

Abstract
Counteraction of the origin and distribution of multidrug-resistant pathogens responsible for intra-hospital infections is a worldwide issue in medicine. In this brief review, we discuss the results of our recent investigations, which argue that many antibiotics, along with inactivation of their traditional biochemical targets, can induce oxidative stress (ROS production), thus resulting in increased bactericidal efficiency. As we previously showed, hydrogen sulfide, which is produced in the cells of different pathogens protects them not only against oxidative stress but also against bactericidal antibiotics. Next, we clarified the interplay of oxidative stress, cysteine metabolism, and hydrogen sulfide production. Finally, demonstrated that small molecules, which inhibit a bacterial enzyme involved in hydrogen sulfide production, potentiate bactericidal antibiotics including quinolones, beta-lactams, and aminoglycosides against bacterial pathogens in in vitro and in mouse models of infection. These inhibitors also suppress bacterial tolerance to antibiotics by disrupting the biofilm formation and substantially reducing the number of persister bacteria, which survive the antibiotic treatment. We hypothesise that agents which limit hydrogen sulfide biosynthesis are effective tools to counteract the origin and distribution of multidrug-resistant pathogens.
Keywords: bacteria, antibiotics, oxidative stress, hydrogen sulfide generation, inhibitors of hydrogen sulfide generation ferments, new class of antimicrobial drugs
INTRODUCTION
The spread of multidrug-resistant bacterial pathogens and nosocomial infections is one of the most serious health problems worldwide. The COVID-19 pandemic has shifted this problem from the category of “important” to the category of “acute.” The slow progress in the development of new antimicrobial drugs and the rapid emergence of resistance to new therapeutic agents cause serious concern and call into question the effectiveness of current methods of treatment of bacterial infections [1–3]. Antibiotic resistance of pathogenic bacteria is the basis of the next pandemic awaiting humanity in the foreseeable future. Currently, approximately 700 000 people worldwide die annually from antibiotic-resistant bacterial infections. With an increase in the number of resistant forms of dangerous bacterial infections, the death rate will amount to millions of victims. The high mortality rate of patients with bacterial infections and economic losses will significantly exceed the negative effect of the COVID-19 pandemic. Forecasts indicate that by 2050, the number of deaths caused by antibiotic-resistant bacterial infections will reach 10 million per year, and the losses to the global economy will amount to an impressive 100 trillion US dollars [4].
The resistance of bacteria to antibiotics, i.e., the hereditary ability to grow and multiply in the presence of high levels of antibiotics [3, 5], is provided by four main mechanisms: (1) modifications of the antibiotic targets because of mutations; (2) reduction of the permeability of the bacterial cell shell for antibiotics; (3) reduction of the process of importing of antibiotics into the cell or increase of the efficiency of their export from the cell, which prevents interaction of the antibiotic with the target; (4) enzymatic degradation or other chemical modifications of the antibiotic, which reduce its affinity for the target [7–12]. Furthermore, bacteria can survive in the presence of antibiotics by halting their growth and reducing the level of metabolism, rather than genetically determined resistance. This condition is called antibiotic tolerance, or persistence. Under these conditions, only a small portion of the bacterial population survives, and the surviving bacteria are called persisters [13]. In addition to antibiotics, the formation of persisters can be induced by other types of stress, and the bacterial population spontaneously generates a small number of persisters to protect against potential genotoxic agents [14].
The problem of antibiotic tolerance attracted the attention of researchers in the early 2000s when it was shown that the resistance of bacterial biofilms to high doses of antibiotics is due to the presence of persisters [15–19]. The formation of biofilms, which accompanies most human infectious diseases, occurs especially often in clinical settings [13, 20]. Some chronic human diseases are accompanied by the generation of persisters, the fight against which is difficult and requires special approaches [14, 21–23]. The role of persisters as precursors of the formation of antibiotic-resistant mutants is becoming more and more obvious [24–27]. To successfully counteract persisters, it is necessary to develop specific compounds, potentiators, which inhibit the formation of persisters in the presence of antibiotics. In this regard, the works aimed at deciphering the mechanisms responsible for the lethal effect of antibiotics are of particular relevance.
Recent studies have shown that in addition to their main activity, many antibiotics cause oxidative stress, which damages cellular macromolecules and promotes the bactericidal activity of antibiotics [28–30]. The involvement of reactive oxygen species (ROS) as a new factor in the bactericidal action of antibiotics has expanded the development of new experimental approaches in the search for compounds with antioxidant properties, which increase the efficiency of antibiotics. We have previously found that bacteria produce hydrogen sulfide (H2S), which reduces oxidative stress and provides significant protection for a wide range of bacterial pathogens from bactericidal antibiotics [31]. Suppression of H2S formation makes bacteria less resistant to the primary action of antibiotics, thus enhancing their bactericidal activity [32]. In almost all bacteria, H2S production involves enzymes orthologous to mammalian enzymes, i.e., cystathionine-γ-synthase (CSE), cystathionine-β-synthase (CBS), and 3-mercaptopyruvate sulfotransferase (3MST) [33, 34]. It has been shown that in some pathogens including Staphylococcus aureus and Pseudomonas aeruginosa, genetic damage to the H2S biosynthesis pathways leads to sensitivity to various classes of antibiotics and the host immune response [35, 36]. Taking these data into account, we chose specific inhibitors of the CSE enzyme and showed their ability to enhance the effect of bactericidal antibiotics on the pathogenic bacteria S. aureus and P. aeruginosa and suppress the formation of bacterial persisters. Before proceeding to the description of the H2S-dependent bacterial protection system against oxidative stress and bactericidal antibiotics, we will consider in more detail the relationship between the lethal effect of antibiotics and oxidative stress.
INTERPLAY OF BACTERICIDAL ACTION OF ANTIBIOTICS AND OXIDATIVE STRESS
The molecular targets of the main classes of bactericidal antibiotics have been studied in sufficient detail. Beta-lactams interfere with cell wall biosynthesis; quinolones inhibit the DNA gyrase of gram-negative bacteria, i.e., one of the key enzymes involved in bacterial chromosome replication at the stage of the tyrosyl phosphate ether bond formation, which leads to a break in the DNA main chain; and aminoglycosides bind to receptors for the 30S subunit of the bacterial ribosome, thus causing translation errors [37]. However, over the past few decades, the accumulated data indicate that the lethal effect of antibiotics cannot be explained solely by their interaction with primary targets; it also depends on metabolic processes accompanied by the generation of ROS [38]. Three types of ROS were found in bacterial cells, i.e., superoxide anion (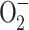 ), hydrogen peroxide (H2O2), and hydroxyl radical (∙OH), which are formed as byproducts of the activity of enzymes in the respiratory system [39]. The bacterial cell contains enzymes, which protect against ROS. SodA, SodB, and SodC superoxide dismutases reduce the superoxide anion to oxygen and hydrogen peroxide, which is further decomposed by katG and KatE catalases and AHPC alkylhydroperoxidase to form water and oxygen [40]. It has been shown that the lethal effect of hydrogen peroxide is associated with the presence of free iron ions Fe2+ in the cell [41, 42]. In this case, hydrogen peroxide turns into a hydroxyl radical ∙OH in the reaction with an iron ion (Fenton reaction):
), hydrogen peroxide (H2O2), and hydroxyl radical (∙OH), which are formed as byproducts of the activity of enzymes in the respiratory system [39]. The bacterial cell contains enzymes, which protect against ROS. SodA, SodB, and SodC superoxide dismutases reduce the superoxide anion to oxygen and hydrogen peroxide, which is further decomposed by katG and KatE catalases and AHPC alkylhydroperoxidase to form water and oxygen [40]. It has been shown that the lethal effect of hydrogen peroxide is associated with the presence of free iron ions Fe2+ in the cell [41, 42]. In this case, hydrogen peroxide turns into a hydroxyl radical ∙OH in the reaction with an iron ion (Fenton reaction):
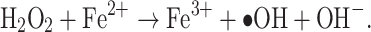 |
The ∙OH radical is characterized by high stability and the ability to cause breaks in the DNA chain [43].
