

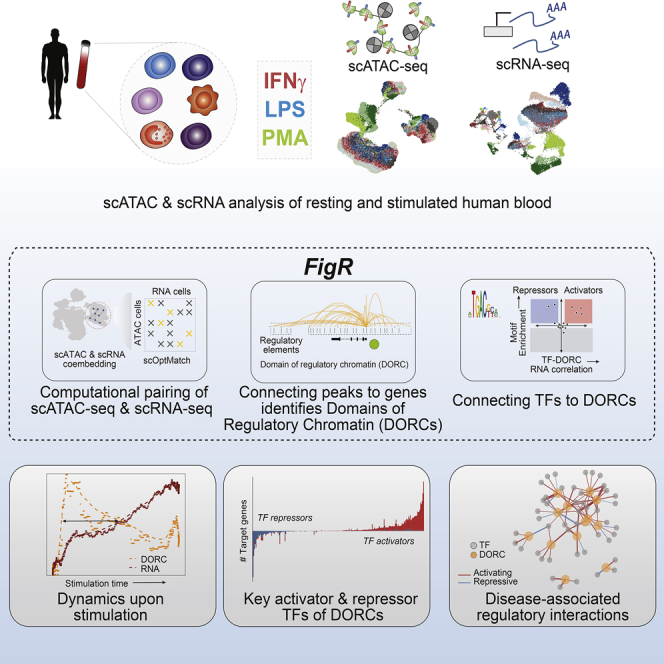
Summary
Cells require coordinated control over gene expression when responding to environmental stimuli. Here we apply scATAC-seq and single-cell RNA sequencing (scRNA-seq) in resting and stimulated human blood cells. Collectively, we generate ∼91,000 single-cell profiles, allowing us to probe the cis-regulatory landscape of the immunological response across cell types, stimuli, and time. Advancing tools to integrate multi-omics data, we develop functional inference of gene regulation (FigR), a framework to computationally pair scATAC-seq with scRNA-seq cells, connect distal cis-regulatory elements to genes, and infer gene-regulatory networks (GRNs) to identify candidate transcription factor (TF) regulators. Utilizing these paired multi-omics data, we define domains of regulatory chromatin (DORCs) of immune stimulation and find that cells alter chromatin accessibility and gene expression at timescales of minutes. Construction of the stimulation GRN elucidates TF activity at disease-associated DORCs. Overall, FigR enables elucidation of regulatory interactions across single-cell data, providing new opportunities to understand the function of cells within tissues.
Keywords: single cell, multi-omics, gene-regulatory networks, gene regulation, stimulus response, scOptMatch, DORC, FigR
Graphical abstract
Highlights
-
•
A multi-omics resource of resting and immunologically stimulated human blood
-
•
Data integration identifies expression-linked regulatory elements across cells
-
•
FigR reveals TF activator and repressor drivers of chromatin accessibility
-
•
Gene-regulatory modeling identifies disease-associated regulatory interactions
Single-cell methods for measuring chromatin accessibility (ATAC-seq) and gene expression (RNA-seq) are rapidly evolving, but tools to integrate data and infer gene-regulatory relationships remain limited. Here we generate multi-omics data of resting and stimulated human blood cells and present a new computational framework for constructing gene-regulatory networks (GRNs). Specifically, we describe functional inference of gene regulation (FigR), a workflow to (1) pair scATAC-seq with scRNA-seq, (2) connect cis-regulatory elements to target genes, and (3) identify TF-gene relationships.
Introduction
Eukaryotic cells have evolved exquisite control to continuously sense and respond to external environmental cues.1, 2, 3, 4 This, in part, involves coordinated changes in signaling dynamics, transcription factor (TF) binding, and, eventually, expression of downstream target genes.3, 4, 5 Immune cells in particular harbor tremendous plasticity in their ability to respond to stimuli, developing diverse and specific functions in response to different pathogenic agents.6 This highly context-specific and often heterogeneous activation of genes promoting the appropriate anti-viral or inflammatory response comprises one of the hallmarks of immunity. Our understanding of immunity has evolved over time; for example, it has been shown that chromatin may prime cells for an immunological response,7,8 leading to exhausted states,9 or further orchestrating activation of surrounding cells through production of key signaling molecules.10
Single-cell genomics methods have greatly advanced our understanding of cellular diversity of immune cells.11, 12, 13 Single-cell RNA sequencing (scRNA-seq) characterizing time- and stimulus-dependent transcriptional signatures in mouse10 and human14 immune cells, for example, has identified distinct transcriptional programs that are activated or repressed over time and highlighted cell-cell variability in response to immunological stimulants.15 Concomitantly, several prior studies have applied chromatin accessibility and gene expression assays to define cis-regulatory atlases across resting12,16,17 and stimulated14,16 immune cell types. Most recently, the coronavirus disease 2019 (COVID-19) pandemic has prompted use of single-cell ATAC-seq and RNA-seq tools to characterize the immunological response to infection.18,19 These diverse efforts have sought to elucidate the epigenetic control of immune cell function; namely, the cellular circuitry that defines the gene-regulatory network (GRN) within the cell.
Although these efforts have resulted in tremendous insights into the transcriptional control of immune cells, these studies are limited by the existing repertoire of computational tools modeling-gene regulatory dynamics among single cells. Advances in constructing GRNs from single-cell data20,21 have facilitated new opportunities to uncover mechanisms of cell function and adaptation after stimulation. However, most approaches that solely utilize co-expression20,22,23 are limited in their ability to (1) define key cis-regulatory elements and (2) elucidate the function of master TF regulators on gene expression. Extensive prior work has demonstrated that epigenomics data can vastly improve the determination of functional GRNs.24, 25, 26 In one example, single-cell genomics,27, 28, 29, 30 bulk epigenomics,17,31,32 and mutagenesis33, 34, 35 studies of non-coding DNA have revealed that certain genes are largely regulated by their enhancer landscape, whereas others are predominantly under promoter control. In another example, scRNA-seq GRN methods that use co-expression rely on the assumption that TFs activate genes; however, extensive functional experiments show that TFs may silence chromatin to repress target genes.36, 37, 38 We reasoned that a computational approach that defines a gene by its connection to distal regulatory elements would fill an unmet need of GRN modeling in single-cell genomics, serving to improve our understanding of the epigenetic mechanisms underlying the function and adaptation upon environmental exposure of eukaryotic cells.
Here, we create an exemplar dataset for construction of immune cell GRNs. To do this, we combine use of multiple stimulus agents with chromatin accessibility and gene expression single-cell analysis to characterize and assess the dynamics of the cis-regulatory landscape linked with immune cell stimulation in human peripheral blood mononuclear cells (PBMCs). We then establish functional inference of gene regulation (FigR), a generalizable approach for independently or concomitantly profiled single-cell ATAC-seq (scATAC-seq) and scRNA-seq, that (1) computationally pairs scATAC-seq and scRNA-seq datasets (when needed), (2) infers cis-regulatory interactions, and (3) defines a TF-gene GRN. Utilizing these integrated data, we establish that changes in chromatin accessibility foreshadow changes in gene expression upon immune stimulation of monocytes. Last, we highlight how this approach can be used to identify key TFs and their relationship to target genes, including stimulus response and disease-associated domains of regulatory chromatin (DORCs).Our work highlights use of blood stimulation combined with high-throughput single-cell multi-omics and advancements in developing enhancer GRNs using FigR as a model to deduce key transcriptional regulatory modules that are required for immune cell activation.
Results
Combined high-throughput single-cell epigenomic and transcriptional profiling of resting and stimulated PBMCs
To characterize the chromatin accessibility and transcriptional landscape associated with host response to stimuli in human blood, we performed droplet-based scATAC-seq and scRNA-seq on resting and stimulated human PBMCs at different time points of stimulus exposure (Figure 1A; STAR Methods). Specifically, cells derived from healthy donors (n = 3 or 4; Table S1) were exposed for 1 or 6 h to stimulants known to elicit antiviral-like or core inflammatory responses, including lipopolysaccharide (LPS; a component of bacterial cell membranes), phorbol myristate acetate (PMA) plus ionomycin (a potent ester that activates nuclear factor B [NF-B] signaling),39 or interferon gamma (IFN-Ɣ; an endogenously produced immunoregulatory cytokine) alongside a DMSO control per time point prior to single-cell profiling (STAR Methods). These stimulants were chosen because they have been shown to induce distinct time- and cell-type-specific changes with unique transcriptional dynamics as part of the host immune response.10,14,39, 40, 41 Additionally, for the 6-h time point using each stimulant, we separately treated cells with a Brefeldin A, a protein secretion inhibitor (Golgi inhibitor [GI]), attenuating paracrine signaling events in immune cells and allowing us to distinguish between primary and secondary stimulation response phenotypes.
Figure 1.

High-throughput single-cell epigenomic and transcriptional profiling of resting and stimulated human blood cells
(A) Schematic highlighting design of stimulation experiment. Human peripheral blood mononuclear cells (PBMCs) were stimulated with DMSO control, lipopolysaccharide (LPS), interferon gamma (IFN-Ɣ), or phorbol myristate acetate (PMA) plus ionomycin for 1 or 6 h with or without a Golgi inhibitor (GI ) for the 6-h treatment condition. Cells were then split and profiled using scATAC-seq and scRNA-seq for each condition and time point considered.
(B) Total number of cells profiled per condition passing quality control filtering for scATAC and scRNA-seq.
(C) Uniform manifold approximation and projection (UMAP) of scATAC-seq cells based on latent semantic indexing (LSI) dimensionality reduction, with cells colored by treatment condition.
(D) UMAP of scRNA-seq cells based on principal-component analysis (PCA) dimensionality reduction, with cells colored by treatment condition.
(E) UMAPs of scATAC-seq cells (top) and scRNA-seq cells (bottom), highlighting individual conditions under control (6 h) and PMA (1 and 6 h) conditions.
(F) Aggregate accessibility profiles for scATAC-seq monocyte cells around genes IFITM3 and HES4.
(G) Distribution of single-cell expression levels based on the imputed scRNA-seq counts for stimulation-specific gene markers shown in (F) per condition for scRNA-seq monocyte cells.
Collectively, we generated over 15 billion reads, resulting in a high-coverage single-cell regulatory atlas comprising of 67,581 scATAC-seq and 23,754 scRNA-seq cells spanning all conditions (Figure 1B) with an average of 8,865.2 (±SD = 4,837) aligned unique nuclear fragments per cell and mean fraction of reads in peaks (FRiP) of 0.6 (±SD = 0.05) for scATAC-seq-profiled cells (Figures S1A and S1B) and averaging 3,021 UMIs (±SD = 425.77) for scRNA-seq-profiled cells (Figures S1C and S1D; STAR Methods). Clustering scATAC-seq and scRNA-seq cells (STAR Methods) yielded discrete cell clusters, largely representing monocytes, T (CD4/CD8) and B lymphocytes, and natural killer (NK) cells, with even distribution of cells from all donors involved per cluster and condition (Figures 1C, 1D, and S1E–S1G). Importantly, each of these broader clusters included sub-clustering of cells by stimulus condition (Figures 1E and S1H).
To formally annotate cell types for scRNA-seq cells, we first aligned cells across batches (here defined as each treatment condition) using a previously described computational approach (in Seurat),42 enabling co-clustering and annotation of scRNA-seq cells across conditions. Clustering of cells using this approach yielded distinct groupings (Figures S2A and S2B) that were enriched for cell type and stimulus-specific gene expression markers and were used to annotate cell types (Figure S2C). Inspection of the myeloid cells for accessibility peaks around gene promoters (scATAC-seq) and gene expression levels (scRNA-seq) confirmed stimulus- and time-specific changes (Figures 1F, 1G, and S2D). Importantly, all major cell types were captured at relatively even proportions across the treatment conditions used (Figure S2E), enabling multi-omics integration of independently assayed chromatin accessibility and gene expression profiles downstream.
A computational cell pairing approach for accurate integration of single-cell chromatin accessibility and gene expression profiles
We reasoned that data from paired contexts may enable determination of GRNs, facilitating interpretation of the key regulatory processes underlying stimulation of immune cells. Current frameworks supporting integration of scATAC and scRNA-seq data29,42,43 rely on identifying “anchor” cells, cells that represent shared biological states in a common lower dimensional space, to then find representative cells from one dataset in the other. Although useful for matching cells of corresponding cell types (i.e., annotation-level pairing), these methods often (1) result in high one-to-many cell barcode matching rates, resulting in overall lower cell usage downstream, or (2) do not adequately address cell type imbalance between datasets.
