

Abstract
Immune checkpoint inhibitors (ICIs) are effective for various types of cancer, and their application has led to paradigm shifts in cancer treatment. While many patients can obtain clinical benefits from ICI treatment, a large number of patients are primarily resistant to such treatment or acquire resistance after an initial response. Thus, elucidating the resistance mechanisms is warranted to improve the clinical outcomes of ICI treatment. ICIs exert their antitumor effects by activating T cells in the tumor microenvironment. There are various resistance mechanisms, such as insufficient antigen recognition by T cells, impaired T‐cell migration and/or infiltration, and reduced T‐cell cytotoxicity, most of which are related to the T‐cell activation process. Thus, we classify them into three main mechanisms: resistance mechanisms related to antigen recognition, T‐cell migration and/or infiltration, and effector functions of T cells. In this review, we summarize these mechanisms of resistance to ICIs related to the T‐cell activation process and progress in the development of novel therapies that can overcome resistance.
Keywords: cancer immunology, exhaustion, immune checkpoint inhibitor, resistance, T cell
While many patients can obtain clinical benefits from immune checkpoint inhibitor treatment, a large number of patients are primarily resistant to such treatment or acquire resistance after an initial response. There are various resistance mechanisms, and we classify them into three main mechanisms related to the T‐cell activation process: resistance mechanisms related to antigen recognition, T‐cell migration and/or infiltration, and effector functions of T cells.
1. INTRODUCTION
Immune checkpoint inhibitors (ICIs), represented by anti‐programmed cell death 1 (PD‐1)/PD‐1 ligand 1 (PD‐L1) monoclonal antibodies (mAbs) and cytotoxic T‐lymphocyte‐associated protein 4 (CTLA‐4) mAbs, have been shown to be effective to a variety of cancers, including melanoma, non–small cell lung cancer, renal cell carcinoma, head and neck cancer, and gastrointestinal cancer. 1 , 2 , 3 , 4 , 5 , 6 However, the efficacy of ICI monotherapy is limited, and the detailed mechanisms are not fully understood, necessitating more basic and clinical researches. 7
In the tumor microenvironment (TME), T cells, especially CD8+ T cells, which reportedly attack cancer cells via recognition of cancer antigens in the context of major histocompatibility complex (MHC) molecules, play a crucial role in antitumor immunity. 7 , 8 , 9 ICIs exert antitumor effects by inhibiting T‐cell suppressive signals and subsequently activating T‐cell–mediated cytotoxicity. 7 , 8 , 9 Somatic mutation‐derived neoantigens, which can be recognized as non‐self antigens, reportedly induce strong immune responses similar to those induced by foreign antigens such as virus antigens. 10 , 11 Thus, neoantigens are believed to induce an inflamed TME characterized by high CD8+ T‐cell infiltration, which is very important for the ICI response, and the number of neoantigens is reportedly correlated with the degree of inflammation in the TME. 11 , 12 , 13 , 14 In addition, CD8+ T‐cell–mediated cytotoxicity reportedly induces PD‐L1 expression via the interferon‐gamma (IFN‐γ) signaling pathway, and the induced PD‐L1 suppresses CD8+ T cells by binding to PD‐1. 15 Therefore, PD‐L1 expression, CD8+ T‐cell infiltration, and tumor mutational burden (TMB) are candidate biomarkers for ICI therapy with close relations to each other. 16 However, there are substantial conflicting data, 17 , 18 , 19 and none of these biomarker candidates seems to be a sufficient predictor of ICI response.
On the other hand, tumors can modify the TME to one that benefits themselves through the process of tumor immune editing. 20 Tumor cells can evade antitumor immunity through various mechanisms even during ICI treatment, such as loss of MHC and impaired IFN‐γ signaling, leading to resistance to ICIs. 21 , 22 Elucidation of primary and acquired resistance mechanisms to ICIs is needed to develop predictive biomarkers for ICIs and/or novel therapies with higher efficacy. Although many studies have shown some representative mechanisms, the complex nature of the mechanisms makes it difficult to be understood sufficiently.
While there are various complicated resistance mechanisms, most of them are related to the T‐cell activation process because T‐cell activation is essential for ICI efficacy. 7 , 8 , 9 For example, MHC loss inhibits T‐cell activation following the loss of antigen presentation. 23 , 24 Reduced chemokines induce a noninflamed TME leading to ICI resistance. 15 , 25 , 26 Suppressive immune checkpoint molecules and immune‐suppressive cells inhibit the effector functions of T cells. 27 , 28 , 29 , 30 In addition, abnormalities in the IFN‐γ signaling pathway are involved in many of these mechanisms. 31 , 32 In this review, we summarize mechanisms of resistance to ICIs related to the process of T‐cell activation and novel therapies to overcome them.
2. THE PROCESS OF T‐CELL ACTIVATION AND THE EFFECTS OF IMMUNE CHECKPOINT MOLECULES
When CD8+ T cells are depleted in mouse models, tumors grow rapidly, and ICIs are completely ineffective. 33 In addition, an inflamed TME characterized by high CD8+ T‐cell infiltration is reportedly associated with ICI response. 8 , 9 Thus, CD8+ T cells are essential for antitumor immunity, and ICIs exert antitumor effects by activating these T cells. 8 , 9 The process in which T‐cell activation leads to killing cancer cells is summarized in seven steps (Figure 1) 34 : (1) cancer antigen release from cancer cells, (2) presentation of cancer antigens via MHC molecules by antigen‐presenting cells (APCs), (3) T‐cell activation via recognition of cancer antigen in the context of MHC molecules (priming phase), (4) migration and (5) infiltration of activated T cells into the TME, (6) T‐cell recognition of cancer antigen presented in the context of MHC molecules of cancer cells, and (7) killing cancer cells by activated T cells (effector phase). During these processes, immune checkpoint molecules modulate the activity of T cells in steps 3 (priming phase) and 7 (effector phase). 34 , 35
FIGURE 1.