The first indications for the interplay between the bactericidal action of antibiotics and ROS generation were obtained in studies which showed that activation of SoxRS regulon led to resistance of bacteria to different classes of antibiotics [44–46]. A similar conclusion was made based on data that showed that the treatment of bacteria with antioxidants, such as vitamin C or glutathione, caused an increase in the minimum inhibitory concentration for antibiotics of the quinolone and aminoglycoside classes [47, 48]. In addition, a statistically significant increase in the level of ROS was recorded in antibiotic-treated bacterial cells compared to the control [49, 50].
Direct evidence of the relationship between the lethal effect of antibiotics and ROS generation was obtained in a series of works by the Collins group. The main observation was that the lethal effect of the norfloxacin, ampicillin, and kanamycin antibiotics was accompanied by a sharp spike in the intracellular level of the hydroxyl radical, which was evaluated using the HPF fluorescent reagent, whereas the use of five bacteriostatic antibiotics did not lead to the appearance of the fluorescent signal [30, 51, 52]. It was shown that the death of bacteria under the action of bactericidal antibiotics decreased with the addition of an iron chelator (2,2-dipyridyl) or a ROS suppressor (thiourea) [30]. It has been also found that the Escherichia coli RecA mutants are characterized by a higher sensitivity to antibiotics, which indicates the involvement of ROS in the DNA damage, which is eliminated using the RecA-dependent repair system. It is important to emphasize that ROSs do not always negatively affect the survival of bacteria. For example, the use of sublethal doses of superoxide anion or inactivation of superoxide dismutase (sodAB) leads to a decrease but not an increase in the lethal effect of antibiotics [53–55].
The important role of ROS in the lethal effect of antibiotics is confirmed by the data that sodA sodB double mutants have greater resistance to antibiotics, whereas genetic damage of catalases/peroxidases (katG katE) increases the lethal effect of antibiotics of all three classes by factors of 10–100 [43]. The data on transposon mutagenesis of the E. coli genome in the presence of antibiotics allowed for an interesting conclusion. It turned out that two-thirds of transposon insertions were localized in genes responsible for electron transfer and oxidative phosphorylation, or genes involved in the formation of iron-sulfur clusters [56]. All these mutants are characterized by a decrease in the pool of enzymes involved in respiration, NADH consumption, or the electron transport chain, i.e., processes which should reduce the generation of ROS [56]. It is noteworthy that the analysis of the sensitivity of the Keio E. coli collection [57] to 22 antibiotics revealed increased sensitivity to bactericidal antibiotics in mutants that contained insertions in the RecA, RecB, or recC loci [58]. Consequently, ROS-induced double-stranded breaks in DNA are one of the factors of the lethal action of antibiotics [59].
It should be emphasized that some changes in the metabolism of bacteria caused by bactericidal antibiotics are in no way related to the functioning of their primary targets. For example, it unexpectedly turned out that the treatment of the S. aureus bacteria with the DNA-damaging agent ciprofloxacin led to the oxidation of fats and guanine bases in DNA with the formation of 8-oxo-dGTP [60]. In addition, it was found that protein aggregation was suppressed with kanamycin under conditions of peroxiredoxin AhpF overproduction [61] or ampicillin- and kanamycin-mediated lethal effects were inhibited in the case of overexpression of the MutT and RibA enzymes involved in the removal of 8-oxo-dGTP, a toxic oxidation product of the GTP pool under the action of ROS [59].
Thus, a set of data on the role of ROS in the action of bactericidal antibiotics allows one to conclude that low concentrations of ROS can be useful to trigger cell defense systems, whereas high concentrations, as a rule, lead to a lethal effect. In this regard, the question arises, how the bacterial cell recognizes these two alternative situations and how the signal is transmitted from the primary stress-causing factor to the ROS-generating system. One of these systems is the MazF/MazE toxin/antitoxin pair. When the bacterial cell is stressed, the MazE antitoxin protein degrades and the mazF toxin protein is released, which promotes the degradation of cellular mRNAs [62, 63]. Some of these RNAs are translated to form shortened improperly folded peptides, which are incorporated into the cell membrane and activate the Cpx regulatory system located in the shell [64–66]. Activation of Cpx induces expression of the YihE protein [67] encoding protein kinase, which is involved in the negative regulation of mazF [64]. The Cpx protein induces other systems, which are involved in the renaturation or degradation of improperly folded proteins, i.e., Cpx performs a protective function in the cell [68]. However, it has been turned out that deletion of the CpxR gene encoding the regulator of the Cpx system leads to the protection of cells from the lethal effects of quinolones, beta-lactams, and aminoglycosides [64, 65]. Thus, the wild-type Cpx system also performs destructive functions, possibly because of the activation of the two-component Arc system involved in the control of the redox balance in the cell [65, 66]. The Arc system participates in the regulation of the activity of components of the transport system, in particular, cytochrome oxidase bd-I [66], and thereby can regulate the intracellular content of ROS.
Thus, the bacterial cell under moderate and short oxidative stress can transform the reactive oxygen species formed and protect itself from the toxic and lethal effects. However, an increase in the amount of ROS in the cell and exposure time leads to a Cpx-dependent change in the activity of the Arc system, which, in turn, leads to an increase in the ROS content to a level exceeding its positive effect on the cell, thus resulting in a lethal effect. Thus, the bacterial cell has several regulatory systems, in which ROS can provide various scenarios of the action of antibiotics on bacteria.
A striking example of the important role of oxidative stress in the lethal effect of antibiotics is the phenomenon of the protective function of hydrogen sulfide in relation to a wide range of antibiotics that we have discovered.
GENETIC CONTROL OF HYDROGEN SULFIDE GENERATION IN BACTERIA
For many decades, hydrogen sulfide (H2S) has been known as a highly toxic poison that suppresses the respiration of organisms because of its ability to effectively reduce and inactivate terminal cytochrome oxidases and other metal-containing enzymes. The ability of bacteria to produce H2S as a byproduct of sulfur metabolism was discovered almost a century ago [69] but the study of its functions in the bacterial cell began only recently. Along with nitrogen oxide (NO) and carbon monoxide (CO), H2S belongs to the group of so-called transmitter gases, which perform (at low concentrations) an important signaling role in the cellular metabolism of eukaryotes [70]. The first transmitter gas detected in bacteria was nitric oxide, the synthesis of which is controlled by nitric oxide synthase (bNOS) [71, 72].