To address this challenge, we developed a method (scOptMatch) that identifies cell pairs between scATAC-seq and scRNA-seq data using a constrained optimal cell mapping approach (Figure 2A). For this approach, we first create a shared co-embedding of scATAC-seq and scRNA-seq cells using canonical correlation analysis (CCA), similar to what has been described previously as a functionality in Seurat.42 Next we address the issues of (1) total cell number imbalance and (2) cell type imbalance between datasets by first sub-clustering the entire cell space and constructing a cell k-nearest neighbor (kNN) graph between ATAC and RNA cells in the co-embedded space, sampling cells from both assays within a given kNN subgraph (STAR Methods). Upon down-sampling to match cell numbers between assays (i.e., scATAC or scRNA) in a given subgraph, cells are paired using a constrained global matching algorithm,44 using the subgraph geodesic distance between ATAC-RNA cells as a cost function. Analogous to the traveling salesman problem, this ensures that resulting ATAC-RNA cell pairs are minimized for the total geodesic distance among all combinations of possible pairs. Importantly, only ATAC-RNA cells within a certain distance (geodesic kNNs) are considered for pairing as a prior, further speeding up computation time relative to if all possible pairs were being considered (STAR Methods).
Figure 2.

Sparse kNN-based ATAC-RNA cell pairing allows optimal pairing and integration of scATAC-seq and scRNA-seq data
(A) Schematic highlighting scOptMatch’s strategy for computational pairing of scATAC-seq and scRNA-seq cells based on geodesic distance kNNs (yellow x marks) within cluster subgraphs (gray x marks).
(B) Schematic depicting experimental bead enrichment of specific immune cell types from human PBMCs.
(C) Distribution of the number of instances of paired RNA cell barcode when using the greedy (left) versus scOptMatch method for the PBMC isolate dataset pairing.
(D) Percentage of total scATAC and scRNA-seq cells paired using the two different pairing strategies.
(E) Accuracy heatmap of scATAC-scRNA-seq pairing between PBMC isolate cell types, colored by percentage of scATAC-seq cells correctly paired with the corresponding scRNA-seq cell type.
(F) UMAP of scRNA-seq stimulated cells shown in Figure 1D, with cells aligned across stimulus conditions to enable cell type annotation, colored by annotated cell type.
(G) UMAP of un-aligned scRNA-seq cells (shown in Figure 1D) colored by annotated cell type (left) and scATAC-seq stimulated cells (shown in Figure 1C) colored by paired scRNA-seq cell annotations (right), enabling downstream data integration for stimulated scATAC- and scRNA-seq-profiled cells.
(H) Pairwise Pearson correlation of aggregate single-cell chromatin accessibility profiles associated with gene promoters (left), distal from the promoter (center) and paired gene expression (right), aggregated by cell type and condition.
To create a reference dataset to benchmark scOptMatch, we isolated cell types within PBMCs and profiled (in separate assays) scRNA-seq and scATAC-seq.13 The complete data reflected scATAC-seq (n = 17,920 cells) and scRNA-seq (n = 8,089 cells) data corresponding to five PBMC sub-populations (Figure 2B; STAR Methods). Using these data, we determined ATAC-RNA cell pairs using (1) the optimal matching described above (scOptMatch) or (2) a “greedy” best match approach (choosing the closest RNA cell for every ATAC cell in CCA space). As expected, we found that scOptMatch results in a significantly larger number of cells being paired from both datasets across all cells (92.06% scATAC and 98.4% scRNA; Figures 2C and S3F), a consequence of fewer ATAC-RNA cell multi-mapping instances (Figures 2D and S3G) compared with the greedy approach (22.2% scRNA). Importantly, the scOptMatch approach also accurately maps cells of the same reference cell type (Figure 2E). To confirm the pairing performance of scOpMatch, we applied it to previously generated SNARE-Seq2 data for primary motor cortex cells (n = 84,178 cells),45 representing a less discrete cellular population with ground truth labels for experimentally paired chromatin accessibility and RNA expression profiles per cell (Figure S3H). Implementing the same integration workflow as with unpaired data resulted in a mean ATAC-RNA mapping rate of 90.05% (±22.3 SD) between cells with shared cell type cluster annotations (n = 10 cluster groups; Figure S3I), despite very few ATAC-RNA matches for the exact same cells (<1%), with mispairings largely occurring for very rare cell types (e.g., L5 ET-2, n = 29 cells) or closely related cluster annotations (e.g., Sngc/Vip cells). Thus, scOptMatch can pair cells of similar cell types across multiple assays using independently generated or multi-modal profiles.
Provided scOptMatch enabled approximately one-to-one pairing of cells of similar annotations between assays, we reasoned that it may facilitate integrative analysis of scATAC-seq and scRNA-seq data generated from matched cellular contexts. We thus sought to apply it to pair our stimulus multi-omics datasets. Pairing of scATAC and scRNA-seq cells per condition using scOptMatch (Figures S3J–S3L), we obtained paired multi-omics data with matching cell numbers across assays (n = 62,219). Importantly, this cell pairing further enabled cell type annotation of scATAC-seq by simply using annotations defined from scRNA-seq gene expression markers (Figures 2F, 2G, and S3M). Aggregating single cells by cell type and condition and filtering for sufficient counts resulted in 139 pseudobulks (averaging a total of 1.94 million RNA and 2.3 million ATAC aggregate counts). Utilizing this high-depth resource, we find that chromatin accessibility at distal peaks is highly cell type specific, even more so than gene expression, whereas promoter accessibility is relatively invariant across cell types and stimulation conditions (Figure 2H), validating prior reports.17,46 Overall, the high quality of these data and the exquisite cell type specificity of distal chromatin accessibility motivated further analysis of the GRN underlying stimulus response. We reasoned that this scOptMatch approach for cell pairing, enabling approximately uniform pairing of scATAC to scRNA profiles, would establish an integrated dataset and could be used for downstream analysis analogous to accessibility and RNA expression profiling concomitantly within the same cell.
Identification of distal peak-gene interactions across stimulation using integrated single-cell data
We next sought to associate changes in cis-regulatory peaks with the expression of genes as a means to prioritize features that are part of the immunological response GRN. To do so, we include as part of the FigR framework a computational approach to determine significant distal peak-to-gene expression interactions, as performed previously on multi-modal data.28 Specifically, we used computationally paired cells (n = 62,219 cells per assay) to correlate accessibility from peaks found within a fixed window (100 kb) around each gene’s transcription start site (TSS) with expression of that gene with permutation-based testing to estimate the statistical significance for a given peak-gene pair (Figure 3A; STAR Methods). In this way, we identified a total of 34,370 unique chromatin accessibility peaks genome wide, showing a significant association with gene expression (permutation p ≤ 0.05), spanning a total of 11,304 genes. Prioritizing genes based on their total number of significantly correlated peaks, we identified a subset of genes associated with a high density of peak-gene interactions, which we recently described as DORCs28 (Figure 3B; n ≥ 7 significant peak-gene associations, n = 1,128 genes, n = 12,583 peaks; Data S1).
Figure 3.

Integrative multi-omics analysis identifies key regulatory modules associated with stimulus response in single cells
(A) Schematic of cis-regulatory analyses for identification of significant chromatin accessibility peak-gene associations using computationally paired scATAC-seq and scRNA-seq stimulation datasets.
(B) Top hits based on the number of significant gene-peak correlations across all cell types and stimulus conditions.
(C) Loop plots highlighting significant peak-gene associations for DORC TRAF1, determined using the approach outlined in (A).
(D) UMAP of DORC accessibility scores (left) and paired RNA expression (right) for TRAF1.
(E) Pairwise Pearson correlation of aggregate DORC accessibility scores and RNA expression of cells per condition per cell type across all DORCs, clustered using hierarchical clustering by DORC score correlations.
(F) Global DORC accessibility (top) and gene expression (bottom) change displayed based on the Pearson correlation coefficient of the aggregate score across DORCs for each stimulation condition versus its corresponding control condition, shown per condition per cell type annotation.
(G) Heatmap showing the mean difference in single-cell DORC accessibility for the union of the top 10 differential DORCs across conditions and cell types (n = 53 genes). The cell type color bar represents the cell group having the most significant change across all conditions for that assay.
The list of DORC-associated genes included many known mediators of immunological response associated with innate and adaptive immune response pathways,10,40,47,48 as also confirmed by gene set enrichment analysis (GSEA) (Figure S4A; Data S1). Notably, among these genes, we see a large fraction of distal cis-regulatory associations (>5 kb away from the gene TSS; Figures 3C, S4B, and S4C). By scoring cells using the total associated peak accessibility signal per DORC (referred to as the DORC accessibility score), we determine correspondence between chromatin accessibility and gene expression across single cells (Figures 3D, S4D, and S4E) or across pseudobulks for each DORC, stimulation condition, and cell type (Figure 3E). Upon comparison with matched control conditions (DMSO controls), we observed the largest effect on DORC accessibility and expression from treatment with PMA, as seen across most cell types, and a more moderate effect with treatment of IFNƔ or LPS, as seen predominantly in monocytes (Figure 3F). Notably, we found that stimulation induces a larger change in the transcriptome of the cells in comparison with chromatin accessibility, but cell types concordantly altered chromatin and expression to induce activation of immunity genes (Figures 3E and 3F). Interestingly, we also find that addition of the GI strongly attenuates the immune response to PMA (CD8, NK, and B cells) and LPS (CD8), likely a consequence of inhibiting paracrine signaling, and in response to IFN (monocytes), likely a consequence of inhibiting autocrine signaling. Surprisingly, only a few cell types, including B and CD8 T lymphocytes, exhibited this dampened response in accessibility and gene expression change after 6 h of PMA exposure when simultaneously treated with the GI, suggesting that, in most contexts, DORCs are intrinsically regulated.
Single-cell differential testing among DORCs identified a number of essential regulators of immunological response (Figures 3G and S4F; Data S2 and S3). This includes shared LPS- and IFN-induced genes (MX1, IFIT3, OAS3, and OASL) and PMA-induced genes associated with cellular apoptosis and survival (NR4A1/2/3, EGR1, REL, and TRAF1). Interestingly, we also observe primary ligand-encoding genes (IL1A, IL1B, and CCL3) and immune inhibitors (CD274 [also known as PDL1], NFKBIA, and TNIP1) among these top differential DORCs. Notably, our cis-regulatory analysis recovers DORCs, the majority of which (∼79%) include genes previously annotated to be linked to super-enhancer regions across diverse cellular contexts31 (STAR Methods; Figures S4G and S4H), the remainder (n = 238 genes) includes several stimulation response genes (IFIT1, MX1, OAS1/3, IL13, IL3RA, and IL27RA) and cell type markers (CD14, NKG7, GZMK, and CD8B). Our approach to identify DORCs uncovers genes under extensive chromatin control, likely a result of immune cells requiring exquisite control of transcription at these genes, reflecting key hubs of immunological response.
Stimulated cells are characterized by early changes in the chromatin accessibility landscape that primes gene expression
Previously, we used multi-modal data to show that DORC accessibility foreshadows gene expression along developmental trajectories and that this activity is predictive of cell state transitions.28 To this end, we sought to use paired multi-omics data to find out whether cells prime for an immunological response through their chromatin accessibility states. Methods to deduce trajectory pseudotime often require definition of a single root cell type. Because we identified 18 discrete cell types, precluding use of pseudotime, we sought to utilize an alternate approach to define trajectories. To do this, we computed a cell nearest neighbor (NN) stimulation time per treatment which represents the weighted average of stimulus exposure time based on experimental treatment labels. Briefly, we take cell-NNs (k = 50) for each cell and compute the average neighborhood for control, 1-h-stimulated, and 6-h-stimulated cells, assigning weights of 0, 1, and 2, respectively (Figures 4A and S5A; STAR Methods). This continuous measure of time allows us to investigate the chromatin accessibility and gene expression dynamics along the stimulation trajectory (Figure S5B).
Figure 4.

Chromatin and gene expression dynamics with respect to stimulus response time
(A) UMAP of scATAC cells colored by estimated NN stimulation (stim) time per stimulus condition.
(B) UpSet plot highlighting overlap of monocyte-constrained DORC genes determined for the three different stimulus conditions.
(C) Heatmaps highlighting smoothed normalized DORC accessibility, RNA expression, and residual (DORC-RNA) levels for DORC genes (n = 38) identified to be associated with LPS NN stimulation time in control (1 h) and stimulated (1 h/6 h) monocytes (n = 1,776 cells).
(D) Chromatin (DORC) versus gene expression (RNA) dynamics of DORCs FOSB (left) and IFIT3 (right) with respect to smoothed PMA and LPS NN stim time, respectively, for control (1 h) and stimulated (1 h/6 h) monocytes (n = 2,002 cells for PMA + control, n = 2,601 cells for IFNƔ + control). A dotted line represents a LOESS fit to the values obtained from a sliding average of DORC accessibility or RNA expression levels (n = 100 cells per sliding window bin). The color bar indicates the most frequent (mode) cell condition within each bin.
