

Abstract
Glutamine transport across cell membranes is performed by a variety of transporters, including the alanine serine cysteine transporter 2 (ASCT2). The substrate binding site of ASCT2 was proposed to be specific for small amino acids with neutral side chains, excluding basic substrates such as lysine. A series of competitive inhibitors of ASCT2 with low μM affinity were developed previously, on the basis of the 2,4-diaminobutyric acid (DAB) scaffold with a potential positive charge in the side chain. Therefore, we tested whether basic amino acids with side chains shorter than lysine can interact with the ASCT2 binding site. Molecular docking of L-1,3-diaminopropionic acid (L-DAP) and L-DAB suggested that these compounds bind to ASCT2. Consistent with this prediction, L-DAP and L-DAB, but not ornithine, lysine or D-DAP, elicited currents when applied to ASCT2-expressing cells. The currents were carried by anions and showed the hallmark properties of ASCT2 currents induced by transported substrates. The L-DAP response could be eliminated by a competitive ASCT2 inhibitor, suggesting that binding occurs at the substrate binding site. The Km for L-DAP was weakly voltage dependent. Further, the pH dependence of the L-DAP response showed that the compound can bind in several protonation states. Together, these results suggest that the ASCT2 binding site is able to recognize L-amino acids with short, basic side chains, such as the L-DAP derivative β-n-methylamino-l-Alanine (BMAA), a well-studied neurotoxin. Our results expand the substrate specificity of ASCT2 to include amino acid substrates with positively charged side chains.
Introduction
The alanine serine cysteine transporter 2 (ASCT2, SLC1A5) is a neutral amino acid transporter that moves amino acids with uncharged side chains into cells, in exchange for intracellular amino acids [1–4]. ASCT2 belongs to the solute carrier 1 (SLC1) family of transport proteins that consists of two members that are specific for neutral amino acid substrates (ASCT1, SLC1A4 [5] and ASCT2, SLC1A5 [6–7]) and five high affinity excitatory amino acid transporters 1–5 (SLC1A1-3 and SLC1A6-7) [8], which recognize acidic amino acid substrates. The subtype ASCT2 has been shown to transport amino acids with small hydrophilic side chains, such as serine, cysteine, asparagine, glutamine, and alanine, which have a shorter hydrophobic side chain [1, 7, 9–10].
ASCT2 has recently received significant attention after it was proposed to be actively involved in the growth of cancer cells, by satisfying specific metabolic and signaling needs [11–15]. ASCT2 is up-regulated in several cancer types, such as glioma [12], hepatoma [16], melanoma [17], neuroblastoma [18], colon [19], breast [20–21], prostate [22] and lung cancer [19, 23], due to high energy demands of rapidly-growing tissue that may not be adequately met by biosynthesis alone. These studies suggest that ASCT2 is an important pharmacological drug target [24]. However, there is limited information on the pharmacology of ASCT2 and small molecule inhibitor binding. The newly solved crystal structures of EAAT1 in outward facing conformations [25], as well as cryo-EM structures of ASCT2 in inward-facing conformations can aid in understanding substrate binding [26–27]. In addition, various mutations of the archaean SLC1 glutamate transporter homolog GltPh (prokaryotic aspartate transporter from Pyrococcus horikoshii) have been made that convert the substrate specificity to recognition of neutral, rather than acidic amino acids [28]. GltPh shares ~23% sequence identity with ASCT2 [29–30]. These studies provide an important understanding of the molecular basis for substrate selectivity in the SLC1 family, which is crucial for the development of high affinity and selective ASCT2 inhibitors [25, 28–29, 31–33].
Recently, a group of amino acid analogs were shown to inhibit ASCT2 with low micromolar affinities [34]. These compounds are based on 2-amino-4-bis(aryloxybenzyl) aminobutanoic acid (most potent inhibitor shown in figure 1A). Under physiological conditions, these compounds may be protonated, thus bearing a positive charge at the side chain nitrogen. However, the parent compound 2,4-diaminobutanoic acid (L-DAB, figure 1B), which has a basic side chain nitrogen, has not been tested for activity with respect to ASCT2. Lysine, another amino acid with basic side chain, failed to inhibit alanine uptake by ASCT2 [1, 3, 6]. Thus, it was concluded that native basic amino acids are not recognized by the ASCT2 binding site.
Figure 1:
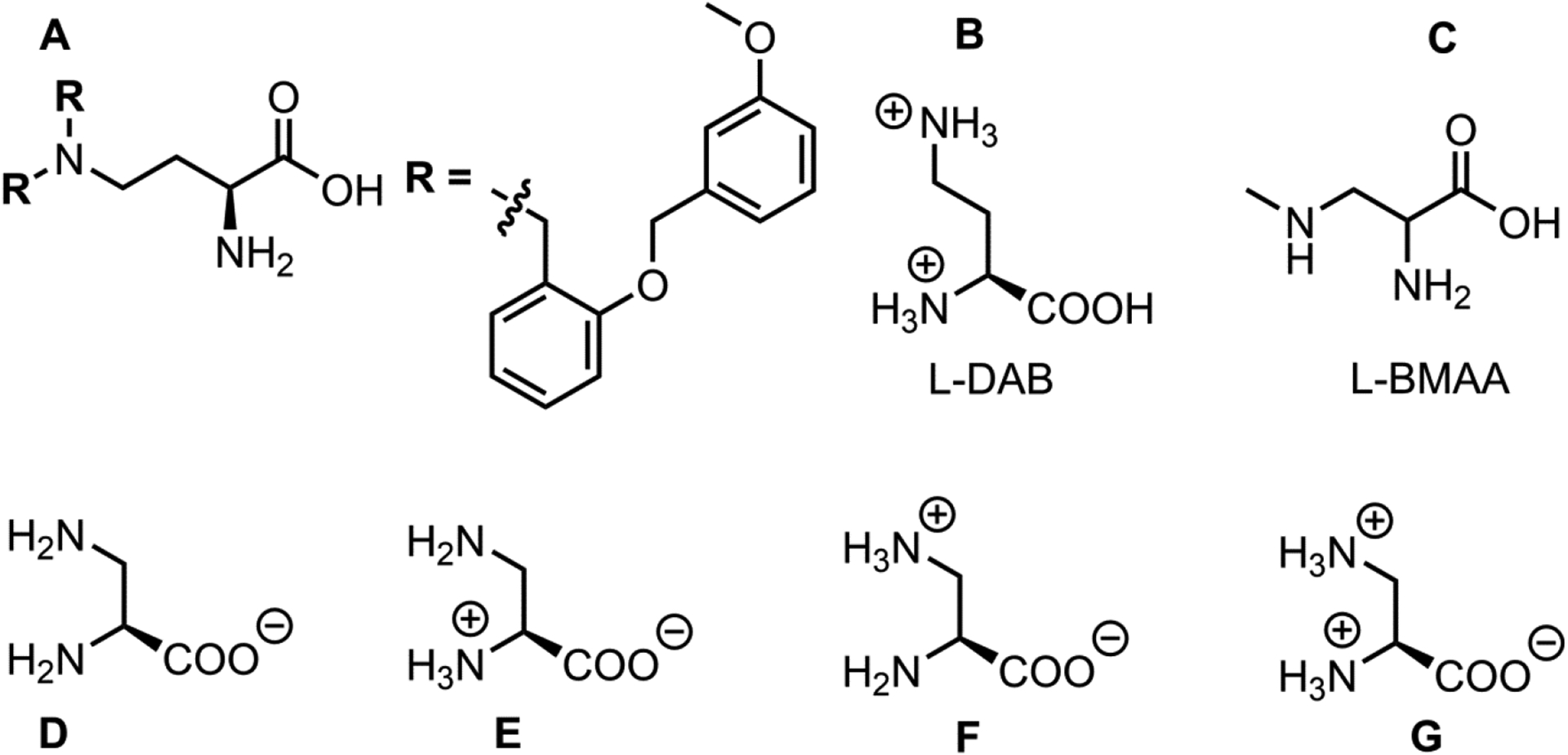
A-C, Derivatives with possible basic side chain. D-G, Major protonation states of L-DAP.
Another amino acid analogue with basic side chain is β-n-Methylamino-l-Alanine (BMAA, figure 1C). BMAA is a l-alanine derivative with a methylamino substitution in the side chain. BMAA is produced by cyanobacteria [35], and has been identified as a neurotoxin and a potential cause of neurodegenerative disease, for example Amyotrophic Lateral Sclerosis (ALS) [36]. BMAA transport was shown to be blocked by the ASCT2 inhibitor benzylserine [37]. Thus, it is possible that ASCT2 contributes to the patho-physiological effects of BMAA [37].
To test whether ASCT2 can recognize amino acid derivatives with basic side chains, we determined activity of ASCT2 in response to application of several amino acid analogues with varying side chain lengths. Interestingly, our results show that ASCT2 binds, and possibly transports small amino acids with an amino group in the side chain, for example L-2,3-diaminopropionic acid (L-DAP). However, consistent with previous results, ASCT2 does not bind or transport lysine [1, 3]. Additionally, we found that apparent affinity increased with decreasing side chain length and L-DAP binding could be inhibited competitively using previously published ASCT2 inhibitors [38]. We propose a mechanism for interaction of ASCT2 with various protonation states of L-DAP (figures 1D–G), based on kinetic modeling as well as molecular docking. Overall, these findings extend the substrate specificity of ASCT2 to include small amino acid substrates with basic side chains.
Materials and Methods
Rat ASCT2 (kindly provided by S. Bröer) and human ASCT2 (Origene) and YFP complementary DNAs were used to transiently co-transfect sub-confluent human embryonic kidney (HEK 293) cells using POLYPLUS Jet-prime transfection reagent according to the instructions of the supplier. Cell analyses were performed 24–30 hours after transfection using electrophysiological techniques. Tested compounds were purchased from Alfa Aesar and BAC unless otherwise stated.
Electrophysiological Techniques
Electrophysiological experiments were performed as described previously in detail [31, 39]. Compounds were dissolved in external buffer with concentrations ranging from in mM 0.05 to 5.00. External buffer consisted of in mM 140 NaCl, 2 CaCl2, 2 MgCl2 and 10 HEPES at pH 7.40 (NaOH). In brief, HEK293 cells immersed in the external buffer and expressing rat/human ASCT2 were suspended from a current recording electrode hosted by a micro glass pipette in whole cell configuration. Then compounds were consequently applied to cells via rapid solution exchange device as described in detail previously [40]. Internal solution in the glass pipette consisted in mM of 130 NaSCN, 2 MgCl2, 10 EGTA, and 10 HEPES, pH 7.40 (NaOH). In voltage jump experiments, external and internal buffers were replaced with 140 mM NaMes and 130 mM NaMes respectively. Series resistance of the open pipette was between 2.9 – 5.0 MΩ. Series resistance was not compensated due to relatively small currents. Currents traces were recorded using an Adams and List EPC7 amplifier and digitized using a Molecular Devices Digidata A/D converter.