Computer analysis of the nucleotide sequences of bacterial genomes revealed potential bNOS analogs in a limited number of gram-positive bacterial species [73]. These data served as a reason for the search for bacterial orthologs of enzymes, which generate H2S in mammals, i.e., cystathionine-β-synthase (CBS, [EC 4.2.1.22]), cystathionine-γ-lyase (CSE, [EC 4.4.1.1]), and ТВ 3-mercaptopyruvate sulfotransferase (3MST [EC 2.8.1.2]). Unlike bNOS, the enzymes, which catalyze the biosynthesis of H2S are very conserved. At least one of the homologs of mammalian enzymes has been found in most analyzed bacterial species, which indicates the important role of hydrogen sulfide in the vital activity of bacteria [31].
Long before the enzymes which catalyze the endogenous synthesis of H2S were discovered, it was known that hydrogen sulfide can be generated in the metabolic pathway of sulfate reduction in anaerobic sulfate-producing bacteria [74]. Some intestinal bacteria can similarly produce H2S by reducing thiosulfate [75].
As noted above, bacteria contain three enzymes involved in the generation of H2S, i.e., CBS, CSE, and 3MST (Fig. 1).
Fig. 1.

Two paths of H2S generation in model organisms. In E. coli cells, cysteine is first converted to 3-mercaptopyruvate with the participation of cysteine aminotransferase (CAT), followed by the formation of H2S, pyruvate, and ammonium under the control of 3-mercaptopyruvate sulfotransferase (3MST). The Bacillus subtilis cells have two ways of generating H2S with the participation of either cystathionine-β-synthase (CBS) or cystathionine-γ-lyase (CSE).
All three enzymes use cysteine as a substrate for the synthesis of H2S, and the CBS/CSE system can also metabolize homocysteine in a series of reactions, thus leading to the formation of H2S and other compounds [74, 76]. CBS and CSE are pyridoxal phosphate-dependent enzymes. The 3MST-dependent reaction requires a preliminary stage, which is controlled by aspartate-aminotransferase with cysteine-aminotransferase activity, with the formation of sulfane, which then is transformed to H2S in the presence of reducing agents [77, 78]. These enzymes are found in the cells of at least four different bacterial pathogens. The Bacillus anthracis, P. aeruginosa, and S. aureus bacteria contain the CBS/CSE enzymes, whereas 3MST is characteristic of E. coli [31]. Other proteins potentially capable of generating H2S are also known, e.g., cysteine desulfurases but their contribution to the generation of hydrogen sulfide has not been proven. Until recently, only one pathway of H2S generation in E. coli cells with the participation of the 3MST enzyme is known [31]. However, the cyuPA operon, which controls the anaerobic degradation of cysteine to hydrogen sulfide, was relatively recently identified in the E. coli and Salmonella enterica genomes [79]. The physiological role of these enzymes is to maintain the intracellular content of cysteine at a level which can restrain the inhibitory effect of cysteine on the expression of some amino acid operons.
HYDROGEN SULFIDE PROTECTS BACTERIA FROM OXIDATIVE STRESS AND THE ACTION OF BACTERICIDAL ANTIBIOTICS
It has been shown that in mammalian organisms, H2S performs the function of a cardioprotector, controls the relaxation of blood vessels and smooth muscles, is involved in neuromodulation and protection of neurons from oxidative stress, and inhibits an anti-inflammatory effect in gastrointestinal tract infections [80, 81]. The protective role of hydrogen sulfide in relation to bactericidal antibiotics we showed for the first time more than ten years ago [31]. It has been turned out that the inactivation of the genes responsible for the generation of H2S in various pathogenic bacteria leads to an increase in the sensitivity of bacteria to a wide range of antibiotics. According to our early studies, nitric oxide (NO) exhibits similar protection of antibiotics in bNOS-containing gram-positive bacteria [82]. Therefore, the first assumption about the possible signaling function of H2S in bacteria was based on our study of the signaling functions of NO.
It is well known that NO is involved in various vital properties of the bacterial cell including resistance to various stresses, virulence, and modulation of the host’s response to stress and cellular communication [72, 73, 82–87]. In 2009, it was shown for the first time that nitric oxide protects bacteria from a wide range of antimicrobial drugs in the bNOS-containing bacterial species [82]. Interestingly, the NO, which is secreted by the B. subtilis and B. anthracis bacteria also protects against the pyocyanin toxin secreted by P. aeruginosa [82]. It is very likely that the use of NO and H2S transmitter gases is a universal strategy used by bacteria to protect against genotoxic agents. Indeed, we have found that a violation of the ability of bacteria to produce H2S as a result of inactivation of the cbs, cse, or mstA genes leads to a sharp increase in the sensitivity of these mutants to a wide range of antibiotics [31].
The addition of an exogenous source of H2S to the growth medium restored the resistance level of these mutants to antibiotics to that of the wild-type strain. It is important to note that among all the mutants we used which were defective in H2S production, only the B. anthracis strain contained active nitric oxide synthase bNOS. However, attempts to combine cbs/cse and ∆nos mutations in one genome were unsuccessful, i.e., the combination of mutations that disrupt the generation of both gases leads to a lethal effect [31]. At the same time, chemical inhibition of bNOS activity in the cbs/cse-defective mutants resulted in a significantly greater sensitivity of these bacteria to antibiotics compared to that of single cbs/cse and ∆nos mutants. These data suggest that bacteria with both the NO and H2S generation systems have a synergistic effect of these gases as protectors from antibiotics.
An important observation indicating the protective effect of H2S at the level of oxidative stress suppression was based on the evaluation of the frequency of hydrogen peroxide-induced (H2O2) double-stranded breaks in the chromosomal DNA of the ∆mstA mutants with impaired H2S production and mutants with overexpression of the mstA gene under the control of the Ptet promoter. It turned out that increasing the level of H2S generation in the Ptet-mstA mutant or the addition of exogenous H2S significantly reduced the number of double-stranded breaks in DNA when treating cells with H2O2 [88]. These data suggest that the protective effect of H2S is caused by reducing the efficiency of the Fenton reaction, which leads to the generation of the toxic hydroxyl radical [89, 90]. To verify this assumption, we introduced an additional mutation into the fur gene in the genome of mutants which contained the ∆mstA deletion. The product of the fur gene is a regulator of enzymes responsible for the assimilation of iron in bacterial cells [91, 92]. It is known that the inactivation of the fur gene leads to an 8-fold increase in the concentration of free iron in the E. coli cells and dramatically enhances the sensitivity of DNA to oxidative stress [93–95]. It has been turned out that the inactivation of the fur gene in the cells of the wild-type strain leads to a 40-fold increase in the sensitivity to H2O2, whereas the survival rate of cells of the strain with the deletion in the mstA gene is reduced by a factor of more than 300. In the cells of the Ptet-mstA mutant, inactivation of the fur gene has virtually no effect on the survival of H2O2-treated bacteria [88]. The results indicate that H2S, which is synthesized endogenously under the control of the mstA gene, protects cells from the bactericidal action of H2O2. The data shows that the protective function of H2S generated under the control of the mstA gene is most pronounced against the background of Δfur gene deletion, i.e., under conditions of high intracellular content of free iron. Therefore, it can be assumed that being a powerful reducing agent, H2S binds free intracellular iron, a substrate of the Fenton reaction, leading to the formation of the hydroxyl radical [88].
