

Abstract
Cancer stem cells (CSCs) are defined as a subpopulation of malignant tumor cells with selective capacities for tumor initiation, self‐renewal, metastasis, and unlimited growth into bulks, which are believed as a major cause of progressive tumor phenotypes, including recurrence, metastasis, and treatment failure. A number of signaling pathways are involved in the maintenance of stem cell properties and survival of CSCs, including well‐established intrinsic pathways, such as the Notch, Wnt, and Hedgehog signaling, and extrinsic pathways, such as the vascular microenvironment and tumor‐associated immune cells. There is also intricate crosstalk between these signal cascades and other oncogenic pathways. Thus, targeting pathway molecules that regulate CSCs provides a new option for the treatment of therapy‐resistant or ‐refractory tumors. These treatments include small molecule inhibitors, monoclonal antibodies that target key signaling in CSCs, as well as CSC‐directed immunotherapies that harness the immune systems to target CSCs. This review aims to provide an overview of the regulating networks and their immune interactions involved in CSC development. We also address the update on the development of CSC‐directed therapeutics, with a special focus on those with application approval or under clinical evaluation.
Keywords: cancer stem cells, CAR‐T therapies, inhibitors, signal pathway
The major signaling pathways that regulate CSCs. Amongst an array of signaling pathways aberrantly activated in cancer Wnt, JAK/STAT, PI3k/Akt/mTOR, Notch, NF‐κB, Hedgehog, and TGF‐β/Smad pathways are crucial for the of self‐renewal, cell growth, metastasis of CSCs, and angiogenesis.

1. INTRODUCTION
The concept of stem cells dates back to the 18th century when scientists tried to elucidate how lower organisms developed tissues and organs. 1 These stem cells produce daughter cells that later undergo different biological processes, either continuous self‐renewal division, or differentiation into specialized cells with a limited lifespan. Normal tissue stem cells provide a life‐long source of cells for self‐renewal of tissues, which leads us to speculate that whether stem cells are capable of deriving a malignant cell population, and this lies the foundation of cancer stem cells (CSCs) theory. CSCs are defined as a subpopulation of malignant tumor cells with selective capacities for tumor initiation, self‐renewal, metastasis, and unlimited growth into bulks. 2
Despite decades of research on cancer treatment, it has been proved extremely challenging to achieve complete remission (CR) in cancer patients. Tumor relapse may be explained by the fact that antitumor therapeutics mainly target proliferative cancer cells but remain ineffective in quiescent CSCs. The role of CSC in tumor initiation was first identified in acute myeloid leukemia (AML). Since its isolation from a number of solid tumors and hematological malignancies, the CSC is believed to form the clonogenic core of these tumors. 3 Growing evidence now suggests that CSCs are responsible for multiple progressive tumor phenotypes, including recurrence, metastasis, and treatment failure. 4 , 5 The intrinsic treatment resistance of tumors has partially attributed to the presence of the CSC subpopulation, 6 , 7 and may also be induced by extrinsic factors, such as treatments and environments. 8 , 9
Major signaling pathways are involved in the maintenance of stem cell properties and survival of CSCs, such as the Notch, Wnt, and Hedgehog (HH) pathways. 10 There is also intricate interplay network between these signal cascades and other oncogenic pathways. 11 , 12 , 13 Thus, targeting pathway molecules that regulate CSCs provides a new option for the treatment of therapy‐resistant or ‐refractory tumors. This review aims to provide an overview of the regulating networks and their immune interactions involved in CSC development. We also summarized the update on the development of CSC‐directed therapeutics, with a special focus on those with application approval or under clinical evaluation.
2. CHARACTERISTICS AND IDENTIFICATION MARKERS OF CSCs
2.1. Characteristics of CSCs
Over the past decades, a wide breadth of literature investigated the biological characteristics of CSCs, with the hope to develop CSC‐targeted strategies that eradicate treatment‐insensitive or ‐refractory tumor cells. The potent self‐renewal ability is probably the best‐characterized property of CSCs and a direct cause of tumor initiation. 14 CSCs divide into daughter cells in a symmetrical splitting manner and ultimately lead to excessive tumor growth. 15 Experimental data revealed the tumorigenic function of CSCs by forming new tumors with CSCs isolated from primary tumor tissues in immunodeficient mice. 16
Another characteristic of CSCs is their differentiation ability. For instance, leukemia stem cells (LSCs), characterized by CD34‐positive expression and deficient CD38 expression, were able to differentiate into multiple cell types in SCID mice. 17 In addition, CSCs isolated from human brains share similar surface markers CD133 and Nestin, with normal neuronal stem cells, and are thus believed to have differentiation capabilities. 18 In normal tissues, the balance between self‐renewal and differentiation of stem cells controls the cell fate, the aberrant regulation of which lead to tumorigenesis. 19 Interestingly, the trans‐differentiation of CSCs into other multilineage cells may also contribute to tumor formation. 20 One such example is the trans‐differentiation of CSCs into vascular endothelial cells, leading to oncogenesis and tumor angiogenesis of glioblastoma, 21 renal, 22 and liver cancer. 23
Tumor heterogeneity is one of the key reasons for therapeutic resistance. 24 The theory of tumor heterogeneity concept dates back to the 1970s when tumors are believed to consist of multiple distinct tumor cell subpopulations. 25 CSCs are believed to contribute to tumor heterogeneity. This cell subpopulation gives rise to cancer cells with diverse differentiation levels, which then go through sporadic mutations and environmental changes for clone selection. 26 The aberrant differentiation programs of CSCs resemble the hierarchical compositions of normal stem cells, which ultimately lead to a hierarchical set of tumor cells.
CSCs have also been identified with surviving and expanding capacity after cytotoxic anticancer treatment, which were recently found to enrich in remaining tumor bulks following chemotherapy treatments. 27 , 28 For instance, after chemotherapy treatment, preleukemic DNMT3Amut hematopoietic stem cells (HSCs) are able to generate a hematopoietic hierarchy that facilitates their survival and expansion. 29 Likewise, the transformation of differentiated tumor cells to glioma stem cells (GSCs) was often observed in temozolomide (TMZ)‐treated glioma. 30 The underlying mechanisms for the chemoresistance caused by CSCs include epithelial‐mesenchymal transition (EMT), dormancy, and tumor environment, which we will discuss in the review. 31 , 32 , 33
2.2. Identification markers of CSCs
The unique gene expression profile of the CSC makes it different from bulk tumor cells, which can be used as CSC‐specific identification markers. Commonly used methodologies that evaluate CSC stemness include the detection of stemness genes, surface proteins, such as CD44 and CD133, intracellular markers, such as aldehyde dehydrogenase (ALDH), and at a more macroscopic level, phenotypic assays, such as tumorsphere formation tests. 34 , 35 , 36 , 37 Table 1 shows the key markers of CSCs in solid tumors and hematological malignancies. A number of stemness genes have been reported to modulate cell stemness in embryos and adults, including the transcription factors POU class 5 homeobox 1 (POU5F1, OCT4), Nanog homeobox (NANOG), Sex‐determining region Y‐box 2 (SOX2), Kruppel‐like factor 4 (KLF4), and MYC proto‐oncogene. 38 , 39
TABLE 1.
Key markers of cancer stem cells in solid tumors and hematological malignancies
| CSC surface marker | Cancer types |
|---|---|
| CD4 | Head and neck squamous cell carcinoma |
| CD9 | Glioblastoma |
| CD10 | Acute myeloid leukemia, head and neck squamous cell carcinoma |
| CD13 | Liver, pancreatic cancer |
| CD15 | Glioblastoma |
| CD19 | Acute myeloid leukemia |
| CD20 | Acute myeloid leukemia, melanoma |
| CD24 | Breast, gastric, liver, colorectal, ovarian cancer |
| CD25 | Chronic myeloid leukemia |
| CD26 | Chronic myeloid leukemia |
| CD29 (ß1 integrin) | Breast cancer |
| CD33 | Chronic/acute myeloid leukemia |
| CD34 | Acute myeloid leukemia |
| CD36 | Chronic myeloid leukemia, glioblastoma |
| CD44 (and its variants) | Breast, lung, gastric, liver, colorectal, prostate, bladder, esophageal, ovarian, pancreatic, cervical cancer, glioblastoma |
| CD47 | Liver cancer |
| CD49f | Breast, gastric, colorectal, cervical cancer, glioblastoma |
| CD54 | Gastric cancer |
| CD61 | Breast cancer |
| CD70 | Breast cancer |
| CD71 | Acute myeloid leukemia |
| CD87 | Lung cancer |
| CD90 | Breast, lung, gastric, liver, esophageal, pancreatic cancer, glioblastoma |
| CD98 | Head and neck squamous cell carcinoma |
| CD117 | Chronic myeloid leukemia, prostate, ovarian, lung cancer |
| CD123 | Chronic/acute myeloid leukemia |
| CD133 | Breast, lung, gastric, liver, colorectal, prostate, ovarian, pancreatic, cervical cancer, melanoma, glioblastoma, head and neck squamous cell carcinoma |
| CD166 | Lung, colorectal, ovarian cancer |
| CD206 | Colorectal, liver cancer |
| CD271 | Melanoma, head and neck squamous cell carcinoma |
| Ov6 | Pancreatic cancer |
| CXCR4 | Breast, gastric, prostate, renal, lung cancer |
| EpCAM | Breast, lung, gastric, liver, colorectal, prostate, pancreatic cancer |
| LGR5 | Breast, gastric, colorectal cancer |
| ProC‐R | Breast cancer |
| IL1RAP | Chronic myeloid leukemia |
| LINGO2 | Gastric cancer |
| CLL‐1 | Acute myeloid leukemia |
| TIM3 | Acute myeloid leukemia |
| L1CAM | Glioblastoma |
| EGFR | Glioblastoma |
| ABCG2 | Lung, cervical cancer |
| CK17 | Cervical cancer |
As for the surface markers that are differentially expressed on CSCs, 40 , 41 some of the markers are not unique to CSCs and may also be present on normal stem cells. 42 , 43 The first reported CSC surface markers were CD34 and CD38 proteins which are used to identify HSCs and LSCs in acute myelogenous leukemia. 44 ABCG2 is a phenotypic marker for CSCs 45 in ovarian, 46 hepatic, 47 breast, 48 lung cancer, 48 and AML. 49 ABCG2 is believed to associate with therapeutic resistance caused by CSC and is highly expressed in side population (SP) cells (defined as the cell fraction that excludes Hoechst DNA binding dye). 50 The fluorescence‐activated cell sorting (FACS)‐based SP sorting method is commonly used for CSCs isolation.
CD133 (Prominin 1), coded by the gene PROM1, was initially considered a surface marker for colorectal CSCs, 51 , 52 and was later confirmed of its expression on CSCs of multiple origins. However, as recent evidence suggested that CD133‐ cell subsets were capable of inducing tumorigenesis, CD133 positivity may not necessarily indicate CSC stemness. 53 In addition, CD133+ cells were also found to promote tumor metastasis. 54 Results from a single‐cell proteomic profiling analysis revealed a higher level of CD133 compared with other stemness markers, such as NANOG and ALDH1A1, in lung cancer cells with EMT phenotypes. 55 Given that epithelial‐mesenchymal plasticity of CSCs is a potential trigger for tumor metastasis, 56 the coexpression of CD133 and other stemness markers may be used to identify cells with stemness characteristics.
CD44, also known as P‐glycoprotein 1, is a transmembrane glycoprotein and cell surface adhesion receptor for hyaluronic acid (HA) and osteopontin (OPN). 57 Accumulating evidence has suggested that stemness markers, including SOX2, NANOG, and OCT4, are highly expressed in CD44+ cell fraction. 58 Though the downstream targets of CD44 remain incompletely defined, known cancer‐associated signaling pathways include Rho GTPases, Ras‐MAPK, and phosphatidylinositol‐3‐kinase (PI3K)/AKT cascades. 59 The alternative splicing of the CD44 gene leads to multiple variants, and among all the variants, CD44v is expressed in epithelial cells and critical for maintaining stemness. 60 Although HA is a ligand for all forms of CD44, OPN only interacts with CD44v rather than CD44s. 61 The crosstalk between HA and CD44 regulates a number of biological processes leading to tumor cell stemness, invasion, and metastasis. For instance, HA binds to CD44 and triggers the NANOG‐STAT3 pathway activation, leading to the self‐renewal of ovarian cancer cells. 62 On the other hand, OPN, enriched in gliomas, has also been found to promote tumor cell stemness via the OPN‐CD44 axis. 63 This evidence suggests that the downstream activities of CD44 might depend on the selection of variants by the ligands. Thus, CD44 should not be simply addressed as a marker for CSCs, the functions of which rely on its preference of variants and ligands present in the microenvironment.
However, the surface markers of CSCs may vary according to tumor types and the cell of tumor origin, demonstrating high heterogeneity between tumors or even among cells within the same tumor. 35 , 64 Examples include breast CSCs, which frequently display different surface marker patterns, such as CD44+, CD24−, and ALDH+, 65 , 66 and melanoma stem cells that can either be CD271− or CD271+. 67 Such heterogeneity of CSC surface markers has also been reported in glioblastoma, 68 prostate cancer, 69 and lung cancer. 70 Moreover, the expression of CSC biomarkers is not constant and may change depending on the external environment. Higher CD133 expression was observed on stem cell‐like pancreatic cancer cells under hypoxia 71 and the enzymatic dissociation alters the retention of surface CD133 on glioma cells. 72
To overcome the challenge caused by constantly changing surface markers for CSCs, researchers then used nonmembrane biomarkers for CSC identification, and the ALDH represents one of its kind. The expression of ALDH is often found in normal stem cells. Highly active ALDH1 can be used to identify CSCs in breast, bladder, lung cancer, embryonal rhabdomyosarcoma, and head and neck squamous cell carcinoma. 73 ALDH1 expression is also related to the therapeutic resistance of cancer cells to chemotherapies. 74 However, subpopulations of breast cancer CSCs appeared to be ALDH‐negative. 75
EMT allows a polarized epithelial cell to escape from its interaction with the basement membrane and transform into mesenchymal cell phenotypes. 76 Thus, cancer cells that undergo EMT are more prone to invasion and metastasis. 77 A subpopulation of breast cancer CSCs showed both EMT and CSC markers CD44, ABCG2, and ALDH1A1/3, which might serve as identification markers for metastasis‐initiating cells. 78 In addition, another CSC subpopulations, which are ITGA6‐positive but deficient in ABCG2 and ALDH1A1/3, are referred to as non‐CSC cells with metastatic abilities but no oncogenic activities. 78 Previous studies reported a fraction of ITGA6+ cells exhibiting epithelial characteristics involved in metastasis. 79 In colon cancer, ITGA6+ cells are included in the CD44+/CD133+ cell population, representing the metastatic non‐CSCs. 80
3. SIGNALING PATHWAYS REGULATING CSCs
The homeostasis of normal stem cells is modulated by an intricate signaling network, the aberrant activation or repression of which promotes oncogenic transformation. These aberrations lead to the self‐renewal and differentiation properties of CSCs, conferring stemness to the cancers. Like their normal counterparts, CSCs also rely on these signaling pathways for survival and stemness maintenance. In this review, we classified these molecular pathways as extrinsic or intrinsic signals (Figure 1).
FIGURE 1.
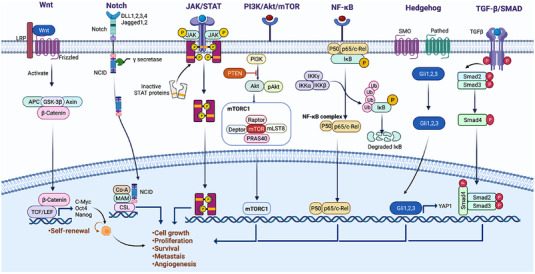
Overview of major signaling pathways that regulate CSCs. Among an array of signaling pathways aberrantly activated in cancer Wnt, JAK/STAT, PI3k/Akt/mTOR, Notch, NF‐κB, Hedgehog, and TGF‐β/Smad pathways are crucial for the self‐renewal, cell growth, metastasis of CSCs, and angiogenesis. (Figures built with biorender.com)
3.1. Intrinsic signaling pathway in CSCs
3.1.1. Wnt signaling
The Wnt pathway is highly conserved and has long been identified as a key regulator of embryonic development and tissue homeostasis. 81 , 82 The abnormal activation of Wnt signaling elements, including adenomatous polyposis coli (APC), Axin, β‐catenin, and Wnt1, is frequently found in a wide range of malignancies, which is related to cancer initiation and progression. 83 , 84 , 85 , 86 , 87 The canonical Wnt pathway is β‐catenin dependent and the noncanonical Wnt pathway does not rely on β‐catenin.
In the context of CSC regulation, the canonical Wnt signaling appears rather important in maintaining stem cell‐like traits of tumor cells. 88 Canonical Wnt signaling is critical for maintaining lung CSC properties, potentially by regulating the expression of CSC marker OCT‐4. 89 Other CSC markers stimulated by the Wnt/β‐catenin pathway include CD24, Prom1, CD44, and ALDH1, thereby enhancing tumor stemness. 90 These CSC markers were also upregulated in Wnt+ glioblastoma cells, indicating the involvement of Wnt/β‐catenin signaling in maintaining the stemness of glioblastomas. 91 The nuclear translocation of β‐catenin also promoted the dedifferentiation of colorectal cancer (CRC) cells, which is a novel index for stemness. 92 , 93 Conversely, ablation of the β‐catenin gene resulted in the loss of CSC populations and complete tumor regression of squamous cell carcinoma. 94
In CRC, both microsatellite instable and microsatellite stable cells demonstrate activated Wnt cascades, especially at the intestinal crypts. 95 This Wnt signal activation is mainly attributed to the functional loss of a negative regulator, APC. 95 Another mechanism for Wnt signal activation is the function loss of RNF43 caused by gene mutations, which delays the removal of Wnt receptors in the intestinal crypt. 96 Though 5‐fluorouracil (5‐FU), the first‐line treatment for CRC, is able to inhibit tumor growth, a recent study revealed that 5‐FU might induce CSC activation via the WNT/β‐catenin signaling pathway and thus cause chemoresistance in CRC patients. 97 Consistent with this finding, Wnt/β‐catenin pathway activation induced by m6A modification‐Sec62‐β‐catenin promotes stemness and chemoresistance of CRC. 98
The activated Wnt/β‐catenin pathway is enriched in more than half of breast cancers and indicates a poor prognosis. 99 The expression of the activated β‐catenin protein is also upregulated in breast CSCs. The inhibition of β‐catenin signaling significantly prevented tumor formation and metastasis in HER2‐overexpressing breast cancer cells. 100
Other cancer types of c regulated by WNT signaling include renal cancer, where the proliferation and self‐renewal of CSCs were significantly impaired by WNT inhibition, 101 lung adenocarcinoma, and were activated by Wnt/β‐catenin and Notch signaling. Hypoxia‐inducible factor‐1ɑ (HIF‐1ɑ)‐regulated miR‐1275 maintains stem cell‐like phenotypes and promotes the progression of LUAD simultaneously. 102 These results collectively suggest the essential role of β‐catenin in sustaining CSC phenotypes.
3.1.2. Notch signaling
Notch signaling is a genetically conserved pathway involved in the embryonic development of the central nervous system, heart, and multiple other organs. 103 , 104 Notch signal is also important to the initiation and progression of cancer. In mammalian cells, four Notch transmembrane receptors (Notch 1–4) and five membrane‐bound cell surface ligands (JAG 1 and 2, DLL 1, 3, and 4) have been reported. 105 Following the ligand‐receptor interaction, the active fragment of Notch receptors, the Notch intracellular domain (NICD), is released via proteolysis. Upon activation, the NICD‐CSL complex was formed following the nuclear translocation of NICD, which then recruits MAML and p300 coactivators, activating the downstream genes of Notch signals Hes‐1, c‐Myc, HER2, NF‐κB, cyclin‐D1, and p21. 106
Hyperactivation of Notch signaling is significantly correlated with the maintenance of CSC characteristics in various cancer types, including breast, 107 , 108 colon, 109 pancreatic, 110 hepatic, 111 cervical, 112 and ovarian cancer. 113 This theory was first established in medulloblastomas, where the tumor‐initiating ability of the CD133+ CSC population largely relied on Notch signal activation. 114 Likewise, the inhibition of Notch2 activation dramatically reduced the number of CSCs, whereas the better‐differentiated cells remained unaffected. 115
In hepatocellular carcinoma (HCC), higher expression of TACE/ADAM17 and Notch1 was found to predict a worse prognosis. 111 Besides, inducible nitric oxide synthase enhances the TACE/ADAM17‐mediated Notch1 signaling, leading to the enrichment of CD24+CD133+ liver CSCs. 111 In breast and pancreatic cancer, there was an increase in the expression levels of JAG1, JAG2, Notch1, Notch3, and Hes1, a downstream gene of Notch signaling. 116 , 117 The hypoxic microenvironment in breast cancer upregulated the expression of HIF‐2α, which then stimulated Notch signaling molecules NICD and promoted stem cell phenotypes, thereby facilitating chemoresistance of breast cancer cells. 107 The hypoxic environment also upregulated Notch1 signaling in glioblastoma and increased its stem cell marker CD133 on the cell surface. 118 Moreover, the heterogeneous metabolic signature of glioblastoma stem cells was also modulated by Notch signaling. 119 Similar findings were obtained from the analyses of CSC populations in pancreatic cancer 120 and myeloid leukemias, 121 which displayed Notch‐mediated chemoresistance. Thus, Notch signaling plays a critical role in promoting therapy‐resistant CSC populations across malignancies. Owing to the significant impact of Notch signaling in CSCs leading to tumor initiation and therapy‐resistance, targeting Notch pathway molecules may be a promising strategy in the wide spectrum of cancers.