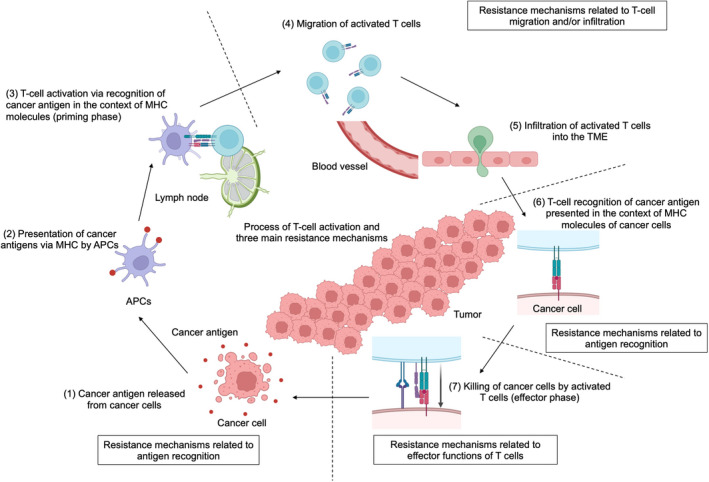
Process of T‐cell activation and main mechanisms of resistance to immune checkpoint inhibitors (ICIs). The seven steps of T‐cell activation that lead to killing cancer cells and the three main mechanisms of resistance to ICIs related to this process are summarized. APC, antigen‐presenting cell; MHC, major histocompatibility complex; TME, tumor microenvironment. This figure was created with BioRender.com
Immune checkpoint molecules, which are mainly expressed on the T‐cell surface, control T‐cell activation by binding to their ligands on APCs and/or cancer cells at the time of T‐cell recognition of cancer antigens. 36 Each immune checkpoint molecule acts through different mechanisms, with some demonstrating inhibitory and others stimulatory effects on T‐cell activation (Figure 2). 36
FIGURE 2.
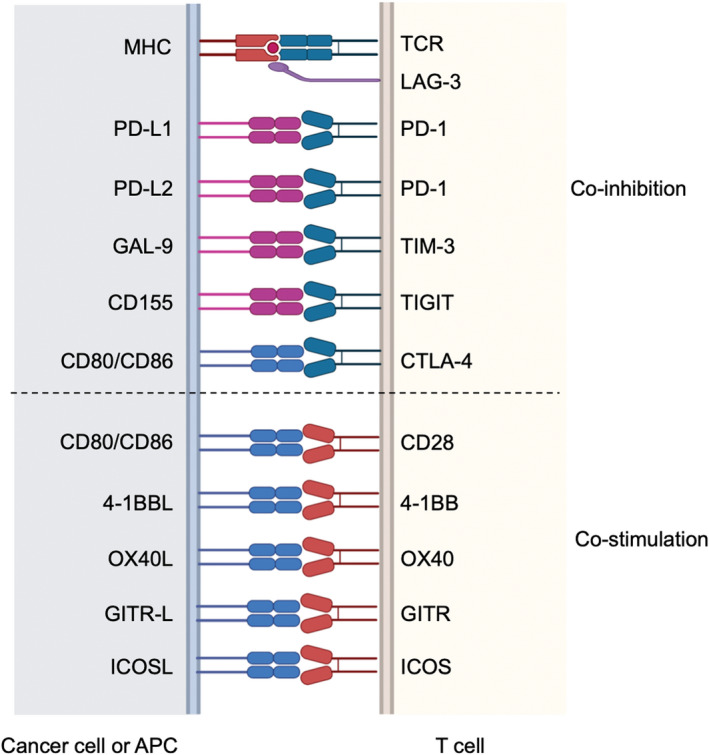
Multiple costimulatory and inhibitory interactions regulate T‐cell responses. Representative coinhibitory and costimulatory immune checkpoint molecules and their ligands are presented. 4‐1BBL, 4‐1BB ligand; CTLA‐4, cytotoxic T‐lymphocyte‐associated protein 4; GAL‐9, galectin 9; GITR, glucocorticoid‐induced tumor necrosis factor receptor; GITR‐L, GITR ligand; ICOS, inducible T‐cell costimulator; ICOSL, ICOS ligand; LAG‐3, lymphocyte activation gene 3; OX40L, OX40 ligand; PD‐1, programmed cell death 1; PD‐L1/2, PD‐ligand 1/2; TIGIT, T‐cell immunoreceptor with immunoglobulin and ITIM domains; TIM‐3, T‐cell membrane protein 3. This figure was created with BioRender.com
CTLA‐4 and PD‐1 are both inhibitory immune checkpoint molecules, and mAbs against them are used clinically (anti‐CTLA‐4 and anti‐PD‐1/PD‐L1 mAbs, respectively). T‐cell receptor (TCR) stimulation with antigen recognition is essential for T‐cell activation, and costimulation is also necessary. 37 A representative type of costimulation is the CD28 signaling pathway, which promotes T‐cell activation mainly by binding to CD80 or CD86. 38 CTLA‐4 suppresses T‐cell activation by binding tightly to CD80/CD86, inhibiting CD28‐CD80/CD86‐mediated costimulation. 38 , 39 In addition, regulatory T cells (Treg cells), one of the representative immune‐suppressive cells, highly express CTLA‐4 on their surface, and Treg cell–expressing CTLA‐4 depletes CD80/CD86 on APCs by trogocytosis, leading to suppression of APCs. 28 As CD80 and CD86 are mainly expressed in APCs, anti‐CTLA‐4 mAbs are expected to work mainly at the T‐cell priming phase (step 3). 36 , 38 , 39 On the other hand, PD‐1 expressed on T cells binds to PD‐L1 or PD‐L2, and PD‐1‐PD‐L1/PD‐L2 mediates inhibitory signals by suppressing TCR signaling pathways. 40 Anti‐PD‐1/PD‐L1 mAbs are considered to work mainly at the effector phase (step 7), whereas PD‐L1/PD‐L2 are expressed on both tumor cells and APCs. 37 Furthermore, it has been recently reported that PD‐1 also exhibits its inhibitory effect by suppressing CD28 signaling. 41 , 42
In addition to PD‐1 and CTLA‐4, many other immune checkpoint molecules regulate T‐cell activation. Coinhibitory immune checkpoint molecules include TIM‐3, LAG‐3, and TIGIT. 36 , 43 On the other hand, costimulatory immune checkpoint molecules that activate immune responses by binding their ligands, like CD28 binds CD80/CD86, include OX40, GITR, 4‐1BB, and ICOS. 36 , 43
3. MECHANISMS OF RESISTANCE TO ICIS
As mentioned above, the process of T‐cell activation is essential for the antitumor effects of ICIs. In cases of resistance, many factors inhibit the T‐cell activation process, which can be classified into three main mechanisms (Figure 1), though there seems to be an overlap among them. The first category is “resistance mechanisms related to antigen recognition,” inhibiting steps 1‐3 and 6; the second is “resistance mechanisms related to T‐cell migration and/or infiltration,” inhibiting steps 4 and 5. The third category is the “resistance mechanisms related to effector functions of T cells,” inhibiting step 7. As these resistance mechanisms can induce both initial and acquired resistance, we will explain them together. 21 , 22 , 44 , 45 , 46
4. RESISTANCE MECHANISMS RELATED TO ANTIGEN RECOGNITION
At the start of T‐cell–mediated antitumor immunity, APCs present cancer antigens on their MHC. T cells with specific TCRs recognize the antigen‐MHC complex on APCs and are stimulated via the TCR signaling pathway, which induces subsequent T‐cell activation. In addition, activated T cells recognize the cancer antigen‐MHC complex on cancer cells in the TME, leading to killing cancer cells. Impairment of any part of the antigen recognition processes can lead to the loss of antitumor immunity. The factors related to these resistance mechanisms include (1) cancer cell antigen loss and (2) reduced antigen presentation.
