, contributes to MRT cell growth and H3K27 methylation. Depletion or selective inhibition of EZH2 led to a compensatory increase in EZH1 expression, and depletion of EZH1 enhanced the effect of EZH2 inhibition. EZH1/2 dual inhibitors suppressed MRT cell growth markedly, reflecting the reduction of H3K27me3 accumulation at one of the EZH1/2 targets, the CDKN2A locus. Dual inhibition of EZH1/2 in vivo suppressed tumor growth completely, with no significant adverse effects. These findings indicate that both EZH1 and EZH2 are potential targets for MRT therapy, and that EZH1/2 dual inhibitors may be promising therapeutic strategies for MRT.
Keywords: EZH1, EZH2, H3K27me3, malignant rhabdoid tumors (MRTs), epigenetic drug, rare cancers
Graphical abstract
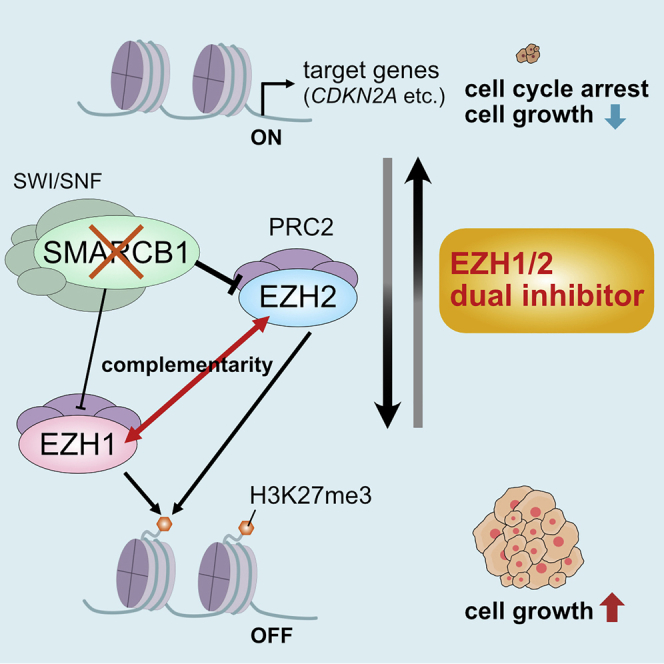
Malignant rhabdoid tumors (MRTs) are pediatric cancers with no standard of care. Both EZH1 and EZH2 contribute to MRT cell growth and H3K27 methylation, and dual inhibition of EZH1/2 strongly suppressed tumor growth of MRT. Our findings suggest that EZH1/2 inhibitors can be a promising therapy for MRT.
Introduction
Malignant rhabdoid tumors (MRTs) are rare and highly aggressive malignancies of infants and young children that occur mainly in the kidney (rhabdoid tumor of the kidney), brain (atypical teratoid rhabdoid tumor [AT/RT]), and soft tissue.1 While multimodal treatment consisting of surgery, chemotherapy, and radiation has brought modest improvements in overall survival, patients with MRT have a very poor prognosis, with a median survival of around 1 year.2, 3, 4 Consequently, new treatment approaches are needed to improve outcomes.
MRTs are characterized by biallelic inactivation of the tumor suppressor gene SMARCB1 (also known as SNF5, INI1, or BAF47),1,5 which encodes a core subunit of the switch/sucrose nonfermentable (SWI/SNF) complex. Loss of SMARCB1 upregulates expression of enhancer of zeste homolog 2 (EZH2).6,7 EZH2 is one of the components of the catalytic subunit of the polycomb repressive complex (PRC2), which catalyzes methylation of lysine 27 in histone H3 trimethylation (H3K27me3), leading to repression of gene expression.8 Depletion and inhibition of EZH2 suppress tumorigenesis in SMARCB1-deficient cells,6,7,9, 10, 11 suggesting that EZH2 inhibition is a promising strategy for anti-tumor therapy.12 Although EZH2-selective inhibitors are under evaluation in clinical trials, favorable responses are not observed in all patients.13
While EZH2 is recognized as an essential epigenetic regulator in various cancers, recent reports revealed that EZH1, the other homolog of EZH, is also involved in abnormal H3K27 methylation.14,15 EZH1 co-localizes with H3K27me3 to silence multiple genes associated with transcriptional regulation, proliferation, and differentiation.14,15 Several small compounds have been developed as EZH1/2 dual inhibitors, and have shown favorable effects against several types of cancer that overexpress or harbor mutations in EZH2 (e.g., adult T cell leukemia-lymphoma, acute myeloid leukemia, and multiple myeloma).15, 16, 17, 18, 19 These reports strongly suggest that EZH1/2 dual inhibitors have efficacy against MRTs; however, the functions of EZH1 in MRT remain unknown. In addition, the therapeutic efficacy of EZH1/2 dual inhibition remains unclear. Here, we investigated the role of EZH1 and EZH2 in MRT and examined the efficacy of EZH1/2 dual inhibitors both in vitro and in vivo.
Results
Function and expression of EZH1 and EZH2 in MRT cells
Previous reports show that EZH2 is required for cell proliferation and tumor formation in SMARCB1-deficient cells6; however, the role of EZH1 in MRT cells has not been clarified. Therefore, to elucidate the function of EZH1 and EZH2 in MRT cells, we examined the effects of knockdown (KD) of EZH1 and EZH2 using short hairpin RNA (shRNA). Double KD of EZH1 and EZH2 reduced H3K27me3 levels more and almost completely and significantly suppressed cell proliferation compared with single KD of each gene (Figures 1A and 1B). These data indicate that both EZH1 and EZH2 are essential for epigenetic regulation and growth of MRT cells. Interestingly, the KD of EZH2 increased the expression level of EZH1 protein in MRT cells (Figure 1B), which occurred independently of the shRNA target sequences (Figure S1C). A strong increase in levels of EZH1 mRNA was also observed in A204.1 cells, but there was a lesser or no increase in G401.6TG and TTC642 cells (Figures S1C and S1D). These data suggest that depletion of EZH2 results in a compensatory increase in EZH1 expression. Based on these findings, we speculated that dual inhibition of EZH1 and EZH2 could efficiently reduce H3K27me3 levels and suppress MRT cell growth.