3.1.3. JAK/STAT signaling
The JAK/STAT signaling pathway comprises various types of ligands, including interleukins, interferons, and hormones, and their corresponding receptors. 122 Upon the binding of ligands to receptors, JAK proteins (JAK1‐3 and TYK2) are activated via phosphorylation, which then phosphorylates the cytoplasmic domain of receptors, recruiting the STAT family proteins. 123 The dimerization and translocation of STATs lead to transcription regulation of downstream target genes.
Similar to Notch and Wnt signaling, JAK/STAT axis is also evolutionarily conserved and facilitates hematopoiesis, neurogenesis, and self‐renewal of normal embryonic stem cells. CSCs from hematological malignancies, such as AML, demonstrated aberrant activation of JAK/STAT signaling. 124 The tumor‐formation ability of AML CSCs was potently inhibited in immunodeficient mice following JAK1/2 inhibitor treatment, 125 reinforcing the promoting effect of JAK/STAT signaling on CSC stemness across a wide panel of cancers. 126
The role of the JAK/STAT pathway in CSC regulation is best characterized by its tumor‐initiating effect in glioblastoma. 127 In a glioblastoma model, the transforming growth factor (TGF)‐β‐activated JAK/STAT pathway induced the self‐renewal capacity and prevented the differentiation of glioma‐initiating cells derived from patient tumors, thereby facilitating tumor formation. 127 Besides, inhibition of STAT3 in CSCs reduced the tumorsphere formation and increased the expression of the neuronal differentiation genes. 128 Similar results were observed in breast CSCs, where STAT3 inhibitors decreased tumor growth and the abundance of CSCs. 129 Recently, JAK‐STAT signaling was reported to modulate stemness and chemoresistance of myxoid liposarcoma. 130 Likewise, JAK2/STAT3/CCND2 signals also control the radioresistance of CRC by regulating its stem cell persistence. 131
The role of STAT3 in determining cell fate is well established. 132 JAK proteins activate STAT3 via phosphorylation at Tyr705 residues. The downstream target genes of STAT3 nuclear translocation include cyclin D1, c‐Myc, and Bcl‐2. STAT3 holds profound importance in governing embryonic and adult stem cells in mice and humans. 133 Apart from its modulation of self‐renewal of both embryonic and CSCs as an element of JAK signaling, STAT3 interacts with Notch ligands DLL1 (Delta‐like 1) to facilitate neocortical development in infancy. 134 STAT3 also interacts with NF‐κB and HIF‐1α to enrich CD133+ cell populations. 135 The activated form of STAT3 (p‐STAT3‐Tyr705) was enriched in ALDH+ and CD44+/CD24+ CSCs, the inhibition of which subsequently reduced stem cell phenotypes of this CSC population. 136
3.1.4. HH signaling
The HH signaling pathway was identified by the Nobel prize winner team in 1980. 137 The HH pathway is critical to the development of multiple organs during embryogenesis. 138 Interestingly, HH signaling remains inactive in all postnatal tissues except central nervous system (CNS), skin, hair, and teeth. 139 The HH pathway is composed of three secreted ligand isoforms–Sonic hedgehog (Shh), Desert hedgehog, and Indian hedgehog, with their corresponding receptors being–Patched, Smoothened (SMO), and three Gli transcription factors (Glis1–3). 140
The aberrant activation of HH signaling has been reported to support the proliferation and stemness maintenance of CSC in various cancer types, such as multiple myeloma, glioma, HCC, and chronic myeloid leukemia (CML). 141 , 142 , 143 HH pathway activation is heterogeneous in multiple myeloma CSCs, with overexpression of the SMO gene and high Gli1 transcriptional activity. 144 The SMO gene encodes Smoothened protein, the chemical inhibition of which reduced stemness and proliferation of multiple myeloma CSCs. 144 In CML, the activation of HH signaling is with early disease progression. 145 The restoration of SMO expression in SMO‐deficient CML animal models promoted tumor growth and led to a four‐fold increase in CSC proportions. 146 Likewise, the overexpression of the HH signaling genes Gli1, SHH, and PATCHED1 was also present in glioma CSCs. 147 HH signaling supports glioma tumor growth in animal models by inducing SMO‐expressing gliomasphere formation. 147 A recent study showed that GLI1 inhibition reduced mammosphere formation of breast cancer cells. Interestingly, GLI1 inhibition resulted in a decrease in expression of YAP1, a Hippo pathway effector, suggesting a regulation activity of HH signaling on the Hippo pathway. 148
The development of treatment resistance to cancer requires the functional support of CSC‐related HH signaling. GLI‐1 regulates oncogenesis and therapeutic resistance of colon rectal cancer, 149 with significantly higher GLI‐1 expression observed in 5‐FU resistant CRC cells than in nonresistant cells. 150 , 151 , 152 The expression of stem cell markers of CRC cells was significantly decreased by GLI‐1 inhibition, and the cell response to 5‐FU, Irinotecan, and Oxaliplatin was also resumed. 153 In gastric adenocarcinoma, the forkhead box C1 (FOXC1) gene mediates the CSC phenotypes and tumor response to chemotherapy by regulating HH signaling. 154
3.1.5. TGF β/SMAD signaling
TGF‐βis a bifunctional regulator in cancer that represents a differentiation signal that potentially inhibits tumor initiation at an early stage. 155 , 156 , 157 In contrast to tumor initiation, TGF‐β promotes the CSC‐like phenotypes of cancer cells by inducing EMT. 158 TGF‐β binds to TGF‐β type I receptor kinase (ALK5) and triggers the Smad‐dependent canonical TGF‐β pathway. 159 TGF‐β–ALK5 activates Smad2/3 via phosphorylation which then forms a complex with Smad4 and regulates gene transcription following nucleus translocation. 160 The crosstalk between TGF‐β and the bioactive lipid mediator sphingosine‐1‐phosphate, a regulator of CSC expansion, is essential for cancer migration and the proliferation of breast CSCs. 161 , 162 , 163
The TGF‐β‐SMAD signaling is implicated in the regulation of CSC‐like properties of CD44+ gastric cancer cells, 164 HCC cells, 165 and cervical cancer cells. 166 In addition, CD44+ breast cancer cells, which are referred to as breast cancer CSCs, are frequently accompanied by activated TGF‐β signaling. 167 TGF‐β induced the expression of EMT‐associated genes Snail and Twist in breast cancer CSCs. 168 A recent study identified the role of TGF‐β‐SMAD signaling in maintaining CSCs in the bone microenvironment. 169
TGF‐β is able to switch non‐CSCs into CSC states via activating ZEB1 and Snail. 170 , 171 , 172 The response of CSCs to chemotherapy is associated with TGF‐β/Smad pathway activation, the suppression of which sensitizes CSCs to chemotherapy. 173 According to a previous study, TGF‐β‐induced chemoresistance is a downstream reaction of the Hh signaling, suggesting the interaction of TGF‐β with Hh signaling. 174 In addition, TGF‐β also demonstrated crosstalk with Notch signaling, with the synergistic promoting effect of TGF‐β and Notch1 on CSC proliferation. 175
The hypoxic microenvironment is a positive regulator of TGF‐β activities in CSC stemness and chemoresistance. 176 HIF‐1αinduces the expression and activation of TGF‐βand COX‐2, thereby promoting CSC enrichment. 177 The positive feedback loop between Snail and TGF‐β is also regulated by hypoxia, which together promotes EMT and recruits CSCs. 178
3.1.6. NF‐κB signaling
There are five members in the NF‐κB protein family: p65 (RelA), RelB, c‐Rel, NF‐κB1 (p105/p50), and NF‐κB2 (p100/p52). 179 , 180 NF‐κB family proteins are present in the cytoplasm of both differentiated cells and stem cells. 181 At the inactive state, NF‐κB are bound to inhibitory IκB proteins, which prevents its nuclear localization. 182 Upon activation by various stimuli, such as lipopolysaccharide, the IkB kinase (IKK) complex (IKKα, IKKβ, and IKKγ) phosphorylates IκB proteins, resulting in their degradation. 183 NF‐κB then translocates into the nucleus and activates the transcription of target genes involved in multiple biological processes.
Abnormal activation of NF‐κB signaling is implicated in the progression of various cancers. The role of the NF‐κB pathway in regulating CSCs was first identified in AML, where the primitive AML cells aberrantly expressing NF‐κB were referred to as potential leukemic stem cells. 184 NF‐κB pathway participates in the viability and self‐renewal of AML stem‐like cells. 185 Since then, growing evidence has shown elevated or constitutive NF‐κB activity in other cancer types. For instance, increased expression of total p65 and downregulation of IκBα expression were found in prostate CSCs. 186 In addition, the CD44+ fraction of ovarian cancer cells displayed higher expression of major stemness genes and NF‐κB signal genes, including RelA, RelB, and IKKα. 187 The loss of the APC gene represents a canonical alteration during tumorigenesis, which promotes the activation of NF‐κB signaling, allowing the expansion of Lgr5+ CSCs. 188
NF‐κB activation mediates the tumorigenesis of glioma. 189 Both adherent and spheroid glioma CSCs exhibited constitutive activation of the STAT3/NF‐κB signaling. 190 Gliomasphere‐forming cells showed increased phosphorylation of p65 and sustained oncogenic activation of NF‐κB signaling. 191 In Her2‐driven breast cancer models, the inactivation of NF‐κB pathways by IκBα‐SR decreased the tumorigenesis of luminal epithelial tumors. 192 A genome‐wide expression analysis suggested that IκBαSR impaired stem cell expansion in breast cancer and CSC markers in transgenic tumors. 193 IKKα activity is required for Her2‐induced oncogenesis, providing self‐renewal signals that maintain mammary tumor‐initiating cells. 194 The underlying mechanism may be the phosphorylation of p27 by IKKα leading to its nuclear export in Her2 breast cancer cells. 195 The expression of Dll1, a Notch ligand, promotes the tumor‐initiating abilities of breast cancer cells. It has been recently reported that NF‐κB activation is a downstream target of Dll1, which collectively contributes to a chemoresistant phenotype of breast cancer CSCs. 196
3.1.7. PI3K/AKT/mammalian target of rapamycin signaling
PI3K is an intracellular phosphatidylinositol kinase composed of a regulatory subunit p85 and a catalytic subunit p110. 197 , 198 AKT is a downstream effector of PI3K and has three isoforms: AKT1, AKT2, and AKT3. 199 As a key member of the PI3K‐associated kinase protein family, mammalian target of rapamycin (mTOR) functions as a nutritional signal sensor and a regulating factor for cell proliferation. 200 There are two protein complexes formed by mTOR. The mTORC1 is composed of mTOR, raptor, mLST8, and two negative regulators, PRAS40 and DEPTOR, 199 , 201 which controls cell growth in response to metabolism and nutrition signals. 202 , 203 The mTORC2 (mTOR complex 2) consists of mTOR, Rictor, mSin1, and mLST8. It is well established that mTORC2 activates Akt via phosphorylation at serine residue 473 and modulates stem‐like properties, 204 whereas mTORC1 and its downstream cascades directly correlate with stem‐like properties. 205
The activation of the PI3K/Akt/mTOR pathway is important to cancer cell growth and its therapeutic resistance. 206 The PI3K/Akt/mTOR pathway can be activated through multiple mechanisms, such as the insulin‐like growth factor (IGF)/IGFR, ErbB, and fibroblast growth factor (FGF)/FGFR signaling. 206 PTEN is known for its negative regulation on PI3K/AKT cascades. Data from several cancer models showed that PTEN depletion resulted in CSC expansion and increased tumor growth in mice. 207 , 208 Importantly, PI3K/AKT/mTOR is critical to maintaining the CSC population in various cancers, including nasopharyngeal carcinoma, 209 glioma, 210 pancreatic cancer, 211 lung cancer, 212 prostate, 213 and breast cancer. 214
In breast cancer, PI3K/Akt/mTOR pathway is required for the colony‐formation capacity of tumor cells and the maintenance of stem‐like properties. 214 One underlying mechanism may be the HIF‐2α‐induced CD44 alteration that promotes CSC activation in triple‐negative breast cancer (TNBC) via PI3K/AKT/mTOR pathway. 215 The transcriptional suppression of negative regulators of mTOR is intrinsic in luminal‐like breast cancer cells, leading to the development of CSC‐like properties. 216 Likewise, the inhibition of mTOR in CRC cells suppressed cell stemness represented by decreased ALDH1 activity. 37 , 217 PI3K/AKT/mTOR signaling pathway also enhances the angiogenesis of CRC and recruitment of tumor‐associated macrophages (TAMs). 218
The chemoresistance of hepatoma is also related to the Akt/mTOR signaling by promoting the expansion of hepatic tumor‐initiating cells. 219 The inhibition of the PI3K/Akt/mTOR pathway overcame the chemoresistance of ovarian cancer by decreasing CSC marker expression. 220 The radioresistance of prostate cancer is significantly associated with PI3K/Akt/mTOR signaling activation via maintaining CSC phenotypes. 221 Moreover, prostate cancer CSCs present a feedback inhibition on AKT signaling through HIF1α, which impairs CSC metabolism and growth. 222
It was previously suggested that CD133 expression was upregulated by mTOR signaling in gastrointestinal cancer. 223 Similar results were obtained from hepatic cancer cells, where mTOR promotes the conversion of CD133‐ to CD133+ cells. 224 Moreover, aberrant activation of the PI3K/Akt/mTOR pathway facilitates the stemness maintenance of NSCLC (non small cell lung cancer) cells by upregulating chemokine (C‐X‐C motif) receptor 4 (CXCR4) and the subsequent CXCR4‐stimulated STAT3 signaling. 225
On the other hand, in gliomas, Akt but not mTOR regulates ATP binding cassette transporters (ABCG2) activity, which is referred to as stemness hallmark. 226 Besides, PI3K inhibition restored the sensitivity to nilotinib of CML stem cells, whereas mTOR inhibition demonstrated no effect on CML. 227
3.1.8. Peroxisome‐proliferator‐activated receptor signaling
The peroxisome‐proliferator‐activated receptor (PPAR) pathway is activated following the binding of the G protein‐coupled receptor with its ligand, stimulating a cascade of signal transducers, such as adenylyl cyclase, cyclic adenosine monophosphate, and protein kinase A, which then induces the translocation of PPAR, a nuclear receptor protein that regulates the target gene expression. 228 , 229 , 230 PPARα, PPARδ, and PPARγ are the three subtypes of PPAR with respective functions. In the context of a tumor, PPARs are involved in the modulation of cell proliferation, apoptosis, and survival of multiple cancers, including prostate cancer, breast cancer, glioblastoma, neuroblastoma, pancreatic cancer, hepatic cancer, leukemia, bladder cancer, and thyroid tumors, with either promoting or inhibitory effects on cancer development. 231 PPARs have also been reported to regulate the EMT process and stem cell‐like properties of CSCs. 232
CPT1A (Carnitine palmitoyl transferase I) and CPT2 (Carnitine palmitoyl transferase II) are two known target genes of PPARα, which increase fatty acid oxidation (FAO) required for the cell metabolism in radioresistant breast cancer cells and radiation‐derived breast CSCs. 233 SCD1 (stearoyl‐CoA desaturase 1) is another functional downstream molecule of PPARα, and the activation of the PPARα‐SCD1 axis is important to the maintenance of CSCs of HCC. 234 PPARα, on the other hand, is considered a downstream molecule of lipid droplet‐derived signaling, which was highly abundant in pancreatic and colorectal CSCs than non‐CSCs. The inhibition of PPARα decreases stemness characteristics of pancreatic and colorectal CSCs. 235 Likewise, PPARδ is involved in the maintenance of HSCs by regulating the FAO pathway. The inhibition of PPAR‐δ or mitochondrial FAO reduced the stemness of HSCs, whereas PPAR‐δ agonists enhanced HSC maintenance. 236
On the contrary, PPARγ is considered a tumor suppressor that reduces the CD49high/CD24+ mesenchymal stem cells (MSCs) and inhibits tumor angiogenesis of breast cancer. 237 The existence of quiescent LSCs may contribute to treatment failure in CML patients. PPARγ agonist glitazones decrease the expression of STAT5 and its downstream targets HIF2α and CITED2, two key genes maintaining the quiescence and stemness of CML LSCs. 238 PPARγ activation decreases the stem cell‐like characteristics of bladder CSCs and accelerates the differentiation of adipocytes. 239 PPARγ also induces the differentiation in osteosarcoma stem cells and melanoma cells by suppressing the transcriptional activity of YAP. 240 , 241 Notably, PTEN is a target gene of PPARγ activation, which in turn blocks the PI3K/Akt/mTOR pathway and prevents the self‐renewal, tumorigenicity, and metastasis of cervical, hepatic, and glioblastoma CSCs. 242 , 243
3.2. Extrinsic signaling pathways that regulate CSCs
The “seed and soil” theory was first brought up in the 19th century, which describes the metastasis of tumor cells to sites with a favorable microenvironment. 244 In this theory, the “seed” refers to the metastatic tumor cells, and “fertile soil” refers to the sites with a microenvironment that favors tumor colonization and growth. In accordance with the “seed and soil” theory, it is widely accepted that CSCs dwell in such “soil,” a specific tumor microenvironment (TME) composed of stroma, immune cells, microvessels, and external regulating signals. 245 This system provides CSCs with a conductive environment via the action of paracrine factors or direct contact with immune cells. 8 , 246
3.2.1. Vascular microenvironments that regulate CSCs
The theory of tumor vascular microenvironment dates back to the 1940s when glioblastoma cells were found to grow into the blood vessels‐enriched sites. 247 The multilineage differentiation capacity of CSCs may allow them to take part in tumor angiogenesis or forming the vascular mimicry (VM) in the TME. Recent research has shed light on the relationship between CSCs and the vascular microenvironment. A typical example is glioblastoma CSC, where the expression of surface marker Nestin is positively related to microvessel density. 248 The vascular endothelium of glioblastoma has similar genomic alterations to CSCs. 21 Neural stem cells cocultured with epithelial cells demonstrated increased self‐renewal and impaired differentiation ability via paracrine signaling, including the Notch pathway and the chemokine axis CXCL12/CXCR4. 249 , 250 , 251
Recently, the CSC surface marker CD44 was reported to promote the VM generation in oral squamous cell carcinoma. 252 The CD44/c‐Met signaling has also been identified as the key regulator for VM in Ewing sarcoma and breast cancers. 253 The presence of VM is also associated with ALDH1 expression in breast cancer. 254 , 255 ALDH+ TNBC cells isolated from FACS initiated VM on matrigel. 256 The increased expression level of VM‐related genes, such as MMP‐2 and MMP‐9, was observed in CD133+ breast cancer cells. 257
The regulation of CSC phenotype by edothelial cells (ECs) can be based on the secretion of soluble factors by ECs. 248 In acute leukemia, bone marrow stromal cells derived from CD133+/CD34+ stem cells secrete IGF‐1, leading to the formation of capillary‐like structures. 258 Shh is a soluble factor secreted by ECs, which enhances CSC properties and stimulates the Hh signaling. 259 , 260 , 261 Hh signaling facilitates the acquisition of CSC self‐renewal in thyroid cancer, via regulating Snail expression. 260 Interestingly, CD133+ GSCs were identified in areas surrounding Shh‐expressing ECs. The depletion of Shh in ECs prevents of promoting effect of ECs on CSC‐like phenotype maintenance. 259
In addition to HH signaling, Notch signal cascades also take an active part in the EC‐mediated regulation of CSCs. 262 , 263 , 264 , 265 In CRCs, the promotion of CSC phenotypes by ECs is dependent on Notch signaling and independent of Shh or Wnt signaling. The knockout of Jagged‐1, a Notch signaling element, in EC impairs its angiocrine effect. 263 The nitric oxide derived from ECs is able to trigger Notch signaling, leading to increased stemness of GSCs and glioma initiation in mice. 264 Furthermore, in SHH‐driven medulloblastomas, the EC‐induced promotion of CSC characteristics additionally requires PI3K/AKT/mTOR signals. 266
3.2.2. Hypoxic microenvironments that regulate CSCs
Hypoxia is a common hallmark of TME in solid tumors. 267 , 268 In solid tumors, the rapidly proliferating tumor cells require a high level of oxygen to meet the expanding demands, resulting in relative hypoxia. 269 , 270 In this sense, an extreme hypoxic environment appears as the natural selection for tumor cells, where aggressive CSCs are more likely to survive and proliferate. 271 , 272 It is thus not surprising that CSCs are more resistant to conventional cancer therapies. 273
Hypoxia leads to the acquisition of CSC phenotypes in breast tumors, which is primarily mediated by HIFs. 274 HIFs (HIF‐1, HIF‐2, and HIF‐3) are key sensors of intracellular oxygen alterations and modulate the transcription of multiple genes at low oxygen levels. 275 , 276 , 277 The Notch signaling is a key regulating pathway for hypoxia response, which can be activated by HIF‐1α and HIF‐2α for the maintenance of CSC stemness. 278 , 279 For instance, the HIF‐2α‐mediated Notch pathway activation promotes the phenotypic transformation of breast cancer cells into breast CSCs and cell resistance to paclitaxel treatment. 107 Likewise, hypoxia‐induced AKT activation contributes to gemcitabine‐induced stemness of pancreatic cancer cells by enhancing downstream Notch1 activity. 280 Another downstream element of PI3K/AKT signaling, mTOR, regulates HIF‐1α activity via phosphorylation of p70 S6Kinase (S6K). 281 However, the inactive state of mTOR also facilitates the maintenance of CSC characteristics, which explains the suboptimal efficacy of mTOR inhibitors in clinical evaluation. 222 The agonist of PTEN, a negative regulator of the PI3K/AKT pathway, provides a new clue to inhibit HIF‐1α activities and thus reduce CSC stemness. 282
Fibroblasts are major components of tumor stroma, which are able to produce various extracellular matrix proteins and growth factors, such as TGF‐β. 283 Hypoxia induces the upregulation in TGF‐β3 expression by promoting the binding of HIF‐1 to the TGF‐β3 gene promoter. 284 It has also been reported that hypoxia increased TGF‐β1 expression in MSCs. 285 The hypoxia‐induced secretion of TGF‐β1 by MSCs in turn enhances tumor progression, 286 potentially by promoting the stabilization of HIFs. 8 Thus, the concomitant inhibition of HIF‐1α and TGF‐β delays tumor initiation and blocks the activity of CSCs. 287
Fibroblasts‐directed CSC reprogramming includes the stimulation of COX‐2 and nuclear factor of κB (NF‐κB). 8 On one hand, hypoxia‐mediated downregulation of dual specificity phosphatase 2 (DUSP2) upregulates COX‐2, leading to increased cancer stemness. 288 On the other hand, HIF‐1α induces COX‐2 expression, which in turn upregulates HIF‐2α expression and enhances treatment resistance of cancer cells. 289
3.2.3. Immune cells that regulate CSCs
As immune evasion and CSCs both substantially contribute to tumor progression, it is widely accepted that there is potential crosstalk between CSCs and immune cells in the TME. A significantly high stemness signature was identified in 21 solid malignancies with a poor immunogenic response. 290 It is thus of paramount importance to elucidate the CSC‐immune cell interactions in cancer, which will facilitate the identification of immunotherapies to eliminate tumor‐promoting CSCs. Figure 2 presents the crosstalk between CSCs and immune cells in the CSC niche, which regulates CSC stemness.
