

Abstract
Tuberculosis (TB) is among the greatest public health and safety concerns in the 21st century, Mycobacterium tuberculosis, which causes TB, infects alveolar macrophages and uses these cells as one of its primary sites of replication. The current TB treatment regimen, which consist of chemotherapy involving a combination of 3-4 antimicrobials for a duration of 6-12 months, is marked with significant side effects, toxicity, and poor compliance. Targeted drug delivery offers a strategy that could overcome many of the problems of current TB treatment by specifically targeting infected macrophages. Recent advances in nanotechnology and material science have opened an avenue to explore drug carriers that actively and passively target macrophages. This approach can increase the drug penetration into macrophages by using ligands on the nanocarrier that interact with specific receptors for macrophages. This review encompasses the recent development of drug carriers specifically targeting macrophages actively and passively. Future directions and challenges associated with development of effective TB treatment is also discussed.
Keywords: Tuberculosis, Drug delivery, Macrophage targeting, Nanomedicine, Nanocarrier
Graphical Abstract
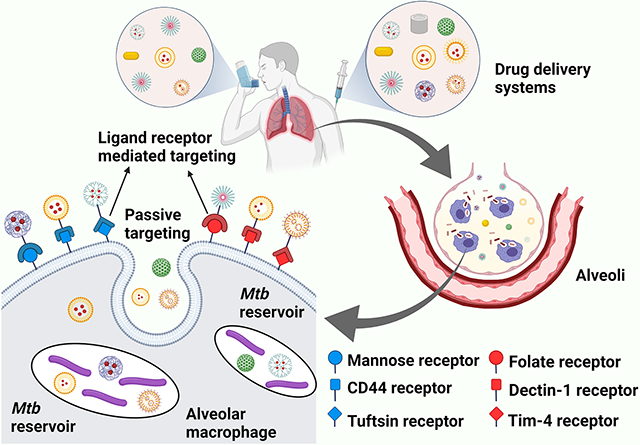
Targeted drug delivery systems have a potential to mitigate many of the problems associated with current TB therapy. Macrophages, which Mtb uses as a reservoir, express various classes of surface receptors which can be actively or passively targeted. By using ligands on the nanocarrier that interact with specific receptors on macrophages, efficiency of TB therapy can be further enhanced.
1. Introduction:
Tuberculosis (TB) is a major bacterial infection caused by Mycobacterium Tuberculosis (Mtb). Until the COVID-19 pandemic, TB was the major cause of death from an infectious disease, overtaking deaths from HIV/AIDS globally.[1] The global TB report published by the World Health Organization (WHO) in 2020 evaluated that there were around 10 million new instances of TB infection in 2019, and 1.2 million cases resulted in deaths.[1] Approximately 3 million TB cases, which sums up to 33% of new cases, remain unreported to the public health system, many of which do not receive appropriate therapy.[2] TB causes about one third of AIDS-related deaths, including 200,000 from HIV positive cases in 2019.[1] Transmission of TB occurs between humans via respiratory route, primarily affecting the lungs (pulmonary TB); however, it can infect any tissue and cause extrapulmonary TB approximately 20% of the time in previously healthy individuals. TB has the ability to transition into a dormant state (latent TB), where infected individuals display no obvious symptoms until later in life. Approximately 10% of latent infections progress to active TB.[2] It is predicted that about 25% of the global population is affected with TB and is at peril of transitioning into active infection.[1] Collectively, these observations demonstrate that TB can elicit a broad range of disease severity, ranging from latent infection to a life-threatening acute disease.[3, 4]
Despite the difficulties associated with containing a highly infectious agent like TB, there has been substantial progress in mitigation and containment, particularly in the last two decades. Between 2000-2019, there was a 29.4% reduction in death from TB in HIV negative cases and a 69.32% reduction in casualties in HIV positive cases.[1] The WHO, with support from the United Nations, is aiming to eradicate TB by 2030.[1] To achieve this goal, the WHO put forward the milestone of a 20% reduction of incidence and 35% reduction in mortalities by 2020, as compared to 2015 TB data. Global TB incidence has declined, but only by 9%, which is not very close to the 2020 milestone of 20%.[1] The most recent global target set by the WHO is to treat 40 million TB patients in the 5-year period of 2018–2022. The inability to reach the 20% reduction may partly be due to unequal distribution of TB in different regions of the world. In 2019, Southeast Asia had the highest incidence, with 44% of global TB cases. In contrast, Europe and the Americas had the lowest with 2.5% and 2.9%, respectively. India had the greatest number of TB cases of any individual country, with 26% of global cases.[1] In order to alleviate the discrepancies in global TB, it is imperative to understand the current treatment and prevention regimen.
Development of a vaccine for TB is likely one of the strongest strategies to combat the TB epidemic. While vaccines have had tremendous impact on health of humans and animals over the last two hundred years, the progress in vaccine development against TB has been relatively modest.[5-10] The current TB vaccine, Bacille Calmette-Guérin (BCG), is weakened Mycobacterium bovis strain and was administered to human infant for the first time in 1921. BCG appears to be the most effective in children, displaying more than 50% protection against pulmonary TB and more than 80% against extra-pulmonary TB.[11] Unfortunately, BCG has exhibited highly inconsistent protection against TB in adolescents and adults.[12]
The current treatment for TB consists of chemotherapy using first-line drugs consisting of ethambutol (EMB), pyrazinamide (PZA), isoniazid (INH), and rifampicin (RIF). Second-line injectable therapeutic agents (amikacin, streptomycin, viomycin, capreomycin, and kanamycin) are used in case of multidrug resistant TB or when first line drugs fail. Fluoroquinolones (moxifloxacin, gatifloxacin, levofloxacin, and ofloxacin ) and other oral agents (para-amino salicylic acid , prothionamide, terizidone cycloserine, and ethionamide) have also been used for TB treatment.[13, 14] A major problem with the current chemotherapy is the long duration of course required, 9-12 months, which often results in poor compliance. Current treatment can also be marred by several adverse side effects. Exposure to antibiotics for long durations can lead to toxicity in some patients.[15] Side effects can include hemolysis, agranulocytosis, optic neuritis, renal failure, ototoxicity, severe hepatitis, and immune thrombocytopaenia.[16] These are some of the main reasons that improvement of TB treatment has garnered a great deal of attention, with a goal of developing a treatment regimen with the shortest duration and lowest toxicity possible.
2. Pathogenesis and host response
Mtb spreads via inhalation of aerosolized particles, often due to coughing or sneezing. The bacteria in these particles travel directly to the lower respiratory tract when breathed in. In the pulmonary alveoli, the pathogen interacts with alveolar macrophages, the first and most critical line of defense at the site of infection in the lung. Alveolar macrophages are also the most significant effector cells with which Mtb interacts following deep lung deposition of pathogenic aerosols.[17] Macrophages internalize the pathogen by phagocytosis mediated by ligand-receptor interactions utilizing numerous different receptors expressed by the macrophages. While this is advantageous against many pathogens, the intracellular parasitic nature of Mtb confers it the ability to avoid most innate immune surveillance mechanisms. Mtb evades the innate immune response within macrophages by preventing acidification of the bacterial vacuole, preventing maturation of their intracellular compartment to a phagolysosome, and by inhibiting the apoptosis and autophagy of infected macrophages.[18-21] Mtb also inhibits the oxidative burst and is relatively resistant to reactive oxygen and reactive nitrogen intermediates within the macrophages.[18, 22, 23] Additionally, phagocytosis of Mtb provides a favorable site for replication, even in presence of a belligerent adaptive cellular immune response.[17] After internalization, Mtb successfully blocks fusion of lysosome with the phagosome, increasing its chances of survival.[24] This leads to replication of Mtb within the alveolar macrophages, making macrophages a safe harbor for the pathogen. After alveolar macrophage is infected in the lower respiratory tract, Mtb invades the lung interstitium, where the infection process progresses. Invasion of pathogens in the lung parenchyma triggers the host immune response, resulting in additional recruitment of T and B cells to the infection site. This recruitment generates a multicellular host response called a granulomatous inflammation.[16] Granulomas are supposed to safely contain the Mtb pathogen at the infection site, thus preventing its transmittance within the host; however, dysfunctional progression of granuloma can lead to pathogen survival, extensive tissue damage, and poor treatment responses.[17] Mtb utilize granulomas as infection sites where phagocytic cells grow in clusters, thus providing ample nutrients for replication. To do this, Mtb uses its ESX-1 secretion system which triggers a type I IFN response. This response elicits the recruitment of a unique CD11b+F4/80+Gr1int myeloid cells to the nascent granuloma that is highly permissive to Mtb infection.[25] Granulomas have a maximum sustainable bacterial population.[26] If infection proceeds beyond the maximal bacterial burden, it can alter the granuloma’s morphology, thereby increasing the probability of extrapulmonary TB infection. Progression of latent infection into an active infection is a complex process and is governed by multiple factors.[27] The manifestation of active TB infection directly correlates with the systematic and local inflammatory response to persistent Mtb and its antigens.[28, 29] The transition from latent to active TB infection is marked by changes in the granulomas present. This normally includes an increase in their size and change in distribution throughout the lungs.[17] In majority of infected hosts with LTBI, the collection of T cells, dendritic cells, and macrophages is enough to suppress the spread of infection. However, in a subset of TB infected patients, the infection can evolve to an active clinical disease. The reasons behind the disease progression from latent to active infection are not completely understood and this progression is marked with widely variable timeframe.[16]
3. Antibiotic resistance: The crux
Propensity of Mtb to develop antibiotic resistance against various antibiotics is one of the primary cruxes in development of an effective anti-TB treatment. The initial instance of antibiotic resistance was first observed in late 1940’s during the initial human trial of anti-TB treatment.[30] Over the following decade, researchers saw increasing cases of various antibiotic resistances develop. Even after 70 years, rise of drug-resistant TB is creating a bottleneck for development of effective TB treatment and management. Drug resistance is likely one of the major hindrances to the WHO’s goal of the TB epidemic’s cessation by 2030.[31]
Like many bacteria, Mtb is known to have different types and extents of antibiotic resistance such as multidrug resistant (MDR) and rifampicin resistant (RR) variants. RR-TB exhibits resistance against rifampicin only, and not towards any other first line or second line drugs such as isoniazid or levofloxacin. MDR-TB strains show resistance towards two or more of the most potent anti-TB drugs, such as rifampicin, ethambutol and isoniazid. Global cases of RR-TB increased from 51% in 2017 to 61% in 2019, while cases of MDR-TB were determined to be around 16.67%.[1] In order to treat MDR-TB, second-line drugs such as injectables and fluoroquinolones are utilized. However, extensive drug resistant (XDR)-TB is a TB variant which has resistances like MDR-TB, but also exhibits resistance to at least one drug from these second-line agents. This makes treating XDR-TB nearly impossible since it does not respond to rifampicin, isoniazid, fluoroquinolones, and to one or more second-line drugs (capreomycin, amikacin, and kanamycin).[32, 33] In some cases, researchers have come across TB infections where the patients have shown resistance against all anti-TB drugs which are typically used.[34] These highly drug resistant strains were termed as totally drug resistant (TDR-TB) strains.
There are various mechanisms by which Mtb shields itself from the detrimental effects of antibiotics. In Mtb, drug resistance evolves through various mechanisms such as epistasis, cell envelope impermeability, efflux pumps, compensatory evolution, target mimicry, phenotypic drug tolerance, clonal interference, and drug degradation and modification.[35, 36] Mtb has intrinsic resistance (natural defense against antibiotics) and can develop extrinsic resistance, such as horizontal gene transfer. Drug resistant strains can evolve due to several factors including perpetual exposure to sub-therapeutic levels of drug during treatment and noncompliance to the entirety of a treatment regimen. As such, drug concentration can be a major driving factor in mutations which result during treatment.[37] Mtb has an inherent resistance against the β-lactam class of antibiotics, conferred by β-lactamases produced by the bacteria.[38-40]
At sub-therapeutic drug concentrations, mutations in resistance genes appear, often at the expense of fitness, which leads to subpar growth, virulence, and survival. But in the case of Mtb, mutations in antibiotic resistant genes are often followed by another mutation, known as a compensatory mutation, which minimizes the detrimental effect on fitness.[41, 42] As a result of this compensatory evolution, Mtb is able to retain its resistant phenotype without losing much viability and/or virulence, as was confirmed by several studies with typical mutations seen in antibiotic resistant Mtb.[41, 42] Identification of such compensatory evolution mechanisms is highly desirable, as suppression of these mechanisms provide a promising strategy for treatment of drug resistant TB.
Presence of broad range of complex lipids confers the mycobacterial cell envelope with extreme thickness and high hydrophobicity. This increased hydrophobicity hampers the diffusion of even hydrophobic drug, including antibiotics such as tetracyclines, macrolides, rifamycins, and fluoroquinolones,.[43-45] Researchers have already demonstrated the efficacy of lipids of mycobacterial cell membrane in imparting inherent drug resistance.[38] Consequently, increased susceptibility to antibiotics was observed in mutants which had defective cell envelope.[45, 46]
Drug molecules which successfully cross the diffusional barriers of Mtb have to face another hindrance. Efflux pumps present in Mtb comprises an active mechanism which effectively removes therapeutic agents penetrating the bacterial cells to the extracellular environment.[38, 44, 47, 48] These efflux pumps are responsible for conferring Mtb with inherent resistance against various anti-TB drugs such as aminoglycosides, tetracycline, and fluoroquinolones. Mtb overexpresses certain efflux pump proteins under antibiotic stress conditions. Researchers have established that efflux pumps have a critical role in isoniazid and rifampicin resistance, notably in a case which involves no mutations.[49]
Drug degradation and modification is another potent tool in the toolbox of intrinsic resistance for Mtb.[37] Mtb produces several enzymes which tend to break down various classes of antibiotics, consisting of β-lactams, aminoglycosides, and macrolides.[38, 45] In addition to degradation, modification by enzymes present in Mtb inactivate antibiotics via the inclusion of chemical moieties on specific antibiotic sites.[38, 44, 45] For example, cyclic peptides/aminoglycosides, which are antibiotics used for MDR-TB treatment, are rendered ineffective by acetyltransferases and phosphotransferases present in Mtb.[50, 51] Along with drug degradation and modification, Mtb has developed a novel mechanism of resistance, target mimicry, that nullifies antibiotics targeting DNA gyrase such as fluoroquinolones.[38, 52] Target mimicry provides an alternate innocuous site of binding for drug molecules, thereby preventing or minimizing the binding to the actual drug target. Target mimicry in Mtb was also found to inhibit effective binding of other antibiotics, including lincosamides, macrolides, and streptomycin.[38, 45, 53]
Mtb is known to show phenotypic drug tolerance. Phenotypic tolerance in Mtb is characterized by reduced physiological or metabolic activities, similar to hibernation. It is unlike intrinsic and acquired drug tolerance since it does not involve direct chromosomal or genetic mutations. A sub-population of Mtb survives the antibiotic regimen by becoming dormant, transitioning into an inactive state, which is marked by minimal or absence of metabolic activity.[37] In its dormant state, Mtb exhibits suspension of physiological functions, tolerance to antibiotics, reversible metabolic shut down, slow growth rate, and triglyceride accumulation inside intracellular lipid bodies.[37] Once the antibiotic stress is removed, the dormant sub-population becomes active and restores its original metabolism.[38, 45, 54, 55] However, efficient targeting of therapeutics directly to macrophages, instead of non-specific localization of therapeutics to all tissues and organs, could be used to increase the local concentrations of therapeutics in the vicinity of Mtb to the point that it is bactericidal for these dormant populations, without impacting toxicity significantly. Higher local concentrations of therapeutics, specifically within the infected cell type, could also prevent development of resistant mutants by ensuring therapeutic levels are reached efficiently. In this way, macrophage targeted therapeutics could dramatically change the landscape of therapeutic strategies for TB, potentially even reducing the duration of sterilizing combination antimicrobial therapy from the current 6-9 months.
4. Drug delivery to alveolar macrophages: A potential solution
Macrophages are integral components of the host defense mechanism and have a pivotal role in engulfing and destroying pathogen through the process of phagocytosis.[56] In phagocytosis, the macrophage engulfs the foreign microbes, thereby leading to destruction and elimination of microbe from the host. However, some pathogenic microorganisms, such as Mtb, have the capability to survive and thrive within these immune cells. This reduces the treatment efficacy for infections occurring from intracellular pathogenic microorganisms.[56] Additionally, ingestion into the macrophage provides an extra diffusional barrier for drug molecule to cross. Antibiotics used to treat intracellular infections often have varying ability to cross the macrophage’s cellular membrane, hence diminishing their bactericidal efficiency. Therefore, drug delivery platforms capable of direct targeting and modulation of macrophages may counteract the ability of intracellular pathogens to evade antibiotic treatments. Such delivery systems would require selectivity towards both macrophages and pathogens and would be invaluable towards improving chemotherapeutic activity. Alveolar macrophages are one known reservoir of Mtb where the bacterial burden is harbored.[57] Hence, direct targeting of alveolar macrophages has good prospects to enhance the effectiveness of current antibiotics, as well as future therapeutics to be used for anti-TB treatment.