(E) Same as in (D) but for TRAF1 with respect to LPS-stimulated and control (1 h) monocytes.
(F) Smoothed accessibility scores for individual cis-regulated elements correlated with TRAF1 expression in control and LPS-stimulated monocytes shown in (D), ordered by LPS NN stim time.
Using these stimulation time definitions, we sought to determine whether chromatin accessibility activates before gene expression to “prime” or “foreshadow” immunological response. For this analysis, we chose to focus on the monocyte cellular population because it is directly activated in response to our inflammatory factors, as described by prior literature and our observations with the GI (Figures 3F and 3G), to assess chromatin and gene expression dynamics with respect to stimulation time. Restricting our peak-gene correlation approach strictly to control 1 h, stimulation 1 h, and stimulation 6 h monocytes, we identified a set of DORC genes associated with LPS (n = 38 genes), IFNƔ (n = 33 genes), or PMA (n = 39 genes) stimulation of monocytes. These DORCs include known expression markers induced upon stimulation in myeloid cells.40 Interestingly, we also found that a small subset of these monocyte-specific DORCs is shared across multiple stimuli (Figure 4B). By averaging single-cell DORC accessibility and RNA levels in response to the NN stimulation (stim) time for each treatment (STAR Methods), we visualize the change of chromatin accessibility and gene expression along the control (0 h) to 6-h stimulation time axis (Figures 4C, S5C, and S5D).
Calculating the difference in chromatin versus RNA (residuals), we predominantly observed that chromatin accessibility change precedes that of expression (high residuals) at early time points. At later time points, we found that residuals were low, reflecting an accumulation of RNA after immune stimulation. These observations were stereotyped by the genes FOSB and IFIT3 with LPS and PMA treatment, respectively (Figures 4C, 4D, and S5E). These changes occurred on relatively fast timescales; for example, chromatin change of FOSB was an early event occurring within the 60-min time point. Notably, these changes in DORC accessibility are constituted by individual cis-regulatory elements where some elements become accessible quickly (i.e., the promoter), whereas others are slow to become accessible (some distal regulatory elements) along the stimulation time axis, as highlighted for the LPS-responsive gene tumor necrosis factor (TNF) receptor-associated factor 1 (TRAF1) (Figures 4E and 4F). Conversely, we note a few exceptions, including the PMA-responsive heat shock protein-encoding genes HSP90AA1 and HSPH1, which exhibit early expression gain compared with the corresponding change in DORC accessibility (Figure S5F). We demonstrate, using computationally paired multi-omics data, the ability to detect activation of chromatin accessibility prior to gene expression associated with stimulation-like cell states.
A computational approach to identify candidate TF regulators of DORC activity
At the core of FigR, we developed a computational approach to define a GRN of immunological response using multi-omics data. At this stage, FigR uses paired scATAC-seq and scRNA-seq data and specifically tests for enrichment of TF motifs among predetermined cis-regulatory elements (i.e., DORCs) as well as correlation of TF expression with the overall accessibility level for a given DORC gene (DORC score) to infer likely TF activators and repressors (Figure 5A). First, for a given DORC gene, we determine a pool of DORC cis-regulatory elements based on its DORC accessibility kNNs. This assumes that DORCs that are co-variable across the entire cell space are co-regulated by shared TFs. We then perform a statistical test for significance (Z test) of TF motif enrichment using the frequency of motif matches across a reference database of TF motifs relative to a background set of permuted peaks matched for GC content and global peak accessibility. Concomitantly, we compute the Spearman correlation coefficient between the TF RNA expression levels and the DORC accessibility score. Last, to determine activators and repressors, we combine significance estimates of relative motif enrichment (Z test P) and RNA expression correlation (Z test P) for a given DORC relative to all TFs, computing a signed probability score we call a “regulation probability” (“regulation score” on −log10 scale), representing the intersection of motif-enriched and RNA-correlated TFs. To enable discovery of new regulators using this approach, we curated an expanded set of unique human (n = 1,141) and mouse (n = 890) TF binding sequence motifs, which extends a previously established database49 (STAR Methods).
Figure 5.

Design and application of FigR’s gene regulatory network (GRN) workflow to identify TF modulators of immune response DORCs
(A) Schematic describing the FigR GRN workflow.
(B) Scatterplot showing all DORC-to-TF associations, colored by the signed regulation score.
(C) Candidate TF regulators of MX1. Highlighted points are TFs with abs(regulation score) ≥ 1 (−log10 scale), with all other TFs shown in gray.
(D) Regulation scores (signed, −log10 scale) for the highlighted TFs in (C).
(E) Mean regulation score (signed, −log10 scale) across all DORCs (n = 1,128) per TF (n = 870), highlighting select TF activators (right skewed) versus TF repressors (left skewed).
(F) Heatmap of DORC regulation scores (left) for all significant TF-DORC enrichments for DORCs implicating GWAS variants (abs(regulation score) ≥ 1.5; n = 89 TFs, n = 73 DORCs). The corresponding minimum GWAS P (right; −log10 scale) for each DORC across all diseases considered is also shown.
(G) TF-DORC network visualization for SLE GWAS SNP-implicated DORCs (orange nodes) and their associated TFs (gray nodes) from (F). Edges are scaled and colored by the signed regulation score.
To demonstrate the utility of the FigR GRN method, we apply it to the paired stimulation scATAC-seq and scRNA-seq data to reveal key regulators of stimulus response. To do this, we begin by testing all stimulus-responsive DORC genes (n = 1,128) and reference TF motifs (Figure 5B; Data S4). Filtering TF-DORC associations using a regulation score threshold (abs(regulation score) ≥ 1), we can then query putative TF regulators for a given DORC (Figures 5C, 5D, S6A, and S6B) as well as sets of DORCs that are potentially driven by a specific TF (Figure S6C). For example, FigR identifies known activators of MX1, including the IRF family of TFs: IRF3, IRF7, IRF9, and STAT2, all belonging to the IFN signaling pathway.50 We generally distinguish TF activators from TF repressors based on their mean regulation score across all DORCs (Figure 5E) or by the fraction of positively and negatively regulated DORCs (Figure S6D). For example, we see SPI1 (PU.1), BACH1, and BCL11A as top transcriptional activators whose roles have been described previously17,51,52 and BCL11B as a top transcriptional repressor (Figures 5E, S6C, and S6D). Importantly, BCL11B has been shown to be a key repressor in T cell maturation.53, 54, 55 Other examples of repressors nominated by FigR include KLF9,56 BACH2,57,58 IKZF1,59,60 and ZBTB4.61 Our approach estimates that 35.6% of TFs associated with DORCs (absolute(regulation score) ≥ 1) to have repressive associations (mean regulation score < 0 across all target DORCs), in line with previous work reflecting the understanding that a large fraction of TFs function as repressors.37,38 Last, demonstrating the value of the unbiased nature of FigR’s GRN inference and utilization of an expanded TF motif database, we identify new regulatory interactions governing immune cell function (Figure S6E). We highlight ZEB2, which has been implicated as a repressor of CD8+ T cell function.62 FigR verifies its repressive activity and identifies downstream target DORCs (n = 132; regulation score < −1.5), including IL7R and TCF7, which are associated with immunological memory in T cells.63 We also identify the activator ZNF467, a completely novel regulator, which regulates 86 genes (regulation score > 1.5). Among the direct targets of ZNF467, we find CD14, a key marker of monocytes and essential for immunological function in monocyte activation and differentiation.64 Importantly, we observe expression of these TFs to be stimulus specific and cell type specific (Figure S6F), suggesting FigR’s ability to leverage covariance in single-cell data to determine context-specific associations.
To highlight the broad generalizability of this approach, we applied our approach to existing multi-modal data from alternative tissues, including SNARE-Seq2 data from the human cortex and SHARE-seq data from murine skin tissue. cis-regulatory correlations for the SNARE-Seq2 brain data (Figure S3H) identified a subset of genes linked to DORCs (n = 432; Figure S7A), including known markers of neuronal (excitatory: FEZF2, RORB; inhibitory: PVALB, LAMP5, GAD1), and non-neuronal (microglia: PAX6, SLC1A3; oligodendrocyte: MBP, FA2H) differentiation (Figures S7B and S7C). Applying FigR’s GRN analysis approach using these cortex-specific DORCs (Figure S7D), we nominated several TF modulators of DORC activity, including the SOX family members SOX10/13, OLIG1/2, POU3F2, and DLX1/2, as TF activators and PAX6 and BATF3 as prominent repressors (Figures S7E–S7G). Next, using predefined peak-gene associations, our GRN inference approach recovered TF regulators of DORCs we previously found to be associated with murine hair follicle differentiation (Figure S7H).28 This includes the activators Lef1, Hoxc13, and Grhl1 and repressors Tcf12 and Pou2f3 (Figure S7I). We determined the activator Dlx365 and repressors Zeb1 and Barx266 to be top TF regulators (Figures S7J and S7K). To assess differences between FigR’s approach of incorporating cis-regulatory elements surrounding a subset of genes (i.e., DORCs) and alternative methods that use scRNA-seq co-expression to derive GRNs, we performed a comparison of prioritized regulators determined using our approach and SCENIC20 when applied to the stimulation PBMC data (STAR Methods). Comparing the number of DORC genes positively regulated by a given TF for either method, we observed a subset of TFs for which a larger number of targets determined via FigR compared with SCENIC were called and vice versa (Figure S8A). Interestingly, interrogating GM12878 chromatin immunoprecipitation sequencing (ChIP-seq) data for the top TFs from either category suggests that SCENIC-prioritized TF regulators tend to bind promoter regions, whereas those prioritized by FigR overlapped regulatory elements mapping to distal enhancers more than expected by baseline (Figure S8B). This suggests that FigR indeed prioritizes TFs that are more enhancer associated. Thus, we show that FigR can exploit diverse single-cell technologies for experimentally paired multi-modal data to derive GRNs using empirically derived peak-to-gene and TF-to-peak motif associations to arrive at candidate TF regulators.
We next wanted to determine whether the inferred stimulus response GRN from FigR may be used to reveal the regulatory mechanisms underlying disease-associated genetic variants and their non-coding regulatory elements. To uncover disease-associated cell states, we scored single cells for accessibility associated with GWAS SNP-overlapping peaks (GWAS, p < 10−7; Figure S8C). We observed stimulus- and cell-type-specific enrichment of chromatin accessibility for different inflammatory diseases tested (Figure S8D), validating prior work16,67 showing that immunological stimulation uncovers regulatory elements enriched for disease GWAS variants. For example, we observed elevated enrichment of GWAS-associated accessibility in LPS- and IFNƔ-stimulated B lymphocyte and monocyte cells for systemic lupus erythematosus (SLE) and IFNƔ- and PMA-stimulated CD4/CD8 lymphocytes for allergies (Figure S8E). We find that our immunological stimulations uncover cell states and their corresponding chromatin accessibility profiles relevant to autoimmunity and associated genetic variation.
Next we reasoned that our GRN-based analysis may identify relevant mechanisms of disease-associated genetic variation. For example, the regulator NF-B is known to function across cell types to promote inflammatory gene expression.68 Indeed, we found NF-B to drive activity of a large fraction of GWAS variant-associated DORCs (Figure 5F). Extending this analysis to all DORCs (n = 77), we uncovered 89 putative TF drivers (abs(regulation score) ≥ 1.5), revealing a combination of lineage-determining as well as stimulus-responsive TFs spanning one or more diseases (Figure 5F). Closer inspection of the subset of SLE-specific DORCs (n = 15 DORCs, n = 48 associated TFs) revealed key regulatory associations, including previously determined SLE genes: BLK, IRF5, IRF8, and NCF2 (Figure 5G). Our approach can prioritize DORCs and their putative TF regulators to dissect the regulatory programs implicated in diverse autoimmune diseases. We include the inferred stimulus-response PBMC GRN, which can be interactively visualized through an RShiny web application (https://buenrostrolab.shinyapps.io/stimFigR/).