This conclusion was confirmed by subsequent experiments with E. coli mutants which contained modifications in the genes responsible for the metabolism and transport of cysteine.
INTERRELATION BETWEEN HYDROGEN SULFIDE GENERATION, CYSTEINE METABOLISM, AND OXIDATIVE STRESS
The high level of resistance to oxidative stress in the Ptet-mstA mutant may be due to not only the binding of free iron in the Fenton reaction but also increased degradation of L-cysteine. It has been shown that L-cysteine can promote the Fenton reaction by reducing trivalent iron (Fe3+) to divalent iron (Fe2+) [96]. It can be assumed that the Fenton reaction undergoes additional repression after intensive degradation of L-cysteine to H2S by the sequential action of the AspC and 3MST enzymes observed in the Ptet-mstA mutant. We hypothesize that the enhanced degradation of L-cysteine in the Ptet-mstA mutant should activate CysB regulon expression. The CysB protein serves as a sensor and regulator of the intracellular content of L-cysteine and sulfur (Fig. 2) [97].
Fig. 2.

Interrelation between H2S generation, serine acetyltransferase activity, and cysB gene expression. The activity of serine acetyltransferase encoded by the cysE gene is subjected to retroinhibition by cysteine. The product of the serine-acetyltransferase reaction is O-acetylserine, which is spontaneously converted into N-acetylserine. N-acetylserine is an inducer of the transcriptional СysB regulator. The CysB protein binds in front of the –35 region of positively regulated promoters and facilitates the formation of the transcription initiation complex in the presence of the inducer. In addition, the cysB gene is subjected to autoregulation by its product, which binds to its promoter as a repressor protein. N-acetylserine stimulates the binding of the cysB protein to sites involved in positive regulation but inhibits binding to the negatively regulated promoter of the cysB gene [88].
To identify the interplay of the H2S generation and degradation of L-cysteine, we evaluated the transcription level of the cysK, cysP, and tau genes, which are regulated by the CysB protein in the ΔmstA and Ptet-mstA strains, by quantitative real-time PCR. It turned out that the transcription level of the cysB, cysK, cysP, and tau genes in the ΔmstA mutant is slightly lower than in the wild-type strain. On the contrary, in the Ptet-mstA mutant, the transcription of the cysK, cysP, and tau genes is significantly increased (by factors of 10.5, 8.2, and 4.8, respectively). The enhanced expression of these genes is most likely due to a 2-fold increase in the expression of the cysB gene because inactivation of this gene reduces their expression to the basal level. Consequently, the induction of the genes of the CysB regulon is caused by the enhanced degradation of L‑cysteine in the cells of the Ptet-mstA mutant. L-cysteine is known to participate in the allosteric inhibition of serine acetyltransferase (the cysE gene), the product of which is O-acetylserine (OAS), which, in turn, is converted into N-acetylserine, an auto-inducer of the transcription regulator (the CysB protein) [98]. Therefore, it is the increased pool of OAS that is most likely the main reason for the increased expression of CysB-regulated genes in the Ptet-mstA mutant, whereas the reduced level of transcription of these genes in the ΔmstA mutant is caused by a violation of the aspC-mstA-dependent pathway of L-cysteine degradation (Fig. 2). According to this assumption, the expression of CysB-regulated genes decreases to the basal level when exogenous L-cysteine is added to the medium with the growing Ptet-mstA mutant. In addition, the transcription levels of the genes of the CysB regulon under oxidative stress were compared in the wild-type strain, ΔmstA deletion mutant, and a Ptet-mstA mutant, a superproducer of H2S. In the wild-type strain, we found a fairly high level of induction of CysB-regulated genes in response to treatment with hydrogen peroxide, which is consistent with the published data. It should be emphasized that the molecular mechanism of the influence of H2O2 on the transcription level of CysB-regulated genes remains undisclosed. Of particular interest are our results, which show the almost complete absence of induction of CysB-dependent genes under the action of H2O2 in the ΔmstA deletion mutant.
We have proposed a model that can explain the unexpected role of H2S in the regulatory response of CysB regulon to oxidative stress (Fig. 3) [88]. According to this model, exogenous hydrogen peroxide, once in the periplasm of the cell, oxidizes L-cysteine to L‑cystine with the release of water. This process should decrease the intracellular pool of L-cysteine, which, in turn, leads to a cessation of autoregulation of cysB gene transcription, the product of which activates the CysB-regulon genes and, in particular, the tcyP gene responsible for the synthesis of the L-cystine transporter. The increased flow of L-cystine/L-cysteine into the cell provides enhanced mstA-dependent generation of H2S, which, by binding to free iron, reduces the effectiveness of the Fenton reaction accompanying the formation of the hydroxyl radical toxic to the cell (∙OH).
Fig. 3.

A model of H2S-mediated protection of E. coli cells from oxidative stress. The fraction of exogenous H2O2 in the cell periplasm reacts with cysteine to form cystine and water. This leads to a decrease in the intracellular cysteine content, followed by autoregulation shutdown of cysB gene and activation of CysB-regulated genes, including tcyP, which controls the transport of cystine from the periplasm to the cytoplasm. The increased flow of cystine/cysteine into the cell leads to an increase in the level of mstA-dependent generation of H2S, which sequesters free iron, inhibits the Fenton reaction, and prevents the formation of the toxic hydroxyl radical [88].
HYDROGEN SULFIDE-GENERATING ENZYMES AS TARGETS FOR ANTIBACTERIAL THERAPY
Despite the potential toxicity at large doses, H2S at physiological concentrations acts as an important signaling molecule, which protects bacteria from oxidative stress, immune attack, and many antibiotics. These results, which support the concept of bactericidal action of antibiotics through oxidative damage, allowed H2S-producing enzymes to be proposed as promising new targets for antimicrobial therapy. A practical task was set for the synthesis of low molecular weight compounds, which would be capable of inhibiting specific enzymes involved in the generation of hydrogen sulfide.
These studies were performed using model pathogenic bacteria, i.e., S. aureus (Gram-positive) and P. aeruginosa (Gram-negative), the main pathogens of nosocomial infections. Using transposon mutagenesis in the genome of these bacteria, the cbs and cse genes encoding the enzymes involved in the production of hydrogen sulfide were inactivated and their contribution to the generation of hydrogen sulfide was assessed [99]. The level of the H2S generation in the mutants was evaluated, and the CSE enzyme was shown to play the main role in the production of hydrogen sulfide in both bacterial species. In addition, the cse deletion mutants demonstrated significantly higher sensitivity to the gentamicin, ampicillin, and norfloxacin antibiotics compared to wild-type strains.