Nano/Micro system-based drug delivery systems present an excellent avenue for direct targeting of alveolar macrophages. In an optimized system for drug delivery, the drug molecule is translocated to the target site to maximize drug absorption and minimize unwanted side effects. Moreover, drug delivery systems safeguard the therapeutic agents from being metabolized and cleared by the host. This increases the quantity of drug delivered to the target and decreases the therapeutic dose.[58, 59] Targeted drug delivery systems offer a methodology to improve availability of anti-TB drug within macrophages. A well-designed drug carrier can demonstrate controlled drug release characteristics in different metabolic and physicochemical responses. Recent progress in domain of nanotechnology has opened new prospects in development of new drug carriers, such as solid-lipid nanoparticles (SLNs), nanospheres, liposomes, micelles, and nano-rods.[56] Owing to their nanoscale dimensions (50-1500 nm) and high surface areas, the nano-based drug carriers have unique biological and chemical characteristics that make them an attractive choice for development of drug delivery platforms.
Broadly speaking, drug delivery efficiencies can be enhanced by two major approaches. One approach is tuning physical properties of drug carriers to improve macrophage internalization and deep lung deposition.[56] The other major approach is targeted delivery of drug. Alveolar macrophages can be targeted by utilizing two separate mechanisms: active targeting and passive targeting. In active targeting, the bulk carrier’s surface is functionalized with ligands which can interact with receptors on a host cell surface, resulting in ligand-receptor mediated phagocytosis.[60] In passive targeting, the drug carrier does not have any host-specific ligand on its surface.
In the past few years, there were only a few publications that have reviewed drug delivery systems for treating tuberculosis. Of these, most have focused on materials and formulations of drug carriers (liposomes, polymeric nanoparticles, etc.); however, one review discussed the different types of macrophage receptors which have been targeted.[56, 61-63] In this article, we will review primarily the recent development of targeting strategies used in drug delivery systems for pulmonary tuberculosis treatment, focusing on the various ligands reported in literature.
5. Tuning physical properties of drug carriers to enhance macrophage uptake and lung deposition
Macrophage-based targeting therapies are an attractive and effective strategy towards the treatment of infection caused by intracellular microbes. The interaction between outer carrier surface and the cell membrane of macrophages can induce the development of plasma membrane surrounded vesicles, subsequently leading to the internalization of drug carriers. There are five known processes for the internalization of nanoparticles, including phagocytosis, macro-pinocytosis, endocytosis uptake moderated by either caveolin or clathrin, and endocytosis that is independent of both.[64]
Internalization of the drug carrier within the macrophage is moderated by variety of factors, which includes physical characteristics and morphology, interaction with plasma protein, physicochemical attributes (surface charge, hydrophobicity/hydrophilicity), and biodistribution.[65] For example, drug carrier with improved hydrophobicity has demonstrated better interaction with the macrophage, thereby enhancing its uptake. Along with carrier surface composition and size, the overall shape is a pivotal parameter which can affect the nanoparticle uptake and efficiency of drug delivery.[66]
In addition to improvements in drug internalization by macrophages, deep lung deposition of inhalable TB drug carriers is a critical process for better targeting of alveolar macrophages. Particle size is an important characteristic that influences the deposition processes.[67] Particle sizes greater than 5 μm will be entrapped in the upper region of respiratory tract. Particles between 1-5 μm can achieve deep lung deposition but are unable to reach the pulmonary alveoli effectively. Particles less than 1 μm in size can reach pulmonary alveoli through diffusion.[62] Drug carriers with diameters between the range of 50 and 200 nm are considered desirable for maximum drug accumulation after administration by inhalation.[68] Particle diameters around 500 nm have also been reported in literature as suitable for passive targeting of alveolar macrophages.[69]
Apart from particle size, zeta potential also affects the identification and phagocytosis of nanoparticles by macrophages, ultimately affecting their distribution within macrophages. Cationic nanoparticles after internalization within the host deliver the payload more readily than non-charged nanoparticles, through the proton sponge effect.[70] In fact, a linear correlation between cellular uptake and zeta potential of the nanoparticles has also been reported in literature.[71] Interestingly, surface charge presents a dichotomous effect on particle internalization. It has been found that nanoparticles with negative zeta potential have tendency to accumulate at the positively charged site because of repulsion by more surface-abundant anions, resulting in better uptake by immune cells.[72-74] This dichotomy stems from variation in other critical parameters, such as surface hydrophobicity or composition, polydispersity, and the cell type variation. Zeta potential’s absolute value is one of the critical factors that influences the nanoparticle phagocytosis. Lower values of absolute zeta potential can actively avoid internalization of nanoparticles by macrophages.[75, 76] In the context of drug delivery platforms for TB treatment, both anionic and cationic systems have been developed successfully.[56]
Hydrophobicity of drug carriers can also influence the internalization processes. Drug carriers with hydrophobic surfaces have shown increased cellular uptake because of higher affinities for the lipid bilayer of macrophages.[56, 63] Hydrophilic modifications have also shown improved performance, mostly on carbohydrate-based drug carriers.[56] On the other hand, PEGylated drug carrier can effectively suppress the macrophage uptake.[77-79] For example, it has been reported that macrophage internalization of PEG functionalized polyelectrolyte nano shells at 24 hours was reduced threefold when compared to either negatively or positively charged nano shells.[80]
6. Passively targeted drug delivery systems
Passive targeting of alveolar macrophages utilizes receptor-independent uptake pathways. As such, it is an alternative for simplified drug delivery systems which do not involve structural modifications of the drug carrier. Numerous drug carriers have been developed so far for passive targeting of alveolar macrophages (Table 1-3). Some common delivery carriers include polymeric particles/micelles and polysaccharide-based particles. Other types of carriers have been used as well, such as liposome, solid lipid nanoparticle (SLN), and gold nanorods. Rifampicin and isoniazid are commonly used drugs in passive targeted drug delivery systems.[81-85] Other therapeutic agents, including gatifloxacin, curcumin, ethionamide, pyrazinoic acid, mycolic acid (for immunization), and siRNA have also been reported in literature.[86-91]
Table 1.
Passively targeted drug delivery system with the polysaccharide-based carriers
| Drug carrier | Drug | Carrier size & zeta potential |
bacteria/target cell/in vivo |
Key findings | Ref |
|---|---|---|---|---|---|
| Carrageenan-chitosan alginate NP | ETH | 320-324nm & −22mV | Mtb H37Ra/ N.A./N.A. | Controlled drug release was observed up to 96 hours. Drug loaded NPs showed antimycobacterial activity against extracellular Mtb. | [88] |
| Chitosan NP | RIF | 124–402nm & +59mV | N.A./ J774/Rat | Inhalable dry powder formulation showed controlled drug release and improved pharmacokinetics. No toxicity was observed against mice organs and J774 macrophages. | [92] |
| chitosan-graft-polymer micelles | RIF | 100-210nm | E. coli, S. aureus/ A549 /N.A. | Formulation exhibited pH dependent in vitro release profile. The drug carrier was effectively internalized by A549 cells. | [93] |
| Inulin | INH | 1-2μm & −29mV | Mtb H37Rv/ RAW264.7/ N.A. | Inulin drug conjugate was internalized by macrophages, and it exhibited a dose-dependent targeting against Mtb-infected macrophages. | [81] |
| Inulin | Pyarzinoic Acid | 1-2μm & +43.8mV | N.A./ RAW264.7/ N.A. | Inulin conjugate showed pH-dependent drug release. Conjugate had good targeting efficacy for RAW 264.7. | [87] |
| Chitosan coated SLN | RIF | 344 ± 11nm & +35mV | N.A./ A549/ N.A. | Chitosan coated SLN showed in vitro mucoadhesive characteristics and a higher permeability in A549 cells than uncoated SLN. | [94] |
| Alginate modified PLGA | AMK & MXF | 312-640nm & −25 ~ +5.4mV | Mtb H37Ra/ THP-1/ N.A. | Co encapsulation of both drugs performed better in inhibiting Mtb. Efficient uptake in macrophages and sustained release was observed. | [95] |
| Chitosan-lipid NP | Bedaquiline | 83~455.6nm & −10~+28mV | Mtb H37Rv/ N.A./ N.A. | Nano formulations had similar MIC when compared to free drug. Minimal or no cytotoxic effects were observed on THP-1, A549 and HepG2 cells by all formulations. | [96] |
| Inulin | RIF | 4.3±0.98nm & −14.8mV | M. smegmatis/ hAM/ N.A. | Inulin was functionalized with vitamin E for carrying hydrophobic drugs. The carrier was further succinylated to enhance the contact with bacterial walls. | [97] |
| Chitosan-grafted micelle | RIF, Ag NP, pyrazinamide | 141.4 ± 1.61nm & −8.44mV | E. coli, K.pneumoniae S.aureus, B. streptococci/ THP-1/ N.A. | Formulation exhibited pH dependent drug release profile. Severe cytotoxicity effect was observed on THP-1 cells. | [98] |
| Chitosan | siRNA | 215.3±4.19nm & +26.75mV | Mtb H37Rv/ mPM | Two-fold down-regulation of the host gene, Bfl1/A1, was observed as compared with untreated controls, when treated with siRNA loaded NPs. | [91] |
| pβCD NP | Blank drug carrier, ETH | 10nm | Mtb H37Rv, B. abortus, B. pertussis/ mAM, BMDM/ Balb/c mice | Blank pβCD had the specific antibacterial activity against Mtb in vivo. pβCD galvanized macrophage apoptosis, and impaired Mtb infection in mouse model. pβCD could also improve antimicrobial activity by the delivery of ETH. | [99] |
| Fucoidan particles | INH, RFB | 3.6~3.9μm† | BCG/ THP-1, NR8383/ N.A. | Drug loaded particle led to 95% inhibition of bacterial burden. The particles were readily internalized by macrophages and induced macrophage activation. | [100] |
Abv: ETH-ethionamide, Mtb- Mycobacterium Tuberculosis, RIF- rifampicin, N.A- not available, AMK-amikacin, MXF-moxifloxacin, hAM- human alveolar macrophages, mPM- mouse peritoneal macrophages, pβCD-poly beta-cyclodextrins, mAM- mouse alveolar macrophages, BCG- Mycobacterium Bovis bacillus Calmette–Guérin , BMDM- bone marrow derived macrophages, INH- isoniazid, RFB-rifabutin, siRNA- small interfering RNA
mass median aerodynamic diameter
Table 3.
Passively targeted drug delivery system with other types of carriers
| Drug carrier |
Drug | Carrier size & zeta potential |
bacteria/target cell/in vivo |
Key findings | Ref |
|---|---|---|---|---|---|
| Lipid-drug conjugate NP | INH | 124nm & −26.6mV | N.A./ THP-1/ N.A. | Slow and sustained release by drug carrier up to 72 hours. Efficient internalization of lipid–drug conjugate nanoparticles by macrophages was observed. | [128] |
| Surfactant-drug complex in mannitol particle | OCT313 | 277.9 ± 56.1nm | N.A./ RAW264.7/ Wistar rat | The formulation resulted in 7.5-fold higher internalization in RAW264.7 cells and 9.1-fold in alveolar macrophages of rats. | [127] |
| Gold nanorod | RIF | length: 25±3nm, width: 5±30.8nm & −21.7mV | Mtb H37Ra/ RAW264.7/ N.A. | Drug conjugated gold nanorods were effectively internalized by macrophages and elicited antimicrobial activity against extracellular and intracellular Mtb. | [82] |
| Liposome, quantum dot, PLGA NP, polymer micelles | RIF | 20 ~703nm | Erdman strain/Zebrafish & C57/BL6 mice | All formulation accumulated in granulomas in zebrafish model and mice model. Higher and rapid uptake by macrophages was observed. | [129] |
| Magnetic NP | P3 (anti-TB drug candidate) | 3.5 ± 0.1μm† | N.A./ THP-1, murine BMDM/ N.A. | External magnetic field actuated the 10-folds more release of drug in macrophages. The formulation was non-toxic to macrophages. | [130] |
| Carbon nanotube | INH | 15~20nm | Mtb S & H37Rv/ N.A./ N.A. | Carbon nanotube conjugated isoniazid had better bactericidal effect on Mtb strains. | [131] |
| BSA | RIF | 232±5.4nm, 3.21±1.2μm† & −30.1mV | Mtb H37Rv/ RAW264.7/ N.A. | The BSA NPs were effectively internalized by the infected macrophages and had better bactericidal effect. | [132] |
| Ca3(PO4)2 | BTZ043 | 30±6nm & −35 ~−55mV | Mtb H37Rv/ mBMDM, hMDM/ N.A. | The carrier effectively transported lipophilic drug to the intracellular pathogen and the encapsulated drug had comparable bactericidal effect on Mtb. H37Rv as that of free drug. | [133] |
| SLN | Streptomycin sulphate | 218.1±15.46nm & - 0.52mV | M.smegmatis, BCG, Mtb H37Rv/ THP-1/Rat | Drug loaded SLN had 20 times more internalization. Encapsulated drug had lower MIC against intracellular pathogen. Oral administration led to higher drug accumulation in rat plasma. | [134] |
Abv- INH: isoniazid, OCT313: 2-acetamido-2-deoxy-b-D-glucopyranosyl N,N-dimethyldithiocarbamate, RIF: rifampicin, PLGA: poly (D, L-lactide-co-glycolide) acid, BSA: bovine serum albumin, BTZ043: 1,3-Benzothiazin-4-one-043, BMDM: bone marrow derived macrophage, SLN: solid lipid nanoparticle
mass median aerodynamic diameter
Polysaccharide-based drug carriers are ubiquitously used in drug delivery platforms, owing to their great compatibility with biological systems and safety.[56] Table 1 lists several polysaccharide-based drug delivery carriers for passive targeting. Interestingly, some of these carriers could be considered as active systems due to potential interactions between their sugar moieties and macrophage receptors. For example, glucose moieties on inulin could interact with dectin-1, a β-glucan receptor. Additionally, fucose moieties on fucoidan could interact with mannose receptors. However, these papers did not specifically investigate the interactions between the carriers and macrophage receptors; therefore, we listed these drug delivery systems as passive.
Polysaccharides could be used to modify surface charges of drug carriers. For instance, chitosan, a polysaccharide with positive charges, has been ubiquitously used in drug delivery. Vieira et al. fabricated a solid lipid nanoparticle coated with chitosan(C-SLN) and encapsulating rifampicin as a model drug.[94] The idea behind chitosan coating of SLNs was to enhance the mucoadhesive properties of the drug delivery system. Improved mucoadhesion would allow drug carriers to avoid mucociliary clearance from the airways.[101] Decoration of SLN with chitosan resulted in a positive zeta potential of 40 mV and a smooth spherical shape with a diameter 245–344 nm. In this study, C-SLNs demonstrated better in vitro mucoadhesive properties and significantly enhanced penetration in A549 (human alveolar basal epithelial cells) when compared with free rifampicin and uncoated SLNs. In addition to C-SLNs, chitosan has been used to decorate polymeric micelles and encapsulate drugs.[91-93, 101] Chitosan-based drug delivery systems have shown improvements in drug delivery efficiency, mitigated unwanted adverse effects, enhanced surface hydrophilicity/hydrophobicity to encapsulate different drugs, and allowed for slow and sustained drug release profiles. The chitosan drug conjugates have showed significant potential for further investigation in clinical applications.[91-94, 98, 101]
Ideally, a drug carrier should not release drug before the desired target site is reached. The lysosomal compartment of the macrophage has an acidic environment (pH ~5), granting it great potential for unique targeting for drug delivery. Few studies have successfully designed a drug carrier which exploits the acidic environment within the macrophage.[81] Afinjuomo et al. developed a hydrazone linked inulin-isoniazid conjugate which targeted Mtb infected murine macrophage cells RAW264.7.78 This conjugate was pH sensitive, which rendered it capable of releasing INH selectively under acidic conditions. After 5 hours, 65%, 77% and 95% of the drug was released at pH 6.0, 5.2 and 4.5 respectively; minimal release of the drug occurred at pH 7.4. The drug carrier also had much higher internalization by RAW264.7 cells when compared with free drug (p<0.0001 across all time points). The inulin-INH conjugate system exhibited dose-dependent killing of intracellular Mtb H37Rv. Despite much higher uptake, the antibacterial activity of inulin-isoniazid conjugate was consistent compared to free isoniazid. In fact, in vitro studies showed that the inulin-INH conjugate was less effective at lower concentrations than free INH. Afinjuomo et al. hypothesized the different diffusion rates of inulin-INH and free INH into cells might be the reason behind low killing activity at low concentrations.[87] The authors suggested that comparing soluble drug against the inulin-INH conjugate made it difficult to evaluate the effectiveness of this targeted delivery system , and thus, in vivo studies would be necessary for further insight. In another interesting study, researchers fabricated an inulin carrier functionalized with vitamin E to enhance the encapsulation of a hydrophobic drug (e.g., rifampicin). The delivery system had a slow and sustained drug release profile, with an MIC similar to that of free rifampicin against M. smegmatis.[97]
Polymers are some of the prominent materials used in drug delivery platforms as they can provide controlled and tunable drug release, and can be utilized for encapsulating hydrophilic and hydrophobic drugs (Table 2). Poly (lactic-co-glycolic acid) (PLGA) is the widely used polymer used for fabrication of anti-TB drug delivery systems.[59, 90, 105, 110, 112, 115] PLGA is an FDA approved biocompatible polymer with very low toxicity. Overall physical properties of the PLGA system can be easily tuned to achieve a desired dosage and release profile by altering parameters such as ratio of lactide to glycolide, molecular weight of the base polymers, and drug concentration.[116] These properties make PLGA an excellent option for fabricating controlled drug delivery carriers. Use of PLGA microspheres to deliver subunit TB vaccine has also been reported in the literature.[117] In a study conducted by Lawlor et al., blank PLGA microparticles were able to activate the innate immunity pathway by increasing the NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells) activity in uninfected macrophages in a size dependent manner.[112] However, treatment with PLGA did not activate the cytokine secretion in uninfected macrophages or change the cytokine profile of macrophages infected with Mtb. This is in contrast with previous studies where the authors observed the stimulation of anti- or pro-inflammatory cytokine secretion after PLGA treatment.[115, 118-121] Even in the absence of any encapsulating drug, PLGA microparticles were able to reduce intracellular pathogen load within infected macrophages, suggesting PLGA may have antimicrobial activity.[112] PLGA treatment also triggered autophagic flux in macrophages infected with Mtb and led to induction of autophagy. Autophagy is a process in which cells recycle cellular components by enveloping them in autophagosomes and fusing them with acidic lysosomal compartments to degrade them.
