

Abstract
Nitric oxide (NO) is a key signaling molecule in health and disease. While nitrite acts as a reservoir of NO activity, mechanisms for NO release require further understanding. A series of electronically varied β-diketiminatocopper(II) nitrite complexes [CuII](κ2-O2N) react with a range of electronically tuned triarylphosphines PArZ3 that release NO with the formation of O=PArZ3. Second-order rate constants are largest for electron-poor copper(II) nitrite and electron-rich phosphine pairs. Computational analysis reveals a transition-state structure energetically matched with experimentally determined activation barriers. The production of NO follows a pathway that involves nitrite isomerization at CuII from κ2-O2N to κ1-NO2 followed by O-atom transfer (OAT) to form O=PArZ3 and [CuI]-NO that releases NO upon PArZ3 binding at CuI to form [CuI]-PArZ3. These findings illustrate important mechanistic considerations involved in NO formation from nitrite via OAT.
Graphical Abstract
INTRODUCTION
Nitric oxide (NO) serves as a signaling molecule involved in a wide array of physiological functions, such as its role as a vasodilator connected with blood pressure and cardiovascular health.1 While endothelial nitrogen oxide synthase (NOS) produces NO from arginine, this enzyme loses activity when the oxygen levels become low.2 Tightly regulated reduction of nitrite to form NO can compensate for this loss of NOS activity under hypoxia.3–5 The enzymes deoxymyoglobin, cytochrome c oxidase, and xanthine oxidase are each able to act as nitrite reductases.6–9
Cu-containing nitrite reductases are common in soil bacteria where they perform a key denitrification step in the nitrogen cycle by converting nitrite to NO. In mammalian biology, Cu has been implicated in the reverse transformation, converting NO to nitrite at the redox-active Cu site of ceruloplasmin.10 Interestingly, a recent report indicates that the ubiquitous carbonic anhydrase II enzyme may act as a nitrite reductase when complexed with Cu rather than Zn, generating NO.11
While one-electron reduction of nitrite to NO may occur in the presence of proton sources to generate water, O-atom transfer (OAT) pathways also exist.12 The former has been well characterized in biological13,14 and biomimetic systems;15–19 however, OAT to nucleophiles is less studied.6,12,20–22 Potential O-atom acceptors such as thiols RSH and dialkyl sulfides RSR′ are readily biologically available and may be oxidized to sulfenic acids RS(O)H and sulfoxides RS(O)R′ (Figure 1A).
Figure 1.
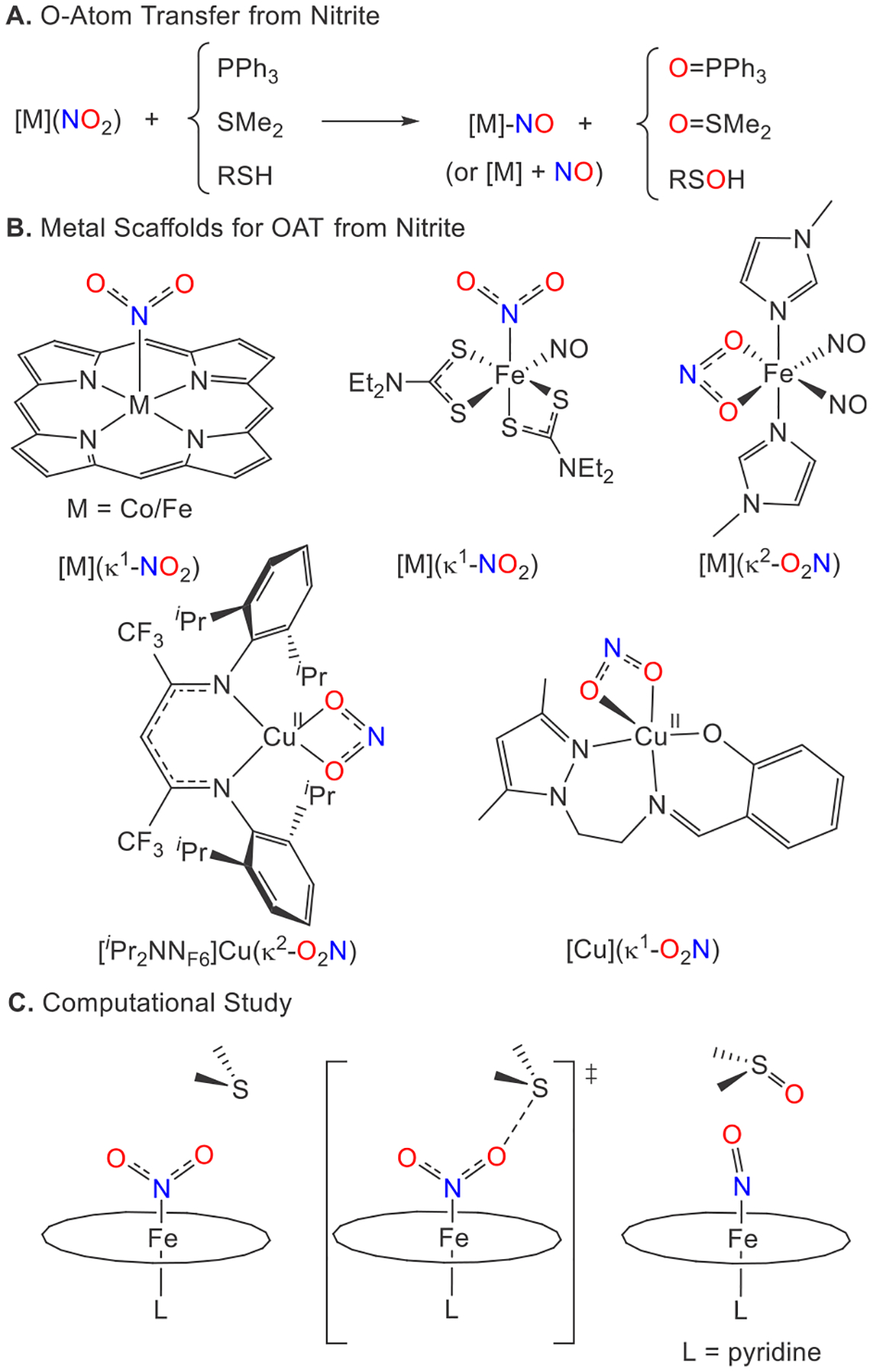
(A) OAT reactions of metal complexes with nitrite. (B) Metal scaffolds supporting OAT from nitrite. (C) Calculated OAT to the SMe2 transition state.
A range of Fe, Co, and Cu biomimetic complexes demonstrate OAT with a range of nucleophiles (Figure 1). The oldest of these models, which dates to 1979, involves a cobalt nitro complex supported by a dianionic tetraazacyle that undergoes reduction by PPh3 to generate the corresponding cobalt nitrosyl and O=PPh3.23 Most model complexes have focused on iron/cobalt porphyrin complexes using a variety of O-atom acceptors,24–33 although other coordination motifs have also been examined (Figure 1B).12,21,34,35 These synthetic models typically possess a nitrite ion bound through the N atom. As shown by computational studies with an iron porphyrin system, [M](κ1-NO2) structures can enable smooth conversion to a metal nitrosyl [M]-NO upon OAT with SMe2 with modest barriers, ΔG⧧(298 K), calculated in either the absence (10.4 kcal mol−1) or presence of an axial ligand L (17.8 kcal mol−1; L = pyridine; Figure 1C).36
Two recent reports document OAT from nitrite at CuII that results in NO loss via OAT to PPh3 with reduction to CuI.12,21 Importantly, these complexes each possess O-bound nitrite with the [CuII](κ2-O2N) binding mode. To better understand OAT from nitrite at Cu, we examine herein the electronic role of the Cu center and incoming O-atom acceptor through kinetic studies supported by computational analysis to develop a reaction pathway for NO release from nitrite at CuII.