4.1. Cancer cell antigen loss
Cancer antigens cause specific antitumor immunity mediated by antigen‐specific T‐cell activation, and the loss of these antigens abolishes specific T‐cell cytotoxicity. In particular, neoantigens, derived from somatic mutations that alter amino acid sequences, are the most characteristic tumor‐specific antigens, leading to strong T‐cell activation as non‐self antigens. 11 , 12 , 13 , 14 Previous reports have shown that ICIs are highly effective for patients with high TMBs. 11 , 12 , 13 , 14 , 47 , 48 In contrast, some patients with low neoantigen levels are primarily resistant to ICIs, and some patients who respond initially acquire resistance to ICIs with the disappearance of neoantigens. 11 , 12 , 13 , 14 , 49 , 50
4.2. Reduced antigen presentation
If any defects in the antigen presentation process occur, ICIs can be ineffective. One such mechanism is dysfunction of APCs. To escape antitumor immunity, cancer cells suppress the antigen‐presenting activity of APCs through various mechanisms. They inhibit APC recruitment into the TME and/or maturation by activating the WNT/β‐catenin signaling pathway or secretion of prostaglandin E2 (PGE2) or vascular endothelial growth factor (VEGF). 51 , 52 , 53 , 54 , 55
Another mechanism is the loss of MHC expression by cancer cells. MHC class I (MHC‐I) on cancer cells is an essential molecule for CD8+ T cells to recognize cancer antigens. 23 , 24 It has been reported that loss of heterozygosity in MHC‐I genes is associated with immune escape in cancer, 56 , 57 which leads to ICI resistance. 58 We recently reported that MHC‐I mutations accumulate in microsatellite instability‐high tumors, suggesting that the accumulation of MHC‐I mutations is an important immune evasion mechanism. 23 Other reports in clinical settings showed that loss or decreased expression of β‐2 macroglobulin (B2M), one of the components of MHC‐I, resulted in defects in antigen presentation and ICI resistance. 59 , 60 We also reported a case of melanoma with ICI resistance due to the loss of the B2M gene, and this resistance was overcome by introducing the B2M gene in vitro. 23 In addition, the loss of MHC‐I in a metastatic lesion but not in other lesions induced a mixed response with a poor prognosis. 61
In addition, it has also been reported that the expression of MHC‐I on cancer cells is decreased by abnormal IFN‐γ signaling caused by genetic abnormalities such as JAK1 and JAK2 abnormalities. NLRC5 and DUX4 are also reported to be involved in IFN‐γ–mediated MHC expression. 31 , 32 , 62 , 63 IFN‐γ, a cytokine secreted mainly from activated T cells, plays an essential role in antitumor immunity. The IFN‐γ signaling pathway induces antitumor effects by promoting MHC expression in cancer cells, chemokine production, cell growth inhibition, and apoptosis. 64 On the other hand, we treated a patient who responded to anti‐PD‐1 mAbs with preserved MHC‐I expression despite defects in IFN‐γ signaling because of the loss of JAK. We experimentally demonstrated that mouse models could be sensitive to treatment despite defects in IFN‐γ signaling. These models had high MHC‐I expression independent of the IFN‐γ signaling pathway. From these findings, we speculate that resistance due to defects in IFN‐γ signaling could be mainly caused by decreased MHC‐I.
Furthermore, downregulation of MHC expression is caused by epigenetic abnormalities such as EZH2 gene mutation and histone deacetylase abnormalities. 65 , 66 Currently, there are attempts to develop drugs that upregulate MHC. One possible approach is to upregulate MHC‐I by inhibiting TRAF3, which negatively regulates the NF‐κB pathway. 67 We also revealed the importance of MHC class II (MHC‐II)‐mediated cytotoxic CD4+ T cells in antitumor immunity even against MHC‐I–negative tumors and suggested them as a potential therapeutic target. 68
5. RESISTANCE MECHANISMS RELATED TO T‐CELL MIGRATION AND/OR INFILTRATION
T cells need to migrate and infiltrate into the TME to attack cancer cells directly. Several kinds of chemokines, such as CXCL9, CXCL10, CXCL11, and CCL5, are necessary for T‐cell migration and infiltration. Cancer cells and surrounding immune cells produce these chemokines, and disruption of the chemokine production process induces ICI resistance. 25 Therefore, abnormalities in the IFN‐γ signaling pathway, which is related to the production of these chemokines, inhibit T‐cell migration and infiltration into the TME.
Several signaling pathways related to carcinogenesis, such as the WNT/β‐catenin, PTEN, LKB1, and EGFR pathways, may also contribute to ICI resistance by suppressing the production of such chemokines. 26 , 51 , 69 , 70 , 71 In addition, epigenetic changes have also been reported to downregulate these chemokines, contributing to resistance to ICIs. 65 , 66
Vascular endothelial growth factor, which directly affects immune cells, as mentioned above, also prevents T‐cell migration and infiltration by inhibiting adhesion between T cells and vascular endothelial cells and suppressing the production of chemokines, such as CXCL10 and CXCL11. 72 , 73 In clinical settings, VEGF inhibitors show efficacy against several types of cancer and are more effective when combined with ICIs. 74 , 75 , 76 We also reported the case of an ICI‐resistant patient who was successfully treated with a VEGF inhibitor. 77
In addition to VEGF, TGF‐β also has immune‐suppressive effects. In addition to directly suppressing T cells, TGF‐β prevents T‐cell infiltration by inducing activation of cancer‐associated fibroblasts (CAFs) and affects CXCR3 expression on T cells, which interferes with chemokine‐induced migration. 78 , 79 Although it has not yet been clinically applied, combination therapy with a TGF‐β–blocking drug is now being developed. 78
6. RESISTANCE MECHANISMS RELATED TO EFFECTOR FUNCTIONS OF T CELLS
This section summarizes some of the significant immunomodulatory factors, including immune‐suppressive cells and other suppressive immune checkpoint molecules, which lead to inadequate effector functions of T cells in the TME. The IFN‐γ, secreted mainly from activated T cells, plays an essential role in antitumor immunity. It has been reported that genetic mutations in JAK cause abnormal IFN‐γ signaling, leading to resistance to ICIs. 31 , 32 Cytotoxic T cells exhibit their effector functions via IFN‐γ by inhibiting cell growth and inducing apoptosis. 64 However, as mentioned above, the resistance due to defects in IFN‐γ signaling could be mainly caused by decreased MHC‐I.