Based on virtual screening (SBVS), three potential inhibitors of the CSE enzyme were selected from ~3 million commercially available small molecules, NL1, NL2, and NL3. Experiments on the co-crystallization of these inhibitors with CSE monomers made it possible to determine the binding sites of all three inhibitors to the enzyme. The ability of selected inhibitors to suppress the activity of the CSE enzyme in vitro and in living cells was assessed. It is important to emphasize that the selected inhibitors show strong specificity to bacterial CSE but do not suppress the activity of CSE activity in mammals including humans. It has been found that NL1, NL2, and NL3 inhibitors suppress the ability of the S. aureus and P. aeruginosa bacteria to generate H2S and significantly increase the sensitivity of these bacteria to the antibiotic gentamicin [99].
The ability of NL1, NL2, and NL3 inhibitors to enhance the effect of gentamicin against S. aureus-induced sepsis and P. aeruginosa-induced lung infection was tested on mouse models of these infections. It has been shown that the combination of the antibiotic gentamicin with an NL1 inhibitor increases the survival rate of sepsis-infected mice and reduces the titer of the P. aeruginosa bacteria in the lung infection. We studied the ability of the selected inhibitors to decrease the proportion of persisters resistant to ciprofloxacin and gentamicin in the population of S. aureus and P. aeruginos bacteria and reduce their effect on the formation of biofilms. It has been shown that the treatment of bacteria with the NL1 inhibitor significantly reduces the proportion of persisters in the population of the S. aureus and P. aeruginosa bacteria. It has also been demonstrated that all three inhibitors suppress the formation of biofilms in the P. aeruginosa bacteria. Thus, it can be concluded that NL1, NL2, and NL3 inhibitors act as highly active antibacterial drugs which enhance the toxic effect of traditional antibiotics [99].
CONCLUSIONS
An infectious disease can often be incurable, even if it is caused by a pathogen sensitive to antibiotics. This is the main paradox of chronic infections. In most cases, chronic infections are accompanied by the formation of persisters and biofilms. Persisters are metabolically inactive nondividing variants of ordinary cells, which randomly or under the influence of stress are formed in microbial populations and have a high tolerance to antibiotics (without acquiring resistance to them). Persisters may be the main reason for the ineffectiveness of therapy for chronic infectious diseases. The periodic use of high doses of bactericidal antibiotics can lead to the selection of strains with an increased level of persisters. This is exactly what happens during the treatment of chronic infections when the patient is periodically exposed to high doses of antibiotics.
Screening knockout libraries did not reveal mutants completely devoid of persisters, which indicates the redundancy of the mechanisms of the formation of dormant cells. This redundancy makes it difficult to find targets to prevent the formation of persisters. We have shown that the production of hydrogen sulfide in the cell is crucial for the formation of a population of persisters. The S. aureus and P. aeruginosa cells with a genetic disorder of the main pathway of hydrogen sulfide formation generate 100 times fewer persisters after treatment with ciprofloxacin at a ten times higher concentration than the minimum inhibitory concentration (MIC). The CSE NL1 inhibitor, which we proposed enhances the action of bactericidal antibiotics, reduced the number of persisters to the same extent as the genetic inactivation of the cse operon.
Persisters generate significantly more H2S than normal cells, which probably leads to controlled self-poisoning and a decrease in ATP synthesis. As a result, metabolism slows down, leading to high tolerance to antibiotics. Another surprising observation is related to pyocyanin, a secondary metabolite, which functions as a signaling molecule and virulence factor in the stationary P. aegidinosa culture. When CSE is inactivated, the color of the P. aeruginosa culture changes from the light yellow characteristic of reduced pyocyanin to green-blue corresponding to oxidized pyocyanin. Since the proportion of persister cells in the wild-type P. aeruginosa culture can increase by a factor of 90 in response to pyocyanin, the absence of redox activity of pyocyanin can explain the antipersister effect of CSE inhibition. Pyocyanin is associated with the formation of biofilms of P. aeruginosa. Inactivation of CSE leads to a sharp change in the morphology of the P. aeruginosa colonies on agar plates and a significant decrease in the formation of static biofilm in P. aeruginosa and S. aureus.
Comparative transcriptomic analysis of P. aeruginosa has shown that the genes involved in biofilm formation (including genes for the biosynthesis of alginate and other exopolysaccharides) are most strongly suppressed in H2S-deficient cells. The discovery of the fact that H2S inhibitors suppress the formation of persisters and biofilms (the two main adaptations of bacteria to the action of antibiotics) opens up new opportunities for further testing our therapeutic approach, which consists of a combination of antibiotics and H2S inhibitors including improved new variants.
ACKNOWLEDGMENTS
The authors thank E.A. Nudler and A.A. Makarov for valuable comments when writing and discussing this review.
FUNDING
The work was supported by the Ministry of Science and Higher Education of the Russian Federation (Contract in the electronic budget system No. 075-10-2021-113, project ID: RF-193021X0001).
COMPLIANCE WITH ETHICAL STANDARDS
The authors state that there is no conflicts of interest. This article does not contain any studies with the use of humans and animals as objects of research.
Footnotes
Translated by A. Levina
REFERENCES
- 1.Payne D.J., Gwynn M.N., Holmes D.J., Pompliano D.L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 2.Ribeiro da Cunha B., Fonseca L.P., Calado C.R.C. 2019. Antibiotic discovery: where have we come from, where do we go? Antibiotics (Basel). 8, 45. [DOI] [PMC free article] [PubMed]
- 3.Schrader S.M., Vaubourgeix J., Nathan C. 2020. Biology of antimicrobial resistance and approaches to combat it. Sci. Transl. Med. 12, eaaz6992. [DOI] [PMC free article] [PubMed]
- 4.O’Neill J. “Tackling Drug-Resistant Infections Globally: Final Report and Recommendations” (Wellcome Trust, 2016). https://wellcomecollection.org/works/thvwsuba.