RESULTS AND DISCUSSION
We previously demonstrated that the β-diketiminatocopper(II) nitrite [iPr2NNF6]CuII (κ2-O2N) reacts with PPh3 to release NO with the formation of [iPr2NNF6]CuI(PPh3) and O=PPh3 .12 To examine the electronic roles of the Cu center and the phosphine that serves as the O-atom acceptor, we sought to employ a range of sterically similar, yet electronically different, β-diketiminatocopper(II) nitrite complexes [CuII](κ2-O2N) and para-substituted triarylphosphines PArZ3. To modulate the electronic environment of the copper(II) nitrite complexes with minimal steric impact, we employed β-diketiminate ligands with backbone substituents X = Me or CF3 along with N-aryl o-Cl or o-Me substituents. The reaction of copper(I) β-diketiminates [CuI] with AgNO2 serves as a reliable route to the corresponding copper(II) nitrites [CuII](κ2-O2N) (1−4; Scheme 1).
Scheme 1.
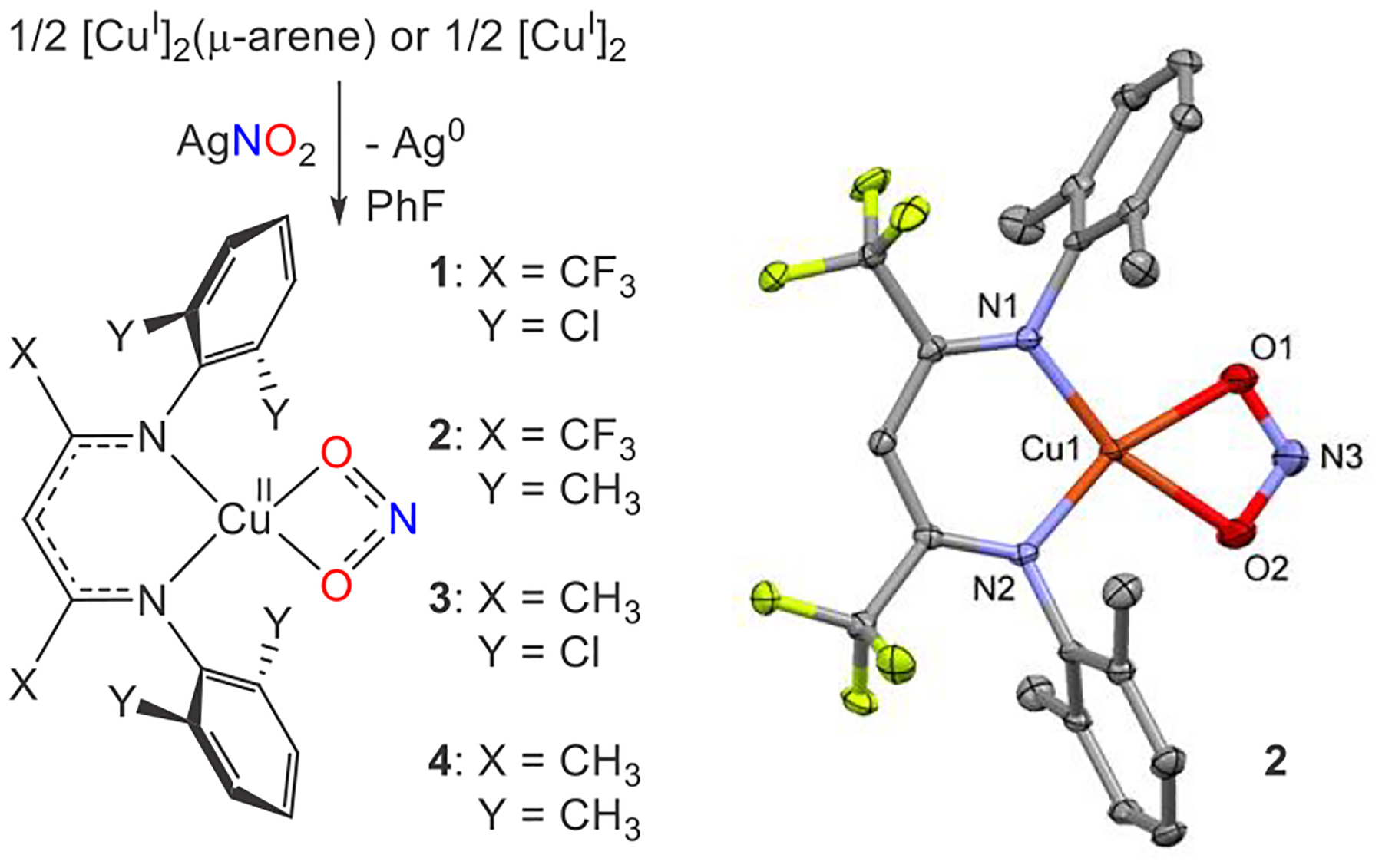
Synthesis of Copper(II) Nitrite Complexes 1–4 and X-ray Structure of Complex 2
Cyclic voltammetry experiments of the copper(II) nitrites 1–4 performed in tetrahydrofuran (THF) reveal quasi-reversible waves that correspond to a range of reduction potentials that span 0.33 V (vs NHE) for 1 (backbone CF3 and N-aryl Cl substituents) to −0.11 V (vs NHE) for 4 (backbone Me and N-aryl Me substituents) (Table 1). Copper(II) nitrite complexes 1–4 are dark green in color, possessing UV–vis spectra in toluene with λmax values around 600 nm. This can be used to follow NO release upon reaction with PArZ3, which results in reduction of the Cu center to give essentially optically silent [CuI]-PArZ3 complexes (Scheme 2). The reaction of 1 with 2 equiv of resulted in a yield of 72% by 19F NMR (Figure S19).
Table 1.
Reduction Potentials and OAT Activation Parameters with for Copper(II) Nitrites 1–4
| [Cu] | E1/2 (V vs NHE) in THF | ΔH⧧ (kcal mol−1) | ΔS⧧ (cal mol−1 K−1) | ΔG⧧(298 K) (kcal mol−1) |
|---|---|---|---|---|
| 1 | 0.3345 | 4.6 ± 0.7 | −38.5 ± 3.1 | 16.0 ± 1.2 |
| 2 | 0.30 | 5.7 ± 0.5 | −34.5 ± 2.4 | 16.0 ± 0.9 |
| 3 | −0.02 | 13.9 ± 0.5 | −13.3 ± 1.8 | 17.9 ± 0.8 |
| 4 | −0.11 | 13.1 ± 0.2 | −18.5 ± 0.8 | 18.6 ± 0.3 |
Scheme 2.