FIGURE 2.

The crosstalk between cancer stem cells (CSCs) and immune cells in the CSC niche via soluble mediators or juxtacrine signals, which regulate CSC stemness. (Figures built with biorender.com)
3.2.3.1. Tumor‐associated macrophage
The critical role of CSCs in monocyte recruitment to tumor sites has been well established as various protumorigenic macrophage factors were increased in supernatant collected from CSC sphere culture, including IL‐13, TGF‐β, and WNT‐induced signaling protein 1. 291 , 292 , 293 Incubation of macrophages with such sphere culture leads to macrophage polarization toward an immunosuppressive phenotype. 294 , 295 , 296
On the other hand, TAMs in turn influence CSC phenotypes by secreting soluble mediators, such as IL‐6, TGF‐β, and WNT ligands, or through juxtacrine signaling. 297 , 298 The direct interactions of CSCs with TAMs activates NF‐κB in CSCs, which stimulates the secretion of cytokines to sustain the stem cell state of breast CSCs. 299 In the pleiotrophin (PTN)‐PTPRZ1 paracrine signaling, which supports glioma progression, PTN released by TAMs binds to its receptor PTPRZ1 on GSCs, suggesting the significance of TAMs as important components of the CSC niche. 300
IL‐6 produced by TAMs promotes the expansion of hepatic CSCs, and the inhibition of IL‐6 with tocilizumab prevents TAM‐stimulated generation of CD44+ cells. 301 In breast cancer, TAM‐produced IL‐6 induces and maintains the CSC characteristics through STAT3. 302 In addition, STAT3 is a transcription factor that could also modulate CSC maintenance in an IL‐6‐independent manner. For instance, the self‐renewal and tumorigenicity of bladder CSCs are regulated by the KMT1A‐GATA3‐STAT3 circuit, which is independent of IL‐6. 303 The STAT3 blockade decreased the expression of PD‐L1 on CD44+ cells in squamous cell carcinoma of the head and neck, a well‐characterized cell population with CSC characteristics, resuming T‐cell‐mediated immunity. 304 These results further justify the development of IL‐6‐ or STAT3‐targeting strategies in cancer treatment.
3.2.3.2. Natural killer cell
Though CSCs were previously believed as less immunogenic than non‐CSCs due to decreased MHC class‐I (MHC I) expression, 305 , 306 growing evidence now suggests that CSCs are preferentially susceptible to natural killer (NK) cell activities. 307 , 308 This vulnerability may be attributed to the activated natural cytotoxicity receptors, particularly NKp30 and NKp44. Though glioblastoma CSCs express deficient MHC I molecules, various ligands that activate NK cell receptors were found on these CSCs, such as PVR and Nectin‐2. 308 Interestingly, CSCs were resistant to NK cells freshly isolated from tumor specimen, but were sensitive to the activities of both allogeneic and autologous IL‐2 or IL‐15‐activated NK cells. 308 In melanoma, both CD133‐ and CD133+ subpopulations are susceptible to the cytotoxicity of IL‐2‐activated allogeneic NK cells. 309 Likewise, the increased sensitivity of breast CSCs to IL‐2 or IL‐15‐activated NK cells, which is potentially mediated by the upregulation of NKG2D ligands ULBP1, ULBP2, and MICA on CD44+CD24− breast CSCs. 310 Similar results were observed in ovarian cancer 311 and CCR7+ melanoma. 312
Notably, an increased frequency of CSCs is often observed following cytotoxic treatments for primary cancers. 313 , 314 A study reported a novel mechanism for the immune escape of breast CSCs from NK cell attack, due to decreased expression of ligands that stimulate NKG2D. 315 The upregulation of the NKG2D stress ligands MICA/B on surviving CSCs following cytotoxic treatments, such as radiotherapy, sensitizes CSCs to NK cell killing. NK cells were recruited to the tumor‐adjacent areas but lost their cytotoxic efficacy in breast tumors due to the altered ligand expression ligands on radioresistant breast CSCs. 316 This evidence further provides a rationale for combining the NK cell‐stimulating factors with conventional therapies.
3.2.3.3. Cancer‐associated fibroblasts and MSCs
The oncogenic effect of cancer‐associated fibroblasts (CAFs) is mostly based on their secretion of a number of paracrine factors, including proinflammatory cytokines, chemokines, prostaglandins, growth factors, and proteases, which collectively promote tumor growth, angiogenesis, and invasion. 317 , 318 , 319 CAFs are also believed to create an immunosuppressive TME by potentiating regulatory T cells, 320 or induce M2‑polarized macrophages. 321 Moreover, CAFs‐derived exosomes lead to treatment resistance of cancer. 322 , 323
Notably, one of the key mechanisms for the CAFs‐mediated tumor promotion is based on their regulation of CSC stemness. 324 The paracrine factors produced by specific CAF subpopulations accelerate the transformation of cancer cells into CSCs and help maintain the stemness properties of existing CSCs. 325 Under the cell stimuli, such as chemotherapy, CAFs acquire a senescence‐like secretory phenotype, and their secretion of prostemness chemokines is further increased, resulting in CSC‐associated chemoresistance. 326
Both resident and recruited MSCs within TME can acquire CAF‐like phenotypes, suggesting that CAFs can be derived from MSC transformation. 327 In pancreatic ductal adenocarcinoma (PDAC) and gastric cancer models, bone marrow‐derived MSCs are recruited to TME in a TGF‐β and CXCL‐12‐dependent manner and differentiate into CAFs. 328 This transformation may be attributed to tumor‐secreted factors, such as the TGF‐β, which activate MSCs into CAFs, further enhancing the cell heterogenicity of the CSC microenvironment. 329 MSCs are stromal cells with multipotent differentiation abilities and can migrate to tumor sites and promote tumor EMT via the secretion of various factors. 330 For example, in gastric cancer, MSCs secret VEGF, macrophage inflammatory protein‐2, TGF‐β1, and the proinflammatory cytokines interleukin IL‐6 and IL‐8, which collectively facilitate tumor growth and angiogenesis. 331
4. THERAPIES TARGETING SIGNALING PATHWAYS OF CSC‐s
Given that CSCs are a major contributing factor to progressive phenotypes of cancer, targeting CSCs in the tumor now appears as a promising strategy against cancer. Numerous efforts have been undertaken these years to identify such therapies, such as kinase inhibitors and antibodies that block CSC‐associated signaling pathway elements, and some of these approaches have already entered the clinical phase. 332 , 333 The ongoing and completed clinical trials on therapies targeting signaling pathways of CSCs are presented in Table 2. Immunotherapies targeting CSCs include MHC‐restricted killing, such as checkpoint inhibitors, and MHC‐unrestricted killing, such as the chimeric antigen receptor (CAR) T‐cell approach. 334 , 335
TABLE 2.
Ongoing and completed clinical trials on therapies targeting signaling pathways of CSCs
| Agents (targets) | Condition | Cotherapy | Phase | NCT number |
|---|---|---|---|---|
| Gamma‐secretase inhibitors (GSIs) | ||||
| RO4929097 (Notch, Aβ40, secretase) | Metastatic pancreas cancer | II | NCT01232829 | |
| Advanced solid tumors | I | NCT01145456 | ||
| Advanced solid tumors | Cediranib maleate | I | NCT01131234 | |
| Advanced solid tumors | I | NCT01096355 | ||
| Refractory NSCLC | II | NCT01070927 | ||
| Metastatic epithelial ovarian cancer, fallopian tube cancer, or primary peritoneal cancer | II | NCT01175343 | ||
| Advanced sarcoma | I/II | NCT01154452 | ||
| Advanced solid tumors | Capecitabine | I | NCT01158274 | |
| Advanced renal cell carcinoma after VEGF/VEGFR therapy failure | II | NCT01141569 | ||
| Advanced solid tumors | I | NCT0121862 | ||
| Advanced solid tumors | Temsirolimus | I | NCT01198184 | |
| Malignant glioma | Temozolomide and radiation therapy | I | NCT01119599 | |
| Metastatic melanoma | Cisplatin, vinblastine, and temozolomide | I/II | NCT01196416 | |
| Metastatic colorectal cancer | II | NCT01116687 | ||
| PF‐03084014 (secretase) | Desmoid tumors | II | NCT01981551 | |
| Advanced cancer and leukemia | I | NCT00878189 | ||
| MK‐0752 (secretase) | Advanced breast cancer | Docetaxel | I/II | NCT00645333 |
| Early‐stage breast cancer | Tamoxifen/letrozole | IV | NCT00756717 | |
| Pancreatic cancer | Gemcitabine hydrochloride | I | NCT01098344 | |
| Advanced cancer | Ridaforolimus | I | NCT01295632 | |
| Advanced breast cancer | I | NCT00106145 | ||
| Pan‐Notch small molecule inhibitor | ||||
| BMS‐906024 (γ‐Secretase and Notch) | Advanced solid tumors | I | NCT01292655 | |
| Advanced solid tumors | Chemotherapy | I | NCT01653470 | |
| Acute T‐cell lymphoblastic leukemia or T‐cell lymphoblastic lymphoma | I | NCT01363817 | ||
| CB‐103 (Notch) | Luminal advanced breast cancer | Nonsteroidal aromatase inhibitor | II | NCT04714619 |
| Advanced solid tumors and hematological malignancies | I/II | NCT03422679 | ||
| Monoclonal antibodies (mAbs) targeting Notch | ||||
| MEDI0639 (Dll4) | Advanced solid tumors | I | NCT01577745 | |
| SIBP‐03 (HER3) | Advanced solid tumors | I | NCT05203601 | |
| OMP‐52M51 (Notch1) | Metastatic colorectal cancer | I | NCT03031691 | |
| Advanced solid tumors | I | NCT01778439 | ||
| Refractory lymphoid malignancies | I | NCT01703572 | ||
| Adenoid cystic carcinoma | NA | NCT02662608 | ||
| OMP‐59R5 (Notch 2/3) | Stage IV pancreatic cancer | Nab‐paclitaxel and gemcitabine | I/II | NCT01647828 |
| Advanced solid tumors | I | NCT01277146 | ||
| SMO inhibitor | ||||
| BMS‐833923 (SMO) | Chronic myeloid leukemia | I/II | NCT01218477 | |
| LEQ506 (SMO) | Advanced solid tumors | I | NCT01106508 | |
| Vismodegib (SMO) | Prostate cancer | I | NCT02115828 | |
| Metastatic colorectal cancer | Chemotherapy | II | NCT00636610 | |
| Ovarian cancer | II | NCT00739661 | ||
| Keratocystic odontogenic tumor | II | NCT02366312 | ||
| Advanced pancreatic cancer | Gemcitabine hydrochloride | I/II | NCT01064622 | |
| II | NCT01195415, NCT01088815 | |||
| Advanced Solid Tumors | I | NCT01546519, NCT03878524, NCT00878163, NCT00607724, NCT01209143, NCT00968981, NCT01537107 | ||
| II | NCT05159245, NCT05238831, NCT05159245, NCT02091141, NCT00959647 | |||
| Basal cell carcinoma | I | NCT01631331, NCT02639117, NCT03158389 | ||
| II | NCT01543581, NCT03035188, NCT00833417, NCT01700049, NCT02667574, NCT01201915, NCT02371967, NCT01367665, NCT01556009, NCT01815840, NCT04416516 | |||
| IV | NCT03610022, NCT02436408 | |||
| Head/neck basal cell carcinoma | Radiation therapy | II | NCT01835626 | |
| Advanced gastric adenocarcinoma | II | NCT03052478 | ||
| Advanced stomach cancer or gastroesophageal junction cancer | Chemotherapy | II | NCT00982592 | |
| Basal cell skin cancer | I/II | NCT02690948 | ||
| Small cell lung carcinoma | Cisplatin and etoposide | II | NCT00887159 | |
| Advanced sarcoma | I/II | NCT01154452 | ||
| Multiple myeloma | I | NCT01330173 | ||
| Recurrent glioblastoma | II | NCT00980343 | ||
| Advanced urothelial carcinoma | II | NCT02788201 | ||
| Advanced malignancies | II | NCT02465060 | ||
| Advanced chondrosarcomas | II | NCT01267955 | ||
| Refractory medulloblastoma | II | NCT01239316, NCT00939484, NCT01878617 | ||
| I | NCT00822458 | |||
| Progressive meningiomas | II | NCT02523014 | ||
| Sonidegib (SMO) | Basal cell carcinoma | NA | NCT01529450 | |
| II | NCT00961896, NCT03534947, NCT01327053, NCT04806646, NCT01350115, NCT00961896 | |||
| Advanced solid tumors | I | NCT00880308, NCT01208831, | ||
| Paclitaxel | I | NCT01954355 | ||
| BKM120 | I | NCT01576666 | ||
| Pembrolizumab | I | NCT04007744 | ||
| Myeloid leukemia | Nilotinib | I | NCT01456676 | |
| Prostate cancer | I | NCT02111187 | ||
| Triple‐negative (TN) advanced breast cancer (ABC) | Docetaxel | I | NCT02027376 | |
| Recurrent ovarian cancer | Paclitaxel | I | NCT02195973 | |
| Extensive stage small cell lung cancer (ES‐SCLC) | Etoposide and cisplatin | I | NCT01579929 | |
| Pancreatic cancer | Gemcitabine and nab paclitaxel | I/II | NCT02358161 | |
| Gemcitabine | I | NCT01487785 | ||
| Chemotherapy | I | NCT01485744 | ||
| Esophageal cancer | Everolimus | I | NCT02138929 | |
| Myeloid malignancies | Azacitidine | I | NCT02129101 | |
| Recurrent brain tumors | I | NCT03434262 | ||
| Multiple myeloma | II | NCT02086552 | ||
| Medulloblastoma | II | NCT01708174, NCT04402073 | ||
| Hepatocellular carcinoma | I | NCT02151864 | ||
| Acute leukemias | II | NCT01826214 | ||
| Chronic myelogenous leukemia | Nilotinib | I | NCT01456676 | |
| Glasdegib (SMO) | Acute myeloid leukemia, chronic myelomonocytic leukemia | Azacitidine | I | NCT02367456 |
| Acute myeloid leukemia | II | NCT01546038, NCT01841333, NCT03226418 | ||
| Soft tissue sarcoma | III | NCT03784014 | ||
| Glioblastoma | Temozolomide and radiotherapy | I/II | NCT03529448 | |
| Inhibitors of WNT pathway elements | ||||
| DKN‐01 (DKK1) | Advanced biliary tract cancer | Nivolumab | II | NCT04057365 |
| Prostate cancer | Docetaxel | I/II | NCT03837353 | |
| Multiple myeloma or advanced solid tumors | I | NCT01457417 | ||
| Epithelial endometrial or epithelial ovarian cancer | Paclitaxel | II | NCT03395080 | |
| Gastric or gastroesophageal cancer | Tislelizumab ± chemotherapy | II | NCT04363801 | |
| Paclitaxel or pembrolizumab | I | NCT02013154 | ||
| Advanced liver cancer | I/II | NCT03645980 | ||
| Cancer of hepatic biliary system or gallbladder | Gemcitabine + cisplatin | I | NCT02375880 | |
| Multiple myeloma | Lenalidomide/dexamethasone | I | NCT01711671 | |
| Vantictumab (FZD receptors) | Metastatic breast cancer | I | NCT01973309 | |
| Pancreatic cancer | Nab‐paclitaxel and gemcitabine | I | NCT02005315 | |
| NSCLC | Docetaxel | I | NCT01957007 | |
| Cirmtuzumab (ROR1) | Metastatic castration‐resistant prostate cancer | II | NCT05156905 | |
| Breast cancer | Cirmtuzumab + paclitaxel | I | NCT02776917 | |
| B‐cell lymphoid malignancies | Ibrutinib | I/II | NCT03088878 | |
| Refractory chronic lymphocytic leukemia | I | NCT02222688 | ||
| II | NCT04501939 | |||
Clinical trial data sources: clinicaltrials.gov.
4.1. Targeting Notch signaling
Tumors with NOTCH1 mutations represent a distinct tumor phenotype with increased activation in Notch1 signaling. NOTCH1‐mutant tumors are often associated with metastasis, poor prognosis, and potential responsiveness to brontictuzumab. 336
4.1.1. Gamma‐secretase inhibitors
A number of Notch‐pathway inhibitors have been developed with different action mechanisms, some of which are currently under clinical evaluation. Gamma‐secretase inhibitors (GSIs) have long been identified as a large family of Notch‐targeted small molecule inhibitors, by blocking the proteolytic cleavage of Notch receptors. GSI PF‐03084014 inhibited tumor growth in a mouse xenograft model of T‐cell acute lymphoblastic leukemia. 337 , 338 GSI MRK‐003 works synergistically with trastuzumab in HER2‐positive breast cancer mouse model. 339 Likewise in NSCLC models, BMS‐906024 has demonstrated potent antitumor efficacy in combination with chemotherapies, such as cisplatin, paclitaxel, docetaxel, and target therapies, such as crizotinib. 340 , 341
The ability of GSIs to block Notch signaling and subsequently reduce CSC burden in preclinical studies has spurred clinical assessment of GSIs in clinical trials. A well‐studied GSI RO4929097 substantially reduced the expression level of stem cell markers on primary melanoma cells and inhibited tumor formation in melanoma xenograft transplants. 342 In a phase II trial, RO4929097 was well tolerated in patients previously treated with PDA, with 25% of patients achieving stable disease. 343 In a phase I trial, four of 24 melanoma patients were reported with clinical benefits from RO4929097 treatments, with one patient achieving a complete response. 344 This encouraged the following phase II trial of RO4929097 in patients with metastatic melanoma with monotherapy. 345 Besides, the combinational treatment of GSIs with other cancer treatments further improved clinical outcomes. One such example is the combination of RO4929097 with bevacizumab in patients with malignant gliomas. 346 These clinical results suggest that GSIs can effectively cross the blood‐brain barrier and reach therapeutic concentrations at tumor sites. However, RO4929097 monotherapy displayed minimal inhibition of neurosphere formation in recurrent glioblastoma samples. 347 Similarly, in metastatic CRCs, the antitumor activity of GSIs, including RO4929097, 344 LY900009, 348 MK‐0752, 349 and BMS‐986115 350 as monotherapies, is suboptimal. 351 A recent phase Ib/II trial evaluated the treatment combination of RO4929097 with vismodegib, an HH inhibitor, in advanced sarcoma, providing a rationale for the synergy of GSIs in this patient population. 352
PF‐03084014 and MK‐0752 are two GSIs frequently used in clinical studies, both of which have been used to treat advanced‐stage solid tumors but failed to reach evident clinical efficacy in patients with lung cancer, breast cancer, or pancreatic cancer as monotherapies. 353 As such, these failures further encouraged combinatorial regimens of GSIs with other anticancer therapies, as evidenced by the fact that PF‐03084014 enhanced the antitumor effect of DOX in prostate cancer stem‐like cells. 354 Moreover, the concomitant use of RO4929097 and cediranib has prolonged disease stabilization in 11 out of 20 patients with advanced solid tumors. 355
The most common dose‐limiting toxicities (DLTs) of GSIs occur in the gastrointestinal system, with secretory diarrhea accounting for 30–60% of all reported DLTs in cancer patients and grade ≥3 diarrhea accounting for around 11%. This may be explained by the fact that inhibition of Notch1 and Notch2 prevents the proliferation of crypt progenitors leading to goblet‐cell metaplasia of the small‐intestinal epithelium. 356 The addiction to glucocorticoids and antiestrogens in the GSI treatment regimens significantly relieved GSI‐induced gastrointestinal toxicities. 338 , 357 Hypophosphatemia is another GSIs‐induced toxicity, which is potentially caused by abnormal gastrointestinal function and can be relieved by oral administration of phosphate replacement. 358
4.1.2. Pan‐Notch small molecule inhibitor
Though Notch1 is the most common activated oncogene in tumors, the coexpression of Notch1 and Notch4 is frequently observed in breast cancer. Moreover, accumulating evidence suggests the Notch3‐mediated progression of cancer. These results collectively reveal the requirement for pan‐Notch inhibition to achieve a broader spectrum of antitumor efficacy. A previous study evaluated the structure–activity relationships in a series of (2‐oxo‐1,4‐benzodiazepin‐3‐yl)‐succinamides as pan‐Notch inhibitors. 359 Among these GSIs, MS‐906024 displayed the broadest spectrum efficacy in multiple in‐vivo tumor models and thus advanced into clinical trials (NCT01292655). 360 BMS‐906024 sensitizes NSCLC to paclitaxel treatment, and patients with wild‐type KRAS and BRAF tumors may have improved response to the BMS‐906024 + paclitaxel combination. 340 It was later reported that BMS‐906024 significantly enhanced the delay in NSCLC tumor spheroid growth delay caused by etoposide and crizotinib, and the most prominent delay in spheroid growth was observed in cells treated with BMS‐906024 + chemoradiation triple combination. 341
CB‐103 is an oral pan‐Notch inhibitor that specifically targets protein–protein interaction by suppressing the Notch transcriptional complex. 361 Another small molecule inhibitor, IMR‐1, inhibits the recruitment of MAML1 to the notch transcriptional complex, thereby preventing its activation. 362 This approach has the advantage of acting downstream of aberrant Notch receptor activation by blocking the assembly of the transcription complex and thereby inhibiting the expression of Notch target genes. A phase I/IIa study is under way to investigate the safety and efficacy of CB‐103 in patients with advanced solid tumors and hematological malignancies (NCT03422679). In a phase II trial, patients with advanced breast cancer will receive the combinational treatment of CB‐103 with NSAI therapy (letrozole or anastrozole, continuing prior therapy) to evaluate the efficacy (NCT04714619).