Discussion
Here we generated a regulatory atlas of immunological stimulation in human blood. This effort was enabled by high-coverage single-cell data and development of a new computational framework supporting multi-omics data integration, cis-regulatory analyses, and construction of an enhancer-aware GRN based on single-cell profiles. In this effort, we overcame 3 key challenges: (1) we implemented an approach to better computationally pair single cells, (2) we associate distal cis-regulatory peaks with target genes, and (3) we associate TFs with target genes. Importantly, the capability of FigR to infer GRNs using independently or concomitantly generated single-cell ATAC/RNA data will broadly enable DORC-GRN analyses across the broad range of scATAC-seq and related multi-omics technologies. Unlike prior methods that solely use co-expression or static measures of co-accessibility, GRN construction using our proposed FigR framework leverages chromatin and RNA dynamics through correlation of these features across single cells, providing a means to identify gene-regulatory relationships spanning cell states. To do this, we utilize an empirical statistical approach to compute the probability of a TF-gene interaction, avoiding use of parameterized machine learning approaches. We observe that this determination of expression-linked cis-regulatory elements using single-cell data inherently selects for genes generally regulated by super-enhancers without the need for independent profiling of histone modifications, as we also confirm in prior work using multi-modal data,28 while harboring context-specific variability across single cells.
Importantly, we also show that the statistical tools in FigR (peak-gene and TF-gene) are generalizable and can be applied to true multi-modal datasets assaying chromatin accessibility and gene expression from the same cell. We believe that a multi-omics-based approach that incorporates both elements that are thought to be functionally relevant to TF activity (i.e., TF RNA expression level together with enrichment of a binding motif within cis-regulatory elements) would reduce potential false positives that can occur from using only ATAC (enhancer-promoter correlation) or RNA (gene-gene co-expression) information alone and can enable unbiased identification of repressive TF-DORC relationships that would not be possible otherwise. In support of this observation, we find that ∼36% of TFs largely function as repressors, consistent with prior functional studies in Drosophila37 and in cell lines38 that report a large fraction of TFs function as repressors. The fact that DORCs contain many associated peaks (mean = 11.15) enables us to determine estimates of enriched TF motifs at these loci. However, a limitation of this approach (and similar methods utilizing single-cell data) is that one can only determine regulatory relationships when they are variable across single cells, constraining GRN models to observed changes across cell states and precluding analysis of “housekeeping” regulators. Our framework may benefit from prior determination of cluster-specific peaks because it may aid identification of rare cell populations and their specific DORCs.
We find that DORCs closely correspond to super-enhancers (78%) and that GWAS variants are enriched within DORCs that respond to immunological stimuli. Thus, defining peak-gene interactions and DORCs provides a useful platform to annotate the function of non-coding genetic variants corresponding to autoimmune and inflammatory conditions. Prior epigenomic studies have extensively utilized bulk analysis of histone modifications, chromatin accessibility, and genome topology to annotate the function of disease-associated non-coding genetic variation.16,31,41,48,69 Advancing beyond these prior studies, our single-cell multi-omics GRN approach provides a framework for associating key disease-associated loci with their target genes and regulating TFs at single-cell resolution, showcasing the utility of this approach using immune cells in response to stimuli.
Generally supporting the hypothesis that chromatin accessibility foreshadows gene expression (chromatin potential28), we find that chromatin accessibility precedes gene expression even with the extraordinarily fast gene expression dynamics associated with immunological stimulation. This, together with a large body of work,1,70,71 upends the notion that chromatin change is “slow” or “stable” and instead paints a picture where chromatin structure is highly dynamic. Future work focused on models distinguishing promoter versus distal enhancer accessibility changes among DORCs and TF drivers of these changes may provide additional insights into the regulatory mechanisms underlying gene priming and activation associated with cell-state-specific changes. We anticipate that our approach for defining GRNs will enable elucidation of latent chromatin states that prime or inhibit cells from diverse environmentally induced stimuli.
We envision that future studies will improve our capacity to predict gene-regulatory relationships using single-cell data. Specifically, because FigR GRN associations rely on correlations, we anticipate that advancements in machine learning-based methods, informed by emerging efforts in large-scale perturbation assays,33,72,73 may be used to more accurately capture regulatory interactions. Higher-coverage multi-omics measurements may enable use of TF footprinting,74,75 providing a direct measurement of TF-peak interactions. These multi-omics single-cell methods and models constructing GRNs advance our ability to nominate essential regulators, defining candidates that may be used for high-throughput perturbation screens76 that reveal the function of genetic variants or TFs that drive cells into new states. We envision a future of single-cell genomics that will shift toward studies of gene-regulatory processes advancing the predictive capability of cells undergoing fate transitions as well as elucidating the latent/primed potential of cells prior to environmental stimulation and their relevance in development and disease.
Limitations of the study
In our framework, we incorporated use of correlation coefficients to capture variability across single-cell chromatin accessibility and coupled gene expression changes together with permutation-based significance testing. This limits applications in cases where the modalities are coupled but the covariance is weak (e.g., testing across a single cell type) or where features can sometimes be dominated by more prevalent cellular populations. There are now multiple tools to assist with pairing of cells across independently assayed modalities, including scATAC-seq and scRNA-seq. These should be evaluated, together with scOptMatch, using a variety of single-cell multi-omics datasets for benchmarking and for potential future incorporation into the FigR framework. Last, further “ground truth” experimental data confirming enhancer-gene or TF-gene relationships will be invaluable to the field in resolving the accuracy of GRN methods, including FigR.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human peripheral blood mononuclear cells (PBMCs), Donor1, 24 yo, Female, 100M cells | Allcells | Cat# LP, CR, MNC, 100M; Lot#3003978 |
| Human peripheral blood mononuclear cells (PBMCs), Donor2, 25 yo, Male, 25M cells | Allcells | Cat# LP, CR, MNC, 25M; Lot#3003790 |
| Human peripheral blood mononuclear cells (PBMCs), Donor3, 26 yo, Male, 25M cells | Allcells | Cat# LP, CR, MNC, 25M; Lot#3003748 |
| Human peripheral blood mononuclear cells (PBMCs), Donor4, 37 yo, Male, 25M cells | Allcells | Cat# LP, CR, MNC, 25M; Lot#3003459 |
| Chemicals, peptides, and recombinant proteins | ||
| DNase I | Thermo Fisher Scientific | Cat# 18047019 |
| 1x PBS | Thermo Fisher Scientific | Cat# 10010023 |
| BSA | MilliporeSigma | Cat# A9418-5G |
| Lipopolysaccharide | Invivogen | Cat# tlrl-3pelps |
| Phorbol 12-myristate 13-acetate | MilliporeSigma | Cat# P8139 |
| Ionomycin calcium salt | MilliporeSigma | Cat# I0634 |
| Interferon gamma | Cell Applications | Cat# RP1077 |
| GolgiPlug | BD Biosciences | Cat# 555029 |
| Tween 20, 10% solution | Teknova | Cat# T0710 |
| Digitonin | Promega | Cat# G9441 |
| Ampure XP beads | Beckman Coulter | Cat# A63880 |
| Bst 2.0 WarmStart | NEB | Cat# M0538S |
| Critical commercial assays | ||
| SureCell ATAC-Seq Library Prep Kit | Bio-Rad | Cat# 17004620 |
| SureCell ddSEQ Index Kit | Bio-Rad | Cat# 12009360 |
| High-Sensitivity DNA kit | Agilent | Cat# 5067-4626 |
| NextSeq High Output Kit (150 cycles) | Illumina | Cat# 20024907 |
| SureCell WTA 3′ Library Prep Kit | Illumina | Cat# 20014280 |
| Deposited data | ||
| All raw and processed scATAC-seq and scRNA-seq data | This paper | GEO:GSE178431 |
| Software and algorithms | ||
| R (v4.0.5) | R Core Team | https://www.R-project.org |
| chromVAR R package (v1.12.0) | Schep et al., 201749 | https://github.com/GreenleafLab/chromVAR |
| motifmatchr R package (v1.12.0) | Schep et al., 201749 | https://github.com/GreenleafLab/motifmatchr |
| hyper R package (v1.7.0) | Federico et al., 202085 | https://github.com/montilab/hypeR |
| Seurat R package (v3.9.9) | Stuart et al., 201942 | https://satijalab.org/seurat/ |
| ArchR R package (v1.0.1) | Granja et al., 202129 | https://www.archrproject.com/ |
| ggplot2 R package (v3.3.3) | Wickham et al., 201686 | https://ggplot2.tidyverse.org/ |
| optmatch R package (v0.9-13) | Hansen and Klopfer, 200644 | https://github.com/markmfredrickson/optmatch |
| SCENIC R package (v1.2.4) | Aibar et al., 201720 | https://scenic.aertslab.org/ |
| FNN R package (v1.1.3) | Beygelzimer et al., 201987 | https://cran.r-project.org/web/packages/FNN/index.html |
| igraph R package (v1.2.6) | Csardi and Nepusz, 200688 | https://igraph.org/r/ |
| ggnet R package (v0.1.0) | Briatte et al., 2016 | https://briatte.github.io/ggnet/ |
| MACS2 (v2.1.2) | Zhang et al., 200878 | https://github.com/taoliu/MACS/ |
| samtools (v1.9) | Li et al., 200989 | http://samtools.sourceforge.net |
| Analysis code generated for this manuscript | This paper |
https://github.com/buenrostrolab/stimATAC_analyses_code/ Zenodo archive: https://zenodo.org/badge/latestdoi/288481382 |
| FigR R package (v1.0.1) | This paper | Github: https://github.com/buenrostrolab/FigR Zenodo archive: https://doi.org/10.5281/zenodo.6795583 |
| cisBP Human and Mouse TF motif position frequency matrix (PFM) lists | This paper | Zenodo archive: https://doi.org/10.5281/zenodo.6814702 |
| Other | ||
| Resource website (R Shiny App) for browsing stimulus multi-omic single cell data and DORC features/GRN | This paper |
https://buenrostrolab.shinyapps.io/stimFigR/ Zenodo archive: https://doi.org/10.5281/zenodo.6820097 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jason D. Buenrostro (jason_buenrostro@harvard.edu).
Materials availability
This study did not generate unique reagents.
Method details
Human peripheral blood mononuclear cells
Cryopreserved human peripheral blood mononuclear cells (PBMCs) and isolated peripheral blood CD4+, CD8+, CD14+, CD19+ and CD56+ cells were purchased from AllCells (see Table S1 for catalog numbers and donor information). Cells were quickly thawed in a 37°C water bath, rinsed with culture medium (Iscove’s Modified Dulbecco’s Medium (IMDM) (ATCC) supplemented with 10% FBS and 1% Pen/Strep) and then treated with 0.2 U/μL DNase I (Thermo Fisher Scientific) in 10mL of culture medium at 37°C for 30 min. After DNase I treatment, cells were washed with medium once and then twice with ice-cold 1x PBS (Gibco) + 0.1% BSA (MilliporeSigma). Cells were then filtered with a 35 μm cell strainer (Corning) and cell viability and concentration were measured with trypan blue on the TC20 Automated Cell Counter (Bio-Rad). Cell viability was greater than 80% for all samples.
Human PBMCs stimulations
PBMCs were quickly thawed in a 37°C water bath, rinsed with culture medium (RPMI 1640 medium supplemented with 15% FBS and 1% Pen/Strep) and then treated with 0.2 U/μL DNase I in 10mL of culture medium at 37°C for 30 min. After DNase I treatment, cells were washed with medium once, filtered with a 35 μm cell strainer and cell viability and concentration were measured with trypan blue on the TC20 Automated Cell Counter. Cell viability was greater than 90% for all samples. Cells were plated at a concentration of 1 × 106 cell/mL, rested at 37°C and 5% CO2 for 1 h and then treated with the specified concentrations of the following stimulants (or DMSO as a control) for either 1h or 6h:
-
1)
20 ng/mL Lipopolysaccharide (LPS) (tlrl-3pelps, Invivogen),
-
2)
50 ng/mL Phorbol 12-myristate 13-acetate (PMA) (P8139, MilliporeSigma) + 250 ng/mL Ionomycin calcium salt (I0634, MilliporeSigma),
-
3)
20 ng/mL Interferon gamma (IFN-Ɣ) (RP1077, Cell Applications)
For the "Golgi Inhibitor" experiments, cells were incubated for 6 h with GolgiPlug (555029, BD Biosciences) at a 1:1000 dilution plus stimulants at the concentrations indicated above (or GolgiPlug only as a control).
After stimulation, cells were washed twice with ice-cold 1x PBS + 0.1% BSA and cell viability and concentration were measured with trypan blue on the TC20 Automated Cell Counter.
scATAC-seq experimental methods
Cell lysis and tagmentation
For a detailed description of tagmentation protocols and buffer formulations refer to the SureCell ATAC-Seq Library Prep Kit User Guide (17004620, Bio-Rad). Harvested cells and tagmentation related buffers were chilled on ice. Lysis was performed simultaneously with tagmentation. Washed and pelleted cells were resuspended in Whole Cell Tagmentation Mix containing 0.1% Tween 20, 0.01% Digitonin, 1x PBS supplemented with 0.1% BSA, ATAC Tagmentation Buffer and ATAC Tagmentation Enzyme (ATAC Tagmentation Buffer and Enzyme are both included in the SureCell ATAC-Seq Library Prep Kit (17004620, Bio-Rad)). Cells were mixed and agitated on a ThermoMixer (5382000023, Eppendorf) for 30 min at 37°C. Tagmented cells were kept on ice prior to encapsulation.