6.1. Other suppressive immune checkpoint molecules
PD‐1, which interacts with PD‐L1, is primarily expressed following the activation of T cells and suppresses T‐cell effector function, causing T cells to fall into a progressive dysfunctional state called exhaustion. 27 In the TME, not all T cells attack cancer cells, as there is a subset of bystander T cells. 80 , 81 Among them, exhausted T cells in the TME directly attack cancer cells, and ICIs exhibit efficacy by reactivating them. 81 , 82
Among exhausted T cells, PD‐1lowCXCR5+TCF1+ progenitor‐exhausted T cells are expected to be reactivated by anti‐PD‐1/PD‐L1 mAbs. In contrast, PD‐1highCXCR5− TCF1− terminally differentiated exhausted T cells are considered to be dysfunctional and not able to be reactivated (Figure 3). 83 , 84 These terminally differentiated exhausted T cells, which express not only PD‐1 but also other inhibitory immune checkpoint molecules, such as LAG‐3, TIM‐3, and TIGIT, are not fully reactivated by blocking only PD‐1, resulting in resistance (Figures 2 and 3). 83 , 84 , 85 A clinical trial of combination therapy with inhibitors of PD‐1 and these immune checkpoint molecules is now underway.
FIGURE 3.
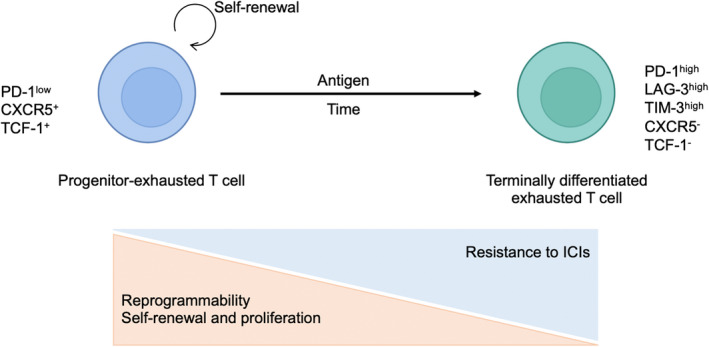
The progressive process of T‐cell exhaustion. Among exhausted T cells, PD‐1lowCXCR5+TCF1+ progenitor‐exhausted T cells are expected to be reactivated by anti‐PD‐1/PD‐L1 mAbs. In contrast, PD‐1highCXCR5−TCF1− terminally differentiated exhausted T cells are considered dysfunctional and incapable of being reactivated. LAG‐3, lymphocyte activation gene 3; PD‐1, programmed cell death 1; TCF‐1, T‐cell factor 1; TIM‐3, T‐cell membrane protein 3. This figure was created with BioRender.com
Using clinical samples and mouse models, we also demonstrated that the TIGIT/CD155 interaction is another mechanism by which melanoma with a high TMB and an inflamed TME can become resistant to therapy, including cases of both primary and acquired resistance. 86 A clinical trial of anti‐TIGIT mAbs combined with anti‐PD‐L1 mAbs for non–small cell lung cancer has been performed, and the combination showed possible efficacy against PD‐L1–high tumors. 87 Another inhibitory molecule, LAG‐3, inhibits T‐cell activation by binding to MHC‐II (Figure 2). We reported that CD4+ T cells in the TME of MHC‐II+ tumors express high LAG‐3 levels and that combination therapy with anti‐PD‐1 and anti‐LAG‐3 mAbs increases CD4+ T‐cell–mediated antitumor immunity. 68 Indeed, anti‐LAG‐3 mAbs have already shown efficacy in combination with anti‐PD‐1 mAbs against malignant melanoma. 88
6.2. Immunosuppressive cells
The ICI resistance mechanisms mediated by various immune‐suppressive cells, such as Treg cells, tumor‐associated macrophages (TAMs), myeloid‐derived suppressor cells (MDSCs), and CAFs, have been reported mainly in mouse models. Therefore, treatment to deplete these cells has been being developed. 28 , 29 , 30 These immune‐suppressive cells also inhibit the antigen presentation of APCs, suggesting that the early‐phase T‐cell activation process may be impaired by these suppressive cells. In addition, these cells secrete various immune‐suppressive factors, such as VEGF and TGF‐β. 73 , 78 , 89 , 90
Treg cells are known to suppress effector T cells and APCs by various mechanisms, such as consuming IL‐2 and binding CD80/CD86 via CTLA‐4. 28 We reported that Treg cells in the TME had high PD‐1 expression and that anti‐PD‐1 mAbs activated PD‐1+ Treg cells as well as PD‐1+CD8+ T cells, which contributed to ICI resistance. 33 Moreover, we also found that hyperprogressive disease can be caused by PD‐1+ Treg cells in the TME activated by anti‐PD‐1 mAbs. 91 In contrast, anti‐CTLA‐4 mAbs are thought to have inhibitory effects on Treg cells, but even when combined with anti‐PD‐1 mAbs, approximately 40%‐50% of treated patients are resistant. 3 , 4 , 5 Further investigations are needed to clarify the effects of ICIs on Treg cells.