Figure 1.

The function and expression of EZH1 and EZH2 in MRT
(A) The functions of EZH1 and EZH2 on cell proliferation in MRT cells. A204.1, TTC642, and G401.6TG cells were infected with retroviral shEZH1 and/or shEZH2 vectors, and transduced cells were selected with 0.5 mg/mL (A204.1) or 0.1 mg/mL (TTC642 and G401.6TG) of G418 for 7 days. The relative cell growth of the transduced cells was measured by WST-8 formazan dye (OD 450) for the indicated durations (n = 3, means ± SD). Differences were statistically evaluated by two-way ANOVA followed by Tukey’s multiple comparisons test. Statistical significance between control and each KD condition (shEZH1, shEZH2, and shEZH1/2) was observed after day 11 in all cell lines. shEZH1 versus control is p < 0.0001, shEZH2 versus control is p < 0.0001, and shEZH1/2 versus control is p < 0.0001 in all cell lines. Statistical significances between single KD and double KD on day 14 are as follows: shEZH1 versus shEZH1/2 is ∗∗∗p < 0.0001 in all cell lines. shEZH2 versus shEZH1/2 is ∗∗∗p < 0.0001 in A204.1 and G401.6TG cells, but p = 0.4519 in TTC642 cells. (B) Expression of EZH1, EZH2, and H3K27me3 proteins in cells single or double KD of EZH1 and EZH2. The expression level of each protein was determined by western blotting analysis. β-actin and H3 were used as the internal controls. The numbers below EZH1, EZH2, and H3K27me3 indicate each band density relative to control (taken as “1”). (C) Expression of EZH1 and EZH2 mRNAs in cells single or double KD. Relative expression level of each mRNA was evaluated by qRT-PCR (n = 3, means ± SD). The expression level of the control is indicated as “1.” ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus control. The KD efficacy was also confirmed by expression levels of proteins (B) and mRNAs (C).
Dual inhibition of EZH1 and EZH2 in MRT cells
To investigate the effects of chemical dual targeting of EZH1 and EZH2 in MRT cells, we treated five MRT cell lines with the EZH1/2 dual inhibitor DS-3201b, the partially selective inhibitors UNC1999 or CPI-360, or the EZH2-selective inhibitors GSK126 or EPZ-6438 (Figure S2A). The half-maximal inhibitory concentration (IC50) values of these EZH inhibitors against EZH1/2 and H3K27me3 were confirmed previously (Figure S2B).9,15,18, 19, 20, 21 Anti-proliferative effects were observed in MRT cells at 7 to 10 days post-exposure to inhibitors in a dose-dependent manner (Figures 2A and S2C). Of note, the EZH1/2 dual inhibitor reduced MRT cell growth more strongly than the EZH2-selective inhibitors or partially selective inhibitors, although there was a difference in sensitivity (Figures 2A and S2C). The IC50 values of these EZH inhibitors for each cell line are shown in Table 1. Sensitivity of each cell line to the EZH1/2 dual inhibitor associated with expression of EZH2 expression in each cell line (Table 1 and Figure 2B). The EZH1 highly expressing cells, such as the A204.1 and G401.6TG cells, showed low sensitivity to the EZH2-selective inhibitors (Table 1 and Figure 2B), suggesting that expression of EZH1 inversely associates with the anti-proliferative effect of EZH2-selective inhibitors.
Figure 2.

Dual inhibition of EZH1 and EZH2 in MRT cells
(A) The effect of EZH1/2 dual inhibitors and EZH2-selective inhibitors on the cell growth. Six MRTs (A204.1, TTC642, G401.6TG, KYM-1, JMU-RTK-2, and TTC549) and SMARCB1 wild-type RMS (RD) cells were treated with DS-3201b, UNC1999, CPI-360, GSK126, or EPZ-6438 for the indicated durations, and relative cell growth was evaluated by a WST-8 assay (n = 3, means ± SD). Differences were statistically evaluated by two-way ANOVA followed by Tukey’s multiple comparisons test. Statistical significance between control and each drug treatment (DS-3201b, UNC1999, CPI-360, GSK126, or EPZ-6438; 100 nM) is p < 0.001 in all MRT cell lines on day 14. (B) Relative expression levels of EZH1 and EZH2 mRNAs in steady state of each cell line (n = 3, means ± SD). (C) The methylation levels of H3K27 after treatment with EZH1/2 dual inhibitors and EZH2-selective inhibitors. Cells were treated with DS-3201b, UNC1999, CPI-360, GSK126, or EPZ-6438 (100 nM each) for 7 days, followed by western blotting analysis. The numbers below H3K27me3 indicate each band density relative to control (taken as “1”). (D and E) Expression of EZH1 and EZH2 proteins (D) and mRNAs (E) in DS-3201b- or EPZ-6438-treated cells. Cells were treated with DS-3201b (100 nM) or EPZ-6438 (100 nM) for 7 days, followed by western blotting and qRT-PCR, respectively. (D) The numbers below EZH1 indicate each band density relative to control (taken as “1”). (E) n = 3, means ± SD. The expression level of the control is indicated as “1.” ∗∗∗p < 0.001 versus control.
Table 1.
IC50 values of EZH inhibitors in MRT and RMS cell lines
| Cell line | Origin | IC50 (nM, day 11) |
||||
|---|---|---|---|---|---|---|
| DS-3201b | UNC1999 | CPI-360 | GSK126 | EPZ-6438 | ||
| TTC642 | soft tissue | 0.19 | 3.77 | 23.21 | 78.7 | 37.1 |
| A204.1 | soft tissue | 0.47 | 12.3 | 33.9 | 749 | 399.7 |
| G401.6TG | kidney | 13.3 | 76.7 | 68.5 | 261.1 | 112.6 |
| KYM-1 | soft tissue | 20.6 | 179.7 | 219.0 | 489.9 | 593.2 |
| JMU-RTK-2 | kidney | 101.3 | 177.3 | 289.7 | 393.4 | 468.7 |
| TTC549 | liver | 526.0 | 900.2 | 772.1 | 1097 | 1233 |
| RD | soft tissue (RMS) | 1303 | 2698 | 1442 | 3942 | 7756 |
We also tested the effect of the EZH inhibitors on SMARCB1 wild-type rhabdomyosarcoma (RMS) cell line RD. The anti-proliferative effect was barely observed in SMARCB1 wild-type RD cells (Figures 2A and S2C). These results suggest that the inhibitors act more selectively in SMARCB1-deficient cells.