Droplet library preparation and sequencing
For a detailed protocol and complete formulations, refer to the SureCell ATAC-Seq Library Prep Kit User Guide (17004620, Bio-Rad). Tagmented cells were loaded onto a ddSEQ Single-Cell Isolator (12004336, Bio-Rad). Single-cell ATAC-seq libraries were prepared using the SureCell ATAC-Seq Library Prep Kit (17004620, Bio-Rad) and SureCell ddSEQ Index Kit (12009360, Bio-Rad). Bead barcoding and sample indexing were performed in a C1000 Touch™ Thermal cycler with a 96-Deep Well Reaction Module (1851197, Bio-Rad): 37°C for 30 min, 85°C for 10 min, 72°C for 5 min, 98°C for 30 s, eight cycles of 98°C for 10 s, 55°C for 30 s, and 72°C for 60 s, and a single 72°C extension for 5 min to finish. Emulsions were broken and products cleaned up using Ampure XP beads (A63880, Beckman Coulter). Barcoded amplicons were further amplified using a C1000 Touch™ Thermal cycler with a 96-Deep Well Reaction Module: 98°C for 30 s, six to nine cycles (cycle number depending on the cell input, Section 4 Table 3 of the User Guide) of 98°C for 10 s, 55°C for 30 s, and 72°C for 60 s, and a single 72°C extension for 5 min to finish. PCR products were purified using Ampure XP beads and quantified on an Agilent Bioanalyzer (G2939BA, Agilent) using the High-Sensitivity DNA kit (5067-4626, Agilent). Libraries were loaded at 1.5 p.m. on a NextSeq 550 (SY-415-1002, Illumina) using the NextSeq High Output Kit (150 cycles; 20024907, Illumina) and sequencing was performed using the following read protocol: Read 1 118 cycles, i7 index read eight cycles, and Read 2 40 cycles. A custom sequencing primer is required for Read 1 (16005986, Bio-Rad; included in the kit).
scRNA-seq experimental methods
Single-cell RNA-seq (scRNA-seq) data for LPS, PMA or IFNƔ-stimulated cells, and isolate (bead-enriched) PBMCs comprising CD19+, CD4+ T-cells, CD8+ T-cells, CD56+ Natural Killer (NK) cells and CD14+ monocytes were generated using the SureCell WTA 3′ Library Prep Kit for the ddSEQ System (20014280, Illumina) with the following modifications. A higher concentration of beads was used to obtain 1,000–2,000 single-cells per emulsion, whilst minimizing the number of droplets with multiple beads to <10%. Furthermore, Bst 2.0 WarmStart (M0538S, NEB) was added to the droplet mix to perform temperature activated second strand synthesis in droplets.
scATAC-seq analysis workflow
Raw read processing, demultiplexing and alignment
Per-read bead barcodes were parsed and trimmed using UMI-TOOLs (https://github.com/CGATOxford/UMI-tools),77 and the remaining read fragments were aligned using BWA (https://github.com/lh3/bwa) on the Illumina BaseSpace online application. Constitutive elements of the bead barcodes were assigned to the closest known sequence allowing for up to one mismatch per 6-mer or 7-mer (mean > 99% parsing efficiency across experiments). All sequence libraries were aligned to the hg19 reference genome. We then used bead-based ATAC-seq data processing (BAP, v0.6.4) (https://github.com/caleblareau/bap)13 to help identify systematic biases (i.e. reads aligning to an inordinately large number of barcodes), barcode-aware deduplication of reads, and to perform merging of multiple bead barcode instances associated with the same cell (barcode merging is necessary due to the nature of the Bio-Rad SureCell scATAC-seq procedure used in this study, which enables multiple beads per droplet). For a detailed description of the bead barcode merging strategy see.13 We ran BAP using a single input alignment (.bam) file for a given experiment with a bead barcode identifier indicated by the SAM tag “DB”, and default parameters.
Chromatin accessibility peak calling
Genome-wide chromatin accessibility peaks were called using MACS v2 (MACS2)78 on the merged aligned scATAC-seq reads per treatment condition, with the following flags explicitly set: --nomodel, --nolambda, --keep-dup all, --call-summits; generating a list of peak summit calls per condition. Summits were then ranked per condition based on their FDR score (from MACS2), and the summit scores rank-normalized such that the normalized summit scores rendered are comparable across conditions, as performed previously.11 Peak summits were then padded by 400 bases on either end (generating 801 bp windows), and overlapping peak windows filtered iteratively such that windows with higher scores were retained at each step. This resulted in a filtered list of disjoint peaks (n = 219,136), which were finally resized to 301 bp (i.e. ± 150 bp from each peak summit) and used for all downstream analyses.
scATAC-seq counts generation and QC
Single cell counts for reads in peaks were generated by intersecting the peak window regions (see previous section) with aligned fragments. First, we offset the start and end coordinates of the aligned fragments to identify Tn5 cut sites by +4 or −5 bp for fragments aligning to the positive or negative strand, respectively. These are then intersected with peak window regions using the findOverlaps function in R, and the total number of unique fragment cut sites overlapping a given peak window tallied for each unique cell barcode detected in the data, producing a matrix of single cell chromatin accessibility counts in peaks (rows) by cells (columns). Only cells with fraction of total reads in peaks (FRIP) ≥ 0.5, a minimum of 2,000 unique nuclear fragments (UN-Fs), and a sequencing library duplication rate ≥0.15 were retained. Cell barcode doublets were inferred and filtered out using ArchR.29 This resulted in a total of n = 67,581 and n = 17,920 cells, for the stimulated and isolate PBMC cells, respectively.
TF motif accessibility scoring
Single cell accessibility scores for TF motifs were computed using chromVAR,49 as also previously described.12,13 For TF motif accessibility scores, the peak by TF motif overlap annotation matrix was generated using a list of human TF motif PWMs (n = 870) from the chromVARmotifs package in R (https://github.com/GreenleafLab/chromVARmotifs), and used along with the scATAC-seq reads in peaks matrix to generate accessibility Z-scores for across all scATAC-seq stimulated cells passing filter.
Gene activity scoring
Single cell gene activity scores were generated using scATAC-seq data based on an exponential decay weighted sum of fragment counts around a given gene TSS as we have previously described79 for all scATAC-seq cells passing filter, using the hg19 reference for gene TSSs. Raw gene scores were then normalized by dividing by the mean gene score per cell.
scATAC-seq cell clustering, visualization and annotation
Single cell clustering of ATAC-seq data was performed using the ArchR framework.29 First, the accessibility counts in a tiled window matrix was determined using default parameters. ArchR’s iterative LSI dimensionality reduction was performed for n = 30 components and n = 2 iterations, taking the top variable 50000 peaks and evaluating resolutions 0.1 to 0.4, sampling 20,000 cells. Cells were projected in 2D space using uniform manifold approximation and projection (UMAP), based on the top 30 LSI components with the addUMAP function (nNeighbors = 50, metric = ”cosine”, min.dist = 0.5). These steps were applied independently for both stimulation cell scATAC-seq cells and PBMC isolate cell scATAC-seq cells passing filters. Annotation of stimulation scATAC-seq cells was obtained using the corresponding annotation of paired scRNA-seq cells (see sections ‘scRNA-seq cell cell clustering, visualization and annotation’ and ‘scRNA-seq and scATAC-seq OptMatch pairing’ below for more details). For isolate PBMC scATAC-seq cell clustering, the same LSI and UMAP parameters were used to obtain 2D clustering of cells based on peak accessibility.
GWAS variant enrichment analyses
Summary statistics for 12 of the 14 GWAS traits were downloaded from sources as previously described.80 The remaining traits were downloaded from the SAIGE resource (Adult/Child onset asthma)81 or the EAGLE consortium (Eczema).82 Raw summary statistics were then reformatted uniformly for downstream analyses and processing, including a per-SNP association p value threshold of p < 10−7 for the list of final variants considered for peak-SNP overlaps. For each trait considered, filtered variant loci were intersected with peaks using the findOverlaps R function, to generate a peak by variant binary overlap matrix. This was then multiplied by a variant by trait binary annotation matrix to yield a peak by trait annotation matrix. This resulting annotation matrix was used, along with scATAC-seq reads in peak counts, as input to chromVAR to generate single cell trait Z-scores based on the relative enrichment of ATAC-seq counts within these trait-associated peaks (used for UMAP visualizations in Figure S6J). For aggregate SNP scores, single cell Z-scores were converted to one-tailed p values using a Z-test, and the resulting p values combined using the Fisher method,83 per condition and cell type, and used for heatmap visualizations (related to Figure S6K).
scRNA-seq analysis workflow
Raw read processing, demultiplexing and alignment
The library preparation for scRNA-seq experiments configures the reads such that read one contains a cell barcode and UMI and read two the cDNA generated from the transcript. Cell barcodes and UMIs were parsed from read one and written into the read name of the corresponding read in the read two fastq. All read two files with valid cell barcodes were aligned using STAR (v2.5.2b) to hg19 (UCSC; PAR masked) reference genome. Reads that aligned to abundant features (chrM, rRNA, and sncRNA) were filtered from the analysis.
scRNA-seq counts generation and QC
Transcript counts per barcode were then generated by counting the number of unique genic UMIs for each read with a minimum mapping quality of 12 that aligned unambiguously to an annotated exon in the RefSeq annotation of hg19. The distribution of unique genic UMI per barcode was then filtered to separate barcodes present in droplets with cells from barcodes present in cell-free droplets. First, a background filtration step was performed to remove barcodes that arose from sequencing errors and empty droplets by computing a background threshold. The background threshold was computed to filter barcodes that arose from sequencing errors and empty droplets by performing a kernel density estimate on the log10 transformed genic UMIs per barcode distribution wherein the largest peak is assumed to be from cell-free droplets. The number of UMIs corresponding to this peak was deemed the “background level”. The half-height of the background peak was calculated by measuring the distance from the top of the background peak to the point on the right where the density dropped to 50% of the peak. The SD of the background peak was then estimated by dividing the half-width by 1.17 under the assumption of the background peak being a normal distribution. Finally, the background threshold was calculated as the background level + 5 ∗ the SD of the background peak. All barcodes below this value were filtered from the analysis.
After background filtration, the remaining barcodes in the genic UMI count distribution were subjected to a “knee calling” algorithm wherein inflection points in kernel density estimate of the log10 transformed UMI count distribution were identified. The leftmost inflection point (= higher genic UMI count) was used to determine the final cell count.
Gene-mapped counts were then loaded into R as a Seurat object42 and used for downstream analysis. Genes with at least one UMI across cells were retained, and cells with number of unique feature counts >200 and <5000 were initially retained. Normalization and scaling of RNA gene expression levels was performed using the SCTransform function. scRNA cell barcode doublets were inferred using DoubletFinder84 and removed.
scRNA-seq cell clustering, visualization and annotation
For the stimulation scRNA-seq cell clustering (shown in Figure 1), PCA was first run on the normalized scRNA-seq counts using the runPCA function in Seurat. The first 30 PCs were then used to run UMAP for single cell 2D projection using Seurat’s RunUMAP function. For stim-corrected clustering of scRNA-seq cells (Figure S2, used for cell type annotation), we followed Seurat’s workflow for integrating batches using canonical correlation analysis (CCA), where we treated each condition (e.g. Control 1h or LPS 6h) as a batch, following the integration protocol steps for finding cell integration anchors with default settings (https://satijalab.org/seurat/archive/v3.1/immune_alignment.html). The corresponding batch-aligned integrated data was scaled, and PCA dimensionality reduction was run. UMAP was used for the final cell projection (top 30 PCs, min.dist = 0.5), and a cell kNN graph was determined using the FindNeighbors function in Seurat (k = 10 cell neighbors). Cells were then grouped into clusters using the FindClusters Seurat function (resolution = 0.8; SLM algorithm), and cluster and cell annotations manually assigned by visualizing the mean and percent expression of cell identity markers within cell clusters (Figure S2). Broader annotations (e.g. monocytes) were determined by merging finer cell groupings (e.g. CD14 and CD16 monocytes). For isolate PBMC scRNA-seq and SNARE-Seq2 (RNA) cell clustering, the same PCA and UMAP parameters were used to obtain 2D clustering of cells (Figure S3).
scRNA-seq and scATAC-seq scOptMatch pairing
Computational pairing of scATAC and scRNA cells was performed either per treatment condition (stimulation data) or across all cells (PBMC isolates) using an approach we refer to throughout as “OptMatch”. First, the union of the top 5,000 variable genes based on genescore (ATAC) and gene expression (RNA) was taken across all cells, determined using Seurat’s FindVariableFeatures function on normalized scATAC genescores and normalized scRNA gene expression. These features were then used to perform a canonical correlation analysis (CCA) using the RunCCA function. The L2-normalized CCA components (n = 30) were then visualized using UMAP to highlight co-embedding of the two assays for the same cellular context (Figure S3). This was done for the PBMC isolate data (n = 17,920 ATAC, n = 8,089 RNA cells), the multi-modal SNARE-Seq2 brain cell data (n = 84,178 cells), as well as the stimulation data (n = 67,581 ATAC, n = 23,754 RNA cells).