Vascular endothelial growth factor has been reported to induce activation of immune‐suppressive cells such as Treg cells, TAMs, and MDSCs and is also secreted by these immune‐suppressive cells. 73 , 89 VEGF inhibitors are expected to improve the TME by increasing cytotoxic T cells and decreasing suppressor cells. 73 , 89 In a phase 2 trial, the combination of a VEGF inhibitor with an ICI in renal cell carcinoma was more effective in TAM‐abundant cancers than TAM‐deficient cancers. 92
6.3. Metabolism in the TME
The TME is known as a hypoxic and low‐glucose environment. In such an environment, the lack of glucose impairs T‐cell activation, limits antitumor immunity, and induces resistance to ICIs. 93 VEGF is known to establish a hypoxic TME, which is expected to be improved by VEGF inhibitors. 73 On the other hand, lactate, abundant in the low‐glucose TME, was recently reported to promote PD‐1 expression in Treg cells and contribute to ICI resistance. 26 In addition, abnormalities of several genes, such as PI3K, LKB1, and MYC, which are involved in cancer metabolism, can also contribute to resistance. 26 , 69 , 71 , 94 Attempts are being made to modulate metabolism with drugs to increase antitumor immunity. 95 , 96
Amino acids play an essential role in T‐cell activation. Serine and arginine are important amino acids for effector T‐cell expansion and antitumor immunity. 97 , 98 The enzyme IDO, which converts tryptophan to the immunosuppressive molecule kynurenine, has been found to be involved in ICI resistance. 99 Although combination therapy with IDO inhibitors and anti‐PD‐1/PD‐L1 mAbs was expected to be efficacious, clinical trials have failed to meet their primary endpoint. 100 , 101 We speculate that the investigators should select an appropriate population based on any biomarkers.
Extracellular adenosine binds to its receptors on T cells and suppresses their function via the subsequent elevation of intracellular cAMP. 102 , 103 Adenosine is produced from ATP through surface CD39 and CD73. Thus, CD73 and CD39 can suppress antitumor immunity, resulting in resistance to ICIs. 80 , 104 Promising drugs that inhibit the binding of adenosine to its receptor have been developed, and clinical trials are underway for combination therapy with PD‐1 inhibitors. 105 , 106 The efficacy of anti‐CD39/CD73 antibodies has also been reported at the preclinical stage, and the results of clinical trials are awaited. 107 , 108
7. OTHER RESISTANCE MECHANISMS
In some patients who are resistant to anti‐PD‐L1 mAbs, PD‐L1 splicing variants are secreted, working as “decoys” of the mAbs. 109 ICI‐neutralizing antibodies can also be produced in resistant patients, which can be related to resistance. 110 , 111 Secreted factors, such as PD‐L1 splicing variants and ICI‐neutralizing antibodies, can disturb ICIs themselves, leading to ICI resistance.
Recently, the relationships between the intestinal microbiota and systemic immune responses and autoimmune diseases have been noted. 112 Metabolites from the microbiota have also been reported to influence immune responses. 113 The intestinal microbiota also affects antitumor immunity, and it has been pointed out that it may be involved in the response to ICIs. 114 , 115 , 116 , 117 , 118 The combination of fecal transplantation and ICIs has already been applied in practice, and some reports indicate that fecal transplants from responders to ICIs can induce ICI responses. 118 , 119 , 120 The detailed mechanism, including the relationships with resistance, is still unclear, so further research is expected.
8. CONCLUSION
Immune checkpoint inhibitors have undoubtedly shifted the cancer therapy paradigm. However, tumors induce a TME that benefits themselves by various mechanisms, and there are still many challenges to be overcome. This review summarizes ICI resistance mechanisms related to T‐cell activation process in three main categories. It is not always possible to identify a single mechanism, such as abnormalities in the IFN‐γ signaling pathway. Various mechanisms are involved in a complex manner, suggesting that resistance occurs via a complex set of processes. However, as ICIs basically exert their effects by activating T cells, many resistance mechanisms can be attributed to the inhibition of T‐cell activation. To overcome resistance, it is important to promote T‐cell activation, and various attempts are being made, including combination therapy. In addition, cell therapy has been proven to be effective in ICI‐resistant hematological tumors, including malignant lymphoma and leukemia, suggesting the possibility of response even in noninflamed tumors. 121 , 122 , 123 Therefore, cell therapy is expected to be applied in solid tumors in the future. 124 , 125
Cancer immunity has been studied mainly in mouse models. However, the mechanisms of resistance to ICIs in clinical settings seem to be very complicated, as we have summarized. Therefore, it can be difficult to understand the detailed mechanisms only in mouse models. We believe it is essential to elucidate the TME using human clinical samples to understand and overcome resistance mechanisms. We should promote translational research with both bedside‐to‐bench approaches and bench‐to‐bedside approaches.
CONFLICT OF INTEREST
Y. T. is a current editorial board member of Cancer Science. Y. T. received research grants and honoraria from Ono Pharmaceutical and Bristol‐Myers Squibb; research grants from KOTAI Biotechnologies, Daiichi‐Sankyo, and KORTUC; and honoraria from Chugai Pharmaceutical and MSD outside this work.
ACKNOWLEDGMENTS
This study was supported by Grants‐in‐Aid for Scientific Research (B, 20H03694) from the Japan Society for the Promotion of Science (JSPS); the Fusion Oriented Research for Disruptive Science and Technology (21‐211033868) from the Japan Science and Technology Agency (JST); the Project for Cancer Research and Therapeutic Evolution (18cm0106340h0001 and 21cm0106383); and the Practical Research for Innovative Cancer Control (19ck0106521h0001 and 22ama221303h0001) from the Japan Agency for Medical Research and Development (AMED).