As described previously [39], non-transfected cells and cells transfected with vector alone showed no significant response to application of alanine and other ASCT2 substrates.
Glutamine uptake fluorescence assay
For the glutamine uptake fluorescence assay, HEK293 cells were transiently transfected with rASCT2 and FLIPQ-TV3.0_100μM glutamine fluorescence sensor DNA and used 2 days post transfection [41]. The FLIPQ-TV3.0_100 μM glutamine fluorescence sensor is an intracellular glutamine FRET sensor consisting of mTFP/Venus as donor and acceptor fluorophores, respectively. We measured emission in only the Venus (YFP) range, which signals “FRET on”. Glutamine binding to the sensor shifts the sensor to “FRET off”, which decreases the fluorescence emission from Venus. The assay utilized a live-cell flow-through imaging chamber (Warner Instruments, Series 20) and an inverted fluorescence microscope (Zeiss Axiovert 25). The light source was X-Cite 120 (Spectra Services), and the images were captured using a Lumenera Infinity3S-1URM microscope camera (Lumenera Corporation). The fluorescence filters used were obtained from Omega Optical. The excitation filter was in the TFP excitation range (420–460 nm; SKU RPB420-460) and the emission filter was in the YFP emission range (530 nm longpass; SKU XF105-2). Bath buffer solution contained 140 mM NaCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), pH 7.3 using NaOH. This solution was used to initially wash the remaining culturing medium from the cell surface. Test solutions contained the 140 mM NaCl solution as a base plus 500 μM glutamine and specified inhibitor (benzylserine or L-DAP). A 140 mM NaCl solution containing 10 mM L-alanine was used to wash out glutamine from cells between test solution exchanges. Solutions were passed through the imaging chamber for 30–45 seconds before recording an image, and the exposure and other camera settings were set constant for each experiment. ImageJ software was used to analyze images by quantifying the fluorescence intensity of 10 cells per image. The % fluorescence change was calculated as follows:
where Finitial was the fluorescence intensity of the image taken after passing the alanine solution directly prior to the test solution and Ffinal was the fluorescence intensity of the same point in the image taken following the test solution.
Radiotracer flux
Uptake studies using 14C BMAA were performed by BioIVT (Optiva Bioctechnology Inc, Santa Clara, CA). In brief, uptake was performed in ASCT2-transfected MDCK-II cells grown as monolayers in 96-well transwell membrane plates. Transport assays were carried out 48 hours after transfection. 2 mM 14C BMAA was added to the apical side of the membrane, after washing. After 5 min incubation, solutions were aspirated at both apical and basolateral side, and both sides were washed. Cells were extracted using acetonitrile:water (50:50) and 30 μl of the extraction mix was added to 200 μl of scintillation fluid. Radioactivity was assayed using a Perkin-Elmer 1450 Microbeta scintillation counter.
Data Analysis
Microcal Origin software was used to fit both linear and nonlinear curves. Standard linear equation (y = a + bx) was used in fitting linear plots. Pearson’s r and adjusted R2 were then obtained from the fitted plot. Non-linear dose response relationships were fitted using Michaelis-Menten-like equation. Km and Imax (current at saturating substrate concentrations) were then obtained from the fitted data. Experiments were performed at least four times and at least four cells per experiment. Error bars in all graphs represent mean ±SD.
Molecular docking
While cryo-EM structures of ASCT2 have been solved in inward facing conformations, the goal of this study is to characterize ligands likely encountering the substrate binding site from the extracellular space. Thus, we focused on docking the ligands to the binding site of our previously published models of ASCT2 based on the outward-occluded and outward-open structures of the human EAAT1 transporter [42]. Docking was performed using Glide from the Schrödinger suite [43]. The models were prepared for docking with the Maestro Protein Preparation Wizard under default parameters. The binding site was defined using the coordinates of a reference ligand from the template structure, EAAT1 (aspartate or (3S)-3-[[3-[[4-(Trifluoromethyl)benzoyl] amino]phenyl]methoxy]-L-aspartic acid, TFB-TBOA) with the Maestro Receptor Grid Generation panel. Constraints were used for docking and varied by conformation. For the outward-occluded model, we used hydrogen bond constraints on S351 and N471. For the outward-open model, we used hydrogen bond constraints at S351 or S353 on hairpin 1 (HP1), as previously described [44]. We also removed UCPH101 prior to assigning the grid for the outward-open model to avoid multiple reference ligands in the box. Small molecules were prepared for docking using LigPrep of the Schrödinger suite [43]. Default settings were used except for the ‘Ionization’ parameter, which was set to ‘Do not ionize’ because ionization states were manually specified per molecule. PyMOL was used to visualize the docking results [45].
Results
Computational analysis of basic amino acid interaction with ASCT2 using docking.
Here, we aimed to investigate ASCT2 amino acid substrate specificity using small molecules with basic amino acid side chains. We used L-2,3-diaminopropionic acid (L-DAP) as the molecule with the shortest basic side chain, docking it to our ASCT2 homology model generated on the basis of the EAAT1 crystal structure in both the outward-occluded (substrate-bound) and outward-open (inhibitor-bound, HP2 open) conformations. Docking was performed in four possible protonation states of L-DAP (figures 1D–G). For the outward-occluded state, we observed interactions between the α-carboxy group of the ligand and the N471 nitrogen atom and S353 backbone nitrogen, as well as the α-amino group of L-DAP with S351 (main chain carbonyl) and the D464 side chain (figure 2). Interactions were also observed with the side chain of T468. These interactions were also found for known amino acid substrates of ASCT2 without basic side chain, for example L-serine and L-asparagine.
Figure 2: Docking poses of L-DAP and L-DAB.
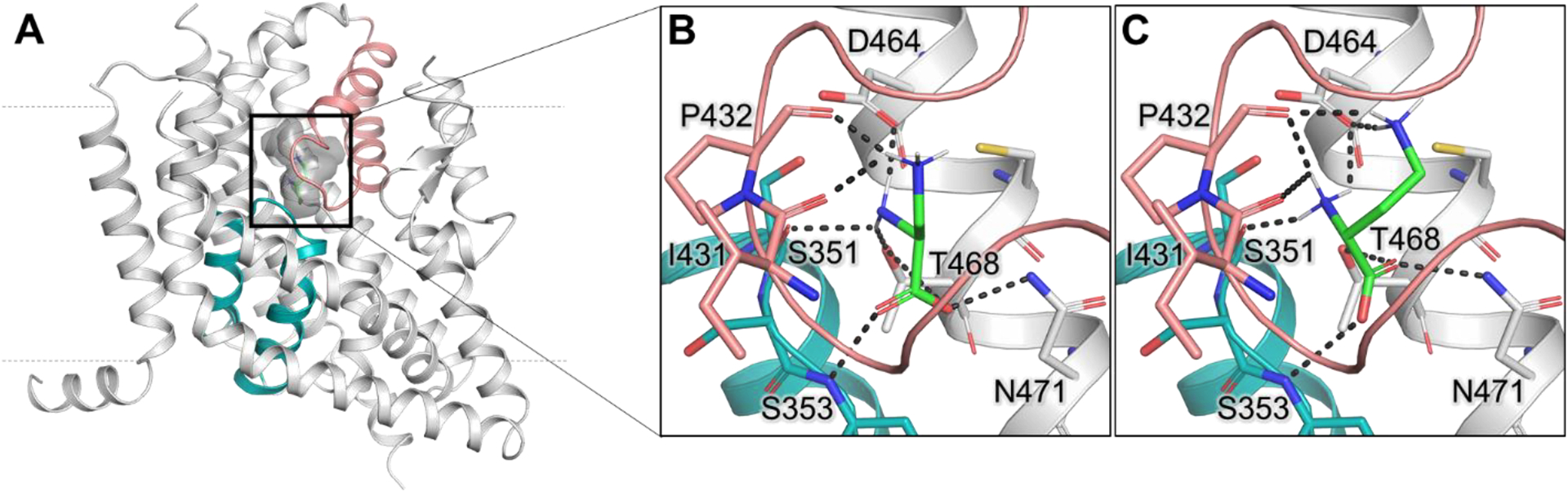
(A) Side view of the ASCT2 homology model in the outward-occluded conformation (gray ribbon) with docked L-DAB (green sticks). The substrate binding site is highlighted with dark gray shadow; HP1 is shown in teal and HP2 in salmon. Broken lines indicate the approximate location of the membrane. (B) Docking pose of L-DAP (Figure 1F) and (C) L-DAB. Residues predicted to make hydrogen bonds (dark gray dashes) are labeled. Oxygen, nitrogen, and sulfur atoms are represented in red, blue, and yellow, respectively.
It should be noted that the neutral, zwitterionic form of L-DAP with the α-amino group deprotonated is the major predicted protonation state at physiological pH (figure 1F), as determined by Hay and Morris (1972) [46]. Additional information about protonation states of L-DAP is provided in the Discussion section. Specifically, in the outward-occluded state, the neutral L-DAP side chain amino group is predicted to be coordinated by the backbone oxygen atoms of P432 and I431 in HP2 (figures 2A and B). Similar interactions were observed for L-2,4-diaminobutyric acid (L-DAB), which has an additional CH2 group in the side chain compared to L-DAP that is predicted to form additional polar contacts with D464 (figure 2C).
The compounds were also docked to the outward-open state, in which the tip of HP2 moves away from the binding site, opening it up to the extracellular side of the membrane. Docking scores were generally lower in this configuration, presumably because of the lack of polar interaction of the side chain amino group with residues in HP2 (supplementary figure 1). In both, outward-occluded and outward-open conformations, docking scores did not vary substantially between the four protonation states that were tested. The neutral L-DAP scored better than other forms in the outward-open state (supplementary table 1).
Amino acids with basic side chains show substrate-like properties.