Next, to (globally) balance ATAC and RNA cell numbers, we first randomly divide the larger (in our case, ATAC) dataset into chunks of cells size equal to the original number of cells in the smaller (in our case, RNA) dataset, re-sampling cells from the RNA cell pool to match the remainder (unsampled) ATAC cells for the final smaller cell chunk. Then, for each generated cell chunk having the same number of sampled ATAC and RNA cells, we rederive a 5D UMAP cell embedding based on the CCA components (1–20; k = 30 cell neighbors) using the uwot R package, and determine for each cell an undirected k-nearest neighbor (kNN) graph (k = 5 cell neighbors) based on the five UMAP embedding dimensions. Using this neighbor graph, we determine the shortest path distance (geodesic distance) between all cells using the shortest.paths function in the igraph R package, using which we divide the cell chunk into connected subgraphs (subclusters with finite non-zero geodesic distance) using the clusters function in the igraph package, only retaining subgraphs of size 50 cells or more. For each subgraph, we then deal with assay cell type imbalance by matching the number of local ATAC/RNA cells through random sampling of the smaller to the larger dataset, without replacement, yielding equal cells for ATAC and RNA in the subgraph.
To greatly reduce computational complexity of optimal matching (traveling salesman problem), we implement a sparse-kNN matching approach by only pairing ATAC-RNA cells that are within a geodesic distance kNN range (kg) from each other in the subgraph, where the threshold kg is set as:
where.
ft = Fraction of total cells in the subgraph to consider as geodesic kNN upper-bound (set to 0.1)
nATAC = # ATAC cells in subgraph
nRNA = # RNA cells in subgraph.
Finally, using the geodesic distance as a cost function, we determine the optimal pairing within the established geodesic ATAC - RNA kNNs subgraph cell space using the fullmatch function in the optmatch R package (https://github.com/markmfredrickson/optmatch),44 setting the following non-default parameters: tol: 0.0001, max_multimatch = 5.
The overall performance of the OptMatch approach described above for pairing single cells across ATAC/RNA datasets was assessed using previously published scATAC-seq data13 from cells sorted for CD19+, CD4+ T-cells, CD8+ T-cells, CD56+ Natural Killer (NK) cells and CD14+ monocytes, and newly generated scRNA-seq data from the same cell pool for each enriched cell population (see scRNA-seq experimental methods). For comparison, we also determined a “greedy” assignment of cell pairs, for which we assigned each cell in the ATAC dataset to the cell with the highest similarity score in the RNA dataset (maximum Pearson correlation based on the first 20 CCA components). Overall performance between the two pairing modes was determined based on the percentage accuracy based on matching concordant cell types across assays for each sorting experiment (e.g. how often a CD19+ cell in the ATAC dataset paired with a CD19+ cell in the RNA dataset), the frequency of ‘multi-matches’ (multiple RNA cells pairing to a single ATAC cell), and the final percentage of paired cells in both ATAC and RNA datasets. Pairs were visualized by picking 300 ATAC-RNA pairs at random, highlighting the corresponding cells in CCA UMAP space.
Aggregate ATAC and RNA profiles
Paired aggregate single cell peak (scATAC-seq) and gene (scRNA-seq) “pseudobulk” counts for different conditions and cell types (see Figure 3E) were obtained by summing the normalized scATAC-seq peak accessibility counts separately for promoter peaks (peak windows found within 1000 bp upstream and 300 bp downstream of each gene’s TSS, using the promoters function in the GRanges package) and distal peaks (peaks found outside defined promoter window), and by summing Seurat-normalized scRNA-seq gene counts across cells per condition and cell type. These pseudobulk counts were then quantile-normalized to adjust for differences in overall cell numbers across groupings.
Peak-gene cis-regulatory correlation analysis
High density domains of regulatory chromatin (DORCs) were determined using scATAC-seq and scRNA-seq data for computationally-paired cells (see section above). Briefly, a 100 kb window was taken around the TSS of all hg19 RefSeq genes that were found to be expressed based on scRNASeq data. Next, peak-gene pairs where peak summits overlapped a given gene TSS window were determined (n = 155,831 peaks and 18,151 genes and a total of 343,640 gene-peak pairs). For each pair, the observed gene-peak correlation coefficient (Spearman ⍴) was determined by correlating the mean-centered scATAC-seq peak counts with the corresponding gene’s expression across all ATAC-RNA paired cells (n = 62,219 cells). Permuted correlation coefficients for each gene-peak pair were calculated using background peaks matched for GC content and total chromatin accessibility levels across cells for each peak tested, determined using chromVAR (n = 100 iterations). Finally, the significance of each gene-peak association was determined using a one-tailed Z-test computed from the observed and permuted coefficients. Only gene-peak associations that show positive correlations and were statistically significant (Z-test permutation p ≤ 0.05) were considered, and used to identify DORCs based on the number of significant peaks associated with each gene (DORCs = genes with n ≥ 7 associated peaks). Single cell DORC scores per gene were calculated as the sum of normalized scATAC-seq reads in peak counts (mean-centered) using the corresponding significantly correlated DORC-peaks for that gene, and smoothed for visualization based on k = 30 cell kNNs derived using the scATAC-seq LSI components.
DORC super-enhancer analysis
To determine overlap of stimulation DORCs and previously annotated super-enhancer regions, we used a previously annotated31 list of genes associated with super-enhancers spanning different cellular contexts (n = 86). We then determined and visualized the cumulative fraction of all stimulus DORCs (n = 1,128) that overlap with each of the different super-enhancer linked gene lists.
Differential DORC analyses
For differential testing of DORC accessibility scores or expression levels, we used normalized single cell DORC scores (paired scATAC-seq cells; n = 62,219), or RNA expression (unpaired scRNA-seq cells; n = 23,754) and performed differential testing using a Wilcoxon rank-sum test per cell type (CD4/CD8 T, B, Monocyte, and NK), comparing each stimulus condition to its corresponding control condition (e.g. IFNƔ 1h vs Control 1h for Monocytes) for all determined DORCs (n = 1,128 genes). FDR was determined to adjust for multiple tests. For visualization, only the union of top 10 genes (ranked by nominal DORC ATAC Wilcoxon test P) per comparison were kept (n = 53 DORCs), and a heatmap of the difference in mean single cell score (DORC accessibility or RNA expression) was used, showing the most significant change across any of the five cell types assessed for each DORC and condition, along with the corresponding cell type which reported the minimum P across any condition for each DORC.
Cell nearest neighbor (NN) stimulation time calculation
Cell NN stimulation time was computed based on a weighted average of cell-nearest neighbor conditions based on their chromatin accessibility profiles. For each scATAC-seq cell in the paired stimulation data (n = 62,219), we used the first 30 LSI components (based on chromatin accessibility single cell peak counts) to derive a k = 50 nearest neighbor (NN) graph as the cell’s nearest condition cells, leaving out the Golgi inhibitor treatment condition. Then, for each cell and its kNNs, we computed the mean stimulation time as the weighted average of its kNNs, using a weight of 0, one and two for Control 1/6 h, stimulation 1h and stimulation 6h time points respectively, done separately for each of the three stimulus conditions (LPS, IFN or PMA). The resulting estimates were then rescaled to fall between 0 and 1, and used for downstream analyses including fitting and visualization of single cell DORC and paired RNA expression values to NN stimulation time.
DORC accessibility and RNA expression dynamics
To visualize dynamics of DORC accessibility and gene expression along the NN stimulation time axis, we took scATAC-seq cells annotated as monocytes pertaining to control 1h, as well as stimulation 1h and stimulation 6h time conditions for LPS (n = 1,776 cells), IFNƔ (n = 2,601 cells) and PMA (n = 2,002 cells). Using a window size of n = 100 cells, we then computed the rolling average DORC accessibility and (paired) gene expression value, which was then min-max normalized to the 1–99 percentile value, respectively. Additionally, we fit a loess smoothing function (loess alpha = 0.1) using the normalized DORC/RNA values to the smoothed (rolling average) NN stimulation time, which was overlaid and visualized.
Motif database
From cisBP (http://cisbp.ccbr.utoronto.ca/index.php), we curated position frequency matrices (PFMs) that represented a total of 113,635 human motifs and 107,308 mouse motifs. We filter motifs to a unique subset, one motif for each TF regulator, resulting in 1,143 unique human or 895 unique mouse TFs and motifs. To do this, we iterated through each unique TF to find all associated motifs from the high-quality motif list (as annotated by cisBP). For these associated high-quality motifs, we computed a similarity matrix using the Pearson correlation of the PFMs. To select the most representative motif for each TF regulator, we found the motif correlated with the most other motifs of the same TF at R > 0.9. If a TF was not represented in the high-quality list, we repeated the process using the medium- and low-quality databases for TF regulators. The final curated motif database contains 1,141 human and 890 mouse unique regulators and motifs, and can be incorporated with FigR and other PFM utilizing packages.
FigR GRN workflow
To associate TF regulators to target DORC gene activity, we deduced a metric that combines the relative enrichment of TF motifs among DORCs and the correlation of TF RNA expression with DORC accessibility. First, for each of our defined DORC genes, we determine a reference pool of expression-correlated chromatin accessibility peaks associated with its k-nearest neighbor DORC genes (default k = 30). Then, for each DORC, we use its pooled peak set to perform an enrichment Z-test of the observed TF motif-to-peak match frequency with respect to each TF in our curated TF motif list described earlier, relative to the expected frequency based on matches to a permuted background peak set matched for GC content and overall accessibility (default n = 50 permutations). We then correlate across all paired cells (n = 62,219) the smoothed DORC accessibility score with the smoothed paired RNA expression levels of all tested TFs (smoothed using k = 30 nearest cell neighbors based on first 30 LSI components), and use the standardized Spearman correlation levels to perform a Z-test to establish significance of correlation. Lastly, we combine the two significance levels (correlation and peak enrichment) and define a “regulation score” in log space as follows:
All regulation scores corresponding to negative TF enrichments (TF enrichment Z score < 0) were set to 0.
For the stimulation GRN, putative regulators of DORCs were defined as TFs that have an absolute regulation score ≥1. SNP-DORC regulatory associations were visualized by taking the list of DORCs whose associated peaks overlap any disease GWAS variant (n = 77 DORCs) and clustering DORC-TF associations with absolute regulation score ≥1.5 (n = 73 DORCs and n = 89 TFs). Network plots for a subset of DORCs (e.g. SLE-specific DORCs) were drawn for the associated filtered edge-node associations using the ggnet R package, and can be further visualized through the R Shiny App. For the murine skin GRN, previously published SHARE-seq data and the corresponding DORC calls were used,28 along with the mouse cisBP TF motif database (n = 797 motifs), with k = 20 DORC kNNs for peak pooling. For the human brain GRN, previously described SNARE-Seq2 human primary motor cortex data45 was used to determine DORCs as detailed above (see Peak-gene cis-regulatory correlation analysis). DORC kNN k = 10 was used for peak pooling while determining peak enrichments against human TF motifs, and the corresponding GRN regulation scores were computed as described earlier.