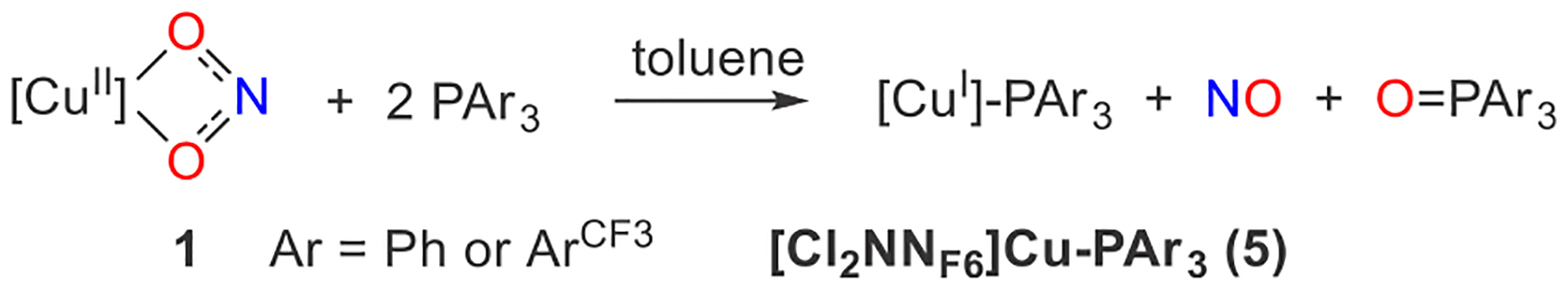
NO Release from CuII-Bound Nitrite 1
Demonstrating NO release, copper(II) nitrite 1 reacts with 2 equiv of PPh3 or in toluene to provide the corresponding [CuI]-PAr3 complex. Capture of NO by the cobalt(II) porphyrin meso-tetra(4-methoxyphenyl)-porphyrincobalt(II) [T(OMe)PP]Co gives the diamagnetic [T(OMe)PP]Co(NO) amenable to 1H NMR analysis, which shows 64% and 59% yield respectively for PPh3 and (Scheme S2).37 Consistent with the 1:2 [CuII](κ2-O2N)/PAr3 stoichiometry, the reaction of (1) with 1 equiv of reduces the yields of and NO to 46% and 51%, respectively. Because the NO that is released can react with copper(I) β-diketiminates [CuI] to reform copper(II) nitrites such as [CuII](O2N)[CuI] and N2O,12 this may represent a pathway that competes with the trapping of [CuI] by PAr3.
To establish the rate law, we followed the reaction of (2; ca. 2.0 mM) with excess in toluene by UV–vis spectroscopy by monitoring the loss of the band at λmax = 600 nm for 2. We chose the electron-poor phosphine to slow the reaction enough to enable a study by straightforward UV–vis kinetics at −60 °C in the presence of 25–50 equiv of phosphine. These studies reveal pseudo-first-order decay of 2 with observed rate constants kobs that vary linearly with (Figures S25 and S26) to give the overall second-order rate law , where k(−60 °C) = 0.27(2) M−1 s−1.
Eyring analysis allowed for quantification of the activation parameters for each electronically different copper nitrite complex 1–4 (Figure 2). We obtained second-order rate constants at various temperatures under pseudo-first-order conditions with 20 equiv of (Figure S27). The very different rates of the reaction require Eyring analysis over different temperature spans in order to conveniently follow these reactions by UV–vis spectroscopy. For instance, we employed a temperature range of −60 to −30 °C in the reaction of 1, while we used a range of 15–45 °C for less reactive 4. These studies reveal that the experimental enthalpy and free energies of activation ΔH⧧ and ΔG⧧(298 K) generally increase with decreasing reduction potential of the copper(II) nitrites 1–4 (Table 1 and Figures S29 and S30). The β-diketiminato backbone substituent exerts the most prominent effect: ΔH⧧ increases from ca. 5 kcal mol−1 (X = CF3) to 13–14 kcal mol−1 (X = Me). We observe a smaller range of free energies of activation ΔG⧧(298 K) = 16–19 kcal mol−1 that reflect more negative entropies of activation observed with X = CF3 versus Me (Figure 2).
Figure 2.
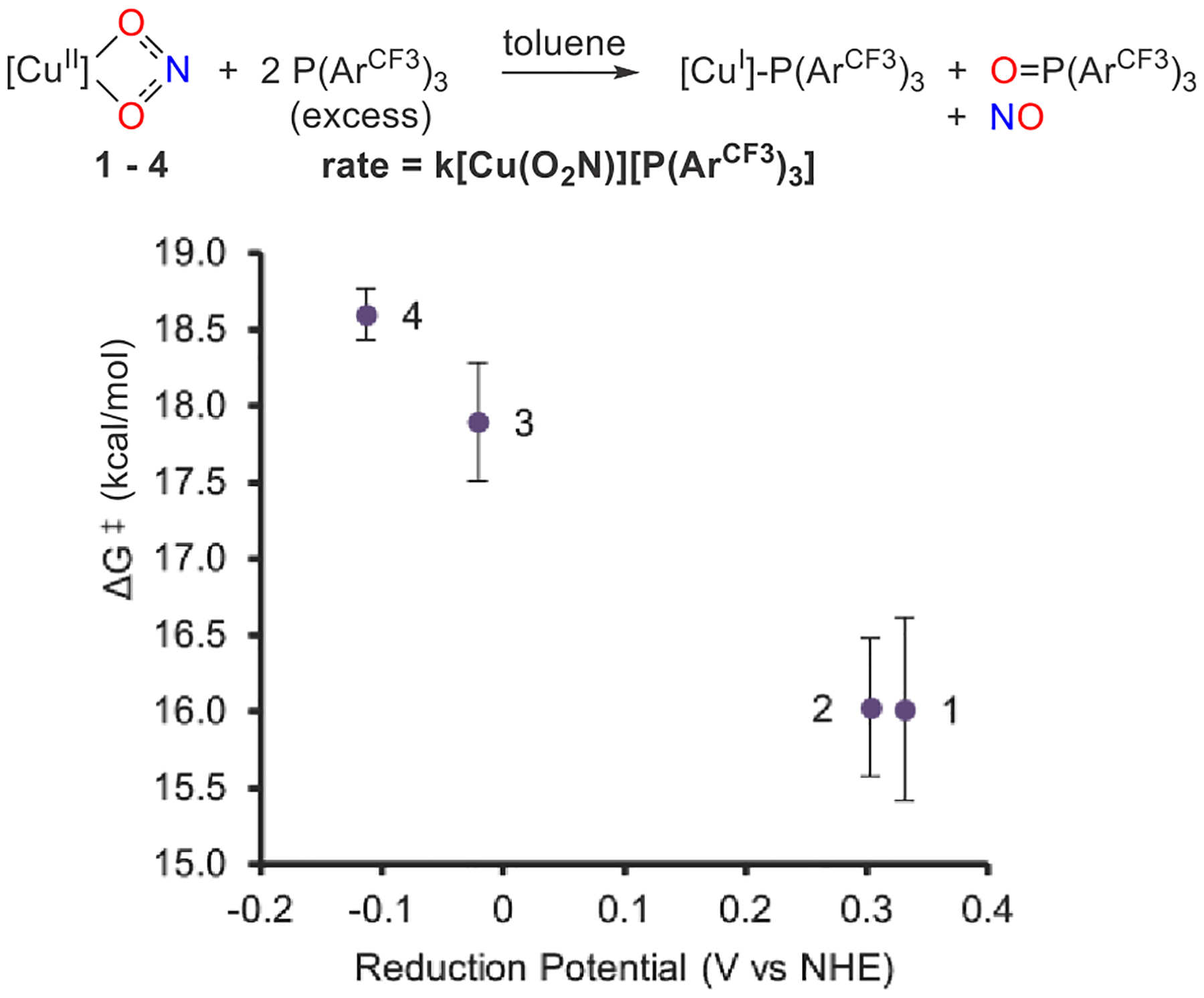
Free energies of activation ΔG⧧ (kcal mol−1) at 298 K for the reaction of 1–4 with excess versus reduction potential of copper(II) nitrite. Bars represent standard errors in ΔG⧧.
Employing compound 3 as a model along with a set of para-substituted phosphines PArZ3, we monitored the electronic influence on the rate of reaction. As can be seen from the Hammett plot (Figure 3), electron-rich phosphines accelerate the reaction [ρ = −1.5(6)]. Thus, the combination of an electron-poor Cu center with an electron-rich phosphine favors nitrite reduction at CuII. Among the phosphines studied, this effect represents a nearly 20-fold increase in the second-order rate constant.