To test the predictions from computational analysis, we performed patch clamp recordings of HEK293 cells over-expressing ASCT2 upon application of amino acids with basic side chains. Interestingly, at pH 7.40 and at 2 mM concentration, L-DAP elicited an inwardly-directed current very similar to the one produced when alanine, an ASCT2 substrate, is applied [1, 3, 31, 44], as shown in figures 3A and B. SCN− at 140 mM concentration was used in the intracellular solution, which flows out of the cell upon substrate application, due to activation of the anion conductance [2, 39]. This anion outflow creates inward current [1, 3, 31, 44]. We also used 10mM alanine and 140 mM Na+ in the internal solutions, to push the transporter to the outward-facing conformations (i.e. outward-occluded or outward-open) best suited for analyzing substrates and inhibitors. In contrast to L-DAP, D-2,3-DAP did not produce any current up to a tested concentration of 4mM, suggesting ASCT2 specificity for L amino acids with basic side chains. L-DAP induced currents increased with increasing concentration and were close to saturation at approximately 2mM concentration. The dose-response relationship of this current could be fitted using a Michaelis-Menten-like equation (see Materials and Methods) to obtain the apparent affinity of L-DAP of 560 ±70 μM, as shown in figure 3C. L-2,4-diaminobutyric acid (L-DAB) also produced an inward current, however, with smaller magnitude than that generated by L-DAP at the same concentration (figure 3B). The L-DAB dose-response relationship curve could not be evaluated, due to much lower affinity as compared to L-DAP (figure 3C). Since no saturation was observed at concentrations up to 5 mM, we were unable to obtain the apparent affinity. However, this affinity is clearly much lower than that for L-DAP.
Figure 3: Basic amino acids generate current responses in ASCT2.
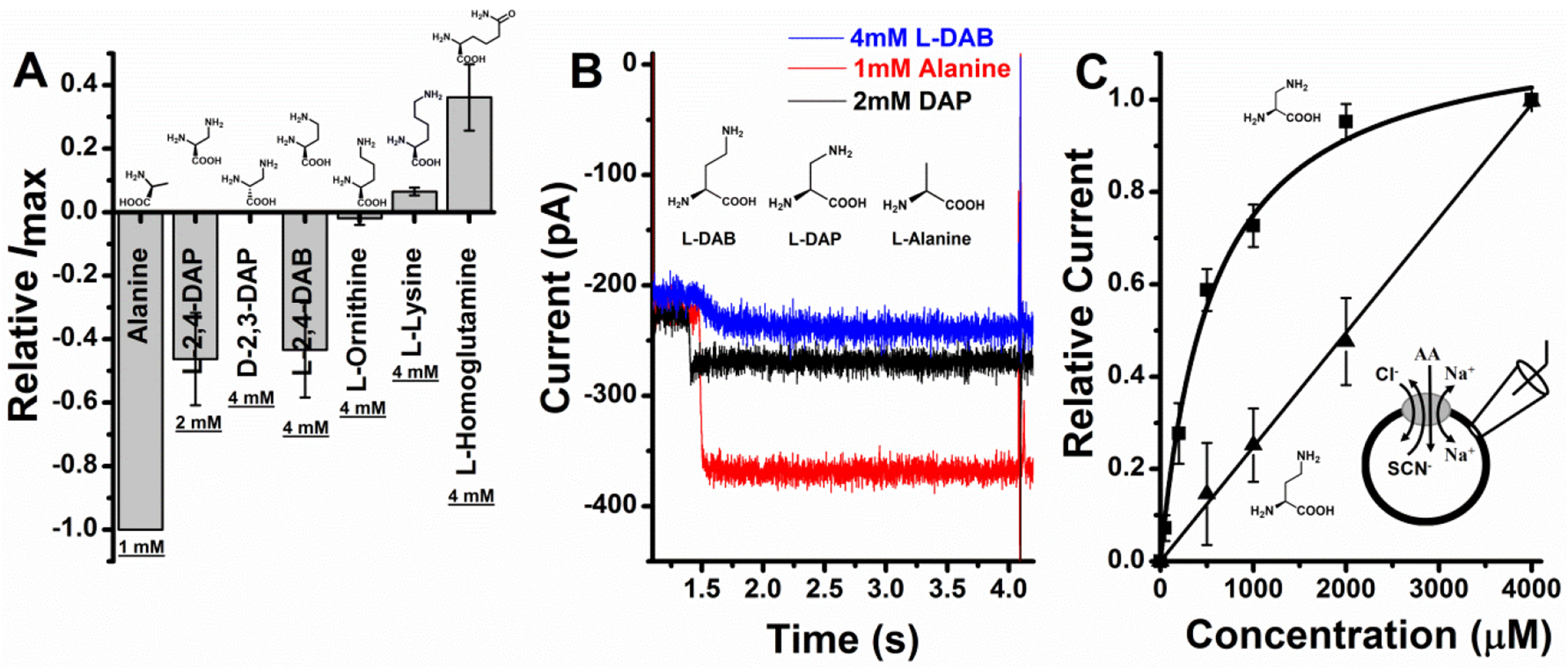
A, Relative Imax of L-DAP, D-DAP, L-DAB, L-ornithine and L-homoglutamine at saturating concentrations normalized to the alanine current at 1 mM. B, 1mM Alanine, 2 mM L-2,3-DAP and 4 mM L-DAB actual traces. C, L-2,3-DAP (squares) and L-DAB (triangles) dose response curves. Binding affinity (Km) for L-DAP was found to be 560 ± 68 μM similar to one reported previously [44]. The almost straight line (triangles) shows L-DAB relative currents normalized to currents at 4 mM. Km couldn’t be obtained at these concentrations since saturation could not be reached. External solution consisted of 140 mM NaCl and intercellular buffer was 130 mM NaSCN at pH 7.40, V = 0 mV.
We extended our scope to molecules with even longer basic side chains. The inwardly directed current decreased with increasing side chain with L-ornithine and L-lysine producing very small currents at 4 mM that were not significantly different from zero, as shown in figure 3A. These results are consistent with previous observations showing that L-lysine is not a transported substrate of ASCT2 [1–3]. Finally, homoglutamine, which has an additional carbon in the side chains compared to glutamine, showed inhibitor-like properties (outward current, figure 3A), suggesting that amino acids with increased chain length, but neutral side chain can bind, but cannot be transported by ASCT2.
L-DAP competes with binding of substrate and a competitive inhibitor.
To investigate further if the compounds with basic side chains were actually binding to the ASCT2 binding site, we conducted competition experiments at varying concentrations of a competitive inhibitor of ASCT2, (R)-γ-(4-biphenylmethyl)-L-proline (BPP). This compound was previously characterized and competes with alanine for the substrate binding site with a Ki of 3μM [38]. Consistent with the hypothesis of L-DAP acting as a substrate, BPP reduced L-DAP induced inward currents at all concentrations and the results could be fitted with a Michaelis-Menten-like relationship to obtain the apparent binding affinity for L-DAP at three different inhibitor concentrations (figures 4A–C). Figure 4D shows the Km for L-DAP plotted vs. the inhibitor concentration and as expected for competitive inhibition, a linear relationship was obtained with a Pearson’s r value of −0.992 and R2 as 0.970 demonstrating very good correlation. These results demonstrate that L-DAP is binding to the same site as the inhibitor, further supporting the idea that DAP is a substrate.
Figure 4: L-DAP binds to the substrate binding site.
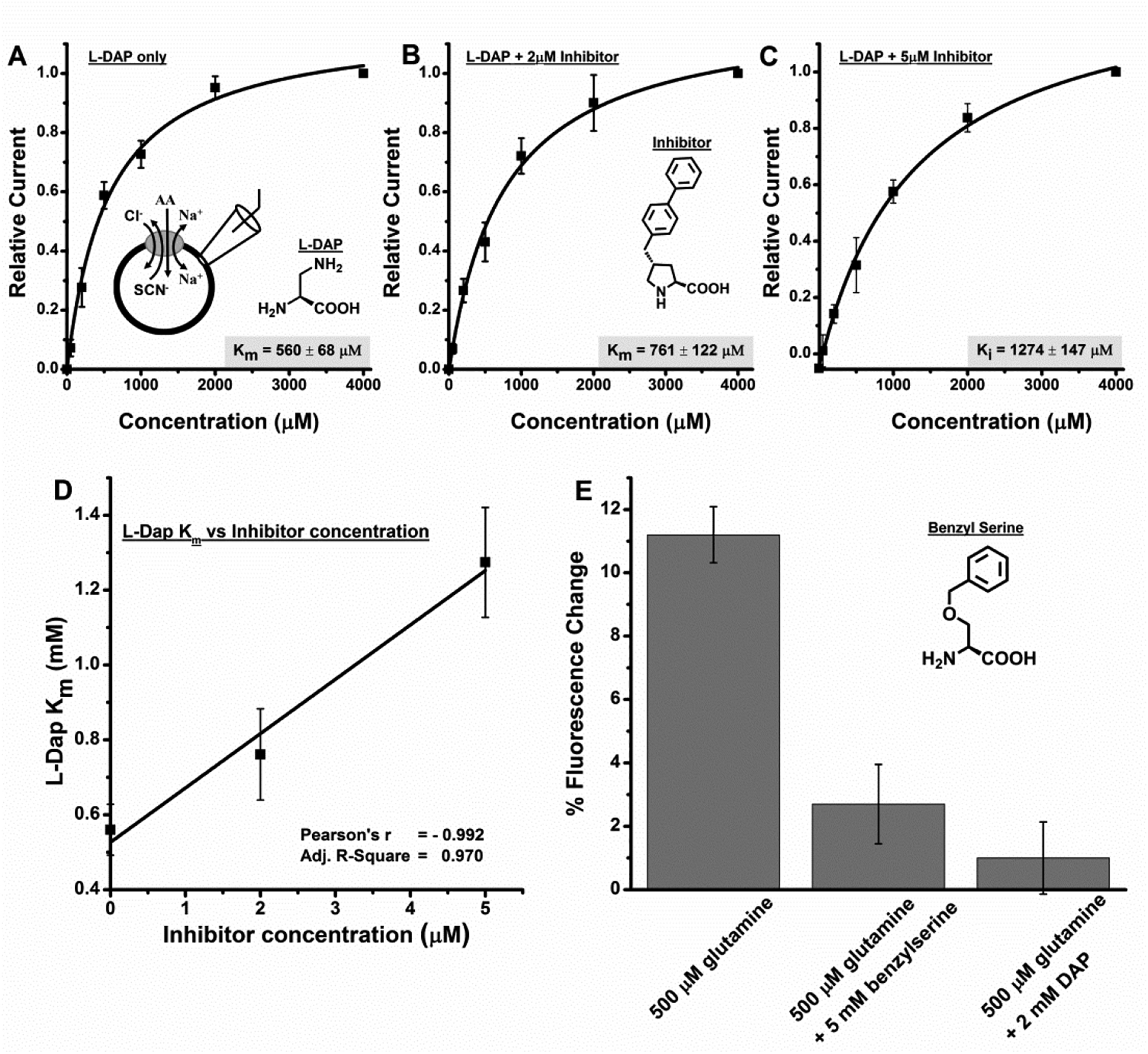
A-C, L-DAP dose response curves with A, no inhibitor, B 2 μM inhibitor [38] and C, 5 μM inhibitor. All experiments were done at 0 mV. D, L-DAP apparent affinity (Km) at different inhibitor concentration showing Pearson’s r as −0.992 and R2 as 0.970 demonstrating very good correlation. E, glutamine influx, as determined through fluorescence change of a glutamine sensor, is inhibited in the presence of L-DAP and the competitive ASCT2 inhibitor benzylserine.