Table 2.
Passively targeted drug delivery system with polymer-based carriers
| Drug carrier |
Drug | Carrier size & zeta potential |
bacteria/target cell/in vivo |
Key findings | Ref |
|---|---|---|---|---|---|
| PLGA NP | Gatifloxacin | 4μm | N.A./RAW264.7/male swiss mice | Surface modification with labrafil improved the uptake rate of PLGA particle by macrophages. | [90] |
| Cu-cluster dendrimer | INH | 900nm & +4.55 ~ +9.69mV | Mtb H37Ra/ N.A /N.A. | Drug carrier exhibited controlled release. Drug loaded nanoclusters provided a synergistic effect. | [85] |
| PES particle | RIF & Curcumin | 400nm & −26.89mV | Mtb H37Rv/ RAW264.7/N.A. | NPs had 1.5-fold higher internalization. RIF-CUR NP had high efficacy against intracellular Mtb at 25× MIC (98% inhibition), and complete clearance above 50× MIC. | [89] |
| PEA NP | RIF | 544.8nm & −8.23mV | M. smegmatis/ NR8383/ N.A. | PEA NP elicited pH dependent release profile and delivered RIF more effectively to macrophages. | [83] |
| PEG-PPS micelles | Mycolic acid | 68.13nm & −16.5mV | N.A./ BMDC, mAM/ hCD1Tg+ mice | Intranasal administration with mycolic acid micelles induced MA-specific T cell responses in the lungs of hCD1Tg mice. Majority of MA-MC were taken up by alveolar macrophage (60%) and remaining by myeloid dendritic cells (40%). | [86] |
| PLGA particles | RIF, MMP (Ag, ZnO) | 1500 ± 620nm & +1.1mV | Mtb H37Ra/ THP-1 | Efficient uptake of MMP by macrophages were observed. MMPs were effective in increasing membrane destruction of extracellular Mtb and increased the potency of the RIF by 76% of intracellular Mtb. | [102] |
| Dendrimer, F-127, P188 | Ga (III), RIF | 305~882nm & +35mV~−10.4mV | Mtb H37 Rv/ THP-1 | Nano formulations with dendrimers encapsulating Ga (III) or rifampicin showed the maximum rate of uptake by macrophage. The nanoparticles colocalized with Mtb containing phagosome. The dendrimer NP showed the most promising anti-tubular activity. | [103] |
| PEG-PLGA | INH & RIF (co-loading) | 187.9 ± 2.68nm & −8.15mV | clinical isolate/ N.A./ N.A. | Approximately 8-fold reduction in MIC was observed. | [104] |
| PLGA NP | TZ & RIF | 249 ± 109nm & −20.7 ~−14.2mV | BCG, Mtb H37Rv/ mBMDM, hMDM/Zebrafish | PLGA rendered TZ non-toxic on cells and zebrafish embryo. The TZ NPs in combination with RIF enhanced embryo survival and reduced the mycobacterial infection. | [105] |
| MPEO-b-PCL NP | RIF | 85nm | Mtb H37Rv/ RAW264.7/ BALB/c mouse | RIF loaded NPs improved the in vivo treatment efficacy by reducing granulomas formation in mouse model. Formulation also led to reduction of Mtb in both spleen and lungs. | [106] |
| ODOC NP | INH & CFZ | 284 ± 11nm & −20mV | Mtb H37Rv, M. marinum/ THP-1 /Zebrafish | The formulation was readily taken up by infected macrophages and elicited enhance killing of intracellular Mtb. Drug carrier efficiently delivered drug to granulomas and extracellular Mtb in an infected zebrafish model. | [107] |
| PLGA particle | HDP with INH | 4780~5640nm | Mtb H37Rv/ RAW264.7/ N.A. | The combination of host defense peptides and INH enhanced the efficacy of the INH. | [108] |
| MPEO-b-PCL NP | RIF | 20~110nm | Mtb H37Rv, M. marinum, M. smegmatis, M. phlei, M. fortuitum, BCG/ RAW264.7/ Zebrafish | Formulation showed good killing efficacy against multiple bacterial strains and shows a significant killing effect of intracellular bacteria. Drug loaded NP significantly improved in vivo survival of zebrafish. | [109] |
| PLGA particle | RIF | 953 ± 20nm & −16mV | N.A./ RAW264.7/ N.A. | Modified fabrication protocol yielded narrower particle size distribution of PLGA particle. Surface modification with polyethyleneimine led to better uptake of nanoparticle by the macrophage. | [110] |
| Polymer micelles | RIF | 107.6±1.16nm & −5.88mV | Mtb ΔRD1/ RAW264.7, THP-1/mice | RIF loaded formulation had 2.5-fold enhancement in in vitro bactericidal activity against intracellular Mtb. Drug carriers remain lung accumulation after 24 h. | [111] |
| PLGA particle | Blank drug carrier | 2.2 ± 0.3μm & −33.4mV | Mtb H37Ra, H37Rv/ THP-1/ N.A. | PLGA microparticle without drugs is potent immunogens and restrict Mtb growth. It increased NFκB activity in macrophages. Induction of autophagy was also observed after treatment with PLGA particle. | [112] |
| PLA/PLGA particle | NTZ, INH, RFB | 6.89 ± 0.62μm† | Mtb H37Rv/ THP-1/Swiss mice | Combination of RFB, NTZ, and INH cleared all bacterial burden from the lungs and spleens and led to more extensive restoration of tissue architecture. | [113] |
| PLGA particle | Rapamycin | 2.88 ± 0.8μm | Mtb H37Rv/ THP-1/ N.A. | Rapamycin, an inducer of autophagy, facilitates killing of intracellular Mtb. PLGA particles were effectively taken up by THP-1 and rapamycin loaded PLGA particle was more efficient in reducing intracellular Mtb burden in THP-1 cells. | [114] |
Abv: PLGA: poly (D, L-lactide-co-glycolide) acid, PLA: poly(lactic acid), INH: isoniazid, RIF: rifampicin, NTZ: nitazoxanide, RFB: rifabutin, PEA: polyester amide, PPS: polypropylene sulfide, MMP: multimeric nanoparticle, PEG: polyethylene glycol, MPEO-b-PCL: methoxy poly (ethylene oxide)-block-poly(ε-caprolactone), ODOC: 1,8-Octanediol-dimethyl 2-oxoglutarate copolymer, CFZ: clofazimine, HDP: host defense peptides, TZ: thioridazine, mAM: murine alveolar macrophages, mBMDM: mouse bone marrow derived macrophage, hMDM: human monocyte-derived macrophages
mass median aerodynamic diameter
Researchers have also developed drug delivery systems with non-traditional anti-TB therapeutic agents.[102, 103] Recently, a PLGA microparticle-based drug delivery system was reported with multimeric silver and zinc oxide nanoparticles as the therapeutic agent.[102] In this study, researchers developed 1.5 μm PLGA carriers which targeted the THP-1 cell line. Silver and zinc oxide multimeric nanoparticles (MMP) improved the efficacy of the model drug rifampicin by disrupting the cellular envelope of Mtb. MMPs increased the efficacy of rifampicin by 76% when subjected to THP-1 macrophages infected with Mtb. Dosing of 60 μg mL−1 MMP (Ag+Zn), MMP(Zn), MMP(Ag), and led to 16.4%, 15.9%, and 4.5% reduction in colony forming units (CFU), respectively.94 The authors also found that co-incubation of subpar therapeutic concentration of rifampicin (1 μg/ml) with MMPs (60 μg/ml), when administered in tandem with Mtb (Multiplicity of infection 1) to THP-1 macrophages, boosted the efficiency of rifampicin. Presence of MMP enhanced the disorderliness of intracellular Mtb membrane, potentially mediating increased penetration of rifampicin.94
In another recent study, Choi et al. explored the anti-tubular potency of gallium (III) meso-tetraphenyl porphyrin chloride (GaTP) using different types of drug carrier.[103] The rationale behind the study was that gallium hinders iron acquisition, which is critical for pathogen proliferation and survival, in various microorganisms including Mtb. [122-125] Even though gallium has chemical properties akin to iron, Ga (III) is unable to gain an electron to become Ga (II), unlike iron which can become Fe (II). Therefore, iron replacement with gallium in biologically important proteins leads to inhibition of critical metabolic pathways. The antimicrobial properties of Ga compounds have already been examined and exhibited against Mtb as well as against various pathogenic bacterias.[122-125] Efficacy of gallium nanoparticle formulations against non-virulent strains of Mtb (Mycobacterium smegmatis and Mtb H37Ra) has already been reported in the literature.[126] Choi et al. developed gallium (III) loaded NP, consisting of commercial F-127 polymeric NP and dendrimers (GaD), to target the virulent Mtb H37Rv residing in THP-1 cells. GaD was internalized faster when compared with other nanoparticle by the THP-1 macrophages. All nanoparticles exhibited significant inhibition in growth of Mtb for up to 15 days after drug delivery to the THP-1 cells, thereby demonstrating extended release and retention of drug from the delivery system within macrophages. Whereas GaTP in form of free drug, did not inhibit the Mtb growth in 5 days within macrophages after incubation with the drug. Interestingly, despite of its relative larger size, the dendrimer nanoparticle loaded with GaTP demonstrated the most promising anti-TB activity. The authors found that gallium nanoparticles were as potent as rifampicin loaded nanoparticles in inhibiting intracellular Mtb in THP-1 macrophages. GaNP resulted in 50 % more co-localization of Mtb in acidic compartments. Authors attributed this to either slowdown of phagosome (endosome) maturation inhibition by the mycobacteria or enhancement of the maturation process.[126]
Other types of drug carriers have been developed as well (Table 3). For instance, Maeda et al. fabricated a dried emulsion of surfactant drug conjugate coated with mannitol (SDC/MAN), loaded with the sugar-based drug OCT313.[127] The SDC was coated with mannitol to make the drug delivery system hydrophilic. The authors found that the SDC/MAN composite had 7.5-fold higher uptake within RAW264.7 cells 4 hours post-treatment when compared against free drug. Maeda et al. also tested the in vivo efficiency of the drug delivery system in wistar rats by administering formulation intratracheally. The in vivo study revealed that SDC/MAN had 9.1-times higher uptake 4 hours post-treatment than that of drug alone in rat alveolar macrophages. SDC/MAN was not tested for its anti-tubular efficacy against Mtb harbored within macrophages.
Apart from using drug delivery systems to transport antibiotics against Mtb, researchers have also utilized nanocarriers to deliver antigen, eliciting a host immune response against Mtb infection.[86] Recently, Shang et al. explored the application of polymeric micelle nanocarriers loaded with mycolic acid as a prospective vaccine candidate for TB. The rationale behind this study is that several of the lipids obtained from the Mtb cell membrane can be identified by CD1-restricted T cells.[135-140] Mycolic acid (MA) is one of the lipids derived from the mycobacterial cell membrane and is a major component of its outer membrane. The CD1 family of antigen presenting molecules is known to present glycolipid/lipid antigens to T cells.[135, 136, 141] These mycolic specific CD1b-restricted T cells are toxic to cells and produce proinflammatory cytokines TNF-α and IFN-γ, crucial cytokines for anti-Mtb immunity.[142, 143] In vitro studies demonstrated higher efficacy of MA loaded polymeric micelles when compared with free MA in activation of MA-specific TCR transgenic T cells. Furthermore, subsequent to intranasal administration during the in vivo study, MA-micelles were majorly internalized up by dendritic cells and alveolar macrophages. In vivo/vitro data collected by Shang et al. demonstrated the efficacy of pulmonary delivery for MA-micelles in order to elicit a potent CD1b-restricted T cell response. The authors championed further exploration of MA-micelles as a potential subunit vaccine against Mtb infection. Another potential positive of this study is that MA-micelles have the potential to treat HIV-TB co-infected patients. Co-infection of HIV and TB often leads to reduction of CD4+ T cells;[144] however, group 1 CD1-restricted T cells are not modulated by HIV infection.[145, 146] Hence, targeting group 1 CD1-restricted T cells by mycolic acid-based subunit vaccines could be specifically instrumental for HIV positive patients co-infected with Mtb.
7. Actively targeted delivery systems
Active targeting of macrophages for delivery of anti-tubular therapeutic agents involves surface modification of the drug carrier with a host-specific ligand. In actively targeted drug delivery systems, therapeutic agents are internalized by macrophage-based ligand receptor mediation. Due to this added layer of interaction, enhancement of macrophage targeting can be attained by functionalization of the drug carrier’s surface with ligands that can be identified by corresponding receptors on macrophages.[60] Various types of phagocytic receptors have been reported in the literature, consisting of mannosyl receptor (CD206), fucosyl receptor, scavenger receptor, Fc (fragment, crystallizable) receptors, folate receptor, hyaluronic acid receptor (CD44), tuftsin receptor, formyl peptide receptor, and other lectin-like receptors.[61, 147-150] Various ligands have been used to target these macrophage receptors.
7.1. Saccharide-based ligand targeting
A commonly adopted methodology for active macrophage targeting is modification of drug carriers with sugars (e.g., polysaccharides or glycoproteins) to target lectin-like receptors (Table 4). Mannose or fucose functional group are commonly used for the binding to mannose receptors, CD206.[151] Apart from utilizing mannose-based ligands, hyaluronic acid (D-glucuronic acid and N-acetyl-D-glucosamine) and curdlan (glucose) have also been reported as targeting ligands in the literature.[152-156] The macrophage-based mannose receptor has the potential to facilitate the pinocytosis of soluble glycoconjugates and the phagocytosis of sugar coated nanoparticles.[157] Researchers have exploited this phenomenon in the fabrication of macrophage-mediated therapies to promote cellular uptake of nanocarrier-based drug delivery systems.[151] Jiang et al. exploited the mannose receptors on antigen presenting cells to increase the cellular uptake by fabricating mannose functionalized chitosan-graft-polyethyleneimine copolymer.[158] Similarly, Ratner et al. reported increased cellular uptake of mannose decorated glycopolymers, when compared to galactose decorated glycopolymers, by the macrophage .[159]
Table 4.