In A204.1 and TTC642 cells, the EZH1/2 dual inhibitor DS-3201b reduced H3K27me3 levels more efficiently than the EZH2-selective inhibitors or partially selective inhibitors (Figure 2C). Because the knockdown of EZH2 led to increased expression of EZH1 protein (Figure 1B), we examined whether chemical inhibition of EZH2 also increased expression of EZH1. EPZ-6438 increased EZH1 protein expression in both A204.1 and TTC642 cells (Figure 2D). Increased expression of EZH1 mRNA was also observed in EPZ-6438-treated A204.1 cells (Figure 2E), which is consistent with the results observed after KD of EZH2 (Figures S1C and S1D). These findings indicate that loss of EZH2 function results in increased expression of EZH1 in MRT cells, and that EZH1/2 dual inhibitors inactivate both EZH1 and EZH2 efficiently, and may be an effective treatment for MRT.
Effect of EZH1 on MRT cell growth under conditions of EZH2 inhibition
Next, we examined the role of EZH1 in MRT cells under conditions of EZH2 inhibition. A204.1 cells expressed high levels of EZH1 (Figure 2B) and exhibited low sensitivity to the EZH2-selective inhibitor (Table 1). To investigate whether EZH1 expression is related to sensitivity to EZH2 inhibitors, A204.1 cells were treated with EPZ-6438 after KD of EZH1. Depletion of EZH1 enhanced the anti-proliferative effect of EPZ-6438 significantly and shifted the IC50 value of EPZ-6438 to 1.98 nM (Figures 3A and 3B). We then overexpressed EZH1 in A204.1 and TTC642 cells using a tetracycline-inducible gene expression system (Tet-On system) and confirmed that doxycycline (Dox) induced expression of both EZH1 protein and mRNA (Figures 3C and 3D). EZH1 overexpression attenuated the anti-proliferative activity of EPZ-6438, even in susceptible TTC642 cells, whereas no significant changes in sensitivity to DS-3201b were observed (Figure 3E). Taken together, these data suggest that EZH1 compensates for the function of EZH2 and supports MRT cell growth under conditions of EZH2 inhibition, and that depletion of EZH1 increases the efficacy of EZH2 inhibition.
Figure 3.

The function of EZH1 on MRT cell growth under the condition of EZH2 inhibition
(A) The effect of EZH1 KD under the condition of EZH2 inhibition on MRT cell growth. A204.1 cells were infected with shEZH1 retrovirus vector or empty vector (shControl), and transduced cells were selected with 1 μg/mL of puromycin for 10 days. The EZH1 KD and control cells were treated with EPZ-6438 (100 nM) for the indicated durations. The relative cell growth of the cells was measured by WST-8 assay (n = 3, means ± SD). Differences were statistically evaluated by two-way ANOVA followed by Tukey’s multiple comparisons test. Statistical significance between shControl and shEZH1 in EPZ-6438 treated cells (shControl plus EPZ-6438 versus shEZH1 plus EPZ-6438) is ∗∗∗p < 0.001. (B) Dose-response curve of EZH1 KD and control cells after treatment with various doses of EPZ-6438 (n = 3, means ± SD). Dose-dependent effects on cell viability at day 11 are shown. (C–E) The overexpression of EZH1 in MRT cells. Cells were infected with retroviral Tet-EZH1 or Tet-empty (as a control) vectors, and transduced cells were selected with 5 μg/mL of blasticidin for 10 days. The cells were incubated with 1 μg/mL of doxycycline (Dox) for 5 days, and GFP+ cells were sorted. (C) Expression of EZH1 protein in the presence or absence of Dox. (D) EZH1 mRNA expression induced by Dox treatment (n = 3, means ± SD). The expression level of the Tet-empty is indicated as “1.” ∗∗∗p < 0.001 versus Tet-empty. (E) The effect of EZH1 overexpression on the anti-proliferative effect of EZH inhibitors. The EZH1 overexpressed cells were treated with DS-3201b (100 nM) or EPZ-6438 (100 nM) for the indicated duration, and relative cell growth was measured by a WST-8 assay (n = 3, means ± SD). Differences were statistically evaluated by two-way ANOVA followed by Tukey’s multiple comparisons test. n.s., not significant, ∗∗∗p < 0.001 versus control. DS-3201b versus control is p < 0.0001, and EPZ-6438 versus control is p = 0.4448 on day 11 in Tet-EZH1 transduced A204.1 cells. DS-3201b versus control is p < 0.0001, and EPZ-6438 versus control is p = 0.4036 on day 11 in Tet-EZH1 transduced TTC642 cells.
Molecular mechanism underlying the anti-proliferative effect of the EZH1/2 inhibitor in MRT cells
To elucidate the molecular mechanism underlying the observed growth inhibition induced by EZH inhibitors in MRT cells, we performed RNA-sequencing (RNA-seq) analysis and compared differentially expressed genes between A204.1 cells treated with DS-3201b treatment or EPZ-6438. The analysis showed that 2,043 genes were altered significantly in DS-3201b-treated cells (upregulated, 1,414; downregulated, 629), and 956 genes were in EPZ-6438-treated cells (upregulated, 802; downregulated, 154). Of these, 584 overlapped between DS-3201b- and EPZ-6438-treated cells, whereas 1,459 were altered specifically in DS-3201b-treated cells (Figure 4A). Gene enrichment analysis revealed that DS-3201b altered genes associated significantly with cell division and cell-cycle processes (Figure 4B), suggesting that DS-3201b affects the cell cycle in MRT cells.
Figure 4.