We also tested whether L-DAP can block substrate flux (figure 4E). To test this possibility, we utilized a fluorescent glutamine sensor as an assay for glutamine influx. HEK293 cells transiently transfected with rASCT2 and the sensor FLIPQ-TV3.0_100 μM (kindly provided by S. Okumoto, [41]) demonstrate that L-DAP and benzylserine block glutamine flux into cells via rASCT2 transport. Cells showed a 11.2 ± 0.89 % decrease in fluorescence emission in the Venus range when 500 μM glutamine was applied externally, indicating glutamine uptake. The same cells showed only a 1.00 ± 1.14 % decrease in fluorescence emission in the Venus range when 500 μM glutamine plus 2 mM L-DAP were applied, demonstrating that L-DAP blocks glutamine uptake by rASCT2. For comparison, 500 μM glutamine plus 5 mM benzylserine [39] produced a 2.70 ± 1.25 % decrease in fluorescence emission in the Venus range.
L-DAP-induced current is carried by anions.
Previous studies have shown that the substrate-induced ASCT2 current is carried by anions [31, 38–39, 47]. To test the ionic basis of the current induced by L-DAP, we tested its voltage dependence. First, 140 mM NaSCN was used as an extracellular buffer and 130 mM NaSCN on the intracellular side. Under these conditions, the reversal potential of the current, if carried by anions, should be very close to 0 mV (−1.9 mV according to the Goldman equation) [39]. Consistent with this expectation, L-DAP and L-DAB-induced currents were inward directed at negative potentials, and outward directed at positive potentials, as shown in figure 5A. When we performed a similar experiment, but replaced the extracellular buffer SCN− with 140 mM Cl−, an anion with low permeability, currents evoked by rapid application of alanine, L-DAP, and L-DAB were inwardly directed at all voltages, but increased with increasingly negative membrane potential (figure 5B). Finally, L-DAP failed to elicit currents in the absence of permeating anions (replacement by methane sulfonate, data not shown). Together, these results are in agreement with the interpretation that L-DAP and L-DAB induced currents are caused by anion conductance that is activated upon binding of these compounds, in analogy to known transported substrates, such as alanine [1–2, 47].
Figure 5: Current induced by basic amino acids is carried by anions.
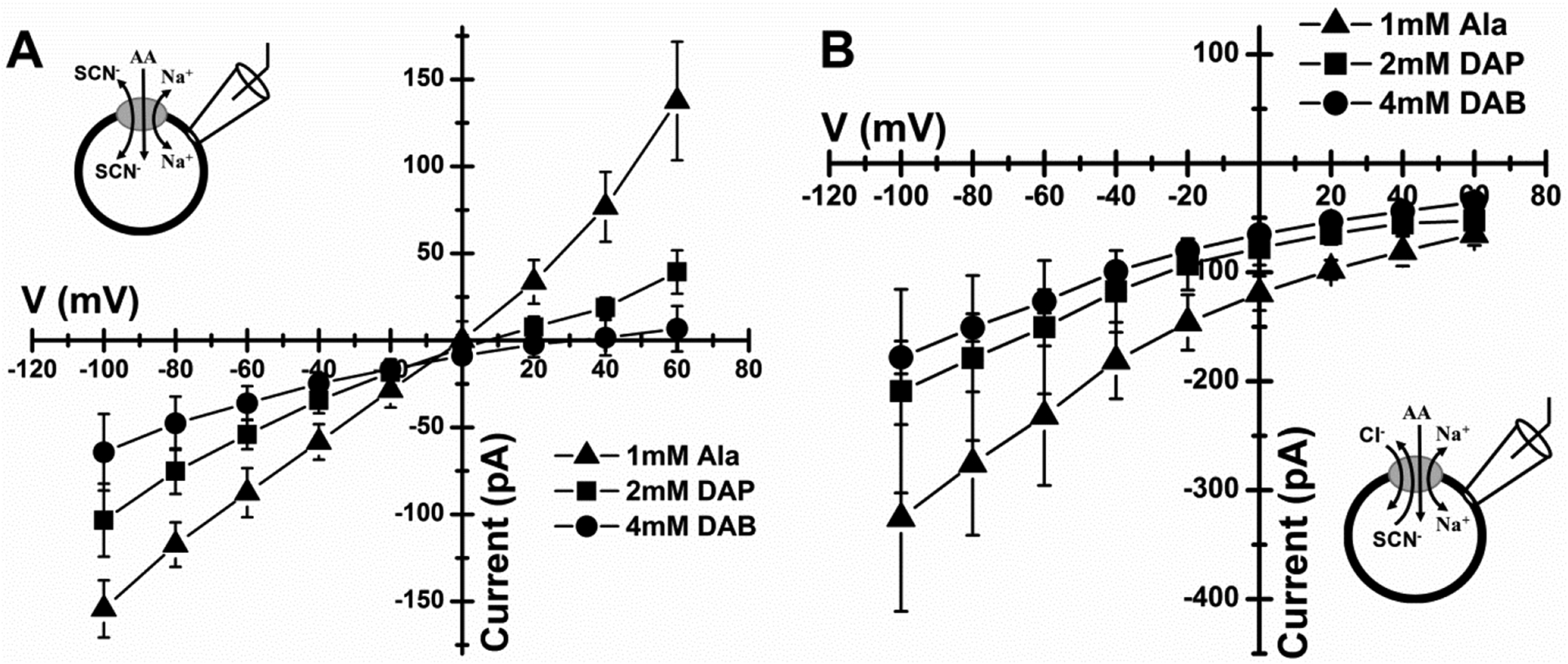
A, Voltage dependence of current generated by the application of 1 mM alanine (triangles), 2 mM L-DAP (squares) and 4 mM L-DAB (circles) to ASCT2-expressing cells in the presence of extracellular 140 mM NaSCN and intracellular 130 mM NaSCN. B, Similar experiment but currents were evoked by application of 4 mM L-DAB (circles), 2 mM L-DAP (squares) and 1mM alanine (triangles). Extracellular solution consisted of 140 mM NaCl while intracellular solution was 130 mM NaSCN.
It should be noted that the anion current carried by SCN− is not a physiological phenomenon. While the anion current may be present in chloride-containing solutions (the major physiological anion), its magnitude is too small to test substrate/inhibitor activity. In fact, the anion selectivity sequence closely matches that found in the related glutamate transporters of the SLC1 family, with P(SCN−) >> P(NO3−) > P(I−) > P(Cl−), as shown by Bröer et. al, ( 2000) [2]. Unfortunately, due to the electroneutral nature of ASCT2 amino acid exchange, the anion conductance is the only possibility to study ASCT2 using electrophysiology.
The apparent affinity of ASCT2 for L-DAP, but not alanine is voltage dependent.
To further investigate the role of the basic side chain of L-DAP, we analyzed apparent affinities of L-DAP at different cell membrane potentials, in comparison to the neutral substrate alanine. As expected, there was almost no change of apparent alanine affinity with voltage (figure 6, circles) because, alanine is a zwitterion and is uncharged at pH 7.4. Conversely, L-DAP showed a slight increase in apparent affinity from positive to negative membrane potentials, suggesting that L-DAP, with a net positive charge, binds more strongly at negative potential (figure 6, squares). The apparent valence calculated from the slope of the log (Km) vs. voltage relationship is 0.21 ± 0.04. Therefore, it is possible that L-DAP senses only a fraction of the membrane electric field while binding to the ASCT2 binding site, or that, at pH 7.4, only a fraction of L-DAP binds in the positively-charged form, or that both of these scenarios apply in combination.
Figure 6: L-DAP binding senses the transmembrane electric field.