Actively targeted drug delivery system using saccharide-based ligands
| Ligand | Drug carrier |
Drug | Carrier size & zeta potential |
bacteria/target cell/in vivo |
Key findings | Ref | |
|---|---|---|---|---|---|---|---|
| Mannose receptor | MP | SLN | RIF | 720 ~ 1380nm & −44.40 ~ −63.7mV | N.A./J774/N.A. | Surface mannosylation provided faster macrophage phagocytosis. | [168] |
| 4-SO4-GalNAc & Man-Lip | Liposome | N.A | 139.4~ 147.4nm & +12.2 ~ +17.3mV | N.A./J774, RAW264.7/ Wistar rat | Uptake of 4-SO4-GalNAc liposome was almost 2-fold higher than Man-Lip liposomes in vitro. | [148] | |
| Locust bean gum | Locust bean gum particle | RFB/INH | 6μm† | Mtb H37Rv/ N.A./BALB/c mice | Locust bean gum contains mannose and galactose. Pulmonary administration led to better treatment efficacy. A negative growth index of Mtb in lungs, spleen, and liver was observed. | [166] | |
| Mannose | Nano-lipid carrier | RFB | 175~ 213nm & +37.6mV | N.A./RAW264.7/N.A. | Drug carrier exhibited pH dependent controlled release. Formulation had no toxicity on RAW264.7, Calu-3, and A529 cells. | [165] | |
| PAM | Spray-dried liposome embedded in microparticle | MXF | Liposome: 244~380nm & −31 ~ +26mV, liposome in particle: 1470~4760nm† | Mtb/J774/albino rats | Deep lung deposition was observed in rats. Charged particles have higher anti-tubercular activity and macrophage uptake. Mannose decoration improved macrophage uptake. | [170] | |
| Mannose | Graphene oxide | RIF | 50~300nm (diameter), 5~10nm (height) & −10mV | Mtb H37Rv/ THP-1/small intestine of Rhesus monkey | Mannosylated durg carrier had enhanced macrophage uptake. Mannosylated graphene oxide exhibited much higher inhibition effects against intracellular Mtb when compared to free rifampicin. | [164] | |
| Mannose | Chitosan NP | INH, selenium | 45nm & +25mV | Mtb H37Rv, BCG/ THP-1/small intestine of Rhesus monkey | Mannosylated NP preferentially entered macrophages and accumulated in lysosomes. The formulation promoted the fusion of Mtb into lysosomes, and induced autophagy leading to Mtb degradation. | [173] | |
| Mannose | SLN | RIF | 252nm & +25.7mV | N.A./ THP-1/ N.A. | Mannosylated drug carrier was less toxic than bare SLN. Mannosylation enhanced macrophage uptake. | [162] | |
| Mannose | F127 | Ga (III) | 309nm & +17mV | Mtb H37Rv & HIV-1/ THP-1/ N.A. | Mannose-Ga (III) formulation was efficiently internalized by macrophages and sustained release was obtained for 15 days. The formulation inhibited the HIV-Mtb coinfection in macrophages. | [174] | |
| mannosyl surfactant | SLN | RIF | 740nm & −35.2mV | N.A./MH-S/N.A. | Lipid corona did not alter the role of mannosylation and rapid drug translocation within macrophage was achieved. | [175] | |
| Mannose | Polymer NP | RIF, Zn | 268nm & −17.59mV | Mtb/ A549/ N.A. | The carrier had pH dependent drug release and low toxicity toward VERO cells. The Zn and RIF-based NP had better bactericidal effect on Mtb as compared with free RIF. | [176] | |
| SAMAN | Lipid carrier | RIF | 240.9nm & −43.3mV, dried: 409.5nm | Bacillus subtilis/ RAW264.7/ N.A. | Mannosylated lipid particle was taken up by the RAW264.7 cells more efficiently than bare lipid particle. | [177] | |
| Mannose | Polymer augmented liposomes | Streptomycin | 117nm | F. novicida U112/ RAW264.7/ N.A. | Targeted liposomes had 2.5-time higher uptake and had better antimicrobial efficacy against intracellular pathogen. | [178] | |
| Mannose | SLN | RIF | 400 ~1430nm & −30.1~−49.73mV | N.A./ J774/ N.A. | Mannosylated SLN entered macrophages more efficiently, compared to 20% free RIF and to 40% non-functionalized SLNs. | [169] | |
| Fucosyl lipid | Lipid carrier | LVX | 108.87nm & −6.05mV | BCG/ MDM, MDDC/ N.A. | Fucosylated carrier effectively transported drug to endosomal compartment of macrophage. The formulation cleared BCG more efficiently than free drugs. | [179] | |
| CD44 | HA | Spray-dried sodium hyaluronate composite | RIF, INH, Verapamil | 940nm | Mtb H37Rv, MDR, XDR strains/ PBMC/ Human blood | The nanocomposite had more than 80% decrement in bacterial viability regardless of profile of the drug resistance. | [155] |
| HA | nanogel | AMP | 533nm & +2.4mV | M. avium, Mtb H37Rv/ mBMDM/ C57BL/6 mice | HA modified nanogel was internalized by macrophage and led to intracellular inhibition of bacteria. The formulation also reduced bacterial level in the lungs in vivo after intratracheal administration. | [154] | |
| HA | Hyaluronan particle | Ofloxacin | 2~5μm | N.A./RAW264.7/ Sprague Dawley rats | HA functionalized microsphere had 2.1- and 1.7-times higher uptake than free drug and non-functionalized microsphere by macrophage. | [153] | |
| HA | Tocopherol succinate micelles | RIF | 212~294.6nm & −23.7 ~ −30.9mV | N.A./ MH-S/ N.A. | HA functionalized micelles had a higher uptake when compared to free RIF, with the maximum uptake at 12 hours. Formulation induced higher Th1 cytokines level than free drug, which enhanced the anti-tuberculosis activity. | [156] | |
| Dectin 1 | Curdlan | Cyclodextrin | RIF & LVX | 523± 126nm & +13.45mV | M. smegmatis/ RAW264.7/ N.A. | The formulation was nontoxic to macrophages and fibroblast cells. Curdlan decorated NP had 1.8 times higher internalization in macrophages as compared with fibroblast cells and resulted in 95% killing of intracellular Mtb in 4 hours. | [152] |
| β-glucan | β-glucan particle | RFB | 2.9 ~ 6.1μm & −9.46mV | N.A./ J774/ N.A. | The nanoparticles were internalized by macrophages in 5 mins of exposure. | [180] | |
| β-glucan | β-glucan particle | RFB | 1~ 4μm | Mtb H37Ra/ J774/ N.A. | Glucan NPs enhanced RFB efficacy 2.5 folds against intracellular Mtb. The exposure of drug carriers enhanced innate immune response including the induction of reactive oxygen and nitrogen species. | [181] | |
| Lectin like receptor | 4-SO4-GalNAc | Chitosan NP | Chitosan | 29nm & 12.2mV | M. smegmatis/ RAW264.7/ N.A. | Chitosan particles serve as an antimicrobial reagent. Acr-1 protein can enhance the antimicrobial activity for intracellular bacteria. 4-SO4-GalNAc ligand enhanced the macrophage uptake and improve the antimicrobial activity in combination with Acr-1. | [182] |
| Hydrolyzed GalMan | Polymer micelles, chitosan NP | RIF | Micelle: 198.7nm &+7.1mV, Chitosan NP: 334.4nm & +18mV | N.A./ RAW264.7/ N.A. | GalMan formulation were taken effectively by macrophages. The modified micelle led to a significant higher intracellular level of RIF in macrophages. | [183] | |
| Transferrin receptor | Transferrin | Ag-quantum dot | RIF-Zn complex | 50~20nm | M. smegmatis, BCG/ RAW264.7/ N.A. | Transferrin decoration led to 10-fold higher antibacterial activity against Mtb. | [184] |
Abv- MP: methyl α-d-manno-pyranoside, SLN: solid lipid nanoparticle, RIF: rifampicin, RFB: rifabutin, INH: isoniazid, PAM: 4-aminophenyl-alpha-d–manno-pyranoside, MXF: moxifloxacin, Ga (III): Ga (III) meso-tetraphenyl porphyrin chloride, SAMAN: N-octadecyl-manno-pyranosylamine, LVX: levofloxacin, HA: hyaluronic acid, AMP: antimicrobial peptide, GalMan: galactomannan
mass median aerodynamic diameter
Saraogi et al. fabricated mannose functionalized gelatin nanoparticles for actively targeted delivery of the isoniazid into macrophages.[160] The authors prepared nanoparticles ranging between 260-380 nm with a maximum drug encapsulation of 40–55%. The efficiency of the delivery system to target macrophages was demonstrated against J774.[160] The same group previously studied the effect of mannosylation on solid lipid nanoparticles encapsulated with rifabutin, and published encouraging data for hematological studies, alveolar macrophage internalization, and drug release.[161]
In the literature, mannose-modified drug carriers are the most common drug delivery platform actively targeting macrophages.[162-169] Mannose can be added to various carriers, including liposomes, SLNs, and polymer micelles. For instance, Pi et al. developed a mannose decorated graphene oxide-based drug delivery system targeting Mtb H37v infected THP-1 macrophages.[164] In this study, mannose was conjugated to graphene oxide by poly-ethylene glycol (GO-PEG-MAN). The in vitro uptake study of GO-PEG and GO-PEG-MAN revealed that mannosylation of GO lead to statistically significant higher uptake in macrophages derived from rhesus monkey infected with Mtb.[164] Macrophages had much higher concentration of rifampicin than that of B and T cells which expressed no or minimal level of mannose receptor. As expected, after encapsulating rifampicin onto mannosylated graphene oxide, the targeted delivery system demonstrated significantly higher inhibition efficacy against intracellular Mtb H37Rv when compared to free rifampicin. These studies show the importance of continued research and effort into the effectiveness of mannosylation of drug delivery systems for enhanced targeting of alveolar macrophages.
Mannose derivatives have also been used for surface functionalization of nanocarriers in the effort to target macrophages better. In a recent study, Hamed et al. decorated the surface of liposomes with 4-aminophenyl α-D-mannopyranoside (PAM) and fabricated a drug delivery system encapsulating moxifloxacin.[170] The authors fabricated multiple liposomes with varying compositions of phospholipids (e.g. PC, PE, DOTAP) and cholesterol. The liposomes were then spray dried to obtain an inhalable powder (SD-PAM-NL). The rationale behind spray drying was to enhance the stability of the liposomes and to make them favorable for direct deep lung deposition. The authors found that mannosylation of liposomes lead to higher uptake of drug carriers in J774A.1 murine macrophages, when compared with non-mannosylated liposomes.[170]
Hyaluronic acid (HA) is another sugar-based ligand for host-cell directed targeting of macrophages. CD44, a hyaluronic acid receptor, is highly expressed on macrophages. HA is also known to interact with toll-like receptors (TLR2 and TLR4), which are abundantly present on macrophages.[171, 172] This makes hyaluronic acid an enticing choice for macrophage targeting. HA has been added to common drug carriers, such as micelles and nanogels. Additionally, the hyaluronic acid polymer could serve as a drug carrier. For example, sodium hyaluronate-based respirable microparticles were reported in the literature to treat drug resistant TB.[155] The authors developed a spray-dried formulation encapsulated with isoniazid, rifampicin, and verapamil. Verapamil is a known efflux pump inhibitor and was added to further enhance the drug concentration inside the macrophages. The spray-dried formulation had a mean volume diameter of 1 μm, which is sufficient for deep lung accumulation and had a slow and sustained in vitro drug release profile. The formulation was able to kill intracellular H37Rv, Mtb1-MDR, and Mtb2-XDR strains, resulting in 80% reduction in bacterial viability.
Similarly, Silva et al. fabricated nanogel which exploited hyaluronic acid’s affinity for macrophages.[154] The nanogel was loaded with antimicrobial peptide (AMP) LLKKK18. Authors were successfully able to reduce the cytotoxicity of AMP against murine bone marrow derived macrophages by encapsulating it into self-assembling hyaluronic acid nanogels. The formulation was effectively internalized by macrophages and co-localized with mycobacteria. Significant reduction in intracellular bacterial load was achieved against M. avium or the pathogenic Mtb H37Rv. In addition, the hyaluronic acid nanogel curtailed the pro-inflammatory cytokine levels of IL-6 and TNF-α. The authors also showed the in vivo efficiency of the hyaluronic acid nanogels using a mouse model. The intratracheal administration of AMP loaded hyaluronic acid nanogels resulted in reduced level of mycobacterium burden in lungs of mice.[154]
β-glucan is another sugar-based ligand which is used for targeting the dectin-1 receptor on macrophages. In a recent study, Basha et al. fabricated a β-cyclodextrin system for actively transporting rifampicin and levofloxacin to a RAW264.7 macrophage cell line infected with M. smegmatis.[152] In this study, β-cyclodextrin (CD), which has a hydrophilic outer surface and hydrophobic inner cavity, was utilized to increase the drug loading. Curdlan, the polymer of β-(1,3)-glucan, was used to achieve active targeting of the dectin-1 receptor. The authors demonstrated that both curdlan-CD nanoparticles and curdlan were internalized by the macrophage via dectin-1-receptor-moderated endocytosis.[152] The results of the intercellular killing study showed that within 4 hours, all drug based nanoparticles led to the reduction of CFU by more than 90%, while on the contrary same concentration of free rifampicin (12 μg/mL) exhibited reduction of only 53% in CFU.[152]
In another interesting study, transferrin was tested as a targeting ligand. Transferrin is a blood-plasma glycoprotein present on macrophages and is responsible for ferric-ion delivery and iron metabolism. Macrophages have an important role in systemic iron trafficking, which is additional to their essential role in immune surveillance and induction of inflammatory response.[185] Macrophages express transferrin receptor 1 (TfR1), which moderates transferrin-bound iron uptake.[185] Pati et al. fabricated a silver quantum dot conjugated with transferrin to test the possibility of active transport of the zinc-rifampicin complex to RAW264.7 cells infected with M. smegmatis and BCG.[184] The authors performed a cytotoxic study and reported that Zn-rifampicin exhibited low cytotoxicity against RAW264.7 macrophages, and had no cytotoxicity at bactericidal levels. The authors also demonstrated that transferrin conjugated quantum dots were efficiently internalized by dendritic cells and peritoneal macrophages, but not by epithelial cells (A549). In addition, they studied the efficacy of the antibacterial efficacy of Zn-rifampicin against BCG and M. smegmatis, and compared it with that of rifampicin and Zn individually. Anti-bacterial studies confirmed that Zn-rifampicin had more potency against both mycobacterial strains than free Zn or rifampicin by themselves. The authors suggested that Zn-rifampicin complex had higher penetration through the bacterial membrane because it had higher lipophilicity than free rifampicin. An in vitro study targeting mycobacteria-infected RAW264.7 cells demonstrated that treatment with Zn-rifampicin encapsulated into transferrin-conjugated quantum dots exhibited at least 10 times more in vitro and intracellular killing efficacy when compared with Zn-rifampicin and free rifampicin, demonstrating that the formation of conjugate led to enhanced antimicrobial activity. The authors attributed this to the targeted delivery of the conjugated quantum dots to the infected phagosomes, accompanied by the sustained and slow release of zinc and rifampicin from the conjugate. Further studies are needed with these complexes, to determine if they can be used for a variety of different diseases and in different tissue types.
Other types of sugar-based ligands have been reported in the literature, but the lectin-like receptors responsible for targeting were not identified.[182, 183] Mubin et.al, decorated chitosan nanoparticles individually with 4SO4-N-Acetyl-Galactosamine (4-SO4-GalNAc) and Acr-1, a protein of Mtb (Mtb protein).[182] The authors did not encapsulate any drug agent within the chitosan; however, chitosan functionalized with 4-SO4-GalNAc exhibited higher binding affinity for macrophages than the bare chitosan particle. Even in the absence of drug, all carriers showed some level of antimicrobial activity. Chitosan functionalized with 4-SO4-GalNAc had better antimicrobial activity against intracellular M. smegmatis than both bare chitosan and chitosan functionalized with Acr-1. In another study, the authors fabricated chitosan nanoparticles and polymeric micelles, and decorated them with hydrolyzed galactomannan to target lectin-like receptors.[182, 183] The hydrolyzed galactomannan formulation was internalized effectively by mouse macrophages and resulted in enhanced intracellular concentration of drug within the macrophages.
7.2. Peptide-based ligand targeting
Peptide-based ligands have also been utilized for targeting macrophages to deliver anti-TB therapeutic agents (Table 5).[186-189] Tuftsin is a natural occurring tetrapeptide created by enzymatic breakdown of the Fc domain of the heavy chain of IgG. It is known to elicit immune response by activating macrophages and other elements of the immune system.[190] Macrophages and monocytes also express tuftsin-specific receptors, and tuftsin is known to stimulate phagocytosis, pinocytosis, and chemotaxis.[190, 191] Synthesis of tuftsin derived carriers has already been reported in literature, where they were found to have chemotactic effect on J774 monocytes.[192]
Table 5.
Actively targeted drug delivery system using other types of ligands
| Ligand | Drug carrier |
Drug | Carrier size & zeta potential |
bacteria/target cell/in vivo |
Key findings | ref | |
|---|---|---|---|---|---|---|---|
| Tuftsin Receptor | Tuftsin derivative | PLGA NP | INH | N. A | Mtb H37Rv/MonoMac6/ Guinea Pig | The formulation had no toxicity on peripheral blood mononuclear cell. After treatment with formulation, no mycobacterial colonies were observed in lungs, spleen and kidney of infected guinea pig. | [189] |
| Tuftsin | PLGA (core), F127 (coating) | TB515 | 200nm | Mtb H37Rv/ MonoMac6/ | Coating NP with pluronic-tuftsin conjugate enhanced the uptake rate and the intracellular activity of the encapsulated drug against Mtb. | [188] | |
| Tuftsin | Lipid carrier | RIF | 285nm & −22mV | Mtb H37Rv/ J774/ N.A. | Formulation elicited slow sustained release. Improvement in MIC was observed with tuftsin functionalized drug carriers. | [187] | |
| Tuftsin | Liposome | RIF | 25~ 65nm | Mtb H37Rv/ N.A./ Swiss albino mice | Tuftsin decorated liposomes was approximately 2,000 times more potent when compared to free drug. | [186] | |
| Tuftsin | N.A. | Salicylanilide | N. A | M. abscessus, Mtb H37Rv, Mtb A8 MDR/ MonoMac6/ N.A. | Conjugation with tuftsin led to better performance of salicylanilide derivatives against intracellular Mtb due to better uptake rate than free drug. | [199] | |
| Folate receptor | Folic acid | Emulsion (oleic acid, Tween 80, chitosan) | RIF | 59.69nm & +0.7mV | N.A./NR8383/Sprague–Dawley rat | Emulsions coated with chitosan folate had higher cellular uptake. Folate-based nano emulsion led to enhanced drug content in lungs in vivo and led to reduction of plasma drug concentration. | [200] |
| Folic acid | liquid-crystalline folate NP | RIF | 110~381nm & −20~−40mV | N.A./ NR8383/ N.A. | RIF loaded folate NP had slow and sustained release for over 25 days. The formulation had low toxicity on alveolar macrophage cell line and was readily taken up by it. | [201] | |
| Folic acid | F127 | Ga (III) | 329nm & +30mV | Mtb H37Rv/ THP-1/ N.A. | Folic acid had better macrophage targeting than mannose. But folate-conjugated NPs have slower drug released than mannose-conjugated NPs. | [103] | |
| Tim-4 | DPPS | Acetalated dextran | Curcumin | 350.5nm &−40.6mV | N.A./RAW264.7/N.A. | DPPS decorated NP had much better uptake in macrophages over epithelial cells. Formulation showed pH dependent controlled release. | [202] |
Abv- PLGA: poly (D, L-lactide-co-glycolide) acid, DPPS: 1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine, Ga (III): Ga (III) meso-tetraphenyl porphyrine chloride, INH: isoniazid, RIF: rifampicin
In a recent study, Carneiro et al. fabricated a rifampicin-encapsulated carrier whose surface was functionalized with tuftsin-modified peptide.[187] The peptide-based carrier was shown to have no cytotoxicity on J774 A.1 macrophages and had a slow, sustained in vitro release profile. The surface modification of these nanoparticles resulted in higher internalization within the macrophages when compared with non-modified nanoparticles. The carrier was stable for at least 60 days with little fluctuation in particle size and zeta potential. The carrier also had good bactericidal activity and was found to be 2-fold more effective against Mtb H37Rv when compared with free rifampicin.[187]
In pursuit to increase the efficacy of isoniazid, Hovarti et al. fabricated a novel lipo-peptide carrier where isoniazid was conjugated with palmitoylated tuftsin derived peptide.[189] The drug conjugate was then encapsulated within Pluronic F127 stabilized PLGA. The PLGA complex exhibited good in vitro activity against Mtb H37Rv and had low cytotoxicity on human cells. In addition, the complex successfully killed intracellular pathogen and had better antibacterial potency when compared to free isoniazid. The authors further demonstrated the efficacy of the complex by testing it on a guinea pig model. In this model, the complex exhibited a good chemotherapeutic effect when orally administered and resulted in decreased bacterial level with no toxicity. The untreated control group showed disease progression, including severe lesions, parenchymal involvement, necrosis, and intralesional mineralization.