- 5.Brauner A., Fridman O., Gefen O., Balaban N.Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 2016;14:320–330. doi: 10.1038/nrmicro.2016.34. [DOI] [PubMed] [Google Scholar]
- 6.Wright G.D. Molecular mechanisms of antibiotic resistance. Chem. Commun. 2011;47:4055. doi: 10.1039/c0cc05111j. [DOI] [PubMed] [Google Scholar]
- 7.Lin J., Nishino K., Roberts M.C., Tolmasky M., Aminov R.I., Zhang L. Mechanisms of antibiotic resistance. Front. Microbiol. 2015;6:34. doi: 10.3389/fmicb.2015.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Webber M., Piddock L. The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 2003;51:9. doi: 10.1093/jac/dkg050. [DOI] [PubMed] [Google Scholar]
- 9.Cox G., Wright G.D. Intrinsic antibiotic resistance: mechanisms, origins, challenges and solutions. Int. J. Med. Microbiol. 2013;303:287. doi: 10.1016/j.ijmm.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Piddock L.J. Multidrug-resistance efflux pumps? Not just for resistance. Nat. Rev. Microbiol. 2006;4:629. doi: 10.1038/nrmicro1464. [DOI] [PubMed] [Google Scholar]
- 11.Wright G.D. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 2007;5:175. doi: 10.1038/nrmicro1614. [DOI] [PubMed] [Google Scholar]
- 12.Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature. 2000;406:775–778. doi: 10.1038/35021219. [DOI] [PubMed] [Google Scholar]
- 13.Balaban N.Q., Helaine S., Lewis K., Ackermann M., Aldridge B., Andersson D., Brynildsen M.P., Bumann D., Camilli A., Collins J., Dehio C., Fortune S., Ghigo J.M., Hardt W.D., Harms A., Heinemann M., Hung D.T., Jenal U., Levin B.R., Michiels J., Storz G., Tan M.W., Nenson T., Van Melderen L., Zinkernagel A. Definitions and guidelines for research on antibiotic persistence. Nat. Rev. Microbiol. 2019;17:441–448. doi: 10.1038/s41579-019-0196-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Michiels J.F., Van den Bergh B., Verstraeten N., Michiels J. Molecular mechanisms and clinical implications of bacterial persistence. Drug Resist. Updat. 2016;29:76–89. doi: 10.1016/j.drup.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Costerton J.W., Stewart P.S., Greenberg E.P. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 16.Kirby A.E., Garner K., Levin B.R. The relative contributions of physical structure and cell density to the antibiotic susceptibility of bacteria in biofilms. Antimicrob. Agents Chemother. 2012;56:2967–2975. doi: 10.1128/AAC.06480-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meredith H.R., Srimani J.K., Lee A.J., Lopatkin A.J., You L. Collective antibiotic tolerance: mechanisms, dynamics and intervention. Nat. Chem. Biol. 2015;11:182–188. doi: 10.1038/nchembio.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dewachter L., Fauvart M., Michiels J. Bacterial heterogeneity and antibiotic survival: understanding and combatting persistence and heteroresistance. Mol. Cell. 2019;76:255–267. doi: 10.1016/j.molcel.2019.09.028. [DOI] [PubMed] [Google Scholar]
- 19.Schulze A., Mitterer F., Pombo J.P., Schild S. Biofilms by bacterial human pathogens: clinical relevance—development, composition and regulation – therapeutical strategies. Microb. Cell. 2021;8:28–56. doi: 10.15698/mic2021.02.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conlon B.P. Staphylococcus aureus chronic and relapsing infections: evidence of a role for persister cells: an investigation of persister cells, their formation and their role in S. aureus disease. BioEssays. 2014;36:991–996. doi: 10.1002/bies.201400080. [DOI] [PubMed] [Google Scholar]
- 21.Donlan R.M., Costerton J.W. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002;15:167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mulcahy L.R., Burns J.L., Lory S., Lewis K. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 2010;192:6191–6199. doi: 10.1128/JB.01651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Percival S.L., Hill K.E., Malic S., Thomas D.W., Williams D.W. Antimicrobial tolerance and the significance of persister cells in recalcitrant chronic wound biofilms. Wound Repair Regen. 2011;19:1–9. doi: 10.1111/j.1524-475X.2010.00651.x. [DOI] [PubMed] [Google Scholar]
- 24.Huemer M., Shambata S.M., Bergada-Pijuana J., Söderholmb S., Boumasmouda M., Vulin C., Gómez-Mejiaa A., Varela M.A., Tripathi V., Götschia S., Maggio E.M., Hasse B., Bruggera S.D., Bumann D., Schuepbach R.F., Zinkernagela A.S. Molecular reprogramming and phenotype switching in Staphylococcus aureus lead to high antibiotic persistence and affect therapy success. Proc. Natl. Acad. Sci. U. S. A. 2021;118:e2014920118. doi: 10.1073/pnas.2014920118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davies J., Davies D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010;74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levin B.R., Rozen D.E. Non-inherited antibiotic resistance. Nat. Rev. Microbiol. 2006;4:556–562. doi: 10.1038/nrmicro1445. [DOI] [PubMed] [Google Scholar]
- 27.Sebastian J., Swaminath S., Nair R.R., Jakkala K., Pradhan A., Ajitkumar P. De novo emergence of genetically resistant mutants of Mycobacterium tuberculosis from the persistence phase cells formed against antituberculosis drugs in vitro. Antimicrob. Agents Chemother. 2017;61:e01343–16. doi: 10.1128/AAC.01343-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Acker H., Coenye T. The role of reactive oxygen species in antibiotic-mediated killing of bacteria. Trends Microbiol. 2017;25:456–466. doi: 10.1016/j.tim.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 29.Dwyer D.J., Collins J.J., Walker G.C. Unraveling the physiological complexities of antibiotic lethality. Annu. Rev. Pharmacol. Toxicol. 2015;55:313–332. doi: 10.1146/annurev-pharmtox-010814-124712. [DOI] [PubMed] [Google Scholar]
- 30.Kohanski M.A., Dwyer D.J., Hayete B., Lawrence C.A., Collins J.J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 31.Shatalin K., Shatalina E., Mironov A., Nudler E. H2S: a universal defense against antibiotics in bacteria. Science. 2011;334:986–990. doi: 10.1126/science.1209855. [DOI] [PubMed] [Google Scholar]
- 32.Luhachack L., Nudler E. Bacterial gasotransmitters: an innate defense against antibiotics. Curr. Opin. Microbiol. 2014;21C:13–17. doi: 10.1016/j.mib.2014.06.017. [DOI] [PubMed] [Google Scholar]
- 33.Kimura H. Signaling of hydrogen sulfide and polysulfides. Antioxid. Redox Signal. 2015;22:347–349. doi: 10.1089/ars.2014.6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szabo C. A timeline of hydrogen sulfide (H2S) research: from environmental toxin to biological mediator. Biochem. Pharmacol. 2018;149:5–19. doi: 10.1016/j.bcp.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nzungize L., Kaisar M., Wang X., Huang X., Yang W., Duan X., Yan S., Li C., Abdalla A.E., Jeyakkumar P., Xie J. Mycobacterium tuberculosis metC (Rv3340) derived hydrogen sulphide conferring bacteria stress survival. J. Drug Target. 2019;27:1004–1016. doi: 10.1080/1061186X.2019.1579820. [DOI] [PubMed] [Google Scholar]