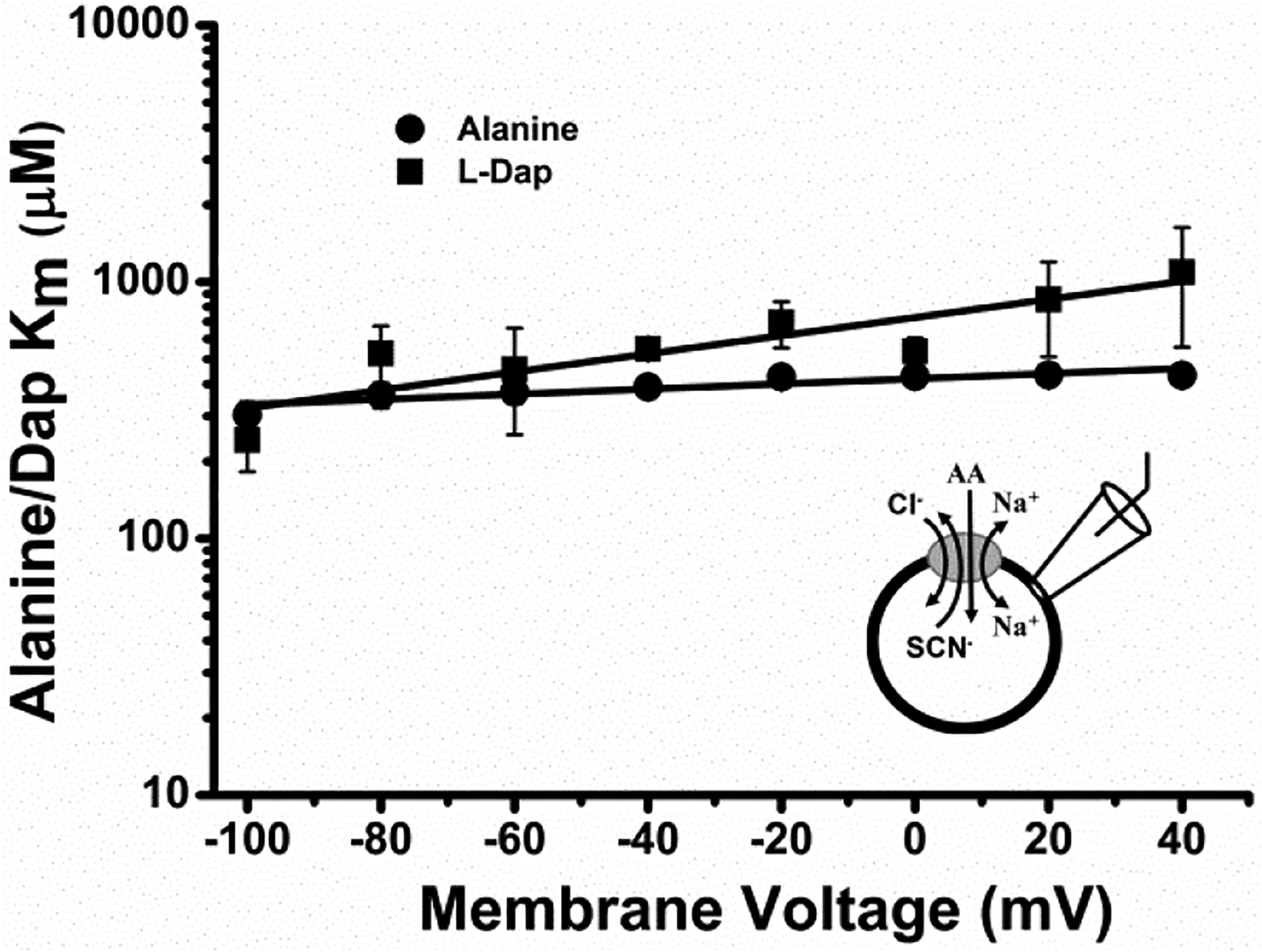
Voltage dependence of alanine and DAP binding affinities (Km) generated by the application of various concentrations of alanine (circles) and L-DAP (squares) to ASCT2-expressing cells in the presence of extracellular 140 mM NaCl and intracellular 130 mM NaSCN. The lines represent the result from a linear regression analysis, with a slope of 0.001 ± 0.0002 / mV for alanine and 0.004 ± 0.0008 / mV for DA. For alanine, Pearson’s r, R2 values were 0.908 and 0.796 respectively while DAP had Pearson’s r, R2 values of 0.886 and 0.748 respectively.
Effect of protonation state of L-DAP on ASCT2 interaction.
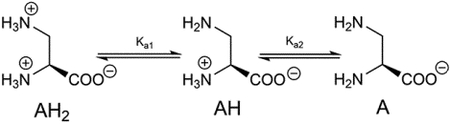
One possibility is that L-DAP binds to ASCT2 in the state with a deprotonated side chain, even though this is only a small fraction of molecules at pH 7.4. To test this possibility, we determined L-DAP currents and apparent affinities as a function of extracellular pH. In all experiments, we kept bath and intracellular solutions at pH 7.4 to maintain cell viability, only switching to pH values different from 7.4 for short periods of time (10 s). A concentration of 1 mM alanine was selected as a control in all experiments because it was shown in the literature to exhibit a Km that is almost independent of the pH [48]. L-DAP induced currents increased with pH, with the largest currents observed at pH 10.4, as shown in figure 7A. At this pH, L-DAP is primarily present in the deprotonated form (figure 1D). Surprisingly at pH below pH 6.50, currents evoked by application of 2mM L-DAP were outwardly directed (figure 7A). At this pH, L-DAP is primarily present in the state that is protonated at both amino groups, as shown in figure 1G. This behavior shows that doubly protonated L-DAP interacts with ASCT2, possibly inhibiting the leak anion current. This behavior is the hallmark of ASCT2 competitive inhibitors [31, 38–39, 44]. L-DAP apparent Km as a function of the pH showed a linear relationship (figure 7C), with the Km increasing with decreasing proton concentration. These results suggest that the protonated form of L-DAP, while behaving as an inhibitor (outward current), binds more strongly to ASCT2 than the deprotonated form, which, on the other hand, behaves analogously to a transported substrate (inward current). Motivated by these results, we propose a mechanism for interaction of ASCT2 with various major protonation states of DAP shown in figure 1D–G. This mechanism allowed us to predict the current/Km vs. pH relationships and is detailed further in the Discussion section. The predicted L-DAP maximum current response as a function of the pH agrees very well with experimental data, figure 7B. The calculated Km vs. pH relationship showed linear increase of Km with pH, in agreement with experimental results. These results demonstrate that L-DAP binds more strongly at lower pH, while possibly becoming an inhibitor in the fully protonated state.
Figure7: L-DAP can interact with ASCT2 in several protonation states.
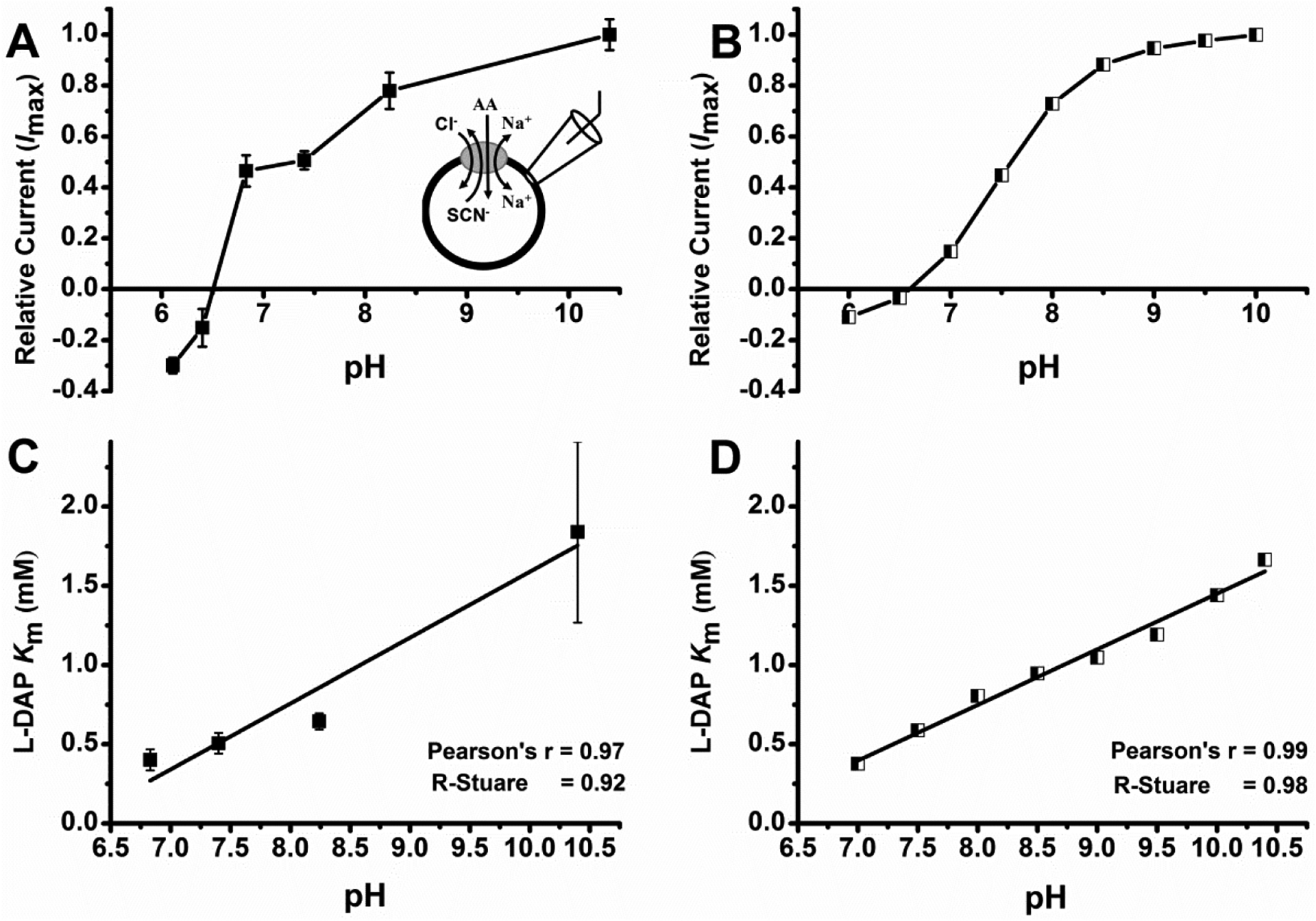
Determinationof pH dependence of L-Dap Km and relative currents. A, L-Dap relative current at different pH. L-DAP currents were normalized to the current response to 1 mM alanine at pH 7.4. Because alanine-induced currents are inward directed at pH 7.4, positive currents are inward, and negative currents are outward. B, calculated relative current vs. corresponding pH. pKa values for the transitions in eqn 1 were 6.8 and 9.8. Relative anion currents for states TA, TAH and TAH2 were 1.05, 0.45, and −0.28. The total anion current was calculated as I(total) = ITA[TA]+ ITAH[TAH]+ ITAH2[TAH2]. C, L-Dap binding affinity (Km) at different pH. D, Calculated L-DAP binding affinity at different pH. In all experiments, external bath solution was maintained at pH 7.40 and contained 140 mM NaCl and internal solution contained 140 mM NaSCN plus 10 mM alanine at pH 7.40. Running buffer, control and other solutions were at specified pH. All experiments were done at V = 0 mV.
The neurotoxin β-n-Methylamino-l-Alanine (BMAA) activates ASCT2 anion current.
We next tested the L-DAP analogue BMAA with ASCT2-expressing HEK293 cells. Like L-DAP, BMAA application elicited inward currents, but of significantly smaller magnitude than L-DAP and alanine (less than 20% of the maximum anion current induced by L-alanine, figures 8A and B). The currents were dose dependent, and almost saturating at highest BMAA concentration used, 2 mM with a Km value of 1400 ± 320 μM (figure 8C). We also tested uptake of 14C BMAA, however, while the uptake at 2 mM concentration (193 ± 24 pmol/min/cm2), was slightly higher in ASCT2-expressing cells compared to control cells (163 ± 36 pmol/min/cm2), this increase was not significant. Most likely, BMAA uptake, if it occurs, is too slow to be detected within the sensitivity of the uptake assay. These results suggest that BMAA shows weak substrate-like properties with respect to ASCT2 anion conductance activation, but may not be a transported substrate, or is transported by ASCT2 at a rate at least 10-fold lower than L-glutamine.