A similar concept was adopted to develop a peptide-Pluronic F127 PLGA complex, where tuftsin derived peptide was conjugated with TB515, a TB drug candidate.[188] Compared to the bare PLGA complex, this peptide complex had significantly higher uptake in MonoMac 6 human monocytes. In vitro inhibition studies of intracellular Mtb H37Rv showed the prowess of the peptide complex over the non-peptide complex and the free drug at all concentration levels.[188]
Ligands targeting the formyl peptide receptor (FPR) provide another good avenue to develop actively targeted therapy against tuberculosis. FPRs are part of a family of chemoattractant receptors primarily expressed on phagocytic immune cells, including neutrophils and macrophages, and have a vital role to play in host defense and inflammation.[193, 194] Peptides containing N-formylated methionine are known to interact with FPR. Several different ligands targeting FPR, apart from N-formyl peptides, have also been reported in literature.[193, 194] Studies exploring the therapeutic use of N-formylmethionyl-leucyl-phenylalanine (fMLF), an FPR agonist, against Mtb infected cells have been reported in literature.[195, 196] Considerable higher amount of reactive oxygen species in murine neutrophils was observed when fMLF was administered alone. Administration of fMLF along with anti-TB drug resulted in higher therapeutic efficacy over anti-TB drug alone by reducing more bacterial load in mice lungs and spleen.[195] The macrophage targeting prowess of fMLF has also been tested with fMLF decorated liposome.[197] N-formyl peptide decoration led to higher internalization of liposome by macrophages and resulted in better targeting of the site of inflammation in a mouse model.[197] Nanocarriers modified with fMLF have also been developed to target the macrophage in order to enhance the potency of HIV therapy.[198] Even though FPR agonists have great potential for targeting of macrophages, they have not yet garnered traction in the field of macrophage targeting against intracellular infections. This presents a fantastic opportunity to explore this option further.
7.3. Phospholipid-based targeting ligand
Along with sugar- and peptide-based ligands, researchers have also explored phospholipids for targeting macrophages.[202] Phospholipids are amphiphilic lipids present in all animal and plant cell membranes, and organized in lipid bilayer arrangement. The phospholipids present in most cell membranes consist of a phosphate group conjugated with glycerol backbone esterified to fatty acids, and a hydrophilic residue (e.g., choline). Phosphatidylserine (PS) is a negatively charged phospholipid that is created and harbored on the inner membrane of healthy cells.[203-206] The onset of apoptosis induces PS transition from the inner layer to the outer layer of the cell membrane.[207] PS transition to the outer layer attracts macrophages, which engulf the presenting cells (phagocytosis) through proteins that have affinity for PS, and are expressed by phagocytes.[203, 206, 208] Although PS is necessary for phagocytosis signaling, it is still debatable if PS exposure alone is enough to mediate the in vitro internalization of apoptotic cells by macrophages.[203, 206, 207] However, in nanocarrier functionalized with PS, specifically 1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine (DPPS), the sole presence of PS enhanced macrophage uptake.[206] Recently, Shah et al. fabricated acetylated dextran nanoparticles coated with DPPS as the targeting ligand.[202] Nanoparticles had zeta potential of −40.6 mV and size of 350.5 nm, and encapsulating curcumin as therapeutic agent. Targeting efficacy was tested on RAW264.7 macrophages with the A549 cell line as a control. The macrophage uptake study revealed that DPPS coated nanoparticles had much higher intracellular presence in RAW264.7 cells when compared with A549 cell line.[202] To verify the role of PS in phagocytosis, DPPC coated nanoparticles were included in the uptake study. It was observed that DPPC coated particles had significantly lower uptake within the RAW264.7 macrophages, affirming the role of PS in active targeting of macrophages.[202]
7.4. Other types of ligands
Folate receptors govern the internalization of folic acid derivatives into the cell by endocytosis. Folate receptor β is expressed by active macrophages and tumor-associated macrophages. This makes folic acid a potential ligand for targeting Mtb infected macrophages. Folic acid-based delivery systems have already proven effective against Pseudomonas aeruginosa infection, where folic acid-based nanoparticles showed better bactericidal properties than free drug or non-folic acid-based carriers.[209] Similarly, folic acid-based carriers have been exploited to deliver therapeutic agents to target viral reservoirs.[210, 211] Folic acid improved the overall efficacy of HIV treatment by improving drug bioavailability, biodistribution, and pharmacodynamics in a murine model of HIV-1 infection.[211] Folate grafted chitosan coating on SLN has also been exploited to improve the bioavailability of therapeutic agents for lung tumor therapy.[212]
Despite utilizing folic acid for active targeting against viral infections, P. aeruginosa, and lung tumors, its use for treating intracellular infections has not been explored much. In a recent study, rifampicin-loaded nanoemulsion coated with chitosan folate was fabricated to develop a respirable formulation for deep lung deposition.[200] The formulation had no cytotoxic effects against alveolar macrophages and resulted in better internalization than formulations without folate modification. The folate formulation also lowered the plasma drug concentration and increased the lung drug content. However, the authors did not test the bactericidal property of this formulation against intra- or extracellular Mtb. In another interesting study focused on TB therapy, Parmar et al. developed a liquid crystalline folate nanoparticle which encapsulated rifampicin.[201] Rather than using folate solely for a targeting purpose, it was used as the building block itself, using calcium or zinc salts for cross linking. The resulting nanoparticles, with size ranging from 100 to 350 nm, showed a slow, sustained in vitro release profile. The folate nanoparticles showed low cytotoxicity to alveolar macrophages and were effectively up taken by NR8383 macrophages. This formulation was not tested against macrophages infected with Mtb; therefore, while folate-based carriers look promising, in vivo and in vitro studies against Mtb infection are necessary.
The thioaptamer that binds to the CD14 moiety has been used to enhance drug accumulation in the infected macrophages.[213] Leonard et al. decorated the thioaptamer on discoidal silicon mesoporous microparticles (SMP) and observed an increase of particle uptake in murine and human Mtb infected macrophages. Thioaptamer-coated SMP efficiently killed the infected cells and reduced the mycobacterial burden. Thioaptamer-coated SMPs also accumulated within the lungs of Mtb infected mice when compared to control treatment. Numerous drugs have been loaded into mesoporous SMP;[214] thus, additional anti-TB drugs should improve the therapeutic efficacy.
8. Future perspective and challenges:
Despite 100 years of BCG vaccine usage, TB is still one of the significant causes of death from any infectious agent. In 2020, TB was still a rampant global public health hazard, leading to heavy social and economic burden.[215] In the past few decades, there has been concerted efforts to fight this disease, much of which is attributed to novel anti-TB drugs.[216, 217] Despite of all these efforts, the latest drugs in the market for public use date back 50 years; the widely used first line anti-TB drugs, isoniazid and rifampicin, were discovered in 1952 and 1965, respectively. Unfortunately, developing new drug molecules for any application is a time and labor-intensive process. In 2019, the US Food and Drug Administration (FDA) approved pretomanid to be administered in conjunction with bedaquiline and linezolid for treating the most drug-resistant forms of tuberculosis. It took pretomanid 15 years to complete the clinical development process, despite being fast tracked by the FDA under the Limited Population Pathway for Antibacterial and Antifungal Drugs (LPAD).
Therefore, developing novel formulations, which repackage existing drugs to improve their efficacy, can have an important role in curbing the TB menace. One of the challenges in developing an efficient anti-TB formulation is its ability to target alveolar macrophages in the lower respiratory tract. Hence, the pulmonary administration of anti-TB drugs seems to be an encouraging strategy to develop an effective anti-TB therapy.[218] Pulmonary administration would bypass the harsh conditions these drug formulations have to face, especially in the acidic stomach environment. Pulmonary administration would also be a good choice for targeted drug delivery systems which require controlled release in the acidic environment within macrophages. Despite of promising results, the pulmonary administration strategies currently used are marred with difficult obstacles, which needs to be overcome. First-line drugs are the most commonly utilized therapeutic agent used in in vitro studies for development of inhalable dry powder formulations.[219-223]. However, no inhalable formulation for the TB treatment has been commercialized so far.[224, 225] One critical aspect associated with development of effective dry powder formulation is that, after inhalation it should be capable of reaching the pulmonary alveoli and deliver the drug to the alveolar macrophages, where most of the Mtb resides. The most common challenges presented by pulmonary administration are the use of accepted and safe excipients, achieving satisfactory drug encapsulation, developing scalable commercial production processes, and developing formulation with proper morphology and particle size for deep lung deposition.[225]
The current trend in anti-TB drug administration is through the oral route. It is the preferred way of drug delivery as it is easy and painless, resulting in improved patient treatment compliance when compared to the parenteral route.[62] Various studies have been reported which deal with fabrication of different oral delivery systems. Designing oral delivery systems involve a specific set of parameters to characterize it, including zeta potential, particle size, polydispersity index, drug loading and in vitro release kinetics. The challenge is to design an efficient drug delivery system in which all of these parameters are desirably tuned. One of the major challenges faced by oral formulation is sustaining harsh physiological conditions present in the stomach.[62] A high acidic environment can easily degrade nano/micro particle formulations, resulting in undesired leakage of drug.
Recently, several studies have reported promising in vitro results of novel nanoparticle formulations, such as silver quantum dots, gold nano rods, and gallium nanoparticles.[82, 103, 184] Future studies are necessary for these formulations in vivo; however, in vitro studies have shown great promise. It is pertinent that these novel nano formulations are well tolerated in animal models, as animal models can help predict the commerciality of the formulation, along with the economic viability of scaling up the fabrication process.
In the last decade, several studies have proven the efficacy of active targeting of macrophages for the better killing of intracellular Mtb. Using saccharide-based ligands to target the lectin-like receptors is a common strategy. Biocompatibility of saccharide-based ligands is one of the advantages.[161] The most popular target for saccharide-based ligands are mannose receptors.[164, 165, 169, 173, 174] Mannose receptors are highly expressed on macrophages and dendritic cells; in addition, it is an endocytic receptor that can facilitate the internalization of mannose-decorated carriers.[62, 161, 226] The ease of mannose conjugation to different type of carriers also makes mannose an attractive choice. Mannose and its derivatives have already been used to modify different types of delivery systems, such as liposomes, solid lipid nanoparticles, graphene oxide, polysaccharide-based nanomaterials, and polymeric nanoparticles.[103, 148, 162, 164, 168, 169, 173, 176] In addition to macrophages, other types of cells, such as endothelia, kidney cells, mesangial cells, trachea smooth muscle cells, and retinal pigment epithelium, also contain mannose receptors. The cross-reaction may influence the biodistribution and targeting specificity.
CD44 and dectin-1 are also popular targets for saccharide-based ligands. CD44 are present in various cell types, including keratinocytes, endothelial cells, epithelial cells, fibroblasts, and macrophages.[227, 228] CD44 is involved in cell adhesion, binding, endocytosis, metabolism of hyaluronan, as well as phagocytosis in macrophages.[229] The high molecular weight HA can bind multivalently to more surface CD44, leading to a high binding avidity.[228] However, the other study has shown that the low molecular weight HA could polarize the macrophage at M1 phenotype, eliciting an inflammatory response.[230] Prior studies show that the macrophage CD44 expression significantly increases during initial phase of an inflammatory response;[231, 232] thus, using HA targeting might be a good strategy to target the activated macrophages. Dectin-1 is a pattern recognition receptor expressed on dendritic cells, macrophages, monocytes, neutrophils, T cells, eosinophils, B cells, and epithelial cells.[233-235] Although dectin-1 binds to β-glucans, the literature shows that short chain β-glucans are not sufficient for binding.[233] Both CD44 and dectin-1 are phagocytic receptors that could assist in internalization of targeted drug carriers. However, in contrast to mannose receptors, long chain oligosaccharides are desired to enhance the targtiong efficiency toward CD44 and dectin-1.
Targeting tuftsin receptors on surface of macrophages presents another opportunity for targeted drug delivery. Tuftsin specifically binds with monocytes, macrophages and polymorphonuclear leukocytes.[236-238] Tuftsin plays a role in regulating motility, pinocytosis, phagocytosis, and bactericidal activities of these immune cells.[236]. The low toxicity associated with tuftsin makes it a good ligand.[236, 239, 240] Drug delivery system decorated with the C terminus modified tuftsin can not only increase the macrophage internalization but also the macrophage activation. [241, 242]. The tetrapeptide structure of the tuftsin can be modified to increase its macrophage targeting efficiency. [188, 192, 243] For example, the pentapeptide derivative of tuftsin has shown higher affinity for tuftsin receptors than the tetrapeptide tuftsin.[243] In future more potent derivatives of tuftsin can be developed and be used for targeting tuftsin receptors on macrophages.
The folate receptor (FR) has already been extensively used to target ovarian carcinomas and activated macrophages in rheumatoid arthritis.[244, 245] One study shows that inflammatory stimuli enhance the expression of folate receptor on the macrophages.[246] The folate receptor expressed after inflammatory stress is fully functional and can be exploited to target the activated macrophages.[246] Folate-based ligands have a high selectivity toward activated pathologic cell type, leaving the majority of healthy macrophages unharmed.[244] Folate-based delivery system offers a good avenue to develop therapies for infections where activated macrophages play a prominent role.
The phospholipid, PS, has been used to target Tim-4 receptors on macrophages. Tim-4 is highly expressed in peripheral lymphoid tissues, and selectively expressed in macrophages, dendritic cells, B1 cells, and natural killer T cells.[247] Tim-4 also mediates phagocytosis in macrophage; however, not all macrophages express Tim-4 receptors.[247-249]
It is vital to find better sets of ligands for targeting the macrophage. A ligand having higher binding affinity for macrophage-based receptors will result in better targeting efficiency; however, such ligands may not exist. Hetero-multivalent targeting (different types of ligands simultaneously binding to different types of macrophage receptors) is an alternative approach to enhance the binding avidity without the necessity of discovering new ligands. The presence of multiple ligands on drug carrier allows exploitation of hetero-multivalent binding phenomenon.[250] Researchers have already shown the in vitro prowess of hetero-multivalent binding against targeting P. aeruginosa using GB3 and lactosyl ceramide (LacCer) as the targeting ligand.[251] Worstell et al. demonstrated the higher efficacy of a multi-ligand liposome (GB3+ LacCer) against mono-ligand liposome (GB3/LacCer) in targeting P. aeruginosa. As of now, we did not come across studies in which researchers have decorated drug carrier with multiple sets of ligands for better targeting to macrophages. It would be interesting to see if hetero-multivalent binding can be utilized to develop more effective anti-TB drug delivery systems by improving the active macrophage targeting.
As of now, most of the in vitro/vivo studies consist of a single drug loaded or conjugated with a drug carrier. In reality, treatment regimens consist of using 2-3 different antibiotics. It will be interesting to see if it is possible to co-load multiple antibiotics (antibiotic cocktail) within a drug carrier with any level of success. Despite all the challenges associated with developing targeted delivery systems, the pursuit of an efficient macrophage-targeted formulation to treat TB should be continued with fervor. Some of the presented results are very promising and have the potential to curb the TB menace, if proven safe for therapeutic use.
In this article we have put forth a review of recent studies pertaining to macrophage targeted drug delivery systems for tuberculosis. Under the current treatment regimen, anti-TB drugs are primarily administered orally. They are often cytotoxic and are marred with poor bioavailability at the site of infection. Current therapies also have limited capability to infiltrate granulomas and attack the Mtb reservoir where dormant pathogen resides.[252] In past decade, a lot of resources have been utilized and many attempts have been made to fabricate new formulations that can improve the bioavailability and reduce the toxicity of current anti-TB drugs. None of the nano- and micro-based formulations which have been explored so far has reached the market, but recent developments in the nanotechnological domain have kindled new optimism for the potential of novel drug delivery systems. However, it should be noted that the process of developing a novel drug delivery system in lab and then commercializing it is a time intensive process. Liposomal drug formulation took 30 years to get FDA approval for commercial use since its discovery in 1965.[253] The success of these targeted delivery platforms will highly depend on the development of economically viable formulations that can mitigate the limitations and drawbacks of current anti-TB therapies, making the therapy more pragmatic and cost effective for all patients, especially those in countries with high proportion of low- and middle-income households.