Nagasaki J, Ishino T, Togashi Y. Mechanisms of resistance to immune checkpoint inhibitors. Cancer Sci. 2022;113:3303‐3312. doi: 10.1111/cas.15497
REFERENCES
- 1. Reck M, Rodríguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;375:1823‐1833. [DOI] [PubMed] [Google Scholar]
- 2. Ferris RL, Blumenschein G, Fayette J, et al. Nivolumab for recurrent squamous‐cell carcinoma of the head and neck. N Engl J Med. 2016;375:1856‐1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Motzer RJ, Tannir NM, McDermott DF, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal‐cell carcinoma. N Engl J Med. 2018;378:1277‐1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Larkin J, Chiarion‐Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hellmann MD, Paz‐Ares L, Bernabe Caro R, et al. Nivolumab plus ipilimumab in advanced non‐small‐cell lung cancer. N Engl J Med. 2019;381:2020‐2031. [DOI] [PubMed] [Google Scholar]
- 6. Kang YK, Boku N, Satoh T, et al. Nivolumab in patients with advanced gastric or gastro‐oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO‐4538‐12, ATTRACTION‐2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet. 2017;390:2461‐2471. [DOI] [PubMed] [Google Scholar]
- 7. Zou W, Wolchok JD, Chen L. PD‐L1 (B7‐H1) and PD‐1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tumeh PC, Harview CL, Yearley JH, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69‐74. [DOI] [PubMed] [Google Scholar]
- 11. Matsushita H, Vesely MD, Koboldt DC, et al. Cancer exome analysis reveals a T‐cell‐dependent mechanism of cancer immunoediting. Nature. 2012;482:400‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science. 2015;348:124‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rooney Michael S, Shukla Sachet A, Wu Catherine J, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hacohen N, Fritsch EF, Carter TA, Lander ES, Wu CJ. Getting personal with neoantigen‐based therapeutic cancer vaccines. Cancer Immunol Res. 2013;1:11‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sugiyama E, Togashi Y, Takeuchi Y, et al. Blockade of EGFR improves responsiveness to PD‐1 blockade in EGFR‐mutated non‐small cell lung cancer. Sci Immunol. 2020;5:eaav3937. [DOI] [PubMed] [Google Scholar]
- 16. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism‐driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McGrail DJ, Pilié PG, Rashid NU, et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol. 2021;32:661‐672. doi: 10.1016/j.annonc.2021.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Voorwerk L, Slagter M, Horlings HM, et al. Immune induction strategies in metastatic triple‐negative breast cancer to enhance the sensitivity to PD‐1 blockade: the TONIC trial. Nat Med. 2019;25:920‐928. [DOI] [PubMed] [Google Scholar]
- 19. Rizvi H, Sanchez‐Vega F, La K, et al. Molecular determinants of response to anti‐programmed cell death (PD)‐1 and anti‐programmed death‐ligand 1 (PD‐L1) blockade in patients with non‐small‐cell lung cancer profiled with targeted next‐generation sequencing. J Clin Oncol. 2018;36:633‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137‐148. [DOI] [PubMed] [Google Scholar]
- 21. Sharma P, Hu‐Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kalbasi A, Ribas A. Tumour‐intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol. 2020;20:25‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Inozume T, Yaguchi T, Ariyasu R, et al. Analysis of the tumor reactivity of tumor‐infiltrating lymphocytes in a metastatic melanoma lesion that lost major histocompatibility complex class I expression after anti‐PD‐1 therapy. J Invest Dermatol. 2019;139:1490‐1496. [DOI] [PubMed] [Google Scholar]
- 24. Kawazu M, Ueno T, Saeki K, et al. HLA class I analysis provides insight into the genetic and epigenetic background of immune evasion in colorectal cancer with high microsatellite instability. Gastroenterology. 2022;162:799‐812. [DOI] [PubMed] [Google Scholar]
- 25. Peng D, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of TH1‐type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kumagai S, Koyama S, Itahashi K, et al. Lactic acid promotes PD‐1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. 2022;40:201‐218.e209. [DOI] [PubMed] [Google Scholar]
- 27. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression – implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16:356‐371. [DOI] [PubMed] [Google Scholar]
- 29. Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour‐associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sahai E, Astsaturov I, Cukierman E, et al. A framework for advancing our understanding of cancer‐associated fibroblasts. Nat Rev Cancer. 2020;20:174‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shin DS, Zaretsky JM, Escuin‐Ordinas H, et al. Primary resistance to PD‐1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7:188‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zaretsky JM, Garcia‐Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med. 2016;375:819‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kumagai S, Togashi Y, Kamada T, et al. The PD‐1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD‐1 blockade therapies. Nat Immunol. 2020;21:1346‐1358. [DOI] [PubMed] [Google Scholar]
- 34. Chen Daniel S, Mellman I. Oncology meets immunology: the cancer‐immunity cycle. Immunity. 2013;39:1‐10. [DOI] [PubMed] [Google Scholar]
- 35. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen L, Flies DB. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat Rev Immunol. 2013;13:227‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Esensten JH, Helou YA, Chopra G, Weiss A, Bluestone JA. CD28 costimulation: from mechanism to therapy. Immunity. 2016;44:973‐988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yokosuka T, Kobayashi W, Takamatsu M, et al. Spatiotemporal basis of CTLA‐4 costimulatory molecule‐mediated negative regulation of T cell activation. Immunity. 2010;33:326‐339. [DOI] [PubMed] [Google Scholar]
- 40. Yokosuka T, Takamatsu M, Kobayashi‐Imanishi W, Hashimoto‐Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hui E, Cheung J, Zhu J, et al. T cell costimulatory receptor CD28 is a primary target for PD‐1‐mediated inhibition. Science. 2017;355:1428‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kamphorst AO, Wieland A, Nasti T, et al. Rescue of exhausted CD8 T cells by PD‐1‐targeted therapies is CD28‐dependent. Science. 2017;355:1423‐1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kraehenbuehl L, Weng C‐H, Eghbali S, Wolchok JD, Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat Rev Clin Oncol. 2022;19:37‐50. [DOI] [PubMed] [Google Scholar]
- 44. Gide TN, Wilmott JS, Scolyer RA, Long GV. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin Cancer Res. 2018;24:1260‐1270. [DOI] [PubMed] [Google Scholar]
- 45. Kawakami Y, Ohta S, Sayem MA, Tsukamoto N, Yaguchi T. Immune‐resistant mechanisms in cancer immunotherapy. Int J Clin Oncol. 2020;25:810‐817. [DOI] [PubMed] [Google Scholar]