4.1.3. Monoclonal antibodies targeting Notch signaling
Brontictuzumab (OMP‐52M51) is a humanized monoclonal antibody (mAb) that selectively targets Notch1 juxtamembrane negative regulatory region and thus inhibits Notch signaling. In a phase II trial of brontictuzumab, six of total 36 (17%) patients with refractory solid tumors demonstrated clinical benefits, with four patients displaying prolonged disease stabilization (NCT01778439). 363 A functional assay evaluated the efficacy of brontictuzumab in a series of glioma stem‐like cell models, supporting brontictuzumab as a promising drug candidate for CNS tumors. 364
Tarextumab (OMP‐59R5) is a human IgG2 antibody with inhibition on both Notch2 and Notch3 and has shown encouraging antitumor efficacy in small cell lung cancer (SCLC). An overall response rate (ORR) of 84% was reported in phase I/II trial when patients received the combination of tarextumab with etoposide and platinum‐based therapies. Meanwhile, tarextumab leads to potent inhibition in Notch signaling, and tarextumab‐induced diarrhea was dose‐limiting above 2.5 mg weekly and 7.5 mg/kg every third week (NCT01277146). The triple combination of tarextumab in combination with gemcitabine plus nab‐paclitaxel resulted in increased inhibition of tumor growth and tumor‐initiating cell frequency compared with the combination of tarextumab with gemcitabine alone. 365 However, a randomized phase II trial found that the addition of tarextumab to nab‐paclitaxel and gemcitabine failed to induce a prolonged overall survival (OS), progression‐free survival (PFS), or ORR in patients with metastatic PDAC. 366 The specific role of Notch signaling in PDAC remains unclear with evidence supporting both its oncogenic and tumor‐suppressive roles. 366 Further research using individual Notch inhibitors and agonists may facilitate the clinical evaluation of Notch‐targeting agents in pancreatic cancer.
4.2. Targeting HH signaling
The pharmacological inhibition targeting the HH pathway in cancer is an active research field, some of which have received regulatory approvals. Major HH pathway antagonists investigated so far include SMO inhibitors and GLI inhibitors. 367
4.2.1. SMO inhibitor
Two SMO antagonists, vismodegib and sonidegib, have been approved by the Food and Drug Administration (FDA) for the treatment of advanced basal cell carcinoma (BCC). Vismodegib (GDC‐0449) was first granted approval following success in clinical trials in 2012, where the independently assessed response rate was 30% in 33 patients with metastatic BCC (NCT00833417). 368 In the subsequent phase II trial, a total number of 1215 patients with advanced BCC were treated with vismodegib (NCT01367665). The response rate of patients with metastatic disease was 36.9% and that in patients with locally advanced disease was 68.5% 29073584. However, no benefits were obtained from the additional use of vismodegib in standard treatment regimens for metastatic CRC, 368 PDAC, 369 gastric cancer, and gastroesophageal junction cancer. 370 Besides, the combinations of erlotinib or chemotherapy with vismodegib were well tolerated but induced no improved outcome in metastatic PDAC or PDA, respectively. 371 Vismodegib also failed to deliver clinical benefits compared with placebo as maintenance therapy for ovarian cancer patients. 372 Vismodegib improved PFS in patients with SHH‐subtype medulloblastoma but not in those with non‐SHH disease subtypes. 373 , 374 , 375 , 376 Recent evidence, however, suggested that the addition of vismodegib to TMZ did not improve PFS even in SHH refractory medulloblastoma. 377 A recent phase II trial suggested that the histopathologic subtypes of BCC had no significant impact on patient response to vismodegib. 378
Sonidegib (LDE225) was approved by FDA in 2015 based on the promising results from a randomized phase II study, where sonidegib at 200 and 800 mg daily induced similar ORR (58% vs. 44%) in patients with locally advanced BCC.135 In the following analyses, sonidegib demonstrated long‐term efficacy and safety profile (30 379 and 42 months 380 ) in patients with advanced BCC. A meta‐analysis showed that ORRs of vismodegib and sonidegib were comparable in locally advanced BCC (69% vs. 57% respectively), whereas the complete response rates of the two drugs were different (31% vs. 3% respectively). 381 Likewise, BCC patients resistant to vismodegib similarly developed resistance with sonidegib. 382 In both mouse models and phase I clinical trial of TNBC, sonidegib decreased CSC markers expression and sensitizes cancer cells to docetaxel chemotherapy. 383
Another key SMO inhibitor, glasdegib (PF‐04449913), received FDA approval in 2018 as a combination partner for cytarabine for the treatment of AML. The addiction of glasdegib to low‐dose cytarabine (LDAC) increased the median OS of newly diagnosed AML patients from 4.9 to 8.8 months. 384 Glasdegib + LDAC continued to induce long‐term survival benefits in patients with AML, especially with secondary AML (NCT01546038). 385 In phase II clinical trial, 46.4% of patients achieved CR after glasdegib + cytarabine and daunorubicin treatment (NCT01546038). 386 The subsequent phase III trial of glasdegib in combination with chemotherapy (7 + 3 schedules) to treat AML patients is currently under way (NCT03416179). 384
4.2.2. Inhibitors of GLI transcriptional activity
GLI‐mediated transcription constitutes the final step of the HH pathway and the inhibition of GLI transcription factors is thus a promising strategy that reduces tumor cell proliferation. 387 Inhibitors of GLI‐mediated transcription, such as GANT58 and GANT61, were first designed to overcome tumor resistance to SMO inhibitors. A wide breadth of literature has described the antitumor activity of these agents in various cancers, including NSCLC, breast cancer, prostate cancer, and rhabdomyosarcoma. 388 , 389 , 390 , 391 , 392 However, neither of these two agents have advanced into clinical trials.
Arsenic trioxide (ATO), an FDA‐approved drug widely accepted in treating acute promyelocytic leukemia, is also a potent inhibitor of GLI1 and GLI2 and inhibits cancer growth by blocking GLI transcription. 392 With its inhibition of GLI transcription activities, ATO inhibits the viability and maintenance of CSCs derived from SCLC 393 and pancreatic cancer. 394 ATO also prevents osteosarcoma growth via DNA damage accumulation. 395 In a phase II study, the concomitant use of ATO and itraconazole was tested in BCC patients who were resistant to SMO inhibitors. Significant alterations in mRNA levels of GLI1 were observed. 396 Given that none of the participants had tumor shrinkage though they experienced SD for 3 months, continuous dosing was later recommended to achieve a better clinical response. 396 Currently, multiple clinical trials of ATO, alone or in synergy with standard therapies in cancer patients, are under way.
4.3. Targeting Wnt signaling
The Wnt pathway inhibitor family is mainly comprised of agents targeting Wnt pathway molecules, Porcupine inhibitors that diminish the ability to secrete Wnt ligands, and inhibitors of downstream β‐catenin‐TCF‐LEF‐dependent transcription. Many of these agents have been extensively studied and are currently under clinical evaluation.
4.3.1. Inhibitors of Wnt pathway elements
DKN‐01 is an IgG4 mAb targeting Dkk1 that suppresses canonical Wnt signaling via negative feedback. 397 Some studies addressed the direct antitumor effects of DKK1 inhibition, 398 whereas some recently reported its indirect antitumor effects via stimulation of immune responses in cancers, including ovarian cancer 399 and prostate cancer. 400 The murine version of DKN‐01 overcomes the DKK1‐mediated immune suppression and improves the efficacy of PD‐1 blockade. 401 Similarly, inhibiting Wnt/β‐catenin signaling by DKN‐01 enhances the antitumor immune infiltration into tumors and improves the response of ovarian tumors to immune checkpoint inhibitors. 399
Multiple clinical trials of DKN‐01 are now carried out across a wide range of cancer types. In a phase I trial, the combination of DKN‐01 with paclitaxel is well tolerated in patients with DKK1‐positive esophageal or gastroesophageal junction tumors (NCT02013154). 402 In a following phase II trial, the combination of DKN‐01 with pembrolizumab was well tolerated in patients with a gastroesophageal junction or gastric cancer, and especially effective in anti‐PD‐1/PD‐L1‐naïve patients with DKK1‐ high tumors. 403 A biomarker analysis revealed that DKN‐01 in combination with chemotherapies potentially led to reduced angiogenesis and inflammation markers in patients with biliary tract cancer (NCT02375880). 404
Vantictumab is a fully human mAb that inhibits Wnt pathway signaling by targeting FZD1, 2, 5, 7, and 8 receptors. Vantictumab decreases the enrichment of CSCs in various tumor types, either alone or in synergy with a chemotherapeutic. 405 A phase I study evaluated the combination of vantictumab with nab‐paclitaxel and gemcitabine in metastatic PDA patients. However, this trial was ultimately terminated due to bone‐related cytotoxicity. 406 Another phase I study assessed the efficacy and safety of the combination of vantictumab with paclitaxel metastatic breast cancer and the further use of this combination was restricted by the frequently occurred fractures. 407
Cirmtuzumab is a humanized mAb that inhibits the activity of ROR1, an oncoembryonic orphan receptor for Wnt5a in CSCs. 408 The antitumor activities of cirmtuzumab are mostly documented in chronic lymphocytic leukemia (CLL), where it inhibits the activation of both NF‐κB and STAT3 in patients. 409 Results from a phase I trial showed that cirmtuzumab is effective in suppressing tumor cell ROR1 signaling in CLL (NCT02222688). 410 Targeting ROR1 with cirmtuzumab may also improve the response of breast cancer patients to chemotherapies. 411 Cirmtuzumab could work synergistically with the Bruton tyrosine kinase inhibitor ibrutinib to treat patients with CLL or other ROR1+ B‐cell malignancies. 412 Currently, a phase Ib/II study is under way to evaluate this combination in patients with CLL, small lymphocytic lymphoma, or mantle cell lymphoma (NCT03088878).
5. CSC‐DIRECTED IMMUNOTHERAPIES
As promising CSC‐directed immunotherapy, CSC‐based dendritic cell (DC) vaccines facilitate tumor cell recognition and eradication by potentiating antigen‐specific T‐cell responses against CSCs. 413 The CSC‐specific T cells can also be produced by CSC priming. CSC lysate‐pulsed DCs stimulate CD8+ T cells, and the generated CSC‐specific T cells induce antitumor immunity by directly targeting CSCs in tumors. 414 , 415 Table 3 summarizes the ongoing and completed clinical trials on CSC‐directed immunotherapies. Bispecific antibodies (BiAbs) targeting CSC‐specific antigens represent another candidate for CSC‐directed immunotherapies. For instance, a BiAb composed of CD133 mAb monomer and a single chain of humanized muromonab‐CD3 targets CD133‐expressing tumor cells by arming activated T cells. 416
TABLE 3.
Ongoing and completed clinical trials on CSC‐directed immunotherapies
| Immunotherapy | Condition | Phase | NCT number |
|---|---|---|---|
| Dendritic cell (DC) vaccines | |||
| Cancer stem cells vaccine | Ovarian cancer | I/II | NCT02178670 |
| Tumor lysate‐pulsed DC vaccine | High‐risk solid tumor | II | NCT00405327 |
| Minor histocompatibility antigens (MiHA)‐loaded PD‐L‐silenced DC vaccination | Hematological malignancies | I/II | NCT02528682 |
| Total tumor RNA (TTRNA)‐loaded‐DCs | Recurrent medulloblastoma and primitive neuroectodermal tumor | II | NCT01326104 |
| CD34+‐derived DCs | Breast neoplasms | I | NCT00197522 |
| Dendritic cell vaccine with mRNA from tumor stem cells | Glioblastoma | I/II | NCT00846456 |
| Dendritic cell fusion vaccine | Multiple myeloma | II | NCT01067287 |
| Autologous DCs with glioma stem‐like cells associated antigens | Glioblastoma | II | NCT01567202 |
| Bispecific antibodies | |||
| Anti‐CD3 × anti‐CD20 bispecific antibody‐armed activated T cells | Multiple myeloma and plasma cell neoplasm | I | NCT00938626 |
| Elranatamab (B‐cell maturation antigen [BCMA] CD3‐targeted bispecific antibody) | Multiple myeloma | III | NCT05317416 |
| Anti‐CD3 × anti‐CD20 bispecific antibody (CD20Bi)‐activated T cells (ATC) | Non‐Hodgkin lymphoma | I | NCT00244946 |
Clinical trial data sources: clinicaltrials.gov.
One of the most studied CSC‐directed immunotherapies that enter clinical trials is the CAR T‐cell transfer, based on the identification of CSC surface antigens by CAR T cells. 417 Though a wide range of CSC‐related antigens are used to design CAR T‐cell therapies, 417 CSC‐targeting CAR T cells to date have been approved by FDA. The largest concern about CAR T‐cell treatment could be its safety profile, cytokine release syndrome, and soluble tumor syndrome. 417 The application of well‐characterized CSC markers in CAR T‐cell design is a promising approach to eliminate CSCs in many cancers, which, however, still requires further investigations to advance CAR T cells into the clinic.
A typical example of CAR T therapies is the CAR T cocktail immunotherapy composed of successive infusions of CART cells targeting epidermal growth factor receptor (EGFR) and CD133, which specifically target CSCs in cholangiocarcinoma (NCT01869166 and NCT02541370). 418 CART‐133 cell therapy patients demonstrate promising antitumor activity. In HCC patients, CAR‐133 cell therapy demonstrated promising efficacy with manageable toxicity. 419 This study also revealed potential biomarkers that predicted patient response to CART‐133 cells (NCT02541370). GBM CSCs are characterized as EGFRVIII+/CD133+ cells with self‐renewal as well as cancer initiation abilities. 420 In the first clinical trial of EGFRVIII‐specific CAR T‐cell infusions, patients with EGFRVIII+ recurrent GBM did not obtain noticeable tumor regression according to MRI. 421 Meanwhile, an additional study suggested that the CAR T‐EGFRVIII cell therapy failed to induce clinical benefits in patients with recurrent GBM. 422 Ongoing and completed clinical trials on CSC‐directed CAR T‐cell therapy are presented in Table 4.
TABLE 4.
Ongoing and completed clinical trials on CSC‐directed chimeric antigen receptor (CAR) T‐cell therapy
| Chimeric antigen receptor (CAR) T cell | Condition | Phase | NCT number |
|---|---|---|---|
| CAR T‐cell therapy | Hematologic neoplasms | NA | NCT04691284 |
| PSCA‐targeted CAR T cells (BPX‐601) | Advanced solid tumors | I/II | NCT02744287 |
| Anti‐CD19 CAR T cells | B‐cell non‐Hodgkin lymphoma | I/II | NCT01318317 |
| B‐cell malignancies | I/II | NCT01475058, NCT01087294, NCT02659943 | |
| Relapsed malignant lymphoma | I | NCT05239676 | |
| CD19CAR/virus‐specific T cells | CD19+ malignancies | I | NCT00840853 |
| CD19+ acute lymphoblastic leukemia or non‐Hodgkin lymphoma | I | NCT03768310 | |
| Ciltacabtagene autoleucel (BCMA‐directed CAR T) | Multiple myeloma | III | NCT04923893, NCT05257083 |
| B‐cell lymphoma | II | NCT04531046 | |
| Anti‐CD19 allo‐CAR T cells | Relapsed B‐cell malignancies | I | NCT04516551 |
| Genetically modified T cells (CMV‐specific CD19‐CAR T cells) | B‐cell non‐Hodgkin lymphoma | I | NCT05432635 |
| CD19+CD22 CAR‐T | B acute lymphoblastic leukemia | I | NCT04626726 |
| CD19CAR‐CD28‐CD3zeta‐EGFRt‐expressing TCM‐enriched T cells | Non‐Hodgkin lymphoma | I | NCT01815749 |
| B‐cell non‐Hodgkin lymphoma | I | NCT02051257 | |
| CD19CAR‐CD28Z T cells | B‐cell lymphoblastic leukemia | I | NCT02050347 |
| CAR T directed against CD19+ B cells | B‐cell non‐Hodgkin lymphoma | I | NCT01840566 |
| Anti‐CD133‐CAR T cells | Advanced malignancies | I/II | NCT02541370 |
| Malignant gliomas | I | NCT03423992 | |
| Sarcoma‐specific CAR T cells | Sarcoma | I/II | NCT03356782 |
| CD44v6‐specific CAR T cells | CD44v6‐positive cancers | I/II | NCT04427449 |
| 4SCAR T cells | Breast cancer | I/II | NCT04430595 |
Clinical trial data sources: clinicaltrials.gov.
The CSC characteristics are associated with an increased level of CD44, 168 which requires the transformation of CD44v to CD44s isoform. 423 , 424 It was reported that 50% of pancreatic cancer tissues were CD44v6‐positive, which indicated a poorer survival in this group of patients. 425 Currently, two phase I/II clinical trials are ongoing to assess the safety and efficacy of CD44v6 CAR‐T‐cell therapy in patients with breast cancer and other CD44v6‐positive tumors. (NCT04430595 and NCT04427449) Recently, a highly specific CAR against CD44v6 was established aiming to eliminate CD44v6‐expressing HNSCC cells. 426
6. CONCLUSION AND FUTURE PERSPECTIVES
In conclusion, CSCs are a subpopulation of malignant tumor cells with selective capacities for tumor initiation, self‐renewal, metastasis, and unlimited growth into bulks. There is intricate signaling network within CSCs that regulates stemness and biological functions. Thus, targeting pathway molecules that regulate CSCs provides a new option for the treatment of therapy‐resistant or ‐refractory tumors. Meanwhile, extracellular regulating factors, including angiogenic microenvironment, hypoxic microenvironment, TAM, fibroblasts, and a series of protumor paracrine factors, collectively provide a fertile soil that favors CSC growth. Numerous efforts have been undertaken these years to identify such therapies, such as kinase inhibitors and antibodies that block CSC‐associated signaling pathway elements, and some of these approaches have already entered the clinical phase. Furthermore, vaccines, antibodies, and CAR T cells have also expanded the range of CSC‐target therapies.
Obstacles remain regarding the design of CSC‐targeted therapies. Though our understanding of CSCs surface biomarkers has been largely improved in recent years, the surface markers of CSCs may vary according to tumor types and the cell of tumor origin, demonstrating high heterogeneity between tumors or even among cells within one tumor. This heterogeneity has highlighted the challenges in identifying and isolating CSC subpopulations from tumors. Thus, functional assays are recommended to more specifically identify CSCs, including sphere formation capacity in vitro and tumor‐initiation of after transplantation in vivo.
Accumulating evidence now suggests that quiescent CSCs contribute to the refraction of cancers to chemotherapies. Thus, therapeutic approaches that merely inhibit CSC stemness might not be sufficient to suppress postchemotherapy recurrence. The TME plays a critical role in CSC regulation, which not only maintains CSC characteristics via various signals but also facilitates the transition of nonstem cells to stem cell states. 427 It is thus conceivable that targeting TME components may be more effective in overcoming treatment resistance than directly inhibiting CSCs stemness. However, the heterogeneity of immune cells across cell types has made it difficult to identify the precise CSC‐immune cell interactions. Recently, single‐cell RNA sequencing is extensively used to identify the altering states of CSCs and immune cells, as well as their interactions under different tumor contexts. Moreover, BiAbs that act on both intrinsic regulating factors of CSCs and the CSC‐immune cell crosstalk are recommended. 31
Finally, in addition to TAMs that have long been identified for their activities in CSC maintenance, recent reports highlight the significance of NK cells in suppressing cancer cell stemness. 290 CSCs are mostly sensitive to NK cell killing, but in some cases, such as GBM, AML, and breast cancer, CSCs may be resistant to activated NK cells. 428 However, the anti‐CSC functions of NK cells are suppressed by TAMs, MDSCs (myeloid‐derived suppressor cells), and T‐reg cells. 429 Future research is required to address the crosstalk between these immune cells in the TME, thereby facilitating the development of more effective CSC‐targeted immunotherapies.
CONFLICT OF INTERESTS
The authors declared no conflict of interests.