Figure 3.
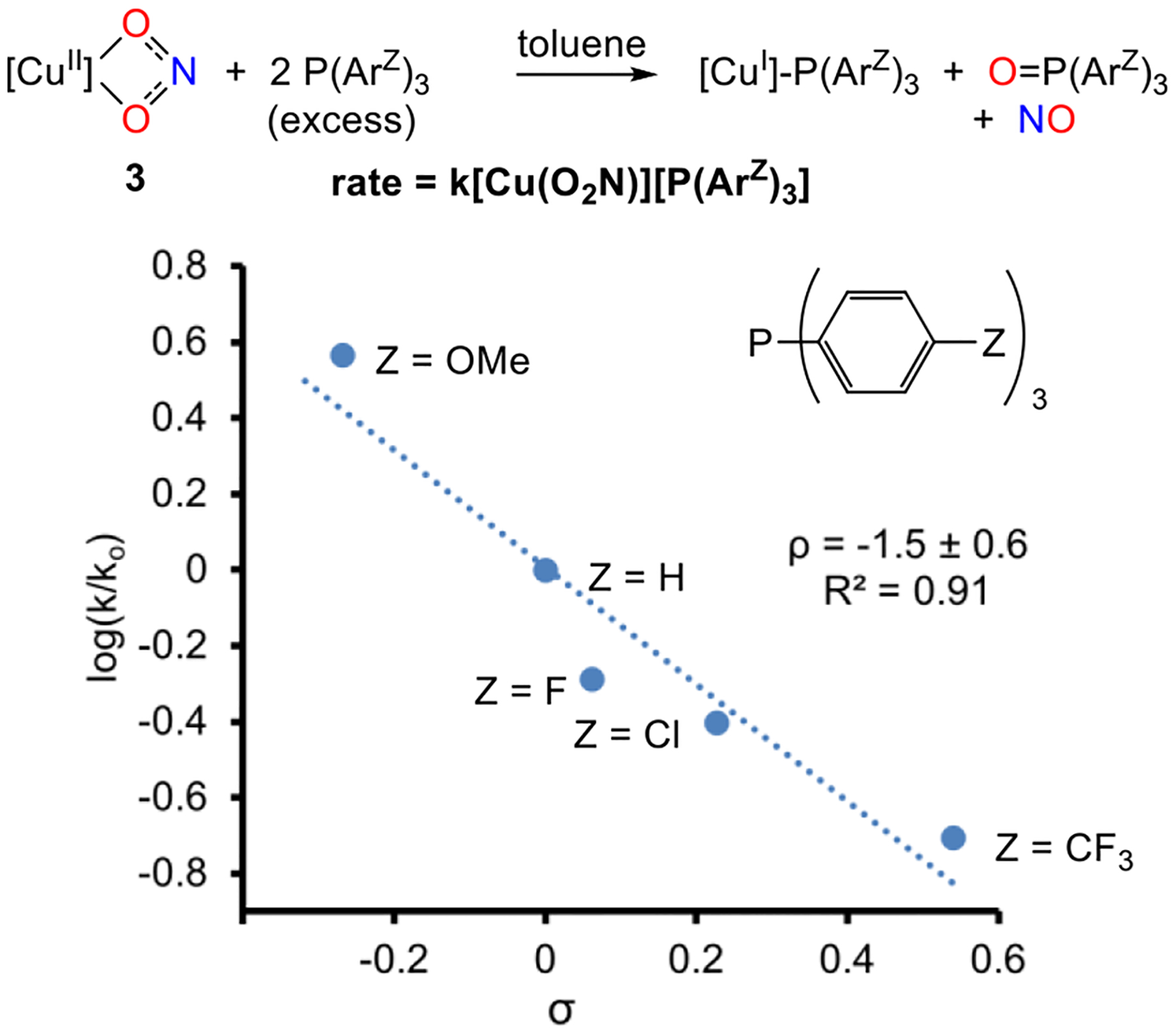
Hammett analysis of the reaction of copper(II) nitrite 3 with para-substituted triarylphosphines P(ArZ)3.
Guided by the experimental rate law and activation parameters in the generation of NO from β-diketiminatocopper(II) nitrites 1–4, we sought to uncover further details of the reaction pathway through density functional theory (DFT) computational analysis. We chose to focus on the synthetic model 4 because its experimental parameters had the lowest estimated errors. In accordance with previous DFT complexes on copper β-diketiminato complexes, we carried out calculations at the BP86+GD3BJ/6–311+ +G(d,p)/SMD-toluene//BP86/6–311+G(d)/gas level of theory.
Direct attack of the phosphine on the copper(II) nitrite 4 with a κ2-O2N nitrite binding mode led to a transition state much higher in energy [ΔG⧧(298 K)calc = 31.1 kcal mol−1] than experimentally determined [ΔG⧧(298 K)exp =18.6(3) kcal mol−1] (Figure S37). By starting with a κ2-O2N bonding mode, OAT to phosphine most directly results in a metastable [Cu]-ON isonitrosyl complex,38,39 calculated to be 20.6 kcal mol−1 higher in free energy than the corresponding three-coordinate nitrosyl [CuI]-NO (Scheme S3).
There are several crystallographically determined binding modes for nitrite at Cu centers, which include κ2-O2N, κ1-ONO, and κ1-NO2 (Figure 4).12,13,40,41 At copper(II) complexes of lower coordination number, nitrite generally prefers the κ2-O2N binding mode in both synthetic complexes21,42 as well as Cu-containing nitrite reductases.22 Tetradentate supporting ligands, however, can lead to the κ1-ONO binding mode.43,44 On the other hand, copper(I) complexes typically display the κ1-NO2 binding mode12,41 because of the availability of back-bonding into the NO2 π* orbitals (Figure S75). While the β-diketiminatocopper(II) nitrites in this study exhibit κ2-O2N binding modes in the solid state,12,45 a recent computational study indicates that both κ1-ONO and κ1-NO2 binding modes are energetically accessible at CuII in the presence of hydrogen bonding.20 Guided by examples in iron porphyrin chemistry for which the [Fe](κ1-NO2) binding mode can enable direct transformation to the corresponding [Fe]-NO nitrosyl complex upon OAT,36 we were eager to consider this κ1-NO2 binding mode as a possible intermediate in OAT from nitrite at CuII.
Figure 4.