- 46. Schoenfeld AJ, Hellmann MD. Acquired resistance to immune checkpoint inhibitors. Cancer Cell. 2020;37:443‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cristescu R, Mogg R, Ayers M, et al. Pan‐tumor genomic biomarkers for PD‐1 checkpoint blockade‐based immunotherapy. Science. 2018;362:eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med. 2014;371:2189‐2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rosenthal R, Cadieux EL, Salgado R, et al. Neoantigen‐directed immune escape in lung cancer evolution. Nature. 2019;567:479‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anagnostou V, Smith KN, Forde PM, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non‐small cell lung cancer. Cancer Discov. 2017;7:264‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Spranger S, Bao R, Gajewski TF. Melanoma‐intrinsic β‐catenin signalling prevents anti‐tumour immunity. Nature. 2015;523:231‐235. [DOI] [PubMed] [Google Scholar]
- 52. Ruiz de Galarreta M, Bresnahan E, Molina‐Sánchez P, et al. β‐Catenin activation promotes immune escape and resistance to anti‐PD‐1 therapy in hepatocellular carcinoma. Cancer Discov. 2019;9:1124‐1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bonavita E, Bromley CP, Jonsson G, et al. Antagonistic inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity. 2020;53:1215‐1229. e1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hangai S, Ao T, Kimura Y, et al. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc Natl Acad Sci USA. 2016;113:3844‐3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ohm JE, Carbone DP. VEGF as a mediator of tumor‐associated immunodeficiency. Immunol Res. 2001;23:263‐272. [DOI] [PubMed] [Google Scholar]
- 56. Shukla SA, Rooney MS, Rajasagi M, et al. Comprehensive analysis of cancer‐associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33:1152‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McGranahan N, Rosenthal R, Hiley CT, et al. Allele‐specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171:1259‐1271. e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee JH, Shklovskaya E, Lim SY, et al. Transcriptional downregulation of MHC class I and melanoma de‐ differentiation in resistance to PD‐1 inhibition. Nat Commun. 2020;11:1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sade‐Feldman M, Jiao YJ, Chen JH, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8:1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gettinger S, Choi J, Hastings K, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017;7:1420‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Morinaga T, Inozume T, Kawazu M, et al. Mixed response to cancer immunotherapy is driven by intratumor heterogeneity and differential inter‐lesion immune infiltration. Cancer Res Commun. 2022. doi: 10.1158//2767-9764.CRC-22-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yoshihama S, Roszik J, Downs I, et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc Natl Acad Sci USA. 2016;113:5999‐6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chew GL, Campbell AE, De Neef E, et al. DUX4 suppresses MHC class I to promote cancer immune evasion and resistance to checkpoint blockade. Dev Cell. 2019;50:658‐671. e657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18:545‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Luo N, Nixon MJ, Gonzalez‐Ericsson PI, et al. DNA methyltransferase inhibition upregulates MHC‐I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat Commun. 2018;9:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ennishi D, Takata K, Béguelin W, et al. Molecular and genetic characterization of MHC deficiency identifies EZH2 as therapeutic target for enhancing immune recognition. Cancer Discov. 2019;9:546‐563. [DOI] [PubMed] [Google Scholar]
- 67. Gu SS, Zhang W, Wang X, et al. Therapeutically increasing MHC‐I expression potentiates immune checkpoint blockade. Cancer Discov. 2021;11:1524‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nagasaki J, Togashi Y, Sugawara T, et al. The critical role of CD4+ T cells in PD‐1 blockade against MHC‐II‐expressing tumors such as classic Hodgkin lymphoma. Blood Adv. 2020;4:4069‐4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Peng W, Chen JQ, Liu C, et al. Loss of PTEN promotes resistance to T cell‐mediated immunotherapy. Cancer Discov. 2016;6:202‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sugiyama E, Togashi Y, Takeuchi Y, et al. Blockade of EGFR improves responsiveness to PD‐1 blockade in EGFR−mutated non–small cell lung cancer. Sci Immunol. 2020;5:eaav3937. [DOI] [PubMed] [Google Scholar]
- 71. Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD‐1 inhibitor resistance in KRAS‐mutant lung adenocarcinoma. Cancer Discov. 2018;8:822‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Voron T, Colussi O, Marcheteau E, et al. VEGF‐A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J Exp Med. 2015;212:139‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Voron T, Marcheteau E, Pernot S, et al. Control of the immune response by pro‐angiogenic factors. Front Oncol. 2014;4:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Socinski MA, Jotte RM, Cappuzzo F, et al. Atezolizumab for first‐line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378:2288‐2301. [DOI] [PubMed] [Google Scholar]
- 75. Rini BI, Plimack ER, Stus V, et al. Pembrolizumab plus Axitinib versus sunitinib for advanced renal‐cell carcinoma. N Engl J Med. 2019;380:1116‐1127. [DOI] [PubMed] [Google Scholar]
- 76. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894‐1905. [DOI] [PubMed] [Google Scholar]
- 77. Fukuoka S, Hara H, Takahashi N, et al. Regorafenib plus nivolumab in patients with advanced gastric or colorectal cancer: an open‐label, dose‐escalation, and dose‐expansion phase Ib trial (REGONIVO, EPOC1603). J Clin Oncol. 2020;38:2053‐2061. [DOI] [PubMed] [Google Scholar]
- 78. Batlle E, Massagué J. Transforming growth factor‐β signaling in immunity and cancer. Immunity. 2019;50:924‐940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gunderson AJ, Yamazaki T, McCarty K, et al. TGFβ suppresses CD8(+) T cell expression of CXCR3 and tumor trafficking. Nat Commun. 2020;11:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Simoni Y, Becht E, Fehlings M, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575‐579. [DOI] [PubMed] [Google Scholar]
- 81. Nagasaki J, Inozume T, Sax N, et al. PD‐1 blockade therapy promotes infiltration of tumor‐attacking exhausted T cell clonotypes. Cell Rep. 2022;38:110331. [DOI] [PubMed] [Google Scholar]
- 82. Oliveira G, Stromhaug K, Klaeger S, et al. Phenotype, specificity and avidity of antitumour CD8+ T cells in melanoma. Nature. 2021;596:119‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Philip M, Schietinger A. Heterogeneity and fate choice: T cell exhaustion in cancer and chronic infections. Curr Opin Immunol. 2019;58:98‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol. 2018;18:340‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD‐1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kawashima S, Inozume T, Kawazu M, et al. TIGIT/CD155 axis mediates resistance to immunotherapy in patients with melanoma with the inflamed tumor microenvironment. J Immunother Cancer. 2021;9:e003134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rodriguez‐Abreu D, Johnson ML, Hussein MA, et al. Primary analysis of a randomized, double‐blind, phase II study of the anti‐TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first‐line (1L) treatment in patients with PD‐L1‐selected NSCLC (CITYSCAPE). J Clin Oncol. 2020;38:9503. [Google Scholar]