Comparison of FigR with SCENIC
The SCENIC R-package (v1.2.4) was used with the normalized stimulation scRNA-seq data as input (n = 23,754 cells), following the recommended workflow as outlined in the application vignette, without modification. This resulted in a table of TF-target gene associations, keeping positive associations with Spearman correlation ≥0.03 (default). For comparisons with FigR, FigR TF-DORC associations filtered to for (-log10 scale) regulation score ≥1 (n = 6,070 total TF-DORC associations). SCENIC hits were filtered for the following criteria to allow comparisons with FigR hits: i) only DORC genes (n = 1,128) that were also tested using FigR were considered; ii) only significant TF-target associations passing the “w0.001” co-expression module threshold category were kept iii) top SCENIC TF-target hits after ranking by decreasing co-expression weight (n = 6,070 hits) were retained to match FigR iv) only TFs that were tested across both methods were kept (n = 91 TFs) to account for different underlying motif databases used by the two methods. After these filters, the total number of target genes were determined for each TF and plotted for either method. For ChIP-seq validation, GM12878 ChIP-seq data was obtained from the ENCODE database. Then, for each TF for which we had ChIP-seq data (n = 84 TFs), we computed the overlap of ChIP-seq narrow peaks (GRCh38-aligned) with our PBMC stimulation scATAC-seq peak ranges (hg19-aligned and lifted over to GRCh38 to allow overlap comparisons using the rtracklayer R package). With this, for each TF we computed the proportion of ChIP-seq peaks that overlap cis-regulatory elements that are either in promoter regions (1 kb upstream and 300 bp downstream of any gene TSS) versus distal enhancer elements (all other regions). Then, the top 10 TFs for either FigR or SCENIC (based on the number of associated targets) were taken, and the corresponding difference in promoter versus enhancer ratios was determined based on the ChIP-seq overlap. The expected difference in promoter versus distal enhancer binding ratio was determined by taking the mean difference in promoter - enhancer ratios across all available GM12878 ChIP-seq TFs.
Quantification and statistical analysis
Details of statistical tests performed, and the number of observations (e.g. number of cells compared, number of features tested etc.) are included under the corresponding sections under the Method details section, and specified in the corresponding Result section where mentioned, as well as in the figure legends where applicable.
Acknowledgments
We thank members of the Buenrostro lab and Bio-Rad team for useful discussions and critical assessment of this work. J.D.B. and the Buenrostro lab acknowledge support from the Gene Regulation Observatory at the Broad Institute of MIT & Harvard, the Chan Zuckerberg Initiative, and the NIH New Innovator Award (DP2).
Author contributions
V.K.K. lead all computational developments and analyses described in this work with contributions from Y.H., S.M., C.A.L., and Z.D.B. V.K.K and A.E. developed the online interactive resources. F.M.D., J.G.C., and A.S.K. generated the data with supervision by J.D.B. and R.L. V.K.K and J.D.B. wrote the manuscript with input from all authors.
Declaration of interests
J.D.B. holds patents related to ATAC-seq and scATAC-seq and serves on the scientific advisory boards of CAMP4 Therapeutics, seqWell, and CelSee. J.G.C., Z.D.B., A.S.K., and R.L. are employees of Bio-Rad.
Published: August 4, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100166.
Supplemental information
Data and code availability
Raw and processed data can be accessed through GEO (GEO ID GSE178431).
Analysis code can be found on GitHub (https://github.com/buenrostrolab/stimATAC_analyses_code), and the FigR workflow can be implemented in R using the downloadable FigR package (https://github.com/buenrostrolab/FigR). The package release has also been published under v1.0.1 on Zenodo (https://doi.org/10.5281/zenodo.6795583), along with the analyses code (https://zenodo.org/badge/latestdoi/288481382). Additionally, gene regulatory networks and single cell profiles (cell metadata, DORC scores and paired RNA expression) for stimulation data can be interactively queried through our R Shiny app (https://buenrostrolab.shinyapps.io/stimFigR/; Zenodo code archive: https://doi.org/10.5281/zenodo.6820097). Newly curated TF motif PFM lists for human and mouse are available for download and use as R PFMList objects on Github and on Zenodo (https://doi.org/10.5281/zenodo.6814702). Normalized aggregate scATAC-seq coverage profiles (BigWig) can be visualized for monocytes per condition (https://genome.ucsc.edu/s/vkartha/stimATAC_CD14_conditions) or for control 1h cells across paired cell type annotations (https://genome.ucsc.edu/s/vkartha/stimATAC_Control1h_cellTypes) using the UCSC genome browser.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Medzhitov R., Horng T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009;9:692–703. doi: 10.1038/nri2634. Available from. [DOI] [PubMed] [Google Scholar]
- 2.Klemm S.L., Shipony Z., Greenleaf W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019;20:207–220. doi: 10.1038/s41576-018-0089-8. [DOI] [PubMed] [Google Scholar]
- 3.Yosef N., Regev A. Writ large: genomic dissection of the effect of cellular environment on immune response. Science. 2016;354:64–68. doi: 10.1126/science.aaf5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fowler T., Sen R., Roy A.L. Regulation of primary response genes. Mol. Cell. 2011;44:348–360. doi: 10.1016/j.molcel.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busslinger M., Tarakhovsky A. Epigenetic control of immunity. Cold Spring Harb. Perspect. Biol. 2014;6 doi: 10.1101/cshperspect.a019307. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen S., Yang J., Wei Y., Wei X. Epigenetic regulation of macrophages: from homeostasis maintenance to host defense. Cell. Mol. Immunol. 2020;17:36–49. doi: 10.1038/s41423-019-0315-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ostuni R., Piccolo V., Barozzi I., Polletti S., Termanini A., Bonifacio S., Curina A., Prosperini E., Ghisletti S., Natoli G. Latent enhancers activated by stimulation in differentiated cells. Cell. 2013;152:157–171. doi: 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 8.Netea M.G., Joosten L.A.B., Latz E., Mills K.H.G., Natoli G., Stunnenberg H.G., O'Neill L.A.J., Xavier R.J. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352:aaf1098. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Virgin H.W., Wherry E.J., Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 10.Shalek A.K., Satija R., Shuga J., Trombetta J.J., Gennert D., Lu D., Chen P., Gertner R.S., Gaublomme J.T., Yosef N., et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510:363–369. doi: 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Satpathy A.T., Granja J.M., Yost K.E., Qi Y., Meschi F., McDermott G.P., Olsen B.N., Mumbach M.R., Pierce S.E., Corces M.R., et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019;37:925–936. doi: 10.1038/s41587-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buenrostro J.D., Corces M.R., Lareau C.A., Wu B., Schep A.N., Aryee M.J., Majeti R., Chang H.Y., Greenleaf W.J. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell. 2018;173:1535–1548.e16. doi: 10.1016/j.cell.2018.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lareau C.A., Duarte F.M., Chew J.G., Kartha V.K., Burkett Z.D., Kohlway A.S., Pokholok D., Aryee M.J., Steemers F.J., Lebofsky R., Buenrostro J.D. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol. 2019;37:916–924. doi: 10.1038/s41587-019-0147-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng Q., Behzadi F., Sen S., Ohta S., Spreafico R., Teles R., Modlin R.L., Hoffmann A. Sequential conditioning-stimulation reveals distinct gene- and stimulus-specific effects of Type I and II IFN on human macrophage functions. Sci. Rep. 2019;9:5288. doi: 10.1038/s41598-019-40503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez-Jimenez C.P., Eling N., Chen H.-C., Vallejos C.A., Kolodziejczyk A.A., Connor F., Stojic L., Rayner T.F., Stubbington M.J.T., Teichmann S.A., et al. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science. 2017;355:1433–1436. doi: 10.1126/science.aah4115. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calderon D., Nguyen M.L.T., Mezger A., Kathiria A., Müller F., Nguyen V., Lescano N., Wu B., Trombetta J., Ribado J.V., et al. Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat. Genet. 2019;51:1494–1505. doi: 10.1038/s41588-019-0505-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshida H., Lareau C.A., Ramirez R.N., Rose S.A., Maier B., Wroblewska A., Desland F., Chudnovskiy A., Mortha A., Dominguez C., et al. The cis-regulatory atlas of the mouse immune system. Cell. 2019;176:897–912.e20. doi: 10.1016/j.cell.2018.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilk A.J., Lee M.J., Wei B., Parks B., Pi R., Martínez-Colón G.J., Ranganath T., Zhao N.Q., Taylor S., Becker W., et al. Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19. J. Exp. Med. 2021;218 doi: 10.1084/jem.20210582. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.You M., Chen L., Zhang D., Zhao P., Chen Z., Qin E.-Q., Gao Y., Davis M.M., Yang P. Single-cell epigenomic landscape of peripheral immune cells reveals establishment of trained immunity in individuals convalescing from COVID-19. Nat. Cell Biol. 2021;23:620–630. doi: 10.1038/s41556-021-00690-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamimoto K., Hoffmann C.M., Morris S.A. CellOracle: dissecting cell identity via network inference and in silico gene perturbation. bioRxiv. 2020 doi: 10.1038/s41586-022-05688-9. https://www.biorxiv.org/content/10.1101/2020.02.17.947416v3.abstract Preprint at. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papili Gao N., Ud-Dean S.M.M., Gandrillon O., Gunawan R. SINCERITIES: inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics. 2018;34:258–266. doi: 10.1093/bioinformatics/btx575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiu X., Rahimzamani A., Wang L., Ren B., Mao Q., Durham T., McFaline-Figueroa J.L., Saunders L., Trapnell C., Kannan S. Inferring causal gene regulatory networks from coupled single-cell expression dynamics using scribe. Cell Syst. 2020;10:265–274.e11. doi: 10.1016/j.cels.2020.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neph S., Stergachis A.B., Reynolds A., Sandstrom R., Borenstein E., Stamatoyannopoulos J.A. Circuitry and dynamics of human transcription factor regulatory networks. Cell. 2012;150:1274–1286. doi: 10.1016/j.cell.2012.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson C.A., Castro D.M., Saldi G.-A., Bonneau R., Gresham D. Gene regulatory network reconstruction using single-cell RNA sequencing of barcoded genotypes in diverse environments. Elife. 2020;9 doi: 10.7554/eLife.51254. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.-K., Cheng C., Mu X.J., Khurana E., Rozowsky J., Alexander R., et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trevino A.E., Müller F., Andersen J., Sundaram L., Kathiria A., Shcherbina A., Farh K., Chang H.Y., Pașca A.M., Kundaje A., et al. Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. Cell. 2021;184:5053–5069.e23. doi: 10.1016/j.cell.2021.07.039. [DOI] [PubMed] [Google Scholar]
- 28.Ma S., Zhang B., LaFave L.M., Earl A.S., Chiang Z., Hu Y., Ding J., Brack A., Kartha V.K., Tay T., et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell. 2020;183:1103–1116.e20. doi: 10.1016/j.cell.2020.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Granja J.M., Corces M.R., Pierce S.E., Bagdatli S.T., Choudhry H., Chang H.Y., Greenleaf W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021;53:403–411. doi: 10.1038/s41588-021-00790-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stuart T., Srivastava A., Madad S., Lareau C.A., Satija R. Single-cell chromatin state analysis with Signac. Nat. Methods. 2021;18:1333–1341. doi: 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.González A.J., Setty M., Leslie C.S. Early enhancer establishment and regulatory locus complexity shape transcriptional programs in hematopoietic differentiation. Nat. Genet. 2015;47:1249–1259. doi: 10.1038/ng.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gasperini M., Hill A.J., McFaline-Figueroa J.L., Martin B., Kim S., Zhang M.D., Jackson D., Leith A., Schreiber J., Noble W.S., et al. A genome-wide framework for mapping gene regulation via cellular genetic screens. Cell. 2019;176:1516. doi: 10.1016/j.cell.2019.02.027. [DOI] [PubMed] [Google Scholar]
- 34.Fulco C.P., Munschauer M., Anyoha R., Munson G., Grossman S.R., Perez E.M., Kane M., Cleary B., Lander E.S., Engreitz J.M. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016;354:769–773. doi: 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gasperini M., Findlay G.M., McKenna A., Milbank J.H., Lee C., Zhang M.D., Cusanovich D.A., Shendure J. CRISPR/Cas9-Mediated scanning for regulatory elements required for HPRT1 expression via thousands of large, programmed genomic deletions. Am. J. Hum. Genet. 2017;101:192–205. doi: 10.1016/j.ajhg.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spitz F., Furlong E.E.M. Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 37.Stampfel G., Kazmar T., Frank O., Wienerroither S., Reiter F., Stark A. Transcriptional regulators form diverse groups with context-dependent regulatory functions. Nature. 2015;528:147–151. doi: 10.1038/nature15545. [DOI] [PubMed] [Google Scholar]