AUTHOR CONTRIBUTIONS
Wu Min offered the main direction and significant guidance of this manuscript. Wang Manni drafted the manuscript and illustrated the figures for the manuscript. All authors have read and approved the final manuscript.
ETHICS APPROVAL
Not applicable.
ACKNOWLEDGMENTS
This work is supported by the “China Postdoctoral Science Foundation” (2021M702347), “Fundamental Research Funds for the Central Universities” (20826041F4164) and The National Natural Science Foundation of China (82203029).
Manni W, Min W. Signaling pathways in the regulation of cancer stem cells and associated targeted therapy. MedComm. 2022;3:e176. 10.1002/mco2.176
Contributor Information
Wang Manni, Email: wangmanni@scu.edu.cn.
Wu Min, Email: min.wu@med.und.edu.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Dinsmore CE. Animal regeneration—from fact to concept. BioScience. 1995;45(7):484‐492. [Google Scholar]
- 2. Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. 2010;120(1):41‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells–old concepts, new insights. Cell Death Differ. 2008;15(6):947‐958. [DOI] [PubMed] [Google Scholar]
- 4. Chang JC. Cancer stem cells: role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore). 2016;95(1 Suppl 1):S20‐S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34(6):732‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756‐760. [DOI] [PubMed] [Google Scholar]
- 7. Li X, Lewis MT, Huang J, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100(9):672‐679. [DOI] [PubMed] [Google Scholar]
- 8. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wiechert A, Saygin C, Thiagarajan PS, et al. Cisplatin induces stemness in ovarian cancer. Oncotarget. 2016;7(21):30511‐30522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saygin C, Matei D, Majeti R, Reizes O, Lathia JD. Targeting cancer stemness in the clinic: from hype to hope. Cell Stem Cell. 2019;24(1):25‐40. [DOI] [PubMed] [Google Scholar]
- 11. Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013;341(1):41‐45. [DOI] [PubMed] [Google Scholar]
- 12. Nishio M, Otsubo K, Maehama T, Mimori K, Suzuki A. Capturing the mammalian Hippo: elucidating its role in cancer. Cancer Sci. 2013;104(10):1271‐1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pelullo M, Zema S, Nardozza F, Checquolo S, Screpanti I, Bellavia D. Wnt, Notch, and TGF‐beta pathways impinge on hedgehog signaling complexity: an open window on cancer. Front Genet. 2019;10:711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJ. Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer. 2005;5(11):899‐904. [DOI] [PubMed] [Google Scholar]
- 15. Hill RP. Identifying cancer stem cells in solid tumors: case not proven. Cancer Res. 2006;66(4):1891‐1895. discussion 1890. [DOI] [PubMed] [Google Scholar]
- 16. Huntly BJ, Gilliland DG. Leukaemia stem cells and the evolution of cancer‐stem‐cell research. Nat Rev Cancer. 2005;5(4):311‐321. [DOI] [PubMed] [Google Scholar]
- 17. Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645‐648. [DOI] [PubMed] [Google Scholar]
- 18. Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100(25):15178‐15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jin X, Jin X, Kim H. Cancer stem cells and differentiation therapy. Tumour Biol. 2017;39(10). [DOI] [PubMed] [Google Scholar]
- 20. Huang Z, Wu T, Liu AY, Ouyang G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget. 2015;6(37):39550‐39563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ricci‐Vitiani L, Pallini R, Biffoni M, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem‐like cells. Nature. 2010;468(7325):824‐828. [DOI] [PubMed] [Google Scholar]
- 22. Bussolati B, Bruno S, Grange C, Ferrando U, Camussi G. Identification of a tumor‐initiating stem cell population in human renal carcinomas. FASEB J. 2008;22(10):3696‐3705. [DOI] [PubMed] [Google Scholar]
- 23. Xiong YQ, Sun HC, Zhang W, et al. Human hepatocellular carcinoma tumor‐derived endothelial cells manifest increased angiogenesis capability and drug resistance compared with normal endothelial cells. Clin Cancer Res. 2009;15(15):4838‐4846. [DOI] [PubMed] [Google Scholar]
- 24. Huang T, Song X, Xu D, et al. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics. 2020;10(19):8721‐8743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23‐28. [DOI] [PubMed] [Google Scholar]
- 26. van Niekerk G, Davids LM, Hattingh SM, Engelbrecht AM. Cancer stem cells: a product of clonal evolution? Int J Cancer. 2017;140(5):993‐999. [DOI] [PubMed] [Google Scholar]
- 27. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755‐768. [DOI] [PubMed] [Google Scholar]
- 28. Quinn HM, Vogel R, Popp O, et al. YAP and beta‐catenin cooperate to drive oncogenesis in basal breast cancer. Cancer Res. 2021;81(8):2116‐2127. [DOI] [PubMed] [Google Scholar]
- 29. Shlush LI, Zandi S, Mitchell A, et al. Identification of pre‐leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Auffinger B, Tobias AL, Han Y, et al. Conversion of differentiated cancer cells into cancer stem‐like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014;21(7):1119‐1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen P, Hsu WH, Han J, Xia Y, DePinho RA. Cancer stemness meets immunity: from mechanism to therapy. Cell Rep. 2021;34(1):108597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen K, Zhang C, Ling S, Wei R, Wang J, Xu X. The metabolic flexibility of quiescent CSC: implications for chemotherapy resistance. Cell Death Dis. 2021;12(9):835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ponomarev A, Gilazieva Z, Solovyeva V, Allegrucci C, Rizvanov A. Intrinsic and extrinsic factors impacting cancer stemness and tumor progression. Cancers (Basel). 2022;14(4):970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ajani JA, Song S, Hochster HS, Steinberg IB. Cancer stem cells: the promise and the potential. Semin Oncol. 2015;42(1):S3‐S17. [DOI] [PubMed] [Google Scholar]
- 35. Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012;22(3):457‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boman BM, Wicha MS. Cancer stem cells: a step toward the cure. J Clin Oncol. 2008;26(17):2795‐2799. [DOI] [PubMed] [Google Scholar]
- 37. Huang EH, Hynes MJ, Zhang T, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009;69(8):3382‐3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122(6):947‐956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA. Circuitry and dynamics of human transcription factor regulatory networks. Cell. 2012;150(6):1274‐1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sangiolo D, Mesiano G, Gammaitoni L, et al. Cytokine‐induced killer cells eradicate bone and soft‐tissue sarcomas. Cancer Res. 2014;74(1):119‐129. [DOI] [PubMed] [Google Scholar]
- 41. Guerra‐Rebollo M, Garrido C, Sanchez‐Cid L, et al. Targeting of replicating CD133 and OCT4/SOX2 expressing glioma stem cells selects a cell population that reinitiates tumors upon release of therapeutic pressure. Sci Rep. 2019;9(1):9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang L, Shi P, Zhao G, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walcher L, Kistenmacher AK, Suo H, et al. Cancer stem cells–origins and biomarkers: perspectives for targeted personalized therapies. Front Immunol. 2020;11:1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730‐737. [DOI] [PubMed] [Google Scholar]
- 45. Moserle L, Ghisi M, Amadori A, Indraccolo S. Side population and cancer stem cells: therapeutic implications. Cancer Lett. 2010;288(1):1‐9. [DOI] [PubMed] [Google Scholar]
- 46. Szotek PP, Pieretti‐Vanmarcke R, Masiakos PT, et al. Ovarian cancer side population defines cells with stem cell‐like characteristics and Mullerian inhibiting substance responsiveness. Proc Natl Acad Sci U S A. 2006;103(30):11154‐11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chiba T, Kita K, Zheng YW, et al. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell‐like properties. Hepatology. 2006;44(1):240‐251. [DOI] [PubMed] [Google Scholar]
- 48. Wang M, Wang Y, Zhong J. Side population cells and drug resistance in breast cancer. Mol Med Rep. 2015;11(6):4297‐4302. [DOI] [PubMed] [Google Scholar]
- 49. Feuring‐Buske M, Hogge DE. Hoechst 33342 efflux identifies a subpopulation of cytogenetically normal CD34(+)CD38(−) progenitor cells from patients with acute myeloid leukemia. Blood. 2001;97(12):3882‐3889. [DOI] [PubMed] [Google Scholar]
- 50. Montanaro F, Liadaki K, Schienda J, Flint A, Gussoni E, Kunkel LM. Demystifying SP cell purification: viability, yield, and phenotype are defined by isolation parameters. Exp Cell Res. 2004;298(1):144‐154. [DOI] [PubMed] [Google Scholar]
- 51. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445(7123):106‐110. [DOI] [PubMed] [Google Scholar]
- 52. Ricci‐Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon‐cancer‐initiating cells. Nature. 2007;445(7123):111‐115. [DOI] [PubMed] [Google Scholar]
- 53. Shmelkov SV, Butler JM, Hooper AT, et al. CD133 expression is not restricted to stem cells, and both CD133+ and CD133– metastatic colon cancer cells initiate tumors. J Clin Invest. 2008;118(6):2111‐2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Min DW, Kim HP, Kim J, et al. Phenotype‐based single cell sequencing identifies diverse genetic subclones in CD133 positive cancer stem cells. Biochem Biophys Res Commun. 2021;558:209‐215. [DOI] [PubMed] [Google Scholar]
- 55. Taverna JA, Hung CN, DeArmond DT, et al. Single‐cell proteomic profiling identifies combined AXL and JAK1 inhibition as a novel therapeutic strategy for lung cancer. Cancer Res. 2020;80(7):1551‐1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Celia‐Terrassa T, Jolly MK. Cancer stem cells and epithelial‐to‐mesenchymal transition in cancer metastasis. Cold Spring Harb Perspect Med. 2020;10(7):a036905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Underhill C. CD44: the hyaluronan receptor. J Cell Sci. 1992;103(Pt 2):293‐298. [DOI] [PubMed] [Google Scholar]
- 58. Gzil A, Zarebska I, Bursiewicz W, Antosik P, Grzanka D, Szylberg L. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol Biol Rep. 2019;46(6):6629‐6645. [DOI] [PubMed] [Google Scholar]
- 59. Al‐Othman N, Alhendi A, Ihbaisha M, Barahmeh M, Alqaraleh M, Al‐Momany BZ. Role of CD44 in breast cancer. Breast Dis. 2020;39(1):1‐13. [DOI] [PubMed] [Google Scholar]
- 60. Negi LM, Talegaonkar S, Jaggi M, Ahmad FJ, Iqbal Z, Khar RK. Role of CD44 in tumour progression and strategies for targeting. J Drug Target. 2012;20(7):561‐573. [DOI] [PubMed] [Google Scholar]
- 61. Katagiri YU, Sleeman J, Fujii H, et al. CD44 variants but not CD44s cooperate with beta1‐containing integrins to permit cells to bind to osteopontin independently of arginine‐glycine‐aspartic acid, thereby stimulating cell motility and chemotaxis. Cancer Res. 1999;59(1):219‐226. [PubMed] [Google Scholar]
- 62. Fang D, Kitamura H. Cancer stem cells and epithelial‐mesenchymal transition in urothelial carcinoma: possible pathways and potential therapeutic approaches. Int J Urol. 2018;25(1):7‐17. [DOI] [PubMed] [Google Scholar]
- 63. Lamour V, Henry A, Kroonen J, et al. Targeting osteopontin suppresses glioblastoma stem‐like cell character and tumorigenicity in vivo. Int J Cancer. 2015;137(5):1047‐1057. [DOI] [PubMed] [Google Scholar]
- 64. Immervoll H, Hoem D, Sakariassen PO, Steffensen OJ, Molven A. Expression of the “stem cell marker” CD133 in pancreas and pancreatic ductal adenocarcinomas. BMC Cancer. 2008;8:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Al‐Hajj M, Wicha MS, Benito‐Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983‐3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ginestier C, Hur MH, Charafe‐Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1(5):555‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Quintana E, Shackleton M, Foster HR, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010;18(5):510‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396‐401. [DOI] [PubMed] [Google Scholar]
- 69. van den Hoogen C, van der Horst G, Cheung H, et al. High aldehyde dehydrogenase activity identifies tumor‐initiating and metastasis‐initiating cells in human prostate cancer. Cancer Res. 2010;70(12):5163‐5173. [DOI] [PubMed] [Google Scholar]
- 70. Zhang WC, Shyh‐Chang N, Yang H, et al. Glycine decarboxylase activity drives non‐small cell lung cancer tumor‐initiating cells and tumorigenesis. Cell. 2012;148(1‐2):259‐272. [DOI] [PubMed] [Google Scholar]
- 71. Hashimoto O, Shimizu K, Semba S, et al. Hypoxia induces tumor aggressiveness and the expansion of CD133‐positive cells in a hypoxia‐inducible factor‐1alpha‐dependent manner in pancreatic cancer cells. Pathobiology. 2011;78(4):181‐192. [DOI] [PubMed] [Google Scholar]
- 72. Lv D, Ma QH, Duan JJ, et al. Optimized dissociation protocol for isolating human glioma stem cells from tumorspheres via fluorescence‐activated cell sorting. Cancer Lett. 2016;377(1):105‐115. [DOI] [PubMed] [Google Scholar]
- 73. Tomita H, Tanaka K, Tanaka T, Hara A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget. 2016;7(10):11018‐11032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Huang CP, Tsai MF, Chang TH, et al. ALDH‐positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. 2013;328(1):144‐151. [DOI] [PubMed] [Google Scholar]
- 75. Luo M, Brooks M, Wicha MS. Epithelial‐mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des. 2015;21(10):1301‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kalluri R, Neilson EG. Epithelial‐mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112(12):1776‐1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kalluri R, Weinberg RA. The basics of epithelial‐mesenchymal transition. J Clin Invest. 2009;119(6):1420‐1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Akrap N, Andersson D, Bom E, Gregersson P, Stahlberg A, Landberg G. Identification of distinct breast cancer stem cell populations based on single‐cell analyses of functionally enriched stem and progenitor pools. Stem Cell Rep. 2016;6(1):121‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ammothumkandy A, Maliekal TT, Bose MV, et al. CD66 and CD49f expressing cells are associated with distinct neoplastic phenotypes and progression in human cervical cancer. Eur J Cancer. 2016;60:166‐178. [DOI] [PubMed] [Google Scholar]
- 80. Haraguchi N, Ishii H, Mimori K, et al. CD49f‐positive cell population efficiently enriches colon cancer‐initiating cells. Int J Oncol. 2013;43(2):425‐430. [DOI] [PubMed] [Google Scholar]
- 81. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13(7):513‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pramanik KC, Fofaria NM, Gupta P, Ranjan A, Kim SH, Srivastava SK. Inhibition of beta‐catenin signaling suppresses pancreatic tumor growth by disrupting nuclear beta‐catenin/TCF‐1 complex: critical role of STAT‐3. Oncotarget. 2015;6(13):11561‐11574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Li J, Ji L, Chen J, Zhang W, Ye Z. Wnt/beta‐catenin signaling pathway in skin carcinogenesis and therapy. Biomed Res Int. 2015;2015:964842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Stewart DJ. Wnt signaling pathway in non‐small cell lung cancer. J Natl Cancer Inst. 2014;106(1):djt356. [DOI] [PubMed] [Google Scholar]
- 86. Li Y, Hu J, Song H, Wu T. Antibiotic anisomycin selectively targets leukemia cell lines and patient samples through suppressing Wnt/beta‐catenin signaling. Biochem Biophys Res Commun. 2018;505(3):858‐864. [DOI] [PubMed] [Google Scholar]
- 87. Chen Z, Zhou L, Wang L, et al. HBO1 promotes cell proliferation in bladder cancer via activation of Wnt/beta‐catenin signaling. Mol Carcinog. 2018;57(1):12‐21. [DOI] [PubMed] [Google Scholar]
- 88. Prasetyanti PR, Zimberlin CD, Bots M, Vermeulen L, Melo Fde S, Medema JP. Regulation of stem cell self‐renewal and differentiation by Wnt and Notch are conserved throughout the adenoma‐carcinoma sequence in the colon. Mol Cancer. 2013;12(1):126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Teng Y, Wang X, Wang Y, Ma D. Wnt/beta‐catenin signaling regulates cancer stem cells in lung cancer A549 cells. Biochem Biophys Res Commun. 2010;392(3):373‐379. [DOI] [PubMed] [Google Scholar]
- 90. de Sousa EMF, Vermeulen L. Wnt signaling in cancer stem cell biology. Cancers (Basel). 2016;8(7):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Liu S, Kin Pong U, Zhang J, et al. R‐spodin2 enhances canonical Wnt signaling to maintain the stemness of glioblastoma cells. Cancer Cell Int. 2018;18:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liu D, Du L, Chen D, et al. Reduced CD146 expression promotes tumorigenesis and cancer stemness in colorectal cancer through activating Wnt/beta‐catenin signaling. Oncotarget. 2016;7(26):40704‐40718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Malta TM, Sokolov A, Gentles AJ, et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell. 2018;173(2):338‐354.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Malanchi I, Peinado H, Kassen D, et al. Cutaneous cancer stem cell maintenance is dependent on beta‐catenin signalling. Nature. 2008;452(7187):650‐653. [DOI] [PubMed] [Google Scholar]
- 95. Vermeulen L, De Sousa EMF, van der Heijden M, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12(5):468‐476. [DOI] [PubMed] [Google Scholar]
- 96. Loregger A, Grandl M, Mejias‐Luque R, et al. The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated beta‐catenin by sequestering TCF4 to the nuclear membrane. Sci Signal. 2015;8(393):ra90. [DOI] [PubMed] [Google Scholar]
- 97. Cho YH, Ro EJ, Yoon JS, et al. 5‐FU promotes stemness of colorectal cancer via p53‐mediated WNT/beta‐catenin pathway activation. Nat Commun. 2020;11(1):5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Liu X, Su K, Sun X, et al. Sec62 promotes stemness and chemoresistance of human colorectal cancer through activating Wnt/beta‐catenin pathway. J Exp Clin Cancer Res. 2021;40(1):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH. Wnt/beta‐catenin pathway activation is enriched in basal‐like breast cancers and predicts poor outcome. Am J Pathol. 2010;176(6):2911‐2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Schade B, Lesurf R, Sanguin‐Gendreau V, et al. Beta‐catenin signaling is a critical event in ErbB2‐mediated mammary tumor progression. Cancer Res. 2013;73(14):4474‐4487. [DOI] [PubMed] [Google Scholar]
- 101. Fendler A, Bauer D, Busch J, et al. Inhibiting WNT and NOTCH in renal cancer stem cells and the implications for human patients. Nat Commun. 2020;11(1):929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jiang N, Zou C, Zhu Y, et al. HIF‐1a‐regulated miR‐1275 maintains stem cell‐like phenotypes and promotes the progression of LUAD by simultaneously activating Wnt/beta‐catenin and Notch signaling. Theranostics. 2020;10(6):2553‐2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Clara JA, Monge C, Yang Y, Takebe N. Targeting signalling pathways and the immune microenvironment of cancer stem cells ‐ a clinical update. Nat Rev Clin Oncol. 2020;17(4):204‐232. [DOI] [PubMed] [Google Scholar]
- 104. Dandawate PR, Subramaniam D, Jensen RA, Anant S. Targeting cancer stem cells and signaling pathways by phytochemicals: novel approach for breast cancer therapy. Semin Cancer Biol. 2016;40‐41:192‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wang Z, Ahmad A, Li Y, Azmi AS, Miele L, Sarkar FH. Targeting notch to eradicate pancreatic cancer stem cells for cancer therapy. Anticancer Res. 2011;31(4):1105‐1113. [PubMed] [Google Scholar]
- 106. Fortini ME. Notch signaling: the core pathway and its posttranslational regulation. Dev Cell. 2009;16(5):633‐647. [DOI] [PubMed] [Google Scholar]
- 107. Yan Y, Liu F, Han L, et al. HIF‐2alpha promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J Exp Clin Cancer Res. 2018;37(1):256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Barnawi R, Al‐Khaldi S, Majed Sleiman G, et al. Fascin is critical for the maintenance of breast cancer stem cell pool predominantly via the activation of the notch self‐renewal pathway. Stem Cells. 2016;34(12):2799‐2813. [DOI] [PubMed] [Google Scholar]
- 109. Qiu L, Yang X, Wu J, Huang C, Miao Y, Fu Z. HIST2H2BF potentiates the propagation of cancer stem cells via notch signaling to promote malignancy and liver metastasis in colorectal carcinoma. Front Oncol. 2021;11:677646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Yu JB, Jiang H, Zhan RY. Aberrant notch signaling in glioblastoma stem cells contributes to tumor recurrence and invasion. Mol Med Rep. 2016;14(2):1263‐1268. [DOI] [PubMed] [Google Scholar]
- 111. Wang R, Li Y, Tsung A, et al. iNOS promotes CD24(+)CD133(+) liver cancer stem cell phenotype through a TACE/ADAM17‐dependent notch signaling pathway. Proc Natl Acad Sci U S A. 2018;115(43):E10127‐E10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bajaj J, Maliekal TT, Vivien E, et al. Notch signaling in CD66+ cells drives the progression of human cervical cancers. Cancer Res. 2011;71(14):4888‐4897. [DOI] [PubMed] [Google Scholar]
- 113. Hopfer O, Zwahlen D, Fey MF, Aebi S. The Notch pathway in ovarian carcinomas and adenomas. Br J Cancer. 2005;93(6):709‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hallahan AR, Pritchard JI, Hansen S, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog‐induced medulloblastomas. Cancer Res. 2004;64(21):7794‐7800. [DOI] [PubMed] [Google Scholar]
- 115. Fan X, Matsui W, Khaki L, et al. Notch pathway inhibition depletes stem‐like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66(15):7445‐7452. [DOI] [PubMed] [Google Scholar]