Figure 1:

Tuberculosis pathogenesis and progression of latent TB to clinical TB
Figure 2:

Drug resistance mechanisms in Mtb
Figure 3:

Targeting schematics for transport of anti-TB drug in infected macrophages.
Figure 4:

Ligands used in literature to target macrophages
Acknowledgements
This work was supported by the funds from National Institutes of Health (award number: R21AI149383). All the figures in the manuscript were created with BioRender.com
Abbreviation list:
- A549
Alveolar basal epithelial cell line
- AIDS
Acquired immunodeficiency syndrome
- AMK
Amikacin
- AMP
Antimicrobial peptide
- AMP
Anti-microbial peptide
- BCG
Mycobacterium Bovis bacillus Calmette–Guérin
- BMDC
Bone marrow-derived dendritic cell
- BMDM
Bone marrow derived macrophage
- BTZ043
1,3-Benzothiazin-4-one-043
- CD
Cyclodextrin
- CFU
Colony forming unit
- CFZ
Clofazimine
- DOTAP
1,2-Dioleoyl-3-trimethylammonium propane
- DPPS
1,2-dipalmitoyl-sn-glycero-3-phospho-L-serine
- DSPC
1,2-Distearoyl-sn-glycero-3-phosphocholine
- DSPE-PEG
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000]
- ETH
Ethionamdide
- fMLF
N-formylmethionyl-leucyl-phenylalanine
- Ga (III)
Ga (III) meso-tetraphenyl porphyrine chloride
- GalMan
Galactomannan
- HA
Hyaluronic Acid
- hAM
Human alveolar macrophages
- HDP
Host defense peptides
- HIV
Human immunodeficiency virus
- INH
Isoniazid
- J774
Murine macrophage cell line
- LVX
Levofloxacin
- mAM
Mouse alveolar macrophages
- mBMDM
Murine Bone Marrow Derived Macrophages
- MDM
Monocyte-derived macrophages
- MIC
Minimum inhibitory concentration
- MMP
Multimeric nanoparticles
- MP
Methyl α-d-mannopyranoside
- MPEO-b-PCL
Methoxy poly(ethylene oxide)-block-poly(ε-caprolactone)
- mPM
Mouse peritoneal macrophages
- Mtb
Mycobacterium tuberculosis
- MXF
Moxifloxacin
- NP
Nanoparticle
- NR8383
Rat alveolar macrophage cell line
- NTZ
Nitazoxanide
- OCT313
2-acetamido-2-deoxy-b-D-glucopyranosyl N,N-dimethyldithiocarbamate
- ODOC
1,8-Octanediol-dimethyl 2-oxoglutarate copolymer
- PAM
4-aminophenyl-alpha-d–manno-pyranoside
- PAMAM
Polyamidoamine
- PC
Phosphocholine
- PDI
Polydispersity index
- PE
Phosphoethanolamine
- PEA
Polyesteramide
- PEG
Polyethylene glycol
- PES
Polyethylene sebacate
- PLGA
Poly (D, L-lactide-co-glycolide) acid
- PPS
Polypropylene sulfide
- pβCD
Poly β-Cyclodextrins
- RAW264.7
Murine macrophage cell line
- RFB
Rifabutin
- RIF
Rifampicin
- SAMAN
N-octadecyl-mannopyranosylamine
- siRNA
Small interfering RNA
- SLN
Solid lipid nanoparticle
- SMP
Silicon mesoporous particle
- TB
Tuberculosis
- TfR1
Transferrin receptor 1
- THP-1
Human monocytic cell line
- Tim
T-cell immunoglobin mucin
- TLR
Toll like receptors
- TZ
Thioridazine
- WHO
World Health Organization
Biography

Anirudh Gairola is currently pursuing Ph.D. in Chemical Engineering from Texas A&M University College Station. His research focuses on developing lipid-based drug delivery systems for targeting bacterial infections. He completed his Masters in Chemical Engineering from Texas A&M University, College Station and earned his Bachelors in Biochemical Engineering from Harcourt Butler Technical University, Kanpur India.

Dr. Aaron Benjamin received his Ph.D. from the Biochemistry Department at Purdue University where he studied acyltransferases via synthetic chemistry and X-ray crystallography in Dr. Jeremy Lohman’s lab. Since then, he has been working in Dr. Cirillo’s laboratory as a postdoctoral research associate, studying therapeutics against a variety of diseases including tuberculosis. His primary interests are in the development and mechanisms of drug design and delivery.

Joshua Weatherston received his Ph.D. in Chemical Engineering from Texas A&M University in 2019. His research focus is investigating the use of nanomaterials for molecular detection of dilute species, especially using Surface-enhanced Raman scattering spectroscopy (SERS). Since receiving his Ph.D. he has dedicated himself to education in the fields of Chemical Engineering and Chemistry.

Dr. Cirillo is interested in the pathogenesis of lung infections including tuberculosis, Staphylococcus, Klebsiella, Pseudomonas, Legionnaires' disease and COVID-19 and has been a tuberculosis researcher for more than 30 years. He is a Regents’ Professor in the Department of Microbial and Molecular Pathogenesis at the Texas A&M Health Science Center and leads the Center for Airborne Pathogens Research and Imaging (CAPRI). He has published more than 140 manuscripts, holds several patents on technologies developed in his laboratory He has been elected a Fellow of the AAAS, a Fellow of the AAM and is on editorial boards for several journals.

Dr. Hung-Jen Wu is an Assistant Professor of Chemical Engineering at Texas A&M University. He received his Ph.D. in Chemical Engineering from Texas A&M University, and completed his postdoctoral training at the University of California, Berkeley and Houston Methodist Hospital Research Institute. Dr. Wu’s research primarily focuses on the development of biosensors, cell membrane physics, drug delivery, and glycobiology.
Footnotes
Declaration of Competing Interest
Authors declare no conflict of interest.
References
- [1].WHO, 2020. [Google Scholar]
- [2].King SB Holmes K, Bloom Barry R., Jha Prabhat, Major Infectious Diseases, 2017. [PubMed] [Google Scholar]
- [3].Esmail H, Barry CE, Young DB, Wilkinson RJ, Philosophical Transactions of the Royal Society B: Biological Sciences 2014, 369, 20130437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Barry CE, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D, Nature Reviews Microbiology 2009, 7, 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, O’Neill LAJ, Xavier RJ, Science 2016, 352, aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nemes E, Geldenhuys H, Rozot V, Rutkowski KT, Ratangee F, Bilek N, Mabwe S, Makhethe L, Erasmus M, Toefy A, Mulenga H, Hanekom WA, Self SG, Bekker L-G, Ryall R, Gurunathan S, DiazGranados CA, Andersen P, Kromann I, Evans T, Ellis RD, Landry B, Hokey DA, Hopkins R, Ginsberg AM, Scriba TJ, Hatherill M, New England Journal of Medicine 2018, 379, 138.29996082 [Google Scholar]
- [7].Izzo AA, Current Opinion in Immunology 2017, 47, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ernst JD, Cell Host & Microbe 2018, 24, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Blok BA, Arts RJW, van Crevel R, Benn CS, Netea MG, Journal of Leukocyte Biology 2015, 98, 347. [DOI] [PubMed] [Google Scholar]
- [10].Greenwood B, Philosophical Transactions of the Royal Society B: Biological Sciences 2014, 369, 20130433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mangtani P, Abubakar I, Ariti C, Beynon R, Pimpin L, Fine PEM, Rodrigues LC, Smith PG, Lipman M, Whiting PF, Sterne JA, Clinical Infectious Diseases 2014, 58, 470. [DOI] [PubMed] [Google Scholar]
- [12].Khader SA, Divangahi M, Hanekom W, Hill PC, Maeurer M, Makar KW, Mayer-Barber KD, Mhlanga MM, Nemes E, Schlesinger LS, van Crevel R, Vankayalapati R, Xavier RJ, Netea MG, The Journal of Clinical Investigation 2020, 129, 3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ahmad S, Mokaddas E, Journal of Infection and Public Health 2014, 7, 75. [DOI] [PubMed] [Google Scholar]
- [14].Zumla JCA, Centis R, D'Ambrosio L, Mwaba P, Bates M, Kapata N, Nyirenda T, Chanda D, Mfinanga S, Hoelscher M, Maeurer M, Migliori GB, Lancet Respiratory Medicine 2015, 3, 220. [DOI] [PubMed] [Google Scholar]
- [15].Saukkonen JJ, Cohn DL, Jasmer RM, Schenker S, Jereb JA, Nolan CM, Peloquin CA, Gordin FM, Nunes D, Strader DB, Bernardo J, Venkataramanan R, Sterling TR, American Journal of Respiratory and Critical Care Medicine 2006, 174, 935. [DOI] [PubMed] [Google Scholar]
- [16].Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, Ginsberg A, Swaminathan S, Spigelman M, Getahun H, Menzies D, Raviglione M, Nature Reviews Disease Primers 2016, 2, 16076. [DOI] [PubMed] [Google Scholar]
- [17].Kiran D, Podell BK, Chambers M, Basaraba RJ, Seminars in immunopathology 2016, 38, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhai W, Wu F, Zhang Y, Fu Y, Liu Z, International Journal of Molecular Sciences 2019, 20, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Keane J, Remold HG, Kornfeld H, The Journal of Immunology 2000, 164, 2016. [DOI] [PubMed] [Google Scholar]
- [20].Armstrong J, Hart PDA, The Journal of experimental medicine 1971, 134, 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mwandumba HC, Russell DG, Nyirenda MH, Anderson J, White SA, Molyneux ME, Squire SB, The Journal of Immunology 2004, 172, 4592. [DOI] [PubMed] [Google Scholar]
- [22].Mishra BB, Lovewell RR, Olive AJ, Zhang G, Wang W, Eugenin E, Smith CM, Phuah JY, Long JE, Dubuke ML, Nature microbiology 2017, 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Köster S, Upadhyay S, Chandra P, Papavinasasundaram K, Yang G, Hassan A, Grigsby SJ, Mittal E, Park HS, Jones V, Proceedings of the National Academy of Sciences 2017, 114, E8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Russell DG, Immunological Reviews 2011, 240, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Antonelli LRV, Gigliotti Rothfuchs A, Gonçalves R, Roffê E, Cheever AW, Bafica A, Salazar AM, Feng CG, Sher A, The Journal of Clinical Investigation 2010, 120, 1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lin PL, Ford CB, Coleman MT, Myers AJ, Gawande R, Ioerger T, Sacchettini J, Fortune SM, Flynn JL, Nature Medicine 2014, 20, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Flynn JL, Chan J, Infection and immunity 2001, 69, 4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].O'Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MP, Annual review of immunology 2013, 31, 475. [DOI] [PubMed] [Google Scholar]
- [29].Flynn JL, Chan J, Annual review of immunology 2001, 19, 93. [DOI] [PubMed] [Google Scholar]
- [30].Daniels M, Hill AB, British medical journal 1952, 1, 1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Raviglione M, Sulis G, Infectious disease reports 2016, 8, 6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Seung KJ, Keshavjee S, Rich ML, Cold Spring Harbor Perspectives in Medicine 2015, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Payam Tabarsi MM, Archives of Clinical Infectious Diseases 2012, 81. [Google Scholar]
- [34].Udwadia ZF, Amale RA, Ajbani KK, Rodrigues C, Clinical Infectious Diseases 2012, 54, 579. [DOI] [PubMed] [Google Scholar]
- [35].Rojas Echenique JI, Kryazhimskiy S, Nguyen Ba AN, Desai MM, PLOS Genetics 2019, 15, e1007958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Al-Saeedi M, Al-Hajoj S, Infection and drug resistance 2017, 10, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Singh R, Dwivedi SP, Gaharwar US, Meena R, Rajamani P, Prasad T, Journal of Applied Microbiology 2020, 128, 1547. [DOI] [PubMed] [Google Scholar]
- [38].Nguyen L, Arch. Toxicol 2016, 90, 1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Müller B, Borrell S, Rose G, Gagneux S, Trends in Genetics 2013, 29, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pinto L, Menzies D, Infection and drug resistance 2011, 4, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Koch A, Mizrahi V, Warner DF, Emerging Microbes & Infections 2014, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dookie N, Rambaran S, Padayatchi N, Mahomed S, Naidoo K, J. Antimicrob. Chemother 2018, 73, 1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gygli SM, Borrell S, Trauner A, Gagneux S, FEMS Microbiology Reviews 2017, 41, 354. [DOI] [PubMed] [Google Scholar]
- [44].Nasiruddin M, Neyaz MK, Das S, Tuberculosis Research and Treatment 2017, 2017, 4920209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Smith T, Wolff KA, Nguyen L, in Pathogenesis of Mycobacterium tuberculosis and its Interaction with the Host Organism, (Eds: Pieters J, McKinney JD), Springer Berlin Heidelberg, Berlin, Heidelberg: 2013, 53. [Google Scholar]
- [46].Nasiri MJ, Haeili M, Ghazi M, Goudarzi H, Pormohammad A, Imani Fooladi AA, Feizabadi MM, Frontiers in Microbiology 2017, 8, 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJV, Nature Reviews Microbiology 2015, 13, 42. [DOI] [PubMed] [Google Scholar]
- [48].Piddock LJV, Clinical Microbiology Reviews 2006, 19, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang J-Y, Sun H-Y, Wang J-T, Hung C-C, Yu M-C, Lee C-H, Lee L-N, PLOS ONE 2015, 10, e0144136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Labby KJ, Garneau-Tsodikova S, Future Medicinal Chemistry 2013, 5, 1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ramirez MS, Tolmasky ME, Drug Resistance Updates 2010, 13, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tao J, Han J, Wu H, Hu X, Deng J, Fleming J, Maxwell A, Bi L, Mi K, Nucleic Acids Res. 2013, 41, 2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Munita JM, Arias CA, Virulence Mechanisms of Bacterial Pathogens 2016, 481. [Google Scholar]
- [54].Zhang Y, Yew WW, Barer MR, Antimicrob. Agents Chemother 2012, 56, 2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Maisonneuve E, Gerdes K, Cell 2014, 157, 539. [DOI] [PubMed] [Google Scholar]
- [56].Mosaiab T, Farr DC, Kiefel MJ, Houston TA, Advanced Drug Delivery Reviews 2019, 151–152, 94. [DOI] [PubMed] [Google Scholar]
- [57].Hokey DA, Misra A, Tuberculosis 2011, 91, 82. [DOI] [PubMed] [Google Scholar]
- [58].Fontana G, Licciardi M, Mansueto S, Schillaci D, Giammona G, Biomaterials 2001, 22, 2857. [DOI] [PubMed] [Google Scholar]
- [59].Toti US, Guru BR, Hali M, McPharlin CM, Wykes SM, Panyam J, Whittum-Hudson JA, Biomaterials 2011, 32, 6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wijagkanalan W, Kawakami S, Higuchi Y, Yamashita F, Hashida M, J. Controlled Release 2011, 149, 42. [DOI] [PubMed] [Google Scholar]
- [61].Baranyai Z, Soria-Carrera H, Alleva M, Millán-Placer AC, Lucía A, Martín-Rapún R, Aínsa JA, de la Fuente JM, Advanced Therapeutics 2020, n/a, 2000113. [Google Scholar]
- [62].Costa A, Pinheiro M, Magalhães J, Ribeiro R, Seabra V, Reis S, Sarmento B, Advanced Drug Delivery Reviews 2016, 102, 102. [DOI] [PubMed] [Google Scholar]