- 36.Toliver-Kinsky T., Cui W., Törö G., Lee S.J., Shatalin K., Nudler E., Szabo C. H2S: a bacterial defense mechanism against the host immune response. Infect. Immun. 2018;87:e00272–18. doi: 10.1128/IAI.00272-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh C. Antibiotics: Actions, Origins, Resistance. Washington, DC: ASM; 2003. [Google Scholar]
- 38.Kohanski M.A., Dwyer D.J., Collins J.J. How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol. 2010;8:423–435. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imlay J.A. Pathways of oxidative damage. Annu. Rev. Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 40.Zhao X., Drlica K. Reactive oxygen species and the bacterial response to lethal stress. Curr. Opin. Microbiol. 2014;21:1–6. doi: 10.1016/j.mib.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imlay J.A., Chin S.M., Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. 1988;240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- 42.Park S., You X., Imlay J.A. Substantial DNA damage from submicromolar intracellular hydrogen peroxide detected in Hpx7 mutants of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2005;102:9317–9322. doi: 10.1073/pnas.0502051102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X., Zhao X. Contribution of oxidative damage to antimicrobial lethality. Antimicrob. Agents Chemother. 2009;53:1395–1402. doi: 10.1128/AAC.01087-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greenberg J.T., Monach P., Chou J.H., Josephy P.D., Demple B. Positive control of a global antioxidant defense regulon activated by superoxide-generating agents in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 1990;87:6181–6185. doi: 10.1073/pnas.87.16.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oethinger M., Podglajen I., Kern W.V., Levy S.B. Overexpression of the marA or soxS regulatory gene in clinical topoisomerase mutants of Escherichia coli. Antimicrob. Agents Chemother. 1998;42:2089–2094. doi: 10.1128/AAC.42.8.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koutsolioutsou A., Martins E.A., White D.G., Levy S.B., Demple B. A soxRS-constitutive mutation contributing to antibiotic resistance in a clinical isolate of Salmonella enterica (serovar typhimurium) Antimicrob. Agents Chemother. 2001;45:38–43. doi: 10.1128/AAC.45.1.38-43.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goswami M., Mangoli S.H., Jawali N. Involvement of reactive oxygen species in the action of ciprofloxacin against Escherichia coli. Antimicrob. Agents Chemother. 2006;50:949–954. doi: 10.1128/AAC.50.3.949-954.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goswami M., Mangoli S.H., Jawali N. Effects of glutathione and ascorbic acid on streptomycin sensitivity of Escherichia coli. Antimicrob. Agents Chemother. 2007;51:1119–1122. doi: 10.1128/AAC.00779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Albesa I., Becerra M.C., Battan P.C., Paez P.L. Oxidative stress involved in the antibacterial action of different antibiotics. Biochem. Biophys. Res. Commun. 2004;317:605–609. doi: 10.1016/j.bbrc.2004.03.085. [DOI] [PubMed] [Google Scholar]
- 50.Becerra M.C., Albesa I. Oxidative stress induced by ciprofloxacin in Staphylococcus aureus. Biochem. Biophys. Res. Commun. 2002;297:1003–1007. doi: 10.1016/S0006-291X(02)02331-8. [DOI] [PubMed] [Google Scholar]
- 51.Lewin C.S., Morrissey I., Smith J.T. The mode of action of quinolones: the paradox in activity of low and high concentrations and activity in the anaerobic environment. Eur. J. Clin. Microbiol. Infect. Dis. 1991;10:240–248. doi: 10.1007/BF01966996. [DOI] [PubMed] [Google Scholar]
- 52.Malik M., Hussain S., Drlica K. Effect of anaerobic growth on quinolone lethality with Escherichia coli. Antimicrob. Agents Chemother. 2007;51:28–34. doi: 10.1128/AAC.00739-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Y., Liu X., Qu Y., Wang X., Li L., Zhao X. Inhibitors of reactive oxygen species accumulation delay and/or reduce the lethality of several antistaphylococcal agents. Antimicrob. Agents Chemother. 2012;56:6048–6050. doi: 10.1128/AAC.00754-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burger R.M., Drlica K. Superoxide protects Escherichia coli from bleomycin mediated lethality. J. Inorg. Biochem. 2009;103:1273–1277. doi: 10.1016/j.jinorgbio.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mosel M., Li L., Drlica K., Zhao X. Superoxide-mediated protection of Escherichia coli from antimicrobials. Antimicrob. Agents Chemother. 2013;57:5755–5759. doi: 10.1128/AAC.00754-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Girgis H.S., Hottes A.K., Tavazoie S. Genetic architecture of intrinsic antibiotic susceptibility. PLoS One. 2009;4:e5629. doi: 10.1371/journal.pone.0005629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006 0008. [DOI] [PMC free article] [PubMed]
- 58.Liu A., Tran L., Becket E., Lee K., Chinn L., Park E., Tran K., Miller J.H. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: generating an antibiotic bar code. Antimicrob. Agents Chemother. 2010;54:1393–1403. doi: 10.1128/AAC.00906-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Foti J.J., Devadoss B., Winkler J.A., Collins J.J., Walker G.C. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science. 2012;336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Becerra M.C., Paez P.L., Larovere L.E., Albesa I. Lipids and DNA oxidation in Staphylococcus aureus as a consequence of oxidative stress generated by ciprofloxacin. Mol. Cell Biochem. 2006;285:29–34. doi: 10.1007/s11010-005-9051-0. [DOI] [PubMed] [Google Scholar]
- 61.Ling J., Cho C., Guo L.T., Aerni H.R., Rinehart J., Soll D. Protein aggregation caused by aminoglycoside action is prevented by a hydrogen peroxide scavenger. Mol. Cell. 2012;48:713–722. doi: 10.1016/j.molcel.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y., Zhang J., Hoeflich K.P., Ikura M., Qing G., Inouye M. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol. Cell. 2003;12:913–923. doi: 10.1016/S1097-2765(03)00402-7. [DOI] [PubMed] [Google Scholar]
- 63.Gerdes K., Christensen S.K., Lobner-Olesen A. Prokaryotic toxin–antitoxin stress response loci. Nat. Rev. Microbiol. 2005;3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 64.Dorsey-Oresto A., Lu T., Mosel M., Wang X., Salz T., Drlica K., Zhao X. YihE kinase is a central regulator of programmed cell death in bacteria. Cell Rep. 2013;3:528–537. doi: 10.1016/j.celrep.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kohanski M.A., Dwyer D.J., Wierzbowski J., Cottarel G., Collins J.J. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008;135:679–690. doi: 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davies B.W., Kohanski M.A., Simmons L.A., Winkler J.A., Collins J.J., Walker G.C. Hydroxyurea induces hydroxyl radical-mediated cell death in Escherichia coli. Mol. Cell. 2009;36:845–860. doi: 10.1016/j.molcel.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pogliano J., Lynch A.S., Belin D., Lin E.C., Beckwith J. Regulation of Escherichia coli cell envelope proteins involved in protein folding and degradation by the Cpx two-component system. Genes Dev. 1997;11:1169–1182. doi: 10.1101/gad.11.9.1169. [DOI] [PubMed] [Google Scholar]