- 116. Abel EV, Kim EJ, Wu J, et al. The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS One. 2014;9(3):e91983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Koury J, Zhong L, Hao J. Targeting signaling pathways in cancer stem cells for cancer treatment. Stem Cells Int. 2017;2017:2925869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Zeng F, Chen H, Zhang Z, et al. Regulating glioma stem cells by hypoxia through the Notch1 and Oct3/4 signaling pathway. Oncol Lett. 2018;16(5):6315‐6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bayin NS, Frenster JD, Sen R, et al. Notch signaling regulates metabolic heterogeneity in glioblastoma stem cells. Oncotarget. 2017;8(39):64932‐64953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Fox RG, Lytle NK, Jaquish DV, et al. Image‐based detection and targeting of therapy resistance in pancreatic adenocarcinoma. Nature. 2016;534(7607):407‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ito T, Kwon HY, Zimdahl B, et al. Regulation of myeloid leukaemia by the cell‐fate determinant Musashi. Nature. 2010;466(7307):765‐768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Rane SG, Reddy EP. Janus kinases: components of multiple signaling pathways. Oncogene. 2000;19(49):5662‐5679. [DOI] [PubMed] [Google Scholar]
- 123. Stine RR, Matunis EL. JAK‐STAT signaling in stem cells. Adv Exp Med Biol. 2013;786:247‐267. [DOI] [PubMed] [Google Scholar]
- 124. de Freitas RM, da Costa Maranduba CM. Myeloproliferative neoplasms and the JAK/STAT signaling pathway: an overview. Rev Bras Hematol Hemoter. 2015;37(5):348‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Cook AM, Li L, Ho Y, et al. Role of altered growth factor receptor‐mediated JAK2 signaling in growth and maintenance of human acute myeloid leukemia stem cells. Blood. 2014;123(18):2826‐2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Chambers I. The molecular basis of pluripotency in mouse embryonic stem cells. Cloning Stem Cells. 2004;6(4):386‐391. [DOI] [PubMed] [Google Scholar]
- 127. Penuelas S, Anido J, Prieto‐Sanchez RM, et al. TGF‐beta increases glioma‐initiating cell self‐renewal through the induction of LIF in human glioblastoma. Cancer Cell. 2009;15(4):315‐327. [DOI] [PubMed] [Google Scholar]
- 128. Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem Cells. 2009;27(10):2383‐2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhou J, Wulfkuhle J, Zhang H, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem‐like cells is required for viability and maintenance. Proc Natl Acad Sci U S A. 2007;104(41):16158‐16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Dolatabadi S, Jonasson E, Linden M, et al. JAK‐STAT signalling controls cancer stem cell properties including chemotherapy resistance in myxoid liposarcoma. Int J Cancer. 2019;145(2):435‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Park SY, Lee CJ, Choi JH, et al. The JAK2/STAT3/CCND2 axis promotes colorectal cancer stem cell persistence and radioresistance. J Exp Clin Cancer Res. 2019;38(1):399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Thomas SJ, Snowden JA, Zeidler MP, Danson SJ. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer. 2015;113(3):365‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Galoczova M, Coates P, Vojtesek B. STAT3, stem cells, cancer stem cells and p63. Cell Mol Biol Lett. 2018;23:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Yoshimatsu T, Kawaguchi D, Oishi K, et al. Non‐cell‐autonomous action of STAT3 in maintenance of neural precursor cells in the mouse neocortex. Development. 2006;133(13):2553‐2563. [DOI] [PubMed] [Google Scholar]
- 135. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15(9):1010‐1012. [DOI] [PubMed] [Google Scholar]
- 136. Lin L, Jou D, Wang Y, et al. STAT3 as a potential therapeutic target in ALDH+ and CD44+/CD24+ stem cell‐like pancreatic cancer cells. Int J Oncol. 2016;49(6):2265‐2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Nusslein‐Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila . Nature. 1980;287(5785):795‐801. [DOI] [PubMed] [Google Scholar]
- 138. Petrova R, Joyner AL. Roles for hedgehog signaling in adult organ homeostasis and repair. Development. 2014;141(18):3445‐3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Matsui WH. Cancer stem cell signaling pathways. Medicine (Baltimore). 2016;95(1 Suppl 1):S8‐S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Kinzler KW, Bigner SH, Bigner DD, et al. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236(4797):70‐73. [DOI] [PubMed] [Google Scholar]
- 141. Li L, Zhao J, Zhang Q, et al. Cancer cell‐derived exosomes promote HCC tumorigenesis through Hedgehog pathway. Front Oncol. 2021;11:756205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Merchant AA, Matsui W. Targeting Hedgehog–a cancer stem cell pathway. Clin Cancer Res. 2010;16(12):3130‐3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Hutchin ME, Kariapper MS, Grachtchouk M, et al. Sustained hedgehog signaling is required for basal cell carcinoma proliferation and survival: conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev. 2005;19(2):214‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Peacock CD, Wang Q, Gesell GS, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A. 2007;104(10):4048‐4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Ghia EM, Rassenti LZ, Neuberg DS, et al. Activation of hedgehog signaling associates with early disease progression in chronic lymphocytic leukemia. Blood. 2019;133(25):2651‐2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458(7239):776‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG‐GLI1 signaling regulates human glioma growth, cancer stem cell self‐renewal, and tumorigenicity. Curr Biol. 2007;17(2):165‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Ko YC, Choi HS, Liu R, Lee DS. Physalin A, 13,14‐seco‐16, 24‐cyclo‐steroid, inhibits stemness of breast cancer cells by regulation of Hedgehog signaling pathway and yes‐associated protein 1 (YAP1). Int J Mol Sci. 2021;22(16):8718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Zhou H, Xiong Y, Peng L, Wang R, Zhang H, Fu Z. LncRNA‐cCSC1 modulates cancer stem cell properties in colorectal cancer via activation of the Hedgehog signaling pathway. J Cell Biochem. 2020;121(3):2510‐2524. [DOI] [PubMed] [Google Scholar]
- 150. Yang Z, Cui Y, Ni W, Kim S, Xuan Y. Gli1, a potential regulator of esophageal cancer stem cell, is identified as an independent adverse prognostic factor in esophageal squamous cell carcinoma. J Cancer Res Clin Oncol. 2017;143(2):243‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Palle K, Mani C, Tripathi K, Athar M. Aberrant GLI1 activation in DNA damage response, carcinogenesis and chemoresistance. Cancers (Basel). 2015;7(4):2330‐2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Tolba MF, Abdel‐Rahman SZ. Pterostilbine, an active component of blueberries, sensitizes colon cancer cells to 5‐fluorouracil cytotoxicity. Sci Rep. 2015;5:15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Usui T, Sakurai M, Umata K, et al. Hedgehog signals mediate anti‐cancer drug resistance in three‐dimensional primary colorectal cancer organoid culture. Int J Mol Sci. 2018;19(4):1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Lu J, Cao LL, Xu Y, et al. FOXC1 modulates stem‐like cell properties and chemoresistance through hedgehog and EMT signaling in gastric adenocarcinoma. Mol Ther. 2021. [DOI] [PubMed] [Google Scholar]
- 155. Huang F, Shi Q, Li Y, et al. HER2/EGFR‐AKT signaling switches TGFbeta from inhibiting cell proliferation to promoting cell migration in breast cancer. Cancer Res. 2018;78(21):6073‐6085. [DOI] [PubMed] [Google Scholar]
- 156. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23(10):1124‐1134. [DOI] [PubMed] [Google Scholar]
- 157. Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol. 2019;234(6):8381‐8395. [DOI] [PubMed] [Google Scholar]
- 158. Nakano M, Kikushige Y, Miyawaki K, et al. Dedifferentiation process driven by TGF‐beta signaling enhances stem cell properties in human colorectal cancer. Oncogene. 2019;38(6):780‐793. [DOI] [PubMed] [Google Scholar]
- 159. Sheen YY, Kim MJ, Park SA, Park SY, Nam JS. Targeting the transforming growth factor‐beta signaling in cancer therapy. Biomol Ther (Seoul). 2013;21(5):323‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Massague J, Wotton D. Transcriptional control by the TGF‐beta/Smad signaling system. EMBO J. 2000;19(8):1745‐1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Hirata N, Yamada S, Yanagida S, Ono A, Yasuhiko Y, Kanda Y. Transforming growth factor beta promotes the expansion of cancer stem cells via S1PR3 by ligand‐independent Notch activation. Biol Pharm Bull. 2022;45(5):649‐658. [DOI] [PubMed] [Google Scholar]
- 162. Miller AV, Alvarez SE, Spiegel S, Lebman DA. Sphingosine kinases and sphingosine‐1‐phosphate are critical for transforming growth factor beta‐induced extracellular signal‐regulated kinase 1 and 2 activation and promotion of migration and invasion of esophageal cancer cells. Mol Cell Biol. 2008;28(12):4142‐4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Hirata N, Yamada S, Shoda T, Kurihara M, Sekino Y, Kanda Y. Sphingosine‐1‐phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand‐independent Notch activation. Nat Commun. 2014;5:4806. [DOI] [PubMed] [Google Scholar]
- 164. Yu D, Shin HS, Lee YS, Lee YC. miR‐106b modulates cancer stem cell characteristics through TGF‐beta/Smad signaling in CD44‐positive gastric cancer cells. Lab Invest. 2014;94(12):1370‐1381. [DOI] [PubMed] [Google Scholar]
- 165. Jiang F, Mu J, Wang X, et al. The repressive effect of miR‐148a on TGF beta‐SMADs signal pathway is involved in the glabridin‐induced inhibition of the cancer stem cells‐like properties in hepatocellular carcinoma cells. PLoS One. 2014;9(5):e96698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Wu L, Han L, Zhou C, et al. TGF‐beta1‐induced CK17 enhances cancer stem cell‐like properties rather than EMT in promoting cervical cancer metastasis via the ERK1/2‐MZF1 signaling pathway. FEBS J. 2017;284(18):3000‐3017. [DOI] [PubMed] [Google Scholar]
- 167. Shipitsin M, Campbell LL, Argani P, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11(3):259‐273. [DOI] [PubMed] [Google Scholar]
- 168. Mani SA, Guo W, Liao MJ, et al. The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Futakuchi M, Lami K, Tachibana Y, Yamamoto Y, Furukawa M, Fukuoka J. The effects of TGF‐beta signaling on cancer cells and cancer stem cells in the bone microenvironment. Int J Mol Sci. 2019;20(20):5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Shao T, Song P, Hua H, et al. Gamma synuclein is a novel Twist1 target that promotes TGF‐beta‐induced cancer cell migration and invasion. Cell Death Dis. 2018;9(6):625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Moon H, Ju HL, Chung SI, et al. Transforming growth factor‐beta promotes liver tumorigenesis in mice via up‐regulation of snail. Gastroenterology. 2017;153(5):1378‐1391.e6. [DOI] [PubMed] [Google Scholar]
- 172. Chaffer CL, Marjanovic ND, Lee T, et al. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013;154(1):61‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Brown JA, Yonekubo Y, Hanson N, et al. TGF‐beta‐induced quiescence mediates chemoresistance of tumor‐propagating cells in squamous cell carcinoma. Cell Stem Cell. 2017;21(5):650‐664.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Gencer S, Oleinik N, Kim J, et al. TGF‐beta receptor I/II trafficking and signaling at primary cilia are inhibited by ceramide to attenuate cell migration and tumor metastasis. Sci Signal. 2017;10(502). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Natsuizaka M, Whelan KA, Kagawa S, et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat Commun. 2017;8(1):1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Tang YA, Chen YF, Bao Y, et al. Hypoxic tumor microenvironment activates GLI2 via HIF‐1alpha and TGF‐beta2 to promote chemoresistance in colorectal cancer. Proc Natl Acad Sci U S A. 2018;115(26):E5990‐E5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Hashemi Goradel N, Najafi M, Salehi E, Farhood B, Mortezaee K. Cyclooxygenase‐2 in cancer: a review. J Cell Physiol. 2019;234(5):5683‐5699. [DOI] [PubMed] [Google Scholar]
- 178. Lee JH, Jung SM, Yang KM, et al. A20 promotes metastasis of aggressive basal‐like breast cancers through multi‐monoubiquitylation of Snail1. Nat Cell Biol. 2017;19(10):1260‐1273. [DOI] [PubMed] [Google Scholar]
- 179. Napetschnig J, Wu H. Molecular basis of NF‐kappaB signaling. Annu Rev Biophys. 2013;42:443‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Oeckinghaus A, Ghosh S. The NF‐kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Widera D, Kaus A, Kaltschmidt C, Kaltschmidt B. Neural stem cells, inflammation and NF‐kappaB: basic principle of maintenance and repair or origin of brain tumours? J Cell Mol Med. 2008;12(2):459‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Hoesel B, Schmid JA. The complexity of NF‐kappaB signaling in inflammation and cancer. Mol Cancer. 2013;12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Schmid JA, Birbach A. IkappaB kinase beta (IKKbeta/IKK2/IKBKB)–a key molecule in signaling to the transcription factor NF‐kappaB. Cytokine Growth Factor Rev. 2008;19(2):157‐165. [DOI] [PubMed] [Google Scholar]
- 184. Guzman ML, Neering SJ, Upchurch D, et al. Nuclear factor‐kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98(8):2301‐2307. [DOI] [PubMed] [Google Scholar]
- 185. Cheng JH, Zhang WJ, Zhu JF, et al. CaMKIIgamma regulates the viability and self‐renewal of acute myeloid leukaemia stem‐like cells by the Alox5/NF‐kappaB pathway. Int J Lab Hematol. 2021;43(4):699‐706. [DOI] [PubMed] [Google Scholar]
- 186. Rajasekhar VK, Studer L, Gerald W, Socci ND, Scher HI. Tumour‐initiating stem‐like cells in human prostate cancer exhibit increased NF‐kappaB signalling. Nat Commun. 2011;2:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Gonzalez‐Torres C, Gaytan‐Cervantes J, Vazquez‐Santillan K, et al. NF‐kappaB participates in the stem cell phenotype of ovarian cancer cells. Arch Med Res. 2017;48(4):343‐351. [DOI] [PubMed] [Google Scholar]
- 188. Myant KB, Cammareri P, McGhee EJ, et al. ROS production and NF‐kappaB activation triggered by RAC1 facilitate WNT‐driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013;12(6):761‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Xu J, Zhang Z, Qian M, et al. Cullin‐7 (CUL7) is overexpressed in glioma cells and promotes tumorigenesis via NF‐kappaB activation. J Exp Clin Cancer Res. 2020;39(1):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Garner JM, Fan M, Yang CH, et al. Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappaB signaling in glioblastoma cancer stem cells regulates the Notch pathway. J Biol Chem. 2013;288(36):26167‐26176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Song L, Liu L, Wu Z, et al. TGF‐beta induces miR‐182 to sustain NF‐kappaB activation in glioma subsets. J Clin Invest. 2012;122(10):3563‐3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Rinkenbaugh AL, Baldwin AS. The NF‐kappaB pathway and cancer stem cells. Cells. 2016;5(2):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Liu M, Sakamaki T, Casimiro MC, et al. The canonical NF‐kappaB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res. 2010;70(24):10464‐10473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Cao Y, Luo JL, Karin M. IkappaB kinase alpha kinase activity is required for self‐renewal of ErbB2/Her2‐transformed mammary tumor‐initiating cells. Proc Natl Acad Sci U S A. 2007;104(40):15852‐15857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Zhang W, Tan W, Wu X, et al. A NIK‐IKKalpha module expands ErbB2‐induced tumor‐initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell. 2013;23(5):647‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Kumar S, Nandi A, Singh S, et al. Dll1(+) quiescent tumor stem cells drive chemoresistance in breast cancer through NF‐kappaB survival pathway. Nat Commun. 2021;12(1):432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Tasian SK, Teachey DT, Rheingold SR. Targeting the PI3K/mTOR pathway in pediatric hematologic malignancies. Front Oncol. 2014;4:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Vanhaesebroeck B, Guillermet‐Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform‐specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329‐341. [DOI] [PubMed] [Google Scholar]
- 199. Wang Q, Chen X, Hay N. Akt as a target for cancer therapy: more is not always better (lessons from studies in mice). Br J Cancer. 2017;117(2):159‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Wei X, Luo L, Chen J. Roles of mTOR signaling in tissue regeneration. Cells. 2019;8(9):1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Sancak Y, Thoreen CC, Peterson TR, et al. PRAS40 is an insulin‐regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25(6):903‐915. [DOI] [PubMed] [Google Scholar]
- 202. Duzgun Z, Eroglu Z, Biray Avci C. Role of mTOR in glioblastoma. Gene. 2016;575(2 Pt 1):187‐190. [DOI] [PubMed] [Google Scholar]
- 203. Yip CK, Murata K, Walz T, Sabatini DM, Kang SA. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell. 2010;38(5):768‐774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Knowles MA, Platt FM, Ross RL, Hurst CD. Phosphatidylinositol 3‐kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009;28(3‐4):305‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Matsubara S, Tsukasa K, Kuwahata T, Takao S. Prevention of Akt phosphorylation is a key to targeting cancer stem‐like cells by mTOR inhibition. Hum Cell. 2020;33(4):1197‐1203. [DOI] [PubMed] [Google Scholar]
- 206. Ghayad SE, Cohen PA. Inhibitors of the PI3K/Akt/mTOR pathway: new hope for breast cancer patients. Recent Pat Anticancer Drug Discov. 2010;5(1):29‐57. [DOI] [PubMed] [Google Scholar]
- 207. Dubrovska A, Kim S, Salamone RJ, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem‐like cell populations. Proc Natl Acad Sci U S A. 2009;106(1):268‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Korkaya H, Paulson A, Charafe‐Jauffret E, et al. Regulation of mammary stem/progenitor cells by PTEN/Akt/beta‐catenin signaling. PLoS Biol. 2009;7(6):e1000121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Yang CF, Yang GD, Huang TJ, et al. EB‐virus latent membrane protein 1 potentiates the stemness of nasopharyngeal carcinoma via preferential activation of PI3K/AKT pathway by a positive feedback loop. Oncogene. 2016;35(26):3419‐3431. [DOI] [PubMed] [Google Scholar]
- 210. Li X, Wu C, Chen N, et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 2016;7(22):33440‐33450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Wolin EM. PI3K/Akt/mTOR pathway inhibitors in the therapy of pancreatic neuroendocrine tumors. Cancer Lett. 2013;335(1):1‐8. [DOI] [PubMed] [Google Scholar]
- 212. Tan AC. Targeting the PI3K/Akt/mTOR pathway in non‐small cell lung cancer (NSCLC). Thorac Cancer. 2020;11(3):511‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Shorning BY, Dass MS, Smalley MJ, Pearson HB. The PI3K‐AKT‐mTOR pathway and prostate cancer: at the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci. 2020;21(12):4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Miricescu D, Totan A, Stanescu S, II, Badoiu SC, Stefani C, Greabu M. PI3K/AKT/mTOR signaling pathway in breast cancer: from molecular landscape to clinical aspects. Int J Mol Sci. 2020;22(1):173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Bai J, Chen WB, Zhang XY, et al. HIF‐2alpha regulates CD44 to promote cancer stem cell activation in triple‐negative breast cancer via PI3K/AKT/mTOR signaling. World J Stem Cells. 2020;12(1):87‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Corominas‐Faja B, Cufi S, Oliveras‐Ferraros C, et al. Nuclear reprogramming of luminal‐like breast cancer cells generates Sox2‐overexpressing cancer stem‐like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle. 2013;12(18):3109‐3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Douville J, Beaulieu R, Balicki D. ALDH1 as a functional marker of cancer stem and progenitor cells. Stem Cells Dev. 2009;18(1):17‐25. [DOI] [PubMed] [Google Scholar]
- 218. Tan X, Zhang Z, Yao H, Shen L. Tim‐4 promotes the growth of colorectal cancer by activating angiogenesis and recruiting tumor‐associated macrophages via the PI3K/AKT/mTOR signaling pathway. Cancer Lett. 2018;436:119‐128. [DOI] [PubMed] [Google Scholar]
- 219. Wen W, Han T, Chen C, et al. Cyclin G1 expands liver tumor‐initiating cells by Sox2 induction via Akt/mTOR signaling. Mol Cancer Ther. 2013;12(9):1796‐1804. [DOI] [PubMed] [Google Scholar]
- 220. Deng J, Bai X, Feng X, et al. Inhibition of PI3K/Akt/mTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial‐mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer. 2019;19(1):618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Chang L, Graham PH, Hao J, et al. Acquisition of epithelial‐mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4:e875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Marhold M, Tomasich E, El‐Gazzar A, et al. HIF1alpha regulates mTOR signaling and viability of prostate cancer stem cells. Mol Cancer Res. 2015;13(3):556‐564. [DOI] [PubMed] [Google Scholar]
- 223. Matsumoto K, Arao T, Tanaka K, et al. mTOR signal and hypoxia‐inducible factor‐1 alpha regulate CD133 expression in cancer cells. Cancer Res. 2009;69(18):7160‐7164. [DOI] [PubMed] [Google Scholar]
- 224. Yang Z, Zhang L, Ma A, et al. Transient mTOR inhibition facilitates continuous growth of liver tumors by modulating the maintenance of CD133+ cell populations. PLoS One. 2011;6(12):e28405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Jung MJ, Rho JK, Kim YM, et al. Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug‐resistant non‐small cell lung cancer cells. Oncogene. 2013;32(2):209‐221. [DOI] [PubMed] [Google Scholar]
- 226. Bleau AM, Hambardzumyan D, Ozawa T, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem‐like cells. Cell Stem Cell. 2009;4(3):226‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Airiau K, Mahon FX, Josselin M, Jeanneteau M, Belloc F. PI3K/mTOR pathway inhibitors sensitize chronic myeloid leukemia stem cells to nilotinib and restore the response of progenitors to nilotinib in the presence of stem cell factor. Cell Death Dis. 2013;4:e827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. You M, Jin J, Liu Q, Xu Q, Shi J, Hou Y. PPARalpha promotes cancer cell Glut1 transcription repression. J Cell Biochem. 2017;118(6):1556‐1562. [DOI] [PubMed] [Google Scholar]