The molecular mechanism underlying the anti-proliferative effect of EZH inhibitors in MRT cells
(A) Venn diagram of the differentially expressed genes (false discovery rate adjusted p < 0.05, and fold-change > 2). A204.1 cells were treated with DS-3201b (100 nM) or EPZ-6438 (100 nM) for 14 days, followed by RNA-sequencing (RNA-seq) analysis. The number in each circle represents the number of differentially expressed genes between DS-3201b treatment and EPZ-6438 treatment. (B) The enrichment analysis of the differentially expressed genes using Metascape. Bar graphs colored by p value show the top 20 enriched terms, specifically changed by DS-3201b treatment (top), overlapped between DS-3201b and EPZ-6438 (middle), and altered by EPZ-6438 treatment (bottom).
To further investigate whether DS-3201b regulates the cell cycle in MRT cells, we analyzed cell-cycle progression in DS-3201b- or EPZ-6438-treated A204.1 cells at days 8 and 14. Although both DS-3201b and EPZ-6438 induced cell-cycle arrest at day 14, the effect was already apparent in DS-3201b-treated cells at day 8 (Figure 5A). DS-3201b increased the percentage of cells in G1 phase, which was concomitant with a decrease in the number of cells in S phase and G2/M phase. No apparent increase in the subG1 fraction was observed at day 8, but DS-3201b increased the percentage of the subG1 fraction at day 14, suggesting that DS-3201b treatment induces apoptosis following cell-cycle arrest (Figure 5A). Gene enrichment analysis revealed apoptotic processes in the list of top 100 terms in DS-3201b-treated cells (Figure S3A). Thus, we assessed apoptosis induction by DS-3201b using Annexin V-APC and DAPI staining. We found that treatment with DS-3201b for 14 days increased the percentage of cells at the early stage of apoptosis (Figure S3B). However, another apoptotic phenotype, measured by cleavage of PARP-1 and caspase-3, was not detectable (Figure S3C), suggesting that the ratio of apoptotic cells was too low to detect signaling proteins. We speculate that the anti-proliferative activity of DS-3201b relies, at least in part, on induction of apoptosis, but that cell-cycle arrest is the most crucial molecular mechanism induced by DS-3201b in MRT cells.
Figure 5.

The reactivation of CDKN2A, one of the EZH1/2 targets, and induction of cell-cycle arrest by EZH1/2 dual inhibition
(A) Representative histograms of cell-cycle analysis. A204.1 cells were treated with DS-3201b (100 nM) or EPZ-6438 (100 nM) for the indicated duration, followed by the cell-cycle analysis with PI-staining. The y axes represent cell numbers, and x axes show DNA content (PI intensity). The numbers indicate the percentages of cells in each cell-cycle phase. (B and C) Heatmaps of differentially expressed genes belonging to the “G1/S transition of mitotic cell cycle” (GO: 0000082; B) and “G2/M transition of mitotic cell cycle” (GO: 0000086; C) terms. Mean values of signal intensity acquired from RNA-seq data are represented by colors (n = 3 each, false discovery rate adjusted p < 0.05, fold-change > 2). (D) Expression of CDKN2A, CDKN2C, and CDKN1A mRNAs in DS-3201b- or EPZ-6438-treated cells. Cells were treated with DS-3201b (DS; 100 nM) or EPZ-6438 (EPZ; 100 nM) for 7 days, followed by qRT-PCR. n = 3, means ± SD. The expression level of the control is indicated as “1.” ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus control. (E) Schematic of the CDKN2A locus and the locations of primer pairs for ChIP-qPCR analysis. (F) H3K27me3 enrichment at the CDKN2A locus. A204.1 cells were treated with DS-3201b (100 nM) for 7 days, followed by ChIP-qPCR analysis. Normal rabbit IgG was used as a negative control for the immunoprecipitation. The primer pair against ACTB was used as a negative control for the qPCR. n = 6, means ± SD. ∗∗∗p < 0.001, as indicated by the bracket.
Next, we sought to identify the molecule responsible for cell-cycle arrest induced by DS-3201b. Comparison of cell-cycle-associated gene expression signatures revealed that CDKN2A was markedly upregulated in DS-3201b-treated cells (Figures 5B and 5C). The qRT-PCR analysis confirmed that DS-3201b increased expression of CDKN2A significantly (Figure 5D). Although expression of other PRC2 target genes CDKN2C and CDKN1A was also upregulated by DS-3201b, the increase in CDKN2A expression was more marked and reflected the anti-proliferative effects of DS-3201b in MRT cells (Figures 5D and S4A). Double KD of EZH1 and EZH2 increased CDKN2A expression to a greater extent than single KD of either gene (Figure S4B), which correlated with the reduction of H3K27me3 levels and suppression of cell proliferation (Figures 1A and 1B). We examined the presence of the H3K27me3 marks at the CDKN2A locus in A204.1 cells by performing chromatin immunoprecipitation (ChIP) coupled with qPCR (ChIP-qPCR). H3K27me3 was more accumulated at the exon 1α (p16-CDKN2A) compared with exon 1β (p14-ARF). DS-3201b reduced the H3K27me3 marks at the CDKN2A locus significantly (Figures 5E and 5F). The reduction in H3K27me3 marks at the CDKN2A locus was induced significantly by single KD of EZH1 or EZH2, although the decrease in H3K27me3 was more marked after double KD of EZH1 and EZH2 (Figure S4C). Collectively, these data indicate that EZH1/2 dual inhibition cancels H3K27me3 accumulation and effectively reactivates EZH1/2 target genes such as CDKN2A in MRT cells.
Morphological changes in MRT cells induced by EZH inhibitors
Gene enrichment analysis also revealed that both DS-3201b and EPZ-6438 significantly altered genes associated with cell growth, cell morphology, and differentiation (Figure 4B). EZH2 maintains the stem cell-associated signature in Smarcb1-deficient mouse embryonic fibroblasts and SMARCB1-deficient human pluripotent stem cells.6,22 EPZ-6438 induces genes responsible for neuronal differentiation and morphological changes in MRT cells,9 which is consistent with our RNA-seq data. Morphological alteration of A204.1 cells was observed after exposure to DS-3201b for 7 days (Figure S5A). EZH inhibitors increased the number of long, thin, spindle-shaped cells. Recent studies show that EZH2 and H3K27me3 are enriched at the loci of differentiation genes.23 Therefore, we confirmed expression of genes reported to be involved in MRT differentiation.9,24,25 Expression of DOCK4 and α-SMA increased in both DS-3201b- and EPZ-6438-treated cells (Figure S5B). Double KD of EZH1 and EZH2 also led to upregulation of these genes (Figure S5C). By contrast, another differentiation-related gene, CD133,9,25 was not detected in A204.1 or TTC642 cells (data not shown). Based on these findings, we speculate that the morphological changes mediated by DS-3201b are partly associated with differentiation.