- 68.Raivio T.L., Silhavy T.J. Periplasmic stress and ECF sigma factors. Annu. Rev. Microbiol. 2001;255:591–624. doi: 10.1146/annurev.micro.55.1.591. [DOI] [PubMed] [Google Scholar]
- 69.Clarke P.H. Hydrogen sulphide production by bacteria. J. Gen. Microbiol. 1953;8:397–407. doi: 10.1099/00221287-8-3-397. [DOI] [PubMed] [Google Scholar]
- 70.Wang R. Wo’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 71.Adak S., Aulak K.S., Stuehr D.J. Direct evidence for nitric oxide production by a nitric-oxide synthase-like protein from Bacillus subtilis. J. Biol. Chem. 2002;277:16167–16171. doi: 10.1074/jbc.M201136200. [DOI] [PubMed] [Google Scholar]
- 72.Gusarov I., Nudler E. NO-mediated cytoprotection: instant adaptation to oxidative stress in bacteria. Proc. Natl. Acad. Sci. U. S. A. 2005;102:13855–13860. doi: 10.1073/pnas.0504307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gusarov I., Starodubtseva M., Wang Z.Q., McQuade L., Lippard S.J., Stuehr D.J., Nudler E. Bacterial nitric-oxide synthases operate without a dedicated redox partner. J. Biol. Chem. 2008;283:13140–13147. doi: 10.1074/jbc.M710178200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 75.Stoffels L., Krehenbrink M., Berks B.C., Unden G. Thiosulfate reduction in Salmonella enterica is driven by the proton motive force. J. Bacteriol. 2012;194:475–485. doi: 10.1128/JB.06014-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mondovi B., Scioscia-Santoro A., Cavallinid D. Further evidence on the identity of cystathionase and cysteine desulfhydrase. Arch. Biochem. Biophys. 1963;101:363–364. doi: 10.1016/S0003-9861(63)80025-9. [DOI] [PubMed] [Google Scholar]
- 77.Mikami Y., Shibuya N., Kimura Y., Nagahara N., Ogasawara Y., Kimura H. Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem. J. 2011;439:479–485. doi: 10.1042/BJ20110841. [DOI] [PubMed] [Google Scholar]
- 78.Singh S., Banerjee R. PLP-dependent H2S biogenesis. Biochim. Biophys. Acta. 2011;1814:1518–1527. doi: 10.1016/j.bbapap.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Loddeke M., Schneider B., Oguri T., Mehta I., Xuan Z., Reitzera L. Anaerobic cysteine degradation and potential metabolic coordination in Salmonella enterica and Escherichia coli. J. Bacteriol. 2017;199:e00117–17. doi: 10.1128/JB.00117-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kimura Y., Goto Y.-I., Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010;12:1–13. doi: 10.1089/ars.2008.2282. [DOI] [PubMed] [Google Scholar]
- 81.Kimura H. Production and physiological effects of hydrogen sulfide. Antioxid. Redox Signal. 2014;20:783–793. doi: 10.1089/ars.2013.5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gusarov I., Shatalin K., Starodubtseva M., Nudler E. Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science. 2009;325:380–1384. doi: 10.1126/science.1175439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kers J.A., Wach M.J., Krasnoff S.B., Widom J., Cameron K.D., Bukhalid R.A., Gibson D.M., Crane B.R., Loria R. Nitration of a peptide phytotoxin by bacterial nitric oxide synthase. Nature. 2004;429:79–82. doi: 10.1038/nature02504. [DOI] [PubMed] [Google Scholar]
- 84.Shatalin K., Gusarov I., Avetissova E., Shatalina Y., McQuade L.E., Lippard S.J., Nudler E. Bacillus anthracis-derived nitric oxide is essential for pathogen virulence and survival in macrophages. Proc. Natl. Acad. Sci. U. S. A. 2008;105:1009–1013. doi: 10.1073/pnas.0710950105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Carlson H.K., Vance R.E., Marletta M.A. H-NOX regulation of c-di-GMP metabolism and biofilm formation in Legionella pneumophila. Mol. Microbiol. 2010;77:930–942. doi: 10.1111/j.1365-2958.2010.07259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bowman L.A., McLean S., Poole R.K., Fukuto J.M. The diversity of microbial responses to nitric oxide and agents of nitrosative stress close cousins but not identical twins. Adv. Microb. Physiol. 2011;59:135–219. doi: 10.1016/B978-0-12-387661-4.00006-9. [DOI] [PubMed] [Google Scholar]
- 87.Gusarov I., Gautier L., Smolentseva O., Shamovsky I., Eremina S., Mironov A., Nudler E. Bacterial nitric oxide extends the lifespan of C. elegans. Cell. 2013;152:818–830. doi: 10.1016/j.cell.2012.12.043. [DOI] [PubMed] [Google Scholar]
- 88.Mironov A., Seregina T., Nagornykh M., Luhachack L., Korolkova L., Errais Lopes L., Kotova V., Zavilgelsky G., Shakulov R., Shatalin R., Nudler E. A mechanism of H2S-mediated protection against oxidative stress in E. coli. Proc. Natl. Acad. Sci. U. S. A. 2017;114:6022–6027. doi: 10.1073/pnas.1703576114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aruoma O.I., Halliwell B., Gajewski E., Dizdaroglu M. Damage to the bases in DNA induced by hydrogen peroxide and ferric ion chelates. J. Biol. Chem. 1989;264:20509–20512. doi: 10.1016/S0021-9258(19)47091-9. [DOI] [PubMed] [Google Scholar]
- 90.Imlay J.A., Chin S.M., Linn S. Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science. 1988;240:640–642. doi: 10.1126/science.2834821. [DOI] [PubMed] [Google Scholar]
- 91.Hantke K. Iron and metal regulation in bacteria. Curr. Opin. Microbiol. 2001;4:172–177. doi: 10.1016/S1369-5274(00)00184-3. [DOI] [PubMed] [Google Scholar]
- 92.Hantke K., Braun V. Bacterial Stress Responses. Washington DC: ASM; 2000. [Google Scholar]
- 93.Touati D., Jacques M., Tardat B., Bouchard L., Despied S. Lethal oxidative damage and mutagenesis are generated by iron in delta fur mutants of Escherichia coli: protective role of superoxide dismutase. J. Bacteriol. 1995;177:2305–2314. doi: 10.1128/jb.177.9.2305-2314.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Keyer K., Imlay J.A. Superoxide accelerates DNA damage by elevating free-iron levels. Proc. Natl. Acad. Sci. U. S. A. 1996;93:13635–13640. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Keyer K., Gort A.S., Imlay J.A. Superoxide and the production of oxidative DNA damage. J. Bacteriol. 1995;177:6782–6790. doi: 10.1128/jb.177.23.6782-6790.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Park S., Imlay J.A. High levels of intracellular cysteine promote oxidative DNA damage by driving the Fenton reaction. J. Bacteriol. 2003;185:1942–1950. doi: 10.1128/JB.185.6.1942-1950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kredich N.M. 1983. Amino acids: biosynthesis and genetic regulation. In: Regulation of Cysteine Biosynthesis in Escherichia coli and Salmonella typhimurium. Ed. Herrmann K.M. Sommervilleth edition. United Kingdom London: Addison-Wesley Publ. Comp. 115–132.
- 98.Kredich N.M. The molecular basis for positive regulation of cys promoters in Salmonella typhimurium and Escherichia coli. Mol. Microbiol. 1992;6:2747–2753. doi: 10.1111/j.1365-2958.1992.tb01453.x. [DOI] [PubMed] [Google Scholar]
- 99.Shatalin K., Nuthanakanti A., Kaushik A., Shishov D., Peselis A., Shamovsky I., Pani B., Lechpammer M., Vasilyev N., Shatalina E., Rebatchouk D., Mironov A., Fedichev P., Serganov A., Nudler E. Inhibitors of bacterial H2S biogenesis targeting antibiotic resistance and tolerance. Science. 2021;372:1169–1175. doi: 10.1126/science.abd8377. [DOI] [PMC free article] [PubMed] [Google Scholar]