- [63].Mehta M, Deeksha, Sharma N, Vyas M, Khurana N, Maurya PK, Singh H, Andreoli de Jesus TP, Dureja H, Chellappan DK, Gupta G, Wadhwa R, Collet T, Hansbro PM, Dua K, Satija S, Chem. Biol. Interact 2019, 304, 10. [DOI] [PubMed] [Google Scholar]
- [64].Sahay G, Alakhova DY, Kabanov AV, J. Controlled Release 2010, 145, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Chellat F, Merhi Y, Moreau A, Yahia LH, Biomaterials 2005, 26, 7260. [DOI] [PubMed] [Google Scholar]
- [66].Tabata Y, Ikada Y, Biomaterials 1988, 9, 356. [DOI] [PubMed] [Google Scholar]
- [67].Sung JC, Pulliam BL, Edwards DA, Trends Biotechnol. 2007, 25, 563. [DOI] [PubMed] [Google Scholar]
- [68].Shegokar R, Al Shaal L, Mitri K, Journal of Pharmacy & Pharmaceutical Sciences 2011, 14, 100. [DOI] [PubMed] [Google Scholar]
- [69].Muttil P, Wang C, Hickey AJ, Pharm. Res 2009, 26, 2401. [DOI] [PubMed] [Google Scholar]
- [70].Yezhelyev MV, Qi L, O’Regan RM, Nie S, Gao X, Journal of the American Chemical Society 2008, 130, 9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Gbadamosi JK, Hunter AC, Moghimi SM, FEBS Lett. 2002, 532, 338. [DOI] [PubMed] [Google Scholar]
- [72].Patil S, Sandberg A, Heckert E, Self W, Seal S, Biomaterials 2007, 28, 4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Wilhelm C, Billotey C, Roger J, Pons JN, Bacri JC, Gazeau F, Biomaterials 2003, 24, 1001. [DOI] [PubMed] [Google Scholar]
- [74].Limbach LK, Li Y, Grass RN, Brunner TJ, Hintermann MA, Muller M, Gunther D, Stark WJ, Environmental Science & Technology 2005, 39, 9370. [DOI] [PubMed] [Google Scholar]
- [75].He C, Hu Y, Yin L, Tang C, Yin C, Biomaterials 2010, 31, 3657. [DOI] [PubMed] [Google Scholar]
- [76].Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS, Biomaterials 2011, 32, 3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Storm G, Belliot SO, Daemen T, Lasic DD, Advanced Drug Delivery Reviews 1995, 17, 31. [Google Scholar]
- [78].Free P, Shaw CP, Lévy R, Chem. Commun 2009, 5009. [DOI] [PubMed] [Google Scholar]
- [79].Nel AE, Mädler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M, Nature Materials 2009, 8, 543. [DOI] [PubMed] [Google Scholar]
- [80].Zahr AS, Davis CA, Pishko MV, Langmuir 2006, 22, 8178. [DOI] [PubMed] [Google Scholar]
- [81].Afinjuomo F BT, Parikh A, Chung R, Song Y, Nagalingam G, Triccas J, Wang L, Liu L, Hayball JD, Petrovsky N, Garg S, Pharmaceutics. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Ali HR, Ali MRK, Wu Y, Selim SA, Abdelaal HFM, Nasr EA, El-Sayed MA, Bioconjugate Chem. 2016, 27, 2486. [DOI] [PubMed] [Google Scholar]
- [83].Amarnath Praphakar R, Munusamy MA, Sadasivuni KK, Rajan M, Int. J. Pharm 2016, 513, 628. [DOI] [PubMed] [Google Scholar]
- [84].Singh H, Jindal S, Singh M, Sharma G, Kaur IP, Int. J. Pharm 2015, 485, 138. [DOI] [PubMed] [Google Scholar]
- [85].Rodrigues B, Shende P, J. Mol. Liq 2020, 304, 112731. [Google Scholar]
- [86].Shang S, Kats D, Cao L, Morgun E, Velluto D, He Y, Xu Q, Wang C-R, Scott EA, Frontiers in Immunology 2018, 9, 2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].F. B. Afinjuomo TG; Parikh A; Song Y; Chung R; Wang L; Liu L; Hayball JD; Petrovsky N; Garg S, Pharmaceutics 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Abdelghany S, Alkhawaldeh M, AlKhatib HS, Journal of Drug Delivery Science and Technology 2017, 39, 442. [Google Scholar]
- [89].Jahagirdar PS, Gupta PK, Kulkarni SP, Devarajan PV, J. Pharm. Sci 2020, 109, 2262. [DOI] [PubMed] [Google Scholar]
- [90].Marcianes P, Negro S, Barcia E, Montejo C, Fernández-Carballido A, AAPS PharmSciTech 2019, 21, 15. [DOI] [PubMed] [Google Scholar]
- [91].Koli U, Nilgiriwala K, Sriraman K, Jain R, Dandekar P, J. Nanopart. Res 2019, 21, 200. [Google Scholar]
- [92].Rawal T, Parmar R, Tyagi RK, Butani S, Colloids and Surfaces B: Biointerfaces 2017, 154, 321. [DOI] [PubMed] [Google Scholar]
- [93].Praphakar RA, Munusamy MA, Rajan M, Int. J. Pharm 2017, 524, 168. [DOI] [PubMed] [Google Scholar]
- [94].Vieira ACC, Chaves LL, Pinheiro S, Pinto S, Pinheiro M, Lima SC, Ferreira D, Sarmento B, Reis S, Int. J. Pharm 2018, 536, 478. [DOI] [PubMed] [Google Scholar]
- [95].Abdelghany S, Parumasivam T, Pang A, Roediger B, Tang P, Jahn K, Britton WJ, Chan H-K, Journal of Drug Delivery Science and Technology 2019, 52, 642. [Google Scholar]
- [96].De Matteis L, Jary D, Lucía A, García-Embid S, Serrano-Sevilla I, Pérez D, Ainsa JA, Navarro FP, de la Fuente JM, Chem. Eng. J 2018, 340, 181. [Google Scholar]
- [97].Tripodo G, Perteghella S, Grisoli P, Trapani A, Torre ML, Mandracchia D, European Journal of Pharmaceutics and Biopharmaceutics 2019, 136, 250. [DOI] [PubMed] [Google Scholar]
- [98].Amarnath Praphakar R, Jeyaraj M, Ahmed M, Suresh Kumar S, Rajan M, Int. J. Biol. Macromol 2018, 118, 1627. [DOI] [PubMed] [Google Scholar]
- [99].Machelart A, Salzano G, Li X, Demars A, Debrie A-S, Menendez-Miranda M, Pancani E, Jouny S, Hoffmann E, Deboosere N, Belhaouane I, Rouanet C, Simar S, Talahari S, Giannini V, Villemagne B, Flipo M, Brosch R, Nesslany F, Deprez B, Muraille E, Locht C, Baulard AR, Willand N, Majlessi L, Gref R, Brodin P, ACS Nano 2019, 13, 3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Cunha L, Rodrigues S, Rosa da Costa AM, Faleiro ML, Buttini F, Grenha A, Polymers 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Islam N, Ferro V, Nanoscale 2016, 8, 14341. [DOI] [PubMed] [Google Scholar]
- [102].Ellis T, Chiappi M, García-Trenco A, Al-Ejji M, Sarkar S, Georgiou TK, Shaffer MSP, Tetley TD, Schwander S, Ryan MP, Porter AE, ACS Nano 2018, 12, 5228. [DOI] [PubMed] [Google Scholar]
- [103].Choi S.-r., Britigan BE, Moran DM, Narayanasamy P, PLOS ONE 2017, 12, e0177987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Rani S, Gothwal A, Khan I, Pachouri PK, Bhaskar N, Gupta UD, Chauhan DS, Gupta U, AAPS PharmSciTech 2018, 19, 3237. [DOI] [PubMed] [Google Scholar]
- [105].Vibe CB, Fenaroli F, Pires D, Wilson SR, Bogoeva V, Kalluru R, Speth M, Anes E, Griffiths G, Hildahl J, Nanotoxicology 2016, 10, 680. [DOI] [PubMed] [Google Scholar]
- [106].Trousil J, Pavliš O, Kubíčková P, Škorič M, Marešová V, Pavlova E, Knudsen KD, Dai YS, Zimmerman M, Dartois V, Fang JY, Hrubý M, J Control Release 2020, 321, 312. [DOI] [PubMed] [Google Scholar]
- [107].Batalha IL, Bernut A, Schiebler M, Ouberai MM, Passemar C, Klapholz C, Kinna S, Michel S, Sader K, Castro-Hartmann P, Renshaw SA, Welland ME, Floto RA, J. Controlled Release 2019, 314, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Sharma A, Vaghasiya K, Ray E, Gupta P, Kumar Singh A, Datta Gupta U, Kumar Verma R, Int. J. Pharm 2019, 558, 231. [DOI] [PubMed] [Google Scholar]
- [109].Trousil J, Syrová Z, Dal N-JK, Rak D, Konefał R, Pavlova E, Matějková J, Cmarko D, Kubíčková P, Pavliš O, Urbánek T, Sedlák M, Fenaroli F, Raška I, Štěpánek P, Hrubý M, Biomacromolecules 2019, 20, 1798. [DOI] [PubMed] [Google Scholar]
- [110].Liu Z, Li X, Xiu B, Duan C, Li J, Zhang X, Yang X, Dai W, Johnson H, Zhang H, Feng X, Colloids and Surfaces B: Biointerfaces 2016, 145, 679. [DOI] [PubMed] [Google Scholar]
- [111].Grotz E, Tateosian NL, Salgueiro J, Bernabeu E, Gonzalez L, Manca ML, Amiano N, Valenti D, Manconi M, García V, Moretton MA, Chiappetta DA, Journal of Drug Delivery Science and Technology 2019, 53, 101170. [Google Scholar]
- [112].Lawlor C, O’Connor G, O’Leary S, Gallagher PJ, Cryan S-A, Keane J, O’Sullivan MP, PLOS ONE 2016, 11, e0149167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Gupta A, Meena J, Sharma D, Gupta P, Gupta UD, Kumar S, Sharma S, Panda AK, Misra A, Molecular Pharmaceutics 2016, 13, 3247. [DOI] [PubMed] [Google Scholar]
- [114].Gupta A, Pant G, Mitra K, Madan J, Chourasia MK, Misra A, Molecular Pharmaceutics 2014, 11, 1201. [DOI] [PubMed] [Google Scholar]
- [115].Pandey R, Sharma A, Zahoor A, Sharma S, Khuller GK, Prasad B, Journal of Antimicrobial Chemotherapy 2003, 52, 981. [DOI] [PubMed] [Google Scholar]
- [116].Makadia HK, Siegel SJ, Polymers 2011, 3, 1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Kirby DJ, Rosenkrands I, Agger EM, Andersen P, Coombes AGA, Perrie Y, Journal of Drug Targeting 2008, 16, 282. [DOI] [PubMed] [Google Scholar]
- [118].Nicolete R, Santos D. F. d., Faccioli LH, International Immunopharmacology 2011, 11, 1557. [DOI] [PubMed] [Google Scholar]
- [119].Sharma R, Yadav AB, Muttil P, Kajal H, Misra A, Tuberculosis 2011, 91, 107. [DOI] [PubMed] [Google Scholar]
- [120].Wang C, Muttil P, Lu D, Beltran-Torres AA, Garcia-Contreras L, Hickey AJ, The AAPS Journal 2009, 11, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Yadav AB, Muttil P, Singh AK, Verma RK, Mohan M, Agrawal AK, Verma AS, Sinha SK, Misra A, Tuberculosis 2010, 90, 188. [DOI] [PubMed] [Google Scholar]
- [122].Gaddy JA, Arivett BA, McConnell MJ, López-Rojas R, Pachón J, Actis LA, Infection and Immunity 2012, 80, 1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Olakanmi O, Schlesinger LS, Ahmed A, Britigan BE, Infection and Immunity 2004, 72, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Olakanmi O, Stokes JB, Britigan BE, Journal of Investigative Medicine 2005, 53, 143. [DOI] [PubMed] [Google Scholar]
- [125].Britigan BE, Rasmussen GT, Olakanmi O, Cox CD, Infection and Immunity 2000, 68, 1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Choi S.-r., Britigan BE, Narayanasamy P, Antimicrob. Agents Chemother 2017, 61, e02505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Maeda R, Ito T, Tagami T, Takii T, Ozeki T, Biol. Pharm. Bull 2019, 42, 1846. [DOI] [PubMed] [Google Scholar]
- [128].Pandit S, Roy S, Pillai J, Banerjee S, ACS Omega 2020, 5, 4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Fenaroli F, Repnik U, Xu Y, Johann K, Van Herck S, Dey P, Skjeldal FM, Frei DM, Bagherifam S, Kocere A, Haag R, De Geest BG, Barz M, Russell DG, Griffiths G, ACS Nano 2018, 12, 8646. [DOI] [PubMed] [Google Scholar]
- [130].Miranda MS, Rodrigues MT, Domingues RMA, Costa RR, Paz E, Rodríguez-Abreu C, Freitas P, Almeida BG, Carvalho MA, Gonçalves C, Ferreira CM, Torrado E, Reis RL, Pedrosa J, Gomes ME, Advanced Healthcare Materials 2018, 7, 1800124. [DOI] [PubMed] [Google Scholar]
- [131].Zomorodbakhsh S, Abbasian Y, Naghinejad M, Sheikhpour M, International journal of nanomedicine 2020, 15, 5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Joshi M, Prabhakar B, Journal of Drug Delivery Science and Technology 2021, 61, 102197. [Google Scholar]
- [133].Rudolph D, Redinger N, Schaible UE, Feldmann C, ChemNanoMat 2021, 7, 7. [Google Scholar]
- [134].Singh M, Schiavone N, Papucci L, Maan P, Kaur J, Singh G, Nandi U, Nosi D, Tani A, Khuller GK, Priya M, Singh R, Kaur IP, Eur J Pharm Biopharm 2021, 160, 100. [DOI] [PubMed] [Google Scholar]
- [135].Sieling PA, Chatterjee D, Porcelli SA, Prigozy TI, Mazzaccaro RJ, Soriano T, Bloom BR, Brenner MB, Kronenberg M, Brennan PJ, a. et, Science 1995, 269, 227. [DOI] [PubMed] [Google Scholar]
- [136].Van Rhijn I. LD, Moody DB, CD1a, CD1b, and CD1c in Immunity Against Mycobacteria, Springer, New York, NY, 2013 [DOI] [PubMed] [Google Scholar]
- [137].Kasmar AG, van Rhijn I, Cheng T-Y, Turner M, Seshadri C, Schiefner A, Kalathur RC, Annand JW, de Jong A, Shires J, Leon L, Brenner M, Wilson IA, Altman JD, Moody DB, Journal of Experimental Medicine 2011, 208, 1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Moody DB, Ulrichs T, Mühlecker W, Young DC, Gurcha SS, Grant E, Rosat J-P, Brenner MB, Costello CE, Besra GS, Porcelli SA, Nature 2000, 404, 884. [DOI] [PubMed] [Google Scholar]
- [139].Beckman EM, Melián A, Behar SM, Sieling PA, Chatterjee D, Furlong ST, Matsumoto R, Rosat JP, Modlin RL, Porcelli SA, The Journal of Immunology 1996, 157, 2795. [PubMed] [Google Scholar]
- [140].Beckman EM, Porcelli SA, Morita CT, Behar SM, Furlong ST, Brenner MB, Nature 1994, 372, 691. [DOI] [PubMed] [Google Scholar]
- [141].Siddiqui S, Visvabharathy L, Wang C-R, Frontiers in Immunology 2015, 6, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Chancellor A, Tocheva AS, Cave-Ayland C, Tezera L, White A, Al Dulayymi J. a. R., Bridgeman JS, Tews I, Wilson S, Lissin NM, Tebruegge M, Marshall B, Sharpe S, Elliott T, Skylaris C-K, Essex JW, Baird MS, Gadola S, Elkington P, Mansour S, Proceedings of the National Academy of Sciences 2017, 114, E10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Van Rhijn I, van Berlo T, Hilmenyuk T, Cheng T-Y, Wolf BJ, Tatituri RVV, Uldrich AP, Napolitani G, Cerundolo V, Altman JD, Willemsen P, Huang S, Rossjohn J, Besra GS, Brenner MB, Godfrey DI, Moody DB, Proceedings of the National Academy of Sciences 2016, 113, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].WHO, WHO, 2016. [Google Scholar]
- [145].Gong J, Stenger S, Zack JA, Jones BE, Bristol GC, Modlin RL, Morrissey PJ, Barnes PF, The Journal of Clinical Investigation 1998, 101, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Montamat-Sicotte DJ, Millington KA, Willcox CR, Hingley-Wilson S, Hackforth S, Innes J, Kon OM, Lammas DA, Minnikin DE, Besra GS, Willcox BE, Lalvani A, The Journal of Clinical Investigation 2011, 121, 2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Lin Y-H, Tsai S-C, Lai C-H, Lee C-H, He ZS, Tseng G-C, Biomaterials 2013, 34, 4466. [DOI] [PubMed] [Google Scholar]
- [148].Singodia D, Verma A, Verma RK, Mishra PR, Nanomedicine: Nanotechnology, Biology and Medicine 2012, 8, 468. [DOI] [PubMed] [Google Scholar]
- [149].Chaubey P, Mishra B, Carbohydr. Polym 2014, 101, 1101. [DOI] [PubMed] [Google Scholar]
- [150].Mosaiab T, Boiteux S, Zulfiker AHM, Wei MQ, Kiefel MJ, Houston TA, ChemBioChem 2018, 19, 1476. [DOI] [PubMed] [Google Scholar]
- [151].Irache JM, Salman HH, Gamazo C, Espuelas S, Expert Opinion on Drug Delivery 2008, 5, 703. [DOI] [PubMed] [Google Scholar]
- [152].Yunus Basha R, T.S SK, Doble M, Carbohydr. Polym 2019, 218, 53. [DOI] [PubMed] [Google Scholar]
- [153].Hwang SM, Kim DD, Chung SJ, Shim CK, J. Controlled Release 2008, 129, 100. [DOI] [PubMed] [Google Scholar]
- [154].Silva JP, Gonçalves C, Costa C, Sousa J, Silva-Gomes R, Castro AG, Pedrosa J, Appelberg R, Gama FM, J. Controlled Release 2016, 235, 112. [DOI] [PubMed] [Google Scholar]