The anti-proliferative effect of the EZH1/2 dual inhibitor in vivo
To confirm the effects of the EZH1/2 dual inhibitor in vivo, we established an A204.1 xenograft mouse model. Oral administration of DS-3201b completely inhibited tumor growth and found a significant difference in tumor size between DS-3201b-treated and untreated mice at day 18 (Figure 6A). No significant body weight loss was observed during treatment (Figure 6B). Tumor size and weight were significantly lower in treated mice than in the control group (Figures 6C and 6D). We sought to compare DS-3201b with EPZ-6438 directly; however, the experiment was not successful due to the difference in solubility and the number of doses a day. Consistent with the investigation in vitro (Figure 2A and Table 1), EPZ-6438 slightly inhibited tumor growth in A204.1 xenografted mice, but the difference is not statistically significant (Figures S6A and S6B). On day 21, the relative tumor volume of DS-3201b-treated mice was 0.018, but that of EPZ-6438-treated mice was 0.88 (Figure S6C). Histologic analysis by H&E staining revealed that tumor tissues from the control mice consisted of densely packed tumor cells with atypical mitotic figures, indicating abnormal cell proliferation (Figure 6E). By contrast, tumor tissues from DS-3201b-treated mice were composed of fewer and loosely distributed tumor cells, and atypical mitosis was observed much more rarely than in control mice (Figure 6E). To validate the mechanism underlying the anti-tumor effects of DS-3201b in vivo, we evaluated expression of H3K27me3 and CDKN2A by immunohistochemistry (IHC). Complete loss of H3K27me3 was observed in DS-3201b-treated tumors (Figure 6F). Expression of CDKN2A in tumors from the DS-3201b-treated mice increased markedly, reflecting the reduction in H3K27me3 levels (Figures 6F and 6G). These results suggest that the potent anti-tumor activity of DS-3201b in vivo is mediated by a reduction in H3K27me3 levels, leading to upregulation of CDKN2A expression. Collectively, our findings indicate that EZH1/2 dual inhibitors efficiently suppress tumor progression and may be promising drugs for successful treatment of MRT.
Figure 6.

The anti-proliferative effect of EZH1/2 dual inhibitor in vivo
(A) The tumor volume of A204.1 xenograft mice. The mice were orally administered once daily with 100 mg/kg DS-3201 (calculated as a free-body DS-3201a, n = 10) or water (as control, n = 10). Differences were statistically evaluated by two-way ANOVA followed by Tukey’s multiple comparisons test. DS-3201 versus control is ∗∗p = 0.003 at day 18. (B) Body weight of the A204.1 xenograft mice during the treatments. (C and D) Representative image (C) and weight (D) of tumors from all of the A204.1 xenograft mice at experimental endpoint (day 22). Differences were statistically evaluated by Mann-Whitney test. ∗∗∗p < 0.001 versus control. (E) Representative images of H&E staining. The tumor tissues from the control group had a high number of mitoses (5–10 per high power field [HPF]) with abnormal mitotic figures, as shown by the arrows. DS-3201-treated tumor tissues rarely had mitosis (0–1 HPF). Scale bars, 50 μm. (F and G) Representative images of immunohistochemical staining of H3K27me3 (F) and CDKN2A (G). The tumors whose volume and weight were closest to the mean were used for immunohistochemistry. Scale bars, 50 μm.
Discussion
MRTs are rare and extremely aggressive pediatric cancers that currently have no consistently efficacious therapeutics. Mutations or deletions in the SMARCB1 gene are observed in virtually all MRTs. Previous studies identified an antagonistic functional relationship between SWI/SNF and PRC2 complexes, and that loss of SMARCB1 results in increased expression of EZH2, widespread accumulation of H3K27me3, repression of PRC2 target genes, and tumor formation.6,7
In this study, we revealed that EZH1, as well as EZH2, also contributes to MRT cell growth and H3K27 methylation. We showed that both EZH1 and EZH2 are responsible for accumulation of H3K27me3 at one of the PRC2 targets, the CDKN2A locus. EZH1 depletion enhances the effect of EZH2 inhibition in MRT cells. These data suggest that inhibition of EZH1 efficiently reduces residual H3K27me3 after EZH2 inhibition, reactivates the PRC2 target genes, and suppresses MRT cell growth. The double KD of EZH1 and EZH2 does not induce the destabilization of the PRC2 complex, at least in the duration we examined (Figure S1E). We also demonstrated that EZH1/2 dual inhibitors markedly suppress the growth of MRT cells even more selectively in SMARCB1-deficient cells, consistent with EPZ-6438.9 Several SWI/SNF complex members are genetically altered in a wide variety of cancers, suggesting that dual inhibition of EZH1/2 may be an effective treatment for these cancers.
We also found that the sensitivities of EZH2-selective inhibitors are inversely correlated with the EZH1 expression. While the biallelic inactivation of SMARCB1 induces the overexpression of EZH2,6,7 the involvement of EZH1 expression has not been clarified. We used previously published datasets from the Gene Expression Omnibus database repository (GEO) and confirmed that expression of EZH2 was significantly higher in tumor tissues regardless of the onset sites, compared with that in corresponding normal tissues (Figure S7A). EZH1 expression was slightly elevated in tumor tissues, but the difference was not statistically significant (Figure S7A). Exogenous expression of SMARCB1 significantly decreased the expression of EZH2 protein and mRNA (Figures S7B and S7C), leading to the growth inhibition of MRT cells (Figure S7D). EZH1 was also reduced, but only a slight change was observed in A204.1 cells (Figures S7B and S7C). EZH1 and EZH2 expressions were increased by KD of SMARCB1 in SMARCB1 wild-type RD cells, promoting cell growth (Figures S7E–S7H). These findings suggest that the loss of SMARCB1 upregulates both EZH1 and EZH2 expression, although SMARCB1 is only partially responsible for the expression of EZH1.