- 229. Hou Y, Moreau F, Chadee K. PPARgamma is an E3 ligase that induces the degradation of NFkappaB/p65. Nat Commun. 2012;3:1300. [DOI] [PubMed] [Google Scholar]
- 230. Zhang W, Xu Y, Xu Q, Shi H, Shi J, Hou Y. PPARdelta promotes tumor progression via activation of Glut1 and SLC1‐A5 transcription. Carcinogenesis. 2017;38(7):748‐755. [DOI] [PubMed] [Google Scholar]
- 231. Yousefnia S, Momenzadeh S, Seyed Forootan F, Ghaedi K, Nasr Esfahani MH. The influence of peroxisome proliferator‐activated receptor gamma (PPARgamma) ligands on cancer cell tumorigenicity. Gene. 2018;649:14‐22. [DOI] [PubMed] [Google Scholar]
- 232. Zhang Y, Zhang X, Wang J, et al. Expression and function of PPARs in cancer stem cells. Curr Stem Cell Res Ther. 2016;11(3):226‐234. [DOI] [PubMed] [Google Scholar]
- 233. Han S, Wei R, Zhang X, et al. CPT1A/2‐mediated FAO enhancement–a metabolic target in radioresistant breast cancer. Front Oncol. 2019;9:1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Ma XL, Sun YF, Wang BL, et al. Sphere‐forming culture enriches liver cancer stem cells and reveals Stearoyl‐CoA desaturase 1 as a potential therapeutic target. BMC Cancer. 2019;19(1):760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Kuramoto K, Yamamoto M, Suzuki S, et al. Inhibition of the lipid droplet‐peroxisome proliferator‐activated receptor alpha axis suppresses cancer stem cell properties. Genes (Basel). 2021;12(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Ito K, Carracedo A, Weiss D, et al. A PML‐PPAR‐delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18(9):1350‐1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Kramer K, Wu J, Crowe DL. Tumor suppressor control of the cancer stem cell niche. Oncogene. 2016;35(32):4165‐4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Prost S, Relouzat F, Spentchian M, et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature. 2015;525(7569):380‐383. [DOI] [PubMed] [Google Scholar]
- 239. Wang Y, Tan H, Xu D, et al. The combinatory effects of PPAR‐gamma agonist and survivin inhibition on the cancer stem‐like phenotype and cell proliferation in bladder cancer cells. Int J Mol Med. 2014;34(1):262‐268. [DOI] [PubMed] [Google Scholar]
- 240. Basu‐Roy U, Han E, Rattanakorn K, et al. PPARgamma agonists promote differentiation of cancer stem cells by restraining YAP transcriptional activity. Oncotarget. 2016;7(38):60954‐60970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Giampietri C, Petrungaro S, Cordella M, et al. Lipid storage and autophagy in melanoma cancer cells. Int J Mol Sci. 2017;18(6):1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Bigoni‐Ordonez GD, Ortiz‐Sanchez E, Rosendo‐Chalma P, Valencia‐Gonzalez HA, Aceves C, Garcia‐Carranca A. Molecular iodine inhibits the expression of stemness markers on cancer stem‐like cells of established cell lines derived from cervical cancer. BMC Cancer. 2018;18(1):928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Liu L, Yang Z, Xu Y, et al. Inhibition of oxidative stress‐elicited AKT activation facilitates PPARgamma agonist‐mediated inhibition of stem cell character and tumor growth of liver cancer cells. PLoS One. 2013;8(8):e73038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Paget S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989;8(2):98‐101. [PubMed] [Google Scholar]
- 245. Liu Q, Zhang H, Jiang X, Qian C, Liu Z, Luo D. Factors involved in cancer metastasis: a better understanding to “seed and soil” hypothesis. Mol Cancer. 2017;16(1):176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Adjei IM, Blanka S. Modulation of the tumor microenvironment for cancer treatment: a biomaterials approach. J Funct Biomater. 2015;6(1):81‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Scherer HJ. Structural development in gliomas. Am J Cancer. 1938;34(3):333‐351. [Google Scholar]
- 248. Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11(1):69‐82. [DOI] [PubMed] [Google Scholar]
- 249. Androutsellis‐Theotokis A, Rueger MA, Park DM, et al. Angiogenic factors stimulate growth of adult neural stem cells. PLoS One. 2010;5(2):e9414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Shen Q, Wang Y, Kokovay E, et al. Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell–cell interactions. Cell Stem Cell. 2008;3(3):289‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Kokovay E, Goderie S, Wang Y, et al. Adult SVZ lineage cells home to and leave the vascular niche via differential responses to SDF1/CXCR4 signaling. Cell Stem Cell. 2010;7(2):163‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Irani S, Dehghan A. The expression and functional significance of vascular endothelial‐cadherin, CD44, and vimentin in oral squamous cell carcinoma. J Int Soc Prev Community Dent. 2018;8(2):110‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Paulis YW, Huijbers EJ, van der Schaft DW, et al. CD44 enhances tumor aggressiveness by promoting tumor cell plasticity. Oncotarget. 2015;6(23):19634‐19646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Zhu B, Zhou L, Yu L, et al. Evaluation of the correlation of vasculogenic mimicry, ALDH1, KAI1 and microvessel density in the prediction of metastasis and prognosis in colorectal carcinoma. BMC Surg. 2017;17(1):47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Xing P, Dong H, Liu Q, et al. ALDH1 expression and vasculogenic mimicry are positively associated with poor prognosis in patients with breast cancer. Cell Physiol Biochem. 2018;49(3):961‐970. [DOI] [PubMed] [Google Scholar]
- 256. Izawa Y, Kashii‐Magaribuchi K, Yoshida K, et al. Stem‐like human breast cancer cells initiate vasculogenic mimicry on matrigel. Acta Histochem Cytochem. 2018;51(6):173‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Liu TJ, Sun BC, Zhao XL, et al. CD133+ cells with cancer stem cell characteristics associates with vasculogenic mimicry in triple‐negative breast cancer. Oncogene. 2013;32(5):544‐553. [DOI] [PubMed] [Google Scholar]
- 258. Mirshahi P, Rafii A, Vincent L, et al. Vasculogenic mimicry of acute leukemic bone marrow stromal cells. Leukemia. 2009;23(6):1039‐1048. [DOI] [PubMed] [Google Scholar]
- 259. Yan GN, Yang L, Lv YF, et al. Endothelial cells promote stem‐like phenotype of glioma cells through activating the Hedgehog pathway. J Pathol. 2014;234(1):11‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Heiden KB, Williamson AJ, Doscas ME, et al. The sonic hedgehog signaling pathway maintains the cancer stem cell self‐renewal of anaplastic thyroid cancer by inducing snail expression. J Clin Endocrinol Metab. 2014;99(11):E2178‐E2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Yu D, Shin HS, Lee YS, Lee D, Kim S, Lee YC. Genistein attenuates cancer stem cell characteristics in gastric cancer through the downregulation of Gli1. Oncol Rep. 2014;31(2):673‐678. [DOI] [PubMed] [Google Scholar]
- 262. Zhu TS, Costello MA, Talsma CE, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self‐renewal of cancer stem‐like cells. Cancer Res. 2011;71(18):6061‐6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Lu J, Ye X, Fan F, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged‐1. Cancer Cell. 2013;23(2):171‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Charles N, Ozawa T, Squatrito M, et al. Perivascular nitric oxide activates notch signaling and promotes stem‐like character in PDGF‐induced glioma cells. Cell Stem Cell. 2010;6(2):141‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Hovinga KE, Shimizu F, Wang R, et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells. 2010;28(6):1019‐1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova‐Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22(4):436‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Chouaib S, Noman MZ, Kosmatopoulos K, Curran MA. Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene. 2017;36(4):439‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Kim H, Lin Q, Glazer PM, Yun Z. The hypoxic tumor microenvironment in vivo selects the cancer stem cell fate of breast cancer cells. Breast Cancer Res. 2018;20(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia‐inducible factor‐1‐dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62(12):3387‐3394. [PubMed] [Google Scholar]
- 270. McIntyre A, Hulikova A, Ledaki I, et al. Disrupting hypoxia‐induced bicarbonate transport acidifies tumor cells and suppresses tumor growth. Cancer Res. 2016;76(13):3744‐3755. [DOI] [PubMed] [Google Scholar]
- 271. Storci G, Bertoni S, De Carolis S, et al. Slug/beta‐catenin‐dependent proinflammatory phenotype in hypoxic breast cancer stem cells. Am J Pathol. 2013;183(5):1688‐1697. [DOI] [PubMed] [Google Scholar]
- 272. Csermely P, Hodsagi J, Korcsmaros T, et al. Cancer stem cells display extremely large evolvability: alternating plastic and rigid networks as a potential mechanism: network models, novel therapeutic target strategies, and the contributions of hypoxia, inflammation and cellular senescence. Semin Cancer Biol. 2015;30:42‐51. [DOI] [PubMed] [Google Scholar]
- 273. Jeong H, Kim S, Hong BJ, et al. Tumor‐associated macrophages enhance tumor hypoxia and aerobic glycolysis. Cancer Res. 2019;79(4):795‐806. [DOI] [PubMed] [Google Scholar]
- 274. Semenza GL. The hypoxic tumor microenvironment: a driving force for breast cancer progression. Biochim Biophys Acta. 2016;1863(3):382‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Barnhart BC, Simon MC. Metastasis and stem cell pathways. Cancer Metastasis Rev. 2007;26(2):261‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Hung SC, Deng WP, Yang WK, et al. Mesenchymal stem cell targeting of microscopic tumors and tumor stroma development monitored by noninvasive in vivo positron emission tomography imaging. Clin Cancer Res. 2005;11(21):7749‐7756. [DOI] [PubMed] [Google Scholar]
- 277. Nishi H, Nakada T, Kyo S, Inoue M, Shay JW, Isaka K. Hypoxia‐inducible factor 1 mediates upregulation of telomerase (hTERT). Mol Cell Biol. 2004;24(13):6076‐6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Schoning JP, Monteiro M, Gu W. Drug resistance and cancer stem cells: the shared but distinct roles of hypoxia‐inducible factors HIF1alpha and HIF2alpha. Clin Exp Pharmacol Physiol. 2017;44(2):153‐161. [DOI] [PubMed] [Google Scholar]
- 279. Seo EJ, Kim DK, Jang IH, et al. Hypoxia‐NOTCH1‐SOX2 signaling is important for maintaining cancer stem cells in ovarian cancer. Oncotarget. 2016;7(34):55624‐55638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Zhang Z, Han H, Rong Y, et al. Hypoxia potentiates gemcitabine‐induced stemness in pancreatic cancer cells through AKT/Notch1 signaling. J Exp Clin Cancer Res. 2018;37(1):291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Qiang L, Wu T, Zhang HW, et al. HIF‐1alpha is critical for hypoxia‐mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ. 2012;19(2):284‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Yang L, Lin C, Wang L, Guo H, Wang X. Hypoxia and hypoxia‐inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp Cell Res. 2012;318(19):2417‐2426. [DOI] [PubMed] [Google Scholar]
- 283. Zeisberg M, Strutz F, Muller GA. Role of fibroblast activation in inducing interstitial fibrosis. J Nephrol. 2000;13(3):S111‐S120. [PubMed] [Google Scholar]
- 284. Nishi H, Nakada T, Hokamura M, et al. Hypoxia‐inducible factor‐1 transactivates transforming growth factor‐beta3 in trophoblast. Endocrinology. 2004;145(9):4113‐4118. [DOI] [PubMed] [Google Scholar]
- 285. Hung SP, Ho JH, Shih YR, Lo T, Lee OK. Hypoxia promotes proliferation and osteogenic differentiation potentials of human mesenchymal stem cells. J Orthop Res. 2012;30(2):260‐266. [DOI] [PubMed] [Google Scholar]
- 286. Hung SP, Yang MH, Tseng KF, Lee OK. Hypoxia‐induced secretion of TGF‐beta1 in mesenchymal stem cell promotes breast cancer cell progression. Cell Transplant. 2013;22(10):1869‐1882. [DOI] [PubMed] [Google Scholar]
- 287. Liu Y, Chen C, Qian P, et al. Gd‐metallofullerenol nanomaterial as non‐toxic breast cancer stem cell‐specific inhibitor. Nat Commun. 2015;6:5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Hou PC, Li YH, Lin SC, et al. Hypoxia‐induced downregulation of DUSP‐2 phosphatase drives colon cancer stemness. Cancer Res. 2017;77(16):4305‐4316. [DOI] [PubMed] [Google Scholar]
- 289. Dong XF, Liu TQ, Zhi XT, et al. COX‐2/PGE2 axis regulates HIF2alpha activity to promote hepatocellular carcinoma hypoxic response and reduce the sensitivity of sorafenib treatment. Clin Cancer Res. 2018;24(13):3204‐3216. [DOI] [PubMed] [Google Scholar]
- 290. Miranda A, Hamilton PT, Zhang AW, et al. Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc Natl Acad Sci U S A. 2019;116(18):9020‐9029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Raggi C, Correnti M, Sica A, et al. Cholangiocarcinoma stem‐like subset shapes tumor‐initiating niche by educating associated macrophages. J Hepatol. 2017;66(1):102‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Tao W, Chu C, Zhou W, et al. Dual role of WISP1 in maintaining glioma stem cells and tumor‐supportive macrophages in glioblastoma. Nat Commun. 2020;11(1):3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Guo X, Pan Y, Gutmann DH. Genetic and genomic alterations differentially dictate low‐grade glioma growth through cancer stem cell‐specific chemokine recruitment of T cells and microglia. Neuro Oncol. 2019;21(10):1250‐1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Zhou W, Ke SQ, Huang Z, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour‐associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17(2):170‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Guo X, Zhao Y, Yan H, et al. Single tumor‐initiating cells evade immune clearance by recruiting type II macrophages. Genes Dev. 2017;31(3):247‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Wu A, Wei J, Kong LY, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12(11):1113‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Fan QM, Jing YY, Yu GF, et al. Tumor‐associated macrophages promote cancer stem cell‐like properties via transforming growth factor‐beta1‐induced epithelial‐mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014;352(2):160‐168. [DOI] [PubMed] [Google Scholar]
- 298. Jinushi M, Chiba S, Yoshiyama H, et al. Tumor‐associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A. 2011;108(30):12425‐12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Lu H, Clauser KR, Tam WL, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. 2014;16(11):1105‐1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Shi Y, Ping YF, Zhou W, et al. Tumour‐associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat Commun. 2017;8:15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Wan S, Zhao E, Kryczek I, et al. Tumor‐associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147(6):1393‐1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121(10):3804‐3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Yang Z, He L, Lin K, et al. The KMT1A‐GATA3‐STAT3 circuit is a novel self‐renewal signaling of human bladder cancer stem cells. Clin Cancer Res. 2017;23(21):6673‐6685. [DOI] [PubMed] [Google Scholar]
- 304. Lee Y, Shin JH, Longmire M, et al. CD44+ cells in head and neck squamous cell carcinoma suppress T‐cell‐mediated immunity by selective constitutive and inducible expression of PD‐L1. Clin Cancer Res. 2016;22(14):3571‐3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17(9):1025‐1036. [DOI] [PubMed] [Google Scholar]
- 306. Codony‐Servat J, Rosell R. Cancer stem cells and immunoresistance: clinical implications and solutions. Transl Lung Cancer Res. 2015;4(6):689‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Tallerico R, Todaro M, Di Franco S, et al. Human NK cells selective targeting of colon cancer‐initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J Immunol. 2013;190(5):2381‐2390. [DOI] [PubMed] [Google Scholar]
- 308. Castriconi R, Daga A, Dondero A, et al. NK cells recognize and kill human glioblastoma cells with stem cell‐like properties. J Immunol. 2009;182(6):3530‐3539. [DOI] [PubMed] [Google Scholar]
- 309. Pietra G, Manzini C, Vitale M, et al. Natural killer cells kill human melanoma cells with characteristics of cancer stem cells. Int Immunol. 2009;21(7):793‐801. [DOI] [PubMed] [Google Scholar]
- 310. Yin T, Wang G, He S, Liu Q, Sun J, Wang Y. Human cancer cells with stem cell‐like phenotype exhibit enhanced sensitivity to the cytotoxicity of IL‐2 and IL‐15 activated natural killer cells. Cell Immunol. 2016;300:41‐45. [DOI] [PubMed] [Google Scholar]
- 311. Patankar MS, Jing Y, Morrison JC, et al. Potent suppression of natural killer cell response mediated by the ovarian tumor marker CA125. Gynecol Oncol. 2005;99(3):704‐713. [DOI] [PubMed] [Google Scholar]
- 312. Cristiani CM, Turdo A, Ventura V, et al. Accumulation of circulating CCR7(+) natural killer cells marks melanoma evolution and reveals a CCL19‐dependent metastatic pathway. Cancer Immunol Res. 2019;7(5):841‐852. [DOI] [PubMed] [Google Scholar]
- 313. Ames E, Canter RJ, Grossenbacher SK, et al. NK cells preferentially target tumor cells with a cancer stem cell phenotype. J Immunol. 2015;195(8):4010‐4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Ames E, Canter RJ, Grossenbacher SK, et al. Enhanced targeting of stem‐like solid tumor cells with radiation and natural killer cells. Oncoimmunology. 2015;4(9):e1036212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Wang B, Wang Q, Wang Z, et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014;74(20):5746‐5757. [DOI] [PubMed] [Google Scholar]
- 316. Jin H, Kim HJ. NK cells lose their cytotoxicity function against cancer stem cell‐rich radiotherapy‐resistant breast cancer cell populations. Int J Mol Sci. 2021;22(17):9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Song M, He J, Pan QZ, et al. Cancer‐associated fibroblast‐mediated cellular crosstalk supports hepatocellular carcinoma progression. Hepatology. 2021;73(5):1717‐1735. [DOI] [PubMed] [Google Scholar]
- 318. Yoon H, Tang CM, Banerjee S, et al. TGF‐beta1‐mediated transition of resident fibroblasts to cancer‐associated fibroblasts promotes cancer metastasis in gastrointestinal stromal tumor. Oncogenesis. 2021;10(2):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Chen Z, Yan X, Li K, Ling Y, Kang H. Stromal fibroblast‐derived MFAP5 promotes the invasion and migration of breast cancer cells via Notch1/slug signaling. Clin Transl Oncol. 2020;22(4):522‐531. [DOI] [PubMed] [Google Scholar]
- 320. Costa A, Kieffer Y, Scholer‐Dahirel A, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33(3):463‐479.e10. [DOI] [PubMed] [Google Scholar]
- 321. Chen S, Morine Y, Tokuda K, et al. Cancer‐associated fibroblast‐induced M2‐polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor‐1 pathway. Int J Oncol. 2021;59(2):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Zhang H, Deng T, Liu R, et al. CAF secreted miR‐522 suppresses ferroptosis and promotes acquired chemo‐resistance in gastric cancer. Mol Cancer. 2020;19(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Liu J, Ren L, Li S, et al. The biology, function, and applications of exosomes in cancer. Acta Pharm Sin B. 2021;11(9):2783‐2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Nallasamy P, Nimmakayala RK, Karmakar S, et al. Pancreatic tumor microenvironment factor promotes cancer stemness via SPP1‐CD44 axis. Gastroenterology. 2021;161(6):1998‐2013.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Li Y, Wang R, Xiong S, et al. Cancer‐associated fibroblasts promote the stemness of CD24(+) liver cells via paracrine signaling. J Mol Med. 2019;97(2):243‐255. [DOI] [PubMed] [Google Scholar]
- 326. Zhao Q, Huang L, Qin G, et al. Cancer‐associated fibroblasts induce monocytic myeloid‐derived suppressor cell generation via IL‐6/exosomal miR‐21‐activated STAT3 signaling to promote cisplatin resistance in esophageal squamous cell carcinoma. Cancer Lett. 2021;518:35‐48. [DOI] [PubMed] [Google Scholar]
- 327. Atiya H, Frisbie L, Pressimone C, Coffman L. Mesenchymal stem cells in the tumor microenvironment. Adv Exp Med Biol. 2020;1234:31‐42. [DOI] [PubMed] [Google Scholar]
- 328. Barcellos‐de‐Souza P, Comito G, Pons‐Segura C, et al. Mesenchymal stem cells are recruited and activated into carcinoma‐associated fibroblasts by prostate cancer microenvironment‐derived TGF‐beta1. Stem Cells. 2016;34(10):2536‐2547. [DOI] [PubMed] [Google Scholar]
- 329. Oya Y, Hayakawa Y, Koike K. Tumor microenvironment in gastric cancers. Cancer Sci. 2020;111(8):2696‐2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Wang Y, Lan W, Xu M, et al. Cancer‐associated fibroblast‐derived SDF‐1 induces epithelial‐mesenchymal transition of lung adenocarcinoma via CXCR4/beta‐catenin/PPARdelta signalling. Cell Death Dis. 2021;12(2):214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Goulet CR, Champagne A, Bernard G, et al. Cancer‐associated fibroblasts induce epithelial‐mesenchymal transition of bladder cancer cells through paracrine IL‐6 signalling. BMC Cancer. 2019;19(1):137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Shibata M, Hoque MO. Targeting cancer stem cells: a strategy for effective eradication of cancer. Cancers (Basel). 2019;11(5):732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Desai A, Yan Y, Gerson SL. Concise reviews: cancer stem cell targeted therapies: toward clinical success. Stem Cells Transl Med. 2019;8(1):75‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Zhang D, Tang DG, Rycaj K. Cancer stem cells: regulation programs, immunological properties and immunotherapy. Semin Cancer Biol. 2018;52(Pt 2):94‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Pan Q, Li Q, Liu S, et al. Concise review: targeting cancer stem cells using immunologic approaches. Stem Cells. 2015;33(7):2085‐2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Ferrarotto R, Mitani Y, Diao L, et al. Activating NOTCH1 mutations define a distinct subgroup of patients with adenoid cystic carcinoma who have poor prognosis, propensity to bone and liver metastasis, and potential responsiveness to Notch1 inhibitors. J Clin Oncol. 2017;35(3):352‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Samon JB, Castillo‐Martin M, Hadler M, et al. Preclinical analysis of the gamma‐secretase inhibitor PF‐03084014 in combination with glucocorticoids in T‐cell acute lymphoblastic leukemia. Mol Cancer Ther. 2012;11(7):1565‐1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Wei P, Walls M, Qiu M, et al. Evaluation of selective gamma‐secretase inhibitor PF‐03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther. 2010;9(6):1618‐1628. [DOI] [PubMed] [Google Scholar]