- [155].Rossi I, Buttini F, Sonvico F, Affaticati F, Martinelli F, Annunziato G, Machado D, Viveiros M, Pieroni M, Bettini R, Pharmaceutics 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Gao Y, Sarfraz MK, Clas S-D, Roa W, Löbenberg R, Journal of Biomedical Nanotechnology 2015, 11, 1312. [DOI] [PubMed] [Google Scholar]
- [157].Chono S, Tanino T, Seki T, Morimoto K, J. Pharm. Pharmacol 2007, 59, 75. [DOI] [PubMed] [Google Scholar]
- [158].Jiang H-L, Kim Y-K, Arote R, Jere D, Quan J-S, Yu J-H, Choi Y-J, Nah J-W, Cho M-H, Cho C-S, Int. J. Pharm 2009, 375, 133. [DOI] [PubMed] [Google Scholar]
- [159].Song E-H, Manganiello MJ, Chow Y-H, Ghosn B, Convertine AJ, Stayton PS, Schnapp LM, Ratner DM, Biomaterials 2012, 33, 6889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Saraogi GK, Sharma B, Joshi B, Gupta P, Gupta UD, Jain NK, Agrawal GP, Journal of Drug Targeting 2011, 19, 219. [DOI] [PubMed] [Google Scholar]
- [161].Nimje N, Agarwal A, Saraogi GK, Lariya N, Rai G, Agrawal H, Agrawal GP, Journal of Drug Targeting 2009, 17, 777. [DOI] [PubMed] [Google Scholar]
- [162].Vieira ACC, Chaves LL, Pinheiro M, Lima SAC, Ferreira D, Sarmento B, Reis S, Artificial Cells, Nanomedicine, and Biotechnology 2018, 46, 653. [DOI] [PubMed] [Google Scholar]
- [163].Bhardwaj A, Grobler A, Rath G, Kumar Goyal A, Kumar Jain A, Mehta A, Current Drug Delivery 2016, 13, 909. [DOI] [PubMed] [Google Scholar]
- [164].Pi J, Shen L, Shen H, Yang E, Wang W, Wang R, Huang D, Lee B-S, Hu C, Chen C, Jin H, Cai J, Zeng G, Chen ZW, Materials Science and Engineering: C 2019, 103, 109777. [DOI] [PubMed] [Google Scholar]
- [165].Pinheiro M, Ribeiro R, Vieira A, Andrade F, Reis S, Drug design, development and therapy 2016, 10, 2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Grenha A, Alves AD, Guerreiro F, Pinho J, Simões S, Almeida AJ, Gaspar MM, European Journal of Pharmaceutics and Biopharmaceutics 2020, 147, 38. [DOI] [PubMed] [Google Scholar]
- [167].Tiwari S, Chaturvedi AP, Tripathi YB, Mishra B, AAPS PharmSciTech 2011, 12, 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Maretti E, Costantino L, Rustichelli C, Leo E, Croce MA, Buttini F, Truzzi E, Iannuccelli V, Int. J. Pharm 2017, 528, 440. [DOI] [PubMed] [Google Scholar]
- [169].Maretti E, Costantino L, Buttini F, Rustichelli C, Leo E, Truzzi E, Iannuccelli V, Drug Delivery and Translational Research 2019, 9, 298. [DOI] [PubMed] [Google Scholar]
- [170].Hamed A, Osman R, Al-Jamal KT, Holayel SM, Geneidi A-S, Journal of Drug Delivery Science and Technology 2019, 51, 513. [Google Scholar]
- [171].Liang J, Jiang D, Noble PW, Advanced Drug Delivery Reviews 2016, 97, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Jiang D, Liang J, Noble PW, Physiological Reviews 2011, 91, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Pi J, Shen L, Yang E, Shen H, Huang D, Wang R, Hu C, Jin H, Cai H, Cai J, Zeng G, Chen ZW, Angew. Chem. Int. Ed 2020, 59, 3226. [DOI] [PubMed] [Google Scholar]
- [174].Choi S.-r., Britigan BE, Narayanasamy P, mSphere 2019, 4, e00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Maretti E, Rustichelli C, Lassinantti Gualtieri M, Costantino L, Siligardi C, Miselli P, Buttini F, Montecchi M, Leo E, Truzzi E, Iannuccelli V, Pharmaceutics 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Praphakar RA, Munusamy MA, Alarfaj AA, Kumar SS, Rajan M, New J. Chem 2017, 41, 11324. [Google Scholar]
- [177].Sansare VA WD, Shinde UA, Journal of Tuberculosis 2020. [Google Scholar]
- [178].Su F-Y, Chen J, Son H-N, Kelly AM, Convertine AJ, West TE, Skerrett SJ, Ratner DM, Stayton PS, Biomaterials Science 2018, 6, 1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Durán V, Grabski E, Hozsa C, Becker J, Yasar H, Monteiro JT, Costa B, Koller N, Lueder Y, Wiegmann B, Brandes G, Kaever V, Lehr CM, Lepenies B, Tampé R, Förster R, Bošnjak B, Furch M, Graalmann T, Kalinke U, J Control Release 2021, 334, 201. [DOI] [PubMed] [Google Scholar]
- [180].Upadhyay TK, Fatima N, Sharma D, Saravanakumar V, Sharma R, Excli j 2017, 16, 210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Upadhyay TK, Fatima N, Sharma A, Sharma D, Sharma R, Artificial Cells, Nanomedicine, and Biotechnology 2019, 47, 427. [DOI] [PubMed] [Google Scholar]
- [182].Mubin N, Umar MS, Zubair S, Owais M, Frontiers in Microbiology 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Moretton MA, Chiappetta DA, Andrade F, das Neves J, Ferreira D, Sarmento B, Sosnik A, Journal of Biomedical Nanotechnology 2013, 9, 1076. [DOI] [PubMed] [Google Scholar]
- [184].Pati R, Sahu R, Panda J, Sonawane A, Scientific Reports 2016, 6, 24184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Gammella E, Buratti P, Cairo G, Recalcati S, Metallomics 2014, 6, 1336. [DOI] [PubMed] [Google Scholar]
- [186].Agarwal A, Kandpal H, Gupta HP, Singh NB, Gupta CM, Antimicrob. Agents Chemother 1994, 38, 588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Carneiro SP, Carvalho KV, de Oliveira Aguiar Soares RD, Carneiro CM, de Andrade MHG, Duarte RS, dos Santos ODH, Colloids and Surfaces B: Biointerfaces 2019, 175, 306. [DOI] [PubMed] [Google Scholar]
- [188].Horváti K, Gyulai G, Csámpai A, Rohonczy J, Kiss É, Bősze S, Bioconjugate Chem. 2018, 29, 1495. [DOI] [PubMed] [Google Scholar]
- [189].Horváti K, Bacsa B, Kiss É, Gyulai G, Fodor K, Balka G, Rusvai M, Szabó E, Hudecz F, Bősze S, Bioconjugate Chem. 2014, 25, 2260. [DOI] [PubMed] [Google Scholar]
- [190].Monika A. G.-K. Siebert; Grzegorz Cholewinski; Krystyna Dzierzbicka, Current Medicinal Chemistry 2017, 24, 3711.28745220 [Google Scholar]
- [191].Siemion IZ, Kluczyk A, Peptides 1999, 20, 645. [DOI] [PubMed] [Google Scholar]
- [192].Mezö G, Kalászi A, Reményi J, Majer Z, Hilbert Á, Láng O, Köhidai L, Barna K, Gaál D, Hudecz F, Biopolymers 2004, 73, 645. [DOI] [PubMed] [Google Scholar]
- [193].Lee HY, Lee M, Bae Y-S, J. Cell. Biochem 2017, 118, 1300. [DOI] [PubMed] [Google Scholar]
- [194].He H-Q, Ye RD, Molecules 2017, 22. [Google Scholar]
- [195].Mir SA, Sharma S, Immunopharmacology and Immunotoxicology 2019, 41, 292. [DOI] [PubMed] [Google Scholar]
- [196].Mir SA, Sharma S, International Immunopharmacology 2014, 18, 298. [DOI] [PubMed] [Google Scholar]
- [197].Qin Y, Li Z.-w., Yang Y, Yu C.-m., Gu D.-d., Deng H, Zhang T, Wang X, Wang A.-p., Luo W.-z., Journal of Drug Targeting 2014, 22, 165. [DOI] [PubMed] [Google Scholar]
- [198].Gunaseelan S, Gunaseelan K, Deshmukh M, Zhang X, Sinko PJ, Advanced Drug Delivery Reviews 2010, 62, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Baranyai Z, Krátký M, Vosátka R, Szabó E, Senoner Z, Dávid S, Stolaříková J, Vinšová J, Bősze S, European Journal of Medicinal Chemistry 2017, 133, 152. [DOI] [PubMed] [Google Scholar]
- [200].Shah K, Chan LW, Wong TW, Drug Delivery 2017, 24, 1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Parmar R, Misra R, Mohanty S, Colloids and Surfaces B: Biointerfaces 2015, 129, 198. [DOI] [PubMed] [Google Scholar]
- [202].Shah NK, Gupta SK, Wang Z, Meenach SA, Journal of Drug Delivery Science and Technology 2019, 50, 57. [Google Scholar]
- [203].Segawa K, Nagata S, Trends in Cell Biology 2015, 25, 639. [DOI] [PubMed] [Google Scholar]
- [204].Lewis RNAH, McElhaney RN, Biophys. J 2000, 79, 2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Matsumoto A, Takahashi Y, Nishikawa M, Sano K, Morishita M, Charoenviriyakul C, Saji H, Takakura Y, J. Pharm. Sci 2017, 106, 168. [DOI] [PubMed] [Google Scholar]
- [206].Ravichandran Kodi S., Immunity 2011, 35, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM, The Journal of Immunology 1992, 148, 2207. [PubMed] [Google Scholar]
- [208].Bagalkot V, Deiuliis JA, Rajagopalan S, Maiseyeu A, Advanced Drug Delivery Reviews 2016, 99, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Wang Y, Yuan Q, Feng W, Pu W, Ding J, Zhang H, Li X, Yang B, Dai Q, Cheng L, Wang J, Sun F, Zhang D, Journal of Nanobiotechnology 2019, 17, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Zhou T, Lin Z, Puligujja P, Palandri D, Hilaire J, Araínga M, Smith N, Gautam N, McMillan J, Alnouti Y, Liu X, Edagwa B, Gendelman HE, Nanomedicine 2018, 13, 871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Puligujja P, Balkundi SS, Kendrick LM, Baldridge HM, Hilaire JR, Bade AN, Dash PK, Zhang G, Poluektova LY, Gorantla S, Liu X.-m., Ying T, Feng Y, Wang Y, Dimitrov DS, McMillan JM, Gendelman HE, Biomaterials 2015, 41, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Rosière R, Van Woensel M, Gelbcke M, Mathieu V, Hecq J, Mathivet T, Vermeersch M, Van Antwerpen P, Amighi K, Wauthoz N, Molecular Pharmaceutics 2018, 15, 899. [DOI] [PubMed] [Google Scholar]
- [213].Leonard F, Ha NP, Sule P, Alexander JF, Volk DE, Lokesh GLR, Liu X, Cirillo JD, Gorenstein DG, Yuan J, Chatterjee S, Graviss EA, Godin B, J. Controlled Release 2017, 266, 238. [DOI] [PubMed] [Google Scholar]
- [214].Fine D, Grattoni A, Goodall R, Bansal SS, Chiappini C, Hosali S, van de Ven AL, Srinivasan S, Liu X, Godin B, Brousseau III L, Yazdi IK, Fernandez-Moure J, Tasciotti E, Wu H-J, Hu Y, Klemm S, Ferrari M, Advanced Healthcare Materials 2013, 2, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Ginsberg AM, Tuberculosis 2010, 90, 162. [DOI] [PubMed] [Google Scholar]
- [216].Sosnik A, Carcaboso ÁM, Glisoni RJ, Moretton MA, Chiappetta DA, Advanced Drug Delivery Reviews 2010, 62, 547. [DOI] [PubMed] [Google Scholar]
- [217].Onozaki I, Raviglione M, Respirology 2010, 15, 32. [DOI] [PubMed] [Google Scholar]
- [218].Pitt JM, Blankley S, McShane H, O'Garra A, Microbial Pathogenesis 2013, 58, 2. [DOI] [PubMed] [Google Scholar]
- [219].Shyamal D, Ian T, Peter S, Current Drug Delivery 2015, 12, 26.25030114 [Google Scholar]
- [220].Chan JGY, Chan H-K, Prestidge CA, Denman JA, Young PM, Traini D, European Journal of Pharmaceutics and Biopharmaceutics 2013, 83, 285. [DOI] [PubMed] [Google Scholar]
- [221].Manion J. a. R., Cape SP, McAdams DH, Rebits LG, Evans S, Sievers RE, Aerosol Sci. Technol 2012, 46, 403. [Google Scholar]
- [222].Hanif SN, Garcia-Contreras L, Frontiers in Cellular and Infection Microbiology 2012, 2, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Son Y-J, McConville JT, European Journal of Pharmaceutics and Biopharmaceutics 2011, 78, 366. [DOI] [PubMed] [Google Scholar]
- [224].Braunstein M, Hickey AJ, Ekins S, Pharmaceutical Research 2019, 36, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Misra A, Hickey AJ, Rossi C, Borchard G, Terada H, Makino K, Fourie PB, Colombo P, Tuberculosis 2011, 91, 71. [DOI] [PubMed] [Google Scholar]
- [226].Azad AK, Rajaram MVS, Schlesinger LS, Journal of cytology & molecular biology 2014, 1, 1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Senbanjo LT, Chellaiah MA, Frontiers in Cell and Developmental Biology 2017, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Jordan AR, Racine RR, Hennig MJP, Lokeshwar VB, Frontiers in Immunology 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Vachon E, Martin R, Plumb J, Kwok V, Vandivier RW, Glogauer M, Kapus A, Wang X, Chow C-W, Grinstein S, Downey GP, Blood 2006, 107, 4149. [DOI] [PubMed] [Google Scholar]
- [230].Ghosh S, Hoselton SA, Dorsam GP, Schuh JM, Immunobiology 2015, 220, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Underhill CB, Nguyen HA, Shizari M, Culty M, Developmental Biology 1993, 155, 324. [DOI] [PubMed] [Google Scholar]
- [232].Sladek Z, Rysanek D, Research in Veterinary Science 2009, 86, 235. [DOI] [PubMed] [Google Scholar]
- [233].Schorey JS, Lawrence C, Current drug targets 2008, 9, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Cohen-Kedar S, Baram L, Elad H, Brazowski E, Guzner-Gur H, Dotan I, European Journal of Immunology 2014, 44, 3729. [DOI] [PubMed] [Google Scholar]
- [235].Lee HM, Yuk JM, Shin DM, Jo EK, J Clin Immunol 2009, 29, 795. [DOI] [PubMed] [Google Scholar]
- [236].Agrawal AK, Gupta CM, Advanced Drug Delivery Reviews 2000, 41, 135. [DOI] [PubMed] [Google Scholar]
- [237].Najjar V, Drugs Future 1987, 12, 147. [Google Scholar]
- [238].Fridkin M, Najjar VA, Crit. Rev. Biochem. Mol. Biol 1989, 24, 1. [DOI] [PubMed] [Google Scholar]
- [239].Khare S, Bhutani LK, Rao DN, Mol. Cell. Biochem 1997, 171, 1. [DOI] [PubMed] [Google Scholar]
- [240].Pawan K, Ivanov BB, Kabilan L, Rao DN, Vaccine 1994, 12, 819. [DOI] [PubMed] [Google Scholar]
- [241].Singh S, Chhabra R, Srivastava V, Experientia 1992, 48, 994. [DOI] [PubMed] [Google Scholar]
- [242].Singhal A, Bali A, Jain RK, Gupta C, FEBS Lett. 1984, 178, 109. [DOI] [PubMed] [Google Scholar]
- [243].Zhang Y, Xiao L, Chordia MD, Locke LW, Williams MB, Berr SS, Pan D, Bioconjugate Chem. 2010, 21, 1788. [DOI] [PubMed] [Google Scholar]
- [244].Paulos CM, Turk MJ, Breur GJ, Low PS, Adv Drug Deliv Rev 2004, 56, 1205. [DOI] [PubMed] [Google Scholar]
- [245].Sudimack J, Lee RJ, Adv Drug Deliv Rev 2000, 41, 147. [DOI] [PubMed] [Google Scholar]
- [246].Xia W, Hilgenbrink AR, Matteson EL, Lockwood MB, Cheng JX, Low PS, Blood 2009, 113, 438. [DOI] [PubMed] [Google Scholar]
- [247].Liu W, Xu L, Liang X, Liu X, Zhao Y, Ma C, Gao L, Frontiers in Immunology 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Min C, Park J, Kim G, Moon H, Lee S-A, Kim D, Moon B, Yang S, Lee J, Kim K, Cho H, Park J, Lee D-H, Lee G, Park D, Cell Death & Disease 2020, 11, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Wong K, Valdez PA, Tan C, Yeh S, Hongo J-A, Ouyang W, Proceedings of the National Academy of Sciences of the United States of America 2010, 107, 8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Krishnan P, Singla A, Lee C-A, Weatherston JD, Worstell NC, Wu H-J, Colloids and Surfaces B: Biointerfaces 2017, 160, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Worstell NC, Singla A, Saenkham P, Galbadage T, Sule P, Lee D, Mohr A, Kwon JS-I, Cirillo JD, Wu H-J, Scientific Reports 2018, 8, 8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Wallis RS, Hafner R, Nature Reviews Immunology 2015, 15, 255. [DOI] [PubMed] [Google Scholar]
- [253].Bulbake U, Doppalapudi S, Kommineni N, Khan W, Pharmaceutics 2017, 9, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]