Figure 8: L-BMAA shows substrate-like properties.
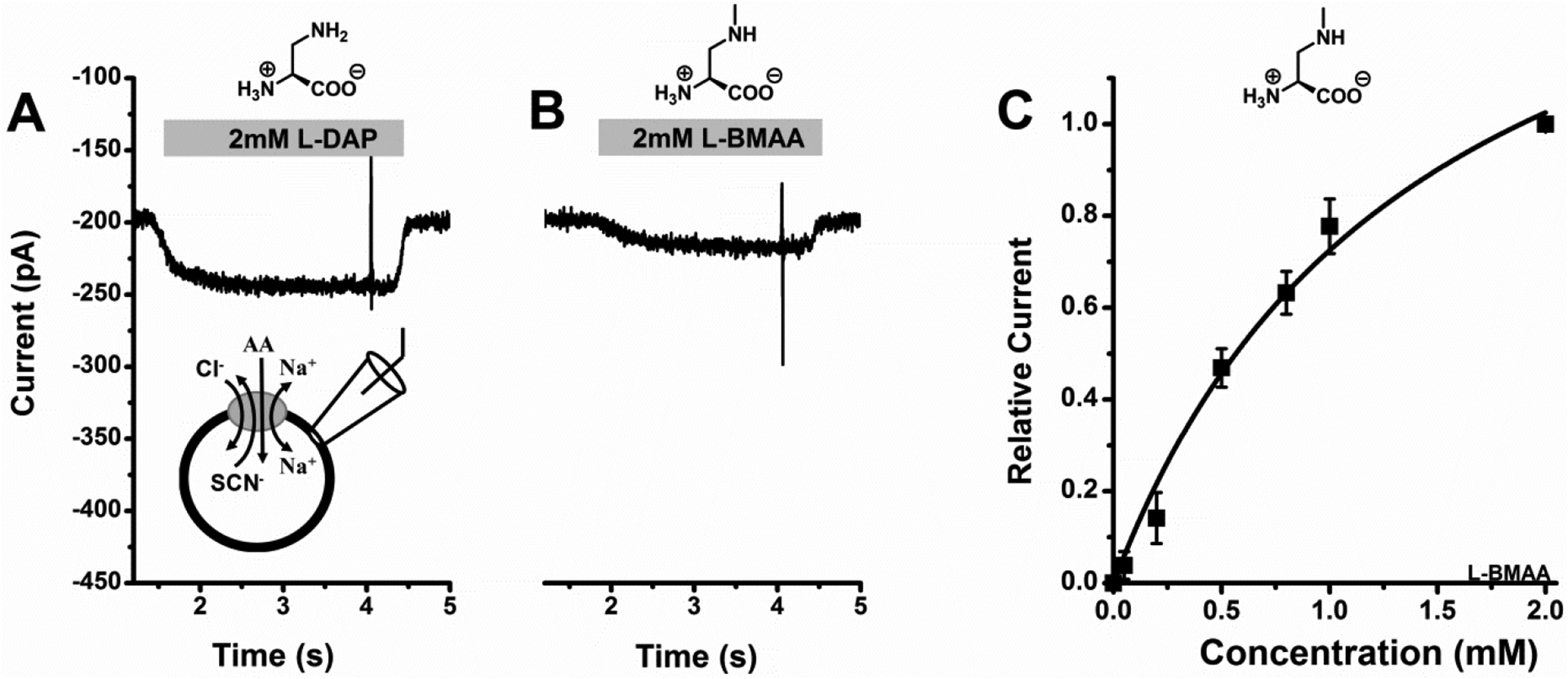
A, 2mM L-DAP-induced original current trace, B, 2 mM L-BMAA-induced original current trace and C, L-BMAA dose response curve with a Km of 1400 ± 330 μM. In all experiments, external solution contained 140 mM NaCl and internal solution contained 140mM NaSCN plus 10 mM alanine at pH 7.40, V = 0 mV.
Discussion
The glutamine transporter ASCT2 has become increasingly important as a potential cancer drug target. Several studies have shown that targeting by siRNA [13, 21, 49–51] and small molecule inhibitors [24, 52], limits cell growth in rapidly-growing tumor tissue. However, there are still substantial gaps with respect to our understanding of substrate and inhibitor specificity and mechanisms of interaction with ASCT2. It has been generally accepted that ASCT2 is a neutral amino acid transporter that cannot transport positively charged amino acids [1, 3]. The major findings from our present work are: (1) amino acids with basic side chains can bind to the ASCT2 substrate binding site, showing properties of transported substrates at physiological pH; (2) the potency of interaction decreases with the length of the basic side chain (ornithine and lysine are excluded from the binding site); (3) the affinity of interaction and ability to induce a current response depends on pH and, thus, protonation state of the basic amino acid, in contrast to interaction of the neutral substrate alanine; and (4) the neurotoxin BMAA, which has been implicated with the development of several neurodegenerative diseases [36], binds to ASCT2 and shows properties of a transported substrate [1–2, 31, 39]. Together, these findings demonstrate that the scope of substrate specificity of ASCTs must be expanded to include amino acids with charged, basic side chains. Interestingly, other amino acid transporters, which were thought to be selective for neutral amino acids, such as LAT1, have been found to transport amino acid substrates with ionizable side chains [53]. Thus, detailed studies of SLC substrate and inhibitor selectivity may uncover previously unknown functional similarities/differences between SLC families and render transporters more promiscuous than previously believed.
Consistent with results previously obtained for ASCT2 substrates, the current evoked by application of basic amino acids is carried by anions. While transported substrates activate the ASCT2 anion conductance, non-transportable inhibitors block the anion conductance [2, 31, 47]. In agreement with this, no measurable currents were generated in the absence of permeating anions, suggesting that extracellular amino acid exchange with intracellular alanine is either electroneutral, or too slow to generate current above the detection limit of electrophysiology (methane sulfonate was used instead of Cl− and SCN− (data not shown)). However, the apparent Km for L-DAP was found to be weakly dependent on the membrane potential, in contrast to the Km for the neutral amino acid substrate alanine [48], which was independent of the voltage. We speculate that the amino acid binding site is located partially within the transmembrane electric field, leading to attraction of a positively-charged substrate to the binding site by an inside-negative membrane potential. Similar conclusions were reached for the binding of negatively-charged amino acids and inhibitors to the acidic amino acid transporters (EAATs) sharing the same solute carrier family with the ASCTs [54]. Thus, it is not unexpected that the binding mechanisms of substrates to the extracellular facing binding site are conserved within the family members.
Additional information about the mechanism of basic amino acid interaction was obtained by characterizing the pH dependence of Km and maximum substrate-induced current, Imax. In previous studies, it was shown that these parameters are largely pH-independent for neutral amino acid substrates, such as alanine, in a pH range in which the neutral zwitterion is the dominant species [48], suggesting that pH-dependent protonation changes of the transporter itself are not responsible for the pH effects on L-DAP Km and Imax. In contrast to neutral substrates like alanine, the Km and Imax for L-DAP were strongly dependent on the pH, indicating that L-DAP protonation state, rather than ASCT2 protonation state of critical residues, causes the functional changes in the pH range from 6.1 to 10.5 Further, varying protonation states of L-DAP interact with ASCT2 with somewhat varying affinities and abilities to induce anion current. L-DAP is an unusual amino acid, because the pKa value of the α-amino group is much lower, 6.7, [46] than for other amino acids (compare to 9.1 for lysine, for example), meaning, this group is only protonated to 20% at pH 7.4, in contrast to 98% protonation of the α-amino group in lysine. Therefore, L-DAP is expected to exist as a mixture of predominately neutral protonation state (positive charge on the side-chain nitrogen, neutral in the α-position, figure 1F, AH in eqn. 1), but partly in the fully protonated, positively-charged form (figure 1G, AH2 in eqn. 1). The neutral protonation state with a positive charge in the α-amino position, but no charge on the β-nitrogen (figure 1E, pKa of 9.6), however, is expected to be present at small concentrations at all pH values tested.
We were able to describe the relationships between Km/Imax and pH with a simplified pre-equilibrium model (eqns. 1–4, appendix), taking into account three major protonation states of L-DAP, A, AH and AH2 (A = L-DAP, eqn.1, appendix).We assume that each protonation state of L-DAP interacts with the ASCT2 substrate binding site with a defined dissociation constant, K1–3 (eqns. 2–4, K1 = 2 mM, K2 =K3 = 200 μM), resulting in L-DAP bound states, TA, TAH, and TAH2 (eqns. 2–4, appendix). It was further assumed that each of these substrate-bound states states catalyzes anion conductance with varying efficiencies (relative conductances listed in the figure legend of figure 7), while the substrate-free state, T, cannot conduct anions. These assumptions are justified by previous reports on the mechanism of the anion conductance of ASCT2 [47] as well the related glutamate transporters [55], showing that transport, i.e. movement of substrate across the membrane, is not necessary for anion current to be activated. This pre-equilibrium approach enabled us to model the populations of the transporter bound to A, AH and AH2 as a function of the pH, reproducing the experimental findings for the pH dependence of the Km for L-DAP (figure 7B), as well as that of the anion current (figure 7D). Irrespective of the exact distribution of these three protonation states, the model allows the following conclusions: (1) The fully deprotonated, negatively-charged state, A, interacts with ASCT2 with low affinity, but with substrate-like behavior (state TA associated with anion current). (2) The fully protonated, positively charged state, AH2, interacts with ASCT2 with higher affinity (200 μM), but displays hallmark properties of an inhibitor, i.e. blockage of the leak anion current [31, 39, 44]. (3) The intermediate, neutral protonation state, AH, binds also with relatively high affinity (200 μM) and activates anion conductance, but less efficiently than the A state. Thus, AH has substrate-like behavior.