- 339. Pandya K, Meeke K, Clementz AG, et al. Targeting both Notch and ErbB‐2 signalling pathways is required for prevention of ErbB‐2‐positive breast tumour recurrence. Br J Cancer. 2011;105(6):796‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Morgan KM, Fischer BS, Lee FY, et al. Gamma secretase inhibition by BMS‐906024 enhances efficacy of paclitaxel in lung adenocarcinoma. Mol Cancer Ther. 2017;16(12):2759‐2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Sosa Iglesias V, Theys J, Groot AJ, et al. Synergistic effects of NOTCH/gamma‐secretase inhibition and standard of care treatment modalities in non‐small cell lung cancer cells. Front Oncol. 2018;8:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Huynh C, Poliseno L, Segura MF, et al. The novel gamma secretase inhibitor RO4929097 reduces the tumor initiating potential of melanoma. PLoS One. 2011;6(9):e25264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. De Jesus‐Acosta A, Laheru D, Maitra A, et al. A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest New Drugs. 2014;32(4):739‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Tolcher AW, Messersmith WA, Mikulski SM, et al. Phase I study of RO4929097, a gamma secretase inhibitor of notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol. 2012;30(19):2348‐2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Lee SM, Moon J, Redman BG, et al. Phase 2 study of RO4929097, a gamma‐secretase inhibitor, in metastatic melanoma: SWOG 0933. Cancer. 2015;121(3):432‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Pan E, Supko JG, Kaley TJ, et al. Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J Neurooncol. 2016;130(3):571‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Peereboom DM, Ye X, Mikkelsen T, et al. A phase II and pharmacodynamic trial of RO4929097 for patients with recurrent/progressive glioblastoma. Neurosurgery. 2021;88(2):246‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Pant S, Jones SF, Kurkjian CD, et al. A first‐in‐human phase I study of the oral Notch inhibitor, LY900009, in patients with advanced cancer. Eur J Cancer. 2016;56:1‐9. [DOI] [PubMed] [Google Scholar]
- 349. Krop I, Demuth T, Guthrie T, et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK‐0752 in adult patients with advanced solid tumors. J Clin Oncol. 2012;30(19):2307‐2313. [DOI] [PubMed] [Google Scholar]
- 350. Aung KL, El‐Khoueiry AB, Gelmon K, et al. A multi‐arm phase I dose escalating study of an oral NOTCH inhibitor BMS‐986115 in patients with advanced solid tumours. Invest New Drugs. 2018;36(6):1026‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Strosberg JR, Yeatman T, Weber J, et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer. 2012;48(7):997‐1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Gounder MM, Rosenbaum E, Wu N, et al. A phase Ib/II randomized study of RO4929097, a gamma‐secretase or Notch inhibitor with or without vismodegib, a Hedgehog inhibitor, in advanced sarcoma. Clin Cancer Res. 2022;28(8):1586‐1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Messersmith WA, Shapiro GI, Cleary JM, et al. A phase I, dose‐finding study in patients with advanced solid malignancies of the oral gamma‐secretase inhibitor PF‐03084014. Clin Cancer Res. 2015;21(1):60‐67. [DOI] [PubMed] [Google Scholar]
- 354. Wang L, Zi H, Luo Y, et al. Inhibition of Notch pathway enhances the anti‐tumor effect of docetaxel in prostate cancer stem‐like cells. Stem Cell Res Ther. 2020;11(1):258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Sahebjam S, Bedard PL, Castonguay V, et al. A phase I study of the combination of ro4929097 and cediranib in patients with advanced solid tumours (PJC‐004/NCI 8503). Br J Cancer. 2013;109(4):943‐949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Kappes DJ, He X, He X. CD4‐CD8 lineage commitment: an inside view. Nat Immunol. 2005;6(8):761‐766. [DOI] [PubMed] [Google Scholar]
- 357. Yun J, Pannuti A, Espinoza I, et al. Crosstalk between PKCalpha and Notch‐4 in endocrine‐resistant breast cancer cells. Oncogenesis. 2013;2:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Kummar S, O'Sullivan Coyne G, Do KT, et al. Clinical activity of the gamma‐secretase inhibitor PF‐03084014 in adults with desmoid tumors (aggressive fibromatosis). J Clin Oncol. 2017;35(14):1561‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Gavai AV, Quesnelle C, Norris D, et al. Discovery of clinical candidate BMS‐906024: a potent pan‐Notch inhibitor for the treatment of leukemia and solid tumors. ACS Med Chem Lett. 2015;6(5):523‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Chan D, Kaplan J, Gordon G, Desai J. Activity of the gamma secretase inhibitor AL101 in desmoid tumors: a case report of 2 adult cases. Curr Oncol. 2021;28(5):3659‐3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Lehal R, Zaric J, Vigolo M, et al. Pharmacological disruption of the Notch transcription factor complex. Proc Natl Acad Sci U S A. 2020;117(28):16292‐16301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Astudillo L, Da Silva TG, Wang Z, et al. The small molecule IMR‐1 inhibits the Notch transcriptional activation complex to suppress tumorigenesis. Cancer Res. 2016;76(12):3593‐3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Ferrarotto R, Eckhardt G, Patnaik A, et al. A phase I dose‐escalation and dose‐expansion study of brontictuzumab in subjects with selected solid tumors. Ann Oncol. 2018;29(7):1561‐1568. [DOI] [PubMed] [Google Scholar]
- 364. Herrera‐Rios D, Li G, Khan D, et al. A computational guided, functional validation of a novel therapeutic antibody proposes Notch signaling as a clinical relevant and druggable target in glioma. Sci Rep. 2020;10(1):16218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Yen WC, Fischer MM, Axelrod F, et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor‐initiating cell frequency. Clin Cancer Res. 2015;21(9):2084‐2095. [DOI] [PubMed] [Google Scholar]
- 366. Hu ZI, Bendell JC, Bullock A, et al. A randomized phase II trial of nab‐paclitaxel and gemcitabine with tarextumab or placebo in patients with untreated metastatic pancreatic cancer. Cancer Med. 2019;8(11):5148‐5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Yang L, Xie G, Fan Q, Xie J. Activation of the Hedgehog‐signaling pathway in human cancer and the clinical implications. Oncogene. 2010;29(4):469‐481. [DOI] [PubMed] [Google Scholar]
- 368. Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal‐cell carcinoma. N Engl J Med. 2012;366(23):2171‐2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Catenacci DV, Junttila MR, Karrison T, et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a Hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol. 2015;33(36):4284‐4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Cohen DJ, Christos PJ, Kindler HL, et al. Vismodegib (V), a Hedgehog (HH) pathway inhibitor, combined with FOLFOX for first‐line therapy of patients (pts) with advanced gastric and gastroesophageal junction (GEJ) carcinoma: a New York Cancer Consortium led phase II randomized study. J Clin Oncol. 2013;31(15):4011. [Google Scholar]
- 371. McCleary‐Wheeler AL, Carr RM, Palmer SR, et al. Phase 1 trial of vismodegib and erlotinib combination in metastatic pancreatic cancer. Pancreatology. 2020;20(1):101‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Kaye SB, Fehrenbacher L, Holloway R, et al. A phase II, randomized, placebo‐controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin Cancer Res. 2012;18(23):6509‐6518. [DOI] [PubMed] [Google Scholar]
- 373. Rudin CM, Hann CL, Laterra J, et al. Treatment of medulloblastoma with Hedgehog pathway inhibitor GDC‐0449. N Engl J Med. 2009;361(12):1173‐1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Robinson GW, Orr BA, Wu G, et al. Vismodegib exerts targeted efficacy against recurrent sonic hedgehog‐subgroup medulloblastoma: results from phase II pediatric brain tumor consortium studies PBTC‐025B and PBTC‐032. J Clin Oncol. 2015;33(24):2646‐2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Gajjar A, Stewart CF, Ellison DW, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res. 2013;19(22):6305‐6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Petrirena GJ, Masliah‐Planchon J, Sala Q, et al. Recurrent extraneural sonic hedgehog medulloblastoma exhibiting sustained response to vismodegib and temozolomide monotherapies and inter‐metastatic molecular heterogeneity at progression. Oncotarget. 2018;9(11):10175‐10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Frappaz D, Barritault M, Montane L, et al. MEVITEM–a phase I/II trial of vismodegib + temozolomide vs temozolomide in patients with recurrent/refractory medulloblastoma with sonic hedgehog pathway activation. Neuro Oncol. 2021;23(11):1949‐1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Fosko SW, Chu MB, Armbrecht E, et al. Efficacy, rate of tumor response, and safety of a short course (12–24 weeks) of oral vismodegib in various histologic subtypes (infiltrative, nodular, and superficial) of high‐risk or locally advanced basal cell carcinoma, in an open‐label, prospective case series clinical trial. J Am Acad Dermatol. 2020;82(4):946‐954. [DOI] [PubMed] [Google Scholar]
- 379. Lear JT, Migden MR, Lewis KD, et al. Long‐term efficacy and safety of sonidegib in patients with locally advanced and metastatic basal cell carcinoma: 30‐month analysis of the randomized phase 2 BOLT study. J Eur Acad Dermatol Venereol. 2018;32(3):372‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Dummer R, Guminksi A, Gutzmer R, et al. Long‐term efficacy and safety of sonidegib in patients with advanced basal cell carcinoma: 42‐month analysis of the phase II randomized, double‐blind BOLT study. Br J Dermatol. 2020;182(6):1369‐1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Xie P, Lefrancois P. Efficacy, safety, and comparison of sonic hedgehog inhibitors in basal cell carcinomas: a systematic review and meta‐analysis. J Am Acad Dermatol. 2018;79(6):1089‐1100.e17. [DOI] [PubMed] [Google Scholar]
- 382. Danial C, Sarin KY, Oro AE, Chang AL. An investigator‐initiated open‐label trial of sonidegib in advanced basal cell carcinoma patients resistant to vismodegib. Clin Cancer Res. 2016;22(6):1325‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Cazet AS, Hui MN, Elsworth BL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9(1):2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Cortes JE, Heidel FH, Hellmann A, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high‐risk myelodysplastic syndrome. Leukemia. 2019;33(2):379‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Heuser M, Smith BD, Fiedler W, et al. Clinical benefit of glasdegib plus low‐dose cytarabine in patients with de novo and secondary acute myeloid leukemia: long‐term analysis of a phase II randomized trial. Ann Hematol. 2021;100(5):1181‐1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Cortes JE, Douglas Smith B, Wang ES, et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high‐risk MDS: phase 2 study results. Am J Hematol. 2018;93(11):1301‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI‐mediated transcription and tumor cell growth by small‐molecule antagonists. Proc Natl Acad Sci U S A. 2007;104(20):8455‐8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Huang L, Walter V, Hayes DN, Onaitis M. Hedgehog‐GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin Cancer Res. 2014;20(6):1566‐1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Didiasova M, Singh R, Wilhelm J, et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017;31(5):1916‐1928. [DOI] [PubMed] [Google Scholar]
- 390. Polydorou C, Mpekris F, Papageorgis P, Voutouri C, Stylianopoulos T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget. 2017;8(15):24506‐24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Zou WJ, Huang Z, Jiang TP, et al. Pirfenidone inhibits proliferation and promotes apoptosis of hepatocellular carcinoma cells by inhibiting the Wnt/beta‐catenin signaling pathway. Med Sci Monit. 2017;23:6107‐6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Beauchamp EM, Ringer L, Bulut G, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J Clin Invest. 2011;121(1):148‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Chang KJ, Yin JZ, Huang H, Li B, Yang MH. Arsenic trioxide inhibits the growth of cancer stem cells derived from small cell lung cancer by downregulating stem cell‐maintenance factors and inducing apoptosis via the Hedgehog signaling blockade. Transl Lung Cancer Res. 2020;9(4):1379‐1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Han JB, Sang F, Chang JJ, et al. Arsenic trioxide inhibits viability of pancreatic cancer stem cells in culture and in a xenograft model via binding to SHH‐Gli. Onco Targets Ther. 2013;6:1129‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Nakamura S, Nagano S, Nagao H, et al. Arsenic trioxide prevents osteosarcoma growth by inhibition of GLI transcription via DNA damage accumulation. PLoS One. 2013;8(7):e69466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Ally MS, Ransohoff K, Sarin K, et al. Effects of combined treatment with arsenic trioxide and itraconazole in patients with refractory metastatic basal cell carcinoma. JAMA Dermatol. 2016;152(4):452‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Wall JA, Klempner SJ, Arend RC. The anti‐DKK1 antibody DKN‐01 as an immunomodulatory combination partner for the treatment of cancer. Expert Opin Investig Drugs. 2020;29(7):639‐644. [DOI] [PubMed] [Google Scholar]
- 398. Betella I, Turbitt WJ, Szul T, et al. Wnt signaling modulator DKK1 as an immunotherapeutic target in ovarian cancer. Gynecol Oncol. 2020;157(3):765‐774. [DOI] [PubMed] [Google Scholar]
- 399. Wall JA, Meza‐Perez S, Scalise CB, et al. Manipulating the Wnt/beta‐catenin signaling pathway to promote anti‐tumor immune infiltration into the TME to sensitize ovarian cancer to ICB therapy. Gynecol Oncol. 2021;160(1):285‐294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Wise DR, Schneider JA, Armenia J, et al. Dickkopf‐1 can lead to immune evasion in metastatic castration‐resistant prostate cancer. JCO Precis Oncol. 2020;4:PO.20.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Haas MS, Kagey MH, Heath H, Schuerpf F, Rottman JB, Newman W. mDKN‐01, a novel anti‐DKK1 mAb, enhances innate immune responses in the tumor microenvironment. Mol Cancer Res. 2021;19(4):717‐725. [DOI] [PubMed] [Google Scholar]
- 402. Ryan DP, Murphy JE, Mahalingam D, et al. Current results of a phase I study of DKN‐01, an anti‐DKK1 antibody, in combination with paclitaxel (P) in patients (pts) with advanced DKK1+ esophageal cancer (EC) or gastro‐esophageal junction tumors (GEJ). J Clin Oncol. 2016;34(15):e15525. [Google Scholar]
- 403. Klempner SJ, Bendell JC, Villaflor VM, et al. Safety, efficacy, and biomarker results from a phase Ib study of the anti‐DKK1 antibody DKN‐01 in combination with pembrolizumab in advanced esophagogastric cancers. Mol Cancer Ther. 2021;20(11):2240‐2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Goyal L, Sirard C, Schrag M, et al. Phase I and biomarker study of the Wnt pathway modulator DKN‐01 in combination with gemcitabine/cisplatin in advanced biliary tract cancer. Clin Cancer Res. 2020;26(23):6158‐6167. [DOI] [PubMed] [Google Scholar]
- 405. Gurney A, Axelrod F, Bond CJ, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. 2012;109(29):11717‐11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406. Davis SL, Cardin DB, Shahda S, et al. A phase 1b dose escalation study of Wnt pathway inhibitor vantictumab in combination with nab‐paclitaxel and gemcitabine in patients with previously untreated metastatic pancreatic cancer. Invest New Drugs. 2020;38(3):821‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Diamond JR, Becerra C, Richards D, et al. Phase Ib clinical trial of the anti‐frizzled antibody vantictumab (OMP‐18R5) plus paclitaxel in patients with locally advanced or metastatic HER2‐negative breast cancer. Breast Cancer Res Treat. 2020;184(1):53‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Karvonen H, Perttila R, Niininen W, et al. Wnt5a and ROR1 activate non‐canonical Wnt signaling via RhoA in TCF3‐PBX1 acute lymphoblastic leukemia and highlight new treatment strategies via Bcl‐2 co‐targeting. Oncogene. 2019;38(17):3288‐3300. [DOI] [PubMed] [Google Scholar]
- 409. Chen Y, Chen L, Yu J, et al. Cirmtuzumab blocks Wnt5a/ROR1 stimulation of NF‐kappaB to repress autocrine STAT3 activation in chronic lymphocytic leukemia. Blood. 2019;134(13):1084‐1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Choi MY, Widhopf GF 2nd, Ghia EM, et al. Phase I trial: cirmtuzumab inhibits ROR1 signaling and stemness signatures in patients with chronic lymphocytic leukemia. Cell Stem Cell. 2018;22(6):951‐959.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Zhang S, Zhang H, Ghia EM, et al. Inhibition of chemotherapy resistant breast cancer stem cells by a ROR1 specific antibody. Proc Natl Acad Sci U S A. 2019;116(4):1370‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Yu J, Chen L, Cui B, et al. Cirmtuzumab inhibits Wnt5a‐induced Rac1 activation in chronic lymphocytic leukemia treated with ibrutinib. Leukemia. 2017;31(6):1333‐1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Xu Q, Liu G, Yuan X, et al. Antigen‐specific T‐cell response from dendritic cell vaccination using cancer stem‐like cell‐associated antigens. Stem Cells. 2009;27(8):1734‐1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Luo H, Zeng C, Fang C, Seeruttun SR, Lv L, Wang W. A new strategy using ALDHhigh‐CD8+T cells to inhibit tumorigenesis. PLoS One. 2014;9(8):e103193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Visus C, Wang Y, Lozano‐Leon A, et al. Targeting ALDH(bright) human carcinoma‐initiating cells with ALDH1A1‐specific CD8(+) T cells. Clin Cancer Res. 2011;17(19):6174‐6184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Zhao L, Yang Y, Zhou P, et al. Targeting CD133high colorectal cancer cells in vitro and in vivo with an asymmetric bispecific antibody. J Immunother. 2015;38(6):217‐228. [DOI] [PubMed] [Google Scholar]
- 417. Guo Y, Feng K, Wang Y, Han W. Targeting cancer stem cells by using chimeric antigen receptor‐modified T cells: a potential and curable approach for cancer treatment. Protein Cell. 2018;9(6):516‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Feng KC, Guo YL, Liu Y, et al. Cocktail treatment with EGFR‐specific and CD133‐specific chimeric antigen receptor‐modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol. 2017;10(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Dai H, Tong C, Shi D, et al. Efficacy and biomarker analysis of CD133‐directed CAR T cells in advanced hepatocellular carcinoma: a single‐arm, open‐label, phase II trial. Oncoimmunology. 2020;9(1):1846926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Emlet DR, Gupta P, Holgado‐Madruga M, et al. Targeting a glioblastoma cancer stem‐cell population defined by EGF receptor variant III. Cancer Res. 2014;74(4):1238‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. O'Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII‐directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Goff SL, Morgan RA, Yang JC, et al. Pilot trial of adoptive transfer of chimeric antigen receptor‐transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. 2019;42(4):126‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Brown RL, Reinke LM, Damerow MS, et al. CD44 splice isoform switching in human and mouse epithelium is essential for epithelial‐mesenchymal transition and breast cancer progression. J Clin Invest. 2011;121(3):1064‐1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Zhao S, Chen C, Chang K, et al. CD44 expression level and isoform contributes to pancreatic cancer cell plasticity, invasiveness, and response to therapy. Clin Cancer Res. 2016;22(22):5592‐5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Gotoda T, Matsumura Y, Kondo H, et al. Expression of CD44 variants and its association with survival in pancreatic cancer. Jpn J Cancer Res. 1998;89(10):1033‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Haist C, Schulte E, Bartels N, et al. CD44v6‐targeted CAR T‐cells specifically eliminate CD44 isoform 6 expressing head/neck squamous cell carcinoma cells. Oral Oncol. 2021;116:105259. [DOI] [PubMed] [Google Scholar]
- 427. de Sousa e Melo F, Kurtova AV, Harnoss JM, et al. A distinct role for Lgr5(+) stem cells in primary and metastatic colon cancer. Nature. 2017;543(7647):676‐680. [DOI] [PubMed] [Google Scholar]
- 428. Sultan M, Coyle KM, Vidovic D, Thomas ML, Gujar S, Marcato P. Hide‐and‐seek: the interplay between cancer stem cells and the immune system. Carcinogenesis. 2017;38(2):107‐118. [DOI] [PubMed] [Google Scholar]
- 429. Ghiringhelli F, Menard C, Martin F, Zitvogel L. The role of regulatory T cells in the control of natural killer cells: relevance during tumor progression. Immunol Rev. 2006;214:229‐238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.