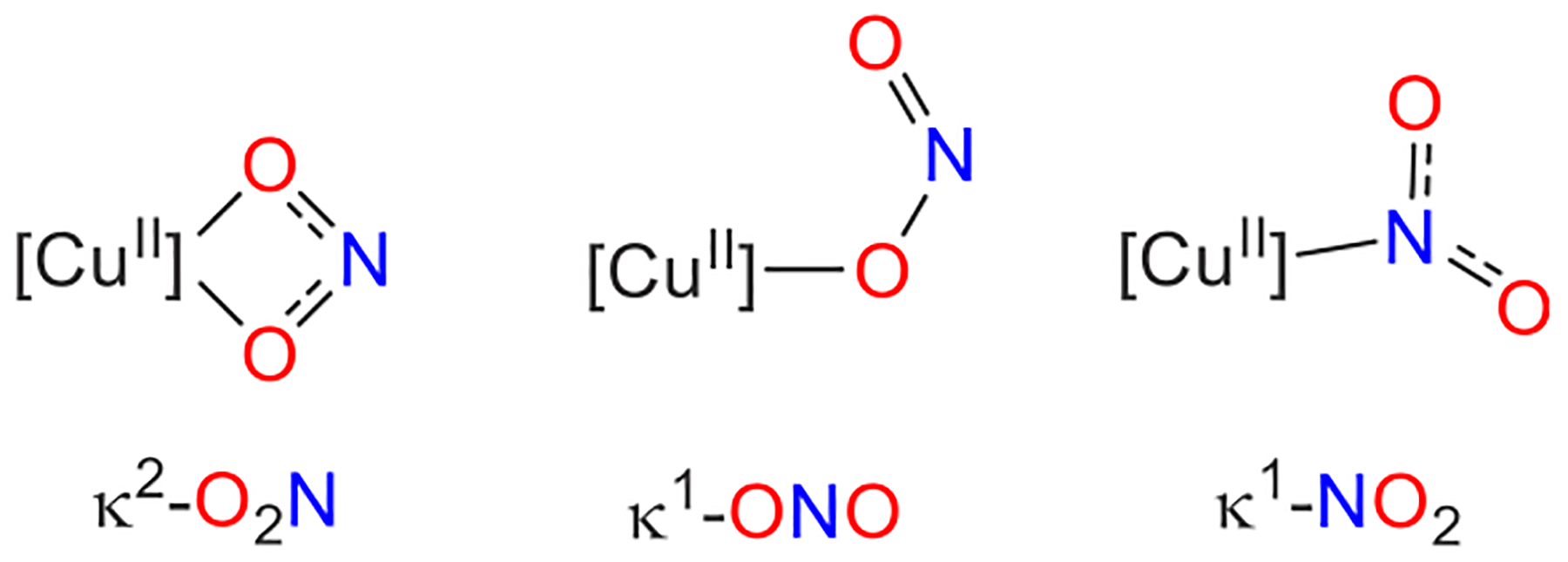
Nitrite bonding modes at Cu.
Computationally examining nitrite isomerization in the copper(II) complex 4, we find that a distorted κ1-NO2 binding mode is only 0.9 kcal mol−1 higher in free energy than the κ2-O2N ground state with a barrier of ΔG⧧(298 K) = 8.1 kcal mol−1. This transition state resembles the κ1-ONO binding mode, which in our model did not result in an optimized stable point (Figure S46). Curiously, this κ1-NO2 binding mode is not symmetrical as found in the crystallographically characterized in which the nitrite ONO plane is orthogonal to the β-diketiminato backbone.12 Rather, this κ1-NO2 binding mode has a close Cu···O contact of 2.440 Å, which results in a highly distorted square-planar coordination. This distortion unequally polarizes a modest amount of unpaired electron density present at nitrite toward the proximal O atom (0.14 e−) at the expense of the distal O atom (0.05 e−) (Figure S74).
A scan of the approach of the phosphine to the O atom with greater spin density led to the optimization of a transition state with ΔG⧧(298 K) = 19.1 kcal mol−1, extremely close to the experimental value of 18.6(3) kcal mol−1. In this transition state, which features κ1-NO2 coordination, the nitrite has twisted to become orthogonal to the β-diketiminato plane. This primes an O atom (O1) for abstraction by the incoming phosphine with N–O1 and P–O1 distances of 1.44 and 1.86 Å, respectively, in the transition state. Developing Cu-NO character is apparent through a shortening of the Cu–N (1.85Å) and N–O2 (1.23 Å) bonds that lead to copper(I) nitrosyl 7 with Cu–N (1.78 Å) and N–O2 (1.19 Å) distances, which are slightly bent orthogonal to the β-diketiminato backbone with a Cu–N–O2 angle of 157.9°. The conversion of copper(II) nitrite 4 and reactants to copper nitrosyl 7 and phosphine oxide is significantly exergonic at −27.2 kcal mol−1. Moreover, the displacement of NO at CuI by phosphine to give the copper(I) phosphine adduct is further downhill by another 6.7 kcal mol−1 in free energy (Figure 5). This is congruent with our experimental observation of [CuI]-PPh3 in the reaction of [CuII](κ2-O2N) complexes with 2 equiv of PPh3 (Scheme 2).12
Figure 5.
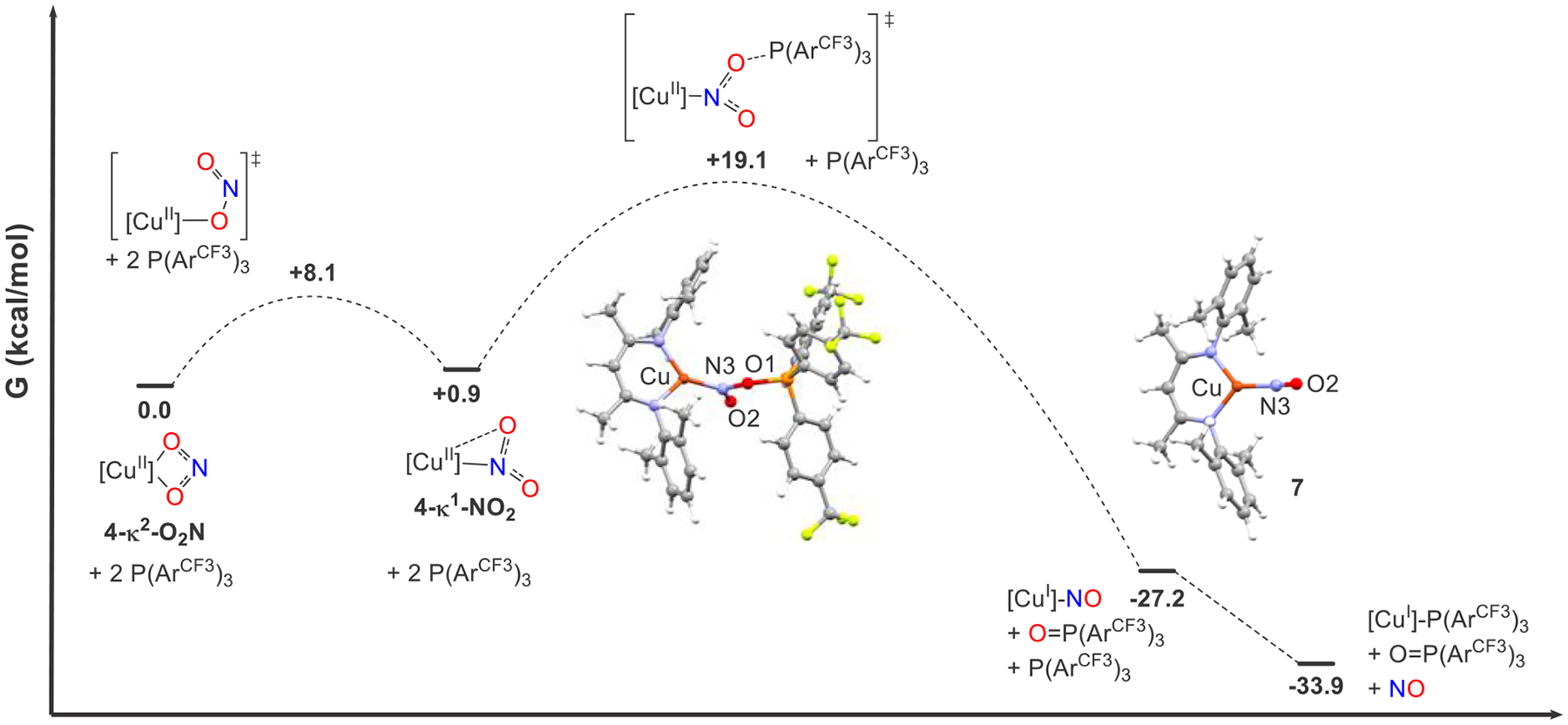
Reaction coordinate diagram of OAT from [Me2NN]Cu(κ2-O2N) (4) to via [Me2NN]Cu-NO (7). Free energies (bold) are in kilocalories per mole at 298 K.