- 88. Lipson EJ, Tawbi HA‐H, Schadendorf D, et al. Relatlimab (RELA) plus nivolumab (NIVO) versus NIVO in first‐line advanced melanoma: primary phase III results from RELATIVITY‐047 (CA224‐047). J Clin Oncol. 2021;39:9503. [Google Scholar]
- 89. Tada Y, Togashi Y, Kotani D, et al. Targeting VEGFR2 with ramucirumab strongly impacts effector/ activated regulatory T cells and CD8(+) T cells in the tumor microenvironment. J Immunother Cancer. 2018;6:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Highfill SL, Cui Y, Giles AJ, et al. Disruption of CXCR2‐mediated MDSC tumor trafficking enhances anti‐PD1 efficacy. Sci Transl Med. 2014;6:237‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kamada T, Togashi Y, Tay C, et al. PD‐1+ regulatory T cells amplified by PD‐1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A. 2019;116:9999‐10008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. McDermott DF, Huseni MA, Atkins MB, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018;24:749‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chang CH, Qiu J, O'Sullivan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229‐1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kumagai S, Togashi Y, Sakai C, et al. An oncogenic alteration creates a microenvironment that promotes tumor progression by conferring a metabolic advantage to regulatory T cells. Immunity. 2020;53:187‐203.e188. [DOI] [PubMed] [Google Scholar]
- 95. Chamoto K, Chowdhury PS, Kumar A, et al. Mitochondrial activation chemicals synergize with surface receptor PD‐1 blockade for T cell‐dependent antitumor activity. Proc Natl Acad Sci USA. 2017;114:E761‐e770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, Udono H. Immune‐mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci USA. 2015;112:1809‐1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Geiger R, Rieckmann JC, Wolf T, et al. L‐arginine modulates T cell metabolism and enhances survival and anti‐tumor activity. Cell. 2016;167:829‐842. e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ma EH, Bantug G, Griss T, et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. 2017;25:345‐357. [DOI] [PubMed] [Google Scholar]
- 99. Uyttenhove C, Pilotte L, Théate I, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3‐dioxygenase. Nat Med. 2003;9:1269‐1274. [DOI] [PubMed] [Google Scholar]
- 100. Long GV, Dummer R, Hamid O, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO‐301/KEYNOTE‐252): a phase 3, randomised, double‐blind study. Lancet Oncol. 2019;20:1083‐1097. [DOI] [PubMed] [Google Scholar]
- 101. Beatty GL, O'Dwyer PJ, Clark J, et al. First‐in‐human phase I study of the Oral inhibitor of indoleamine 2,3‐Dioxygenase‐1 Epacadostat (INCB024360) in patients with advanced solid malignancies. Clin Cancer Res. 2017;23:3269‐3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer‐derived adenosine: new therapeutic approaches. Cancer Discov. 2014;4:879‐888. [DOI] [PubMed] [Google Scholar]
- 103. Togashi Y, Nishikawa H. Suppression from beyond the grave. Nat Immunol. 2017;18:1285‐1286. [DOI] [PubMed] [Google Scholar]
- 104. Sade‐Feldman M, Yizhak K, Bjorgaard SL, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2019;176:404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Naing A, Gainor JF, Gelderblom H, et al. A first‐in‐human phase 1 dose escalation study of spartalizumab (PDR001), an anti‐PD‐1 antibody, in patients with advanced solid tumors. J Immunother Cancer. 2020;8:e000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Fong L, Hotson A, Powderly JD, et al. Adenosine 2A receptor blockade as an immunotherapy for treatment‐refractory renal cell cancer. Cancer Discov. 2020;10:40‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Perrot I, Michaud HA, Giraudon‐Paoli M, et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 2019;27:2411‐2425. e2419. [DOI] [PubMed] [Google Scholar]
- 108. Allard D, Allard B, Stagg J. On the mechanism of anti‐CD39 immune checkpoint therapy. J Immunother Cancer. 2020;8:e000186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Gong B, Kiyotani K, Sakata S, et al. Secreted PD‐L1 variants mediate resistance to PD‐L1 blockade therapy in non‐small cell lung cancer. J Exp Med. 2019;216:982‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kverneland AH, Enevold C, Donia M, Bastholt L, Svane IM, Nielsen CH. Development of anti‐drug antibodies is associated with shortened survival in patients with metastatic melanoma treated with ipilimumab. Onco Targets Ther. 2018;7:e1424674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Enrico D, Paci A, Chaput N, Karamouza E, Besse B. Antidrug antibodies against immune checkpoint blockers: impairment of drug efficacy or indication of immune activation? Clin Cancer Res. 2020;26:787‐792. [DOI] [PubMed] [Google Scholar]
- 112. Ruff WE, Greiling TM, Kriegel MA. Host‐microbiota interactions in immune‐mediated diseases. Nat Rev Microbiol. 2020;18:521‐538. [DOI] [PubMed] [Google Scholar]
- 113. Rooks MG, Garrett WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol. 2016;16:341‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Vétizou M, Pitt JM, Daillère R, et al. Anticancer immunotherapy by CTLA‐4 blockade relies on the gut microbiota. Science. 2015;350:1079‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Routy B, Le Chatelier E, Derosa L, et al. Gut microbiome influences efficacy of PD‐1‐based immunotherapy against epithelial tumors. Science. 2018;359:91‐97. [DOI] [PubMed] [Google Scholar]
- 116. Matson V, Fessler J, Bao R, et al. The commensal microbiome is associated with anti‐PD‐1 efficacy in metastatic melanoma patients. Science. 2018;359:104‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gopalakrishnan V, Spencer CN, Nezi L, et al. Gut microbiome modulates response to anti‐PD‐1 immunotherapy in melanoma patients. Science. 2018;359:97‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Gopalakrishnan V, Helmink BA, Spencer CN, Reuben A, Wargo JA. The influence of the gut microbiome on cancer, immunity, and cancer immunotherapy. Cancer Cell. 2018;33:570‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Davar D, Dzutsev AK, McCulloch JA, et al. Fecal microbiota transplant overcomes resistance to anti‐PD‐1 therapy in melanoma patients. Science. 2021;371:595‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Baruch EN, Youngster I, Ben‐Betzalel G, et al. Fecal microbiota transplant promotes response in immunotherapy‐refractory melanoma patients. Science. 2021;371:602‐609. [DOI] [PubMed] [Google Scholar]
- 121. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21:145‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T‐cell therapy in refractory large B‐cell lymphoma. N Engl J Med. 2017;377:2531‐2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Schuster SJ, Svoboda J, Chong EA, et al. Chimeric antigen receptor T cells in refractory B‐cell lymphomas. N Engl J Med. 2017;377:2545‐2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. 2019;36:471‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Qi C, Gong J, Li J, et al. Claudin18.2‐specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat Med. 2022;28:1189‐1198. doi: 10.1038/s41591-022-01800-8 [DOI] [PMC free article] [PubMed] [Google Scholar]