EZH1 compensates for the genetic deletion of Ezh2 in embryonic stem cells.14 While the enzymatic EZH2 inhibition removes EZH2 from its target loci, redeployment of EZH1 occurs at the EZH2-free loci, which prevents reduction of H3K27me3 and gene reactivation in adult T cell leukemia-lymphoma cells,15 indicating that EZH1 has a compensatory function in response to EZH2 inhibition. In exploring the role of EZH1 in MRT cells, we discovered that the KD of EZH2 or the selective inhibition of EZH2 increased the expression level of EZH1 protein. ChIP-qPCR analysis reveals that H3K27me3 was generally downregulated after treatment with either DS-3201b or EPZ-6438 (Figure S8), suggesting that increased expression of EZH1 is independent of the H3K27me3 reduction. Although the expression of EZH1 mRNA differed depending on each cell line, the expression level of EZH1 protein was consistently increased in all cell lines tested. These findings suggest that transcriptional regulation may be partly involved in the increased expression of EZH1, while other conceivable hypotheses are that the protein modification pathway or translational regulation is related to the expression or stability of EZH1 protein. As shown in Figure 4B, genes associated with protein modification are specifically enriched in EPZ-6438-treated cells. Previous reports indicate that the translation of EZH2 is regulated by microRNAs (miRNAs), such as miR-101.26 Based on the TargetScan database (http://www.targetscan.org/), the miRNAs predicted to be associated with EZH1 expression are entirely different from those targeting EZH2. The depletion or inhibition of EZH2 may mediate the miRNAs targeting EZH1, resulting in the increased expression of EZH1 protein. Although there are several plausible mechanisms, further investigation is needed to elucidate the regulation of EZH1 expression under the condition of EZH2 depletion or inhibition.
We investigated the mode of action underlying the MRT cell growth inhibition of the EZH1/2 dual inhibitor and found that the CDKN2A-mediated cell-cycle arrest is one of the most crucial molecular mechanisms. The induction of apoptosis and cell differentiation by EZH1/2 dual inhibitors also has been reported in hematologic malignancies.15, 16, 17 Our data reveal that DS-3201b treatment induces the morphological changes of MRT cells and increases the expression of DOCK4 and α-SMA. Although these genes are reported to be involved in MRT differentiation, whether the EZH1/2 dual inhibitor induces differentiation in MRT cells cannot be concluded. As reported previously,9 morphological alteration is not observed in all cell lines. Various genes have been proposed as differentiation markers, whereas all marker genes are not expressed in all MRT cells. MRT can arise throughout the body, and the expressions of marker genes are not necessarily reflected in a clinical case. Further study about differentiation markers of MRT may be helpful to investigate the detailed mechanism of EZH1/2 dual inhibitors.
We showed the efficacy of the EZH1/2 dual inhibitor in the xenograft mice model. Our data strongly suggest that DS-3201b effectively suppresses tumor growth without significant adverse effects. Repeated dose toxicity study also demonstrated that DS-3201b shows no critical or severe toxicity.19 Phase I investigator-initiated study of DS-3201b in pediatric, adolescent, and young adult patients with malignant solid tumors (ELEPHANT trial, NCCH1904/MK007 trial) is now undergoing. Collectively, this study provides evidence that EZH1 and EZH2 are effective targets in MRT cells, and our findings support further studies of EZH1/2 dual inhibitors as a potential therapeutic option for patients with MRT.
Materials and methods
Cell culture and treatment
Human MRT cell lines A204.1, TTC642, G401.6TG, and TTC549 were described elsewhere.27,28 The cells were cultured in RPMI-1640 medium (30264-56; Nacalai Tesque, Kyoto, Japan) supplemented with 10% heat-inactivated FBS (10270106; Thermo Fisher Scientific, Waltham, MA, USA) and 1% penicillin/streptomycin (P/S, 26253-84; Nacalai Tesque). JMU-RTK-2 (JCRB1484) was purchased from the Japanese Collection Research Bioresources Cell Bank (Osaka, Japan). JMU-RTK-2 cells were cultured in DMEM (08456-36; Nacalai Tesque) plus 10% FBS and 1% P/S. KYM-1 (JCRB0627) and human rhabdomyosarcoma cell line RD (JCRB9072) were obtained as gifts from Prof. Yukihiro Akao, Gifu University.29 RD cells were cultured in Eagle’s minimum essential medium (M4655; Merck, Darmstadt, Germany) with 10% FBS and 1% P/S, and KYM-1 cells were cultured in a 1:1 mixture of DMEM and Ham’s F12 medium (17458-65; Nacalai Tesque) supplemented with 10% FBS and 1% P/S. All cell lines were maintained at 37°C with 5% CO2. The cells were tested for mycoplasma contamination using an e-Myco Plus Mycoplasma PCR Detection Kit (25,237; iNtRON Biotechnology, Burlington, MA, USA).
For in vitro experiments, an EZH1/2 dual inhibitor, DS-3201b (also called valemetostat, HY-109108A; MedChemExpress, Monmouth Junction, NJ, USA), partially selective inhibitors, UNC1999 (S7165; Selleck, Houston, TX, USA), and CPI-360 (S7656; Selleck), and EZH2-selective inhibitors, GSK126 (S7061; Selleck), and EPZ-6438 (namely tazemetostat, E7438; Selleck) were dissolved in DMSO and added to the cell culture medium at a final concentration of DMSO <0.1%; this concentration showed no significant effect on the growth and differentiation of the cells (data not shown). The relative cell growth was measured by the absorption spectrum of WST-8 formazan dye (OD 450). The detailed information of the cell growth assay is shown in the supplemental methods.
Protein extraction and western blotting
Whole cell lysate preparations and western blotting experiments were carried out as described previously.30 Histone proteins were isolated by the acid extraction method.31 The primary antibodies used for western blotting are listed in the supplemental methods.