The identification of a DFT transition state structure for OAT from the CuII-bound nitrite to a phosphine that matches the experimental activation energies encourages the consideration of biologically relevant O-atom acceptors. For instance, methionine residues are often found in the vicinity of Cu active sites.46–49 Employing SMe2 as a simple model, experimentally we find that it does not undergo OAT with β-diketiminatocopper(II) nitrites at room temperature or even with modest heating. DFT analysis that involves a κ2-O2N-to-κ1-NO2 isomerization of copper(II) nitrites 1–4 prior to OAT to SMe2 reveals calculated free energies of activation that range from 33.0 to 42.1 kcal mol−1 for the four copper nitrite complexes (Scheme 3 and Figure S38). Thus, the ease of oxidation of the incoming O-atom acceptor is a crucial feature of OAT from nitrite at CuII.
Scheme 3. Considering OAT from Copper(II) Nitrites 1–4 to SMe2a.
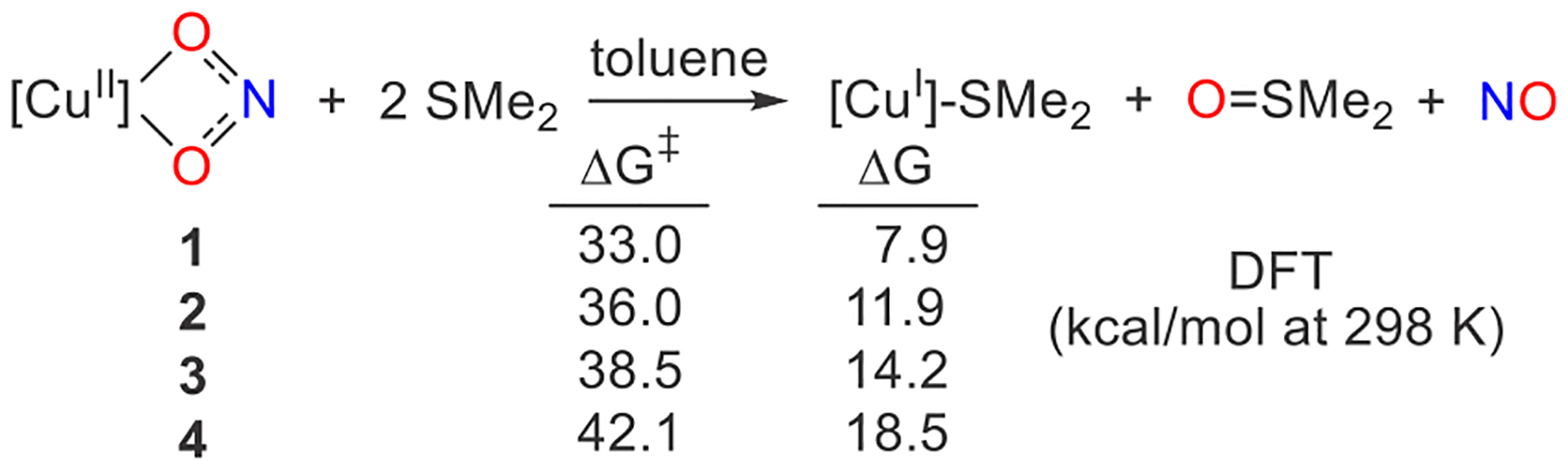
aCalculated free energies are in kilocalories per mole at 298 K.
CONCLUSIONS
Cu-containing nitrite reductases have reduction potentials in the range of 0.17–0.28 V (vs NHE) at the type 2 Cu site,50–54 similar to both copper(II) models 1 and 2, which exhibit reduction potentials of 0.33 and 0.31 V (vs NHE) in THF. This study reveals that increasing the reduction potential of a copper(II) nitrite facilitates OAT from nitrite, a feature that controls the rate of reaction at roughly isosteric models. Moreover, the combination of an electron-poor Cu center with an electron-rich O-atom acceptor proves optimal for OAT. DFT studies benchmarked on the experimental free energy of activation reveal that OAT requires isomerization of the nitrite to a κ1-NO2 binding mode, which enables efficient transfer of a nitrite O atom from N to P to form O=PAr3 along with the copper nitrosyl [Cu]-NO. In contrast to the strong binding of NO at iron(II) porphyrins, the more labile Cu–NO interaction16 results in NO release, with an additional equivalent of the incoming nucleophile that binds to the CuI center.
These studies reveal higher thermodynamic and kinetic barriers for OAT from copper(II) nitrites to more modest O-atom acceptors such as dialkyl sulfides. Nonetheless, turning on the OAT pathways from nitrite to dialkyl sulfides such as methionine commonly found within the coordination sphere of copper enzymes could represent a pathway to connect nitrite and NO with oxidative methionine signaling, a post-translational means to control protein activity.55–57
Supplementary Material
ACKNOWLEDGMENTS
T.H.W. acknowledges funding from the National Institutes of Health (Grant R01GM126205).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c00625.
Experimental, characterization, and computational details (PDF)
Accession Codes
CCDC 1875639, 1983021, and 2081113 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Contributor Information
Molly Stauffer, Department of Chemistry, Georgetown University, Washington, D.C. 20057, United States.
Zeinab Sakhaei, Department of Chemistry, Georgetown University, Washington, D.C. 20057, United States.
Christine Greene, Department of Chemistry, Georgetown University, Washington, D.C. 20057, United States.
Pokhraj Ghosh, Department of Chemistry, Georgetown University, Washington, D.C. 20057, United States.
Jeffery A. Bertke, Department of Chemistry, Georgetown University, Washington, D.C. 20057, United States.
Timothy H. Warren, Department of Chemistry, Georgetown University, Washington, D.C. 20057, United States.