- 38.Tycko J., DelRosso N., Hess G.T., Aradhana B.A., Mukund A., Van M.V., Ego B.K., Yao D., Spees K., et al. High-throughput discovery and characterization of human transcriptional effectors. Cell. 2020;183:2020–2035.e16. doi: 10.1016/j.cell.2020.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phanstiel D.H., Van Bortle K., Spacek D., Hess G.T., Shamim M.S., Machol I., Love M.I., Aiden E.L., Bassik M.C., Snyder M.P. Static and dynamic DNA loops form AP-1-bound activation hubs during macrophage development. Mol. Cell. 2017;67:1037–1048.e6. doi: 10.1016/j.molcel.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Das A., Yang C.-S., Arifuzzaman S., Kim S., Kim S.Y., Jung K.H., Lee Y.S., Chai Y.G. High-resolution mapping and dynamics of the transcriptome, transcription factors, and transcription Co-factor networks in classically and alternatively activated macrophages. Front. Immunol. 2018;9:22. doi: 10.3389/fimmu.2018.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fairfax B.P., Humburg P., Makino S., Naranbhai V., Wong D., Lau E., Jostins L., Plant K., Andrews R., McGee C., Knight J.C. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science. 2014;343:1246949. doi: 10.1126/science.1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang C., Sun D., Huang X., Wan C., Li Z., Han Y., Qin Q., Fan J., Qiu X., Xie Y., et al. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020;21:198. doi: 10.1186/s13059-020-02116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hansen B.B., Klopfer S.O. Optimal full matching and related designs via network flows. J. Comput. Graph Stat. 2006;15:609–627. [Google Scholar]
- 45.Bakken T.E., Jorstad N.L., Hu Q., Lake B.B., Tian W., Kalmbach B.E., Crow M., Hodge R.D., Krienen F.M., Sorensen S.A., et al. Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature. 2021;598:111–119. doi: 10.1038/s41586-021-03465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corces M.R., Buenrostro J.D., Wu B., Greenside P.G., Chan S.M., Koenig J.L., Snyder M.P., Pritchard J.K., Kundaje A., Greenleaf W.J., et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allen N.C., Philip N.H., Hui L., Zhou X., Franklin R.A., Kong Y., Medzhitov R. Desynchronization of the molecular clock contributes to the heterogeneity of the inflammatory response. Sci. Signal. 2019;12 doi: 10.1126/scisignal.aau1851. Available from. [DOI] [PubMed] [Google Scholar]
- 48.Bentham J., Morris D.L., Graham D.S.C., Pinder C.L., Tombleson P., Behrens T.W., Martín J., Fairfax B.P., Knight J.C., Chen L., et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015;47:1457–1464. doi: 10.1038/ng.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schep A.N., Wu B., Buenrostro J.D., Greenleaf W.J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods. 2017;14:975–978. doi: 10.1038/nmeth.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Platanias L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 51.Marecki S., Riendeau C.J., Liang M.D., Fenton M.J. PU.1 and multiple IFN regulatory factor proteins synergize to mediate transcriptional activation of the human IL-1 beta gene. J. Immunol. 2001;166:6829–6838. doi: 10.4049/jimmunol.166.11.6829. [DOI] [PubMed] [Google Scholar]
- 52.Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C.L., et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 53.Avram D., Califano D. The multifaceted roles of Bcl11b in thymic and peripheral T cells: impact on immune diseases. J. Immunol. 2014;193:2059–2065. doi: 10.4049/jimmunol.1400930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Longabaugh W.J.R., Zeng W., Zhang J.A., Hosokawa H., Jansen C.S., Li L., Romero-Wolf M., Liu P., Kueh H.Y., Mortazavi A., Rothenberg E.V. Bcl11b and combinatorial resolution of cell fate in the T-cell gene regulatory network. Proc. Natl. Acad. Sci. USA. 2017;114:5800–5807. doi: 10.1073/pnas.1610617114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cismasiu V.B., Adamo K., Gecewicz J., Duque J., Lin Q., Avram D. BCL11B functionally associates with the NuRD complex in T lymphocytes to repress targeted promoter. Oncogene. 2005;24:6753–6764. doi: 10.1038/sj.onc.1208904. [DOI] [PubMed] [Google Scholar]
- 56.Ying M., Tilghman J., Wei Y., Guerrero-Cazares H., Quinones-Hinojosa A., Ji H., Laterra J. Kruppel-like factor-9 (KLF9) inhibits glioblastoma stemness through global transcription repression and integrin α6 inhibition. J. Biol. Chem. 2014;289:32742–32756. doi: 10.1074/jbc.M114.588988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Richer M.J., Lang M.L., Butler N.S. T cell fates zipped up: how the Bach2 basic leucine zipper transcriptional repressor directs T cell differentiation and function. J. Immunol. 2016;197:1009–1015. doi: 10.4049/jimmunol.1600847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsukumo S.-I., Unno M., Muto A., Takeuchi A., Kometani K., Kurosaki T., Igarashi K., Saito T. Bach2 maintains T cells in a naive state by suppressing effector memory-related genes. Proc. Natl. Acad. Sci. USA. 2013;110:10735–10740. doi: 10.1073/pnas.1306691110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J., Jackson A.F., Naito T., Dose M., Seavitt J., Liu F., Heller E.J., Kashiwagi M., Yoshida T., Gounari F., et al. Harnessing of the nucleosome-remodeling-deacetylase complex controls lymphocyte development and prevents leukemogenesis. Nat. Immunol. 2011;13:86–94. doi: 10.1038/ni.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ng S.Y.-M., Yoshida T., Zhang J., Georgopoulos K. Genome-wide lineage-specific transcriptional networks underscore Ikaros-dependent lymphoid priming in hematopoietic stem cells. Immunity. 2009;30:493–507. doi: 10.1016/j.immuni.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Filion G.J.P., Zhenilo S., Salozhin S., Yamada D., Prokhortchouk E., Defossez P.-A. A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol. Cell Biol. 2006;26:169–181. doi: 10.1128/MCB.26.1.169-181.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dominguez C.X., Amezquita R.A., Guan T., Marshall H.D., Joshi N.S., Kleinstein S.H., Kaech S.M. The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J. Exp. Med. 2015;212:2041–2056. doi: 10.1084/jem.20150186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Omilusik K.D., Best J.A., Yu B., Goossens S., Weidemann A., Nguyen J.V., Seuntjens E., Stryjewska A., Zweier C., Roychoudhuri R., et al. Transcriptional repressor ZEB2 promotes terminal differentiation of CD8+ effector and memory T cell populations during infection. J. Exp. Med. 2015;212:2027–2039. doi: 10.1084/jem.20150194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Landmann R., Knopf H.P., Link S., Sansano S., Schumann R., Zimmerli W. Human monocyte CD14 is upregulated by lipopolysaccharide. Infect. Immun. 1996;64:1762–1769. doi: 10.1128/iai.64.5.1762-1769.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hwang J., Mehrani T., Millar S.E., Morasso M.I. Dlx3 is a crucial regulator of hair follicle differentiation and cycling. Development. 2008;135 doi: 10.1242/dev.022202. https://pubmed.ncbi.nlm.nih.gov/18684741/ Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olson L.E., Zhang J., Taylor H., Rose D.W., Rosenfeld M.G. Barx2 functions through distinct corepressor classes to regulate hair follicle remodeling. Proc. Natl. Acad. Sci. USA. 2005;102:3708. doi: 10.1073/pnas.0500519102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Farh K.K.-H., Marson A., Zhu J., Kleinewietfeld M., Housley W.J., Beik S., Shoresh N., Whitton H., Ryan R.J.H., Shishkin A.A., et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ray J.P., de Boer C.G., Fulco C.P., Lareau C.A., Kanai M., Ulirsch J.C., Tewhey R., Ludwig L.S., Reilly S.K., Bergman D.T., et al. Prioritizing disease and trait causal variants at the TNFAIP3 locus using functional and genomic features. Nat. Commun. 2020;11:1237. doi: 10.1038/s41467-020-15022-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Armache A., Yang S., Martínez de Paz A., Robbins L.E., Durmaz C., Cheong J.Q., Ravishankar A., Daman A.W., Ahimovic D.J., Klevorn T., et al. Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature. 2020;583:852–857. doi: 10.1038/s41586-020-2533-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beagan J.A., Pastuzyn E.D., Fernandez L.R., Guo M.H., Feng K., Titus K.R., Chandrashekar H., Shepherd J.D., Phillips-Cremins J.E. Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression. Nat. Neurosci. 2020;23:707–717. doi: 10.1038/s41593-020-0634-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dixit A., Parnas O., Li B., Chen J., Fulco C.P., Jerby-Arnon L., Marjanovic N.D., Dionne D., Burks T., Raychowdhury R., et al. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell. 2016;167:1853–1866.e17. doi: 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rubin A.J., Parker K.R., Satpathy A.T., Qi Y., Wu B., Ong A.J., Mumbach M.R., Ji A.L., Kim D.S., Ch S.W., et al. Coupled single-cell CRISPR screening and epigenomic profiling reveals causal gene regulatory networks. Cell. 2019;176:361–376.e17. doi: 10.1016/j.cell.2018.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bentsen M., Goymann P., Schultheis H., Klee K., Petrova A., Wiegandt R., Fust A., Preussner J., Kuenne C., Braun T., et al. ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat. Commun. 2020;11:4267. doi: 10.1038/s41467-020-18035-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vierstra J., Lazar J., Sandstrom R., Halow J., Lee K., Bates D., Diegel M., Dunn D., Neri F., Haugen E., et al. Global reference mapping of human transcription factor footprints. Nature. 2020;583:729–736. doi: 10.1038/s41586-020-2528-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Doench J.G. Am I ready for CRISPR? A user’s guide to genetic screens. Nat. Rev. Genet. 2018;19:67–80. doi: 10.1038/nrg.2017.97. [DOI] [PubMed] [Google Scholar]
- 77.Smith T., Heger A., Sudbery I. UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017;27:491–499. doi: 10.1101/gr.209601.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.LaFave L.M., Kartha V.K., Ma S., Meli K., Del Priore I., Lareau C., Naranjo S., Westcott P.M.K., Duarte F.M., Sankar V., et al. Epigenomic state transitions characterize tumor progression in mouse lung adenocarcinoma. Cancer Cell. 2020;38:212–228.e13. doi: 10.1016/j.ccell.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Soskic B., Cano-Gamez E., Smyth D.J., Rowan W.C., Nakic N., Esparza-Gordillo J., Bossini-Castillo L., Tough D.F., Larminie C.G.C., Bronson P.G., et al. Chromatin activity at GWAS loci identifies T cell states driving complex immune diseases. Nat. Genet. 2019;51:1486. doi: 10.1038/s41588-019-0493-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou W., Nielsen J.B., Fritsche L.G., Dey R., Gabrielsen M.E., Wolford B.N., LeFaive J., VandeHaar P., Gagliano S.A., Gifford A., et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 2018;50:1335–1341. doi: 10.1038/s41588-018-0184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Paternoster L., Standl M., Waage J., Baurecht H., Hotze M., Strachan D.P., Curtin J.A., Bønnelykke K., Tian C., Takahashi A., et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet. 2015;47:1449–1456. doi: 10.1038/ng.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dai H., Leeder J.S., Cui Y. A modified generalized Fisher method for combining probabilities from dependent tests. Front. Genet. 2014;5:32. doi: 10.3389/fgene.2014.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Federico A., Monti S. hypeR: an R package for geneset enrichment workflows. Bioinformatics. 2020 Feb 15;36(4):1307-1308. 10.1093/bioinformatics/btz700. [DOI] [PMC free article] [PubMed]
- 86.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York: 2016. https://ggplot2.tidyverse.org [Google Scholar]
- 87.Beygelzimer A., Kakadet S., Langford J., Arya S., Mount D., and Li S. (2019). FNN: Fast Nearest Neighbor Search Algorithms and Applications. R package version 1.1.3.
- 88.Csardi G. and Nepusz T. (2006). “The igraph software package for complex network research.” InterJournal, Complex Systems, 1695. https://igraph.org.
- 89.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009 Aug 15;25(16):2078-9. 10.1093/bioinformatics/btp352. Epub 2009 Jun 8. PMID: 19505943; PMCID: PMC2723002. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed data can be accessed through GEO (GEO ID GSE178431).
Analysis code can be found on GitHub (https://github.com/buenrostrolab/stimATAC_analyses_code), and the FigR workflow can be implemented in R using the downloadable FigR package (https://github.com/buenrostrolab/FigR). The package release has also been published under v1.0.1 on Zenodo (https://doi.org/10.5281/zenodo.6795583), along with the analyses code (https://zenodo.org/badge/latestdoi/288481382). Additionally, gene regulatory networks and single cell profiles (cell metadata, DORC scores and paired RNA expression) for stimulation data can be interactively queried through our R Shiny app (https://buenrostrolab.shinyapps.io/stimFigR/; Zenodo code archive: https://doi.org/10.5281/zenodo.6820097). Newly curated TF motif PFM lists for human and mouse are available for download and use as R PFMList objects on Github and on Zenodo (https://doi.org/10.5281/zenodo.6814702). Normalized aggregate scATAC-seq coverage profiles (BigWig) can be visualized for monocytes per condition (https://genome.ucsc.edu/s/vkartha/stimATAC_CD14_conditions) or for control 1h cells across paired cell type annotations (https://genome.ucsc.edu/s/vkartha/stimATAC_Control1h_cellTypes) using the UCSC genome browser.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.