We recently published ASCT2 homology models based on the EAAT1 structure in both outward-occluded and outward-open conformations [42, 44]. In the outward-open state, HP2 shows a more pronounced opening, generating a larger binding site suitable for the binding of inhibitors. It is assumed that inhibitor binding locks the transporter in the outward-open conformation, which is unable to undergo conformational changes needed for substrate transport [31]. While crystal structures of ASCT2 in substrate-bound and substrate-free conformations are known, they are in the inward-facing conformation and, thus, not suitable for docking potential binders that act from the extracellular side [26, 56]. It should be noted that while this work was in progress, an outward-occluded crystal structure with bound glutamine was published [27]. This structure confirms the molecular interactions with substrates in the binding site predicted from our EAAT1 homology model [42, 44], but is not available for docking analysis. The docking results with L-DAP in four protonation states in both outward-occluded and outward-open states show similar coordination of the α-carboxy and amino groups and confirm the general findings from the experiment data, namely that L-DAP can interact with ASCT2, and the three relevant protonation states interact with the binding site with docking scores in the same range.
It is noteworthy that in the outward-open state [56] , a L-DAP configuration is predicted, in which the positions of the α and β-amino groups are switched, resembling a β-amino acid binding to the ASCT2 binding site. Although, we have found no evidence in the literature for the effect of β-amino acids on ASCT2, we speculate that it is this β-configuration that gives rise to the inhibitory effect of the fully-protonated species at low pH. Second, our docking, as well as experimental results indicate that the α-amino group does not necessarily need to be protonated for interaction with ASCT2, especially if there is polar interaction in the side chain. This finding may be important, possibly allowing us in the future to extend the pharmacology of ASCT2 inhibitors by altering the α-NH2 function.
In addition to the structure-function implications of ASCT2 substrate specificity, our results have implications on the interaction of the non-proteinogenic amino acid BMAA with ASCT2 in the central nervous system. BMAA is a derivative of L-DAP, differing by only a methyl group on the β-amino group. BMAA, and to a lesser extent L-DAB, have been characterized as potent neurotoxins, potentially acting on neurotransmitter receptors in the mammalian brain. Both compounds are found in samples of cyanobacteria [57], which during algae blooms can contaminate food sources and move through the food chain into food products. Transport of BMAA and L-DAB by ASCT2 would be a compelling mechanism for the appearance of these compounds in the brain, as well as their transport into cells and eventual incorporation into proteins. Although the involvement of ASCT2 in blood brain barrier (BBB) transport of amino acids is less than clear, one report provides evidence for its expression in capillary endothelial cells, a model system of the BBB [58]. Therefore, it is possible that BMAA gets access to the cerebrospinal fluid through ASCT2, in exchange for other amino acids, although other transport pathways at the BBB cannot be excluded. More direct evidence has been obtained from mammary gland cells, which allow movement of neurotoxic BMAA from the mother’s blood stream to the milk during lactation, and, thus transfer to the off-spring [59] it was shown that benzylserine [39] inhibits BMAA transport in a mammary cell line, pointing to involvement of ASCT2. While our results are consistent with these findings, uptake of 14C BMAA by ASCT2 was not significant over control, non-ASCT2 expressing cells. Therefore, it is likely that uptake, if it occurs, is at a rate at least 10-fold lower than that of transported substrates, such as glutamine.
In conclusion, the work presented here provides direct evidence that amino acids with basic side chains can interact with the neutral amino acid transporter ASCT2, expanding the scope of molecular interactions in the substrate binding site. Like the transported substrate alanine, these basic amino acids elicit anion current, but in contrast to alanine bind in a voltage dependent manner. In addition to model compounds, the neurotoxin BMAA was found to be an activator of ASCT2 anion conductance, although uptake could not be ascertained within the sensitivity of the uptake assay. Further work will be required to study BMAA distribution in organisms and how it is affected by blocking ASCT2.
Supplementary Material
Acknowledgements
The authors are grateful to Dr. Stefan Bröer for providing rat ASCT2 cDNA, and to Dr. Sakiko Okumoto for providing the fluorescent glutamine sensor FLIPQ-TV3.0_100 μM.
Funding
This study was supported by a grant from the National Institutes of Health (http://www.nih.gov) (R01 GM108911) to A.S. and C.G. and T32 CA078207 to R.A.G., and by the National Science Foundation Grant 1515028 awarded to C.G.
Abbreviations
- ALS
Amyotrophic Lateral Sclerosis
- ASCT1
Alanine serine cysteine transporter 1
- ASCT2
Alanine serine cysteine transporter 2
- BBB
Blood brain barrier
- BMAA
β-N-methylamino-L-Alanine
- BPP
(biphenylmethyl)-L-proline
- DAB
2,4-diaminobutyric acid
- DAP
1,3-diaminopropionic acid
- EAAT1
Excitatory amino acid transporter 1
- EGTA
ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEK
Human embryonic kidney
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- HP1
Hairpin 1
- HP2
Hairpin 2
- LAT1
Large amino acid transporter 1
- SCN
Thiocyanate
- SLC1
Solute Carrier 1
- TFB-TBOA
Trifluoromethyl)benzoyl] amino]phenyl]methoxy]-L-aspartic acid
- YFP
Yellow fluorescent protein
Appendix
Equations
 |
eqn 1 |
| eqn 2 |
| eqn 3 |
| eqn 4 |
| eqn 5 |
| eqn 6 |
| eqn 7 |
| eqn 8 |
| eqn 9 |
| eqn10 |
| eqn 11 |
Footnotes
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Arriza JL, Kavanaugh MP, Fairman WA, Wu YN, Murdoch GH, North RA and Amara SG (1993) Cloning and expression of a human neutral amino acid transporter with structural similarity to the glutamate transporter gene family. J Biol Chem. 268, 15329–15332 [PubMed] [Google Scholar]
- 2.Broer A, Wagner C, Lang F and Broer S (2000) Neutral amino acid transporter ASCT2 displays substrate-induced Na+ exchange and a substrate-gated anion conductance. Biochem J. 346 Pt 3, 705–710 [PMC free article] [PubMed] [Google Scholar]
- 3.Utsunomiya-Tate N, Endou H and Kanai Y (1996) Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J Biol Chem. 271, 14883–14890 [DOI] [PubMed] [Google Scholar]
- 4.Kanai Y and Hediger MA (2004) The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflugers Arch. 447, 469–479 [DOI] [PubMed] [Google Scholar]
- 5.Zerangue N and Kavanaugh MP (1996) ASCT-1 is a neutral amino acid exchanger with chloride channel activity. J Biol Chem. 271, 27991–27994 [DOI] [PubMed] [Google Scholar]
- 6.Kanai Y (1996) [Na+ -dependent amino acid transporters: their structure and function]. Nihon Rinsho. 54, 638–645 [PubMed] [Google Scholar]
- 7.Broer A, Brookes N, Ganapathy V, Dimmer KS, Wagner CA, Lang F and Broer S (1999) The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J Neurochem. 73, 2184–2194 [PubMed] [Google Scholar]
- 8.Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP and Amara SG (1994) Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 14, 5559–5569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shafqat S, Tamarappoo BK, Kilberg MS, Puranam RS, McNamara JO, Guadano-Ferraz A and Fremeau RT Jr. (1993) Cloning and expression of a novel Na(+)-dependent neutral amino acid transporter structurally related to mammalian Na+/glutamate cotransporters. J Biol Chem. 268, 15351–15355 [PubMed] [Google Scholar]
- 10.Pingitore P, Pochini L, Scalise M, Galluccio M, Hedfalk K and Indiveri C (2013) Large scale production of the active human ASCT2 (SLC1A5) transporter in Pichia pastoris--functional and kinetic asymmetry revealed in proteoliposomes. Biochim Biophys Acta. 1828, 2238–2246 [DOI] [PubMed] [Google Scholar]
- 11.Broer S and Broer A (2017) Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem J. 474, 1935–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuchs BC and Bode BP (2005) Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol. 15, 254–266 [DOI] [PubMed] [Google Scholar]
- 13.Fuchs BC, Finger RE, Onan MC and Bode BP (2007) ASCT2 silencing regulates mammalian target-of-rapamycin growth and survival signaling in human hepatoma cells. Am J Physiol Cell Physiol. 293, C55–63 [DOI] [PubMed] [Google Scholar]
- 14.Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, Blonska M, Lin X and Sun SC (2014) Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 40, 692–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pochini L, Scalise M, Galluccio M and Indiveri C (2014) Membrane transporters for the special amino acid glutamine: structure/function relationships and relevance to human health. Front Chem. 2, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Namikawa M, Kakizaki S, Kaira K, Tojima H, Yamazaki Y, Horiguchi N, Sato K, Oriuchi N, Tominaga H, Sunose Y, Nagamori S, Kanai Y, Oyama T, Takeyoshi I and Yamada M (2015) Expression of amino acid transporters (LAT1, ASCT2 and xCT) as clinical significance in hepatocellular carcinoma. Hepatol Res. 45, 1014–1022 [DOI] [PubMed] [Google Scholar]
- 17.Wang Q, Beaumont KA, Otte NJ, Font J, Bailey CG, van Geldermalsen M, Sharp DM, Tiffen JC, Ryan RM, Jormakka M, Haass NK, Rasko JE and Holst J (2014) Targeting glutamine transport to suppress melanoma cell growth. Int J Cancer. 135, 1060–1071 [DOI] [PubMed] [Google Scholar]
- 18.Ren P, Yue M, Xiao D, Xiu R, Gan L, Liu H and Qing G (2015) ATF4 and NMyc coordinate glutamine metabolism in MYCN-amplified neuroblastoma cells through ASCT2 activation. J Pathol. 235, 90–100 [DOI] [PubMed] [Google Scholar]
- 19.Hassanein M, Hight MR, Buck JR, Tantawy MN, Nickels ML, Hoeksema MD, Harris BK, Boyd K, Massion PP and Manning HC (2016) Preclinical Evaluation of 4-[18F]Fluoroglutamine PET to Assess ASCT2 Expression in Lung Cancer. Mol Imaging Biol. 18, 18–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim S, Jung WH and Koo JS (2013) The expression of glutamine-metabolism-related proteins in breast phyllodes tumors. Tumour Biol. 34, 2683–2689 [DOI] [PubMed] [Google Scholar]