REFERENCES
- (1).Nitric Oxide: Biology and Pathobiology, 2nd ed.; Ignarro LJ, Ed.; Elsevier/Academic Press: Amsterdam, The Netherlands, 2010. [Google Scholar]
- (2).De Pascali F; Hemann C; Samons K; Chen C-A; Zweier JL Hypoxia and Reoxygenation Induce Endothelial Nitric Oxide Synthase Uncoupling in Endothelial Cells through Tetrahydrobiopterin Depletion and S-Glutathionylation. Biochemistry 2014, 53, 3679–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gladwin MT; Schechter AN; Kim-Shapiro DB; Patel RP; Hogg N; Shiva S; Cannon RO; Kelm M; Wink DA; Espey MG; Oldfield EH; Pluta RM; Freeman BA; Lancaster JR; Feelisch M; Lundberg JO The Emerging Biology of the Nitrite Anion. Nat. Chem. Biol 2005, 1, 308–314. [DOI] [PubMed] [Google Scholar]
- (4).Hematian S; Kenkel I; Shubina TE; Dürr M; Liu JJ; Siegler MA; Ivanovic-Burmazovic I; Karlin KD Nitrogen Oxide Atom-Transfer Redox Chemistry; Mechanism of NO(g) to Nitrite Conversion Utilizing μ-oxo Heme-FeIII−O−CuII(L) Constructs. J. Am. Chem. Soc 2015, 137, 6602–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hematian S; Garcia-Bosch I; Karlin KD Synthetic Heme/Copper Assemblies: Toward an Understanding of Cytochrome c Oxidase Interactions with Dioxygen and Nitrogen Oxides. Acc. Chem. Res 2015, 48, 2462–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Basu S; Azarova NA; Font MD; King SB; Hogg N; Gladwin MT; Shiva S; Kim-Shapiro DB Nitrite Reductase Activity of Cytochrome c. J. Biol. Chem 2008, 283, 32590–32597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang Z; Naughton D; Winyard PG; Benjamin N; Blake DR; Symons MCR Generation of Nitric Oxide by a Nitrite Reductase Activity of Xanthine Oxidase: A Potential Pathway for Nitric Oxide Formation in the Absence of Nitric Oxide Synthase Activity. Biochem. Biophys. Res. Commun 1998, 249, 767–772. [DOI] [PubMed] [Google Scholar]
- (8).Cosby K; Partovi KS; Crawford JH; Patel RP; Reiter CD; Martyr S; Yang BK; Waclawiw MA; Zalos G; Xu X; Huang KT; Shields H; Kim-Shapiro DB; Schechter AN; Cannon RO; Gladwin MT Nitrite Reduction to Nitric Oxide by Deoxyhemoglobin Vasodilates the Human Circulation. Nat. Med 2003, 9, 1498–1505. [DOI] [PubMed] [Google Scholar]
- (9).Shiva S; Huang Z; Grubina R; Sun J; Ringwood LA; MacArthur PH; Xu X; Murphy E; Darley-Usmar VM; Gladwin MT Deoxymyoglobin Is a Nitrite Reductase That Generates Nitric Oxide and Regulates Mitochondrial Respiration. Circ. Res 2007, 100, 654–661. [DOI] [PubMed] [Google Scholar]
- (10).Maia LB; Moura JJG How Biology Handles Nitrite. Chem. Rev 2014, 114, 5273–5357. [DOI] [PubMed] [Google Scholar]
- (11).Andring JT; Kim CU; McKenna R Structure and Mechanism of Copper–Carbonic Anhydrase II: A Nitrite Reductase. IUCrJ 2020, 7, 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sakhaei Z; Kundu S; Donnelly JM; Bertke JA; Kim WY; Warren TH Nitric Oxide Release via Oxygen Atom Transfer from Nitrite at Copper(II). Chem. Commun 2017, 53, 549–552. [DOI] [PubMed] [Google Scholar]
- (13).Rose SL; Antonyuk SV; Sasaki D; Yamashita K; Hirata K; Ueno G; Ago H; Eady RR; Tosha T; Yamamoto M; Hasnain SS An Unprecedented Insight into the Catalytic Mechanism of Copper Nitrite Reductase from Atomic-Resolution and Damage-Free Structures. Sci. Adv 2021, 7, eabd8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Fukuda Y; Tse KM; Nakane T; Nakatsu T; Suzuki M; Sugahara M; Inoue S; Masuda T; Yumoto F; Matsugaki N; Nango E; Tono K; Joti Y; Kameshima T; Song C; Hatsui T; Yabashi M; Nureki O; Murphy MEP; Inoue T; Iwata S; Mizohata E Redox-Coupled Proton Transfer Mechanism in Nitrite Reductase Revealed by Femtosecond Crystallography. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 2928–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Cioncoloni G; Roger I; Wheatley PS; Wilson C; Morris RE; Sproules S; Symes MD Proton-Coupled Electron Transfer Enhances the Electrocatalytic Reduction of Nitrite to NO in a Bioinspired Copper Complex. ACS Catal. 2018, 8, 5070–5084. [Google Scholar]
- (16).Merkle AC; Lehnert N Binding and Activation of Nitrite and Nitric Oxide by Copper Nitrite Reductase and Corresponding Model Complexes. Dalton Trans. 2012, 41, 3355–3368. [DOI] [PubMed] [Google Scholar]
- (17).Moore CM; Szymczak NK Nitrite Reduction by Copper through Ligand-Mediated Proton and Electron Transfer. Chem. Sci 2015, 6, 3373–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Matson EM; Park YJ; Fout AR Facile Nitrite Reduction in a Non-Heme Iron System: Formation of an Iron(III)-Oxo. J. Am. Chem. Soc 2014, 136, 17398–17401. [DOI] [PubMed] [Google Scholar]
- (19).Sanders BC; Hassan SM; Harrop TC NO2− Activation and Reduction to NO by a Nonheme Fe(NO2)2 Complex. J. Am. Chem. Soc 2014, 136, 10230–10233. [DOI] [PubMed] [Google Scholar]
- (20).Cheng R; Wu C; Cao Z; Wang B QM/MM MD Simulations Reveal an Asynchronous PCET Mechanism for Nitrite Reduction by Copper Nitrite Reductase. Phys. Chem. Chem. Phys 2020, 22, 20922–20928. [DOI] [PubMed] [Google Scholar]
- (21).Maria S; Chattopadhyay T; Ananya S; Kundu S Reduction of Nitrite to NO at a Mononuclear Copper(II)-Phenolate Site. Inorg. Chim. Acta 2020, 506, 119515. [Google Scholar]
- (22).Horrell S; Kekilli D; Strange RW; Hough MA Recent Structural Insights into the Function of Copper Nitrite Reductases. Metallomics 2017, 9, 1470–1482. [DOI] [PubMed] [Google Scholar]
- (23).Tovrog BS; Diamond SE; Mares F Oxygen Transfer from Ligands: Cobalt Nitro Complexes as Oxygenation Catalysts. J. Am. Chem. Soc 1979, 101, 270–272. [Google Scholar]
- (24).Khin C; Heinecke J; Ford PC Oxygen Atom Transfer from Nitrite Mediated by Fe(III) Porphyrins in Aqueous Solution. J. Am. Chem. Soc 2008, 130, 13830–13831. [DOI] [PubMed] [Google Scholar]
- (25).He C; Howes BD; Smulevich G; Rumpel S; Reijerse EJ; Lubitz W; Cox N; Knipp M Nitrite Dismutase Reaction Mechanism: Kinetic and Spectroscopic Investigation of the Interaction between Nitrophorin and Nitrite. J. Am. Chem. Soc 2015, 137, 4141–4150. [DOI] [PubMed] [Google Scholar]
- (26).Heinecke J; Ford PC Formation of Cysteine Sulfenic Acid by Oxygen Atom Transfer from Nitrite. J. Am. Chem. Soc 2010, 132, 9240–9243. [DOI] [PubMed] [Google Scholar]
- (27).Heinecke JL; Khin C; Pereira JCM; Suárez SA; Iretskii AV; Doctorovich F; Ford PC Nitrite Reduction Mediated by Heme Models. Routes to NO and HNO? J. Am. Chem. Soc 2013, 135, 4007–4017. [DOI] [PubMed] [Google Scholar]
- (28).Goodwin J; Kurtikyan T; Standard J; Walsh R; Zheng B; Parmley D; Howard J; Green S; Mardyukov A; Przybyla DE Variation of Oxo-Transfer Reactivity of (Nitro)Cobalt Picket Fence Porphyrin with Oxygen-Donating Ligands. Inorg. Chem 2005, 44, 2215–2223. [DOI] [PubMed] [Google Scholar]