RNA isolation and RT-PCR
Total RNA was isolated from cells by using a NucleoSpin RNA Plus kit (740984; Takara, Otsu, Japan) according to the manufacturer’s protocol. To synthesize cDNA, total RNA was reverse transcribed with SuperScript IV VILO (11766050; Invitrogen, Carlsbad, CA, USA). Quantitative PCR was then performed using real-time PCR system (QuantStudio 12K Flex; Applied Biosystems, Waltham, MA, USA) with Taqman probes (Applied Biosystems) or specific primers and SYBRGreen (Roche). The probes and primers used in this study are summarized in the supplemental methods.
In vivo xenograft studies
BALB/c nu/nu nude mice were obtained from Japan SLC (Hamamatsu, Japan). After 1 week of preliminary care, mice were used for experiments. Human MRT A204.1 cells (2 × 106 cells) were suspended in 100 μL of 50% Matrigel (#356234; Corning, NY, USA) prepared in PBS and subcutaneously inoculated into the left flank of 5-week-old female mice. After the mean tumor volume had reached approximately 100 mm3, the mice were randomized and separated into two groups. The grouping day was set as day 0, and treatment was started at day 1. DS-3201b (valemetostat tosylate) was suspended in the sterile purified water and administered orally once daily at a dose of 100 mg/kg (calculated as a free-body DS-3201a). EPZ-6438 was suspended in 5% DMSO, 40% PEG300, 5% Tween 80, and 50% sterile purified water. Tumor size was monitored by measuring the length (L) and width (W), and the volumes (V) were estimated according to the following formula: V = (L × W2) × 0.5. The day after the final administration, the mice were killed for the assessment of tumor tissues.
Statistical analysis
Each experiment was performed in triplicate. Data are presented as the mean ± SD. Unless stated otherwise, differences were statistically evaluated by one-way ANOVA followed by the t test or Dunnett’s multiple comparison test. Statistic evaluation was performed using GraphPad 6.0 (GraphPad Software, La Jolla, CA, USA) and JMP 14 (SAS Institute, Cary, NC, USA) software. The level of significance was set at p < 0.05.
Study approval
All animal experimental protocols were approved by the Institutional Animal Care and Use Committee at the National Cancer Center. Animal experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals. Each experiment was carried out in a specific pathogen-free environment at the animal facility of the National Cancer Center, in accordance with institutional guidelines.
Acknowledgments
The authors thank Yukihiro Akao for providing the cell lines. We are grateful to Kazuki Heishima for providing expert advice and for histology. This work was supported by the National Cancer Center Research and Development Fund (2020-S-6 to H.S.) and the Rare Cancer Grant II in the National Cancer Center Japan (G018 to H.S.).
Author contributions
H.S. designed the study and performed most of the experiments, and data collection and analysis, as well as writing and final approval of the manuscript. R.S., M.N., A.H., K.Y., K.T., J.I., Y.K., T.O., and C.O. provided technical support for experiments and approved the final version of the manuscript. I.K. helped to design the experiments, helped to write the manuscript, and assisted with final approval of the manuscript.
Declaration of interests
I.K. received research grants from Daiichi Sankyo Company (CH24043) and Sumitomo Dainippon Pharma (C2018-189).
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2022.09.006.
Supplemental information
Data availability
The gene expression data generated during this study are deposited in GEO with accession numbers GSE205086.
References
- 1.Biegel J.A., Tan L., Zhang F., Wainwright L., Russo P., Rorke L.B. Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin. Cancer Res. 2002;8:3461–3467. [PubMed] [Google Scholar]
- 2.Dufour C., Beaugrand A., Le Deley M.C., Bourdeaut F., André N., Leblond P., Bertozzi A.I., Frappaz D., Rialland X., Fouyssac F., et al. Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer. 2012;118:3812–3821. doi: 10.1002/cncr.26684. [DOI] [PubMed] [Google Scholar]
- 3.Farber B.A., Shukla N., Lim I.I.P., Murphy J.M., La Quaglia M.P. Prognostic factors and survival in non-central nervous system rhabdoid tumors. J. Pediatr. Surg. 2017;52:373–376. doi: 10.1016/j.jpedsurg.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng H., Yang S., Cai S., Ma X., Qin H., Zhang W., Fu L., Zeng Q., Wen M., Peng X., et al. Clinical and prognostic characteristics of 53 cases of extracranial malignant rhabdoid tumor in children. A single-institute experience from 2007 to 2017. Oncologist. 2019;24:e551–e558. doi: 10.1634/theoncologist.2018-0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Versteege I., Sévenet N., Lange J., Rousseau-Merck M.F., Ambros P., Handgretinger R., Aurias A., Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- 6.Wilson B.G., Wang X., Shen X., McKenna E.S., Lemieux M.E., Cho Y.J., Koellhoffer E.C., Pomeroy S.L., Orkin S.H., Roberts C.W. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18:316–328. doi: 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helming K.C., Wang X., Roberts C.W.M. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell. 2014;26:309–317. doi: 10.1016/j.ccr.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Margueron R., Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knutson S.K., Warholic N.M., Wigle T.J., Klaus C.R., Allain C.J., Raimondi A., Porter Scott M., Chesworth R., Moyer M.P., Copeland R.A., et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. USA. 2013;110:7922–7927. doi: 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurmasheva R.T., Sammons M., Favours E., Wu J., Kurmashev D., Cosmopoulos K., Keilhack H., Klaus C.R., Houghton P.J., Smith M.A. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer. 2017;64 doi: 10.1002/pbc.26218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torchia J., Golbourn B., Feng S., Ho K.C., Sin-Chan P., Vasiljevic A., Norman J.D., Guilhamon P., Garzia L., Agamez N.R., et al. Integrated (epi)-genomic analyses identify subgroup-specific therapeutic targets in CNS rhabdoid tumors. Cancer Cell. 2016;30:891–908. doi: 10.1016/j.ccell.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim K.H., Roberts C.W. Targeting EZH2 in cancer. Nat. Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Italiano A., Soria J.C., Toulmonde M., Michot J.M., Lucchesi C., Varga A., Coindre J.M., Blakemore S.J., Clawson A., Suttle B., et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19:649–659. doi: 10.1016/S1470-2045(18)30145-1. [DOI] [PubMed] [Google Scholar]