- 21.van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, Ritchie W, Feng Y, Bailey CG, Deng N, Harvey K, Beith JM, Selinger CI, O’Toole SA, Rasko JE and Holst J (2016) ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. 35, 3201–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Q, Tiffen J, Bailey CG, Lehman ML, Ritchie W, Fazli L, Metierre C, Feng YJ, Li E, Gleave M, Buchanan G, Nelson CC, Rasko JE and Holst J (2013) Targeting amino acid transport in metastatic castration-resistant prostate cancer: effects on cell cycle, cell growth, and tumor development. J Natl Cancer Inst. 105, 1463–1473 [DOI] [PubMed] [Google Scholar]
- 23.Shimizu K, Kaira K, Tomizawa Y, Sunaga N, Kawashima O, Oriuchi N, Tominaga H, Nagamori S, Kanai Y, Yamada M, Oyama T and Takeyoshi I (2014) ASC amino-acid transporter 2 (ASCT2) as a novel prognostic marker in non-small cell lung cancer. Br J Cancer. 110, 2030–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, Kondo J, Coffey RJ, Johnson MO, Rathmell JC, Sharick JT, Skala MC, Smith JA, Berlin J, Washington MK, Nickels ML and Manning HC (2018) Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med. 24, 194–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Canul-Tec JC, Assal R, Cirri E, Legrand P, Brier S, Chamot-Rooke J and Reyes N (2017) Structure and allosteric inhibition of excitatory amino acid transporter 1. Nature. 544, 446–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garaeva AA, Oostergetel GT, Gati C, Guskov A, Paulino C and Slotboom DJ (2018) Cryo-EM structure of the human neutral amino acid transporter ASCT2. Nat Struct Mol Biol. 25, 515–521 [DOI] [PubMed] [Google Scholar]
- 27.Yu X, Plotnikova O, Bonin PD, Subashi TA, McLellan TJ, Dumlao D, Che Y, Dong YY, Carpenter EP, West GM, Qiu X, Culp JS and Han S (2019) Cryo-EM structures of the human glutamine transporter SLC1A5 (ASCT2) in the outward-facing conformation. Elife. 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scopelliti AJ, Font J, Vandenberg RJ, Boudker O and Ryan RM (2018) Structural characterisation reveals insights into substrate recognition by the glutamine transporter ASCT2/SLC1A5. Nat Commun. 9, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scopelliti AJ, Ryan RM and Vandenberg RJ (2013) Molecular determinants for functional differences between alanine-serine-cysteine transporter 1 and other glutamate transporter family members. J Biol Chem. 288, 8250–8257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yernool D, Boudker O, Jin Y and Gouaux E (2004) Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 431, 811–818 [DOI] [PubMed] [Google Scholar]
- 31.Albers T, Marsiglia W, Thomas T, Gameiro A and Grewer C (2012) Defining substrate and blocker activity of alanine-serine-cysteine transporter 2 (ASCT2) Ligands with Novel Serine Analogs. Mol Pharmacol. 81, 356–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colas C, Grewer C, Otte NJ, Gameiro A, Albers T, Singh K, Shere H, Bonomi M, Holst J and Schlessinger A (2015) Ligand Discovery for the Alanine-Serine-Cysteine Transporter (ASCT2, SLC1A5) from Homology Modeling and Virtual Screening. PLoS Comput Biol. 11, e1004477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oppedisano F, Catto M, Koutentis PA, Nicolotti O, Pochini L, Koyioni M, Introcaso A, Michaelidou SS, Carotti A and Indiveri C (2012) Inactivation of the glutamine/amino acid transporter ASCT2 by 1,2,3-dithiazoles: proteoliposomes as a tool to gain insights in the molecular mechanism of action and of antitumor activity. Toxicol Appl Pharmacol. 265, 93–102 [DOI] [PubMed] [Google Scholar]
- 34.Schulte ML, Khodadadi AB, Cuthbertson ML, Smith JA and Manning HC (2016) 2-Amino-4-bis(aryloxybenzyl)aminobutanoic acids: A novel scaffold for inhibition of ASCT2-mediated glutamine transport. Bioorg Med Chem Lett. 26, 1044–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cox PA, Banack SA, Murch SJ, Rasmussen U, Tien G, Bidigare RR, Metcalf JS, Morrison LF, Codd GA and Bergman B (2005) Diverse taxa of cyanobacteria produce beta-N-methylamino-L-alanine, a neurotoxic amino acid. Proc Natl Acad Sci U S A. 102, 5074–5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Munck E, Munoz-Saez E, Miguel BG, Solas MT, Ojeda I, Martinez A, Gil C and Arahuetes RM (2013) beta-N-methylamino-l-alanine causes neurological and pathological phenotypes mimicking Amyotrophic Lateral Sclerosis (ALS): the first step towards an experimental model for sporadic ALS. Environ Toxicol Pharmacol. 36, 243–255 [DOI] [PubMed] [Google Scholar]
- 37.Andersson M, Ersson L, Brandt I and Bergstrom U (2017) Potential transfer of neurotoxic amino acid beta-N-methylamino-alanine (BMAA) from mother to infant during breast-feeding: Predictions from human cell lines. Toxicol Appl Pharmacol. 320, 40–50 [DOI] [PubMed] [Google Scholar]
- 38.Singh K, Tanui R, Gameiro A, Eisenberg G, Colas C, Schlessinger A and Grewer C (2017) Structure activity relationships of benzylproline-derived inhibitors of the glutamine transporter ASCT2. Bioorg Med Chem Lett. 27, 398–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grewer C and Grabsch E (2004) New inhibitors for the neutral amino acid transporter ASCT2 reveal its Na+-dependent anion leak. J Physiol. 557, 747–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grewer C, Watzke N, Wiessner M and Rauen T (2000) Glutamate translocation of the neuronal glutamate transporter EAAC1 occurs within milliseconds. Proc Natl Acad Sci U S A. 97, 9706–9711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gruenwald K, Holland JT, Stromberg V, Ahmad A, Watcharakichkorn D and Okumoto S (2012) Visualization of glutamine transporter activities in living cells using genetically encoded glutamine sensors. PLoS One. 7, e38591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garibsingh RA, Otte NJ, Ndaru E, Colas C, Grewer C, Holst J and Schlessinger A (2018) Homology Modeling Informs Ligand Discovery for the Glutamine Transporter ASCT2. Front Chem. 6, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schrodinger L (2018) Schrodinger Release 2018–4: Glide. In, New York, NY. [Google Scholar]
- 44.Ndaru E, Garibsingh RA, Shi Y, Wallace E, Zakrepine P, Wang J, Schlessinger A and Grewer C (2019) Novel alanine serine cysteine transporter 2 (ASCT2) inhibitors based on sulfonamide and sulfonic acid ester scaffolds. J Gen Physiol. 151, 357–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schrodinger L (2018) The PyMOL Molecular Graphics System, Version 2.0.
- 46.Hay RW and Morris PJ (1972) Proton lonisation Constants and Kinetics of Base Hydrolysis of Some a-Amino-acid Esters in Aqueous Solution. Part 111.l Hydrolysis and Intramolecular Aminolysis of àw-Diamino-acid Methyl Esters J. Chem. Soc., Perkin Trans 2. 8, 9 [Google Scholar]
- 47.Zander CB, Albers T and Grewer C (2013) Voltage-dependent processes in the electroneutral amino acid exchanger ASCT2. J Gen Physiol. 141, 659–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grewer C, Watzke N, Rauen T and Bicho A (2003) Is the glutamate residue Glu-373 the proton acceptor of the excitatory amino acid carrier 1? J Biol Chem. 278, 2585–2592 [DOI] [PubMed] [Google Scholar]
- 49.Fuchs BC, Perez JC, Suetterlin JE, Chaudhry SB and Bode BP (2004) Inducible antisense RNA targeting amino acid transporter ATB0/ASCT2 elicits apoptosis in human hepatoma cells. Am J Physiol Gastrointest Liver Physiol. 286, G467–478 [DOI] [PubMed] [Google Scholar]
- 50.Hassanein M, Qian J, Hoeksema MD, Wang J, Jacobovitz M, Ji X, Harris FT, Harris BK, Boyd KL, Chen H, Eisenberg R and Massion PP (2015) Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int J Cancer. 137, 1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Q, Hardie RA, Hoy AJ, van Geldermalsen M, Gao D, Fazli L, Sadowski MC, Balaban S, Schreuder M, Nagarajah R, Wong JJ, Metierre C, Pinello N, Otte NJ, Lehman ML, Gleave M, Nelson CC, Bailey CG, Ritchie W, Rasko JE and Holst J (2015) Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J Pathol. 236, 278–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li YX, Yang JY, Alcantara M, Abelian G, Kulkarni A, Staubli U and Foster AC (2018) Inhibitors of the Neutral Amino Acid Transporters ASCT1 and ASCT2 Are Effective in In Vivo Models of Schizophrenia and Visual Dysfunction. J Pharmacol Exp Ther. 367, 292–301 [DOI] [PubMed] [Google Scholar]
- 53.Chien H-C, Colas C, Finke K, Springer S, Stoner L, Zur AA, Venteicher B, Campbell J, Hall C, Flint A, Augustyn E, Hernandez C, Heeren N, Hansen L, Anthony A, Bauer J, Fotiadis D, Schlessinger A, Giacomini KM and Thomas AA (2018) Reevaluating the Substrate Specificity of the L-Type Amino Acid Transporter (LAT1). J Med Chem. 61, 7358–7373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zielewicz L, Wang J, Ndaru E and Grewer CT (2019) Transient Kinetics Reveal Mechanism and Voltage Dependence of Inhibitor and Substrate Binding to Glutamate Transporters. ACS Chem Biol. 14, 1002–1010 [DOI] [PubMed] [Google Scholar]
- 55.Wadiche JI and Kavanaugh MP (1998) Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J Neurosci. 18, 7650–7661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garaeva AA, Guskov A, Slotboom DJ and Paulino C (2019) A one-gate elevator mechanism for the human neutral amino acid transporter ASCT2. Nat Commun. 10, 3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Manolidi K, Triantis TM, Kaloudis T and Hiskia A (2019) Neurotoxin BMAA and its isomeric amino acids in cyanobacteria and cyanobacteria-based food supplements. J Hazard Mater. 365, 346–365 [DOI] [PubMed] [Google Scholar]
- 58.Tetsuka K, Takanaga H, Ohtsuki S, Hosoya K and Terasaki T (2003) The l-isomer-selective transport of aspartic acid is mediated by ASCT2 at the blood-brain barrier. J Neurochem. 87, 891–901 [DOI] [PubMed] [Google Scholar]
- 59.Andersson M (2017) Potential transfer of neurotoxic amino acid β- N -methylamino-alanine (BMAA) from mother to infant during breast-feeding: Predictions from human cell lines. Toxicology and Applied Pharmacology. 320, 11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.