- (29).O’Shea SK; Wall T; Lin D Activation of Nitrite Ion by Biomimetic Iron(III) Complexes. Transition Met. Chem 2007, 32, 514–517. [Google Scholar]
- (30).Kurtikyan TS; Hovhannisyan AA; Iretskii AV; Ford PC Six-Coordinate Nitro Complexes of Iron(III) Porphyrins with Trans S-Donor Ligands. Oxo-Transfer Reactivity in the Solid State. Inorg. Chem 2009, 48, 11236–11241. [DOI] [PubMed] [Google Scholar]
- (31).Goodwin J; Bailey R; Pennington W; Rasberry R; Green T; Shasho S; Yongsavanh M; Echevarria V; Tiedeken J; Brown C; Fromm G; Lyerly S; Watson N; Long A; De Nitto N Structural and Oxo-Transfer Reactivity Differences of Hexacoordinate and Pentacoordinate (Nitro)(Tetraphenylporphinato)Cobalt(III) Derivatives. Inorg. Chem 2001, 40, 4217–4225. [DOI] [PubMed] [Google Scholar]
- (32).Munro OQ; Scheidt WR (Nitro)Iron(III) Porphyrins. EPR Detection of a Transient Low-Spin Iron(III) Complex and Structural Characterization of an O Atom Transfer Product. Inorg. Chem 1998, 37, 2308–2316. [DOI] [PubMed] [Google Scholar]
- (33).Castro CE; O’Shea SK Activation of Nitrite Ion by Iron(III) Porphyrins. Stoichiometric Oxygen Transfer to Carbon, Nitrogen, Phosphorus, and Sulfur. J. Org. Chem 1995, 60, 1922–1923. [Google Scholar]
- (34).Tsai F-T; Kuo T-S; Liaw W-F Dinitrosyl Iron Complexes (DNICs) Bearing O-Bound Nitrito Ligand: Reversible Transformation between the Six-Coordinate {Fe(NO)2}9 [(1-MeIm)2(H2-ONO)Fe(NO)2] (g = 2.013) and Four-Coordinate {Fe(NO)2}9 [(1-MeIm)(ONO)Fe(NO)2] (g = 2.03). J. Am. Chem. Soc 2009, 131, 3426–3427. [DOI] [PubMed] [Google Scholar]
- (35).Tsai F-T; Lee Y-C; Chiang M-H; Liaw W-F Nitrate-to-Nitrite-to-Nitric Oxide Conversion Modulated by Nitrate-Containing {Fe(NO)2}9 Dinitrosyl Iron Complex (DNIC). Inorg. Chem 2013, 52, 464–473. [DOI] [PubMed] [Google Scholar]
- (36).Conradie J; Ghosh A Iron(III)–Nitro Porphyrins: Theoretical Exploration of a Unique Class of Reactive Molecules. Inorg. Chem 2006, 45, 4902–4909. [DOI] [PubMed] [Google Scholar]
- (37).Sanders BC; Hassan SM; Harrop TC NO2− Activation and Reduction to NO by a Nonheme Fe(NO2)2 Complex. J. Am. Chem. Soc 2014, 136, 10230–10233. [DOI] [PubMed] [Google Scholar]
- (38).Bitterwolf TE Photochemical Nitrosyl Linkage Isomerism/Metastable States. Coord. Chem. Rev 2006, 250, 1196–1207. [Google Scholar]
- (39).De La Cruz C; Sheppard N A Structure-Based Analysis of the Vibrational Spectra of Nitrosyl Ligands in Transition-Metal Coordination Complexes and Clusters. Spectrochim. Acta, Part A 2011, 78, 7–28. [DOI] [PubMed] [Google Scholar]
- (40).Boulanger MJ; Murphy MEP Directing the Mode of Nitrite Binding to a Copper-Containing Nitrite Reductase from Alcaligenes Faecalis S-6: Characterization of an Active Site Isoleucine. Protein Sci. 2003, 12, 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chandra Maji R; Mishra S; Bhandari A; Singh R; Olmstead MM; Patra AK A Copper(II) Nitrite That Exhibits Change of Nitrite Binding Mode and Formation of Copper(II) Nitrosyl Prior to Nitric Oxide Evolution. Inorg. Chem 2018, 57, 1550–1561. [DOI] [PubMed] [Google Scholar]
- (42).Lehnert N; Cornelissen U; Neese F; Ono T; Noguchi Y; Okamoto K; Fujisawa K Synthesis and Spectroscopic Characterization of Copper(II)–Nitrito Complexes with Hydrotris(Pyrazolyl)-Borate and Related Coligands. Inorg. Chem 2007, 46, 3916–3933. [DOI] [PubMed] [Google Scholar]
- (43).Hematian S; Siegler MA; Karlin KD Nitric Oxide (NO) Generation from Heme/Copper Assembly Mediated Nitrite Reductase Activity. JBIC, J. Biol. Inorg. Chem 2014, 19 (0), 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Hematian S; Garcia-Bosch I; Karlin KD Synthetic Heme/Copper Assemblies: Toward an Understanding of Cytochrome c Oxidase Interactions with Dioxygen and Nitrogen Oxides. Acc. Chem. Res 2015, 48, 2462–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kundu S; Kim WY; Bertke JA; Warren TH Copper(II) Activation of Nitrite: Nitrosation of Nucleophiles and Generation of NO by Thiols. J. Am. Chem. Soc 2017, 139, 1045–1048. [DOI] [PubMed] [Google Scholar]
- (46).Davis AV; O’Halloran TV A Place for Thioether Chemistry in Cellular Copper Ion Recognition and Trafficking. Nat. Chem. Biol 2008, 4, 148–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Liu J; Chakraborty S; Hosseinzadeh P; Yu Y; Tian S; Petrik I; Bhagi A; Lu Y Metalloproteins Containing Cytochrome, Iron–Sulfur, or Copper Redox Centers. Chem. Rev 2014, 114, 4366–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wilson TD; Yu Y; Lu Y Understanding Copper-Thiolate Containing Electron Transfer Centers by Incorporation of Unnatural Amino Acids and the CuA Center into the Type 1 Copper Protein Azurin. Coord. Chem. Rev 2013, 257, 260–276. [Google Scholar]
- (50).Suzuki S; Kataoka K; Yamaguchi K; Inoue T; Kai Y Structure–Function Relationships of Copper-Containing Nitrite Reductases. Coord. Chem. Rev 1999, 190–192, 245–265. [Google Scholar]
- (51).Suzuki S; Deligeer; Yamaguchi, K.; Kataoka, K.; Kobayashi, K.; Tagawa, S.; Kohzuma, T.; Shidara, S.; Iwasaki, H. Spectroscopic characterization and intramolecular electrontransfer processes of native and type 2 Cu-depleted nitrite reductases. JBIC, J. Biol. Inorg. Chem 1997, 2, 265–274. [Google Scholar]
- (52).Farver O; Eady RR; Abraham ZH; Pecht I The intramolecular electron transfer between copper sites of nitrite reductase: a comparison with ascorbate oxidase. FEBS Lett. 1998, 436, 239–242. [DOI] [PubMed] [Google Scholar]
- (53).Kobayashi K; Tagawa S; Deligeer; Suzuki, S. The pH-Dependent Changes of Intramolecular Electron Transfer on Copper-Containing Nitrite Reductase. J. Biochem 1999, 126, 408–412. [DOI] [PubMed] [Google Scholar]
- (54).Pinho D; Besson S; Brondino CD; de Castro B; Moura I Copper-containing nitrite reductase from Pseudomonas chlororaphis DSM 50135. Eur. J. Biochem 2004, 271, 2361–2369. [DOI] [PubMed] [Google Scholar]
- (55).Lin S; Yang X; Jia S; Weeks AM; Hornsby M; Lee PS; Nichiporuk RV; Iavarone AT; Wells JA; Toste FD; Chang CJ Redox-Based Reagents for Chemoselective Methionine Bioconjugation. Science 2017, 355, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Henry C; Loiseau L; Vergnes A; Vertommen D; Mérida-Floriano A; Chitteni-Pattu S; Wood EA; Casadesús J; Cox MM; Barras F; Ezraty B Redox Controls RecA Protein Activity via Reversible Oxidation of Its Methionine Residues. eLife 2021, 10, e63747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ezraty B; Gennaris A; Barras F; Collet J-F Oxidative Stress, Protein Damage and Repair in Bacteria. Nat. Rev. Microbiol 2017, 15, 385–396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.