- 14.Shen X., Liu Y., Hsu Y.J., Fujiwara Y., Kim J., Mao X., Yuan G.C., Orkin S.H. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell. 2008;32:491–502. doi: 10.1016/j.molcel.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamagishi M., Hori M., Fujikawa D., Ohsugi T., Honma D., Adachi N., Katano H., Hishima T., Kobayashi S., Nakano K., et al. Targeting excessive EZH1 and EZH2 activities for abnormal histone methylation and transcription network in malignant lymphomas. Cell Rep. 2019;29:2321–2337.e7. doi: 10.1016/j.celrep.2019.10.083. [DOI] [PubMed] [Google Scholar]
- 16.Fujita S., Honma D., Adachi N., Araki K., Takamatsu E., Katsumoto T., Yamagata K., Akashi K., Aoyama K., Iwama A., et al. Dual inhibition of EZH1/2 breaks the quiescence of leukemia stem cells in acute myeloid leukemia. Leukemia. 2018;32:855–864. doi: 10.1038/leu.2017.300. [DOI] [PubMed] [Google Scholar]
- 17.Nakagawa M., Fujita S., Katsumoto T., Yamagata K., Ogawara Y., Hattori A., Kagiyama Y., Honma D., Araki K., Inoue T., et al. Dual inhibition of enhancer of zeste homolog 1/2 overactivates WNT signaling to deplete cancer stem cells in multiple myeloma. Cancer Sci. 2019;110:194–208. doi: 10.1111/cas.13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu B., On D.M., Ma A., Parton T., Konze K.D., Pattenden S.G., Allison D.F., Cai L., Rockowitz S., Liu S., et al. Selective inhibition of EZH2 and EZH1 enzymatic activity by a small molecule suppresses MLL-rearranged leukemia. Blood. 2015;125:346–357. doi: 10.1182/blood-2014-06-581082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Honma D., Kanno O., Watanabe J., Kinoshita J., Hirasawa M., Nosaka E., Shiroishi M., Takizawa T., Yasumatsu I., Horiuchi T., et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017;108:2069–2078. doi: 10.1111/cas.13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradley W.D., Arora S., Busby J., Balasubramanian S., Gehling V.S., Nasveschuk C.G., Vaswani R.G., Yuan C.C., Hatton C., Zhao F., et al. EZH2 inhibitor efficacy in non-Hodgkin's lymphoma does not require suppression of H3K27 monomethylation. Chem. Biol. 2014;21:1463–1475. doi: 10.1016/j.chembiol.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 21.McCabe M.T., Ott H.M., Ganji G., Korenchuk S., Thompson C., Van Aller G.S., Liu Y., Graves A.P., Della Pietra A., 3rd, Diaz E., et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 22.Terada Y., Jo N., Arakawa Y., Sakakura M., Yamada Y., Ukai T., Kabata M., Mitsunaga K., Mineharu Y., Ohta S., et al. Human pluripotent stem cell-derived tumor model uncovers the embryonic stem cell signature as a key driver in atypical teratoid/rhabdoid tumor. Cell Rep. 2019;26:2608–2621.e6. doi: 10.1016/j.celrep.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 23.Erkek S., Johann P.D., Finetti M.A., Drosos Y., Chou H.C., Zapatka M., Sturm D., Jones D.T.W., Korshunov A., Rhyzova M., et al. Comprehensive analysis of chromatin states in atypical teratoid/rhabdoid tumor identifies diverging roles for SWI/SNF and polycomb in gene regulation. Cancer Cell. 2019;35:95–110.e8. doi: 10.1016/j.ccell.2018.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato H., Ohta S., Koshida S., Narita T., Taga T., Takeuchi Y., Sugita K. Expression of pericyte, mesangium and muscle markers in malignant rhabdoid tumor cell lines: differentiation-induction using 5-azacytidine. Cancer Sci. 2003;94:1059–1065. doi: 10.1111/j.1349-7006.2003.tb01401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gadd S., Sredni S.T., Huang C.C., Perlman E.J. Rhabdoid tumor: gene expression clues to pathogenesis and potential therapeutic targets. Lab. Invest. 2010;90:724–738. doi: 10.1038/labinvest.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao P., Deng Z., Wan M., Huang W., Cramer S.D., Xu J., Lei M., Sui G. MicroRNA-101 negatively regulates Ezh2 and its expression is modulated by androgen receptor and HIF-1alpha/HIF-1beta. Mol. Cancer. 2010;9:108. doi: 10.1186/1476-4598-9-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuwahara Y., Charboneau A., Knudsen E.S., Weissman B.E. Reexpression of hSNF5 in malignant rhabdoid tumor cell lines causes cell cycle arrest through a p21(CIP1/WAF1)-dependent mechanism. Cancer Res. 2010;70:1854–1865. doi: 10.1158/0008-5472.CAN-09-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuwahara Y., Wei D., Durand J., Weissman B.E. SNF5 reexpression in malignant rhabdoid tumors regulates transcription of target genes by recruitment of SWI/SNF complexes and RNAPII to the transcription start site of their promoters. Mol. Cancer Res. 2013;11:251–260. doi: 10.1158/1541-7786.MCR-12-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugito N., Taniguchi K., Kuranaga Y., Ohishi M., Soga T., Ito Y., Miyachi M., Kikuchi K., Hosoi H., Akao Y. Cancer-specific energy metabolism in rhabdomyosarcoma cells is regulated by MicroRNA. Nucleic Acid Ther. 2017;27:365–377. doi: 10.1089/nat.2017.0673. [DOI] [PubMed] [Google Scholar]
- 30.Shinohara H., Kumazaki M., Minami Y., Ito Y., Sugito N., Kuranaga Y., Taniguchi K., Yamada N., Otsuki Y., Naoe T., et al. Perturbation of energy metabolism by fatty-acid derivative AIC-47 and imatinib in BCR-ABL-harboring leukemic cells. Cancer Lett. 2016;371:1–11. doi: 10.1016/j.canlet.2015.11.020. [DOI] [PubMed] [Google Scholar]
- 31.Shechter D., Dormann H.L., Allis C.D., Hake S.B. Extraction, purification and analysis of histones. Nat. Protoc. 2007;2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The gene expression data generated during this study are deposited in GEO with accession numbers GSE205086.